
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-doped carbon dots with enhanced Fenton reaction activity for rhodamine B degradation†
Zhiru
Jin‡
ac,
Qiuying
Li‡
a,
Peiduo
Tang‡
b,
Ganfeng
Li
a,
Li
Liu
a,
Dong
Chen
b,
Ji
Wu
*c,
Zhihui
Chai
*b,
Gang
Huang
 *b and
Xing
Chen
*a
*b and
Xing
Chen
*a
aSchool of Public Health, Guangxi Medical University, Nanning, 530021, China. E-mail: chenx63@163.com
bState Key Laboratory of Non-Food Biomass and Enzyme Technology, Guangxi Academy of Sciences, Nanning, 530007, China. E-mail: czh1716302002@163.com; wangyi.07@163.com
cDepartment of Ultrasonic Medicine, First Affiliated Hospital of Guangxi Medical University, Nanning, 530021, China. E-mail: gxnnwuji@126.com
First published on 8th June 2022
Abstract
The Fenton reaction has attracted extensive attention due to its potential to be a highly efficient and environmentally friendly wastewater treatment technology. Noble copper-doped carbon dots (CuCDs) are prepared through a simple one-step hydrothermal method with 3,4-dihydroxyhydrocinnamic acid, 2,2′-(ethylenedioxy)bis(ethylamine) and copper chloride, endowing the Fenton reaction with enhanced catalytic activity for rhodamine B (RhB) degradation. The effects of the concentration of CuCDs, temperature, pH, oxygen (O2), metal ions and polymers on the catalytic activity of CuCDs are investigated. It is worth noting that electron transfer happening on the surface of CuCDs plays a vital role in the RhB degradation process. As evidenced by radical scavenger experiments and electron spin resonance (ESR) studies, CuCDs significantly boost the formation of hydroxyl radicals (˙OH) and singlet oxygen (1O2), facilitating the Fenton reaction for RhB degradation. Due to the strong oxidation of ROS generated by the Fe2+ + H2O2 + CuCD system, RhB degradation may involve the cleavage of the chromophore aromatic ring and the de-ethylation process. Additionally, the toxicity of RhB degradation filtrates is assessed in vitro and in vivo. The as-prepared CuCDs may be promising catalytic agents for the enhancement of the Fenton reaction.
1. Introduction
To address a great threat to human health and the environment posed by wastewater, various methods and strategies have been applied for the removal of pollutants, such as adsorption,1,2 biological degradation3 and advanced oxidation.4–6 Among them, Fenton oxidation is one of the green and effective advanced oxidation processes and plays an important role in the degradation of organic dyes.7 The homogeneous Fenton reaction is composed of Fe2+/H2O2, where the hydroxyl radical (˙OH) generated by the reaction of Fe2+ with H2O2 attacks dye molecules and then converts them to low-toxicity or non-toxic small-molecular substances.8 Several drawbacks, such as low utilization of H2O2, strict pH requirement and iron sludge induced by slow conversion of Fe3+/Fe2+, are not still addressed, limiting the development of the homogeneous Fenton reaction.9,10 Considerable efforts have been devoted to enhancing the catalytic reactivity of the homogeneous Fenton reaction, e.g. fabrication of heterogeneous catalysts,11 addition of reducing agents,12,13 and varying experimental means (electro-Fenton, photo-Fenton).14,15Carbon dots (CDs) are rapidly boosted in the aspect of synthesis and application owing to their small size, easy synthesis, excellent biocompatibility and unique optical and physical properties, thus being investigated in numerous potential applications, such as bioimaging,16 drug delivery,17 and sensing applications.18–20 CDs can act as excellent potential catalysts thanks to their excellent intrinsic electron transfer characteristics.21 CDs constructed with citric acid and thiourea are used as electron donors due to the high density of oxygen groups during the catalytic process. CDs prepared using H. undatus as the carbon source act as electron acceptors for catalytic reduction.22
Heteroatom-doping is an effective method to enhance the fluorescence intensity and the density of active sites.23–25 Among heteroatom-doped CDs, inorganic doping affects the electrochemical properties of materials.26,27 Copper-doped carbon dots (CuCDs), as inorganic doped CDs, have attracted considerable attention in the fields of drug delivery28 and fluorescence probes.29,30 Apart from those, CuCDs are employed as catalytic materials for the catalytic reaction of p-phenylenediamine31 and p-nitrophenol.32 Additionally, CuCDs prepared from ascorbic acid and Na2[Cu(EDTA)] through pyrolysis possess reductive properties associated with the unsaturated valence and Cu+, differentiating from other CuCDs. Thus, CuCDs exhibit enhanced catalytic activity in the Fenton system,33 which establishes that the reducing agents are conducive to the Fenton process.12,13 However, it is unclear whether the other novel CuCDs tailored by altering the carbon source enhance the catalytic activity of the Fenton reaction.
In this work, CuCDs fabricated by a hydrothermal method were beneficial to accelerate RhB degradation in the Fenton process. The effects of the concentration of CuCDs, temperature, pH, O2, metal ions and polymers on the catalytic reactivity of CuCDs were assessed. The reactive oxygen species (ROS) were identified by the scavenger test and ESR to elucidate the roles of ROS in the case of Fe2+ + H2O2 + CuCDs. On the basis of the results of liquid chromatography-mass spectrometry (LC-MS), a possible RhB degradation pathway was proposed. Furthermore, the toxicity of RhB degradation filtrates was investigated in vitro and in vivo.
