
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrosynthetic C–F bond cleavage
Johannes L.
Röckl
a,
Emma L.
Robertson
b and
Helena
Lundberg
 *a
*a
aDepartment of Chemistry, KTH Royal Institute of Technology, SE-100 44, Stockholm, Sweden. E-mail: hellundb@kth.se
bNovogensys Ltd, Dunnet, Kirrenleigh, Thurso KW14 8YD, UK
First published on 3rd August 2022
Abstract
Fluorinated organic compounds are common among pharmaceuticals, agrochemicals and materials. The significant strength of the C–F bond results in chemical inertness that, depending on the context, is beneficial, problematic or simply a formidable synthetic challenge. Electrosynthesis is a rapidly expanding methodology that can enable new reactivity and selectivity for cleavage and formation of chemical bonds. Here, a comprehensive overview of synthetically relevant electrochemically driven protocols for C–F bond activation and functionalization is presented, including photoelectrochemical strategies.
 Johannes L. Röckl | Johannes L. Röckl is currently a Senior Scientist at RISE, Research Institutes of Sweden in Södertälje, Sweden. He received his Ph.D. under supervision of Prof. Dr Siegfried R. Waldvogel at Johannes Gutenberg University Mainz in Germany and Prof. Dr Bill Morandi at ETH Zurich in Switzerland, working mainly on novel electro-synthetic and catalytic transformations. In 2021, he joined the group of Dr Helena Lundberg at KTH Royal Institute of Technology in Stockholm in Sweden, focusing on kinetic analysis and novel electroreductive reactions. |
 Emma L. Robertson | Emma L. Robertson studied medicinal and biological chemistry at Edinburgh University, Scotland and ETH Zurich, Switzerland. After internships in medicinal chemistry at Roche in Basel, Switzerland and agrochemistry at BASF SE in Ludwigshafen, Germany, Emma worked as an Editor for Wiley VCH, Weinheim in Germany. She pursues a career as a publications specialist and medical writer in the pharmaceutical industry. |
 Helena Lundberg | Helena Lundberg is employed as Assistant Professor in Organic Chemistry at KTH Royal Institute of Technology in Stockholm, Sweden. She received her Ph.D. degree under the guidance of Professor Hans Adolfsson at Stockholm University, after which she carried out postdoctoral research at the same institution with Professor Fahmi Himo. In 2017, she joined Professors Donna G. Blackmond and Phil Baran as a postdoctoral fellow at Scripps Research in La Jolla, USA. Helena's research group at KTH focuses on activation and functionalization of strong polarized σ-bonds using catalysis and electrosynthesis. |
1. Introduction
Fluorinated organic compounds have widespread use as pharmaceutical motifs, agrochemicals and materials due to their unique physical, chemical, and biological properties. For example, incorporation of fluorine in molecules can result in increased lipophilicity, bioavailability and metabolic stability.1 Fluorine has the highest electronegativity of all elements in the periodic table2 and forms highly polarized bonds with carbon, similar to its heavier halogen congeners. However, the C–F bond is significantly stabilized by the electrostatic attraction between the positively polarized carbon and the negatively polarized fluoride.3 This stabilization is reflected in the considerably higher dissociation energies of C–F bonds4,5 compared to other carbon-halide bonds (Fig. 1) and affects the kinetics for their cleavage, resulting in low reactivity of fluorocarbons.6 This relative chemical inertness is evident in everything from classic polar nucleophilic substitution reactions of alkyl halides3 to formation of carbon centered radicals via reductive dissociative electron transfer in e.g. cross-electrophile coupling (XEC) reactions7 and halogen atom transfer (XAT) processes8 as well as in transfer halogenation of alkenes.9 Depending on the context, the chemical inertness of the C–F bond can be viewed as beneficial (e.g. metabolic stability of pharmaceuticals), problematic (e.g. persistence of per- and polyfluoroalkyl substances – PFAS) or simply an intriguing synthetic challenge.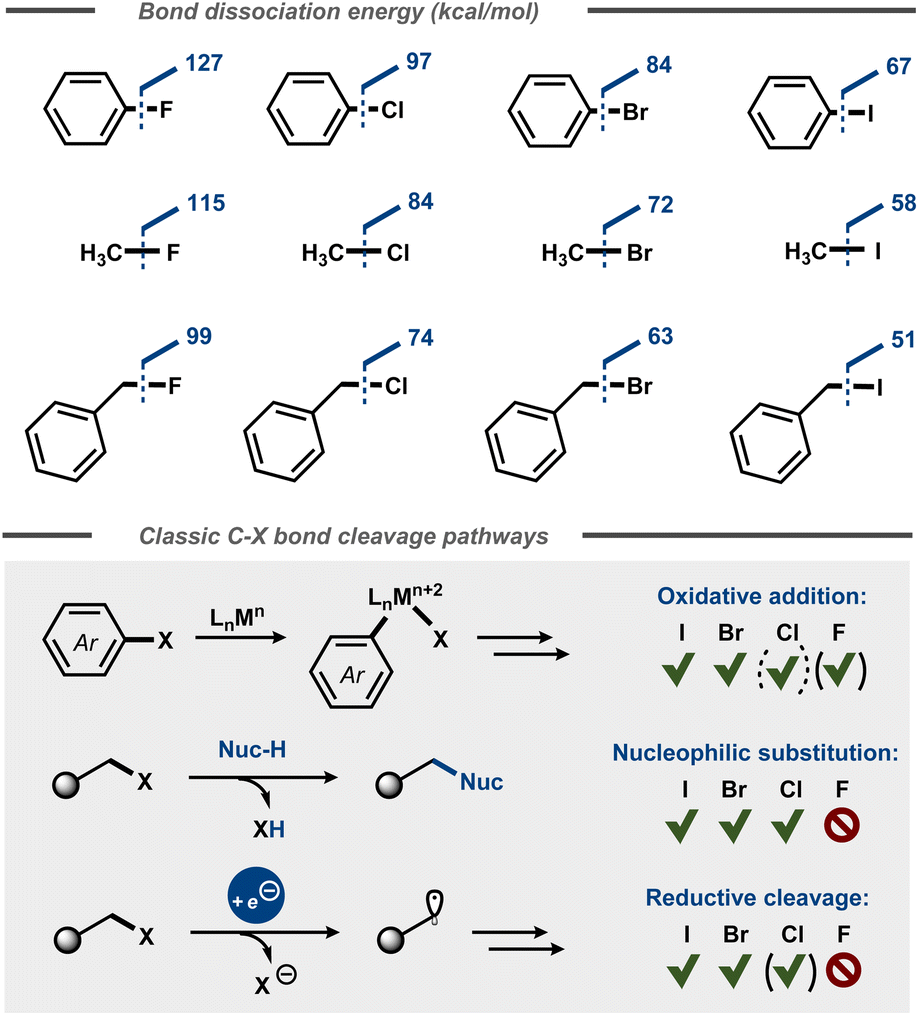 | ||
| Fig. 1 Top: Bond dissociation energies for aromatic and aliphatic C–X bonds.4 Bottom: Oxidative addition, nucleophilic substitution, and reductive cleavage of C–X bonds. | ||
While fluorides are typically introduced into a compound for the macroscopic properties they give rise to, selective defluorination can be of high synthetic benefit. For example, such a strategy can enable the use of trifluoromethyl groups as precursors for difluoromethyl (CF2H) groups that have received significant attention for medicinal chemistry applications.10 Despite the low reactivity of C–F bonds, synthetic strategies are available for their cleavage. The most common approach relies on elimination through an E1cB mechanism, including nucleophilic aromatic substitutions.3,11 In these processes, an inductively stabilized anion is formed adjacent to a fluoride and subsequently undergoes an irreversible elimination step to form a double bond with release of the halide. In addition, C–F bond activation via a variety of mechanisms has been reported using e.g. transition metal catalysis,12 Lewis acids based on main group elements,6,13 early transition metals and lanthanides13,14 and low-valent metal reagents.10,15 Recent developments in radical synthesis have also delivered new methods for C–F bond cleavage, including photo- and electrochemical strategies.16 The latter is becoming an increasingly popular synthetic approach, due to the prospects of new reactivity and selectivity, as well as resource friendly synthesis that follows with electricity as terminal reagent.17 While excellent reviews summarizing the principles of and synthetic possibilities for electrosynthesis have been published lately,18 a comprehensive overview of electrochemical protocols for C–F bond cleavage is missing in the literature to date. To address this gap, this review discusses synthetically relevant protocols for electrochemically promoted C–F bond cleavage from the 1950s to present, along with mechanistic details, with the overarching aim of promoting further developments in the field. Electrochemically driven degradation of PFAS was recently surveyed and is thus not included in the present review.19,20 In all figures throughout this review, the anode is found to the left and the cathode to the right when depicted above a reaction arrow.
2. Cleavage of C(sp2)–F bonds
Electroanalytical studies of C–F bond cleavage in fluorobenzenes were first reported more than 50 years ago,21,22 establishing that the standard reduction potential for fluorobenzene was close to −3 V vs. SCE.23,24 One of the first synthetic protocols for hydrodefluorination of fluoroarenes was reported by Kariv-Miller et al.25 Using a Hg pool cathode in the presence of dimethylpyrrolidinium tetrafluoroborate ((DMP)BF4), selective monodefluorination of 1,3-difluorobenzene to fluorobenzene was achieved in 85% yield using a divided cell setup (Fig. 2, top). A constant current of 1.25 mA cm−2 at 2 °C and 2 F mol−1 was found optimal for the monodefluorination, whereas higher current densities resulted in over-reduction to benzene. While the selectivity for monodefluorination was kept intact, the concentration of both water and (DMP)BF4 was found to influence the yield due to competing hydrogen evolution. The optimized conditions were also amenable for reduction of fluorobenzene to benzene in 75% yield. The mechanism was probed using cyclic voltammetry, revealing an increase of cathodic current and decrease of anodic current for (DMP)BF4 in the presence of substrate, indicative of catalysis. Supported by separate studies,26 the authors proposed that a catalytically active “amalgam” of DMP+ and mercury forms upon single electron reduction at the cathode. This mediating surface was claimed to react with the fluorinated substrate to form the corresponding radical anion that, in turn, decomposes to an aryl radical with release of fluoride (Fig. 2, bottom). A second single electron transfer to the aryl radical followed by protonation would thereafter form the hydrodefluorinated arene product. This mechanistic proposal was further supported by studies on defluorinative cyclization of o-(3-butenyl)fluorobenzene at Hg and Pb cathodes in the presence of catalytic amounts of DMP+.27 From experiments with cyclic voltammetry and preparative electrolysis, it was found that the DMP+ ion enabled a shift in reduction potential from −2.9 V to −2.75 V vs. SCE for reduction of the substrate, thus supporting the idea of its mediating effect for the electron transfer. In addition, a selectivity enhancement for cyclization product 1-methylindane over hydrodefluorination product 3-butenylbenzene was observed in the presence of DMP+ (Fig. 2, middle). The cyclization pathway was proposed to proceed via the same aryl radical intermediate as the hydrodefluorination product (Fig. 2, bottom), followed by rapid intramolecular cyclization and subsequent single electron reduction/protonation to afford the defluorinated indane product. The solvent, residual moisture or the tetrabutylammonium cation of the supporting electrolyte was hypothesized to serve as proton source. | ||
| Fig. 2 Top: Selective monodefluorination of 1,3-difluorobenzene.25 Middle: Defluorinative reductive cyclization.27 Bottom: Mechanistic proposal for reductive hydrodefluorination.25–27 | ||
Hydrodefluorination of a limited number of 7-piperazinoquinolones under electroreductive conditions was reported by Albini and co-workers (Fig. 3).28 Similar to Kariv-Miller's findings,25 selective monodefluorination of position 8 in 6,8-difluorinated lomefloxacin was achieved after 2 F mol−1 and defluorination of the monofluoride-containing enoxacin was observed. In contrast, norfloxacin resulted in a mere 10% yield of defluorinated product along with side-products. The higher yields and selectivities for enoxacin and lomefloxacin compared to norfloxacin correlated with the reduction potentials of the compounds observed by differential pulse voltammetry. While distinct reduction waves were observed for the former two, the cathodic wave of the latter was found very close to that of the electrolyte solution. This difference was argued to result from the substitution pattern of the heterocyclic compounds, resulting in greater or lesser stabilization of the hypothesized intermediate aryl radical and, hence, a more or less selective reaction. Selective monodefluorination of methyl-2,6-difluorobenzoate was also reported by Périchon and co-workers in the presence of 10 mol% SmCl3.29 While no mechanistic details were conveyed, the catalyst afforded a selectivity switch from ester reduction to defluorination. Defluorination of the monofluoride product was not observed under the applied conditions.
