
Room-temperature Barbier single-atom polymerization induced emission as a versatile approach for the utilization of monofunctional carboxylic acid resources†
Quan-Xi
Shi
ab,
Qian
Li
ab,
Hang
Xiao
bc,
Xiao-Li
Sun
 c,
Hongli
Bao
b and
Wen-Ming
Wan
*ab
c,
Hongli
Bao
b and
Wen-Ming
Wan
*ab
aCollege of Chemistry, Fuzhou University, Fuzhou, 350108, P. R. China. E-mail: wanwenming@fjirsm.ac.cn
bState Key Laboratory of Structural Chemistry, Key Laboratory of Coal to Ethylene Glycol and Its Related Technology, Center for Excellence in Molecular Synthesis Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, 155 West Yangqiao Road, Fuzhou 350002, P. R. of China
cCollege of Environmental Science and Engineering, Engineering Research Center of Polymer Green Recycling of Ministry of Education, Fujian Key Laboratory of Pollution Control & Resource Reuse, Fujian Normal University, Fuzhou, 350007, P. R. China
First published on 27th December 2021
Abstract
Carboxylic acids are widely available from both biomass and fossil sources on the Earth. In comparison with multifunctional carboxylic acid containing chemicals that have been comprehensively used as building blocks of polymer materials, monofunctional carboxylic acid resources exhibit broader availability but are rarely utilized as monomers for polymerization, attributed to their monofunctionality. Here, we demonstrate a Barbier single-atom polymerization (SAP) as a versatile approach for the utilization of monofunctional carboxylic acid resources, where they act as a carbon source to contribute only one carbon atom for the construction of the polymer main chain. The key point for the polymerization of the monofunctional carboxylic acid resource is to difunctionalize it, which is realized through two Barbier additions between bifunctional aromatic halides and monofunctional peroxyester in the presence of Mg. Prior to the Barbier SAP, monofunctional phenylcarboxylic acid is converted into dibenzoyl peroxide (BPO) with higher reactivity. Through the Barbier SAP of BPO at room temperature, a series of nonconjugated polytriphenylmethanols (PTPMs) were prepared as polymerization-induced emission luminogens (PIEgens) with structure-specific nonconjugated luminescence including aggregation-caused quenching (ACQ) and aggregation induced emission (AIE) characteristics, where starting monomers and repeating units are nonemissive. Further applications of PIEgens were carried out for an artificial light-harvesting system with an antenna effect of over 18.5 and explosive detection at the ppm level in solution and ng level on test paper. This work therefore opens a new avenue for the design of nonconjugated luminescence by utilizing Earth's monofunctional carboxylic acid resources sufficiently.
1. Introduction
Staudinger, a famous organic chemist and the father of modern polymer science, proposed the concept of polymerization in 1920, which successfully opened the prelude of polymer science.1 In the past 100 years, generations of polymer chemists have spent considerable efforts to develop different kinds of polymerization methods based on the corresponding organic reactions, resulting in prosperous polymer science with abundant synthetic polymer materials in the form of plastics, fibers, rubbers, etc., which have played important roles in human life.2–11 For example, polyesterification and polyamidation are based on the corresponding esterification and amidation reactions, resulting in significant polyesters and polyamides, e.g., polyethylene terephthalate (PET) and nylon.12–14 A series of coupling polymerizations are based on the corresponding Stille-, Suzuki-, Kumada-coupling reactions, etc.15–17 Living ring-opening metathesis polymerization is based on the olefin metathesis reaction.18–20 Click polymerization, Michael polyaddition, and multicomponent polymerizations are based on the corresponding click reaction, Michael addition, and multicomponent reactions respectively.21–36 Living radical polymerizations, also named reversible-deactivation radical polymerizations including atom transfer radical polymerization and reversible addition–fragmentation chain transfer polymerization, are based on the corresponding reversible-deactivation radical reactions as well.2,3,37–41Accompanied by the developments of different polymerization methods, different chemical moieties, including dicarboxylic acids, diols, diamines, alkenes, alkynes, thiols, etc., have been successfully developed as building blocks of polymer materials.42–47 The variety and availability of monomers are important factors for evaluating polymerization methods. Carboxylic acid containing compounds are widely available chemicals from both biomass and fossil sources on the Earth, and have been comprehensively used in organic synthesis for centuries.48,49 In polymer science, multifunctional carboxylic acid containing chemicals have been comprehensively used as building blocks of polyesters and polyamides, e.g., PET and nylon, through polyesterification and polyamidation. Compared with these widely used multifunctional carboxylic acid containing chemicals, monofunctional carboxylic acid containing chemicals exhibit broader availability from both biomass and fossil sources on the Earth, e.g., phenylcarboxylic acid can be widely available from benzoin resin, black cherry bark, and the chemical industry. However, more than two functionalities are required for the molecular design of monomers through polyesterification and polyamidation. Monofunctional carboxylic acid chemicals are therefore rarely utilized as monomers for polymerization due to their monofunctionality. Investigation of the possibility of utilizing monofunctional carboxylic acid chemicals as potential monomers for polymerization will expand the monomer library of polymerization and is therefore highly desirable.
