
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ionic liquid-assisted synthesis of mesoporous polymers and carbon materials: the self-assembly mechanism†
Yaoguang
Song
 a,
Fraser
Norris
b,
Daryl
Hinchcliffe
a,
Yong
Xu
c,
Xiaolei
Zhang
*b and
Peter
Nockemann
*a
a,
Fraser
Norris
b,
Daryl
Hinchcliffe
a,
Yong
Xu
c,
Xiaolei
Zhang
*b and
Peter
Nockemann
*a
aThe QUILL Research Centre, School of Chemistry and Chemical Engineering, Queen's University Belfast, BT9 5AG, Belfast, UK. E-mail: p.nockemann@qub.ac.uk
bDepartment of Chemical and Process Engineering, University of Strathclyde, G1 1XJ, Glasgow, UK. E-mail: xiaolei.zhang@strath.ac.uk
cJiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, College of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, People's Republic of China
First published on 15th September 2022
Abstract
Soft-templating synthesis has been widely employed to fabricate ordered mesoporous polymer and carbon materials with effectively tuneable pore sizes. However, the commonly used templating agents, block copolymers, are normally decomposed during the process, thus are barely recyclable; this increases the costs and hampers the scale-up feasibility. Therefore, it becomes imperative to seek promising alternatives; amphiphilic ionic liquids (ILs) are excellent candidates due to their good recyclability. This study explored the templating behaviour of IL templates for preparing mesoporous polymers and carbons. In details, the self-assembly of ternary systems (comprising of IL templates, precursors and solvent) were investigated by a combination of coarse-grained molecular dynamics (CGMD) simulations, density function theory (DFT) calculations and experimental techniques. The results indicate that the morphologies of IL templates are tuneable not only by the adjustment of water content in the mixture but also by the selection of suitable precursors. Material precursors containing increasing numbers of hydroxyl moieties also induce various precursor-template spatial correlations, resulting in different topological structures of nanomaterials. This work presents a fundamental investigation into the mechanisms of templating synthesis with amphiphilic ILs as recyclable templates and gives insight into the effective design of coveted carbon nanomaterials for targeted applications.
Introduction
In the burgeoning research area of nanoscience, porous materials have gained growing attention; their applications have expanded from catalysis and molecule separation in the industry to cutting-edge research fields such as energy storage and drug delivery.1,2 Ordered mesoporous polymers are one of the most attractive materials due to the uniformity and tuneability of their pore structures. Notably, they are also ideal precursors for mesoporous carbon materials via one additional step of calcination. However, challenges still exist during their preparation, as well as in the fabrication of other nanomaterials, especially on how to create and implement function-led design of nanostructures for specific applications. This can be achieved by precisely manipulating nanostructures on a molecular level leading to the manifestation of desired properties which can then be investigated regarding their processability at industrial scale.Templating synthesis is an essential technique to achieve controlled structural manipulation of nanomaterials. Within all the templating methodologies, soft-templating synthesis, based on the self-assembly of amphiphiles, can effectively tune the structure while eliminating the template easily thus is one of the most adopted strategies to fabricate ordered mesoporous polymer and carbon materials.3–7 Commercial block copolymers such as Pluronic F127 are typical soft templates which are widely used. However, they are generally decomposed via thermal treatment during the pore structures generating process. The consumption of block copolymer templates has heightened the barrier of scaling-up production of ordered mesoporous polymers and carbons at industrial scale due to the relatively costly nature of these templates. Therefore, there is an imperative to seek for more promising alternatives that have excellent recyclability without sacrificing templating effectiveness.
A good option is long-chain ionic liquids (ILs) as they have excellent amphiphilicity, which is one key criteria during the selection of appropriate soft templates. Lyotropic liquid crystals (LLCs), micelles, and (micro) emulsions formed by ILs have already drawn much attention in templating synthesis of nanomaterials. Hejazifar et al.8 reviewed three types of IL-based microemulsions that are widely used for polymerisation and material preparations. Kang et al.9 summarised recent explorations on employing ILs as templates, as well as synthetic media and precursors, to fabricate various dimensional nanomaterials, including zero-dimensional nanoparticles, one-dimensional nanowires/nanotubes/nanorods, two-dimensional nanosheets, and three-dimensional porous materials. The utilisation of ILs as templates (or structure-directing agents) for material preparation is still continuously growing.9–17 In general, the rationale for achieving diverse structures lies in confining IL templates during self-assembly into various morphologies, mainly: spherical, columnar, and lamellar.
The use of ILs as templates to prepare porous polymer and carbon materials still remains at the initial stage, despite their growing popularity. Nevertheless, one notable advantage of ILs as the template is that they can be recycled.11,18–21 This makes ILs more promising as the preparation costs can be potentially reduced. So far, no highly ordered pore structures have been reported as of yet. Xie et al.21 employed a reusable IL 1-butyl-3-methylimidazolium tetrachloroferrate(III) to prepare hierarchical porous carbons via ionothermal carbonisation of carbohydrates. The IL played a triple role: soft template, solvent, and catalyst, although no obvious ordered mesoporosity was obtained. Yang et al.22 prepared porous resorcinol-formaldehyde polymer gels and carbon aerogels employing ILs with various cation chain lengths (2–8 carbon atoms) as templates. As the length of alkyl chains increased, resultant porous aerogels showed increased specific surface area. However, IL templates were decomposed during pyrolysis and the resultant pore structures were disordered. Nagy et al.11 reported the use of ILs as recyclable templates and metal-free catalyst for polymerisation to synthesise porous polymer gels from phenolic compounds. The ILs were extracted out from polymer–ILs composites using water and acetone in sequence. The influence of different cations and anions on the porosity of the polymers was studied. The ILs employed in their works were less amphiphilic as the longest chain length of cation only reached 4 carbon atoms. Generally, ILs show noticeable amphiphilicity when the alkyl side chain has over 6 carbon atoms,23–26 though microphase separation could occur with shorter alkyl chains.27 The absence of ordered pore structures in aforementioned explorations can be associated with: (1) the use of less amphiphilic ILs as templates which are unable to form long range ordered mesostructures, and (2) the lack of fundamental understanding on how to effectively manipulate the morphologies of templates in systems containing both IL templates and precursors.
Typically, during a soft-templating synthesis, the self-assembly of soft templates separates the mixture into two phases, hydrophobic cores of the micelles and hydrophilic corona parts. The two key factors that eventually determine the topological structures of mesoporous polymer and carbon materials are the morphology of templates and the spatial correlation between precursor and the hydrophilic parts of the template.3,28 Therefore, when employing ILs as the template, the successful implement requires the thorough understanding of both the morphology evolutions of ILs in mixture and the spatial distributions of precursors and IL templates.
Although there has been numerous research, both computational and experimental, which has investigated the phase behaviour of amphiphilic ILs, most of the published work focused on the micellization/aggregation of ILs in the presence of another single component, especially IL aqueous binary mixtures. Multiple factors were identified as being able to facilitate the formation of IL micelle clusters, including: the water concentration, the length cationic side chain, solvation ability of the anion, and the influence of temperature.17,22–25,28–32 However, in practical templating fabrication, three or more chemical species would inevitably be encompassed in total, including, but not limited to: IL templates, material precursors and solvents. In such scenarios, there is a lack of detailed theory whether the presence of material precursors will influence the self-assembly of IL templates, particularly their morphologies, and the precursor-template spatial correlations. Consequently, it adds to the challenge to effectively design polymer and carbon materials with coveted structures aiming for specific applications.
This paper investigates the self-assembly behaviour of ternary mixtures comprising of IL templates, material precursors, and solvent through a combination of computational and experimental techniques. The overall aim is to shed light on the templating mechanism of long-chain IL templates in the presence of material precursors and gives insight into the effective design of coveted carbon nanomaterials for various applications. To achieve this, two crucial factors during soft templating synthesis were explored, (1) the morphology of the IL templates and (2) the spatial correlation between precursor and template. The morphological evolution process was studied under varying water contents, different precursors, as well as various temperatures, to understand how IL templates assemble into certain morphologies. The influence of different material precursors on the template-precursor spatial correlation was investigated, which provided key information on precursors selection towards desired nanostructured materials. Interestingly, the precursors were found to play a crucial role in both the morphologies of IL templates and template-precursor spatial correlations. This, to the best of our knowledge, has not been reported before. More importantly, this study provides a fundamental understanding on the mechanism of soft-templating synthesis of nano-structured polymer and carbon materials by employing amphiphilic ILs as recyclable templates and gives insight into how the structures of the resultant materials could be manipulated aiming for various applications.