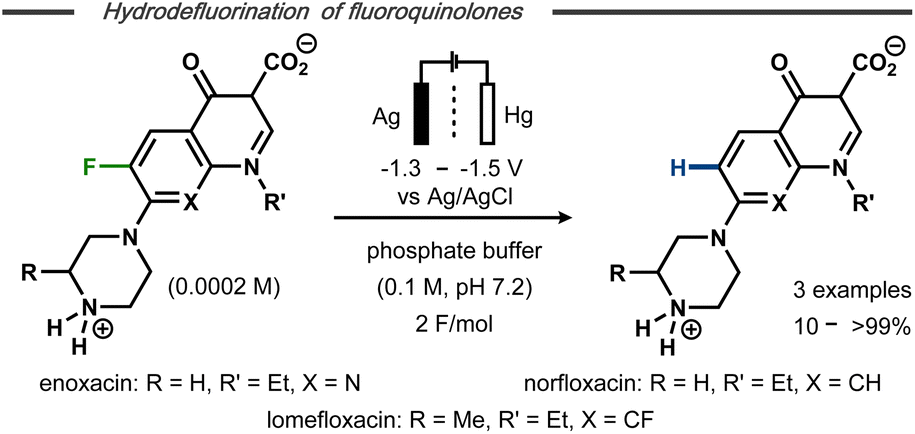 | ||
| Fig. 3 Electrochemical hydrodefluorination of fluoroquinolones.28 | ||
Wu et al. described electrochemical hydrodefluorination of fluoroaromatic compounds in the presence of borohydride reagents.30 Using platinum electrodes in an undivided cell, the electrolysis was carried out in diglyme or N-methylpyrrolidine (NMP) at constant current to provide 15 examples of the corresponding hydrodefluorinated products of mono- and perfluorinated arenes in good to excellent yields (up to 98%) (Fig. 4). Labelling experiments revealed that deuterium incorporation occurred to only a minor extent in the presence of sodium borodeuteride, whereas GC-MS analysis indicated the presence of tributylamine in the crude reaction mixture when Bu4NBF4 was used as electrolyte. Based on these findings, it was hypothesized that the tetrabutylammonium cation serves as hydrogen source rather than the hydride reagent. Similar to Kariv-Miller's proposal,25,26 the cathodic defluorination was suggested to proceed via stepwise dissociative electron transfer with a radical anion intermediate that decomposes to the corresponding fluoride and an open shell aryl species. Reductive radical-polar crossover31 by a second SET to the aryl radical forms the corresponding aryl anion and, subsequently, the final product upon protonation.
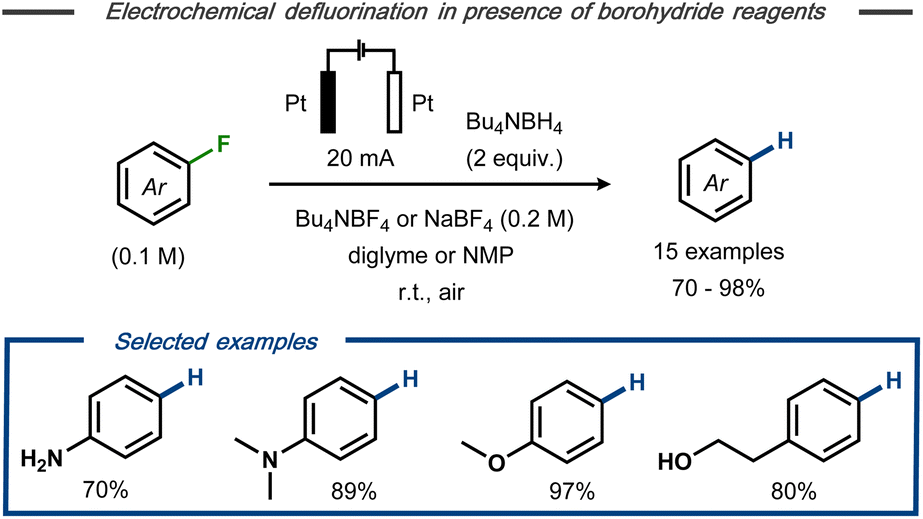 | ||
| Fig. 4 Electrochemical hydrodefluorination of aryl fluorides in the presence of borohydride reagents.30 | ||
The formation of aryl radicals via stepwise dissociative electron transfer with an intermediate radical anion is a commonly envoked mechanism for electrochemical C(sp2)–F bond cleavage in aryl fluorides (Fig. 5, top).25,28,30,32 Nevertheless, alternative mechanisms have been proposed in certain cases, suggesting that an initially formed arylfluoride radical anion can undergo chemical steps prior to C–F bond cleavage.22,33 An interesting case of a substrate-dependent mechanistic switch was reported by Muthukrishnan and Sangaranarayanan for electrochemical defluorination of fluorobenzoates (Fig. 5, bottom).34 Voltammetric studies of methyl-2-fluorobenzoate at high scan rates revealed a reversible electron transfer, indicative of a stable radical anion with decomposition to the corresponding aryl radical and fluoride being rate limiting. In contrast, methyl-4-fluorobenzoate was found to undergo electro-dimerization rather than hydrodefluorination. After systematic voltammetric studies and with support by analogous dimerization of 4-fluorobenzonitrile35 and pentafluoronitrobenzene,36 the authors concluded that the dimerization likely occurs between initially formed radical anions, followed by C–F bond cleavage. DFT calculations for both fluorobenzoate substrates indicated that the π* orbital of the extended aromatic system is the initial electron acceptor, with the charge subsequently being transferred to the C–F σ* bond and resulting bond cleavage.
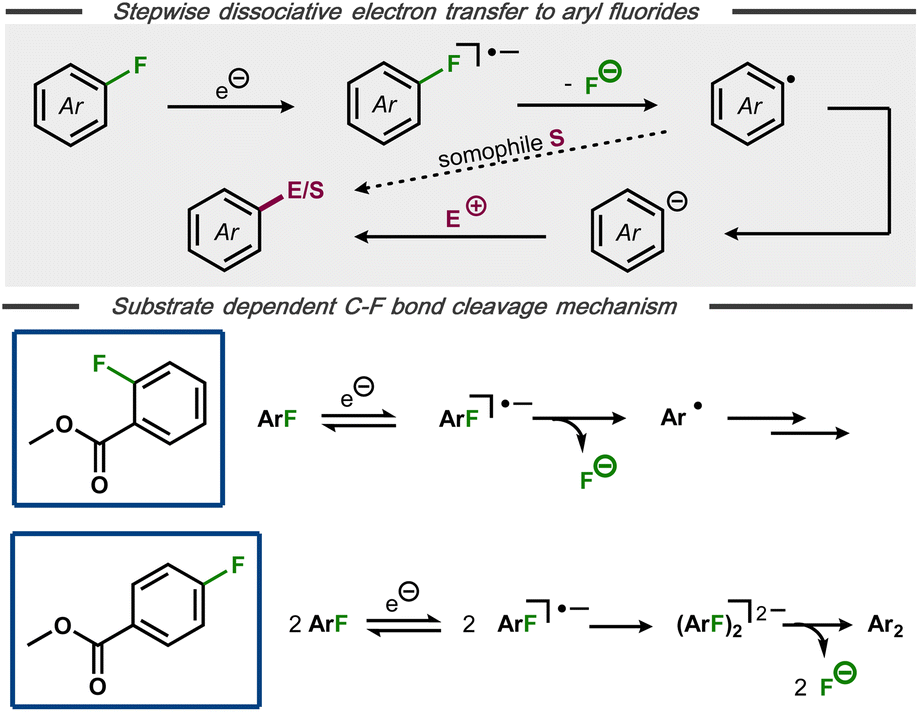 | ||
| Fig. 5 Top: Mechanism for stepwise defluorination of aryl fluorides. Bottom: Substrate dependent pathways for electroreductive defluorination of methylfluorobenzoates.34 | ||
Electroreductive polymerization of polyhalogenated arenes with concomitant C–X bond cleavage was recently reported by Ekinci and co-workers.37 In this reductive synthesis, polyaromatic compounds were formed on the surface of silicon or gold cathodes from polyhalogenated starting materials, including hexafluorobenzene, and were subsequently treated thermally to form graphene-like films. Mechanistically, it was proposed that the coupling reaction either occurs between two radical anions, similar to the mechanism envoked for dimerization of methyl-4-fluorobenzoate,34 or via dimerization of neutral aryl radicals. The latter coupling type is commonly deemed unfavorable due to the propensity of aryl radicals to undergo further electron transfers or hydrogen atom transfer events.38,39 However, voltammetric studies of hexafluorobenzene suggested that the dissociative electron transfer may take place in a concerted rather than a stepwise manner under the applied conditions. Alternative pathways, such as intermediate aryl anions taking part in nucleophilic aromatic substitutions (SNAr), were not discussed.
Senboku et al. reported a defluorinative electrocarboxylation protocol for polyfluorinated arenes with carbon dioxide.40 Constant current electrolysis in an undivided cell equipped with a Pt cathode and a Mg anode at −40 °C resulted in mono-carboxylated polyfluorobenzoic acids as products in moderate to good yields and high regio- and chemoselectivity (Fig. 6, top). Lower yields were obtained using either higher reaction temperature or higher current density. Methoxy and acetoxy groups, as well as a benzylic alcohol protected as the tetrahydropyran (THP) acetal, were tolerated under the applied conditions and resulted in selective carboxylation in para position (Fig. 6, bottom). The transformation was proposed to proceed via stepwise dissociative electron transfer, followed by radical-polar crossover to form the aryl anion (see Fig. 5, top). This species would, in turn, react with CO2 to form the magnesium carboxylate by interaction with the ions from the dissolving metal anode and, eventually, the polyfluorinated carboxylic acid products upon acidic workup.
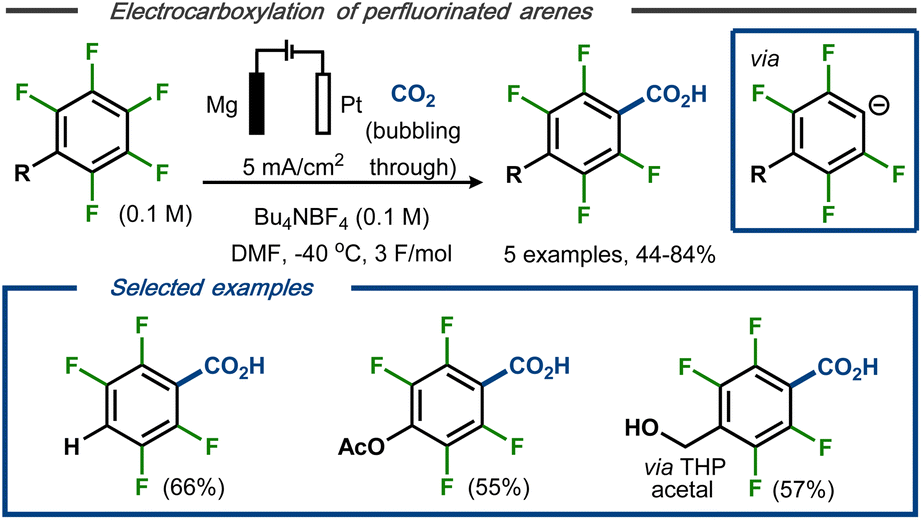 | ||
| Fig. 6 Defluorination/carboxylation of polyfluorinated arenes.40 | ||
Electrocarboxylation with C(sp2)–F bond cleavage was reported by Xie et al. using gem-difluoroalkenes as starting material.41 Under constant current electrolysis in an undivided cell, 20 α-fluoroacrylic acids were demonstrated with up to 83% yield and 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Z:E selectivity (Fig. 7, top). The protocol displayed tolerance towards functional groups such as carboxylic esters and amides, sulfones, nitriles, and alkynes and allowed for selective dehalogenation of the olefinic fluorides, whereas both aromatic and benzylic fluorides remained intact. Based on cyclic voltammetry studies, and in contrast to Senboku's anionic electrocarboxylation mechanism,40 it was proposed that the initially formed radical anion is involved in the C–C bond formation prior to C–F bond cleavage (Fig. 7, bottom).22,33–36 Single electron reduction of the resulting C-centered radical followed by fluoride displacement via an E1cB mechanism would thereafter furnish the product as carboxylate. Dissolution of the nickel anode was proposed to serve as the oxidative counter reaction in the process. For synthetic ease, the carboxylate products were converted to the corresponding methyl esters as the final step to facilitate isolation and analysis.
