
Recent progress in polydiacetylene mechanochromism†
Bratati
Das
 ,
Seiko
Jo
,
Jianlu
Zheng
,
Jiali
Chen
and
Kaori
Sugihara
*
,
Seiko
Jo
,
Jianlu
Zheng
,
Jiali
Chen
and
Kaori
Sugihara
*
Institute of Industrial Science, The University of Tokyo, 4-6-1 Komaba Meguro-Ku, Tokyo 153-8505, Japan. E-mail: kaori-s@iis.u-tokyo.ac.jp
First published on 19th January 2022
Abstract
Polydiacetylenes (PDAs) are a family of mechanochromic polymers that change color from blue to red and emit fluorescence when exposed to external stimuli, making them extremely popular materials in biosensing. Although several informative reviews on PDA biosensing have been reported in the last few years, their mechanochromism, where external forces induce the color transition, has not been reviewed for a long time. This mini review summarizes recent progress in PDA mechanochromism, with a special focus on the quantitative and nanoscopic data that have emerged in recent years.
 Bratati Das | Bratati Das obtained her doctoral degree in 2019 from the Department of Physics, Visva-Bharati University, India. She is currently a postdoctoral fellow under the guidance of Dr Kaori Sugihara in the Laboratory of Biophysical Engineering, Institute of Industrial Science at the University of Tokyo. Her research interests mainly focus on the development of Bio-MEMS based on a mechanochromic polymer. |
 Seiko Jo | Seiko Jo received her BSc degree in Chemical Engineering from the University of Tokyo, Japan, in 2021. She is now pursuing her MS degree under the supervision of Dr Kaori Sugihara in the School of Chemical Engineering at the University of Tokyo, Japan. Her research focuses on polydiacetylene mechanochromism. |
 Jianlu Zheng | Jianlu Zheng obtained her MS degree in Materials Engineering from Shenzhen University (China) in 2019. She is currently a PhD student under the guidance of Prof. Kaori Sugihara in the Department of Chemical System Engineering at the University of Tokyo. Her research interests mainly focus on the development of nano-force sensors operated under liquid conditions. |
 Jiali Chen | Jiali Chen received her BSc degree in Chemical Engineering and Technology from Nanjing Tech University, China, in 2021. She is currently a graduate student under the supervision of Prof. Kaori Sugihara in the Institute of Industrial Science at the University of Tokyo, Japan. Her current research interests focus on polydiacetylene mechanochromism at the sub-crystal level. |
 Kaori Sugihara | Kaori Sugihara received her Dr Sc. at ETH Zurich (2012). After being a postdoctoral fellow at Max Planck Institute for Intelligent Systems (2012–2014) and an assistant professor at the University of Geneva (2014–2020), she joined the Institute of Industrial Science at the University of Tokyo as a lecturer in 2020. Her main research interest is electrical and mechanical characterization of self-assembled lipid nanostructures. |
1. Introduction
Mechanochromic polymer polydiacetylenes (PDAs) have garnered attention due to their potential for applications in chemo-biosensing and their unique force-sensing mechanism that can be used to detect forces impossible to quantify by conventional techniques such as piezoresistive1,2 or capacitive tactile sensors.3,4 They were first reported by Wegner in 1969.5 Their mechanosensitive conjugated backbone with customizable side chains reacts to a specific type of external perturbation, which turns their original blue color to red and causes them to fluoresce. They are frequently fabricated using lipid monomers, where a polar group is attached on one side of the carbon chains, which then self-assemble into vesicles,6,7 supported bilayers,8,9 and monolayers.10 Upon ultraviolet (UV) irradiation, they polymerize into PDA (Fig. 1).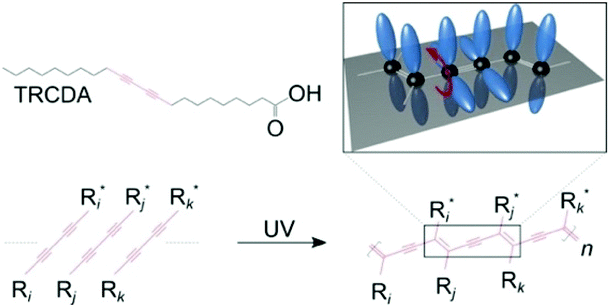 | ||
| Fig. 1 Chemical structures of the 10,12-tricosadiynoic acid (TRCDA) monomers, their polymerization by UV light, and a 3D scheme, where the p-orbitals of the carbon atoms (black) are colored in blue. This figure has been reproduced from ref. 61 with permission from the American Chemical Society, copyright 2020. | ||
Many papers have been published in the past few decades, where researchers have attempted to use PDAs as chromic and fluorescent sensors for the detection of temperature,11 pH changes,12 mechanical stimuli,13 ions,14 solvents,15 light,16 surfactants,17 bacteria,18 and other biomolecules19 such as peptides,20,21 as well as for other non-sensing applications.22–31 Significant progress has been made to improve their specificity by synthesizing monomers with different headgroups.32–34 For example, a PDA with an epoxy headgroup was shown to present selectivity toward mercury(II).32 The recent introduction of a differential approach, in which the color change of several types of PDAs was used as a fingerprint to identify solvents,15 has further improved their selectivity. These promising results have paved the way for overcoming the rather weak specificity of PDAs and the development of a convenient colorimetric sensor.
The mechanochromic properties of PDAs have been extensively characterized by the Langmuir–Blodgett technique,35 electron paramagnetic resonance (EPR),36 X-ray crystallography,37 ultraviolet-visible (UV-vis) spectroscopy,38 fluorescence spectroscopy,20 infrared (IR) spectroscopy,39 Raman spectroscopy,40 nuclear magnetic resonance (NMR) spectroscopy,41 scanning tunneling microscopy (STM),42 atomic force microscopy (AFM),43 fluorescence microscopy,44 and electrical conductivity measurements.45 These experimental efforts, combined with theoretical studies,46 have provided a mechanistic model where the torsion of the sidechains shortens the π conjugation in the backbone, thus broadening the bandgap to cause a blueshift in the absorption.
Despite our general understanding of their mechanism and the large number of reports in biosensing, the lack of a quantitative understanding of the color-changing mechanism of PDAs at the nanoscale has hindered further development until recently, when AFM was introduced.44,47,48 The recent technological advancement of AFM could shift the status of PDA as a quantitative mechanosensor at the nanoscale, which may lead to another leap in application development. Although there have been many informative reviews on PDAs, especially from the perspective of biosensing,49–59 their mechanochromism, where external forces stimulate the blue-to-red transition, has not been reviewed since a brief report by Burns et al. in 2004.60 In this mini review, we summarize the recent progress in PDA mechanosensing.