The Barbier reaction, a named reaction for the utilization of carbonyl compounds as electrophiles for the construction of new chemicals through carbon–carbon bond formation, has been comprehensively used and has played important roles in organic chemistry for more than one century.50,51 Recently, based on the Barbier reaction, the Barbier polymerization has been successfully developed through A2 + B2, AB, AB2 and A2 + B types of one-pot polyadditions between organohalides and carbonyls in the presence of an activator at temperatures ranging from 40–80 °C, resulting in a series of alcohol-containing polymers that are difficult to synthesize by other polymerization methods.52–54 The Barbier polymerization exhibits intriguing polymerization-induced emission, where nonconjugated emission has been created from nonemissive monomers and repeating units.55 Generally, the formation of a Grignard reagent is arduous, especially for inactive organohalides, where a higher temperature (e.g., refluxing) and an activator (e.g., 1,2-dibromoethane and I2) are required.56,57 The room-temperature Barbier polymerization without an activator simplifies the process and saves energy and is therefore highly desirable.
Herein, a room-temperature Barbier polymerization without an activator is demonstrated as a versatile approach for the utilization of Earth's monofunctional carboxylic acid resources. This Barbier polymerization exhibits characteristics such as SAP and PIE, and results in a series of nonconjugated emissive polytriphenylmethanols as PIEgens. Further potential applications of PIEgens were investigated for an artificial light-harvesting system and explosive detection.
2. Results and discussion
2.1 Room-temperature Barbier single atom polymerization
In our previous studies, carbonyl groups in the form of ketones, esters and amides have been successfully polymerized through the Barbier polymerization (Scheme 1, Previous work). Earth's carbonyl resources in the form of monofunctional carboxylic acid and peroxyester haven't been realized yet. To realize the polymerization of monofunctional carboxylic acid resources, the key point is to difunctionalize monofunctional carboxylic acid, which can be realized through two Barbier additions between bifunctional aromatic halides and monofunctional peroxyester in the presence of Mg. The Barbier SAP is therefore proposed, where carboxylic acid resources act as a carbon source to contribute only one carbon atom for the construction of the polymer main chain. Prior to the Barbier SAP, monofunctional carboxylic acid needs to be converted into peroxyester with higher reactivity,58 which is supposed to lower the barrier energy of polymerization and realize polymerization at room temperature without an activator (Scheme 1, This work). Meanwhile, in order to prevent potential side reactions, e.g., intramolecular cyclization and the coupling reaction derived from Grignard reagents, monomers with rigid phenyl rings are specifically used under one-pot Barbier conditions. | ||
| Scheme 1 Typical Barbier polymerization strategy and monomers for the Barbier polymerization (previous work). Room-temperature Barbier single atom polymerization (this work). | ||
To verify the above hypothesis, phenyl carboxylic acid was taken as an example and converted into BPO with higher reactivity prior to the Barbier SAP (Fig. S1†). The one-pot Barbier SAP of BPO with a series of dibromobenzene was carried out at room temperature without any activator in the presence of Mg for 24 h (Table 1). Through two Barbier polyadditions between bromobenzene and the carbonyl group that results in the transition of an A2 + B type monomer into an A2 + B2* type monomer, a series of polytriphenylmethanols were prepared. Taking the Barbier SAP of BPO and p-dibromobenzene for the synthesis of para-polytriphenylmethanol (p-PTPM) as an example, the disappearance of the characteristic sharp monomer 1H NMR signals (Fig. 1A1 and A2) and the appearance of the characteristic broader aromatic polymer signals at 7.8–7.0 ppm and the hydroxide proton at 3.8–3.3 ppm (Fig. 1B) confirm the successful polymerization. p-PTPM was further confirmed by gel permeation chromatography (GPC) characterization with an Mw of 6700 and a PDI of 1.21 (Fig. 1C). This slightly higher molecular weight than that obtained in our previous work might be due to the higher reactivity of BPO than those of previous monomers. Fourier transform infrared (FT-IR) characterization (Fig. 1D) and polymerization tracking (Fig. S2†) were carried out to verify the chemical structure of p-PTPM. In the FT-IR spectrum, the characteristic peaks for C–O and O–H were observed at 1154 cm−1 and 3450 cm−1, respectively. The existence of C–OH was further confirmed from the 13C NMR spectrum at 82 ppm (Fig. S3†), which verified the chemical structure of p-PTPM. Other examples of the Barbier SAP of different monomers and the corresponding polymers are listed in Table 1. From the comparison of 1H NMR spectra of these monomers and the corresponding products (Fig. S9–S11†), it is found that the products clearly exhibit much broader signals for aromatic protons at ∼8.5–6.5 ppm and the hydroxide proton at ∼4.0–2.5 ppm, which are typical evidence of successful polymer synthesis. These successful Barbier SAPs are further confirmed by GPC and FT-IR characterization (Fig. S9–S11† and Table 1).