:1 Z:E selectivity (Fig. 7, top). The protocol displayed tolerance towards functional groups such as carboxylic esters and amides, sulfones, nitriles, and alkynes and allowed for selective dehalogenation of the olefinic fluorides, whereas both aromatic and benzylic fluorides remained intact. Based on cyclic voltammetry studies, and in contrast to Senboku's anionic electrocarboxylation mechanism,40 it was proposed that the initially formed radical anion is involved in the C–C bond formation prior to C–F bond cleavage (Fig. 7, bottom).22,33–36 Single electron reduction of the resulting C-centered radical followed by fluoride displacement via an E1cB mechanism would thereafter furnish the product as carboxylate. Dissolution of the nickel anode was proposed to serve as the oxidative counter reaction in the process. For synthetic ease, the carboxylate products were converted to the corresponding methyl esters as the final step to facilitate isolation and analysis.
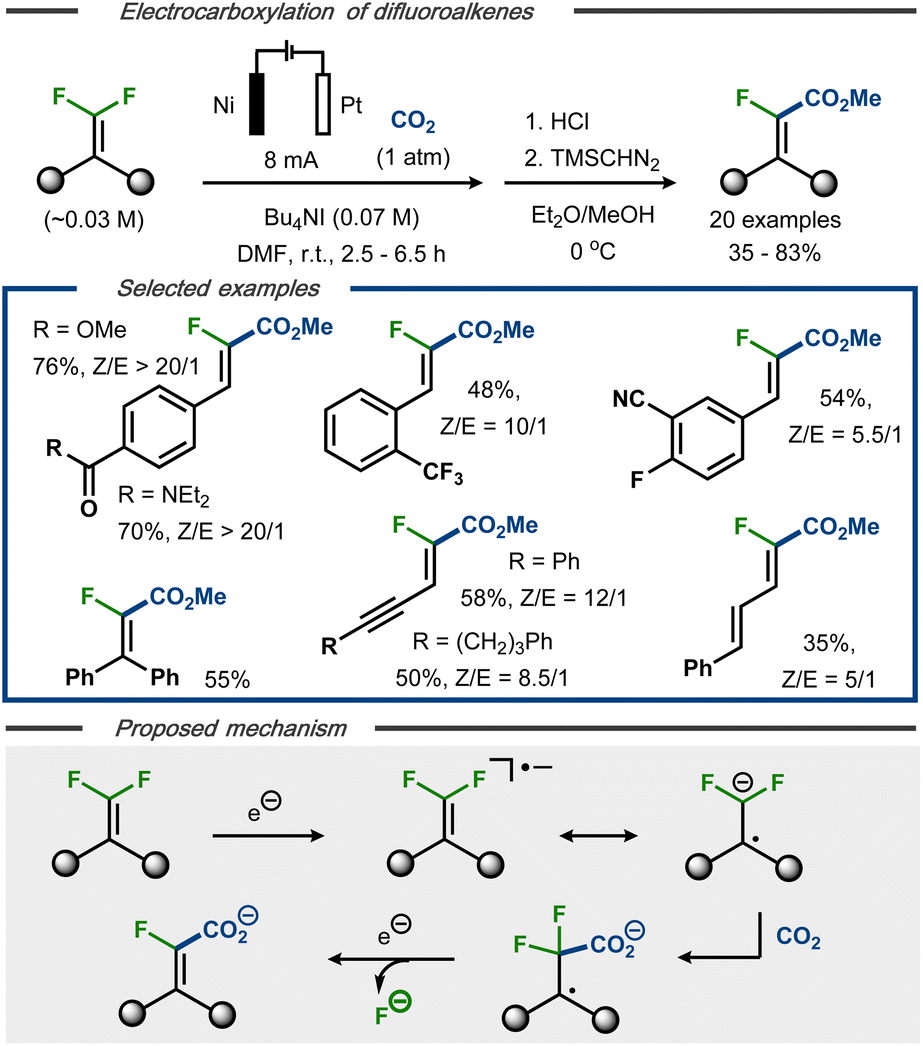 | ||
| Fig. 7 Top: Defluorination/carboxylation of gem-difluoroalkenes.41 Bottom: Proposed mechanism. | ||
Xu et al. reported on electrocatalytic hydrodefluorination protocols for fluorinated aromatic compounds.42,43 In a divided cell equipped with either a Rh– or a Rh–Pd alloy-modified Ni foam cathode with phosphate buffer (pH ∼3) and a graphite anode at constant current electrolysis, hydrodefluorinated products were formed in high yields. In some cases, the aromatic hydrodefluorinated products underwent further electroreduction to afford saturated counterparts. For example, 4-fluorophenol was transformed into phenol (∼5%), cyclohexanone (∼25%) and cyclohexanol (∼70%) as end products using the Rh–Pd alloy-modified Ni cathode (Fig. 8).42 Such reduction of the aromatic ring had previously been reported by Trnková and co-workers for fluorobenzene using a polycrystalline Pt cathode in aqueous H2SO4.44 Mechanistic studies and control experiments indicated that the hydrodefluorination on the modified Ni foam was likely to follow an indirect hydrogenation mechanism with in situ formed metal hydrides on the cathode surface, similar to hydrogenations using late transition metal catalysts and hydrogen gas.45
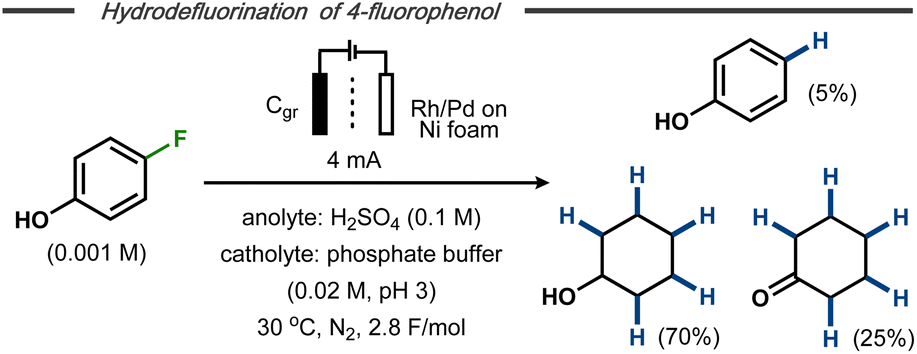 | ||
| Fig. 8 Hydrodefluorination/hydrogenation of fluorophenol on transition metal modified cathode.43 | ||
Lambert and Huang recently developed an electrophotocatalytic SNAr reaction of non-activated aryl fluorides and nucleophiles to forge C–N and C–O bonds.46 Using 10 mol% 2,3-dichloro-5,6-dicyanoquinone (DDQ) as photocatalyst under blue light irradiation at constant potential, 40 examples of defluorinated coupling products were formed in moderate to high yields (Fig. 9, top). In contrast to fully electrochemical protocols, this photoelectrocatalytic protocol was claimed to undergo an oxidative pathway to afford the C–F bond cleavage (Fig. 9, bottom). Mechanistically, photoexcitation of DDQ results in an excited catalyst species able of oxidizing the fluoroarene. Nucleophilic attack on the resulting radical cation intermediate furnishes a radical that, in turn, undergoes a single-electron reduction to the corresponding anion. Re-aromatization with expulsion of the fluoride results in the SNAr product. Due to the poor reducing power of the radical anion of DDQ, this species was deemed insufficient to mediate the reduction of the aromatic radical intermediate and, hence, that this step as well as the re-oxidization of the photocatalyst are accomplished electrochemically.
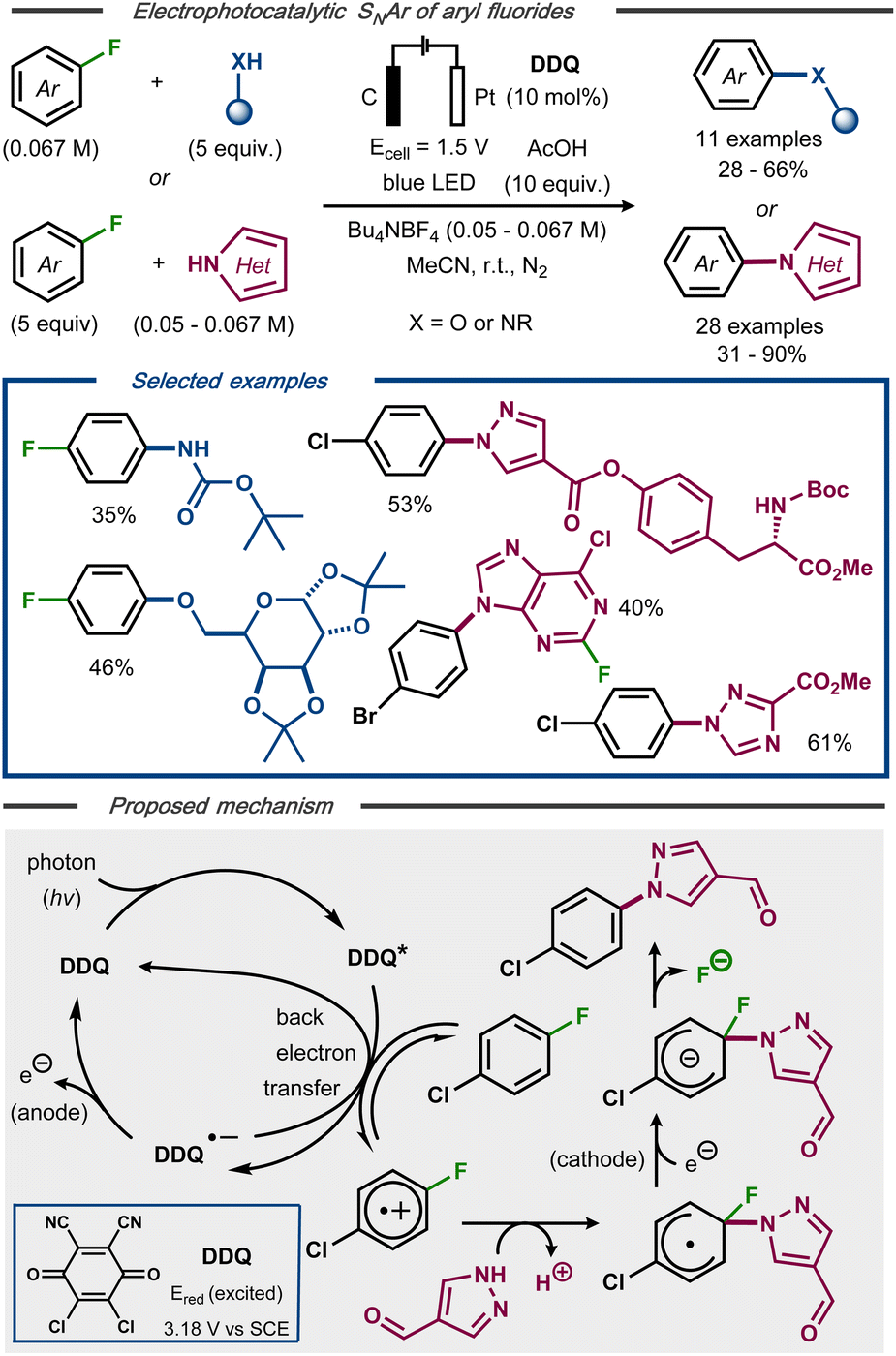 | ||
| Fig. 9 Top: Electrophotocatalytic nucleophilic aromatic substitution of aryl fluorides.46 Bottom: Proposed mechanism. | ||
3. Cleavage of C(sp3)–F bonds
C(sp3)–F bonds have lower bond dissociation energies (BDE) compared to their C(sp2)–F counterparts, with C–F bonds in π-activated positions being weaker compared to fully aliphatic counterparts (Fig. 1). The latter property is reflected in the number of synthetic protocols available for cleavage of the different aliphatic fluorides using any defluorination method, including electrosynthesis. The BDE of C(sp3)–F bonds increase with the degree of fluorination of the carbon. For example, the BDE has been determined to increase from 99 kcal mol−1 to ∼115 kcal mol−1 for the C–F bond when going from α-fluorotoluene to α,α,α-trifluorotoluene.47 As a result, selective partial defluorination of polyfluorinated sites is challenging since the C–F bonds in the products of the sequential defluorination are more easily cleaved compared to those in the starting material.3.1. Benzylic and allylic C(sp3)–F bonds