2. Experimental section
2.1 Materials
RhB was bought from Tokyo Chemical Industry (TCI). 3,4-Dihydroxyhydrocinnamic acid (DHCA), 2,2′-(ethylenedioxy)bis(ethylamine) (EDA), iron(II) chloride tetrahydrate (FeCl2·4H2O), iron(III) chloride hexahydrate (FeCl3·6H2O), copper(II) chloride dihydrate (CuCl2·2H2O) and calcium chloride dihydrate (CaCl2·2H2O) were purchased from Energy Chemical Co. Ltd. Other chemicals were used without further purification. The 4T1 cell line and 293T cell line were obtained from the Cell Bank of Chinese Academy of Sciences. Kunming mice were obtained from Guangxi Medical University.2.2 Preparation of CuCDs
364.3 mg (2 mmol) of DHCA and 341 mg (2 mmol) of CuCl2·2H2O were dissolved in 30 mL of deionized water and stirred for 5 min. Afterward, 0.584 mL of EDA was added and continuously stirred for 5 min at room temperature before transferring to an autoclave. After heating to 180 °C for 5 h, it was cooled to room temperature, centrifuged for 20 min (6000 rpm), and filtered three times with a 0.22 μm membrane. The filtrate was dialyzed against deionized water for 72 h (molecular weight cut-off (MWCO) = 500 D) before lyophilization, with changing the deionized water every 8 h during dialysis, and the resulting compound was denoted as CuCDs. Additionally, two other different proportions of CuCDs were prepared by a similar method but varying the Cu proportion (1 mmol of CuCl2·2H2O and 3 mmol of CuCl2·2H2O), and the resulting compounds were denoted as 0.5CuCDs and 1.5CuCDs.2.3 Characterization
Fourier-transform infrared (FT-IR) spectra were recorded on a Thermo Scientific Nicolet iS10 spectrometer. X-ray photoelectron spectroscopy (XPS) was performed using a Kratos Axis Ultra Dld. The UV-vis absorbance spectra were measured on an Agilent Cary 60 spectrophotometer. The crystal structure of the CuCDs was determined on an X-ray diffractometer (XRD) with a Cu–Kα radiation source operated at 40 kV, at a 10° min−1 scan rate, and 2θ from 0° to 80°. The morphology of the CuCDs was observed using a high-resolution transmission electron microscope (HRTEM) using a FEI Tecnai G2F20. Electron spin resonance (ESR) measurements were conducted using a Bruker A300. The copper content of the CuCDs was quantified by Thermo Fisher iCAP Qnova Series inductively coupled plasma mass spectrometry (ICP-MS). The elemental distribution of CuCDs was analyzed via scanning electron microscopy (SEM) with an energy dispersive spectrometer (EDS) on a Hitachi s-3400N. The fluorescence lifetime was measured on an Edinburgh Instruments FLS980. The quantum yield of CuCDs in deionized water was determined relative to a standard of quinine sulfate (a quantum yield of 0.54 in 0.1 M H2SO4). A series of CuCD solutions and quinine sulfate solution were prepared. The emission spectra of CuCDs or quinine sulfate solution between 400 nm and 650 nm were recorded, and the excitation wavelength was set at 350 nm. The integrated emission of solutions was plotted against the absorbance at 350 nm. The quantum yield of CuCDs (Φx) was calculated using the following formula:where Φst represents the quantum yield of quinine sulfate, Kx is the slope of CuCDs, Kst represents the slope of quinine sulfate, nx represents the refractive index of the solvent for CuCDs, and nst is the refractive index of 0.1 M H2SO4, the solution for quinine sulfate.
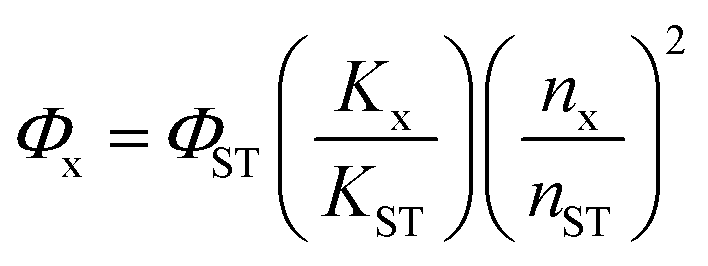
2.4 Evaluation of RhB degradation by accelerated Fenton reaction
1.5 mL of 1% H2O2 (V/V), 0.2 mL of 0.01 mol L−1 FeSO4 solution, and 0.08 mL of 1 mg mL−1 RhB solution were added to deionized water in the absence and presence of CuCDs (10 mg mL−1, 20 μL) within 90 min. The total volume of the mixture solution was 4 mL. Three sample solutions were prepared and 2.5 mL of sample solution was taken out for each measurement. The RhB degradation ratio was calculated using the following formula:where C and C0 are the observed concentration at various time intervals and the initial concentration of RhB, and A and A0 are the observed and the initial absorbance of the RhB solution at 550 nm, respectively.