 | ||
| Fig. 1 Characterization of p-PTPM. 1H NMR spectra of benzoyl peroxide (A1), p-dibromobenzene (A2) and p-PTPM (B) in CDCl3, GPC curve of p-PTPM in DMF (C), and FT-IR spectrum of p-PTPM (D). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)






All the above characterization studies verify the proposed polymerization mechanism (Scheme 1, This work). To further verify this proposed mechanism, a model reaction between BPO and bromobenzene or p-methylbromobenzene was carried out under Barbier reaction conditions at room temperature without an activator for 12 h. Triphenylmethanol and 4-4′-dimethyl triphenylmethanol were successfully obtained and confirmed by 1H and 13C NMR (Fig. S5–S8†). These model reactions confirm the two Barbier additions between organohalides and BPO, which further confirm the above proposed polymerization mechanism (Scheme 1, This work). This Barbier SAP involves two nucleophilic polyadditions of organohalides to BPO, where the carboxylic carbon resource provides only a single carbon atom to construct the polymer main chain, resulting in a series of corresponding PTPMs. This Barbier SAP therefore enables the polymerization of carbonyl resources in the form of monofunctional carboxylic acid and peroxyester, which expands the monomer library of the polymerization method.
2.2 PIE and photophysical properties of polyphenylmethanols
As is known, nonconjugated moieties are weak/not emissive. Through the Barbier SAP, an intriguing PIE phenomenon is observed (Fig. 2). Before polymerization, all monomers are nonemissive. After polymerization, the prepared PTPMs become emissive, while the repeating unit of triphenylmethanol is weak/not emissive. Meanwhile, PTPMs exhibit structure-specific nonconjugated luminescence including both ACQ and AIE characteristics, depending on the connecting mode of monomers. When the dibromo monomer is para-substituted, e.g., p-dibromobenzene and p-dibromobiphenyl, the prepared p-PTPM and para-polyphenyltriphenylmethanol (p-PPTPM) are AIE type PIEgens, where all phenyl groups in the polymer chain are rotatable (Fig. 2A and B). When the dibromo monomer is ortho- or meta-substituted, e.g., o-dibromobenzene and m-dibromobenzene, the prepared o-PTPM and m-PTPM are ACQ type PIEgens, where some phenyl groups in the polymer chain are rotation forbidden (Fig. 2C and D). All these ACQ and AIE type luminescence characteristics are similar to our previous work, which confirms that these nonconjugated luminescence characteristics of PTPM are structure dependent rather than synthetic route dependent. To investigate the mechanism of PIE, time-dependent density functional theory (TD-DFT) calculations were carried out (Fig. 3 and S15†). With the increase of the polymer chain length, significant differences in the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO) and band gap are observed, even though the repeating unit remains the same. | ||
| Fig. 2 Different emission types of PIEgens (under UV irradiation at 365 nm). | ||
 | ||
| Fig. 3 Electron cloud distributions and energy levels (eV) of TPM, dimer, trimer, and tetramer in the geometry-optimized S1 state calculated using the TD-DFT B3LYP/6-311G*, Gaussian 09 program. | ||
These changes indicate that the polymerization will affect the molecular orbitals and narrow the corresponding bandgap, causing through-space conjugation and an intramolecular charge transfer effect under the constraint of polymer chains, resulting in nonconjugated luminescence eventually, which is similar to the PIE literature.52,55–57,59–62 Meanwhile, the structure-specific luminescence of PIE obeys the restriction of intramolecular rotation theory, which is similar to the AIE literature.63–65 It is worth mentioning that PIEgens can be easily adjusted between ACQ and AIE by simply adjusting the connection mode of the phenyl containing monomer, which enables easier molecular design of PIEgens.