An early electrochemical reduction of a benzylic CF3-group was reported by Lund et al. with polarography studies as well as preparative electrolysis with a dropping mercury cathode.48 The potentiostatic hydrodefluorination of 1,1-dioxo-6-(trifluoromethyl)-3,4-dihydro-2H-1λ6,2,4-benzothiadiazine-7-sulfonamide was performed in 30% aqueous methanol and resulted in 72% yield of the desired product after 6.3 F of charge at a potential of −1.70 V vs. SCE (Fig. 10).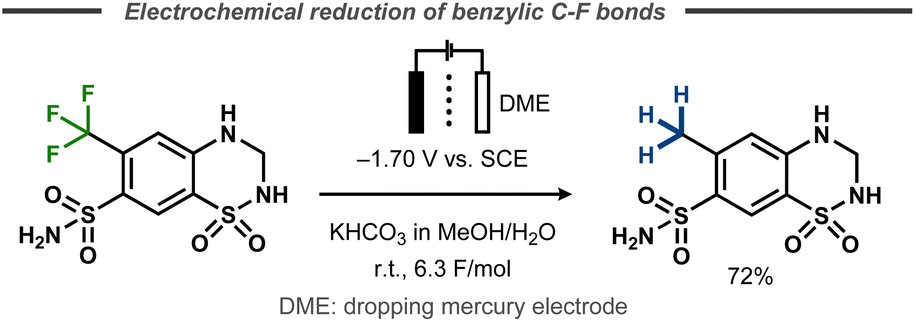 | ||
| Fig. 10 Defluorination of benzylic fluorides.48 | ||
In contrast to the stepwise reduction of other trihalomethyl compounds,48 Lund et al. proposed that the cleavage of the three C–F bonds takes place in one single step. This claim was not supported by subsequent studies, that instead suggest that benzylic C–F bonds in p-substituted trifluoromethyl benzenes take place in a stepwise manner in polar aprotic solvents like DMF with cathode materials such as Hg, Pt and Pb.49–55 Similar to the postulated stepwise dissociative electron transfer for many aryl fluorides (Fig. 5, top), initial single electron reduction of the trifluoromethyl benzene is generally believed to form a stabilized radical anion that decomposes to a benzylic radical with release of fluoride. Reductive radical-polar crossover upon a second electron transfer results in a benzylic anion, which can be quenched by a proton (Fig. 11)50–52 or other electrophiles.56,57 In the case of 4-(trifluoromethyl)acetophenone, hydrodefluorinated pinacol products were observed by Liotier et al. along with the hydrodefluorinated alcohol in aqueous alkaline medium (pH 10).58
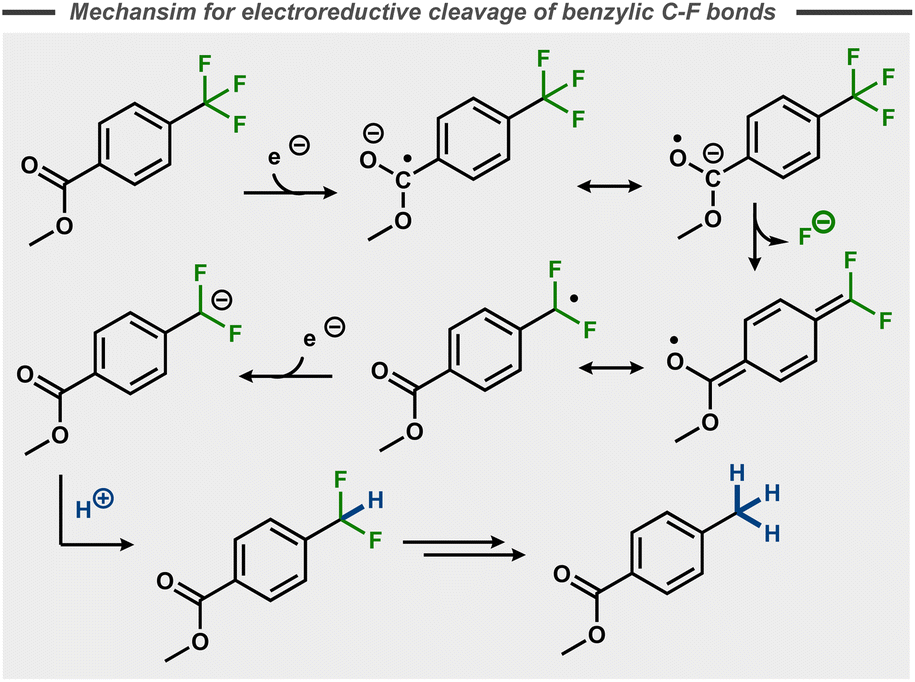 | ||
| Fig. 11 Reductive C–F bond cleavage mechanism for a trifluoromethyl arene.50 | ||
One example of benzylic defluorination was reported by Périchon and co-workers using a SmCl3-catalyzed electrochemical hydrodehalogenation protocol in an undivided cell using a nickel foam cathode and a sacrificial magnesium anode.29 In this case, α,α,α-trifluorotoluene was transformed into a mixture of α,α-difluorotoluene, α-fluorotoluene and toluene and in 50% yield (Fig. 12). Addition of D2O did not result in deuterium incorporation for a benchmark chloride substrate and it was concluded that the hydrogen in the product originates from either the tetraalkylammonium ion in the supporting electrolyte or the solvent. While cyclization experiments using 1-allyloxy-2-chlorobenze suggested that a radical mechanism was operating in the presence of samarium, no mechanistic details or discussion on the valence state of the catalyst were provided.
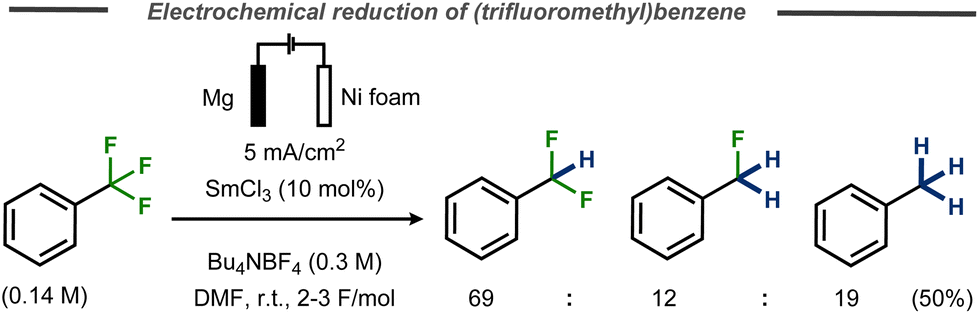 | ||
| Fig. 12 Samarium-catalyzed C–F bond cleavage in trifluoromethyl benzene.29 | ||
Périchon and co-workers demonstrated that electroreductive defluorination could be coupled with C–C bond formation using electrophiles such as CO2, DMF or acetone to form carboxylic acids, aldehydes, or tertiary alcohols.57 The electrolyses were carried out at room temperature in DMF containing the corresponding trifluoromethyl aryl compound, using a stainless steel cathode and a sacrificial anode (magnesium or aluminium rod) (Fig. 13, top). For synthesis of tertiary alcohols, acetone was used as a co-solvent (10 vol%) to furnish the products in yields up to 80% yield, whereas up to 62% yield was achieved in formylation reactions with DMF. In these cases, the crude reactions were poured into acetic anhydride and hydrolyzed to furnish diacetate products. For electrocarboxylation, carbon dioxide was bubbled through the solution at atmospheric pressure to provide benzoic acid products in up to 70% yield. The same strategy was used by Senboku and co-workers to trap benzylic anionic intermediates bearing benzylic pentafluoroethyl groups in the synthesis of tetrafluorinated analogues of the NSAIDs fenoprofen and ketoprofen (Fig. 13, bottom).59 In this case, the electrochemical reduction was conducted in DMF using a Pt cathode and a Mg anode in an undivided cell with CO2 bubbled through the reaction medium.
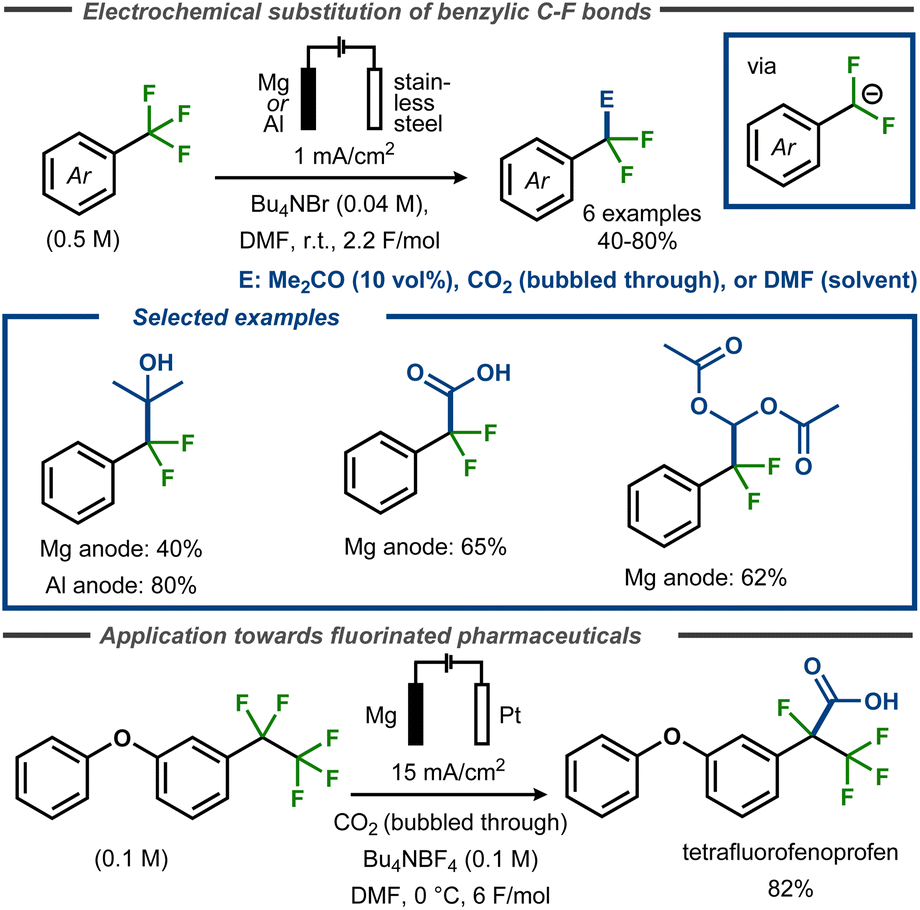 | ||
| Fig. 13 Top: Defluorinative functionalization of benzyl fluorides.57 Bottom: Defluorinative electrocarboxylation for polyfluorinated pharmaceutical analogues.59 | ||
Benzylic C–F bond cleavage with trapping of the intermediate anion has also been achieved with TMSCl as electrophile, forming trimethylsilyldifluoromethylbenzenes.55,56 Marzouk et al. demonstrated that fluorides in α,α,α-trifluorotoluene substituents can subsequently be replaced by TMS groups in a THF/DMPU mixture, thus avoiding carcinogenic HMPA, by simply increasing the equivalents of charge applied.56 As such, the synthesis of corresponding mono-, bis- or tris-trimethylsilyl derivatives using aluminium as a sacrificial anode was enabled in yields up to 70%, 60% and 50% respectively (Fig. 14). This reaction was scaled up to 1 mol of starting material, using a tubular flow cell.