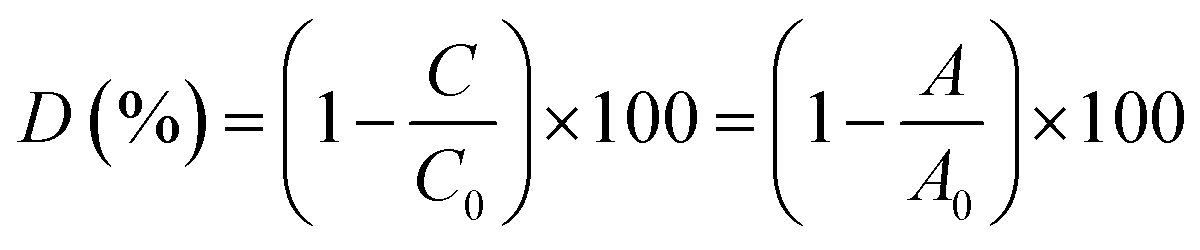
The effects of temperature, pH, concentration of CuCDs, O2, metal ions and polymers on RhB degradation by accelerated Fenton reaction were evaluated. For example, 1.5 mL of 1% H2O2 (V/V), 0.2 mL of FeSO4 solution (0.01 mol L−1), 0.08 mL of RhB solution (1 mg mL−1) and 20 μL of CuCDs (10 mg mL−1) were added to the deionized water and the solution volume was 6 mL. Three sample solutions were prepared and 2.5 mL of sample solution was taken out for each measurement. The above solutions were incubated at every temperature (25, 30, 40, 50, 60, 70, and 80 °C) for 5 min and then the absorbance of the solution was recorded at 550 nm. Similarly, the effect of pH was tailored by altering pH via addition of HCl or NaOH solution under negligible total volume change. The effect of concentrations of CuCDs was evaluated under similar conditions except for addition of CuCDs in the range of 0–20 μL. To assess the effect of O2, the reaction mixture (Fe2+ + H2O2 + CuCDs + RhB) was pumped with N2 or O2 gas for 30 min, while the original reaction mixture was used as the control. The effects of metal ions and polymers were evaluated through addition of NaCl, KCl, FeCl3, CuCl2, CaCl2, CdCl2, AlCl3, CrCl3, polyethylene glycol (PEG, Mn = 200), polyethyleneimine (PEI, Mn = 800) and poly(acrylic acid) (PAA, Mn = 3000).
To investigate the reproducibility of CuCDs, successive experiments were performed. Briefly, 0.09 mL of 1% H2O2 (V/V), 0.3 mL of FeSO4 solution (0.01 mol L−1), 0.12 mL of RhB (1 mg mL−1), 0.03 mL of CuCDs (10 mg L−1) and the deionized water were mixed and the total volume was 6 mL. The RhB absorbance was recorded after 30 min incubation. Then 0.09 mL of H2O2 and 0.12 mL of RhB were added to the above solution. After 30 min incubation, the RhB absorbance was also recorded. Similarly, 0.09 mL of H2O2 and 0.12 mL of RhB were added each time in the next three replicates and the RhB absorbance was recorded.
2.5 Toxicity assay in vitro and in vivo
After RhB degradation, the mixture was subjected to centrifugal separation for 20 min (6000 rpm). 0.4 g of NaCl, 0.01 g of KCl, 0.182 g of Na2HPO4·12H2O, and 0.012 g of KH2PO4 were dissolved in 50 mL of the RhB degradation filtrates followed by sterilization, and the resulting solution was denoted as the RhB degradation buffer solution.2.6 Statistical methods
All the data in this work were calculated using SPSS 26.0 software and the results were expressed by the average value ± standard deviation (SD). The measurement data were compared by analysis of variance. A one-way analysis of variance (ANOVA) was performed when the variances were uniform. P < 0.05 was defined as statistically significant.2.7 Live subject statement
All experiments were approved by the Ethics Committee of Guangxi Medical University and carried out in strict compliance with the relevant laws and institutional guidelines of Guangxi Medical University, Nanning, China.3. Results and discussion
3.1 Characterization of CuCDs
The FT-IR spectra of CuCDs showed that the broad peaks in the range from 3826 to 2221 cm−1 were attributed mostly to carboxylic acid groups (–COOH) and hydroxyl groups (–OH), while the absorption band at 2881 cm−1 was characteristic of –CH2–CH2– stretching vibrations (Fig. S1a†). The characteristic absorption peaks at 1554, 1450, 1396, 1101 and 1025 cm−1 were attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–N, and C–OH stretching vibrations and symmetric C–O–C stretching.34,35 The XPS results further confirmed chemical compositions of CuCDs. The chemical elements C, O and N co-existed on the surface of CuCDs as shown in the XPS survey spectrum of CuCDs (Fig. S1b†). The high-resolution XPS spectrum of C1s revealed three dominant peaks at 283.4, 284.9 and 286.7 eV, attributed to the presence of C–C/CC groups, C–N bonds and C–O/CO/CN groups, respectively (Fig. S1c†).36 The two fitted peaks at about 529.3 and 531 eV corresponded to CO and OH, respectively (Fig. S1d†).37,38 In the N1s spectrum, the peaks at 398.2 and 399.9 eV were assigned to C–N/OC–N and NH2 (Fig. S1e†).39 Unfortunately, the Cu2p characteristic peaks were not observed in the XPS spectra due to the low amount of Cu in CuCDs (only 0.0584% Cu), which was confirmed by ICP-MS. EDS results showed that the elemental composition of the CuCD surface was C (66.23%), O (27.4%), and N (6.37%), and it was difficult for EDS to show Cu deposition on the CuCD surface (Fig. S5 and Table S1†). Similarly, the Cu diffraction peak was not revealed in the XRD pattern (Fig. S1f†). A weak diffraction peak at 2θ = 23.5° indicated that CuCDs generated an amorphous carbon structure, similar to the graphene sheet structure.40 To assess the thermal stability of CuCDs, TGA was performed under N2 or oxygen (O2) (Fig. S1g†). CuCDs showed a rapid decomposition, about 60% of weight loss at 420 °C in a N2 atmosphere, whereas CuCDs were completely destroyed after 546 °C in an O2 atmosphere. CuCDs exhibited a negative zeta potential (−5.56 ± 1.46 mV) and a spherical shape with an average diameter of 2.6 nm and a lattice fringe distance of 0.14 nm in the HRTEM images (Fig. S1h and j†).