The investigations on the photophysical properties of the prepared PTPMs were further carried out to demonstrate these PIEgens. All these PIEgens exhibit strong absorbance at about 250 nm and relatively weak absorbance at the 300–400 nm region, resulting in yellowish color of these polymers under sunlight (Fig. 4A and Fig. S12–S14 and Table S1†). The differences between absorption and excitation spectra also confirm the specificity of these PIEgens as untraditional luminogens. For para-substituted PIEgens, e.g., p-PTPM and p-PPTPM, they are not emissive in solution but highly emissive in the solid state triggered by the rotation forbidden phenyl rings in the aggregated state, which are typical AIE behaviors (Table S1, entries 1 and 2 and Fig. S12†). For ortho- or meta-substituted polymers, e.g., o-PTPM and m-PTPM, they are highly emissive in solution because of their rotation forbidden phenyl groups, which is a traditional luminescence behavior, in comparison with AIE type luminescence. Meanwhile, they exhibit weak emission in the aggregated state, which is similar to the traditional luminescence with an ACQ effect in the literature (Table S1, entries 3 and 4 and Fig. S13 and S14†). For better understanding AIE behaviors of p-PTPM and p-PPTPM, further detailed characterization studies of their luminescence properties were carried out (Fig. 4, Fig. S12 and Table S1,† entries 1 and 2). Taking p-PTPM as an example, it exhibits a maximum emission wavelength of 468 nm and a maximum excitation wavelength of 386 nm (Fig. 4A). Its AIE behavior is further illustrated by adding water into its solution in THF. As the water content increases, an increased emission intensity is observed (Fig. 4B and G). When the water fraction is lower than 30 vol% (Fig. 4B, D, E and G), the p-PTPM solution is transparent and exhibits weak luminescence. As the water fraction increases from 30% to 90%, the p-PTPM chain starts to phase separate with the gradual decrease of transmittance from 100 to 22.78%, accompanied by a fast increase of the luminescence intensity. Its AIE behavior is further illustrated by placing a drop of p-PTPM solution on a thin layer chromatography plate (Fig. 4F). As the solvent gradually evaporates, p-PTPM begins to aggregate in the solution, accompanied by a gradual increase of cyan luminescence on the thin layer chromatography plate. According to the Commission Internationale deL'Eclairage chromaticity diagram, the emission color coordinate of p-PTPM is (0.17, 0.24) (Fig. 4C).
 | ||
| Fig. 4 Luminescence properties of p-PTPM. Normalized excitation (red line) and emission (blue line) spectra of p-PTPM in the solid state and normalized absorption spectra (black line) of p-PTPM in THF (A). Emission spectra of p-PTPM (0.1 mg mL−1) in water/THF mixtures with different water volume fractions (fw, vol%) (excited at 386 nm) (B). CIE coordinates of p-PTPM (C). Plots of emission intensities of p-PTPM (0.1 mg mL−1) in water/THF mixtures with different water volume fractions (fw, vol%) (excited at 386 nm) (D). Transmittance of p-PTPM (0.1 mg mL−1) in water/THF mixtures with different water volume fractions (fw, vol%) at 500 nm (E). Digital photos of one drop of p-PTPM solution (10 mg mL−1 in THF) on a thin-layer chromatography plate at different times (time unit is second) of evaporation (under UV irradiation at 365 nm) (F). Digital photos of p-PTPM (0.1 mg mL−1) in water/THF mixtures with different water volume fractions (fw, vol%) (under sunlight and UV irradiation at 365 nm) (G). | ||
2.3 Application of p-PTPM for the facile fabrication of an artificial light-harvesting system