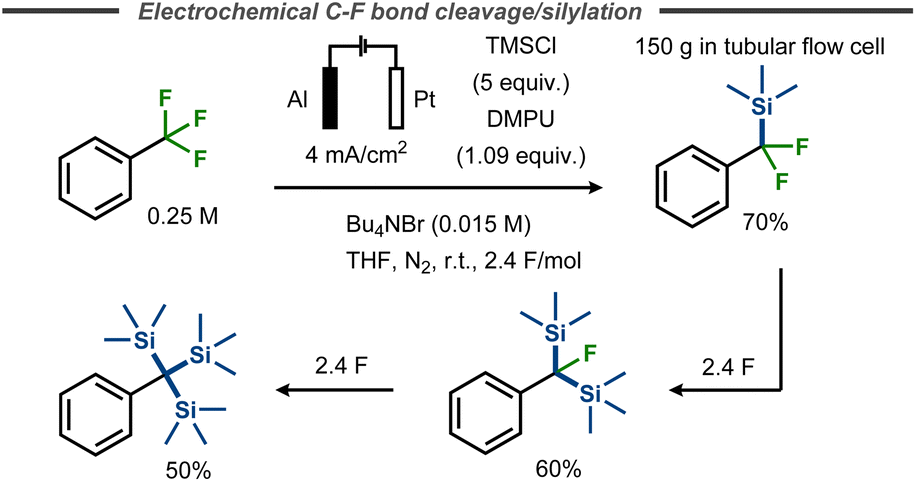 | ||
| Fig. 14 Defluorinative silylation of benzyl fluorides with TMSCl.56 | ||
Recently, a facile electroreductive system for the removal of various functional groups was developed by Xia and co-workers, using triethylamine (Et3N) as sacrificial reductant in combination with inert platinum electrodes in an undivided cell at constant current.60 Amongst examples such as reduction of cyano groups and azoles, the authors reported complete reduction of benzylic CF3 – groups in yields up to 89% (Fig. 15). Similarly, Huang and co-workers provided one example of C–F bond cleavage in trifluoromethylbenzene in a moderate yield of 35% at low conversion using platinum electrodes in diglyme and Bu4NBF4 (0.2 M) under air at room temperature with NaBH4 as additive.61
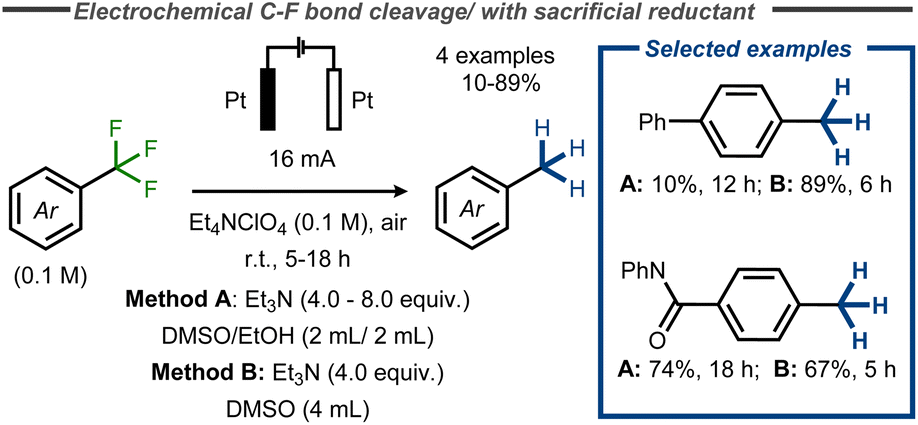 | ||
| Fig. 15 Electroreductive C–F bond cleavage in benzyl fluorides with Et3N as sacrificial reductant.60 | ||
3.2. C–F bond cleavage in α-CF3 alkenes
Recently, Cheng and co-workers developed a defluorinative method to convert α,α,α-trifluoromethyl cinnamates to gem-difluorostyrenes in moderate to good yields, using an electroreductive approach (Fig. 16).62 The reactions were performed in acetonitrile with controlled potential, using graphite felt electrodes under an ammonia atmosphere that acted as proton donor. The method was demonstrated to proceed well with electron-rich substrates e.g. methoxy groups (89%) or large π-systems (90%), whereas moderate yields were observed for substrates with functional groups such as chloride (52%). Selective defluorination was observed for substrates with fluoride and trifluoromethyl substituents, furnishing the difluoroalkene products in moderate yields. The mechanism was investigated using cyclic voltammetry and square wave voltammetry, indicating a net two-electron transfer. The first electron transfer was proposed to convert the substrate to the corresponding radical anion that, upon protonation, forms the corresponding radical. A second electron transfer furnishes the benzylic anion, which undergoes an E1cB elimination of fluoride to form the desired product (Fig. 16, bottom).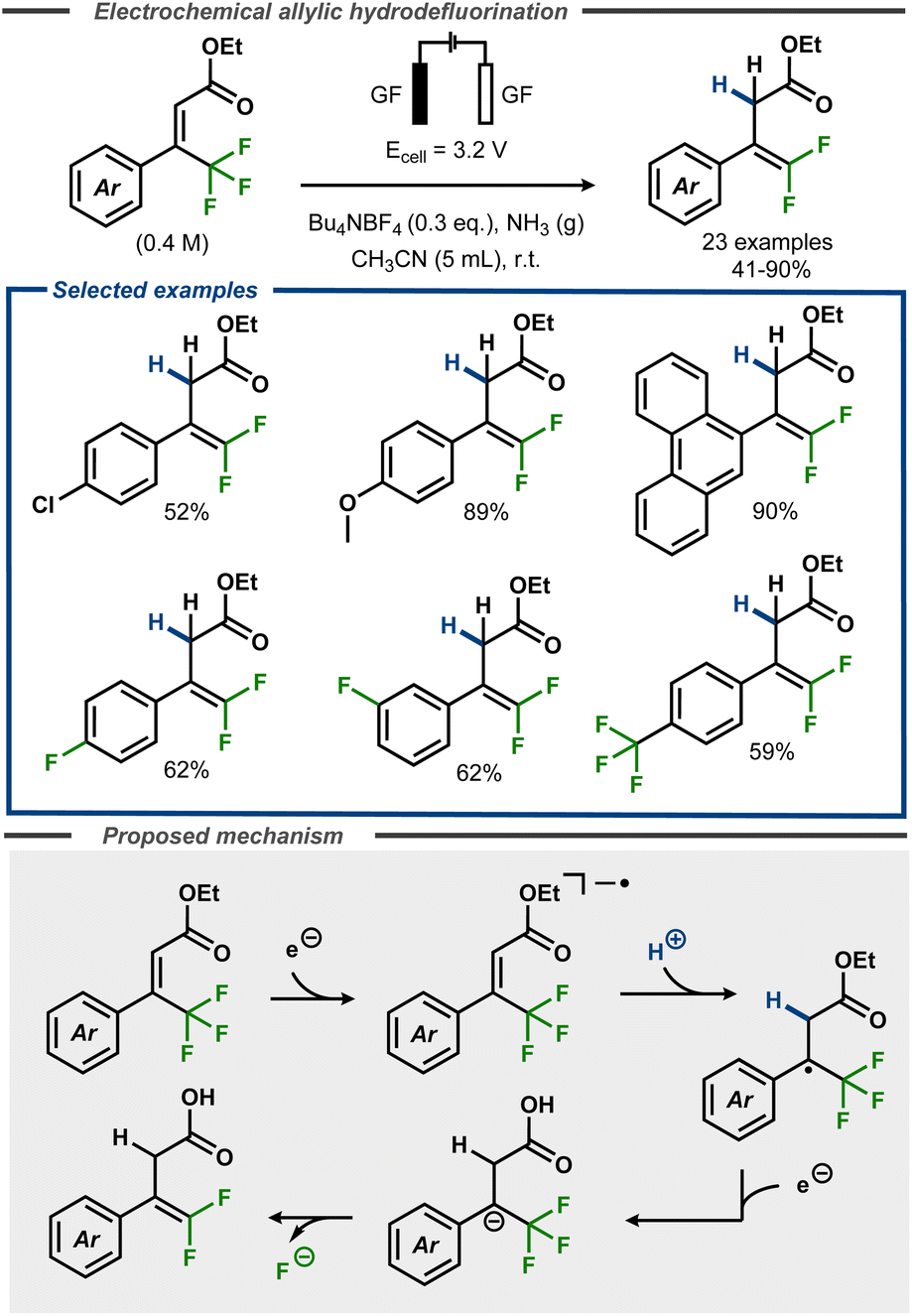 | ||
| Fig. 16 Electroreductive formation of gem-difluorostyrenes from α,α,α-trifluoromethyl cinnamates.62 | ||
Along the same lines, Gao et al. recently developed a regioselective electrochemical γ-carboxylation of α-CF3 alkenes using CO2 under constant current conditions.63 The transformation was carried out in DMF with Bu4NClO4 as supporting electrolyte, using platinum electrodes (Fig. 17). The method proceeded with a good functional group tolerance, with yields up to 84%, including examples such as sugars and steroids. Supported by cyclic voltammetry studies, radical trap experiments, as well as DFT calculations, the reaction mechanism was believed to be of a similar type of that proposed by Cheng and co-workers (Fig. 16).62 After an initial reduction of the trifluoromethyl alkene to the corresponding radical anion, addition of CO2 would take place. The resulting radical carboxylate would thereafter undergo a second electron transfer to the corresponding enolate that decomposes to the gem-difluoroalkene product with release of fluoride (Fig. 17, bottom). The anodic counter reaction was proposed to be oxidation of either DMF or water.
 | ||
| Fig. 17 Electroreductive formation of carboxylated gem-difluorostyrenes from α-trifluoromethyl styrenes.63 | ||
Electroreductive C–F bond cleavage and concomitant functionalization of α-trifluoromethyl styrenes to gem-difluoroalkenes have recently been reported using radical precursors such as alkyl halides, redox active esters and Katritzky salts, as well as aryl halides (Fig. 18).64–68 Ni, Guo and Wang and co-workers demonstrated that Katritzky salts could be successfully used as coupling partners in DMSO under galvanostatic conditions with a sacrificial Zn anode and a Ni foam cathode, furnishing products in up to 85% yield (Fig. 18, Method A).64 Masson, Claraz and co-workers demonstrated coupling of Katritzky salts as well as NHP esters in yields up to 94%, including amino acid derived alkyl NHP esters (Fig. 18, Method B).65 Similarly, Xia, Guo and co-workers demonstrated that α-trifluoromethyl styrene substrates react with NHP esters and Katritzky salts, as well as alkyl halides, in yields up to 76% (Fig. 18, Method C).66 In the cases where a Fe sacrificial anode was not used, amine bases such as triethylamine or DABCO were employed as sacrificial reductants, similar to what was previously reported for electroreduction of trifluoromethyl arenes.60 Qi, Huang and Lei and co-workers reported yields up to 99% with alkyl halides as coupling partners, using a direct electrolysis method for alkyl iodides (Fig. 18, Method D) or an indirect electrolysis method with a Ni(bpy)-complex in catalytic amounts for alkyl bromides.67 In addition, the former method was also demonstrated using one Katritzky salt and one NHP ester as coupling partner to furnish the desired product in moderate to good yield, whereas the latter method resulted in moderate yield using an alkyl chloride as partner. Oxidation of the phosphine or amine additives were postulated as the oxidative counter reactions for the process. A fully catalytic system was reported by Ni and Wang and co-workers, who demonstrated that alkyl and aryl halides react with α-trifluoromethyl styrene substrates using a Ni catalyst with a terpyridine ligand, furnishing products in yields up to 93% (Fig. 18, Method E).68
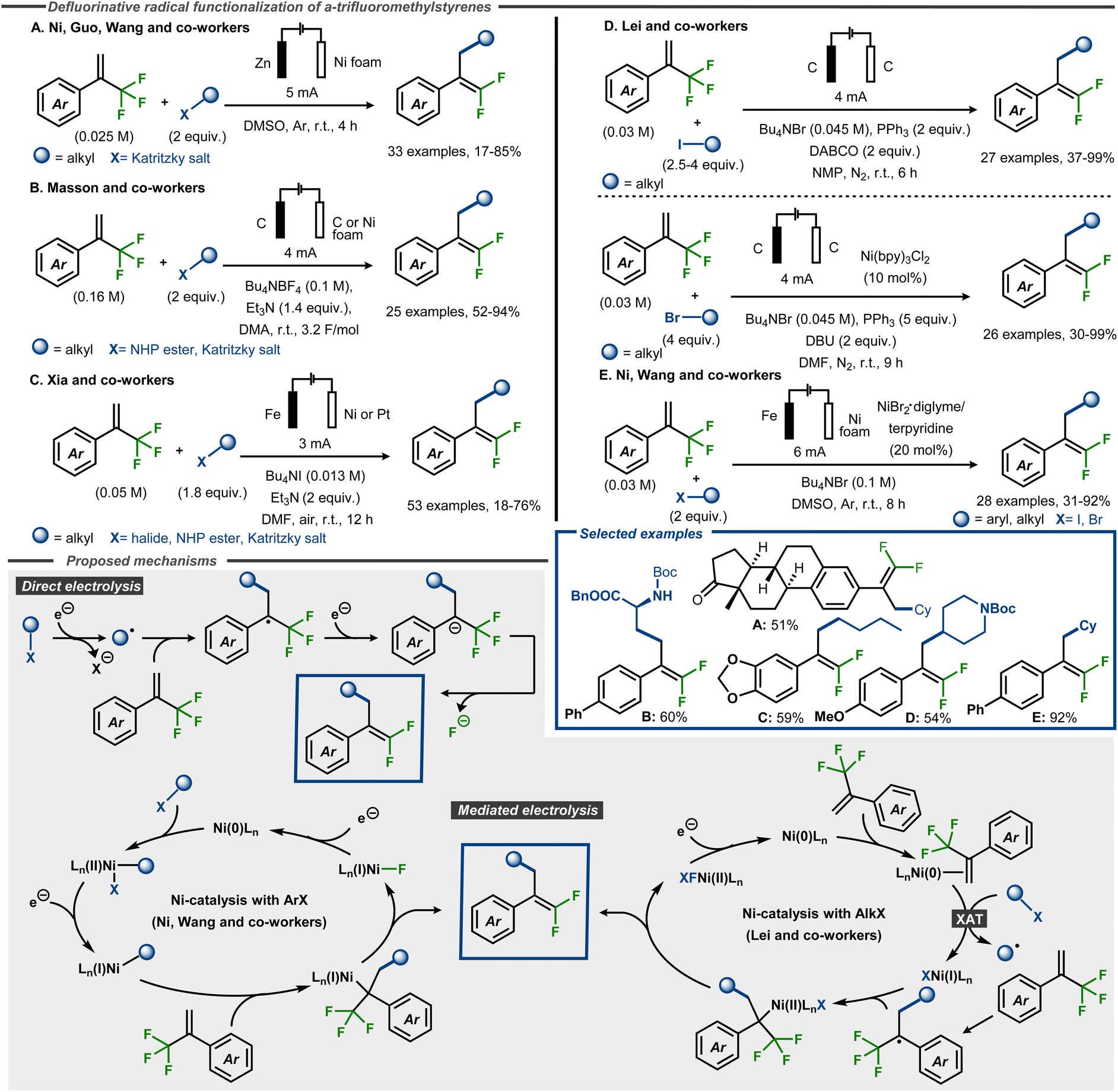 | ||
| Fig. 18 Electroreductive formation of γ-functionalized gem-difluorostyrenes from α-trifluoromethyl styrenes and radical precursors.64–68 | ||
Mechanistically, the direct electrolytic methods are thought to be initiated by cathodic dissociative electron transfer to the radical precursor to provide an aliphatic carbon-centered radical intermediate. This radical would thereafter attack the α-trifluoromethyl alkene to form a C–C bond and a benzylic stabilized α-CF3 carbon radical. Cathodic reduction of this radical furnishes α-CF3 carbanion that undergoes a β-fluoride elimination to afford the gem-difluoromethyl alkene product (Fig. 18). For the Ni-catalyzed protocols, the Lei group and Ni and Wang and co-workers propose different mechanisms. The former group utilizes alkyl bromides as coupling partners and suggest, supported by DFT calculations, that the Ni(0) catalyst initially coordinates the alkene to thereafter interact with the alkyl bromide in an XAT event to form an alkyl radical and a Ni(I)–Br complex. This open-shell species adds to the CF3-alkene substrate analogous to the indirect mechanism, whereas the Ni(I) species traps the resulting benzylic radical to form a Ni(II)-complex. After a two-electron reduction and β-fluoride elimination, the product is released and the Ni(0) catalyst is regenerated (Fig. 18, bottom). In contrast, Ni and Wang and co-workers do not speculate about the mechanism for alkyl halides but focuses on aryl halides as coupling partners. In this case, the Ni(0) catalyst is proposed to undergo an oxidative addition with the aryl halide, thereby forming a Ni(II)ArX complex. This species undergoes a single electron reduction at the cathode to furnish the corresponding Ni(I)Ar complex. Migratory insertion of the trifluoromethyl styrene derivative generates a Ni(I)R species that is expected to undergo rapid β-fluoride elimination to deliver the desired product. Another cathodic reduction would regenerate the Ni(0) species from Ni(I)F to close the catalytic cycle (Fig. 18, bottom).