O, C–N, and C–OH stretching vibrations and symmetric C–O–C stretching.34,35 The XPS results further confirmed chemical compositions of CuCDs. The chemical elements C, O and N co-existed on the surface of CuCDs as shown in the XPS survey spectrum of CuCDs (Fig. S1b†). The high-resolution XPS spectrum of C1s revealed three dominant peaks at 283.4, 284.9 and 286.7 eV, attributed to the presence of C–C/CC groups, C–N bonds and C–O/CO/CN groups, respectively (Fig. S1c†).36 The two fitted peaks at about 529.3 and 531 eV corresponded to CO and OH, respectively (Fig. S1d†).37,38 In the N1s spectrum, the peaks at 398.2 and 399.9 eV were assigned to C–N/OC–N and NH2 (Fig. S1e†).39 Unfortunately, the Cu2p characteristic peaks were not observed in the XPS spectra due to the low amount of Cu in CuCDs (only 0.0584% Cu), which was confirmed by ICP-MS. EDS results showed that the elemental composition of the CuCD surface was C (66.23%), O (27.4%), and N (6.37%), and it was difficult for EDS to show Cu deposition on the CuCD surface (Fig. S5 and Table S1†). Similarly, the Cu diffraction peak was not revealed in the XRD pattern (Fig. S1f†). A weak diffraction peak at 2θ = 23.5° indicated that CuCDs generated an amorphous carbon structure, similar to the graphene sheet structure.40 To assess the thermal stability of CuCDs, TGA was performed under N2 or oxygen (O2) (Fig. S1g†). CuCDs showed a rapid decomposition, about 60% of weight loss at 420 °C in a N2 atmosphere, whereas CuCDs were completely destroyed after 546 °C in an O2 atmosphere. CuCDs exhibited a negative zeta potential (−5.56 ± 1.46 mV) and a spherical shape with an average diameter of 2.6 nm and a lattice fringe distance of 0.14 nm in the HRTEM images (Fig. S1h and j†).
Two characteristic absorption peaks at 278 nm and 344 nm were observed in UV-vis spectra (Fig. S2a†). The peak centered at 278 nm was ascribed to the π–π* transitions of CC in the conjugated double bond, and the other absorption peak at 344 nm was attributed to the n–π* transition of CO.41 The maximum emission wavelength was 443 nm when the excitation wavelength was set at 365 nm (Fig. S2a†). Additionally, CuCDs exhibited excitation-dependent fluorescence properties (Fig. S2b†). Interestingly, the fluorescence intensity of CuCDs was related to the concentrations of CuCDs and the maximum intensity was at 0.2 mg mL−1 (Fig. S2c†). No obvious fluorescence intensity changes were observed upon either the elevated temperature (Fig. S2d†) or the radiation of the UV lamp at 365 nm (Fig. S2e†). The fluorescence lifetime of CuCDs was calculated as 7.42 ns on the basis of the decay curve (Fig. S3a†) and the quantum yield was about 1.35% using quinine sulfate as a reference (Fig. S3b†).42
3.2 Catalytic reactivity of CuCDs