The artificial light-harvesting system has attracted widespread attention because of its high contribution to basic scientific research and the sustainable use of light energy.66 In light-harvesting systems, inhibition of the self-quenching effect of energy acceptors and energy donors is a decisive factor for improving the energy conversion efficiency and achieving high antenna effects. Due to the non-self-quenching AIE characteristics of p-PTPM and p-PPTPM and their good film-forming performance, their applications as potential energy donor materials for the design of artificial light-harvesting systems were therefore carried out. Because the absorption of the commercially available dye Nile Red (NiR) has a good overlap with the emission of p-PTPM or p-PPTPM (Fig. 5C and Fig. S16†) and the well-known ACQ effect of NiR,67 NiR was chosen as an energy acceptor to prepare the artificial light-harvesting system, by simply casting p-PTPM or p-PPTPM solution containing a tiny amount of NiR to form a film on the substrate (Fig. 5A). The luminescence results of the fabricated light-harvesting film were tested and are shown in Fig. 5E and Fig. S15.† With the increase of the loading of the NiR acceptor from 0.1–0.3%, the emission of NiR around 620 nm gradually increases under 386 nm excitation, while the emission of p-PTPM at 468 nm gradually decreases, indicating that the energy of the p-PTPM donor is successfully transferred to the NiR acceptor. With the increase of the NiR content from 0.1–0.3%, the antenna effect of the artificial light-harvesting film increases from 7.8–18.5 (Table S2†). When the content of NiR is 0.3 wt%, the energy transfer efficiency of the artificial light-harvesting film reaches 66.6% and 50.1% and the antenna effect is 18.5 and 18.1 for p-PTPM and p-PPTPM, respectively (Fig. S16 and Tables S2 and S3†). At the same time, the emission intensity of p-PTPM with 0.3 wt% NiR excited at 468 nm is lower than that excited at 386 nm, confirming the energy transfer process from the p-PTPM donor at the excited state to the NiR acceptor at the ground state with the formation of NiR* at the excited state (Fig. 5D). The photophysical process of this artificial light-harvesting system is therefore proposed (Fig. 5B). | ||
| Fig. 5 Artificial light-harvesting system application of p-PTPM. (A) Schematic illustration of the artificial light-harvesting system. (B) Schematic diagram of the energy transfer mechanism. (C) Absorption spectra of NiR and emission spectra of p-PTPM (ex = 386 nm). (D) Emission spectra of the artificial light-harvesting system using p-PTPM as an energy donor and NiR as an energy acceptor. (E) Emission spectra of p-PTPM with different amounts of NiR, excitation at 386 nm. (F) Emission intensities (at 620 nm) of the artificial light-harvesting system. GS, ground state; ES, excited state; ET, energy transfer. | ||
When the artificial light-harvesting system absorbs light energy at 386 nm, p-PTPM at the ground state will be excited to the excited state in the form of p-PTPM*, and p-PTPM* will return to the ground state by radiation and emit at 468 nm. However, after adding the NiR acceptor, the resonant energy transfer from p-PTPM* will take place to the ground state NiR to form NiR* in the excited state. Finally, NiR* returns to the ground state with an emission wavelength of 620 nm radiatively.
2.4 Application of PTPM for explosive detection
Due to the charge transfer interaction between electron-deficient 2,4,6-trinitrotoluene (TNT) and electron-rich p-PTPM that results in emission quenching, its application for explosive detection was carried out.68–70p-PTPM solution (0.1 mg mL−1 in a 90% water/THF mixture (vol%)) emits strong cyan fluorescence (Fig. 6A). With the addition of TNT, the luminescence intensity of the p-PTPM solution decreased rapidly (Fig. 6B). The fluorescence quenching rate reached near 80% with 22.5 ppm TNT (Fig. 6D), indicating the rapid and sensitive explosive detection at the ppm level, which is similar to the previous report and indicates that the functionality of PTPM is structure dependent rather than synthetic route dependent.52 In order to verify the practical application of p-PTPM in explosive detection, the p-PTPM solution was dropped on filter paper to prepare the test paper, and then dried in air, showing cyan luminescence under a 365 nm ultraviolet lamp. When the solution containing the residual amount of TNT was dropped on the test paper, efficient explosive detection of the residual TNT at 100 ng can be realized, accompanied by the disappearance of luminescence on the test paper (Fig. 6C).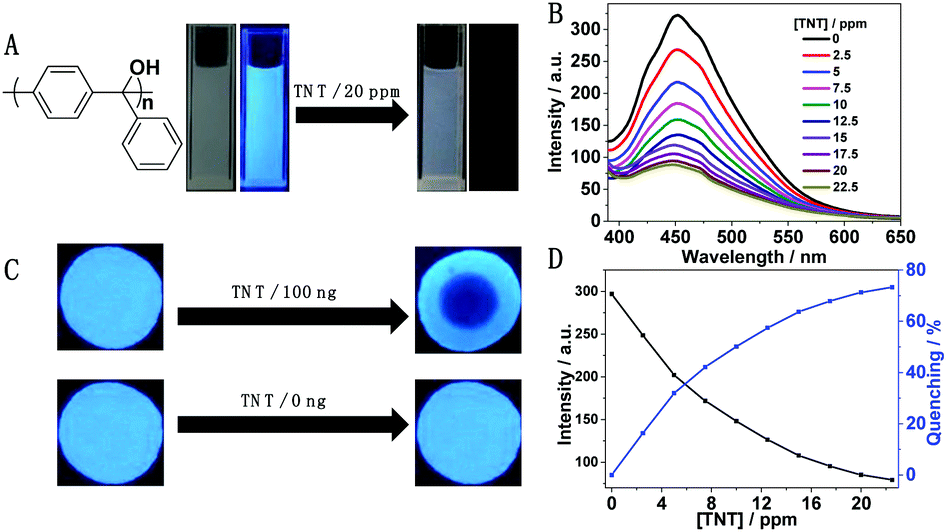 | ||