2. Polydiacetylene mechanosensing
2.1 Surface pressure
Tomioka et al.10 investigated the dependence of the excitonic absorption on the surface pressure by observing the in situ reflection spectra of PDA made of poly-(heptacosa-10,12-diynoic acid) at the N2 gas–water interface. A reversible color change was observed when the PDA monolayer was compressed using a Langmuir–Blodgett trough at pressures between 3 and 25 mN m−1. This reversible color change was reproducible after several cycles of compression and re-expansion. The fact that thermochromically irreversible poly-(heptacosa-10,12-diynoic acid) presented reversibility in its mechanochromism indicates that the type of stimuli or the assembled polymer structure also affects the reversibility of PDA.2.2 Tensile strain
Reversible tensile strain, or (stretch)-induced phase transitions, was observed by Nallicheri et al.13 in segmented polyurethanes containing a small fraction of PDAs made of 2,4-hexadiyne-l,6-diol and 5,7-dodecadiyne-1,12-diol in hard-segmented structures. Polyurethanes (PUs) are a class of segmented copolymers composed of soft and hard segments. The diacetylene groups were linked to the hard segments via a chain extender. When the strain levels were greater than 250%, an irreversible hard-domain disruption was observed. From visible absorption spectroscopy, it was confirmed that during tensile elongation, stress was transmitted from the soft segments to the hard domains, where the hard segments were oriented perpendicular to the stretch direction. This resulted in tensile (or shear) stress on PDAs oriented along the stretch direction, causing the color transformation.2.3 Compression
Nakamitsu et al.64 prepared a highly sensitive compression stress sensor by integrating a stimuli-responsive layered PDA and dry liquids (DLs) on a filter paper substrate to induce a “response cascade” (Fig. 2a). Macroscopic compression stresses induced the collapse of the DLs, which were micrometer-sized particles that consisted of liquid droplets covered by solid powders, starting the response cascade. As a consequence, the DLs released their interior liquid, a polyethyleneimine (PEI) solution, in response to the mechanical stress. The subsequent PEI–PDA interaction induced color changes in the layered PDA. As the strength and duration of the compression increased, the intensity of the red color was enhanced. This compression stress sensor visualized weak compression stresses on the order of 3.9 Pa–4.9 kPa, comparable to those upon the impact of an object. The device has the potential for the visualization and measurement of weak mechanical stresses in the biomedical and healthcare fields.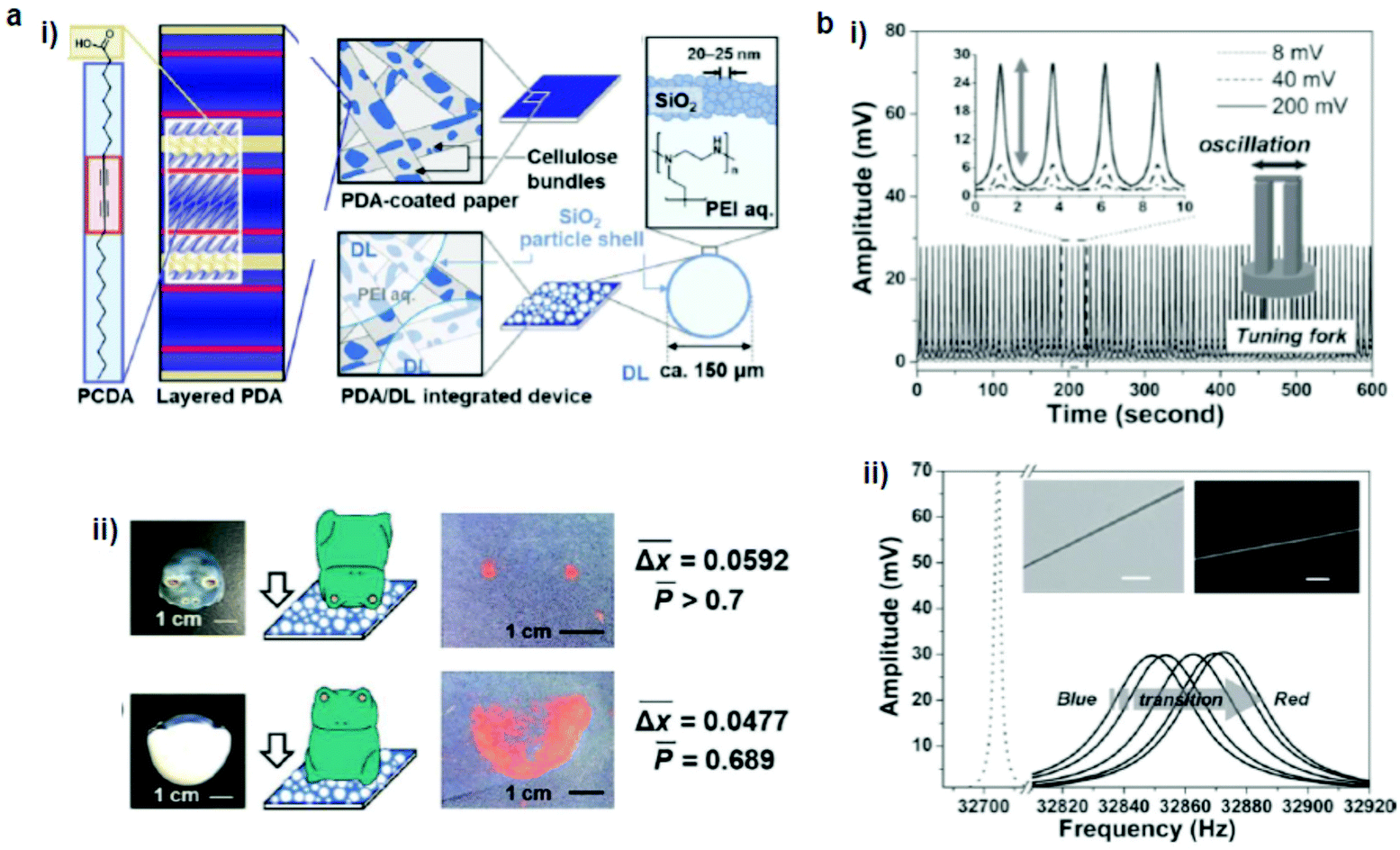 | ||
| Fig. 2 (a) (i) Schematic illustration of a PDA/dry liquid-integrated paper device. (ii) Two different settings, where the compression stresses were applied using a ceramic frog ornament. (b) A hybrid mechanoresponsive polymer wire fabricated by mixing 10,12-pentacosadiynoic acid and a poly(ethylene oxide) matrix. (i) Time evolution of applied AC voltage, which vibrated the tuning fork to produce a force pulse stimulus. (ii) Oscillation amplitude vs. frequency curves show the transduction process from the blue phase to the red phase. The inset shows the bright-field and fluorescence images of the hybrid wire. Panel (a) has been adapted from ref. 62 with permission from the Wiley Online Library, copyright 2021. Panel (b) has been adapted from ref. 63 with permission from the Wiley Online Library, copyright 2013. | ||
2.4 Shear