3.3. Cleavage of C(sp3)–F bonds in α-position to carbonyls
The first mechanistic insights into cleavage of C–F bonds in α-position to carbonyl carbons were reported by Elving and Leone in 1956.69 In this case, polarographic studies of the reduction of 2-fluoroacetophenone indicated two cathodic waves that were interpreted as initial cleavage of the C–F bond, and subsequent reduction of the resulting acetophenone to the corresponding alcohol. Stocker and Jenevein reported that initial defluorination of α,α,α-trifluoroacetophenone to acetophenone was followed by pinacolization (Fig. 19, top) with the selectivity being dependent on applied potential and reaction time.70 However, later cyclic voltammetry studies by Yang et al. suggested that the fluorinated pinacol forms first and is subsequently reduced (Fig. 19, bottom).71 Mechanistically, it was suggested that the pinacol formation occurs via radical–radical coupling between one protonated and one unprotonated electrochemically formed ketyl radical, whereas certain substrates were proposed to undergo a second electron transfer and two protonations to form the corresponding alcohol.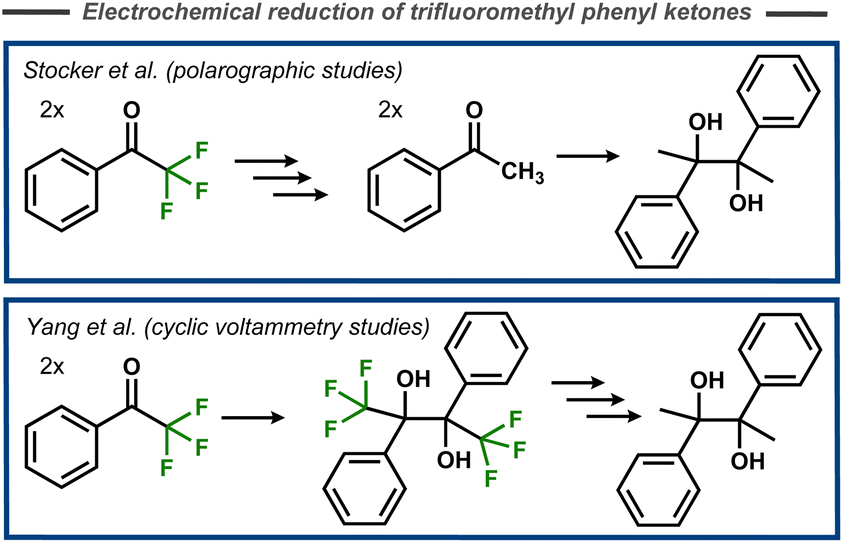 | ||
| Fig. 19 Electroreductive pinacolization of α,α,α-trifluoroacetophenone.70,71 | ||
Evans and Kopilov reported that defluorination of α-fluoroacetanilide took place during their attempts to electrochemically remove the electron-poor amide protecting group.72 Instead of furnishing the free aniline, potentiostatic electrolysis of α,α,α-trifluoroacetanilide in a divided cell with a mercury pool cathode resulted in a mixture of 65% α-fluoroacetanilide and 35% acetanilide (Fig. 20).
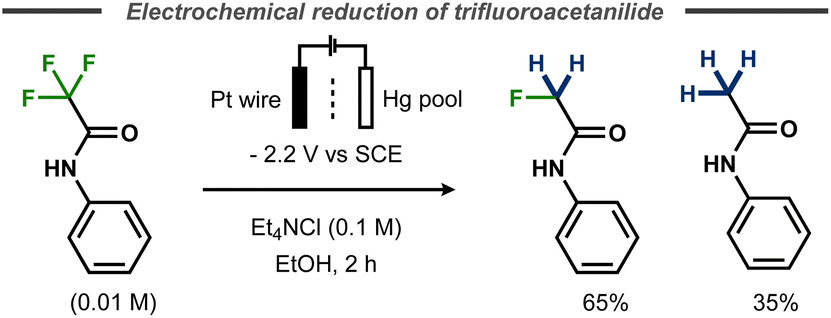 | ||
| Fig. 20 Electrochemical defluorination of α,α,α-trifluoroacetanilide.72 | ||
Uneyama et al. demonstrated successful trapping of the intermediate after the initial hydrodefluorination of trifluoromethyl ketones as well as trifluoroketimines with silyl chloride reagents, isolating difluoro silyl enol ethers and difluoro silyl enamines in yields up to 85% and 78%, respectively (Fig. 21).73,74 In the former case, a Pb cathode and carbon anode was utilized to achieve electroreductive defluorination of a range of trifluoromethyl ketones in anhydrous acetonitrile using an H-type divided cell (Fig. 21, top). The reaction was reported to proceed efficiently with aromatic substrates, resulting in yields up to 85%, whereas aliphatic substrates furnished the corresponding products in yields up to 55%. Similar conditions were used for the electroreductive defluorination of ketimines (Fig. 21, bottom). The authors did not provide any mechanistic details for the transformations.
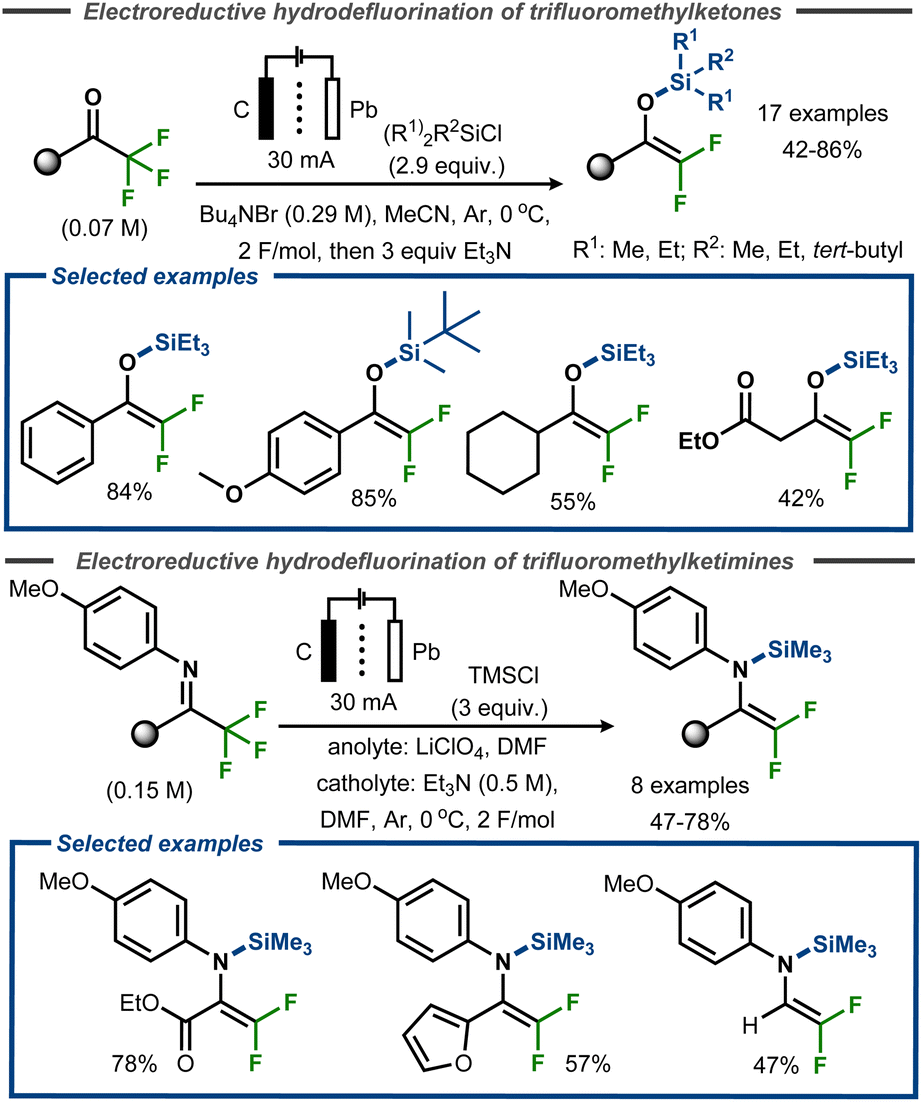 | ||
| Fig. 21 Electroreductive defluorinative silylation of trifluoromethyl ketones.73 | ||
The methodology was expanded to trifluoroacetic acid derivatives to yield 2,2-difluoro-2-trimethylsilylacetate and difluoroketene silyl (O,O-, O,S-, and O,N-) acetals.75 These findings stood in contrast to those of Stepanov et al. who reported Claisen adducts under similar conditions.76 After a thermal isomerization with silyl migration from oxygen to carbon, the corresponding products were successfully coupled with a range of electrophiles (aldehydes, ketones, imines, acyl- and alkylhalides) to give α-alkylated-α,α-difluoroacetates in good to excellent yields (Fig. 22). This isomerization could either be carried out in a stepwise procedure with electrolysis carried out at low reaction temperature, or direct by electrolysis at 50 °C.