Results and discussion
1-Alkyl-3-methylimidazolium-based ILs are the most studied family for templating synthesis.9–17,23 We chose 1-decyl-3-methylimidazolium acetate ([C10MIM][OAc]) due to the following reasons: (1) [C10MIM]+ as cation has desired amphiphilicity, and (2) the acetate anion not only has a strong dissolution ability33–37 and is slightly basic, but also makes the process greener by being halogen-free. Phenolic compounds such as phenol, resorcinol, and phloroglucinol that have been used extensively to prepare porous polymer/carbon materials were selected as precursors and were added equimolarly as IL templates. The detailed methodology can be found in the ESI.†The morphological evolution process of amphiphilic IL templates in ternary mixtures is first emphasised under varying water content, precursors, and temperature. The template-precursor spatial correlation is subsequentially highlighted, which together with template morphology determine the resultant topological structures of nanomaterials. Finally, potential consequences of this work are briefly discussed with possible promising syntheses routes proposed for some nanomaterials targeting particular applications.
Influence of water content on the morphology of IL templates
The self-assembly process of [C10MIM][OAc]/water/phenol ternary systems with varying water contents at 298.15 K was first studied via coarse-grained molecular dynamics (CGMD) simulations coupled with experimental techniques. CG models in this study are available in ESI and depicted in Fig. S1.†All ternary systems with corresponding morphologies obtained from CGMD simulations are summarised in Table S1.†Fig. 1 shows three typical morphologies formed by [C10MIM]+ cations in the presence of phenol as the precursor. With the decreasing water content (H2O wt% = m(H2O)/(m(H2O) + m(ILs)), excluding phenolic precursor) in the mixture, spherical, columnar, and lamellar phases formed sequentially once the IL concentration exceeded the critical micelle concentration (CMC). Fig. 1a presents a disordered configuration of spherical micelle clusters formed when the relative water content is 50.5 wt%. As the water content decreases, spherical clusters slowly grow longer into rod-like clusters. Eventually long rod-like clusters reorganise and evolve into honeycomb-like hexagonal columnar phase with long-range order (Fig. 1b); corresponding water content ranges from 29.3–44.0 wt%. After a short transition stage, lamellar sheet-like arrays as shown in Fig. 1c are formed as the water content reaches smaller than 25.0 wt%.
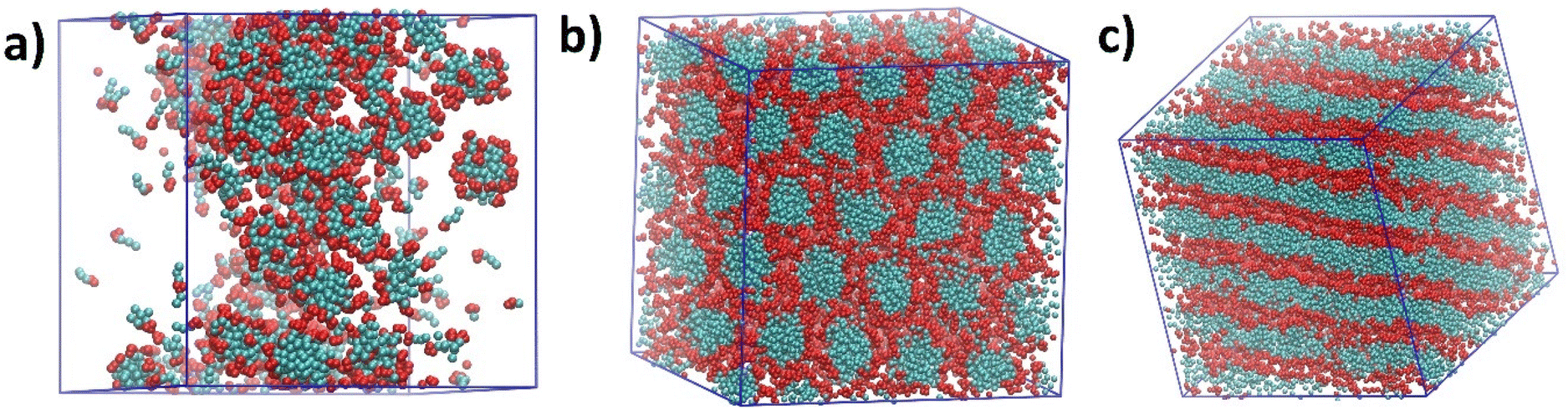 | ||
| Fig. 1 Snapshots of [C10MIM][OAc]/water/phenol ternary systems at water contents around: (a) 50 wt%, (b) 34 wt%, and (c) 25 wt%. Red and cyan beads represent imidazolium rings and alkyl chains of IL cations, respectively. Phenol, water, and anions are not shown for clarity. | ||
During a typical morphology evolution process in IL/water binary mixtures, the formation of LLCs or lyotropic gels indicates that the mixture is in a lyotropic phase. Different texture patterns from polarised optical microscopy (POM) can be linked to certain morphologies, such as a hexagonal columnar or smectic phase.38–44 POM was also performed to observe [C10MIM][OAc]/water/phenol ternary mixtures. Fig. S2a† shows a characteristic fan-like pattern of [C10MIM][OAc]/water binary gels with a water content around 18 wt%, indicating the presence of hexagonal liquid crystalline structures, while ternary mixture containing phenol exhibited a clear liquid phase (Fig. S2b†), although hexagonal columnar and smectic lamellar morphologies are observable in CGMD simulations.
However, the absence of LLCs or lyotropic gels on a macroscopic aspect does not necessarily indicate that the IL templates in ternary mixtures are not confined in hexagonal or lamellar arrays. In neat long-chain 1-alkyl-3-methylimidazolium salts, the crystalline phase exhibited highly ordered lamellar arrays at lower temperatures but could directly turn into isotropic liquids after melting.45 Analogous temperature-dependent mesophase behaviours were also seen in IL/water binary mixtures.42,43 This is possibly because heating could cause an increased kinetic energy and faster molecule motion, which destabilises the periodic three-dimensional H-bonding lattices. Although the liquid crystalline phases disappeared after melting, the array of long-chain cations still maintained a dynamic short-range structural ordering.45 The disappear of LLCs or gels in ternary mixture is likely due to the addition of phenol precursor.
Small-angle X-ray scattering (SAXS) measurements were carried out to seek for experimental evidence of the morphologies of both binary and ternary mixtures. As shown in Fig. S3a,† the SAXS profiles of [C10MIM][OAc]/water binary mixtures containing 18–50 wt% of water indicate the dominating hexagonal phase, evidenced by the characteristic scattering vector ratio q1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :q2:q3 = 1:√3:√4.41,43 CGMD simulation also pointed out an ordered hexagonal structure when the water content reached 18 wt% (Fig. S3b†). Moreover, with the rise of temperature, these sharp characteristic scattering peaks become less intense even unobservable due to higher kinetic energy. Only a single broad q1 peak was detected for [C10MIM][OAc]/water binary mixtures containing 34 wt% of water when temperature reached to 358.15 K (Fig. S3c†). At this point, the lyotropic mesophase reversibly melted into an isotropic liquid phase and the long-range liquid crystallinity was disturbed, resulting in a dynamic short-range structural ordering of IL cations.
:q2:q3 = 1:√3:√4.41,43 CGMD simulation also pointed out an ordered hexagonal structure when the water content reached 18 wt% (Fig. S3b†). Moreover, with the rise of temperature, these sharp characteristic scattering peaks become less intense even unobservable due to higher kinetic energy. Only a single broad q1 peak was detected for [C10MIM][OAc]/water binary mixtures containing 34 wt% of water when temperature reached to 358.15 K (Fig. S3c†). At this point, the lyotropic mesophase reversibly melted into an isotropic liquid phase and the long-range liquid crystallinity was disturbed, resulting in a dynamic short-range structural ordering of IL cations.
Fig. 2 shows SAXS spectra for ternary mixtures containing phenol where only broad peaks were observed rather than sharp intense peaks. The SAXS profile for ternary mixture containing 18 wt% of water shows a single broad peak centred at 0.24 Å−1, which could be associated with the relative structural ordering in short-range. With the increase in water content from 30 wt%, the q1 peak gets lower and shifts to lower q range, while a second broad peak q2 gets higher and centred around 0.38 Å−1. When the water content exceeds 50 wt%, both peaks become weaker then merge into a much broader peak for mixture containing 90 wt% of water. Owing to the less intense nature of these scattering peaks, it is challenging to interpret morphological information for IL templates in ternary mixture. However, when the mixtures were quenched down to 263.15 K, more recognisable scattering peaks are manifested, which is probably because lower temperature results in slower molecule motion due to lower kinetic energy. As shown in Fig. S4,† two scattering peaks at 0.22 and 0.44 Å−1 are observed for a ternary mixture containing 18 wt% of water; the ratio q1:q2 = 1:2 indicates the formation of lamellar bilayer array with a repeating distance d = 2π/q = 28.5 Å. At 44 wt% of water, a hexagonal phase is detected with a scattering vector ratio of q1:q2 = 1:√3. The broad peak observed from ternary mixture containing 90 wt% of water indicates that IL cations are dominatingly assembled into spherical clusters.