The catalytic reactivity of CuCDs was assessed in terms of the RhB degradation evaluation. Fig. S3c† shows that (a) Fe2+, (b) H2O2, (c) CuCDs, (d) Fe2+ + CuCDs, and (e) H2O2 + CuCDs removed less than 1% in 30 min, indicating that the contribution of these materials was negligible for RhB degradation. However, significant acceleration was achieved by addition of Fe2+ + H2O2 or Fe2+ + H2O2 + CuCDs. To further elucidate the influence of CuCDs on Fenton processes, RhB degradation processes with and without CuCDs were compared. It was established that the RhB degradation ratio was 80.99 ± 2.90% within 18 min and reached 92.52 ± 1.68% within 30 min (Fig. 1a). In the presence of CuCDs the RhB degradation ratio could reach 80.20 ± 2.73% using the Fe2+ + H2O2 system within 10 min (Fig. 1a), implying that the addition of CuCDs was conducive to enhancing the Fenton reaction rate. In addition, three different proportions of CuCDs were prepared through varying reacted Cu proportions and the corresponding Cu levels were 0.035%, 0.0584%, and 0.032% via ICP-MS. As depicted in Fig. 1b, the RhB degradation ratios were 53.53 ± 8.21%, 79.25 ± 3.17%, and 63.86 ± 5.97% during 10 min using various CuCDs. After 30 min, the RhB degradation ratios were 75.96 ± 6%, 93.83 ± 0.614%, and 86.26 ± 3.14%, illustrating that the CuCDs with 0.0584% Cu mass exhibited excellent RhB degradation performance among them and their RhB degradation performances were attributed to their composition. In this work, we chose the CuCDs with 0.0584% Cu mass as the main research object. The concentration of CuCDs was 20 mg L−1, and the RhB degradation ratio was 52.62 ± 2.42% within 5 min. When the amount of CuCDs was 60 mg L−1, the RhB degradation ratio increased to 68.62 ± 3.48%. Such a significant increase of degradation ratio was observed by varying concentrations of CuCDs (Fig. 1c), suggesting that CuCDs played an important role in RhB degradation.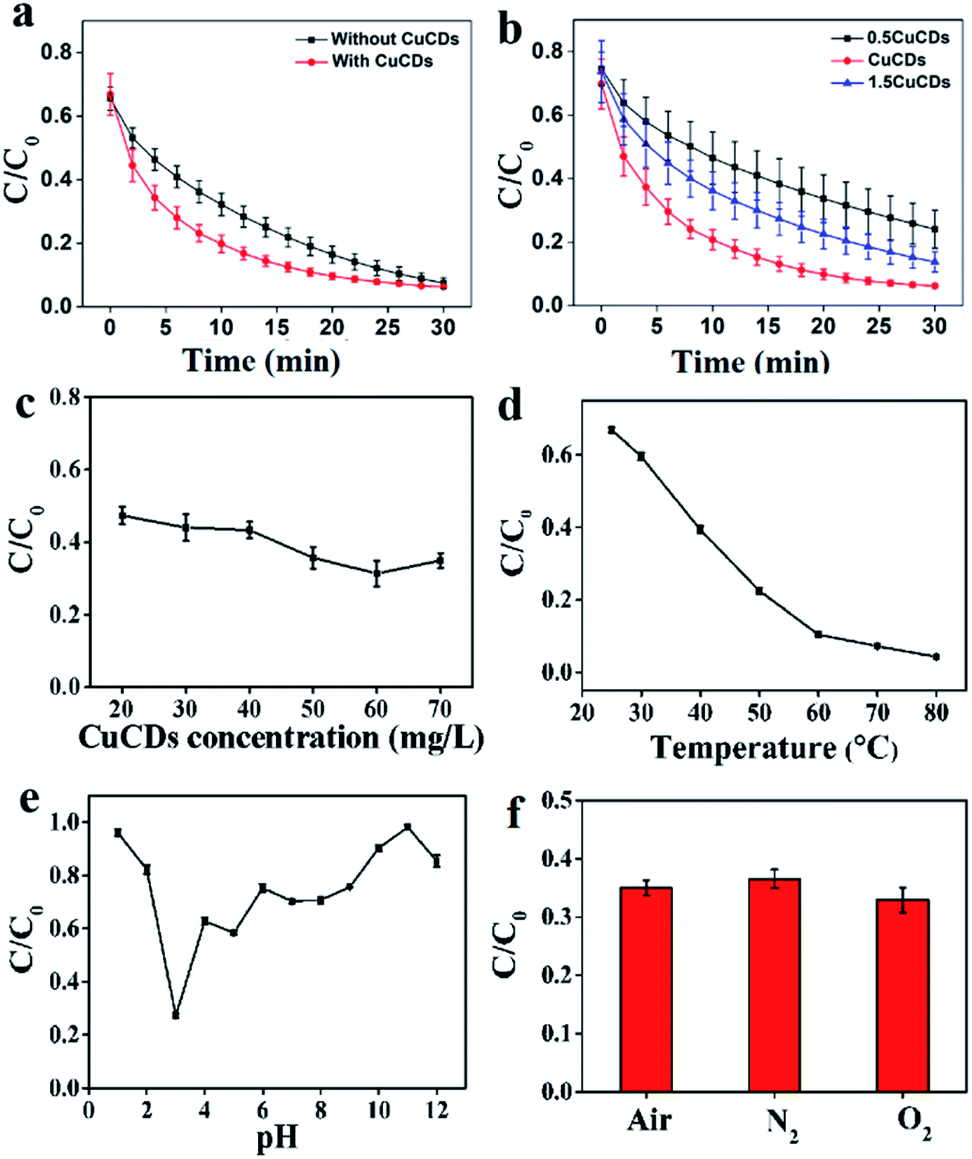 | ||
| Fig. 1 The effects on RhB degradation upon the Fenton reaction: (a) the absence and presence of CuCDs; (b) different proportions of CuCDs; (c) CuCD concentrations; (d) temperatures, (e) pH values, and (f) the absence/presence of O2. | ||
3.3 Analysis of factors influencing the catalytic reactivity of CuCDs
The factors influencing the catalytic performance of CuCDs on RhB degradation were further evaluated, including temperature, pH, O2, metal ions and polymers. As temperature was increased, the RhB degradation ratio gradually increased (Fig. 1d), indicating that the Fenton reaction was accelerated in the presence of CuCDs, which may be related to the fact that more ROS were yielded with increased temperature under protection of CuCDs, thus leading to more effective collision between ROS and RhB. As depicted in Fig. 1e, the Fe2+ + H2O2 + CuCD system showed the highest catalytic reactivity at pH = 3, because it tended to promote generation of ROS in an acidic environment.43 To understand the role of O2, either N2 or O2 was pumped into the Fe2+ + H2O2 + CuCD system. The result showed that it was unable to significantly tune the RhB degradation ratio (Fig. 1f).The effects of metal ions and polymers on RhB degradation rates were investigated. Under the same experimental conditions, KCl, NaCl, CaCl2, CdCl2, AlCl3 and CrCl3 were respectively added to the Fe2+ + H2O2 + CuCD solution, which were unable to induce obvious changes in RhB degradation rates (Fig. 2a). Yet the RhB degradation ratio was increased upon addition of FeCl3, FeSO4 and CuCl2 (Fig. 2a), which was responsible for the electron transfer. Generally, the fluorescence of CDs is quenched once electron transfer occurs. Hence, the fluorescence intensity of CuCDs was assessed upon