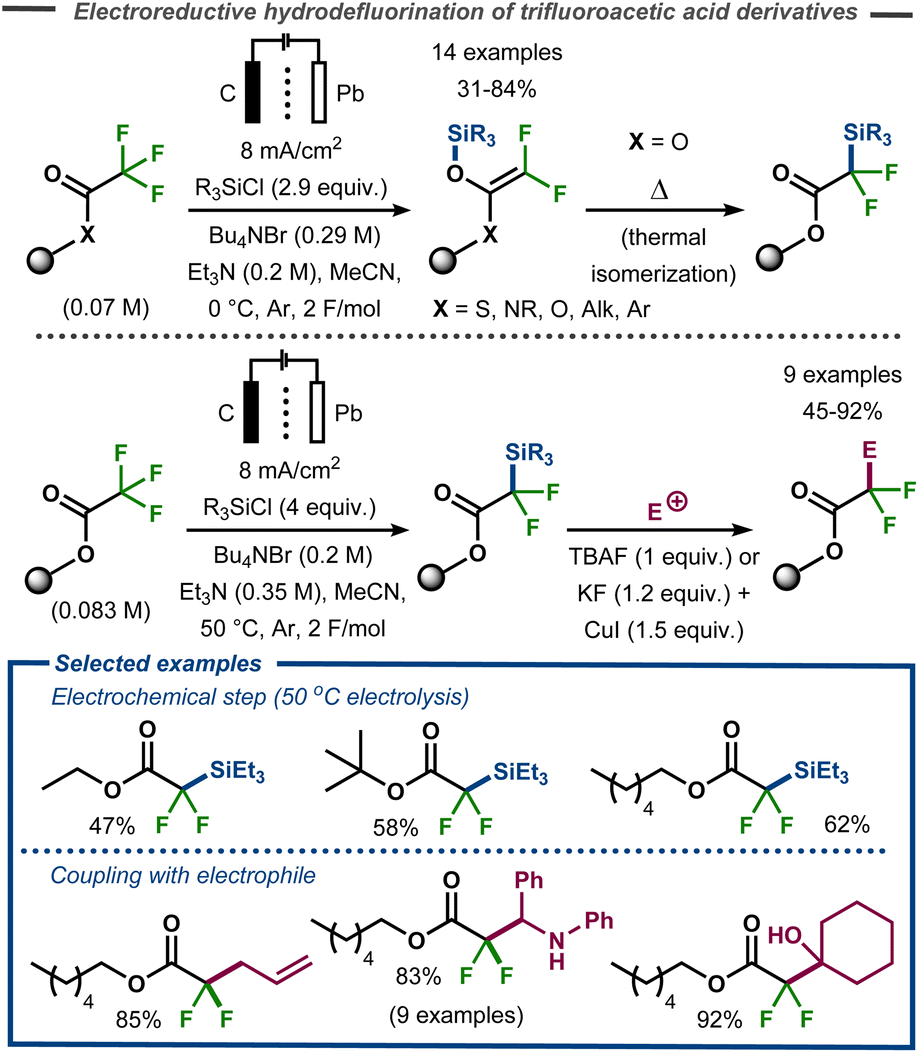 | ||
| Fig. 22 Electrochemical defluorinative C–Si bond formation and subsequent functionalization.75 | ||
Recently, Lennox et al. demonstrated selective monodefluorination of α,α,α-trifluoromethylketones in a single step to access a broad scope of difluoromethylketones (Fig. 23).77 Using a divided cell setup, the reaction was reported to proceed efficiently on platinum or nickel cathodes with TMSCl present as a radical anion trapping agent via a proposed silyl enol ether intermediate of Uneyama-type.73 The transformation tolerated heterocyclic groups such as indoles as well cyclopropanes or even aryl fluorides incorporated in the substrates. The reaction performs best with electron-rich and simple alkyl substrates (up to 95%), whereas unprotected indoles as well as protected indoles bearing cyano groups resulted in lower yields (45 and 42%, respectively). Similar to other electrochemical hydrodefluorination protocols, tetraalkylammonium salts were found to act as reductively stable, masked proton sources upon Hoffmann elimination. Supported by DFT calculations, the transformation was proposed to proceed via cathodic reduction of the substrate to furnish a transient ketyl radical anion that is trapped with TMSCl to furnish an O-TMS protected benzylic radical. A second electron transfer furnishes the corresponding anion that undergoes loss of fluoride to form the O-TMS protected gem-difluoroalkene that, in turn, furnishes the hydrodefluorinated product upon protonation. Voltammetric studies suggested that oxidation of bromide to tribromide was the anodic counter reaction.
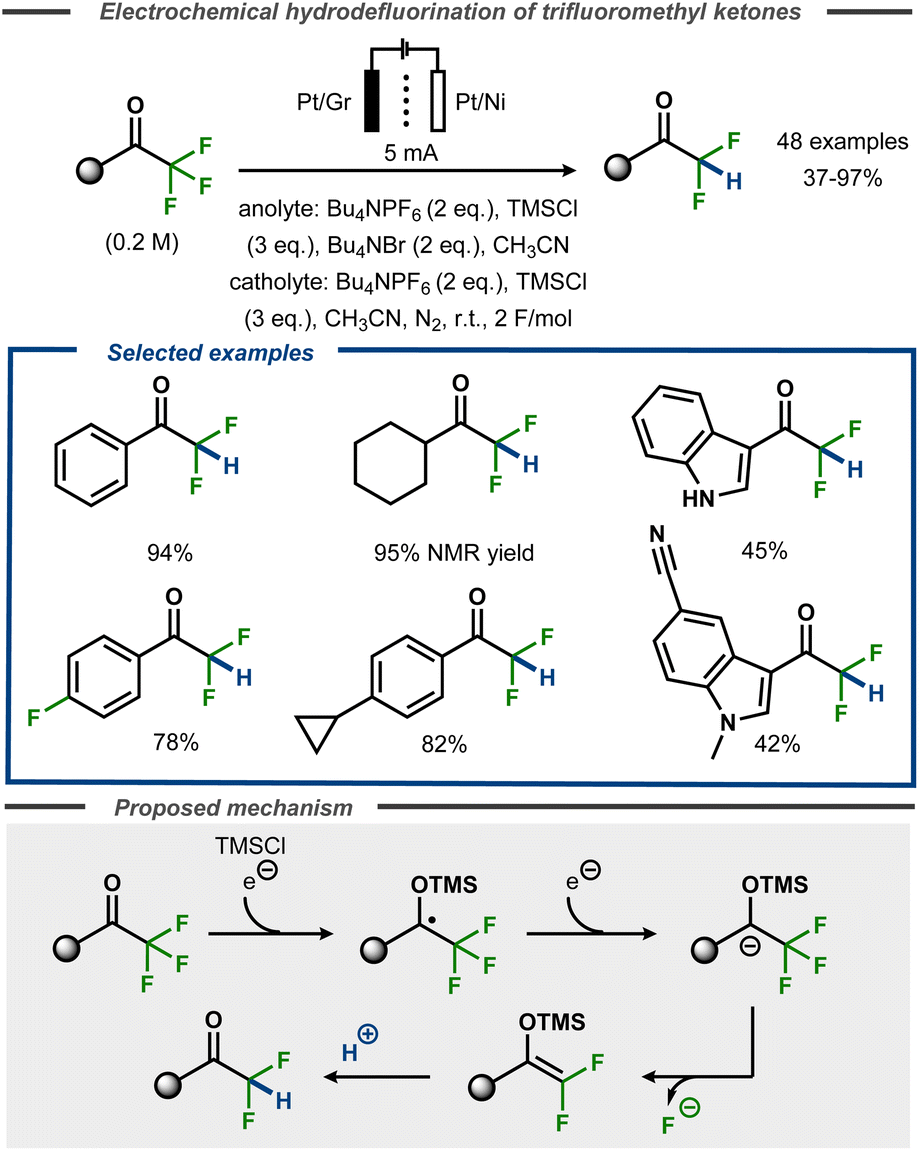 | ||
| Fig. 23 Selective monoreduction of α,α,α-trifluoromethyl ketones.77 | ||
4. Conclusions
In contrast to their heavier congeners, fluorocarbons are challenging from a reactivity perspective for any synthetic strategy. This challenge is present also using an electrochemical approach, as evident from the limited number of synthetically relevant examples of C–F bond cleavage for both sp2- and sp3-hybridized carbon centers. With only a few exceptions, available electrosynthetic protocols are only amenable for cleaving C–F bonds in close proximity to π-systems via similar reductive mechanisms. While a limited number of chemical protocols are at hand for C(sp3)–F bond activation in fully aliphatic positions, electrochemical variants for selective activation/functionalization are yet to be developed. In depth mechanistic understanding, alternative activation modes and mediated (photo)electrolysis are likely keys to enable new fluoroselective synthetic methodology that can open new reactivity space and retrosynthetic disconnections, as well as facilitate efficient remediation of fluorinated organic pollutants.19,20,78Author contributions
All authors contributed to the writing of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Swedish Research Council (grant 2021-05551), Formas (grant 2021-00678), the Swedish Foundation for Strategic Research (grant FFL21-0005), Stiftelsen Olle Engkvist Byggmästare, Magnus Bergvalls stiftelse, Stiftelsen Lars Hiertas Minne, and KTH Royal Institute of Technology is gratefully acknowledged.References
- (a) M. Shimizu and T. Hiyama, Angew. Chem., Int. Ed., 2005, 44, 214–231 CrossRef CAS PubMed; (b) M. Schlosser, Angew. Chem., Int. Ed., 2006, 45, 5432–5446 CrossRef CAS PubMed; (c) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (d) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed; (e) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed; (f) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467–101467 CrossRef CAS PubMed; (g) P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed; (h) R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC; (i) R. Ragni, A. Punzi, F. Babudri and G. M. Farinola, Eur. J. Org. Chem., 2018, 3500–3519 CrossRef CAS.
- L. Pauling, The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, Cornell University Press, Ithaca, NY, 1939 Search PubMed.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- C. Cao, Sci. China, Ser. B: Chem., 2009, 52, 943–951 CrossRef CAS.
- T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578–1587 CrossRef CAS.
- D. A. Everson and D. J. Weix, J. Org. Chem., 2014, 79, 4793–4798 CrossRef CAS PubMed.
- F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed.
- X. Dong, J. L. Röckl, S. R. Waldvogel and B. Morandi, Science, 2021, 371, 507–514 CrossRef CAS PubMed.
- F. Zhao, W. Zhou and Z. Zuo, Adv. Synth. Catal., 2022, 364, 234–267 CrossRef CAS.
- T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390–402 CrossRef CAS PubMed.
- (a) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (b) H. Torrens, Coord. Chem. Rev., 2005, 249, 1957–1985 CrossRef CAS; (c) M. K. Whittlesey and E. Peris, ACS Catal., 2014, 4, 3152–3159 CrossRef CAS; (d) H. Kameo, H. Yamamoto, K. Ikeda, T. Isasa, S. Sakaki, H. Matsuzaka, Y. García-Rodeja, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2020, 142, 14039–14044 CrossRef CAS PubMed.
- (a) Q. Shen, Y.-G. Huang, C. Liu, J.-C. Xiao, Q.-Y. Chen and Y. Guo, J. Fluor. Chem., 2015, 179, 14–22 CrossRef CAS; (b) H.-J. Ai, X. Ma, Q. Song and X.-F. Wu, Sci. China: Chem., 2021, 64, 1630–1659 CrossRef CAS; (c) K. Balaraman and C. Wolf, Sci. Adv., 2022, 8, eabn7819 CrossRef CAS PubMed.
- (a) W. D. Jones, Dalton Trans., 2003, 3991–3995 RSC; (b) A. G. M. Barrett, M. R. Crimmin, M. S. Hill, P. B. Hitchcock and P. A. Procopiou, Angew. Chem., Int. Ed., 2007, 46, 6339–6342 CrossRef CAS PubMed; (c) M. Klahn and U. Rosenthal, Organometallics, 2012, 31, 1235–1244 CrossRef CAS; (d) T. L. Gianetti, R. G. Bergman and J. Arnold, Chem. Sci., 2014, 5, 2517–2524 RSC; (e) A. M. Träff, M. Janjetovic, L. Ta and G. Hilmersson, Angew. Chem., Int. Ed., 2013, 52, 12073–12076 CrossRef PubMed; (f) W. Huang and P. L. Diaconescu, Organometallics, 2017, 36, 89–96 CrossRef CAS.
- (a) H. Amii, T. Kobayashi, Y. Hatamoto and K. Uneyama, Chem. Commun., 1999, 1323–1324 RSC; (b) K. Matsubara, T. Ishibashi and Y. Koga, Org. Lett., 2009, 11, 1765–1768 CrossRef CAS PubMed; (c) C. J. Burns and R. A. Anderson, J. Chem. Soc., Chem. Commun., 1989, 136–137 RSC; (d) G. B. Deacon and P. I. MacKinnon, Tetrahedron Lett., 1984, 25, 783–784 CrossRef CAS; (e) P. L. Watson, T. H. Tulip and I. Williams, Organometallics, 1990, 9, 1999–2009 CrossRef CAS; (f) M. Janjetovic, A. M. Träff, T. Ankner, J. Wettergren and G. Hilmersson, Chem. Commun., 2013, 49, 1826–1828 RSC.