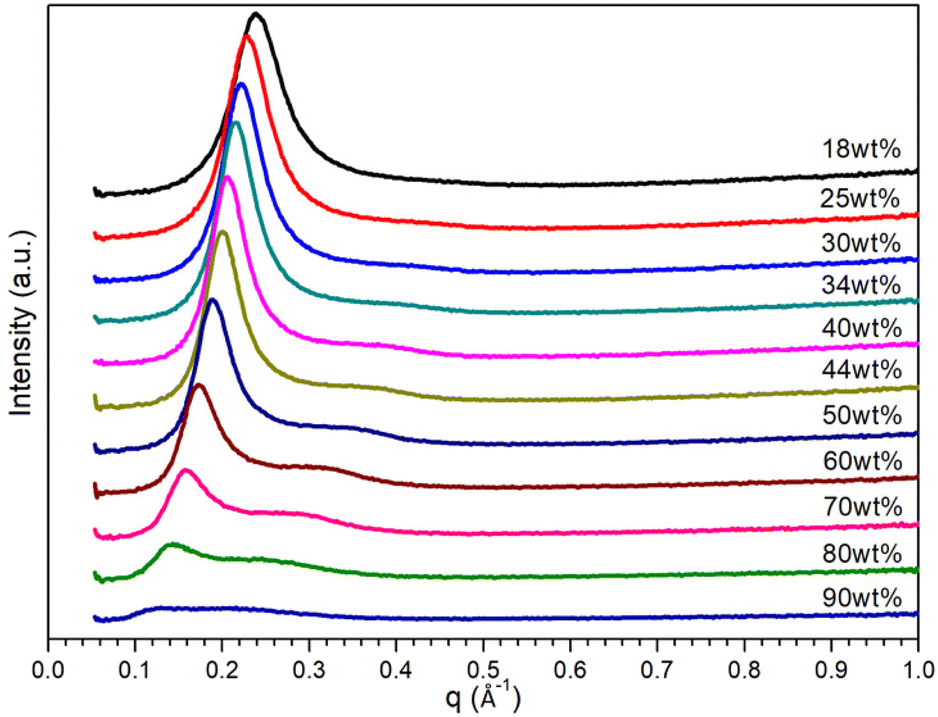 | ||
| Fig. 2 SAXS patterns of [C10MIM][OAc]/water/phenol ternary mixtures with varying water contents at 298.15 K. | ||
The SAXS result implies that the introduction of phenol can destabilise the H-bonding lattice between IL and water hence the weakened scattering intensity of characteristic peaks for ternary mixtures. This is likely attributed to the hydroxyl group in phenol that enables phenol to interact with ILs and water via H-bonding.
The detailed interaction energies for ternary systems are also obtained from DFT calculations with a single ion pair and varying water numbers (Fig. 3). The overall system interaction energy shows a steady growth from 619.2 to 1439.8 kJ mol−1 when the number of water molecules increases in the system. This growth is mostly contributed by the interactions of water–IL pair (from 78.2 to 425.8 kJ mol−1) and water–water pair (from 0 to 566.2 kJ mol−1). Noticeably, the interaction within [C10MIM][OAc] molecules becomes weaker, evidenced by the declining interaction energy from 430.9 to 315.4 kJ mol−1. Since H-bonding is the predominating interaction pathway between [C10MIM][OAc] and water, adding more water into the ternary system essentially enlarges the H-bonding probability of IL with water and initiates the morphological evolution of micelles during self-assembly. In the presence of a precursor, phenol shows a significantly strong H-bonding interaction with [C10MIM][OAc], especially via acetate⋯H-phenol bond. Consequently, the H-bonding lattices between ILs and water are destroyed by the competitional interaction between IL with phenol, hence the disappear of the liquid crystalline mesophase.
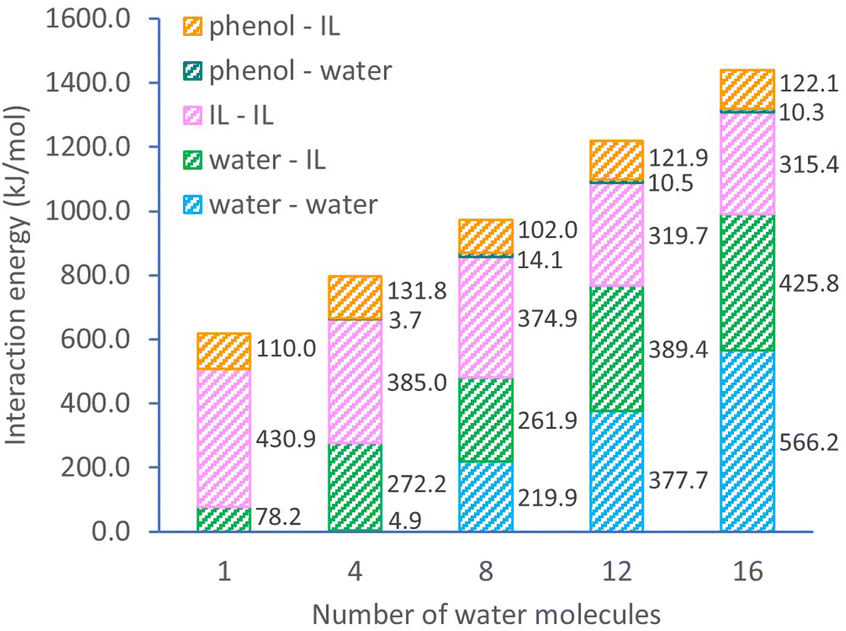 | ||
| Fig. 3 Energy changes with increasing water concentration in ternary systems. | ||
Influence of different precursors on the morphology of IL templates
Beside of phenol, resorcinol and phloroglucinol are also extensively used precursors for porous polymer and carbon materials. CG-MD simulations were performed for ternary mixtures containing resorcinol and phloroglucinol individually to reveal whether different precursor could influence the self-assembly of ILs during templating synthesis. A benzene based ternary mixture was studied for comparison. Due to their accumulative hydroxyl groups from 0 to 3, these four aromatic compounds possess increasing hydrophilicity and hydrogen bonding ability in the order of benzene < phenol < resorcinol < phloroglucinol. The mixing ratio in ternary systems remained equal, so the only variable was the increasing hydroxyl functionality and consequent H-bonding ability.The eventual configurations of ternary mixtures at 18 wt% of water are shown in Fig. 4. The benzene based ternary mixture also presents a planar lamellar phase like their phenol counterpart. In comparison with [C10MIM][OAc]/water binary mixture where dominating hexagonal columnar phase is observable, the addition of equimolar of phenol or benzene has changed the morphology of [C10MIM]+ micelles. Most noticeably, when using resorcinol and phloroglucinol as precursors, the morphology of the [C10MIM][OAc] became hexagonal columnar, which is expected to appear at higher water contents for phenol-based cases. This indicates that the introduction of more hydroxyl functionalities also plays an analogous role as increasing water ratio in mixture. One hydroxyl functionality on a phenolic compound can act as one H-bond donor and acceptor while per water molecule has double the H-bonding capacity of phenol; phloroglucinol is able to form H-bonds quantitively equal that of phenol “plus” water at maximum. This may lead to the morphological transition when using phloroglucinol to replace phenol.
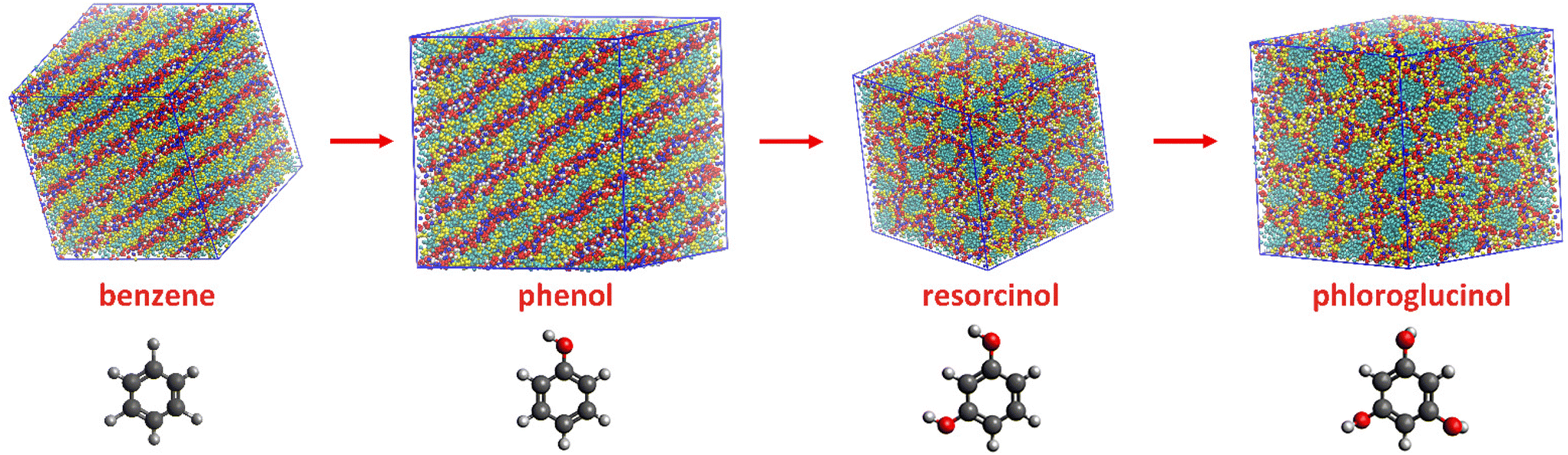 | ||
| Fig. 4 Morphology evolution under increasing hydroxyl groups in precursors at the same mixing ratio (red: imidazolium ring, cyan: alkyl chain, blue: acetate, yellow: phenolic compounds, white: water). | ||
The SAXS profiles of the ternary mixtures with the four different precursors are shown in Fig. 5. Only broad peaks were observed for ternary mixtures, except that containing benzene where its scattering vector ratio q1:q2 = 1:2 signifies the presence of smectic lamellar phase. With the increase in hydroxyl groups, the broad scattering q1 peak gets stronger from ternary mixture containing phenol. The absence of the second scattering peak q2 makes it more challenging to interpret the morphological structures of [C10MIM]+ cations in a ternary mixture containing resorcinol or phloroglucinol. Unlike their phenol counterpart, lower temperatures do not make the interpretation of SAXS profiles more straightforward as there is still only one broad scattering peak observable (Fig. S5†).
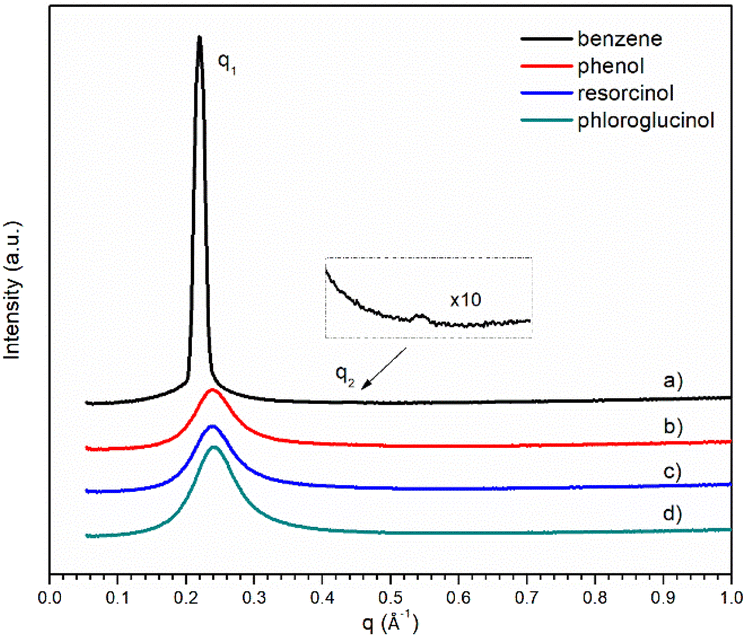 | ||
| Fig. 5 SAXS pattern of ternary mixtures containing 18 wt% of water with different precursors: (a–d) benzene, phenol, resorcinol, and phloroglucinol, respectively. | ||
POM was performed for ternary mixtures containing resorcinol and phloroglucinol. No lyotropic gels or LLCs were observed, although a hexagonal array was predicted by the CGMD simulation. The absence of a lyotropic gel phase did not apply to the [C10MIM][OAc]/water/benzene system, the cross pattern of which could be associated with the formation of a lamellar phase (Fig. S6a†).41,42,44 Liquid crystalline phases were also reported observable in [C16MIM][Br]/water/p-xylene ternary mixtures.41 This is because neither benzene nor p-xylene has functional moieties containing hetero-atoms to serve as H-bond donors and acceptors, therefore benzene and p-xylene are chemically stable in mixtures and unlikely to destabilise the H-bonding interactions between IL and water. However, when resorcinol or phloroglucinol were added in molar quantities lower than the concentration of [C10MIM][OAc], lyotropic gels were observed for ternary mixtures; the representative fan-like pattern exhibits the characteristics of hexagonal liquid crystalline lattice41,42,44 (Fig. S6b†). This phenomenon confirmed that phenolic compounds compete with water to form H-bonds with ILs, particularly acetate anion. When the molar concentration of the ILs is higher than that of the precursors, phenolic compounds are likely confined in dynamic hexagonal arrays via H-bonds with [C10MIM][OAc] and water, and excessive concentrations of [C10MIM][OAc] form liquid-crystalline H-bonding interactions with water generating lyotropic gels on a macroscopic aspect. When resorcinol or phloroglucinol was added equimolarly with [C10MIM][OAc], the strong scattering q1 peak in their SAXS profiles could be resulted from the formation of hexagonal columnar arrays.
To reveal how hydroxyl moieties on polymer precursors influence the morphology of IL templates, DFT calculations were performed for ternary systems containing different precursors at the same mixing ratio ([C10MIM][OAc]:water:phenolic compound = 1:1:4, at which the weighted content of water is approximately 18 wt%). Fig. 6a shows the overall interaction energies of four ternary mixtures. It was found that non-hydrogen bonding is the major interaction for the benzene-based ternary system, within which the van der Waals forces and hydrophobic interactions likely dominate the self-assembly. Upon the addition of phenolic compounds containing hydroxyl groups, H-bonding interactions become the major driving force for self-assembly. The increase in the number of hydroxyl groups leads to a steady growth in H-bonding energies from 329.2 to 539.5 kJ mol−1. Conversely, the non-hydrogen bonding interaction energy declines gradually from 383.9 to 252.8 kJ mol−1. This trend becomes more evident in a ternary system containing phloroglucinol, within which the H-bonding energy is more than twice that of non-H-bonding interactions.
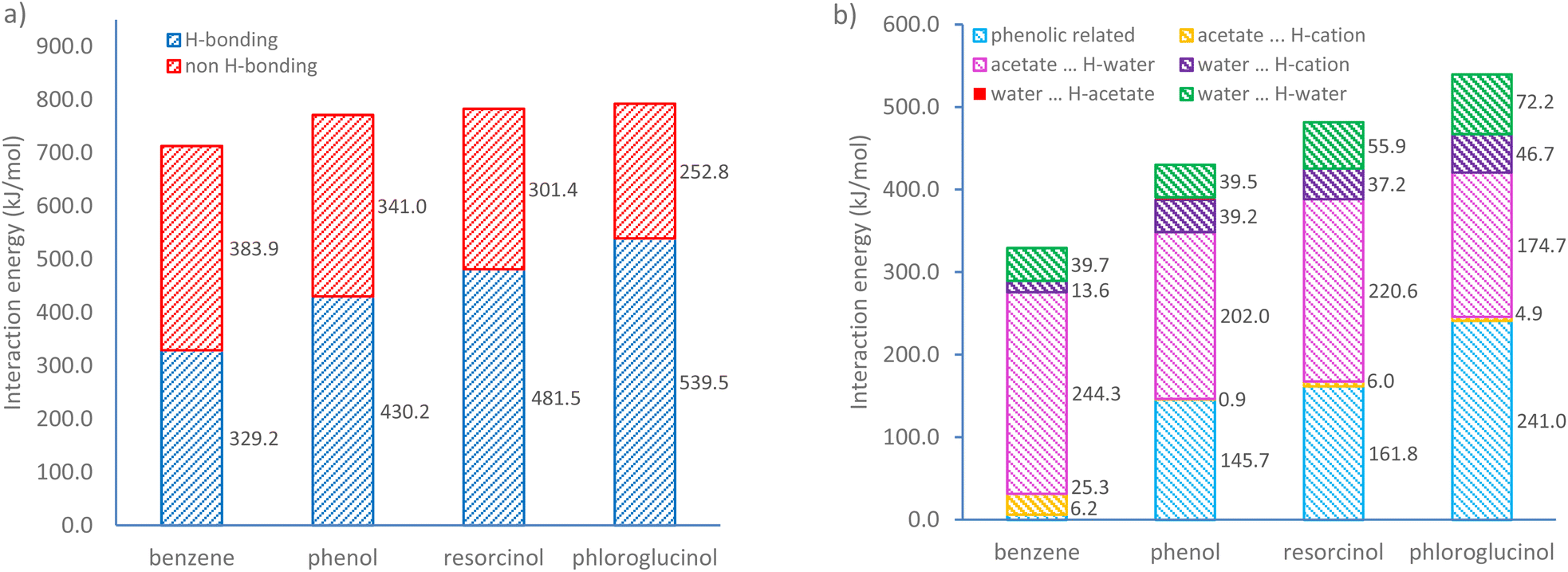 | ||
| Fig. 6 (a) Total interaction energy and (b) H-bonding interaction energy changes with increasing hydroxyl group numbers obtained by natural bond orbital (NBO) analysis. | ||
To further investigate the role of increasing numbers of hydroxyl groups, H-bonds were classified in two types: the H-bonds associated with phenolic compounds (i.e. phenolic⋯H-acetate, phenolic⋯H-water, phenolic⋯H-cation, water⋯H-phenolic, and acetate⋯H-phenolic), the H-bonds involving the [C10MIM]+ cation, acetate anion, and water. As shown in Fig. 6b, benzene barely forms H-bonds with the other two species and the only H-bonds exist between ILs and water, mainly acetate⋯H-water. Detailed H-bonding interaction energy and individual significant H-bond within four different ternary systems can be found in Tables S2, 3 and Fig. S7.† In the presence of a growing number of hydroxyl groups, the H-bonding energy associated with phenolic compound sees a steady rise from 145.7 kJ mol−1 in ternary mixture containing phenol to 241.0 kJ mol−1 in that with phloroglucinol. An obvious diminishing effect in acetate⋯H-water interactions is shown as the hydroxyl group triples. This is because the acetate⋯H-phenolic bond and water⋯H-phenolic bond are two dominating H-bonds, contributing to over 92% of total H-bonding energy involving phenolic precursors, especially the former. When phenolic compounds compete in their interactions with water, the number of H-bonds formed between acetate and water becomes less. Consequently, more water molecules tend to form H-bonds with themselves via O⋯H–O bond, whose energy value climbs from 39.5 (in phenol case) to 72.2 kJ mol−1 (in phloroglucinol case) with an increment around 16.3 kJ per mol per hydroxyl group. This also elucidates how phenolic compounds could destabilise the periodic H-bonds lattice between ILs and water, hence the disappear of lyotropic gels.
H-bonding in systems incorporating ions is likely to correlate with charge transfer between ion pairs; this brings difficulties in estimating the H-bond energy.46 We studied the charge transfer from acetate to [C10MIM]+ cation within ternary systems with a single isolated ion-pair; corresponding values are 0.228e, 0.229e, 0.257e, and 0.230e for mixtures containing benzene, phenol, resorcinol, and phloroglucinol, respectively. Considering that the values for individual per anion in bulk systems are expected to be slightly smaller,46 the charge transfer values within ternary systems with different phenolic precursors are quite comparable. Therefore, the increase in H-bonding interaction energy values is attributed primarily to the growing hydroxyl group number.
Influence of temperature on the morphology of IL templates
Temperature is also a typical factor that influences the phase behaviour of amphiphiles in binary mixture.30 During templating synthesis, the cross-linking of phenolic precursors is normally carried out above 343.15 K, where morphology evolution might be different with that at lower temperatures.As shown in Table S1 and Fig. S8,† CGMD simulation results indicate that the morphology evolution at 358.15 K, a typical temperature for cross-linking, follows the same trend with that at lower temperature, but the phase transition points (or CMCs) varies slightly. For IL/water/phenol ternary mixture with 44 wt% of water, IL templates present spherical structures at higher temperature which then evolve into ordered hexagonal columnar phases after temperature quenching. Morphological evolution has not taken place at higher temperature. Similarly, the third CMC with morphology transiting from hexagonal columnar to smectic lamellar happens at a higher water content at 358.15 K than that at 298.15 K. This is possibly because the larger kinetic energy at a higher temperature is accelerating the molecule motions, which makes it easier to cross the energy barrier to form a more stable structure.
Polymer precursor – IL template spatial correlation analysis
In a practical templating synthesis of mesoporous polymer and carbon materials, precursors are expected to be first attached and subsequently being cross-linked in the amphiphilic part of the templates.3,5,28 Therefore, it is important to understand the spatial correlations between precursors and IL templates.In Fig. 4, the distribution difference of various phenolic precursors in mixtures can be observed. For ternary mixtures containing 18 wt% of water, phenol (yellow particles), as well as benzene, mostly resides in the hydrophobic cores formed by the aggregation of alkyl chain of [C10MIM]+ cations (cyan particles); resorcinol and phloroglucinol are dispersed toward the outer layer, the aqueous phase. The increasing number of hydroxyl groups in phenolic precursors are likely to not only influence the morphology of IL templates, also the distribution of phenolic precursors within the multiple-species systems.
To acquire a more detailed distributional preference, centre of mass (COM) radial distribution functions (RDF) were extracted from the simulation. The anion-water pair is the most prominent correlation throughout four ternary systems (Fig. S9†). This might be attributed to the strong H-bonding ability between acetate and water, where RCOO− is expected to form 6 H-bonds with water molecules on average.47 For ternary mixtures containing benzene or phenol, the subsequent strongest correlations are cation–anion and cation-water pairs while the weakest correlation is found between the precursor with the ILs and water (Fig. S9a and b†). As the number of hydroxyl groups increases in the precursor, the peaks for the resorcinol-anion correlation and resorcinol-water correlation grow higher, leaving the cation-resorcinol correlation as the weakest (Fig. S9c†). The correlation of the phloroglucinol-acetate pair becomes the second strongest, followed by the phloroglucinol-water pair. The presence of more hydroxyl groups to polymer precursors seemingly favours the interaction between precursor and aqueous phase.
Fig. 7 shows the detailed site–site RDFs between phenolic precursors and [C10MIM]+ cation, which plays the role of a template (beads cation 1–3 and 4–6 represent the imidazolium ring and alkyl chain, respectively; see Fig. S1†). Phenol, as well as benzene, has the strongest correlation with bead cation 4, followed by cation 5 and 6 (Fig. 7b). This indicates phenol has a distributional preference towards the area where the alkyl chain attaches to the imidazolium ring. The interactions between resorcinol and the imidazolium ring of the cation become stronger, whereas the correlation for cation 4-resorcinol pair remains the strongest (Fig. 7c). Compared with phenol, the two hydroxyl groups in resorcinol double its H-bonding ability. Consequently, resorcinol is dispersed towards the imidazolium ring where there is a higher possibility to interact with acetate and water via H-bonds. In ternary mixture containing phloroglucinol, the strongest site–site correlations are found in phloroglucinol with cation 1 and cation 2, as opposed to cation 4 as witnessed previously. Since phloroglucinol has mostly dispersed into the hydrophilic phase, the outer layer of template clusters, its correlations with alkyl chain become the weakest.