| Fig. 6 TNT sensory application of p-PTPM. (A) Photographic images of p-PTPM formed in 0.1 mg mL−1 90% water/THF mixtures (vol%) under irradiation with a UV lamp@365 nm, before and after adding 20 ppm amounts of TNT, respectively. (B) Emission spectra and (D) intensities and quenching ratio of p-PTPM in 0.1 mg mL−1 90% water/THF mixtures (vol%) upon the addition of TNT, excitation wavelength = 386 nm. (C) Photographic image of TNT test strips (up: with residual TNT under irradiation with a UV lamp@386 nm, down: blank, respectively). | ||
3. Conclusions
In conclusion, a room-temperature Barbier SAP is demonstrated as a versatile approach for the utilization of Earth's monofunctional carboxylic acid resources in the form of carboxylic acid and peroxyester, which is realized through two Barbier polyadditions between bifunctional aromatic halides and monofunctional peroxyester in the presence of Mg. Attributed to the reactivity improvement from phenylcarboxylic acid to BPO, the Barbier polymerization conditions are simplified, i.e., without an activator and room temperature. Through the Barbier SAP, a series of PTPMs have been prepared, where the carboxylic acid resource acts as a carbon source to contribute only one carbon atom for the construction of the polymer main chain. This Barbier SAP also exhibits intriguing structure-specific PIE characteristics, where the luminescence type can be adjusted from ACQ to AIE by simply adjusting the connection modes of the phenyl ring of monomers. When all phenyl rings of PIEgens are rotatable, they exhibit AIE behaviours. When all phenyl rings of PIEgens are rotation forbidden, they exhibit ACQ behaviours. TD-DFT calculations reveal the nonconjugated PIE mechanism, where through-space conjugation and an intramolecular charge transfer effect under the constraint of polymer chains play important roles. Further applications of AIE type PIEgens are illustrated for the fabrication of artificial light-harvesting systems with an antenna effect of up to 18.5 and sensitive TNT detection at the ppm level in solution and ng level on test paper. This work therefore expands the monomer library of the polymerization method and opens a new avenue for the design of nonconjugated luminescence by utilizing Earth's monofunctional carboxylic acid resources functionally and sufficiently.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding support from the NSFC (21971236, 21922112, 21672213 and 21871258), the State Key Laboratory of Molecular Engineering of Polymers (Fudan University) (K2020-27), the National Key R&D Program of China (Grant No. 2017YFA0700103), and the Haixi Institute of CAS (Grant No. CXZX-2017-P01).Notes and references
- H. Staudinger, Ber. Dtsch. Chem. Ges., 1920, 53, 1073–1085 CrossRef.
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087–1097 RSC.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- L. Tao, G. Mantovani, F. Lecolley and D. M. Haddleton, J. Am. Chem. Soc., 2004, 126, 13220–13221 CrossRef CAS PubMed.
- E. Jellema, A. L. Jongerius, J. N. H. Reek and B. de Bruin, Chem. Soc. Rev., 2010, 39, 1706–1723 RSC.
- A. J. C. Walters, O. Troeppner, I. Ivanovic-Burmazovic, C. Tejel, M. P. del Rio, J. N. H. Reek and B. de Bruin, Angew. Chem., Int. Ed., 2012, 51, 5157–5161 CrossRef CAS PubMed.
- D. Huang, Y. Liu, A. Qin and B. Tang, Polym. Chem., 2018, 9, 2853–2867 RSC.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 29212990 CrossRef PubMed.
- C. W. Bielawski and R. H. Grubbs, Angew. Chem., Int. Ed., 2000, 39, 2903–2906 CrossRef CAS PubMed.
- N. Zhu, M. F. Chiou, H. Xiong, M. Su, M. Su, Y. Li, W. M. Wan and H. Bao, iScience, 2020, 23, 100902 CrossRef CAS PubMed.
- R. Saint-Loup, T. Jeamnaire, J. J. Robin and B. Boutevin, Polymer, 2003, 44, 3437–3449 CrossRef CAS.
- Q. Cai, X. Li and W. Zhu, Macromolecules, 2020, 53, 2177–2186 CrossRef CAS.
- M. Luo, Y. Li, Y. Y. Zhang and X. H. Zhang, Polymer, 2016, 82, 406–431 CrossRef CAS.
- A. F. Littke, C. Y. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef CAS.
- X. Yin, F. Guo, R. A. Lalancette and F. Jäkle, Macromolecules, 2016, 49, 537–546 CrossRef CAS.
- S. Tasler and B. H. Lipshutz, J. Org. Chem., 2003, 68, 1190–1199 CrossRef CAS PubMed.
- T. L. Choi and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 1743–1746 CrossRef CAS PubMed.
- F. R. Fischer and C. Nuckolls, Angew. Chem., Int. Ed., 2010, 49, 7257–7260 CrossRef CAS PubMed.
- J. Z. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799–5867 CrossRef CAS PubMed.
- H. K. Li, J. Wang, J. Z. Sun, R. R. Hu, A. J. Qin and B. Z. Tang, Polym. Chem., 2012, 3, 1075–1083 RSC.
- A. Qin, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2010, 39, 2522–2544 RSC.
- A. Qin, J. W. Y. Lam and B. Z. Tang, Macromolecules, 2010, 43, 8693–8702 CrossRef CAS.
- W. Yang and C. Y. Pan, Macromol. Rapid Commun., 2009, 30, 2096–2101 CrossRef CAS PubMed.
- L. Xu, K. Yang, R. Hu and B. Z. Tang, Synlett, 2018, 2523–2528 CAS.
- W. Z. Li, X. Y. Wu, Z. J. Zhao, A. J. Qin, R. R. Hu and B. Z. Tang, Macromolecules, 2015, 48, 7747–7754 CrossRef CAS.
- T. Tian, R. R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2018, 140, 6156–6163 CrossRef CAS PubMed.
- O. Kreye, T. Toth and M. A. R. Meier, J. Am. Chem. Soc., 2011, 133, 1790–1792 CrossRef CAS PubMed.
- A. J. Qin, J. W. Y. Lam, L. Tang, C. K. W. Jim, H. Zhao, J. Z. Sun and B. Z. Tang, Macromolecules, 2009, 42, 1421–1424 CrossRef CAS.
- B. Wei, W. Li, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 5075–5084 CrossRef CAS PubMed.
- B. He, J. Zhang, J. Wang, Y. Wu, A. Qin and B. Z. Tang, Macromolecules, 2020, 53, 5248–5254 CrossRef CAS.
- C. Qi, C. Zheng, R. Hu and B. Z. Tang, ACS Macro Lett., 2019, 8, 569–575 CrossRef CAS.
- H. Xue, Y. Zhao, H. Wu, Z. Wang, B. Yang, Y. Wei, Z. Wang and L. Tao, J. Am. Chem. Soc., 2016, 138, 8690–8693 CrossRef CAS PubMed.
- X. X. Deng, L. Li, Z. L. Li, A. Lv, F. S. Du and Z. C. Li, ACS Macro Lett., 2012, 1, 1300–1303 CrossRef CAS.
- Z. h. Huang, Y. y. Zhou, Z. m. Wang, Y. Li, W. Zhang, N. c. Zhou, Z. b. Zhang and X. l. Zhu, Chin. J. Polym. Sci., 2017, 35, 317–341 CrossRef CAS.
- J. Zhao, Y. Zhou, Y. Zhou, N. Zhou, X. Pan, Z. Zhang and X. Zhu, Polym. Chem., 2016, 7, 1782–1791 RSC.
- W. M. Wan, C. Y. Hong and C. Y. Pan, Chem. Commun., 2009, 5883–5885 RSC.
- G. Moad, J. Chiefari, Y. K. Chong, J. Krstina, R. T. A. Mayadunne, A. Postma, E. Rizzardo and S. H. Thang, Polym. Int., 2000, 49, 993–1001 CrossRef CAS.
- W. M. Wan and C. Y. Pan, Macromolecules, 2007, 40, 8897–8905 CrossRef CAS.
- J. Q. Meng, F. S. Du, Y. S. Liu and Z. C. Li, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 752–762 CrossRef CAS.
- Y. L. Cao, Y. Shi, X. H. Wu and L. Q. Zhang, Chin. Chem. Lett., 2020, 31, 1660–1664 CrossRef CAS.
- C. Chen, T. Wang, Y. Fu and M. Liu, Chem. Commun., 2016, 52, 1381–1384 RSC.
- D. M. Hunsicker, B. C. Dauphinais, S. P. Mc Ilrath and N. J. Robertson, Macromol. Rapid Commun., 2012, 33, 232–236 CrossRef CAS PubMed.
- C. Chen, H. Fu, R. Baumgartner, Z. Song, Y. Lin and J. Cheng, J. Am. Chem. Soc., 2019, 141, 8680–8683 CrossRef CAS PubMed.