Lee et al.68 examined the shear-induced color transition of PDA vesicles in three polymeric solutions dissolved in water using a rheometer: 2% poly(vinyl alcohol) + 1% sodium borate (PVA/B), 15% PVA and 1% hyaluronic acid (HA). In the PVA/B solution at 47 °C, shear at 100 Pa was sufficient to induce a blue–red transition in the PDA vesicles made of 10,12-pentacosadiynoic acid (PCDA). In contrast, there was no color change in the PVA or HA solutions. The authors interpreted the color change as a result of the alteration of the structure of the PDA polymeric backbone owing to perturbation. However, the experimental setup used was not able to distinguish between the effect of the shear and the increased temperature due to mixing, which made it difficult to conclude whether the shear itself induced the color change.2.5 Oscillation
Feng et al.63 fabricated a hybrid mechanoresponsive polymer wire by mixing PDA made of 10,12-pentacosadiynoic acid into poly(ethylene oxide) (PEO) as a polymer matrix (Fig. 2b). The color change was induced by high-frequency mechanical oscillations controlled by AC voltages. The efficiency of the color transition increased by decreasing the diameter of the wires, because smaller diameters minimize the defect density and thus improve the mechanical transduction. This process was characterized using Raman spectroscopy and the resonant frequency shifts of the hybrid wire. Such a combination of cost-effective, easily processed polymers with PDA may be useful as a force sensor for detecting stresses and damages at the microscale.2.6 Swelling
Park et al.65 developed a PDA–polydimethylsiloxane (PDMS) composite sensor, which underwent a blue-to-red colorimetric transition during swelling upon exposure to different solvents (Fig. 3a). The composite sensor was easy to fabricate by the mixing–irradiation–curing method. The rate of swelling and the color change depend on the alkyl chain length of the saturated aliphatic hydrocarbons. The mechanism of this color transition is complicated. The authors explained that the swelling of PDMS induced mechanical strain on the embedded PDA, which exposed the unreacted monomers present in the PDA crystals to the solvent and dissolved them, creating voids in the PDA structures. This combined effect of mechanical strain and void creation caused a decrease in the interchain interactions in PDA and served as a driving force for a PDA phase transition. The same group prepared a PDMS microbead–PDA composite sensor.66 This sensor underwent a blue-to-red color change in response to the hydrocarbons of the shorter alkyl chains and enabled visual differentiation between n-pentane, n-heptane, n-nonane, and n-undecane (Fig. 3b). The degree of swelling of the PDA–PDMS composite beads in these hydrocarbons was inversely correlated with the length of the alkyl chains.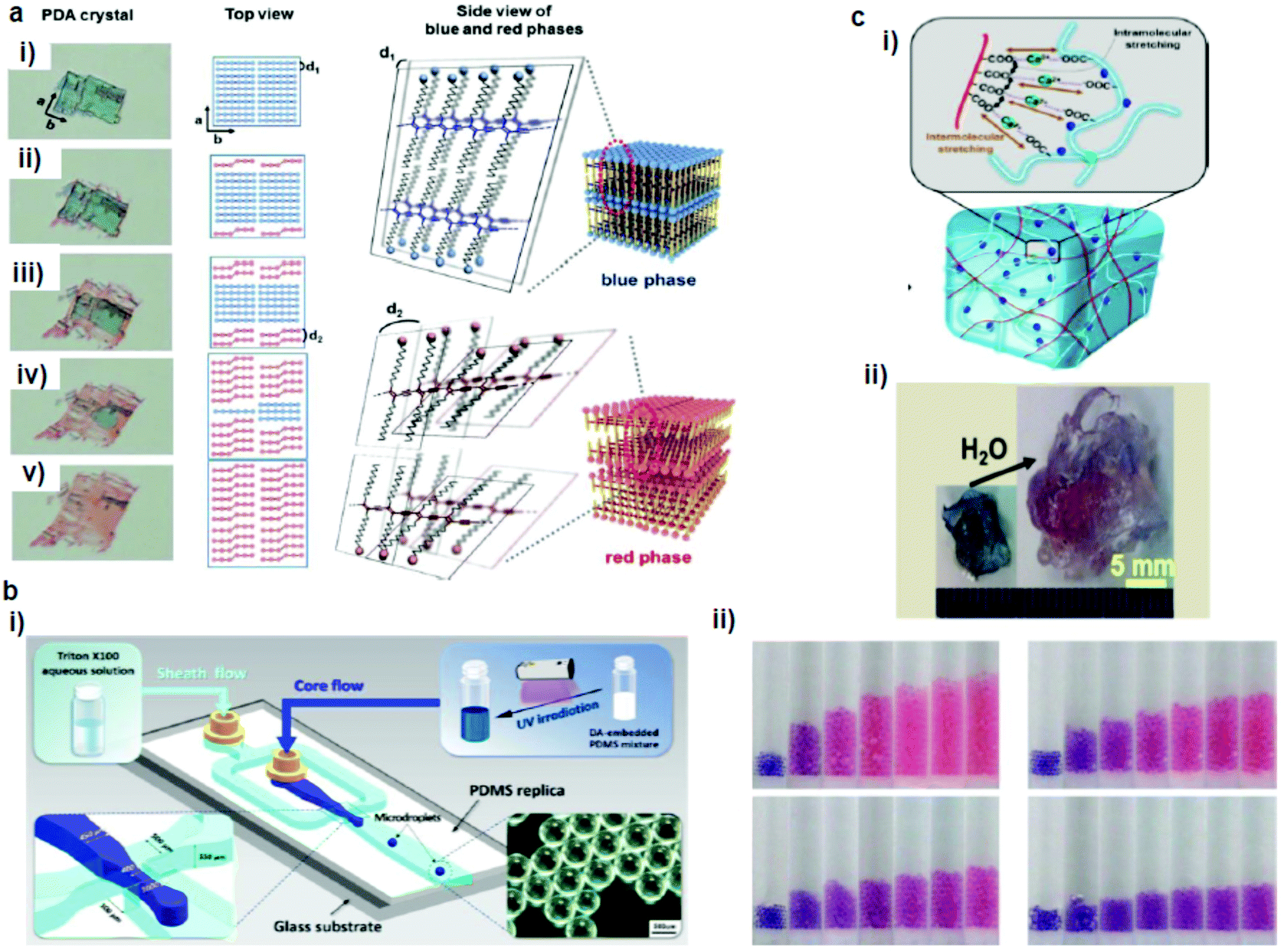 | ||
| Fig. 3 (a) A PDA microcrystal embedded in PDMS experienced drastic color and shape changes during PDMS swelling in octane at (i) 0, (ii) 5, (iii) 10, (iv) 15, and (v) 23 min. (b) Schematic diagram of a microfluidic chip used for the generation of PDA-embedded PDMS microdroplets and an optical microscopy image of the microdroplets. (ii) Photographs of the tubes that contain the PDA-embedded PDMS microbeads upon exposure to pentane (upper left), heptane (upper right), nonane (lower left), and undecane (lower right). (c) (i) Schematic diagram of alginate hydrogel-assisted PDA and (ii) its colorimetric response upon water absorption. Panel (a) has been adapted from ref. 65 with permission from the Wiley Online Library, copyright 2014. Panel (b) has been adapted from ref. 66 with permission from the American Chemical Society, copyright 2015. Panel (c) has been adapted from ref. 67 with permission from the American Chemical Society, copyright 2015. | ||
Seo et al.67 prepared a sensory system in which one-dimensional (1D) PDA nanofibers were integrated into the three-dimensional (3D) matrix of a hygroscopic alginate hydrogel to detect water (Fig. 3c). Alginate has a dramatic volume swelling property when it absorbs water. This volumetric expansion induced mechanical stress on the PDA nanofibers, which deformed the PDA backbone, leading to the color transition.