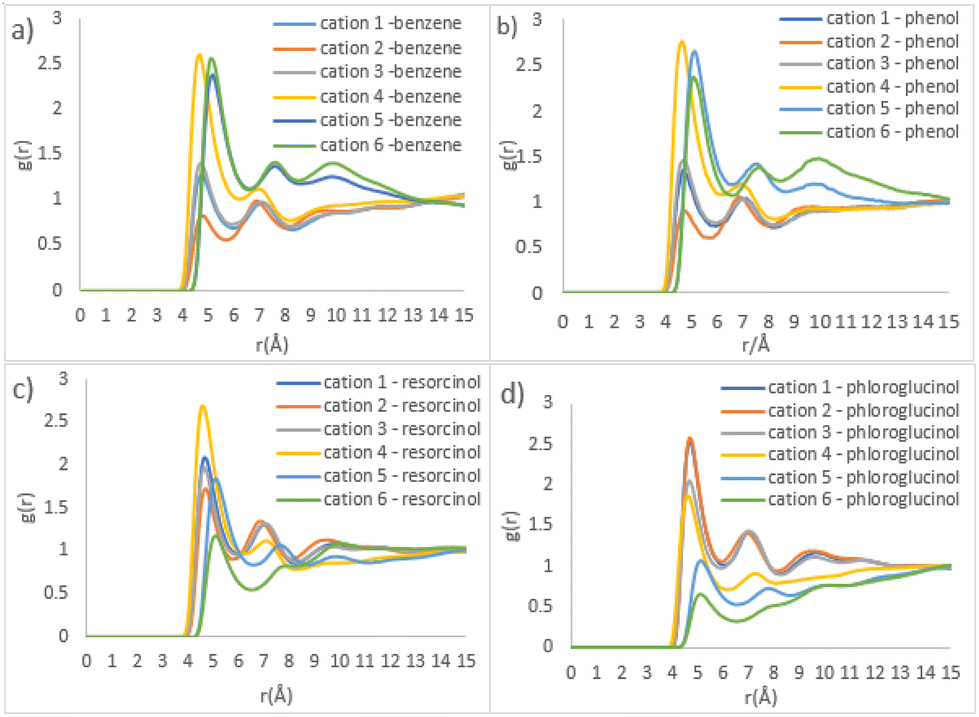 | ||
| Fig. 7 Site–site RDFs of IL templates and various precursors (a) benzene, (b) phenol, (c) resorcinol, and (d) phloroglucinol, respectively. | ||
According to the RDF analyses, involving more hydroxyl moieties in the precursor favours the interaction with the amphiphilic part of template, leaving the hydrophobic part as potential pore structures. To confirm this, the structures of ternary mixtures containing a single ion pair were optimised by DFT calculations to manifest the dominant configurations within the dynamic systems. As shown in Fig. 8, reference distances between terminal carbon (C10) and the projection point of precursor geometric centre onto the alkyl chain were calculated based on Table S4† to show the degree of dispersion. A clear trend is found that the reference distance becomes larger from benzene to phloroglucinol as the hydroxyl groups increase. Benzene and phenol have the least distance with C5–C7, C2–C5 of the alkyl chain, respectively. Resorcinol is distributed around C1 and C2, and phloroglucinol around N atom linked to the methyl side group of the cation. This result coincides well with the strongest pair spatial correlations in CGMD simulations.
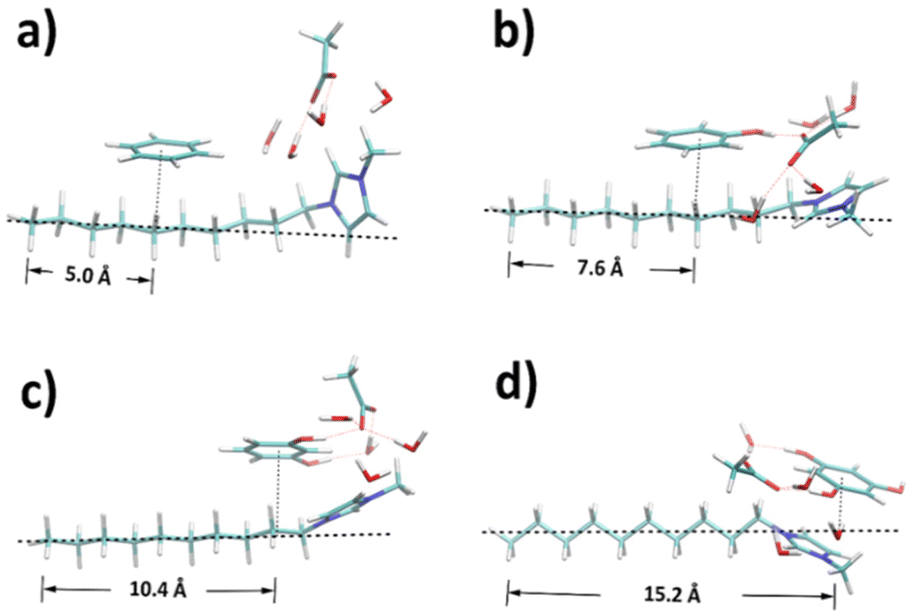 | ||
| Fig. 8 Structures optimised by DFT for ternary mixtures containing (a) benzene, (b) phenol, (c) resorcinol, and (d) phloroglucinol, respectively. | ||
Experimental evidence was acquired by 2D NOESY NMR spectroscopy. Protons close to each other in space (with a distance less than 3.5 Å) are likely to show stable cross peaks in the 2D spectra. A NOESY experiment for ternary mixture containing benzene was performed with deuterated oxide as internal reference due to the formation of lyotropic gels. From Fig. 9, the protons on benzene (7.0 ppm) only show cross peaks with H atoms linked to C3–C9 in the alkyl chain (broad peak from 0.9 to 1.0 ppm), therefore benzene is mostly distributed near the alkyl chain. As aforementioned, benzene barely interacts with [C10MIM][OAc] and water so non-H-bonding interactions such as the van der Waals force and hydrophobic interaction play major roles in the distribution of benzene. H atoms on phenol (6.5, 6.3, and 6.1 ppm) also present weak cross peaks with methyl group linked to the imidazolium ring, which infers that a small proportion of phenol remains in the aqueous phase. The cross peaks between protons on resorcinol (6.2, 5.8, and 5.7 ppm) and the imidazolium ring (6.7 and 6.6 ppm) confirm that some resorcinol molecules have started to disperse into the aqueous phase. When the hydroxyl groups are tripled, cross peaks resulting from the interactions between phloroglucinol (5.0 ppm) and the aqueous phase (mainly water at 6.0 ppm and acetate at 1.1 ppm) become more evident due to their strong interaction with the hydrophilic aqueous phase. Moreover, the peak for water shifts to high field with a growing number of hydroxyl group, from 5.5 ppm in the case of phenol to 6.0 ppm in the case of phloroglucinol, which can be attributed to the growing H-bonding interactions between precursor and water.
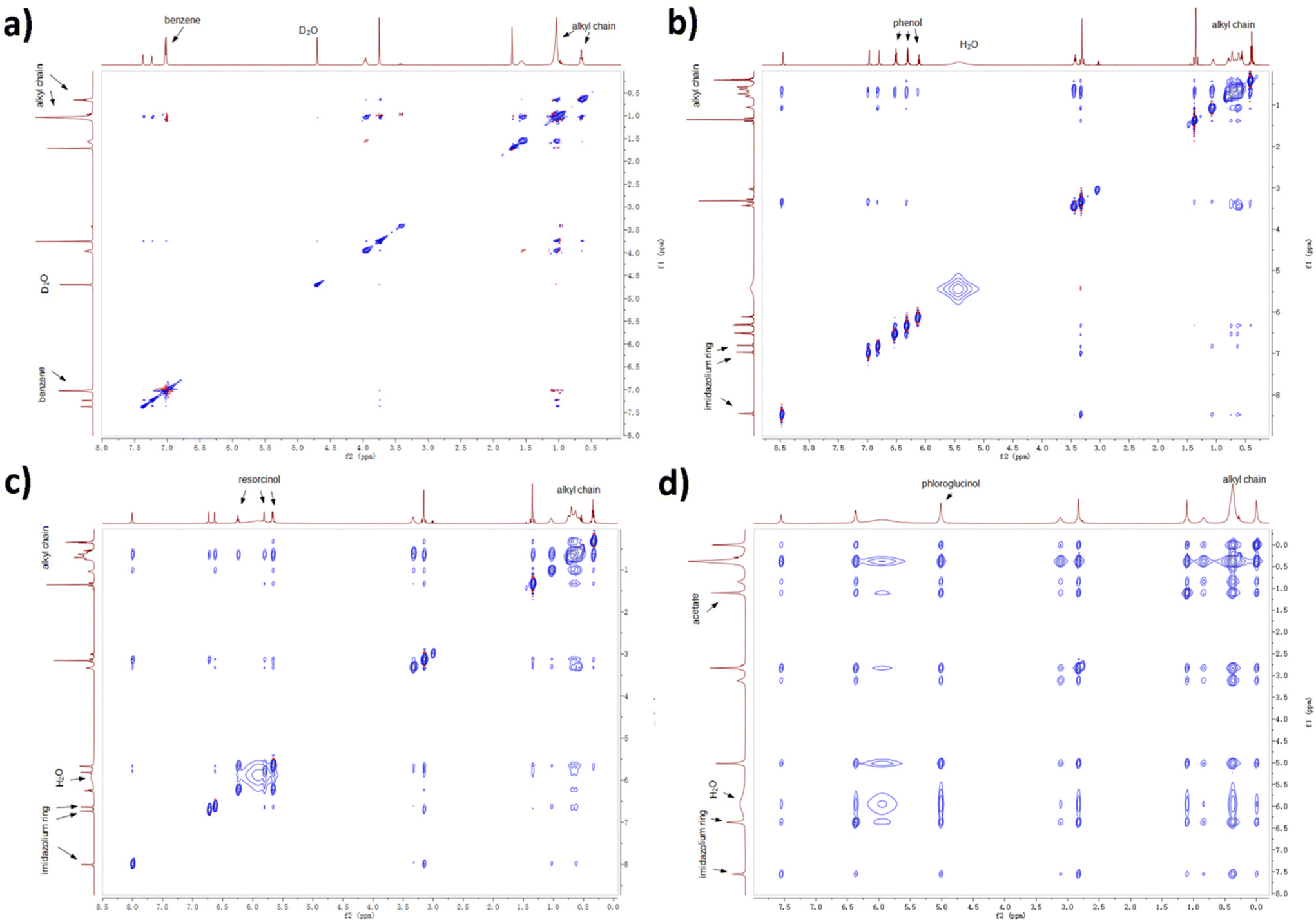 | ||
| Fig. 9 2D NOESY experiments for ternary mixtures containing (a) benzene, (b) phenol, (c) resorcinol, and (d) phloroglucinol, respectively. | ||
The results above also provide explanations on why phenolic compounds with different hydroxyl groups present different extraction efficiencies during extraction from waste water by employing imidazolium-based ILs.48–50 The extraction percentage for phenol generally exceeds 90%, while that for resorcinol is smaller than 60%.48 Phenol resides mostly in the hydrophobic phase of the IL cation, but its H-bonding ability via –OH results in a small ratio of phenol remaining in the aqueous phase. Resorcinol, due to the doubled hydroxyl groups, is observed mostly near the C1 atom of the IL alkyl chain thus has a poorer extraction efficiency.