- (a) S. Li and W. Shu, Chem. Commun., 2022, 58, 1066–1077 RSC; (b) G. Yan, K. Qiu and M. Guo, Org. Chem. Front., 2021, 8, 3915–3942 RSC; (c) J. Wang, H. Gao, C. Shi, G. Chen, X. Tan, X. Chen, L. Xu, X. Cai, B. Huang and H. Li, Chem. Commun., 2021, 57, 12203–12217 RSC; (d) G. Yan, Chem. – Eur. J., 2022, 28, e202200231 CAS; (e) Z. Wang, Y. Sun, L.-Y. Shen, W.-C. Yang, F. Meng and P. Li, Org. Chem. Front., 2022, 9, 853–873 RSC.
- (a) E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS PubMed; (b) B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC; (c) A. Wiebe, T. Gieshoff, S. Mohle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed; (d) A. Shatskiy, H. Lundberg and M. D. Kärkäs, ChemElectroChem, 2019, 6, 4067–4092 CrossRef CAS; (e) C. Margarita and H. Lundberg, Catalysts, 2020, 10, 982–1006 CrossRef CAS.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (b) R. D. Little and K. D. Moeller, Chem. Rev., 2018, 118, 4483–4484 CrossRef CAS PubMed; (c) C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404–6422 RSC; (d) R. Francke and R. D. Little, ChemElectroChem, 2019, 6, 4373–4382 CrossRef CAS; (e) R. D. Little, J. Org. Chem., 2020, 85, 13375–13390 CrossRef CAS PubMed; (f) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed; (g) E. C. R. McKenzie, S. Hosseini, A. G. C. Petro, K. K. Rudman, B. H. R. Gerroll, M. S. Mubarak, L. A. Baker and R. D. Little, Chem. Rev., 2022, 122, 3292–3335 CrossRef CAS PubMed.
- S. Sharma, N. P. Shetti, S. Basu, M. N. Nadagouda and T. M. Aminabhavi, Chem. Eng. J., 2022, 430, 13289–132911 Search PubMed.
- E. Mousset and K. Doudrick, Curr. Opin. Electrochem., 2020, 22, 221–227 CrossRef CAS.
- S. H. Langer and S. Yurchak, J. Electrochem. Soc., 1969, 116, 1228–1229 CrossRef.
- B. H. Campbell, Anal. Chem., 1972, 44, 1659–1663 CrossRef CAS.
- C. P. Andrieux, C. Blocman and J. M. Savéant, J. Electroanal. Chem. Interfacial Electrochem., 1979, 105, 413–417 CrossRef CAS.
- A. Kakhr, Y. Mugnier and E. Laviron, Electrochim. Acta, 1983, 28, 1897–1898 CrossRef.
- E. Kariv-Miller and Z. Vajtner, J. Org. Chem., 1985, 50, 1394–1399 CrossRef CAS.
- E. Kariv-Miller and R. Andruzzi, J. Electroanal. Chem. Interfacial Electrochem., 1985, 187, 175–186 CrossRef CAS.
- D. M. Loffredo, J. E. Swartz and E. Kariv-Miller, J. Org. Chem., 1989, 54, 5953–5957 CrossRef CAS.
- A. Profumo, E. Fasani and A. Albini, Heterocycles, 1999, 51, 1499–1502 CrossRef CAS.
- H. Hebri, E. Duñach and J. Périchon, Synth. Commun., 1991, 21, 2377–2382 CrossRef CAS.
- W. B. Wu, M. L. Li and J. M. Huang, Tetrahedron Lett., 2015, 56, 1520–1523 CrossRef CAS.
- L. Pitzer, J. L. Schwarz and F. Glorius, Chem. Sci., 2019, 10, 8285–8291 RSC.
- A. Muthukrishnan and M. V. Sangaranarayanan, Chem. Phys. Lett., 2007, 446, 297–303 CrossRef CAS.
- D. B. Denney, D. Z. Denney and S. P. Fenelli, Tetrahedron, 1997, 53, 9835–9846 CrossRef CAS.
- A. Muthukrishnan and M. V. Sangaranarayanan, Electrochim. Acta, 2010, 55, 1664–1669 CrossRef CAS.
- K. J. Houser, D. E. Bartak and M. D. Hawley, J. Am. Chem. Soc., 1973, 95, 6033–6040 CrossRef CAS.
- C. P. Andrieux, A. Batlle, M. Espin, I. Gallardo, Z. Jiang and J. Marquet, Tetrahedron, 1994, 50, 6913–6920 CrossRef CAS.
- Z. Kudaş, E. Gür and D. Ekinci, Langmuir, 2018, 34, 7958–7970 CrossRef PubMed.
- H. Lund and O. Hammerich, Organic Electrochemistry, Marcel Dekker, New York, 4th edn, 2001 Search PubMed.
- C. P. Andrieux, C. Blocman, J. M. Dumas-Bouchiat and J. M. Saveant, J. Am. Chem. Soc., 1979, 101, 3431–3441 CrossRef CAS.
- H. Senboku, K. Yoneda and S. Hara, Electrochemistry, 2013, 81, 380–382 CrossRef CAS.
- S. L. Xie, X. T. Gao, H. H. Wu, F. Zhou and J. Zhou, Org. Lett., 2020, 22, 8424–8429 CrossRef CAS PubMed.
- Y. H. Xu, H. X. Ma, T. J. Ge, Y. Q. Chu and C. A. Ma, Electrochem. Commun., 2016, 66, 16–20 CrossRef CAS.
- Y. Xu, T. Ge, H. Ma, X. Ding, X. Zhang and Q. Liu, Electrochim. Acta, 2018, 270, 110–119 CrossRef CAS.
- N. Musilova-Kebrlova, P. Janderka and L. Trnkova, Collect. Czech. Chem. Commun., 2009, 74, 611–625 CrossRef CAS.
- M. K. Whittlesey and E. Peris, ACS Catal., 2014, 4, 3152–3159 CrossRef CAS.
- H. Huang and T. H. Lambert, Angew. Chem., Int. Ed., 2020, 59, 658–662 CrossRef CAS PubMed.
- H. Dang, A. M. Whittaker and G. Lalic, Chem. Sci., 2016, 7, 505–509 RSC.
- H. Lund, I. Fischmeister, E. Stenhagen, G. Andersson and H. Palmstierna, Acta Chem. Scand., 1959, 13, 192–194 CrossRef CAS.
- A. I. Cohen, B. T. Keeler, N. H. Coy and H. L. Yale, Anal. Chem., 1962, 34, 216–219 CrossRef CAS.
- J. P. Coleman, H. G. Gilde, J. H. P. Utley and B. C. L. Weedon, J. Chem. Soc., Chem. Commun., 1970, 12, 738–739 RSC.
- J. P. Coleman, Naser-ud-din, H. G. Gilde, J. H. P. Utley, B. C. L. Weedon and L. Eberson, J. Chem. Soc., Perkin Trans. 2, 1973, 14, 1903–1908 RSC.
- H. Lund, N. J. Jensen, S.-O. Almqvist, C. R. Enzell, A. Taticchi and B. Mannervik, Acta Chem. Scand., 1974, 28b, 263–265 CrossRef.
- C. Combellas, F. Kanoufi and A. Thiébault, J. Electroanal. Chem., 1996, 407, 195–202 CrossRef.
- C. P. Andrieux, C. Combellas, F. Kanoufi, J.-M. Savéant and A. Thiébault, J. Am. Chem. Soc., 1997, 119, 9527–9540 CrossRef CAS.
- P. Clavel, G. Lessene, C. Biran, M. Bordeau, N. Roques, S. Trevin and D. de Montauzon, J. Fluor. Chem., 2001, 107, 301–310 CrossRef CAS.
- P. Clavel, M. P. Léger-Lambert, C. Biran, F. Serein-Spirau, M. Bordeau, N. Roques and H. Marzouk, Synthesis, 1999, 829–834 CrossRef CAS.
- C. Saboureau, M. Troupel, S. Sibille and J. Perichon, J. Chem. Soc., Chem. Commun., 1989, 16, 1138–1139 RSC.
- E. Liotier, G. Mousset and C. Mousty, Can. J. Chem., 1995, 73, 1488–1496 CrossRef CAS.
- Y. Yamauchi, K. Sakai, T. Fukuhara, S. Hara and H. Senboku, Synthesis, 2009, 3375–3337 CAS.
- B. B. Huang, L. Guo and W. J. Xia, Green Chem., 2021, 23, 2095–2103 RSC.
- W. B. Wu, M. L. Li and J. M. Huang, Tetrahedron Lett., 2015, 56, 1520–1523 CrossRef CAS.
- J. Sheng, N. Wu, X. Liu, F. Liu, S. Liu, W. Ding, C. Liu and X. Cheng, Chin. J. Org. Chem., 2020, 40, 3873–3880 CrossRef CAS.
- X. T. Gao, Z. Zhang, X. Wang, J. S. Tian, S. L. Xie, F. Zhou and J. Zhou, Chem. Sci., 2020, 11, 10414–10420 RSC.
- Y. Liu, X. Tao, Y. Mao, X. Yuan, J. Qiu, L. Kong, S. Ni, K. Guo, Y. Wang and Y. Pan, Nat. Commun., 2021, 12, 6745–6752 CrossRef CAS PubMed.
- A. Claraz, C. Allain and G. Masson, Chem. – Eur. J., 2022, 28, e202103337 CrossRef CAS PubMed.
- H. Zhang, M. Liang, X. Zhang, M.-K. He, C. Yang, L. Guo and W. Xia, Org. Chem. Front., 2022, 9, 95–101 RSC.
- X. Yan, S. Wang, Z. Liu, Y. Luo, P. Wang, W. Shi, X. Qi, Z. Huang and A. Lei, Sci. China: Chem., 2022, 65, 762–770 CrossRef CAS.
- W. Chen, S. Ni, Y. Wang and Y. Pan, Org. Lett., 2022, 24, 3647–3651 CrossRef CAS PubMed.
- P. J. Elving and J. T. Leone, J. Am. Chem. Soc., 1957, 79, 1546–1550 CrossRef CAS.
- J. H. Stocker and R. M. Jenevein, Chem. Commun., 1968, 16, 934–935 RSC.
- J.-S. Yang, K.-T. Liu and Y. O. Su, J. Phys. Org. Chem., 1990, 3, 723–731 CrossRef CAS.
- J. Kopilov and D. H. Evans, J. Electroanal. Chem. Interfacial Electrochem., 1990, 280, 435–438 CrossRef CAS.
- K. Uneyama, K. Maeda, T. Kato and T. Katagiri, Tetrahedron Lett., 1998, 39, 3741–3744 CrossRef CAS.
- K. Uneyama and T. Kato, Tetrahedron Lett., 1998, 39, 587–590 CrossRef CAS.
- K. Uneyama, G. Mizutani, K. Maeda and T. Kato, J. Org. Chem., 1999, 64, 6717–6723 CrossRef CAS PubMed.
- A. A. Stepanov, T. V. Minyaeva and B. I. Martynov, Tetrahedron Lett., 1999, 40, 2203–2204 CrossRef CAS.
- J. R. Box, A. P. Atkins and A. J. J. Lennox, Chem. Sci., 2021, 12, 10252–10258 RSC.
- (a) J. Niu, H. Lin, J. Xu, H. Wu and Y. Li, Environ. Sci. Technol., 2012, 46, 10191–10198 CrossRef CAS PubMed; (b) H. Lin, J. Niu, J. Xu, H. Huang, D. Li, Z. Yue and C. Feng, Environ. Sci. Technol., 2013, 47, 13039–13046 CrossRef CAS PubMed; (c) Y. Su, U. Rao, C. M. Khor, M. G. Jensen, L. M. Teesch, B. M. Wong, D. M. Cwiertny and D. Jassby, ACS Appl. Mater. Interfaces, 2019, 11, 33913–33922 CrossRef CAS PubMed; (d) E. Mousset, N. Oturan and M. A. Oturan, Appl. Catal., B, 2018, 226, 135–146 CrossRef; (e) Y. Su, U. Rao, C. M. Khor, M. G. Jensen, L. M. Teesch, B. M. Wong, D. M. Cwiertny and D. Jassby, ACS Appl. Mater. Interfaces, 2019, 11, 33913–33922 CrossRef CAS PubMed; (f) J. X. Zhu, Y. H. Chen, Y. R. Gu, H. Ma, M. Y. Hu, X. L. Gao and T. Z. Liu, J. Hazard. Mater., 2022, 422, 126953–126962 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |