
Direct C(sp3)–H difluoromethylation via radical–radical cross-coupling by visible-light photoredox catalysis†
Wei
Xiong‡
,
Wen-Bing
Qin‡
,
Ya-Shi
Zhao
,
Kai-Zhong
Fu
and
Guo-Kai
Liu
 *
*
School of Pharmaceutical Sciences, Shenzhen University Health Science Centre, Shenzhen University, 3688 Nanhai Ave., Shenzhen, 518060, Guangdong, China. E-mail: gkliu@szu.edu.cn
First published on 3rd March 2022
Abstract
Herein, a radical–radical cross-coupling strategy for direct difluoromethylation of the C(sp3)–H bond is reported. This transformation was readily accomplished under transition metal-free photoredox catalysis in the presence of 3 mol% of an organic photocatalyst, allowing the direct difluoromethylation of C(sp3)–H of a wide variety of 1,4-dihydroquinoxalin-2-ones in good yields under mild reaction conditions. Moreover, various 3-difluoromethyl quinoxalin-2-ones were also easily furnished in a one-pot manner through this radical difluoromethylation protocol. Mechanistic studies clearly reveal that the radical–radical cross-coupling between a difluoromethyl radical and a C(sp3) radical is responsible for this transformation. To the best of our knowledge, this is the first example of radical–radical cross-coupling difluoromethylation of the C(sp3)–H bond, which not only provides a promising strategy for a straightforward installation of a CF2H group at a C(sp3) center but would also promote the development of other new fluorination and fluoroalkylation methods.
Introduction
Nowadays, the incorporation of one or more fluorine atoms into bioactive compounds has become a routine strategy for the development of pharmaceuticals and agrochemicals.1 Amongst various fluorine-containing units, the CF2H group can endow the parent molecule with significantly improved physicochemical and biological properties due to its unique instincts.2 For example, the CF2H group can serve as a competent lipophilic hydrogen bond donor3 as well as act as metabolically stable bioisosteres of alcohol, thiol, or amine groups.4 Therefore, the development of strategies and synthetic versions for assembling the CF2H unit has attracted considerable attention. Amongst the existing strategies, the difluoromethylation reaction is the most straightforward and efficient approach to access CF2H-containing compounds. Consequently, for this purpose, continuous efforts on difluoromethylating reagents and thereby difluoromethylation protocols have been increasingly made in the past decade.5 Notably, the C(sp3)–CF2H moiety is of great interest in pharmaceutical chemistry because it is a bioisosteric replacement for aliphatic alcohols and thiols, two prevalent functional groups in drug design and discovery.6 As one of the most straightforward and general reactions for the rapid and efficient installation of CF2H to a C(sp3) center, the direct C(sp3)–H activation difluoromethylation represents a greatly reliable and ideal method for the construction of C(sp3)–CF2H bonds in the late-stage modification of bioactive molecules. However, to date, a direct C(sp3)–H difluoromethylation method has been rarely studied and a great challenge. The related research is mainly focused on the electrophilic difluoromethylation of highly activated aliphatic-carbon acids with difluorocarbene in the presence of a strong base due to the fact that a great variety of difluorocarbene precursors were developed by Hu, Shibata, Shen, and our group (Scheme 1a).7–11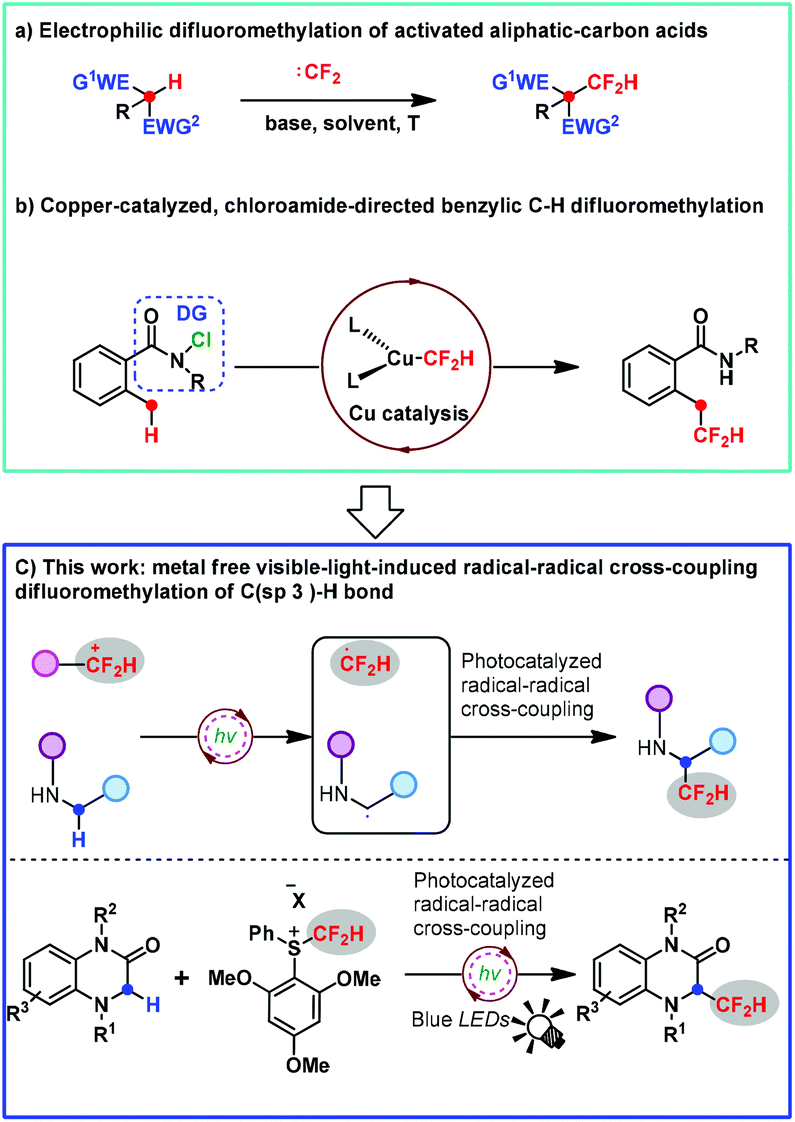 | ||
| Scheme 1 Strategies for difluoromethylation of C(sp3)–H bonds. | ||
However, these methods usually suffer from limitations such as requirement of highly activated substrates, requirement of excess reagents and strong bases, and undesirable regioselectivity, thus significantly narrowing their practicability and further application. In 2019, Liu and co-workers reported the first copper-catalyzed benzylic C–H difluoromethylation. However, this transformation was accomplished with the aid of a directing group chloroamide to generate a benzylic radical (Scheme 1b).12
In spite of the aforementioned preliminary progress, the direct C(sp3)–H difluoromethylation remains largely underdeveloped and highly desirable as one of the most straightforward and promising strategies. To address this topic and challenge, we decided to develop an elegant and highly efficient solution by using our bench-stable S-(difluoromethyl)diarylsulfonium salts 2,10 which have proved to be good difluoromethyl radical precursors under photoredox catalysis,13 as well as electrophilic difluoromethylating reagents and difluorocarbene precursors.10,14 On the other hand, the photoredox-catalyzed reaction is becoming a more and more popular synthetic version for incorporating the fluoroalkyl group into various organic molecules via a radical pathway.15,16 The direct C(sp3)–H radical difluoromethylation remains currently unknown except the copper-catalyzed benzylic C–H difluoromethylation reported by Liu.12 Consequently, we question whether 2 can achieve the direct C(sp3)–H difluoromethylation, serving as a difluoromethyl radical precursor via a photoredox-catalyzed radical–radical cross-coupling reaction. In particular, amines could be oxidized to generate N-radicals under photoredox catalysis, thus allowing the generation of α-C radical species via single electron transfer (SET) from C to N atoms (Scheme 1c, for the detailed information, see the ESI S2–S4†).17 Bearing this in mind, we reasoned that reagent 2 and 1,4-dihydroquinoxalin-2-ones would lead to a radical–radical cross-coupling reaction under photoredox catalysis. As a result, we report herein a transition metal-free visible-light-induced direct C(sp3)–H difluoromethylation approach via the radical–radical cross-coupling reaction in this article (Scheme 1c). To the best of our knowledge, this transformation is the first example involving a radical–radical cross-coupling difluoromethylation of the C(sp3)–H bond, allowing for the direct C3–H radical difluoromethylation of a wide variety of 1,4-dihydroquinoxalin-2-ones, which is of great interest to pharmaceuticals and drug candidates as a prevalent scaffold frequently existing in a lot of biologically active compounds.
Results and discussion
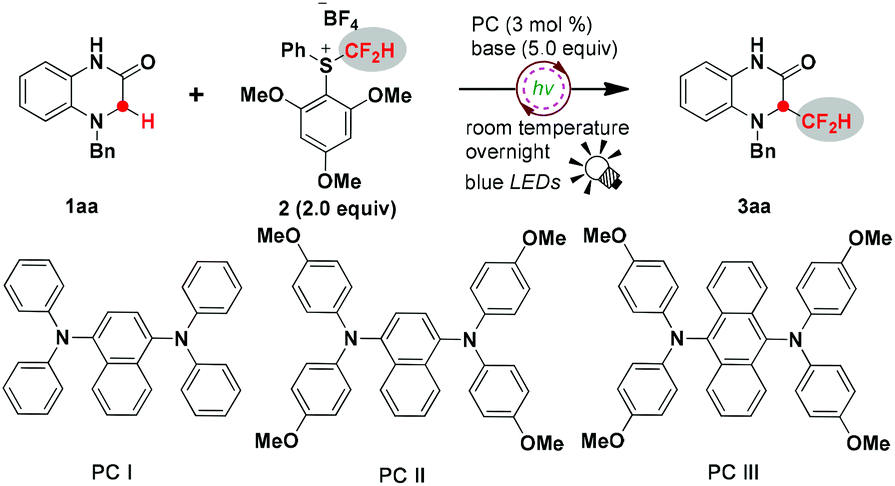
Initially, we started our investigation employing 4-benzyl-1,4-dihydroquinoxalin-2-one 1aa as a model substrate to optimize the reaction conditions (Table 1). Gratifyingly, the reaction proceeded smoothly under the irradiation of blue light overnight in the presence of 3 mol% photocatalyst PCI, 5.0 equivalents of LiOH and 2.0 equivalents of S-(difluoromethyl) sulfonium salt 2, affording 3-difluoromethylated 1,4-dihydroquinoxalin-2-one 3aa in 63% isolated yield (Table 1, entry 1). Inspired by this exciting result, other conveniently available common photosensitizers were further examined, and all the organic photosensitizers exhibited high catalytic activity to promote this reaction, leading to moderate to good yields; PCII achieved 74% isolated yield of the desired product (Table 1, entries 2–4). A metal photosensitizer Ir(dFppy)3 was also compatible with this reaction to deliver the product in 52% isolated yield (Table 1, entry 5), whereas Ru(bpy)3Cl2 did not work (Table 1, entry 6). Predictably, the reaction was shut down in the absence of light or the photocatalyst (Table 1, entries 7 and 8), hence suggesting that the process might proceed through a radical pathway. Next, screening of various bases was carried out. Compared to inorganic bases, organic bases were clearly less effective to drive this reaction (Table 1, entries 9 and 10). Interestingly, alkaline lithium seems to be superior to other inorganic bases. Li2CO3 and Li3PO4 also gave the products in good yields of 60% and 69%, respectively (Table 1, entries 15 and 16), whereas dramatically diminished yields were obtained when KOH, NaOH, CsOH and K2CO3 were used (Table 1, entries 11–14). The effect of solvents on the transformation efficiency was also surveyed. Thus, the use of CH3CN instead of AcOEt decreased the yield to 57% (Table 1, entry 17). THF and CH2Cl2 also led to inferior results (Table 1, entries 18 and 19). The reduction of the amount of LiOH or the difluoromethylating reagent was obviously harmful to the yields of the desired product (Table 1, entries 20 and 21). The conversion efficiency was also eroded when the reaction time was shortened to 3 hours (Table 1, entry 22).|
|
||||
|---|---|---|---|---|
| Entry | PC (3 mmol%) | Base (5.0 equiv.) | Solvent | Yieldb (%) |
| a Reaction conditions (unless otherwise specified): 1aa (0.1 mmol, 1.0 equiv.), photocatalyst (3 mol%), reagent 2 (0.2 mmol, 2.0 equiv.), base (0.5 mmol, 5.0 equiv.), solvent (2.0 mL), room temperature, overnight. b Isolated yields. c No blue light irradiation. d LiOH (0.3 mmol) was used. e Reagent 2 (0.15 mmol) was used. f The reaction time was shortened to 3 hours. | ||||
| 1 | PC I | LiOH | AcOEt | 63 |
| 2 | PC II | LiOH | AcOEt | 74 |
| 3 | PC III | LiOH | AcOEt | 73 |
| 4 | Perylene | LiOH | AcOEt | 55 |
| 5 | Ir(dFppy)3 | LiOH | AcOEt | 52 |
| 6 | Ru(bpy)3Cl2 | LiOH | AcOEt | 0 |
| 7 | — | LiOH | AcOEt | Trace |
| 8c | PC II | LiOH | AcOEt | 0 |
| 9 | PC II | DBU | AcOEt | 10 |
| 10 | PC II | Et3N | AcOEt | 0 |
| 11 | PC II | NaOH | AcOEt | 5 |
| 12 | PC II | CsOH·H2O | AcOEt | 8 |
| 13 | PC II | KOH | AcOEt | 43 |
| 14 | PC II | K2CO3 | AcOEt | 18 |
| 15 | PC II | Li2CO3 | AcOEt | 60 |
| 16 | PC II | Li3PO4 | AcOEt | 69 |
| 17 | PC II | LiOH | CH3CN | 57 |
| 18 | PC II | LiOH | THF | 8 |
| 19 | PC II | LiOH | CH2Cl2 | 17 |
| 20d | PC II | LiOH | AcOEt | 55 |
| 21e | PC II | LiOH | AcOEt | 56 |
| 22f | PC II | LiOH | AcOEt | 51 |
To showcase the generality of this radical–radical cross-coupling difluoromethylation protocol, we further explored the substrate scope under the optimized reaction conditions (Table 1, entry 3). As illustrated in Table 2, a wide range of N4-benzyl-1,4-dihydroquinoxalin-2-ones were readily transformed to the desired products in moderate to good yields. The structure of 3aa was confirmed by X-ray crystallographic analysis. Notably, many functional groups were tolerated, regardless of their electron-donating and electron-withdrawing properties, including halogens (F, Cl, and Br), CF3O, CH3, CH3O, C(CH3)3, etc. Most substitution sites, i.e., C6, C7 and C8, were compatible with the reaction conditions. However, C5-substituents led to significantly low reactivity (3ae and 3am). Next, the influence of protection groups on N4 was investigated. Various benzyls were well tolerated to give the corresponding products in good yields, and the electronic properties of para-substituents on benzyls did not show a noticeable influence on the transformation efficiency (3aa, 3aq, and 3ar). N4-methyl and N4-allyl-protected 1,4-dihydroquinoxalin-2-ones also reacted well to give good yields (3as and 3at). However, electron-withdrawing benzoyl on N4 shut down the desired transformation (3au). Moreover, N1 and N4 di-substituted substrates were also suitable substrates and provided 3av and 3aw in moderate yields. A tertiary C(sp3)–H bond, e.g., 3-methyl-1,4-dihydroquinoxalin-2-one, also smoothly underwent the reaction to give the desired product, albeit in a lower isolated yield of 38% (3ax), because its poor solubility eroded the reaction efficiency and a large amount of 1ax was recovered. Remarkably, N4-unprotected substrates were also compatible with this transformation, furnishing the corresponding difluoromethylated products in moderate to good yields (3ba–3be). Finally, a larger scale transformation of 1ak (2.0 mmol) was demonstrated with 60% isolated yield.
| a Reaction conditions (unless otherwise specified): 1 (0.1 mmol, 1.0 equiv.), PCII (3 mol%), reagent 2 (0.2 mmol, 2.0 equiv.), LiOH (0.5 mmol, 5.0 equiv.), AcOEt (2.0 mL), room temperature, overnight. Isolated yields. b 2.0 mmol scale. c Yield was calculated according to the 19F NMR ratio of 3 and 4; 4 refers to Table 3. d CH3CN was used as the solvent. |
|---|

|
Interestingly, when N4-unprotected substrates 1,4-dihydroquinoxalin-2-ones 1ba–1bd were subjected to this reaction, oxidation products 3-difluoromethyl-quinoxalin-2-ones 4a–4d were detected as the minor products (Table 2). It is reasonable to presume that 4a–4d might be derived from the oxidation of the corresponding desired 3ba–3bd. We therefore further expanded the application of this method for the synthesis of 3-difluoromethyl functionalized quinoxalin-2-ones 4, which belong to a class of pharmaceutically important compounds and were hitherto difficult to synthesize, although many methods were reported for the functionalization of quinoxalin-2-ones.18 Thus, as shown in Table 3, various 3-difluoromethyl-quinoxalin-2-ones were readily accessed in moderate to good yields via a facile one-pot process with the addition of an oxidant DDQ.
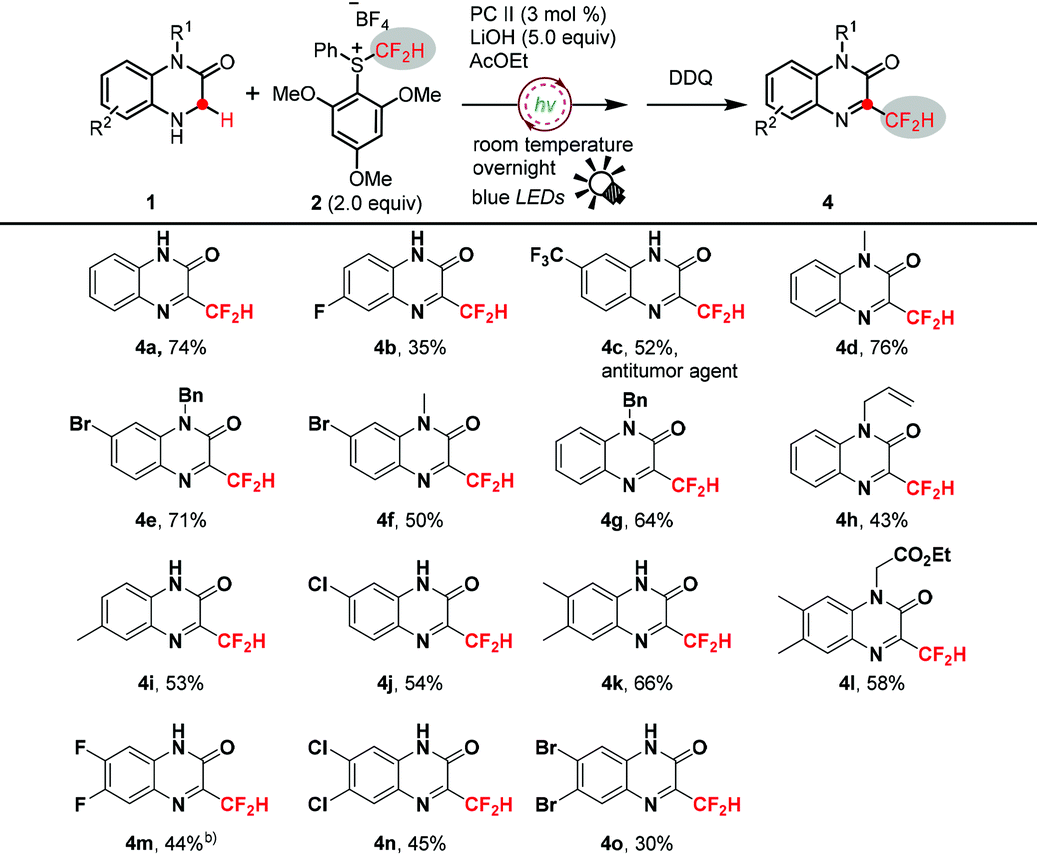
Notably, in these cases of low yields, i.e.4b and 4o, the poor solubility of substrates 1bb and 1bo significantly diminished the desired transformation, and the unreacted starting materials were oxidized to the corresponding quinoxalin-2-ones as the main side-products after adding DDQ. Importantly, 4c, an antitumor agent,19 was conveniently synthesized from 1bc in 52% isolated yield with this simple one-pot method.
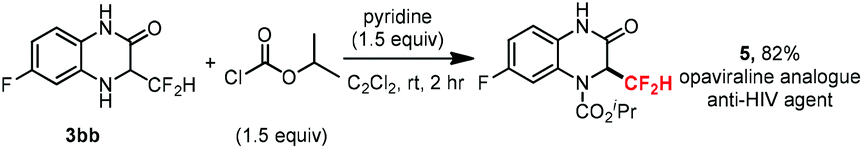
Quinoxalinones constitute a prevalent skeleton, frequently occurring in many biologically active compounds and pharmaceuticals, and possessing a wide variety of biological activities such as antiviral, anti-inflammatory, antidiabetic, antimicrobial, and anticancer properties. In order to further demonstrate the potential application of this significant method, we herein explored a rapid synthesis of difluoromethyl modified opaviraline (GW420867X) 5 from 3bb in 82% yield (Scheme 2), which is an anti-HIV-1 reverse transcriptase inhibitor.20 This process further demonstrates the synthetic utility of this radical–radical cross-coupling difluoromethylation reaction in the late-stage CF2H-modification of drugs and biologically active compounds.
 | ||
| Scheme 2 Synthesis of the opaviraline analogue. | ||
The mechanistic insight into this reaction was preliminarily studied. Thus, only a trace of 3aa was found under the standard reaction conditions in the presence of 1,4-dinitrobenzene (Scheme 3a), suggesting the existence of difluoromethyl radical species. Furthermore, a radical clock experiment was also conducted (Scheme 3a). The yield of the desired product 3aa was downgraded to 10%, along with the rearranged product 7 as the major product in 34% isolated yield. Moreover, in the low-yield case of 3am (22%), the dimer product 8 was isolated as the major product in 42% yield (Scheme 3c), which is strong evidence for obtaining the α-C radical from 1,4-dihydroquinoxalin-2-ones under photoredox catalysis conditions. Consequently, these experimental results above confirmed that a radical–radical cross-coupling process is involved during the reaction.
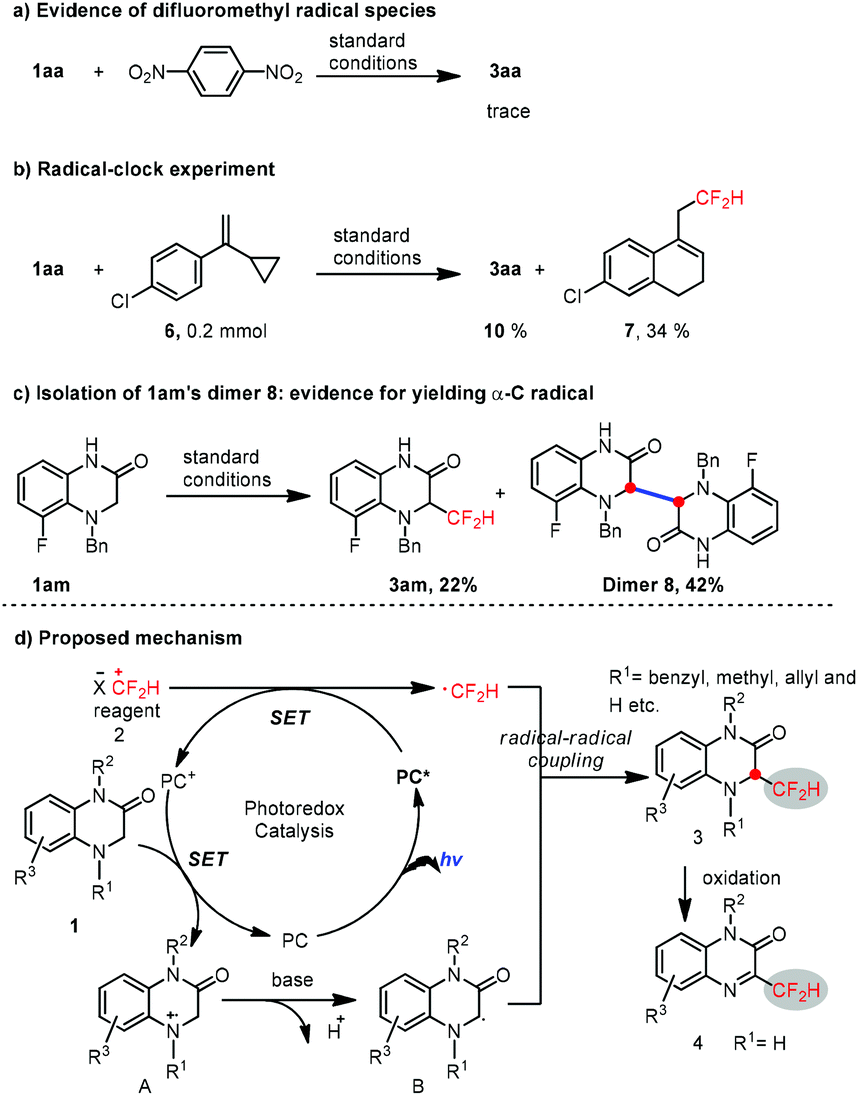 | ||
| Scheme 3 Mechanistic studies and the proposed mechanism. | ||
Based on the results of these mechanistic experiments, the radical–radical cross-coupling reaction mechanism was proposed as depicted in Scheme 3d. Initially, the S-(difluoromethyl)diarylsulfonium salt 2 is reduced by the excited PC* to generate the ˙CF2H radical along with PC+, followed by single electron transfer between substrate 1 and PC+ to render a cationic nitrogen radical A. A is delivered to the sp3 carbon radical Bvia deprotonation in the presence of a base, thus allowing for a radical–radical cross-coupling between the ˙CF2H radical and sp3 carbon radical B to generate the desired product 3, which can be readily oxidized to 4 for the N4-unprotected 1,4-dihydroquinoxalin-2-ones.
Conclusions
In conclusion, we have developed the first approach for a direct difluoromethylation of the C(sp3)–H bond via a radical–radical cross-coupling strategy, featuring transition metal-free photoredox catalysis, broad substrate scope and functional group tolerance, and mild reaction conditions. A wide variety of 1,4-dihydroquinoxalin-2-ones were smoothly converted to 3-difluoromethyl-1,4-dihydroquinoxalin-2-ones and 3-difluoromethylquinoxalin-2-ones in moderate to good yields. Employing this protocol, pharmacologically antiviral agent 5 and anticancer agent 4c were readily accessed in good yields, proving its potential utility in medicinal chemistry. Furthermore, mechanistic studies support the radical–radical cross-coupling process between a difluoromethyl radical and a C(sp3) radical involved in this reaction. This pioneering radical–radical cross-coupling strategy for significantly efficient C(sp3)–H difluoromethylation not only provides a powerful tool for the straightforward assembly of CF2H-containing bioactive molecules in the late-stage synthesis but would also promote the development of other new fluorination and fluoroalkylation methods, which is of great interest in synthetic chemistry and pharmaceutical chemistry.Experimental
General information
1H NMR spectra were recorded on either a Bruker Ascend 400 MHz (400 MHz) spectrometer, a Bruker Ascend 500 MHz (500 MHz) spectrometer or a Bruker Ascend 600 MHz (600 MHz) spectrometer at ambient temperature unless otherwise indicated. Data were reported as follows: chemical shifts in ppm from tetramethylsilane as an internal standard in CDCl3, integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet-doublet, m = multiplet, and br = broad), coupling constants (Hz), and assignment. 13C NMR spectra were recorded on either a Bruker Ascend 500 MHz (126 MHz) spectrometer or a Bruker Ascend 600 MHz (151 MHz) spectrometer at ambient temperature and were proton decoupled. Chemical shifts are reported in ppm from tetramethylsilane on the scale with the solvent resonance employed as the internal standard. 19F NMR spectra were recorded on a Bruker Ascend 400 MHz (377 MHz) spectrometer or a Bruker Ascend 500 MHz (471 MHz) spectrometer at ambient temperature. Chemical shifts are reported in ppm from CFCl3 as the internal standard. Single crystal X-ray diffraction data for the compounds were collected on a Rigaku Oxford Diffraction SuperNova dual source at 100 K using Cu-Kα radiation. ESI-MS analysis was performed in the positive ionization mode on an Agilent 1260-Infinity LC/MSD resolution mass spectrometer. All high-resolution mass spectra were obtained on a Thermo Scientific Q-Exactive (HR/AM) Orbitrap mass spectrometer. Commercially available reagents were used as received. Reactions were monitored by TLC (detection with UV light). Flash chromatography: silica gel (300–400 mesh). Visible light irradiation was performed using blue LED lamps (3 W × 4; λ = 450 nm) for the preparative scale.General procedure for the visible-light photoredox catalyzed direct C(sp3)–H difluoromethylation of 3,4-dihydroquinoxalin-2(1H)-ones
To a 25 mL Schlenk tube equipped with a magnetic stir bar were added the derivative of 3,4-dihydroquinoxalin-2(1H)-one 1 (0.1 mmol, 1.0 equiv.), 2 (83.0 mg, 0.2 mmol, 2.0 equiv.), PCII (1.7 mg, 0.003 mmol, 3 mol%), and LiOH (12.0 mg, 0.5 mmol, 5.0 equiv.). Then the flask was flushed with argon, followed by the addition of EtOAc (2 mL). The tube was placed at a distance of ∼2 mm away from (3 W × 4) blue LED lamps (λ = 450 nm), and the reaction mixture was stirred under the irradiation of blue LEDs. After stirring overnight, the solvent was evaporated in vacuo and the residue was purified by flash column chromatography on silica gel (petrol ether/ethyl acetate) to afford product 3.General procedure for the synthesis of 3-difluoromethylquinoxalin-2-ones
To a 25 mL Schlenk tube equipped with a magnetic stir bar were added the derivative of 3,4-dihydroquinoxalin-2(1H)-one 1 (0.1 mmol, 1.0 equiv.), 2 (83.0 mg, 0.2 mmol, 2.0 equiv.), PCII (1.7 mg, 0.003 mmol, 3 mol%), and LiOH (12.0 mg, 0.5 mmol, 5.0 equiv.). Then the flask was flushed with argon, followed by the addition of EtOAc (2 mL). The tube was placed at a distance of ∼2 mm away from 3 W blue LED lamps (λ = 450 nm), and the reaction mixture was stirred under the irradiation of blue LEDs. After stirring overnight, DDQ (45.5 mg, 0.2 mmol, 2.0 equiv.) was added and the mixture was stirred at 80 °C for another 2 hours; then the solvent was evaporated in vacuo and the residue was purified by flash column chromatography on silica gel (petrol ether/ethyl acetate) to afford product 4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the NSFC (92056201), the Guangdong Basic and Applied Basic Research Foundation (2020A1515010874), and the SZU Top Ranking Project (86000000210). We would like to thank the Instrumental Analysis Center of Shenzhen University for analytical work.Notes and references
- (a) R. E. Banks, B. E. Smart and J. C. Tatlow, Organofluorinechemistry: nomenclature and historical landmarks, Plenum, New York, 1994 Search PubMed; (b) P. Jeschke, The Unique Role of Fluorine in the Design of Active Ingredients for Modern Crop Protection, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed; (c) K. MÜller, C. Faeh and F. Diederich, Fluorine in pharmaceuticals: looking beyond Intuition, Science, 2007, 317, 1881–1886 CrossRef PubMed; (d) M. Cametti, B. Crousse, P. Metrangolo, R. Milani and G. Resnati, The fluorous effect in biomolecular applications, Chem. Soc. Rev., 2012, 41, 31–42 RSC; (e) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, Applications of fluorine in medicinal chemistry, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed; (f) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, Current contributions of organofluorinecompounds to the agrochemical industry, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- (a) W. K. Hagmann, The Many Roles for Fluorine in Medicinal Chemistry, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed; (b) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011), Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (c) N. A. Meanwell, Fluorine and fluorinated motifs in the design and application of bioisosteres for drug design, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- (a) J. A. Erickson and J. I. McLoughlin, Hydrogen Bond Donor Properties of the Difluoromethyl Group, J. Org. Chem., 1995, 60, 1626–1631 CrossRef CAS; (b) C. D. Sessler, M. Rahm, S. Becker, J. M. Goldberg, F. Wang and S. J. Lippard, CF2H, a hydrogen bond donor, J. Am. Chem. Soc., 2017, 139, 9325–9332 CrossRef CAS PubMed; (c) Y. Zafrani, D. Yeffet, G. Sod-Moriah, A. Berliner, D. Amir, D. Marciano, E. Gershonov and S. Saphier, Difluoromethyl bioisostere: examining the “lipophilic hydrogen bond donor” concept, J. Med. Chem., 2017, 60, 797–804 CrossRef CAS PubMed; (d) Y. Zafrani, G. Sod-Moriah, D. Yeffet, A. Berliner, D. Amir, D. Marciano, S. Elias, S. Katalan, N. Ashkenazi, M. Madmon, E. Gershonov and S. Saphier, CF2H, a Functional Group-Dependent Hydrogen-Bond Donor: Is It a More or Less Lipophilic Bioisostere of OH, SH, and CH3?, J. Med. Chem., 2019, 62, 5628–5637 CrossRef CAS PubMed; (e) Y. Zafrani, S. Saphier and E. Gershonov, Utilizing the CF2H moiety as a H-bond-donating group in drug discovery, Future Med. Chem., 2020, 12(5), 361–365 CrossRef CAS PubMed.
- (a) I. Ojima, Fluorine in medicinal chemistry and chemical biology, Blackwell, Oxford, 2009 CrossRef; (b) N. A. Meanwell, Synopsis of some recent tactical application of bioisosteres in drug design, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed; (c) M. D. Martínez, L. Luna, A. Y. Tesio, G. E. Feresin, F. J. Durán and G. Burton, Antioxidant properties in a non-polar environment of difluoromethyl bioisosteres of methyl hydroxycinnamates, J. Pharm. Pharmacol., 2016, 68(2), 233–244 CrossRef PubMed; (d) C. R. Rodriguez, M. C. del Fueyo, V. J. Santillán, D. M. Virginia, A. S. Veleiro, O. A. Castro and G. Burton, Synthesis and biological activity of fluorinated analogues of the DAF-12 receptor antagonist 24-hydroxy-4-cholen-3-one, Steroids, 2019, 151, 108469 CrossRef CAS PubMed.
- For reviews on difluoromethylating reagents and difluoromethylation reactions, see: (a) J. Hu, W. Zhang and F. Wang, Selective difluoromethylation and monofluoromethylation reactions, Chem. Commun., 2009, 7465–7478 RSC; (b) W.-B. Qin, J.-Y. Chen, W. Xiong and G. K. Liu, Recent advance in development and application of electrophilic difluoromethylating reagents, Chin. J. Org. Chem., 2020, 40, 3177–3195 CrossRef CAS; (c) Y.-L. Liu, J.-S. Yu and J. Zhou, Catalytic Asymmetric Construction of Stereogenic Carbon Centers that Feature a gem-Difluoroalkyl Group, Asian J. Org. Chem., 2013, 2, 194–206 CrossRef CAS; (d) C.-P. Zhang, Q.-Y. Chen, Y. Guo, J.-C. Xiao and Y.-C. Gu, Progress in fluoroalkylation of organic compounds via sulfinatodehalogenation initiation system, Chem. Soc. Rev., 2012, 41, 4536–4559 RSC; (e) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Advances in Catalytic Enantioselective Fluorination, Mono-, Di-, and Trifluoromethylation, and Trifluoromethylthiolation Reactions, Chem. Rev., 2015, 115, 826–870 CrossRef CAS PubMed; (f) C. Ni, M. Hu and J. Hu, Good Partnership between Sulfur and Fluorine: Sulfur-Based Fluorination and Fluoroalkylation Reagents for Organic Synthesis, Chem. Rev., 2015, 115, 765–825 CrossRef CAS PubMed; (g) D. Alexander and V. Levin, Difluorocarbene as a Building Block for Consecutive Bond-Forming Reactions, Acc. Chem. Res., 2018, 51, 1272–1280 CrossRef PubMed; (h) M. Reichel and K. Karaghiosoff, Reagents for Selective Fluoromethylation: A Challenge in Organofluorine Chemistry, Angew. Chem., Int. Ed., 2020, 59, 12268–12281 CrossRef CAS PubMed; (i) J. B. I. Sap, C. F. Meyer, N. J. W. Straathof, N. Iwumene, C. W. Ende, A. A. Trabanco and V. Gouverneur, Late-stage difluoromethylation: concepts, developments and perspective, Chem. Soc. Rev., 2021, 50, 8214–8247 RSC; (j) R. Britton, V. Gouverneur, J.-H. Lin, M. Meanwell, C. Ni, G. Pupo, J.-C. Xiao and J. Hu, Contemporary synthetic strategies in organofluorine chemistry, Nat. Rev. Methods Primers, 2021, 1, 47 CrossRef CAS; (k) C. F. Meyer, S. M. Hell, A. Misale, A. A. Trabanco and V. Gouverneur, Hydrodifluoromethylation of Alkenes with Difluoroacetic Acid, Angew. Chem., Int. Ed., 2019, 58, 8829–8833 CrossRef CAS PubMed; (l) Q.-W. Gui, F. Teng, Z.-C. Li, Z.-Y. Xiong, X.-F. Jin, Y.-W. Lin, Z. Cao and W.-M. He, Visible-light-initiated tandem synthesis of difluoromethylated oxindoles in 2-MeTHF under additive-, metal catalyst-, external photosensitizer-free and mild conditions, Chin. Chem. Lett., 2021, 32, 1907–1910 CrossRef CAS; (m) Q.-W. Gui, F. Teng, H. Yang, C. Xun, W.-J. Huang, Z.-Q. Lu, M.-X. Zhu, W.-T. Ouyang and W.-M. He, Visible-Light Photosynthesis of CHF2/CClF2/CBrF2-Substituted Ring-fused Quinazolinones in Dimethyl Carbonate, Chem. – Asian J., 2022, 17, e202101139 CrossRef CAS PubMed.
- N. Malquin, K. Rahgoshay, N. Lensen, G. Chaume, E. Miclet and T. Brigaud, CF2H as a hydrogen bond donor group for the fine tuning of peptide bond geometry with difluoromethylated pseudoprolines, Chem. Commun., 2019, 55, 12487–12490 RSC.
- Q. Xie, Z. Zhu, L. Li, C. Ni and J. Hu, A General protocol for C−H difluoromethylation of carbon acids with TMSCF2Br, Angew. Chem., Int. Ed., 2019, 58, 6405–6410 CrossRef CAS PubMed.
- (a) X. Wang, G. Liu, X.-H. Xu, N. Shibata, E. Tokunaga and N. Shibata, S-((phenylsulfonyl)difluoromethyl)thiophenium salts: carbon-selective electrophilic difluoromethylation of β-ketoesters, β-diketones, and dicyano alkylidenes, Angew. Chem., Int. Ed., 2014, 53, 1827–1831 CrossRef PubMed; (b) J. Wang, E. Tokunaga and N. Shibata, Highly C-selective difluoromethylation of β-ketoesters by using TMSCF2Br/lithium hydroxide/N,N,N-trimethylhexadecan-1-ammonium bromide, Chem. Commun., 2018, 54, 8881–8884 RSC.
- J. Zhu, H. Zheng, X.-S. Xue, Y. Xiao, Y. Liu and Q. Shen, Carbon-selective difluoromethylation of soft carbon nucleophiles with difluoromethylated sulfonium ylide, Chin. J. Chem., 2018, 36, 1069–1074 CrossRef CAS.
- S.-L. Lu, X. Li, W.-B. Qin, J.-J. Liu, Y.-Y. Huang, H. N. C. Wong and G.-K. Liu, Air- and Light-Stable S-(Difluoromethyl)sulfonium Salts: C-Selective Electrophilic Difluoromethylation of β-Ketoesters and Malonates, Org. Lett., 2018, 20, 6925–6929 CrossRef CAS PubMed.
- L. Peng, H. Wang and C. Guo, Copper-Catalyzed Enantioselective Difluoromethylation of Amino Acids via Difluorocarbene, J. Am. Chem. Soc., 2021, 143, 6376–6381 CrossRef CAS PubMed.
- X. Zeng, W. Yan, M. Paeth, S. B. Zacate, P.-H. Hong, Y. Wang, D. Yang, K. Yang, T. Yan, C. Song, Z. Cao, M.-J. Cheng and W. Liu, Copper-catalyzed, chloroamide-Directed benzylic C–H difluoromethylation, J. Am. Chem. Soc., 2019, 141, 19941–19949 CrossRef CAS PubMed.
- (a) W.-B. Qin, W. Xiong, X. Li, J.-Y. Chen, L.-T. Lin, H. N. C. Wong and G.-K. Liu, Visible-light-driven difluoromethylation of isocyanides with S-(difluoromethyl) diarylsulfonium Salt: Access to a wide variety of difluoromethylated phenanthridines and isoquinolines, J. Org. Chem., 2020, 85, 10479–10487 CrossRef CAS PubMed; (b) W.-B. Qin, W. Xiong, Y.-S. Zhao, K.-Z. Fu, L. Su and G.-K. Liu, Difluoromethyl radical triggered tandem reaction of N-Allylamides to difluoromethylated β-amino alcohols by photoredox catalysis, Org. Lett., 2021, 23, 8482–8487 CrossRef CAS PubMed.
- (a) G.-K. Liu, X. Li, W.-B. Qin, X.-S. Peng, H. N. C. Wong, L. Zhang and X. Zhang, Facile difluoromethylation of aliphatic alcohols with an S-(difluoromethyl)sulfonium salt: reaction, scope and mechanistic study, Chem. Commun., 2019, 55, 7446–7449 RSC; (b) G.-K. Liu, W.-B. Qin, X. Li, L.-T. Lin and H. N. C. Wong, Difluoromethylation of phenols and thiophenols with the S-(difluoromethyl)sulfonium salt: reaction, scope, and mechanistic study, J. Org. Chem., 2019, 84, 15948–15957 CrossRef CAS PubMed; (c) G.-K. Liu, X. Li, W.-B. Qin, W.-F. Lin, L.-T. Lin, J.-Y. Chen and J.-J. Liu, Selective O-difluoromethylation of 1,3-diones using S-(difluoromethyl) sulfonium salt, Chin. Chem. Lett., 2019, 30, 1515–1518 CrossRef CAS.
- For reviews on photoredox-catalyzed fluoroalkylation, see: (a) T. Koike and M. Akita, Trifluoromethylation by Visible-Light-Driven Photoredox Catalysis, Top. Catal., 2014, 57, 967–974 CrossRef CAS; (b) S. Barata-Vallejo, S. M. Bonesi and A. Postigo, Photocatalytic fluoroalkylation reactions of organic compounds, Org. Biomol. Chem., 2015, 13, 11153–11183 RSC; (c) X. Pan, H. Xia and J. Wu, Recent advances in photoinduced trifluoromethylation and difluoroalkylation, Org. Chem. Front., 2016, 3, 1163–1185 RSC; (d) T. Chatterjee, N. Iqbal, Y. You and E.-J. Cho, Controlled fluoroalkylation reactions by visible-light photoredox catalysis, Acc. Chem. Res., 2016, 49, 2284–2294 CrossRef CAS PubMed; (e) J. Rong, C. Ni, Y. Wang, C. Kuang, Y. Gu and J. Hu, Radical fluoroalkylation of aryl alkenes with fluorinated sulfones by visible-light photoredox catalysis, Acta Chim. Sin., 2017, 75, 105–109 CrossRef CAS; (f) T. Koike and M. Akita, New Horizons of Photocatalytic Fluoromethylative Difunctionalization of Alkenes, Chem., 2018, 4, 409–437 CrossRef CAS; (g) E. H. Oh, H. J. Kim and S. B. Han, Recent Developments in Visible-Light-Catalyzed Multicomponent Trifluoromethylation of Unsaturated Carbon–Carbon Bonds, Synthesis, 2018, 50, 3346–3358 CrossRef CAS; (h) A. Lemos, C. Lemaire and A. Luxen, Adv. Synth. Catal., 2019, 361, 1500–1537 CrossRef CAS PubMed; (i) T. Koike and M. Akita, Recent progress in photochemical radical di- and mono-fluoromethylation, Org. Biomol. Chem., 2019, 17, 5413–5419 RSC; (j) T. Koike, Frontiers in Radical Fluoromethylation by Visible-Light Organic Photocatalysis, Asian J. Org. Chem., 2020, 9, 529–537 CrossRef CAS.
- For selected examples of photoredox-catalyzed fluoroalkylation, see: (a) D. A. Nagib and D. W. C. MacMillan, Trifluoromethylation of arenes and heteroarenes by means of photoredox catalysis, Nature, 2011, 480, 224–228 CrossRef CAS PubMed; (b) X.-J. Tang and W. R. Dolbier Jr., Efficient Cu-catalyzed Atom Transfer Radical Addition Reactions of Fluoroalkylsulfonyl Chlorides with Electron-deficient Alkenes Induced by Visible Light, Angew. Chem., Int. Ed., 2015, 54, 4246–4249 CrossRef CAS PubMed; (c) L. Li, X. Mu, W. Liu, Y. Wang, Z. Mi and C.-J. Li, Simple and Clean Photoinduced Aromatic Trifluoromethylation Reaction, J. Am. Chem. Soc., 2016, 138, 5809–5812 CrossRef CAS PubMed; (d) Q.-Y. Lin, X.-H. Xu, K. Zhang and F.-L. Qing, Visible-Light-Induced Hydrodifluoromethylation of Alkenes with a Bromodifluoromethylphosphonium Bromide, Angew. Chem., Int. Ed., 2016, 55, 1479–1483 CrossRef CAS PubMed; (e) J. Rong, L. Deng, P. Tan, C. Ni, Y. Gu and J. Hu, Radical Fluoroalkylation of Isocyanides with Fluorinated Sulfones by Visible-Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2016, 55, 2743–2747 CrossRef CAS PubMed; (f) N. Noto, Y. Tanaka, T. Koike and M. Akita, Strongly Reducing (Diarylamino)anthracene Catalyst for Metal-Free Visible-Light Photocatalytic Fluoroalkylation, ACS Catal., 2018, 8, 9408–9419 CrossRef CAS; (g) V. Bacauanu, B. Cardinal, M. Yamauchi, M. Kondo, D. F. Fernández, R. Remy and D. W. C. MacMillan, Metallaphotoredox Difluoromethylation of Aryl Bromides, Angew. Chem., Int. Ed., 2018, 57, 12543–12548 CrossRef CAS PubMed; (h) W. Zhang, X. X. Xiang, J. Chen, C. Yang, Y.-L. Pan, J.-P. Cheng, Q. Meng and X. Li, Direct C–H difluoromethylation of heterocycles, Nat. Commun., 2020, 11, 638 CrossRef CAS PubMed.
- (a) C. Wang, M. Guo, R. Qi, Q. Shang, Q. Liu, S. Wang, L. Zhao, R. Wang and Z. Xu, Visible-Light-Driven, Copper-Catalyzed Decarboxylative C(sp3)−H Alkylation of Glycine and Peptides, Angew. Chem., Int. Ed., 2018, 57, 15841–15846 CrossRef CAS PubMed; (b) P. S. Akula, B. C. Hong and G. H. Lee, Visible-light-induced C(sp3)–H activation for a C–C bond forming reaction of 3,4-dihydroquinoxalin-2(1H)-one with nucleophiles using oxygen with a photoredox catalyst or under catalyst-free conditions, RSC Adv., 2018, 8, 19580–19584 RSC; (c) J. Rostoll-Berenguer, G. Blay, J. R. Pedro and C. Vila, Photocatalytic Giese addition of 1,4-dihydroquinoxalin-2-ones to electron-poor alkenes using visible light, Org. Lett., 2020, 22, 8012–8017 CrossRef CAS PubMed; (d) C. Wang, R. Qi, H. Xue, Y. Shen, M. Chang, Y. Chen, R. Wang and Z. Xu, Visible-light-promoted C(sp3)−H alkylation by intermolecular charge transfer: preparation of unnatural α-amino acids and late-stage modification of peptides, Angew. Chem., Int. Ed., 2020, 59, 7461–7466 CrossRef CAS PubMed; (e) I. Fleming, Molecular Orbitals and Organic ChemicalReactions, John Wiley & Sons, 2009, pp. 67–69 CrossRef.
- K. Sun, F. Xiao, B. Yu and W.-M. He, Photo–/electrocatalytic functionalization of quinoxalin–2(1H)–ones, Chin. J. Catal., 2021, 32, 1921–1943 CrossRef.
- P. Sanna, A. Carta, M. Loriga, S. Zanetti and L. Sechi, Preparation and biological evaluation of 6/7-trifluoromethyl(nitro)-, 6,7-difluoro-3-alkyl (aryl)-substituted-quinoxalin-2-ones, Farmaco, 1999, 54, 169–177 CrossRef CAS PubMed.
- (a) J. Balzarini, E. De Clercq, A. Carbonez, V. Burt and J. P. Kleim, Long-Term Exposure of HIV Type 1-Infected Cell Cultures to Combinations of the Novel Quinoxaline GW420867X with Lamivudine, Abacavir, and a Variety of Nonnucleoside Reverse Transcriptase Inhibitors, AIDS Res. Hum. Retroviruses, 2000, 16, 517–528 CrossRef CAS PubMed; (b) L. M. Cass, K. H. P. Moore, N. S. Dallow, A. E. Jones, J. R. Sisson and W. T. J. Prince, The Bioavailability of the Novel Nonnucleoside Reverse Transcriptase Inhibitor GW420867X Is Unaffected by Food in Healthy Male Volunteers, Clin. Pharmacol., 2001, 41, 528–535 CrossRef CAS PubMed; (c) K. H. P. Moore, L. M. Cass, N. Dallow, T. C. Hardman, A. Jones, M. Boyce and W. T. Prince, Pharmacokinetics and safety of escalating single and repeat oral doses of GW420867X, a novel non-nucleoside reverse transcriptase inhibitor, Eur. J. Clin. Pharmacol., 2001, 56, 805–811 CrossRef CAS PubMed; (d) K. Arasteh, R. Wood, M. Muller, W. Prince, L. M. Cass, K. H. Moore, N. Dallow, A. Jones, A. Klein, V. Burt and J. P. Kleim, GW420867X administered to HIV-1-infected patients alone and in combination with lamivudine and zidovudine, HIV Clin. Trials, 2001, 2(4), 307–316 CrossRef CAS PubMed; (e) L. Fabiana, M. T. Porro, N. Gómez, M. Salvatori, G. Turk, D. Estrin and A. Moglioni, Design, synthesis and biological evaluation of quinoxaline compounds as anti-HIV agents targeting reverse transcriptase enzyme, Eur. J. Med. Chem., 2020, 188, 111987 CrossRef PubMed; (f) J. Ren, C. E. Nichols, P. P. Chamberlain, K. L. Weaver, S. A. Short, J. H. Chan, J. P. Kleim and D. K. Stammers, Relationship of Potency and Resilience to Drug Resistant Mutations for GW420867X Revealed by Crystal Structures of Inhibitor Complexes for Wild-Type, Leu100Ile, Lys101Glu, and Tyr188Cys Mutant HIV-1 Reverse Transcriptases, J. Med. Chem., 2007, 50, 2301–2309 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2112795. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2qo00192f |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2022 |