- A. D. Asandei, O. I. Adebolu, C. P. Simpson and J.-S. Kim, Angew. Chem., Int. Ed., 2013, 52, 10027–10030 CrossRef CAS PubMed.
- M. Bednarek, React. Funct. Polym., 2013, 73, 1130–1136 CrossRef CAS.
- B. Zhao, H. L. Ma, C. G. Wang, Z. K. Shang, Y. Ding and A. G. Hu, Macromolecules, 2020, 53, 240–248 CrossRef CAS.
- J. B. White, P. R. Blakemore, C. C. Browder, J. Hong, C. M. Lincoln, P. A. Nagornyy, L. A. Robarge and D. J. Wardrop, J. Am. Chem. Soc., 2001, 123, 8593–8595 CrossRef CAS PubMed.
- G. Zhao, X. W. Sun, H. Bienayme and J. P. Zhu, J. Am. Chem. Soc., 2001, 123, 6700–6701 CrossRef CAS PubMed.
- C. J. Li and W. C. Zhang, J. Am. Chem. Soc., 1998, 120, 9102–9103 CrossRef CAS.
- W. C. Zhang and C. J. Li, J. Org. Chem., 1999, 64, 3230–3236 CrossRef CAS PubMed.
- X. L. Sun, D. M. Liu, D. Tian, X. Y. Zhang, W. Wu and W. M. Wan, Nat. Commun., 2017, 8, 1210 CrossRef PubMed.
- M. Arslan, Macromol. Chem. Phys., 2018, 219, 1800087 CrossRef.
- M. Su, T. Li, Q. X. Shi, H. Xiao, H. Bao and W. M. Wan, Macromolecules, 2021, 54, 9919–9926 CrossRef CAS.
- Y. N. Jing, S. S. Li, M. Su, H. Bao and W. M. Wan, J. Am. Chem. Soc., 2019, 141, 16839–16848 CrossRef CAS PubMed.
- S. S. Li, Y. N. Jing, H. Bao and W. M. Wan, Cell Rep. Phys. Sci., 2020, 1, 100116 CrossRef.
- S. S. Li, N. Zhu, Y. N. Jing, Y. Li, H. Bao and W. M. Wan, iScience, 2020, 23, 101031 CrossRef CAS PubMed.
- H. Tian, W. Xu, Y. Liu and Q. Wang, Chem. Commun., 2019, 55, 14813–14816 RSC.
- B. Liu, H. Zhang, S. Liu, J. Sun, X. Zhang and B. Z. Tang, Mater. Horiz., 2020, 7, 987–998 RSC.
- W. B. Wu and B. Liu, Mater. Horiz., 2022, 9, 99–111 RSC.
- B. Song, X. Li, A. Qin and B. Z. Tang, Macromolecules, 2021, 54, 9019–9026 CrossRef CAS.
- W. B. Liu, X. H. Xu, S. M. Kang, X. Song, L. Zhou, N. Liu and Z.-Q. Wu, Macromolecules, 2021, 54, 3158–3168 CrossRef CAS.
- H. Zhang, X. Zheng, N. Xie, Z. He, J. Tiu, N. L. C. Leung, Y. Niu, X. Huang, K. S. Wong, R. T. K. Kwok, H. H. Y. Sung, I. D. Williams, A. Qin, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16264–16272 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- S. Tang, T. Yang, Z. Zhao, T. Zhu, Q. Zhang, W. Hou and W. Z. Yuan, Chem. Soc. Rev., 2021, 50, 12616–12655 RSC.
- T. X. Xiao, W. W. Zhong, L. Zhou, L. X. Xu, X. Q. Sun, R. B. P. Elmes, X. Y. Hu and L. Y. Wang, Chin. Chem. Lett., 2019, 30, 31–36 CrossRef CAS.
- L. Wong, C. Hu, R. Paradise, Z. Zhu, A. Shtukenberg and B. Kahr, J. Am. Chem. Soc., 2012, 134, 12245–12251 CrossRef CAS PubMed.
- M. Su, Y. N. Jing, H. Bao and W. M. Wan, Mater. Chem. Front., 2020, 4, 2435–2442 RSC.
- K. Zhang, H. Zhou, Q. Mei, S. Wang, G. Guan, R. Liu, J. Zhang and Z. Zhang, J. Am. Chem. Soc., 2011, 133, 8424–8427 CrossRef CAS PubMed.
- W. M. Wan, D. Tian, Y. N. Jing, X. Y. Zhang, W. Wu, H. Ren and H. L. Bao, Angew. Chem., Int. Ed., 2018, 57, 15510–15516 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01493e |
| This journal is © The Royal Society of Chemistry 2022 |