2.7 Macroscopic scratch and rubbing
Chae et al.69 prepared a directly writable crayon-like PDA–wax composite sensor by embedding different PDAs into paraffin wax (melting temperature 58–62 °C) (Fig. 4a). The wax mixed with 2% PCDA showed irreversible thermochromism, whereas that with PCDA-mBzA presented a reversible color change. A mechanically drawn PDA image showed a colorimetric transition from blue to red upon heating or soft rubbing. Optical microscopy showed that diacetylene and the wax formed a complex at the single-crystal level, where the wax molecules intercalated between the diacetylene crystals. Upon heating, the PDA crystals underwent significant shrinkage because of the release of unreacted diacetylene monomers and embedded wax molecules from the crystals. The release of the wax molecules caused a distortion of the arrayed p-orbitals and thus the blue-to-red color transition. The same group developed a reversible mechanochromic PDA by the self-assembly of diphenyldisulfide-containing bisdiacetylene (PCDA-4APDS).72 The temperature of the powder remained below the thermochromic transition temperature (80 °C) of PDA during grinding, which proves that the color change was caused by mechanical stress. The mechanical energy produced from grinding transferred to the alkyl chain of PDA, which caused a partial distortion of the conjugated ene–yne backbone, shortened the effective conjugation length of PDA, and thus induced the color transition.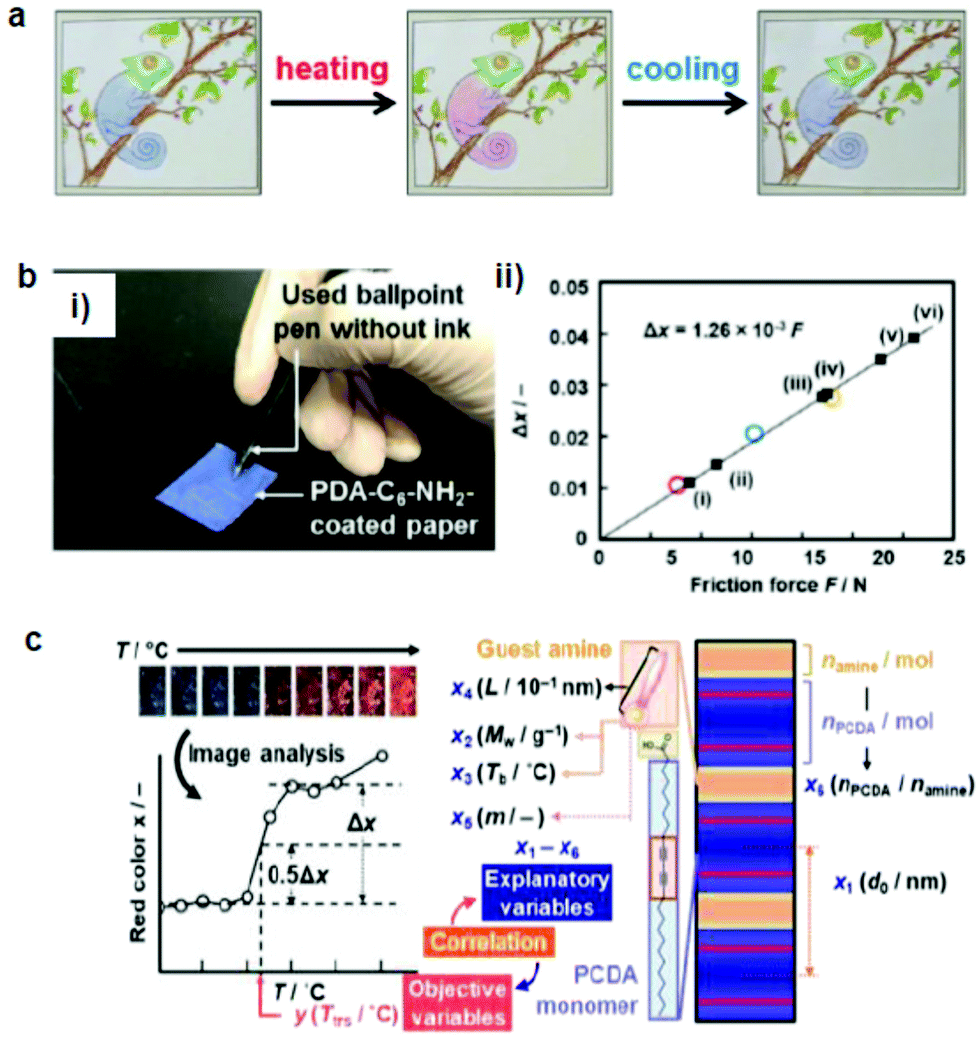 | ||
| Fig. 4 (a) A hand-writable PDA sensor prepared by using the mixing–molding polymerization process. Drawings based on these pens responded to heating, cooling, and rubbing. (b) (i) PDA-C6-NH2-coated paper was tested against writing pressure. (ii) The intensity of the red color (Δx) vs. the applied friction force. (c) Sparse modeling toward the prediction of the color-transition temperatures based on the sample image analysis. Panel (a) has been adapted from ref. 69 with permission from the Wiley Online Library, copyright 2016. Panel (b) has been adapted from ref. 70 with permission from the Wiley Online Library, copyright 2018. Panel (c) has been adapted from ref. 71 with permission from the Royal Society of Chemistry, copyright 2020. | ||
Ishijima et al.73 prepared an organic-layered material using PDA with the intercalation of guest organic amines, that is, alkyl amines and diamines. The amine-intercalated PDA showed a tunable temperature (46–106 °C) and a mechanoresponsive color change. Rubbing gradually changed the PDA color, which indicated that force was the reason for the color change rather than the heat during rubbing. The same group fabricated a paper-based friction detector based on the amine-intercalated PDA (Fig. 4b).70 The composite was formed through self-organization followed by polymerization, which was then homogeneously coated on a sheet of paper. The application of friction force by a cage showed a force-dependent color change with a detectable range of 7.6–23.0 N. The properties were tuned by using various types of guest ions and amines without a complex synthesis. Measurement of the writing pressure was demonstrated using a paper device. Weak, moderate, and strong writing forces in the range of 8.80–31.1 N were visualized and quantitatively detected by the friction force. The group further developed a layered PDA/PEI composite-coated paper device to detect weak forces (Fig. 4c).71 In this work, tooth-brushing forces in the range of 0.91–6.60 N were used as a model to create weak forces. Soft, normal, and hard brushing forces were applied to the paper-based PDA device, which showed a blue-to-red color change. Sparse modeling was used to analyze the PDA color images for selecting the best interlayer guest.
2.8 Atomic force microscopy
PDA studies by AFM were pioneered by Burns et al.74 They observed an irreversible blue-to-red transformation by applying shear forces with an AFM tip. Out-of-plane rotations of the side chains caused by the tip–PDA interactions disrupted the π-orbital overlap, causing the mechanochromic transition. Although this work marked an important step toward PDA mechanochromism research at the nanoscale, the shear force used was only shown qualitatively since standard AFM can only quantify forces vertical to the substrate. The same group also prepared ultrathin PDA films using the horizontal Langmuir deposition technique from two diacetylene monomers, PCDA(I) and N-(2-ethanol)-10,12-pentacosadiynamide (PCEA) (II).60 AFM or near-field scanning optical microscopy (NSOM) tips were used to apply qualitative shear forces. Their data revealed that the friction depends on the angle between the polymer backbone and the scanning direction; the maximum occurred when the scanning direction was perpendicular to the backbones. In particular, PCEA exhibited a threefold friction anisotropy depending on the scanning direction relative to the polymer backbone. These studies revealed the anisotropic friction of PDA for the first time based on scanning microscopy.Polacchi et al.48 prepared a PDA derivative with an amplified fluorescence response by covalently linking a tetrazine fluorophore to diacetylene (poly-TzDA, Fig. 5). The fluorescence emission wavelength from tetrazine matched with the absorption of blue PDA, causing an energy transfer only when the system was in the blue phase. Therefore, in the monomer state, the sample fluoresced from tetrazine, which was quenched during the polymerization owing to the increased amount of blue PDA.
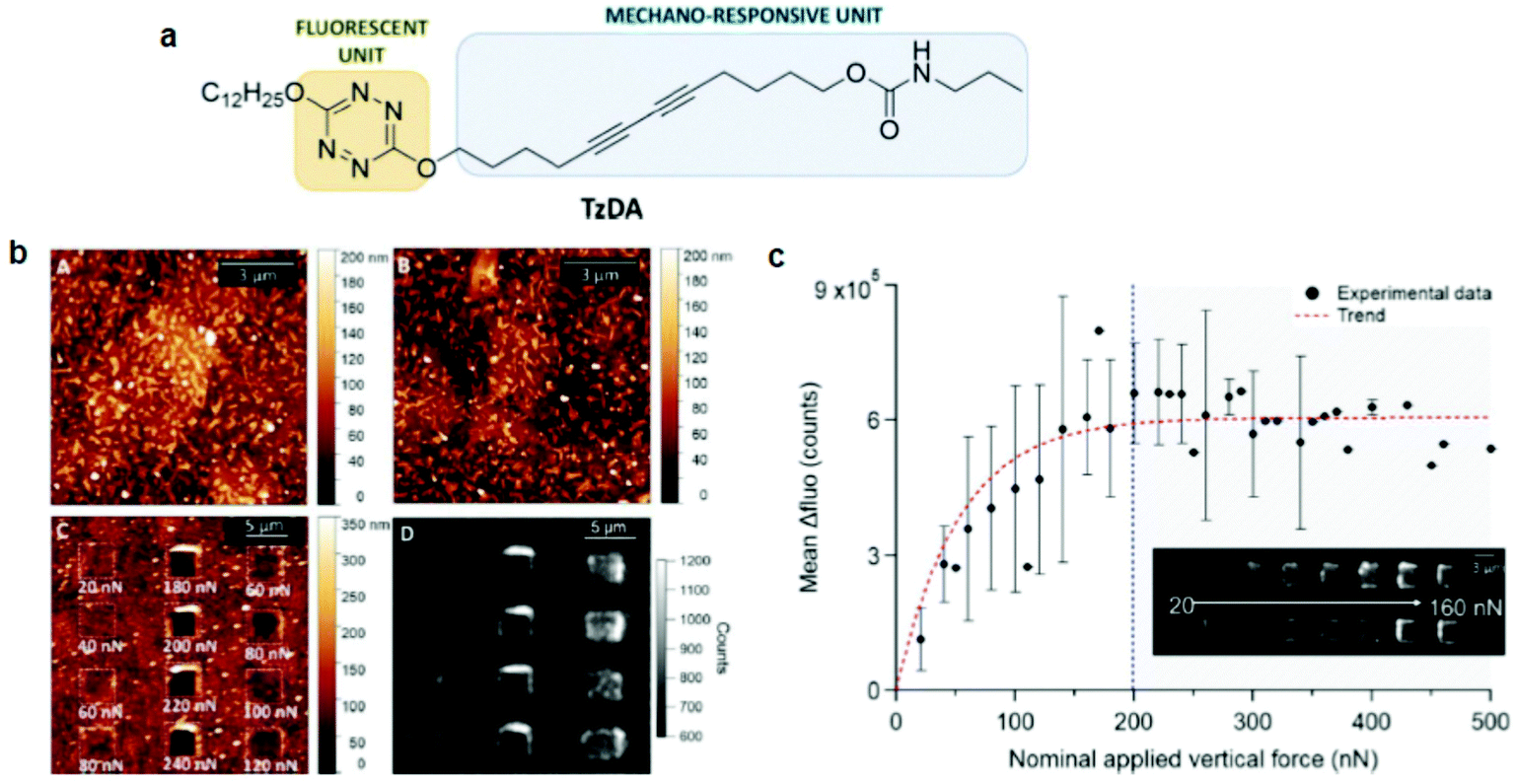 | ||
| Fig. 5 (a) Schematic diagram of a molecular design of TzDA. (b) AFM morphology images of the (A) monomer, (B) polymer, and (C) polymer after mechanical stimulation. (D) Fluorescence image of poly-TzDA after the application of various forces. (c) A plot showing the recovered fluorescence signal vs. the applied vertical force. This figure has been adapted from ref. 48 with permission from the Royal Society of Chemistry, copyright 2019. | ||
During the blue-to-red transition, the energy transfer was again weakened and the tetrazine fluorescence was efficiently restored. Poly-TzDA was further characterized using AFM coupled with fluorescence microscopy. A vertical force in the range of 20–500 nN was applied to the tip with a scanning speed of 6.1 μm s−1. Fluorescence images showed that poly-TzDA restored its fluorescence emission locally from the scratched part of the film. Such a fluorescence resonance energy transfer (FRET)-based enhancement of the PDA fluorescence may be used to improve its sensitivity toward sensing applications.
Although the above mentioned studies showed that PDA is mechanochromic at the nanoscale and that shear force is the key to inducing the blue-to-red transition, they failed to quantify the shear forces required to activate PDAs. This is because standard AFM can measure and manipulate only the forces vertical to the substrate. In 2021, we overcame this bottleneck by utilizing quantitative friction force microscopy, which measures lateral forces (Fig. 6).47 Friction force microscopy is an AFM-based technique that enables the quantification of forces lateral to the substrate by calibrating lateral laser deflection into forces.75,76 The use of this experimental technique was partially enabled by our recent identification of an error source in the wedge calibration method over the nanonewton range.77 Quantitative friction force microscopy combined with fluorescence microscopy confirmed that PDA reacts only to lateral forces, F∥, as the lateral force presents a perfect correlation with Δfluorescence, whereas the vertical force does not (Fig. 6a). The setup also disproved the previously claimed hypothesis that the edges of the polymer crystals exhibit higher force sensitivity than the rest of the crystal. This was accomplished by correlating Δfluorescence and lateral forces at the edge and the bulk separately. In addition, we reported a link between mechanochromism and thermochromism, which can be attributed to the fact that both work and heat are different means of providing the same transition energy (Fig. 6b and c).47 These data provide the first insight into quantitative, anisotropic PDA mechanochromism at the nanoscale, where the crystal-to-amorphous transition of the PDA structure seems to play an important role. Friction force microscopy combined with fluorescence microscopy can be also used to characterize other mechanosensitive polymers78–80 and mechanophores81 in the future.
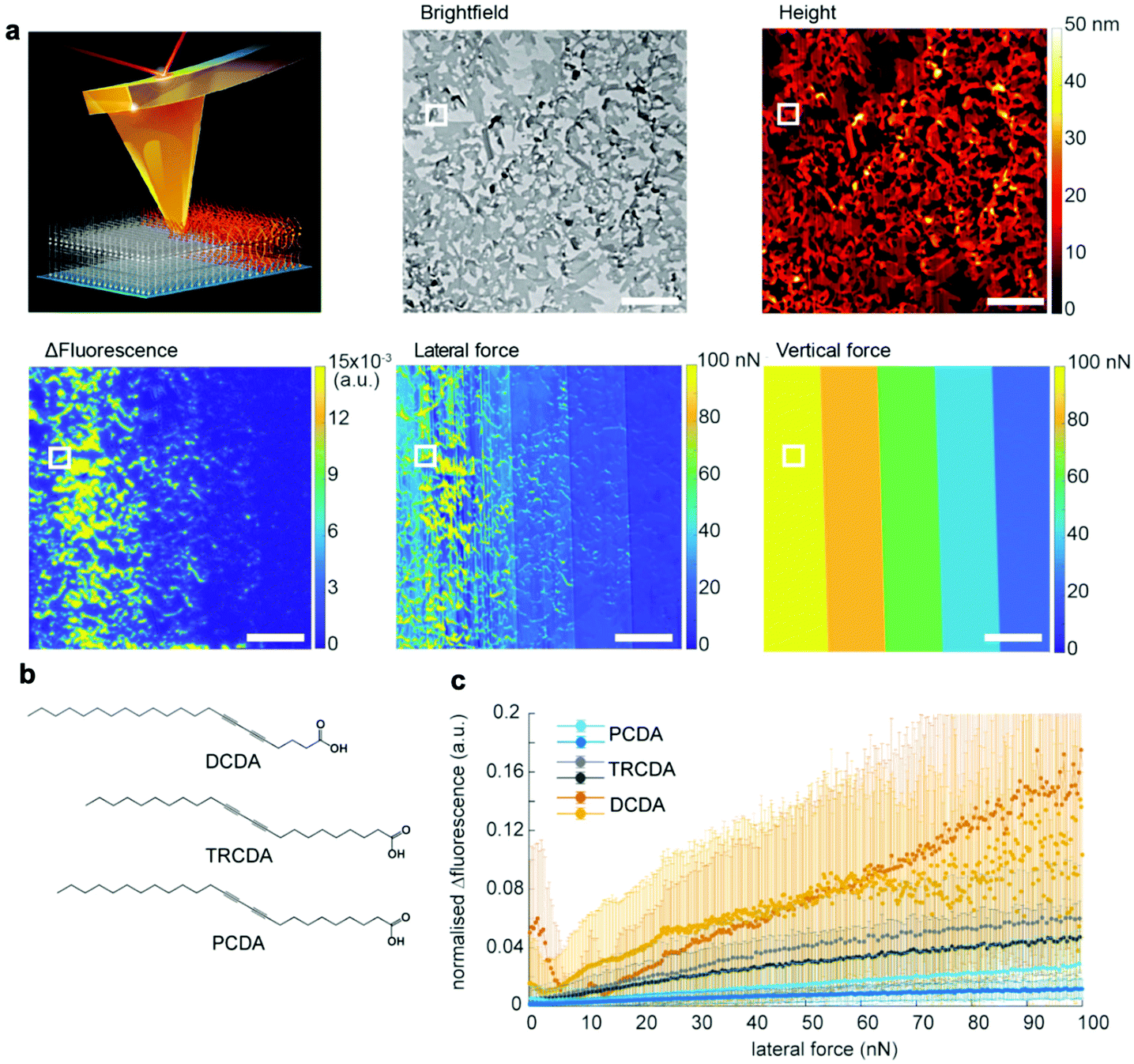 | ||
| Fig. 6 Characterization of 5,7-docosadiynoic acid (DCDA) Langmuir–Blodgett films by lateral force microscopy coupled with fluorescence microscopy. (a) Scheme of the experiment, a bright-field microscopy image, an AFM height image, Δfluorescence before and after scratching, a lateral force map, and a vertical force map. The scale bar is 10 μm. (b) Chemical structures of the used monomers with different color transition temperatures (DCDA: 50 °C, TRCDA: 55 °C, and PCDA: 65 °C). (c) Fluorescence increase vs. lateral force. This figure has been adapted from ref. 47 with permission from the American Chemical Society, copyright 2021. | ||
3. Conclusions and future perspective
This mini review provides an overview of the recent progress in PDA mechanochromism, where various forces can be used as stimuli to induce the color change. The quantitative and anisotropic force–fluorescence correlation at the nanoscale obtained in recent years has just begun to provide a new perspective on its mechanism. In biosensing, bound ligands are expected to exert forces on PDAs, as in the case of an AFM cantilever, yet how each molecule does that is unclear in many cases. Ligands are often surface-bound,82 deep-bound (where the receptor is embedded within PDAs),83 inserted,84 or aggregated85 to initiate the color transition (Fig. 7; also see the categorization shown in Table S1†). Nevertheless, when binding to a receptor exposed on a PDA headgroup (surface-bound), there is no obvious reason why the ligand should apply force to PDA except that some receptors undergo a conformational change after binding that induces stress on PDA; alternatively, the ligands may eventually insert into the PDA matrix, where the role of binding was to elevate the local ligand concentration right above the PDA surface. Some binding events may not be interpreted as force application. For example, we previously reported that antimicrobial peptides are “partially melting PDAs” (inducing the solid-to-liquid phase transition of side chains) by increasing disorder in the system, which turns their blue color to red.61 In such a case, although the peptide–PDA mechanical interaction must occur, it is difficult to attribute a certain amount of force to the change in the color. As a future perspective, we believe that learning from the quantitative information obtained in the mechanochromism studies and applying them in biosensing research to understand the detailed mechanism of how each ligand induces the color change will be useful for improving the sensitivity and selectivity of PDA sensors.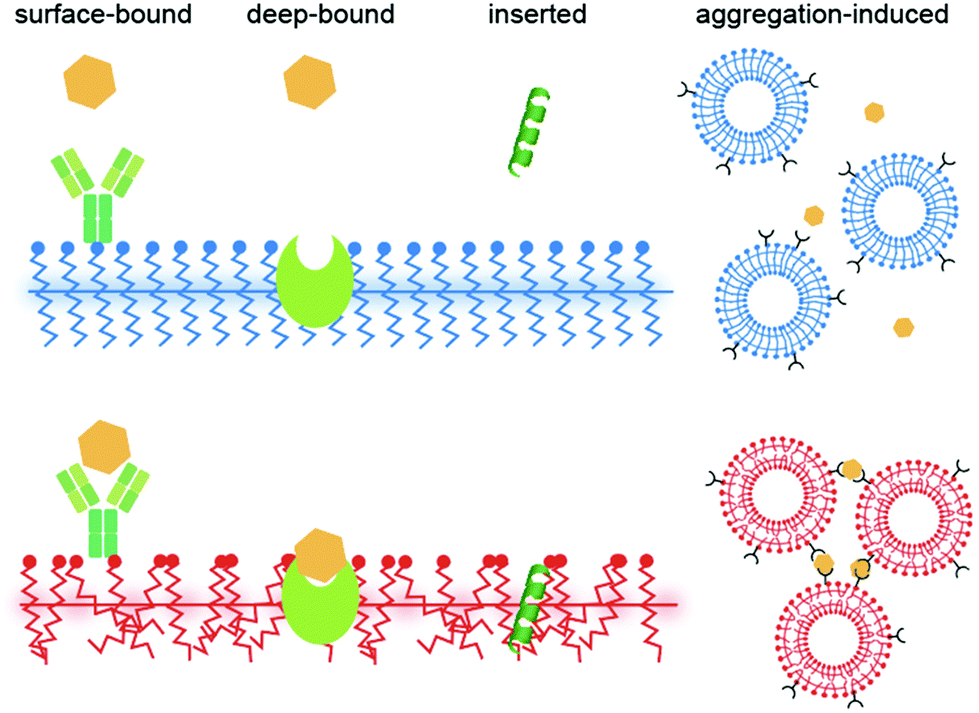 | ||
| Fig. 7 Categories of ligand–PDA interactions in biosensing. | ||
Author contributions
K. S. initiated the entire work and wrote the article with B. D. S. J., J. Z., and J. C. contributed to the literature search, the investigation of the current trends and making Table S1 in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of the research leading to these results has received funding from the Shiseido Female Researcher Science Grant, the UTEC-UTokyo FSI Research Grant Program, the FY 2020 University of Tokyo Excellent Young Researcher, the Japan Society for the Promotion of Science (JP20K22324), the Takeda Science Foundation, the Mitsubishi Foundation, the Inoue Foundation for Science, the Naito Foundation and the Kanamori Foundation.Notes and references
- Y. L. Tai and Z. G. Yang, J. Mater. Chem. B, 2015, 3, 5436–5441 RSC.
- N. Hu, Y. Karube, M. Arai, T. Watanabe, C. Yan, Y. Li, Y. L. Liu and H. Fukunaga, Carbon, 2010, 48, 680–687 CrossRef CAS.
- S. C. B. Mannsfeld, B. C. K. Tee, R. M. Stoltenberg, C. V. H. H. Chen, S. Barman, B. V. O. Muir, A. N. Sokolov, C. Reese and Z. N. Bao, Nat. Mater., 2010, 9, 859–864 CrossRef CAS PubMed.
- C. Ge and E. Cretu, J. Micromech. Microeng., 2017, 27(4), 045002 CrossRef.
- G. Wegner, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1969, 24, 824–832 CrossRef CAS.
- O. Yarimaga, J. Jaworski, B. Yoon and J. M. Kim, Chem. Commun., 2012, 48, 2469–2485 RSC.
- B. Yoon, S. Lee and J. M. Kim, Chem. Soc. Rev., 2009, 38, 1958–1968 RSC.
- R. Volinsky, S. Kolusheva, A. Berman and R. Jelinek, Langmuir, 2004, 20, 11084–11091 CrossRef CAS PubMed.
- R. Volinsky, M. Kliger, T. Sheynis, S. Kolusheva and R. Jelinek, Biosens. Bioelectron., 2007, 22, 3247–3251 CrossRef CAS PubMed.
- Y. Tomioka, N. Tanaka and S. Imazeki, J. Chem. Phys., 1989, 91, 5694–5700 CrossRef CAS.
- C. Girard-Reydet, R. D. Ortuso, M. Tsemperouli and K. Sugihara, J. Phys. Chem. B, 2016, 120, 3511–3515 CrossRef CAS PubMed.
- S. J. Kew and E. A. H. Hall, Anal. Chem., 2006, 78, 2231–2238 CrossRef CAS PubMed.
- R. A. Nallicheri and M. F. Rubner, Macromolecules, 1991, 24, 517–525 CrossRef CAS.
- Q. Xu, S. Lee, Y. Cho, M. H. Kim, J. Bouffard and J. Yoon, J. Am. Chem. Soc., 2013, 135, 17751–17754 CrossRef CAS PubMed.
- S. Dolai, S. K. Bhunia, S. S. Beglaryan, S. Kolusheva, L. Zeiri and R. Jelinek, ACS Appl. Mater. Interfaces, 2017, 9, 2891–2898 CrossRef CAS PubMed.
- X. Chen, L. Hong, X. You, Y. L. Wang, G. Zou, W. Su and Q. J. Zhang, Chem. Commun., 2009, 1356–1358, 10.1039/b820894h.
- X. Q. Chen, J. Lee, M. J. Jou, J. M. Kim and J. Yoon, Chem. Commun., 2009, 3434–3436, 10.1039/b904542b.
- Z. F. Ma, J. R. Li, M. H. Liu, J. Cao, Z. Y. Zou, J. Tu and L. Jiang, J. Am. Chem. Soc., 1998, 120, 12678–12679 CrossRef CAS.
- J. M. Kim, Y. B. Lee, D. H. Yang, J. S. Lee, G. S. Lee and D. J. Ahn, J. Am. Chem. Soc., 2005, 127, 17580–17581 CrossRef CAS PubMed.
- R. D. Ortuso, U. Cataldi and K. Sugihara, Soft Matter, 2017, 13, 1728–1736 RSC.
- S. Kolusheva, L. Boyer and R. Jelinek, Nat. Biotechnol., 2000, 18, 225–227 CrossRef CAS PubMed.
- L. L. Li, X. Q. An and X. J. Yan, Colloids Surf., B, 2015, 134, 235–239 CrossRef CAS PubMed.
- M. Ripoll, P. Neuberg, A. Kichler, N. Tounsi, A. Wagner and J. S. Remy, ACS Appl. Mater. Interfaces, 2016, 8, 30665–30670 CrossRef CAS PubMed.
- E. Morin, M. Nothisen, A. Wagner and J. S. Remy, Bioconjugate Chem., 2011, 22, 1916–1923 CrossRef CAS PubMed.
- H. Jiang, X. Y. Hu, S. Schlesiger, M. Li, E. Zellermann, S. K. Knauer and C. Schmuck, Angew. Chem., Int. Ed., 2017, 56, 14526–14530 CrossRef CAS PubMed.
- V. Haridas, S. Sadanandan, P. Y. Collart-Dutilleul, S. Gronthos and N. H. Voelcker, Biomacromolecules, 2014, 15, 582–590 CrossRef CAS PubMed.
- M. A. Desta, C. W. Liao and S. S. Sun, Chem. – Asian J., 2017, 12, 690–697 CrossRef CAS PubMed.
- J. Nishide, T. Oyamada, S. Akiyama, H. Sasabe and C. Adachi, Adv. Mater., 2006, 18, 3120–3124 CrossRef CAS.
- M. Ulaganathan, R. V. Hansen, N. Drayton, H. Hingorani, R. G. Kutty, H. Joshi, S. Sreejith, Z. Liu, J. L. Yang and Y. L. Zhao, ACS Appl. Mater. Interfaces, 2016, 8, 32643–32648 CrossRef CAS PubMed.
- Y. K. Choi, H. J. Kim, S. R. Kim, Y. M. Cho and D. J. Ahn, Macromolecules, 2017, 50, 3164–3170 CrossRef CAS.
- K. Morigaki, T. Baumgart, U. Jonas, A. Offenhausser and W. Knoll, Langmuir, 2002, 18, 4082–4089 CrossRef CAS.
- J. Lee, H. Jun and J. Kim, Adv. Mater., 2009, 21, 3674–3677 CrossRef CAS.
- E. Y. Park, J. W. Kim, D. J. Ahn and J. M. Kim, Macromol. Rapid Commun., 2007, 28, 171–175 CrossRef CAS.
- H. O. Yoo, S. K. Chae, J. M. Kim and D. J. Ahn, Macromol. Res., 2007, 15, 478–481 CrossRef CAS.
- Y. Lifshitz, A. Upcher, O. Shusterman, B. Horovitz, A. Berman and Y. Golan, Phys. Chem. Chem. Phys., 2010, 12, 713–722 RSC.
- H. Sixl, W. Neumann, R. Huber, V. Denner and E. Sigmund, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 142–148 CrossRef CAS PubMed.
- Y. Lifshitz, Y. Golan, O. Konovalov and A. Berman, Langmuir, 2009, 25, 4469–4477 CrossRef CAS PubMed.
- R. R. Chance and G. N. Patel, J. Polym. Sci., Polym. Phys. Ed., 1978, 16, 859–881 CrossRef CAS.
- R. D. Ortuso, N. Ricardi, T. Bürgi, T. A. Wesolowski and K. Sugihara, Spectrochim. Acta, Part A, 2019, 219, 23–32 CrossRef CAS PubMed.