Furthermore, template-precursor spatial correlations may also influence the morphological evolution of IL templates. At around 18 wt% of water, [C10MIM][OAc]/water binary mixture shows a hexagonal columnar phase. When phenol or benzene is added, the precursors enrich the hydrophobic phase by a space-occupying effect due to their spatial preference to the IL alkyl chains. The morphology of [C10MIM]+ cations has transformed into lamellar bilayer array, which tends to appear at more reduced water contents, i.e. the reduced hydrophilic phase. Phloroglucinol and resorcinol tend to strongly correlate with the aqueous phase thus could enrich the hydrophilic phase by a space-occupying effect. Consequently, spherical clusters are observed at 33.8 wt% of water for a ternary mixture containing resorcinol, and at 25.0 wt% for their phloroglucinol counterpart (Table S1†), where the binary mixtures based on Fig. S3a† still remain in the hexagonal phase.
Proposed synthesis routes for nanomaterials and applications
This work systematically investigated the morphological evolutions of ILs as the recyclable template and the spatial correlation between IL templates and precursors; the use of recyclable IL templates delivers increasing sustainability to the preparation process of nanomaterials by potentially lowering economic cost. The findings from this study provide significant guidance on seeking promising synthetic routes of tailored porous nanomaterials aimed for specific applications.Based on the difference in spatial distribution, the selection of precursors could lead to diverse nanostructures for various applications (Table 1). For example, the correlation preference of phenol to hydrophobic phase of IL templates enables them for preparing 0-D carbon nanospheres, 1-D nanowires, and 2-D nanoplates/nanosheets when the morphologies of the IL templates are spherical, columnar, and lamellar, respectively. Resorcinol and phloroglucinol preferentially reside in the hydrophilic aqueous phase, leaving the hydrophobic alkyl chain phase to generate pores after removal of the IL templates. Therefore, they could be promising candidates for hollow spheres or porous materials, ordered mesoporous polymer/carbon materials when spherical, hexagonal columnar micelle clusters form, especially when using phloroglucinol as a precursor.
| Precursor | Water contenta (wt%) | Morphologies of [C10MIM][OAc] templates | Potential nanomaterials | Representative down-stream applications |
|---|---|---|---|---|
| a Means relative water content: m(H2O)/(m(H2O) + m(ILs)), excluding the precursor. | ||||
| Phenol | 50–90 | Spherical | Carbon nanoparticles/nanosphere | Catalysis, oxygen reduction reactions |
| 30–45 | Hexagonal columnar | Carbon nanowires/fibres | Energy storage, electrocatalysis | |
| 18–25 | Lamellar bilayer | Nanoplates, nanosheets | Photocatalysis, energy storage | |
| Resorcinol | >34 | Spherical | Hollow spheres, porous carbons/polymers | Water treatment, heterogeneous catalysis |
| 18–25 | Hexagonal columnar | Ordered mesoporous polymer/carbon | Energy storage, drug delivery, catalysis | |
| Phloroglucinol | >25 | Spherical | Hollow spheres, porous carbons/polymers | Separation, water treatment, catalysis |
| <18 | Hexagonal columnar | Ordered mesoporous polymer/carbon | Biosensor, energy storage, drug delivery | |
However, the influence of cross-linkers on the self-assembly needs to be stressed. Phenolic precursors are expected to be cross-linked suitably after the self-assembly process to obtain a stable polymer structure. But commonly used cross-linkers such as formaldehyde, glyoxal and glyoxylic acid, may introduce hydroxyl groups into mixture, which can potentially affect both the template morphologies and precursor-template spatial correlations. Different precursor/template mixing ratios and chain lengths of IL cations may also lead to different results. These could be further investigated in the future by performing CGMD simulations, which in this work presented great reliability as confirmed by the experimental results. Nevertheless, the current version of Martini models typically underestimates the stacking distance of ring structures, resulting in lower densities than found as experimental values.51–54 Therefore, CGMD simulations might be more competent to emphasise the mesoscopic self-assembly behaviour rather than quantifying statistic properties such as density and atomistic interaction energies.
Conclusions
The self-assembly process in a soft-templating synthesis of nano-structured polymers and carbon materials was systematically investigated, whereby long-chain imidazolium-based ILs were employed as recyclable templates. The two crucial factors that determine the eventual structure of nanomaterials were highlighted particularly as (1) the morphology of IL templates and (2) the precursor-template spatial correlations.The morphologies of amphiphilic IL templates are not only tuneable through the adjustment of water content in mixture but also by the selection of the precursors. With the increase of water content in the mixture, the morphology of IL templates undergoes smectic lamellar bilayer, hexagonal columnar and spherical structures. At the same mixing ratio, introducing more hydroxyl groups to the material precursor also changes the morphology of IL templates, such as from a lamellar bilayer to a hexagonal columnar phase. The rationale of both approaches lies in adjusting the H-bonding probability of IL templates in the mixture.
Material precursors with different hydroxyl moieties lead to different precursor-template spatial correlations. More hydroxyl groups in the polymer precursor could increase the H-bonding ability and disperse precursors from a hydrophobic phase to a hydrophilic phase.
This work presents a theoretical basis on the mechanism of soft-templating synthesis with amphiphilic ILs as recyclable templates and gives insight into how to design coveted nanomaterials for targeting applications. Consequently, special attention is expected to be paid to the precise manipulation of templates into desired morphologies by adjusting mixing ratios and the rational selection of material precursors based on template-precursor spatial correlations.
Disclaimer
The views and opinions expressed in this work do not necessarily reflect those of the European Commission or the Special EU Programmes Body (SEUPB).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to acknowledge the funding from The Bryden Centre, which is supported by the European Union's INTERREG VA Programme, managed by the Special EU Programmes Body (SEUPB), and additional support from EPSRC First Grant (EP/R010986/1), Leverhulme Trust Research Grant (RPG-2017-254), Royal Society International Exchanges 2021 Cost Share (IEC\NSFC\211370) that combines with the National Natural Science Foundation of China (32211530071). Computational work was performed on High-Performance Computers, Kelvin2 funded by EPSRC (EP/T022175) at Queen's University Belfast and ARCHIE-WeSt at University of Strathclyde. The authors also want to thank Dr Sam Burholt and Dr Nick Terrill at the DL-SAXS facility (funded by EPSRC, grant no. EP/R042683/1) at the Diamond Light Source, to provide useful training on the instrument and support with the data collection and processing.References
- M. S. Attia, M. Y. Hassaballah, M. A. Abdelqawy, M. Emad-Eldin, A. K. Farag, A. Negida, H. Ghaith and S. E. Emam, Drug Dev. Ind. Pharm., 2021, 47, 1029–1037 CrossRef CAS PubMed.
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1, 16023 CrossRef CAS.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- X. Zhang and A. Lu, in Materials for Carbon Capture, Wiley, 2020, pp. 29–95 Search PubMed.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CrossRef CAS PubMed.
- A. Eftekhari and Z. Fan, Mater. Chem. Front., 2017, 1, 1001–1027 RSC.
- D. Wu, F. Xu, B. Sun, R. Fu, H. He and K. Matyjaszewski, Chem. Rev., 2012, 112, 3959–4015 CrossRef CAS PubMed.
- M. Hejazifar, O. Lanaridi and K. Bica-Schröder, J. Mol. Liq., 2020, 303, 112264 CrossRef CAS.
- X. Kang, X. Sun and B. Han, Adv. Mater., 2016, 28, 1011–1030 CrossRef CAS PubMed.
- J. Xia, J. Di, H. Li, H. Xu, H. Li and S. Guo, Appl. Catal., B, 2016, 181, 260–269 CrossRef CAS.
- B. Nagy, E. Geissler and K. László, Microporous Mesoporous Mater., 2020, 294, 109888 CrossRef CAS.
- Z. L. Xie and D. S. Su, Eur. J. Inorg. Chem., 2015, 2015, 1137–1147 CrossRef CAS.
- M. Zhen, J. Yu and S. Dai, Adv. Mater., 2010, 22, 261–285 CrossRef PubMed.
- Q. Yang, Z. Zhang, X. G. Sun, Y. S. Hu, H. Xing and S. Dai, Chem. Soc. Rev., 2018, 47, 2020–2064 RSC.
- B. G. Trewyn, C. M. Whitman and V. S. Y. Lin, Nano Lett., 2004, 4, 2139–2143 CrossRef CAS.
- Z. Chen, T. L. Greaves, R. A. Caruso and C. J. Drummond, J. Mater. Chem., 2012, 22, 10069–10076 RSC.
- H. Kaper and B. Smarsly, in Zeitschrift fur Physikalische Chemie, De Gruyter, 2006, vol. 220, pp. 1455–1471 Search PubMed.
- M. Murakami, Y. Kaneko and J. Kadokawa, Carbohydr. Polym., 2007, 69, 378–381 CrossRef CAS.
- P. Snedden, A. I. Cooper, K. Scott and N. Winterton, Macromolecules, 2003, 36, 4549–4556 CrossRef CAS.
- K. Matsumoto and T. Endo, Macromolecules, 2008, 41, 6981–6986 CrossRef CAS.
- Z. L. Xie, R. J. White, J. Weber, A. Taubert and M. M. Titirici, J. Mater. Chem., 2011, 21, 7434–7442 RSC.
- H. Yang, X. Cui, Y. Deng and F. Shi, J. Mater. Chem., 2012, 22, 21852–21856 RSC.
- J. M. Vicent-Luna, J. M. Romero-Enrique, S. Calero and J. A. Anta, J. Phys. Chem. B, 2017, 121, 8348–8358 CrossRef CAS PubMed.
- Y. Zhao, S. Gao, J. Wang and J. Tang, J. Phys. Chem. B, 2008, 112, 2031–2039 CrossRef CAS PubMed.
- J. N. A. Canongia Lopes and A. A. H. Pádua, J. Phys. Chem. B, 2006, 110, 3330–3335 CrossRef CAS.
- I. Goodchild, L. Collier, S. L. Millar, I. Prokeš, J. C. D. Lord, C. P. Butts, J. Bowers, J. R. P. Webster and R. K. Heenan, J. Colloid Interface Sci., 2007, 307, 455–468 CrossRef CAS PubMed.
- S. S. Sarangi, B. L. Bhargava and S. Balasubramanian, Phys. Chem. Chem. Phys., 2009, 11, 8745–8751 RSC.
- D. Saha, R. Zacharia and A. K. Naskar, Soft-Templated Mesoporous Carbons: Chemistry and Structural Characteristics, in Polymer Precursor-Derived Carbon, ACS Symposium Series, 2014, 1173, 61–83 Search PubMed.
- R. Vanyúr, L. Biczók and Z. Miskolczy, Colloids Surf., A, 2007, 299, 256–261 CrossRef.
- E. A. Crespo, N. Schaeffer, J. A. P. Coutinho and G. Perez-Sanchez, J. Colloid Interface Sci., 2020, 574, 324–336 CrossRef CAS PubMed.
- K. Dong, X. Liu, H. Dong, X. Zhang and S. Zhang, Chem. Rev., 2017, 117, 6636–6695 CrossRef CAS.
- M. A. Firestone, P. G. Rickert, S. Seifert and M. L. Dietz, Inorg. Chim. Acta, 2004, 357, 3991–3998 CrossRef CAS.
- I. Kilpeläinen, H. Xie, A. King, M. Granstrom, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142–9148 CrossRef.
- B. G. Janesko, Phys. Chem. Chem. Phys., 2011, 13, 11393–11401 RSC.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- Y. Zhang, H. He, K. Dong, M. Fan and S. Zhang, RSC Adv., 2017, 7, 12670–12681 RSC.
- A. Casas, J. Palomar, M. V. Alonso, M. Oliet, S. Omar and F. Rodriguez, Ind. Crops Prod., 2012, 37, 155–163 CrossRef CAS.
- P. A. Heiney, in Handbook of Liquid Crystals, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2014, pp. 1–47 Search PubMed.
- J. Grolik, Ł. Dudek and J. Eilmes, Tetrahedron Lett., 2012, 53, 5127–5130 CrossRef CAS.
- J. Kirres, K. Schmitt, I. Wurzbach, F. Giesselmann, S. Ludwigs, M. Ringenberg, A. Ruff, A. Baro and S. Laschat, Org. Chem. Front., 2017, 4, 790–803 RSC.
- J. Zhang, B. Dong, L. Zheng, N. Li and X. Li, J. Colloid Interface Sci., 2008, 321, 159–165 CrossRef CAS.
- T. Inoue, B. Dong and L. Q. Zheng, J. Colloid Interface Sci., 2007, 307, 578–581 CrossRef CAS PubMed.
- N. Goujon, M. Forsyth, L. F. Dumée, G. Bryant and N. Byrne, Phys. Chem. Chem. Phys., 2015, 17, 23059–23068 RSC.
- X. W. Li, J. Zhang, B. Dong, L. Q. Zheng and C. H. Tung, Colloids Surf., A, 2009, 335, 80–87 CrossRef CAS.
- A. E. Bradley, C. Hardacre, J. D. Holbrey, S. Johnston, S. E. J. McMath and M. Nieuwenhuyzen, Chem. Mater., 2002, 14, 629–635 CrossRef CAS.
- P. A. Hunt, C. R. Ashworth and R. P. Matthews, Chem. Soc. Rev., 2015, 44, 1257–1288 RSC.
- M. V. Fedotova and S. E. Kruchinin, J. Mol. Liq., 2011, 164, 201–206 CrossRef CAS.
- O. G. Sas, I. Domínguez, B. González and Á. Domínguez, J. Environ. Manage., 2018, 228, 475–482 CrossRef CAS PubMed.
- A. H. Turner, E. L. Byrne, T. Pereira and J. D. Holbrey, Phys. Chem. Chem. Phys., 2020, 22, 10219–10226 RSC.
- E. J. González, I. Díaz, M. Gonzalez-Miquel, M. Rodríguez and A. Sueiras, Sep. Purif. Technol., 2018, 201, 214–222 CrossRef.
- X. Periole and S.-J. Marrink, The Martini Coarse-Grained Force Field, in Biomolecular Simulations, Humana Press, Totowa, NJ, 2013, pp. 533–565 Search PubMed.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. De Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS PubMed.
- C. A. López, A. J. Rzepiela, A. H. de Vries, L. Dijkhuizen, P. H. Hünenberger and S. J. Marrink, J. Chem. Theory Comput., 2009, 5, 3195–3210 CrossRef.
- R. Alessandri, J. J. Uusitalo, A. H. De Vries, R. W. A. Havenith and S. J. Marrink, J. Am. Chem. Soc., 2017, 139, 3697–3705 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Methodologies and supplementary results. See DOI: https://doi.org/10.1039/d2nr02875a |
| This journal is © The Royal Society of Chemistry 2022 |