- P. E. Schoen and P. Yager, J. Polym. Sci., Polym. Phys. Ed., 1985, 23, 2203–2216 CrossRef CAS.
- H. Tanaka, M. A. Gomez, A. E. Tonelli and M. Thakur, Macromolecules, 1989, 22, 1208–1215 CrossRef CAS.
- Y. Okawa and M. Aono, J. Phys. Chem., 2001, 115, 2317–2322 CrossRef CAS.
- A. Burns, R. Carpick, D. Sasaki, J. Shelnutt and R. Haddad, Tribol. Lett., 2001, 10, 89–96 CrossRef CAS.
- R. W. Carpick, D. Y. Sasaki and A. R. Burns, Langmuir, 2000, 16, 1270–1278 CrossRef CAS.
- Y. Okawa and M. Aono, Surf. Sci., 2002, 514, 41–47 CrossRef CAS.
- V. Dobrosavljević and R. M. Stratt, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 2781–2794 CrossRef PubMed.
- L. Juhasz, R. D. Ortuso and K. Sugihara, Nano Lett., 2021, 21, 543–549 CrossRef CAS PubMed.
- L. Polacchi, A. Brosseau, R. Metivier and C. Attain, Chem. Commun., 2019, 55, 14566–14569 RSC.
- X. M. Sun, T. Chen, S. Q. Huang, L. Li and H. S. Peng, Chem. Soc. Rev., 2010, 39, 4244–4257 RSC.
- A. C. D. Pires, N. D. F. Soares, L. H. M. da Silva, N. J. de Andrade, M. F. A. Silveira and A. F. de Carvalho, Food Bioprocess Technol., 2010, 3, 172–181 CrossRef.
- J. P. Huo, Q. J. Deng, T. Fan, G. Z. He, X. H. Hu, X. X. Hong, H. Chen, S. H. Luo, Z. Y. Wang and D. C. Chen, Polym. Chem., 2017, 8, 7438–7445 RSC.
- E. Lebegue, C. Farre, C. Jose, J. Saulnier, F. Lagarde, Y. Chevalier, C. Chaix and N. Jaffrezic-Renault, Sensors, 2018, 18, 599 CrossRef PubMed.
- J. T. Wen, J. M. Roper and H. Tsutsui, Ind. Eng. Chem. Res., 2018, 57, 9037–9053 CrossRef CAS.
- X. M. Qian and B. Stadler, Chem. Mater., 2019, 31, 1196–1222 CrossRef CAS.
- Z. J. Zhang, F. Wang and X. Q. Chen, Chin. Chem. Lett., 2019, 30, 1745–1757 CrossRef CAS.
- F. Fang, F. L. Meng and L. Luo, Mater. Chem. Front., 2020, 4, 1089–1104 RSC.
- M. Weston, A. D. Tjandra and R. Chandrawati, Polym. Chem., 2020, 11, 166–183 RSC.
- Q. Huang, W. Wu, K. L. Ai and J. H. Liu, Front. Chem., 2020, 8, 565782 CrossRef CAS PubMed.
- E. Cho and S. Jung, Molecules, 2018, 23, 107 CrossRef PubMed.
- R. W. Carpick, D. Y. Sasaki, M. S. Marcus, M. A. Eriksson and A. R. Burns, J. Phys.: Condens. Matter, 2004, 16, R679–R697 CrossRef CAS.
- J. Nuck and K. Sugihara, Macromolecules, 2020, 53, 6469–6475 CrossRef CAS.
- M. Nakamitsu, K. Oyama, H. Imai, S. Fujii and Y. Oaki, Adv. Mater., 2021, 33, 2008755 CrossRef CAS PubMed.
- H. B. Feng, J. Lu, J. H. Li, F. Tsow, E. Forzani and N. J. Tao, Adv. Mater., 2013, 25, 1729–1733 CrossRef CAS PubMed.
- M. Nakamitsu, K. Oyama, H. Imai, S. Fujii and Y. Oaki, Adv. Mater., 2021, 33, 2008755 CrossRef CAS PubMed.
- D. H. Park, J. Hong, I. S. Park, C. W. Lee and J. M. Kim, Adv. Funct. Mater., 2014, 24, 5186–5193 CrossRef CAS.
- J. Hong, D. H. Park, S. Baek, S. Song, C. W. Lee and J. M. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 8339–8343 CrossRef CAS PubMed.
- S. Seo, J. Lee, M. S. Kwon, D. Seo and J. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 20342–20348 CrossRef CAS PubMed.
- S. S. Lee, E. H. Chae, D. J. Ahn, K. H. Ahn and J. K. Yeo, Korea-Aust. Rheol. J., 2007, 19, 43–47 Search PubMed.
- S. Chae, J. P. Lee and J. M. Kim, Adv. Funct. Mater., 2016, 26, 1769–1776 CrossRef CAS.
- H. Terada, H. Imai and Y. Oaki, Adv. Mater., 2018, 30, 1801121 CrossRef PubMed.
- K. Watanabe, H. Imai and Y. Oaki, J. Mater. Chem. C, 2020, 8, 1265–1272 RSC.
- J. P. Lee, H. Hwang, S. Chae and J. M. Kim, Chem. Commun., 2019, 55, 9395–9398 RSC.
- Y. Ishijima, H. Imai and Y. Oaki, Chem., 2017, 3, 509–521 CAS.
- A. R. Burns, R. W. Carpick, D. Y. Sasaki, J. A. Shelnutt and R. Haddad, Tribol. Lett., 2001, 10, 89–96 CrossRef CAS.
- M. Varenberg, I. Etsion and G. Halperin, Rev. Sci. Instrum., 2003, 74, 3362–3367 CrossRef CAS.
- D. F. Ogletree, R. W. Carpick and M. Salmeron, Rev. Sci. Instrum., 1996, 67, 3298–3306 CrossRef CAS.
- R. D. Ortuso and K. Sugihara, J. Phys. Chem. C, 2018, 122, 11464–11474 CrossRef CAS.
- Q. Z. Yang, Z. Huang, T. J. Kucharski, D. Khvostichenko, J. Chen and R. Boulatov, Nat. Nanotechnol., 2009, 4, 302–306 CrossRef CAS PubMed.
- Y. J. Lin, H. R. Hansen, W. J. Brittain and S. L. Craig, J. Phys. Chem. B, 2019, 123, 8492–8498 CrossRef CAS PubMed.
- K. L. Berkowski, S. L. Potisek, C. R. Hickenboth and J. S. Moore, Macromolecules, 2005, 38, 8975–8978 CrossRef CAS.
- T. Seki, N. Tokodai, S. Omagari, T. Nakanishi, Y. Hasegawa, T. Iwasa, T. Taketsugu and H. Ito, J. Am. Chem. Soc., 2017, 139, 6514–6517 CrossRef CAS PubMed.
- Y. J. Zhang, B. L. Ma, Y. J. Li and J. H. Li, Colloids Surf., B, 2004, 35, 41–44 CrossRef CAS PubMed.
- S. Kolusheva, T. Shahal and R. Jelinek, J. Am. Chem. Soc., 2000, 122, 776–780 CrossRef CAS.
- S. Kolusheva, T. Shahal and R. Jelinek, Biochemistry, 2000, 39, 15851–15859 CrossRef CAS PubMed.
- R. R. Adhikary, O. Koppaka and R. Banerjee, Nanoscale, 2020, 12, 8898–8908 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr07129g |
| This journal is © The Royal Society of Chemistry 2022 |