the addition of various metal ions. There were no obvious changes in the fluorescence intensity of CuCDs upon addition of KCl, NaCl, CaCl2, CdCl2, AlCl3 or CrCl3 to the Fe2+ + H2O2 + CuCD system (Fig. S2f†). These results indicated that the electron transfer did not significantly occur among CuCDs and metallic salts (KCl, CaCl2, CdCl2, AlCl3, NaCl, and CrCl3) in the Fe2+ + H2O2 + CuCD system. However, the maximum fluorescence quenching of CuCDs occurred in the presence of Fe3+, Fe2+ and Cu2+ (Fig. S2f†), which indicated that electron transfer occurred between CuCDs and metal ions (Fe3+, Fe2+ and Cu2+). Therefore, with the help of electron transfer, the RhB degradation ratio was improved after addition of FeCl3, FeSO4 and CuCl2. It was worth mentioning that electron transfer played a vital role in the RhB degradation process. The effects of polymers with different electric charges on the RhB degradation rates were investigated. PEG, PEI, and PAA were used as a nonionic polymer, a cationic polymer and an anionic polymer in aqueous solution after hydrolysis. Similar to the aforementioned conclusion that the electron transfer was inhibited due to high medium concentration, the RhB degradation rates decreased in the presence of polymers because the polymers hindered the collision between ˙OH and RhB. Interestingly, the RhB degradation ratio in the case of adding PAA was superior to those of addition of PEG or PEI, and it was lowest upon addition of PEI (Fig. 2b), which indicated that a nonionic polymer and a cationic polymer had greater effects on the RhB degradation ratio than an anionic polymer. In this work, the enhancement of RhB degradation with the help of CuCDs was attributable to the facilitation of electron transfer among Fe2+, H2O2 and CuCDs in the Fenton process. Based on this, the anionic PAA was repelled by the CuCDs with negative charges because of electrostatic interactions, which did not entirely hinder the electron transfer among Fe2+, H2O2, CuCDs and RhB. On the other hand, the cationic PEI was attracted to the surface of the CuCDs, impeding electron transfer. But for nonionic PEG, the RhB degradation ratio declined because the high concentration of PEG made the collision between ˙OH and RhB inactive.
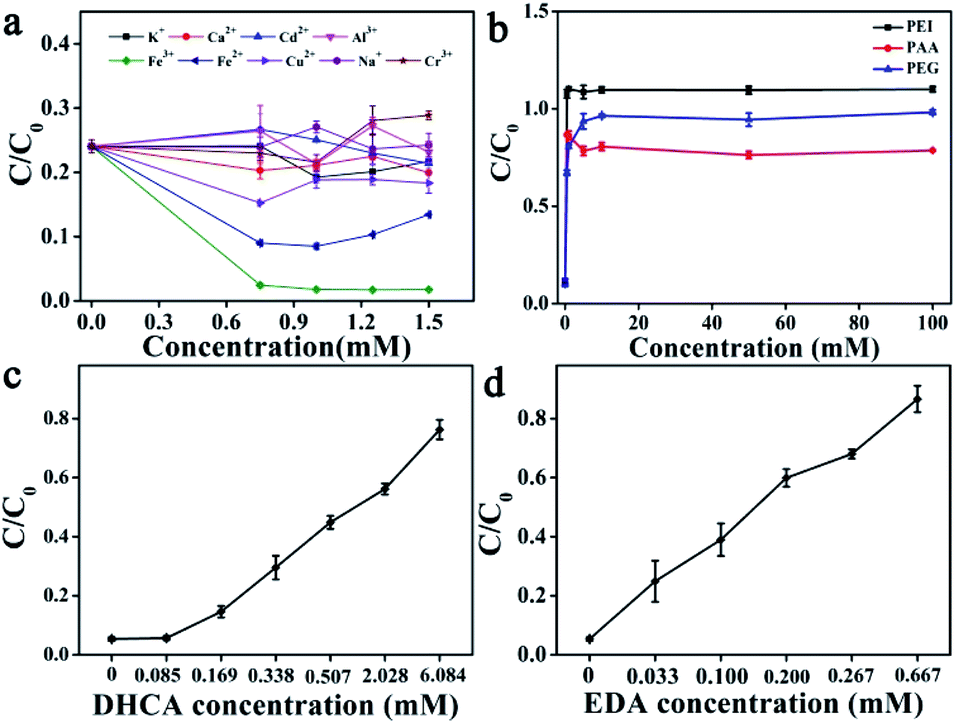 | ||
| Fig. 2 The effects on RhB degradation in the case of Fe2+ + H2O2 + CuCDs: (a) metal ions; (b) polymers; substrate concentrations: (c) DHCA; (d) EDA. | ||
Besides, it was established that the reducing agents accelerated the Fenton reaction. In our work, the addition of CuCDs was conducive to enhancing the Fenton reaction rate, inspiring us to reveal whether the substrates (DHCA, EDA, and CuCl2) or CuCDs acting as the reducing agents induced acceleration of the Fenton reaction in the case of Fe2+ + H2O2 + CuCDs. As displayed in Fig. 2c and d, the RhB degradation rates decreased with the increase of the concentrations of DHCA or EDA. The results implied that these substrates were unable to serve as the reducing agents to accelerate the Fenton reaction. As is well known, copper ions were able to accelerate the Fenton reaction. However, the CuCDs were dialyzed for 72 h and the water was changed every 8 h during the purification process to remove the copper ions. Previous studies indicated that the fluorescence of CDs was quenched by copper ions and recovered after addition of ethylenediaminetetraacetic acid (EDTA).44 EDTA was able to readily form extremely stable chelating complexes with copper ions, resulting in that copper ions were away from the CuCDs. Thus the fluorescence of CuCDs was assessed in the absence/presence of EDTA (Fig. S3d†). As expected, the fluorescence of CuCDs was quenched by Cu2+ while it was recovered when EDTA was added. However, no obvious change in the fluorescence of CuCDs was observed after addition of EDTA, implying that a few copper ions were adsorbed on the surface, probably embedded inside the CuCDs. Thus the substrates did not serve as reducing agents to accelerate the Fenton reaction. Yet it was established that CDs with the hydroxyl groups exhibited reducing activity.45 In this work, hydroxyl groups were introduced into CuCDs as evidenced by FT-IR and XPS, implying that the microstructure of CuCDs presumably served as the reducing agent to accelerate the Fenton reaction. In comparison to the RhB degradation based on CuCDs and Cu2+ with the same quantity of Cu, CuCDs offered higher catalytic activity (Fig. S3e†). Additionally, Table S2† summarizes the catalytic activity of conventional CDs toward other pollutants with various catalytic methods. Some CDs possessed inherent optical properties and were applied for the removal of RhB or methyl blue upon the photo-Fenton reaction. The Fenton reaction is commonly performed in a limited pH range. However, the addition of CDs didn't allow the Fenton reaction to get rid of the limitation of being performed in an acidic solution. In this work, CuCDs were able to help the Fenton reaction accelerate the degradation of RhB under neutral conditions without light irradiation, attributable to the role of CuCDs as co-catalysts. The degradation performance of CuCDs was superior to those of the other CDs.
3.4 Identification of ROS
Although ˙OH was the major active species, 1O2 and ˙O2− were accompanied during the Fenton reaction. To further understand the roles of ROS in the catalytic process, various radical scavengers were utilized including isopropanol (IPA), 1,3-diphenylisobenzofuran (DPBF) and nitroblue tetrazolium (NBT) for ˙OH, 1O2 and ˙O2−, respectively.46,47 For the Fe2+ + H2O2 + CuCDs system, over 85% of RhB could be degraded after 32 min in the absence of IPA. With the addition of IPA to the Fe2+ + H2O2 + CuCD system, the RhB degradation ratio sharply decreased and became only 20.7 ± 0.04% after 32 min. This indicated that once ˙OH was quenched by IPA, the catalytic activity of the Fe2+ + H2O2 + CuCDs system was dramatically reduced (Fig. 3a). The characteristic absorption peak of DPBF at 415 nm significantly decreased with time upon the addition of DPBF, indicating that 1O2 was generated in the degradation system (Fig. 3b). Yet no obvious change in RhB degradation was observed despite the addition of NBT (Fig. 3c). These results indicated that ˙OH and 1O2 acted as the major ROS in the Fe2+ + H2O2 + CuCD system.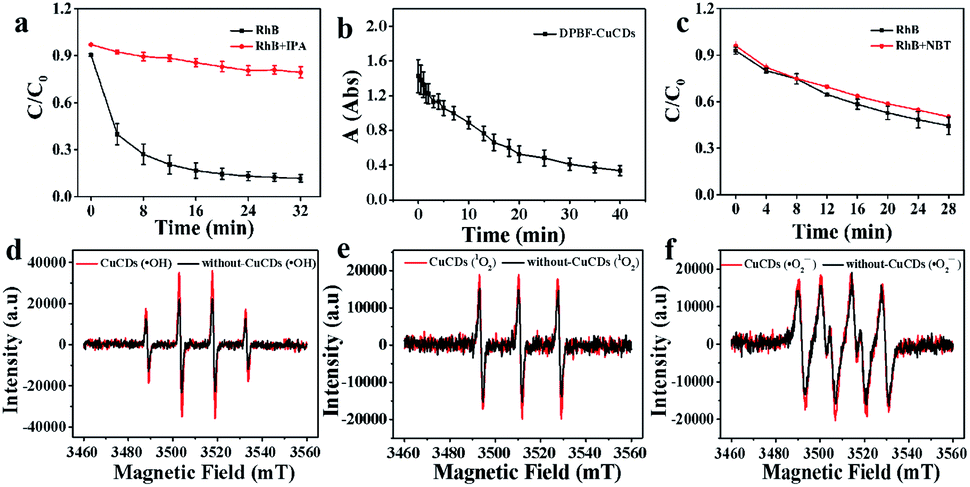 | ||
| Fig. 3 The radical scavenger experiments: (a) IPA; (c) NBT probe as a radical scavenger; (b) UV-vis absorption of DPBF at different times in the presence of CuCDs in solution; ESR signals generated by the Fenton reagent with or without CuCDs in solutions: (d) ˙OH radicals; (e) 1O2; (f) ˙O2− radicals. | ||
ESR, with 5,5-dimethyl-L-pyrroline-N-oxide (DMPO) and 4-amino-2,2,6,6-tetramethylpiperidine (TEMP) as the trapping agents for ˙OH, ˙O2− and 1O2, was performed to verify the species of ROS in the Fe2+ + H2O2 + CuCD system. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 four-wire signal characteristic peak was obtained in the absence of CuCDs, which corresponded to DMPO-˙OH (Fig. 3d). Upon the addition of CuCDs, the intensity of the DMPO-˙OH peak became stronger. The three-line ESR spectrum with equal intensities (1:1:1), corresponding to TEMP-1O2, was also slightly increased after addition of CuCDs (Fig. 3e). Nevertheless, the quadruple signal characteristic peak of DMPO-˙O2− with the ratio of 1:1:1:1 had a negligible change despite the addition of CuCDs (Fig. 3f). The ESR results demonstrated that ˙OH mainly contributed to RhB degradation, 1O2 was secondary and ˙O2− was the weakest among them, which were consistent with the results of the scavenger test. It was reported that the ˙O2− derived from the ˙OH48 and 1O2 was produced from ˙O2−.49 Negligible changes of ˙O2− were observed, along with subsequent upregulation of 1O2 levels, suggesting that CuCDs could directly or indirectly induce a higher level of 1O2. The speculation was shown as below:
:2:2:1 four-wire signal characteristic peak was obtained in the absence of CuCDs, which corresponded to DMPO-˙OH (Fig. 3d). Upon the addition of CuCDs, the intensity of the DMPO-˙OH peak became stronger. The three-line ESR spectrum with equal intensities (1:1:1), corresponding to TEMP-1O2, was also slightly increased after addition of CuCDs (Fig. 3e). Nevertheless, the quadruple signal characteristic peak of DMPO-˙O2− with the ratio of 1:1:1:1 had a negligible change despite the addition of CuCDs (Fig. 3f). The ESR results demonstrated that ˙OH mainly contributed to RhB degradation, 1O2 was secondary and ˙O2− was the weakest among them, which were consistent with the results of the scavenger test. It was reported that the ˙O2− derived from the ˙OH48 and 1O2 was produced from ˙O2−.49 Negligible changes of ˙O2− were observed, along with subsequent upregulation of 1O2 levels, suggesting that CuCDs could directly or indirectly induce a higher level of 1O2. The speculation was shown as below:
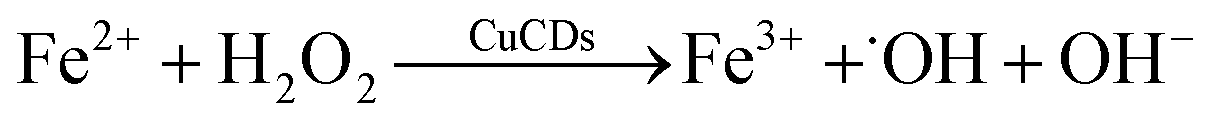 | (1) |
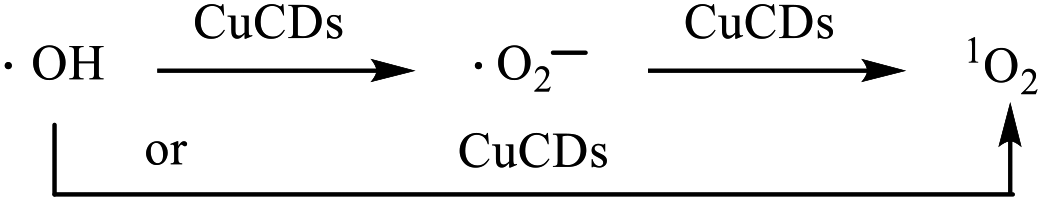 | (2) |
3.5 Optimization of the H2O2/Fe2+ molar ratio and degradation kinetics
With increasing the H2O2/Fe2+ molar ratio, the RhB degradation ratio was initially increased and then slightly decreased for the Fe2+ + H2O2 + CuCD system, in which the optimum H2O2/Fe2+ molar ratio was approximately 10:1 in the RhB concentration range from 20 to 40 mg L−1 (Fig. 4a). Additionally, the catalytic activity for the Fe2+ + H2O2 + CuCD system was further evaluated at different RhB concentrations (Fig. 4b). The degradation ratios were over 94.32 ± 1.58% in the RhB concentration range from 10 to 50 mg L−1, suggesting that RhB was almost destructed by ROS. Yet at a higher RhB concentration (>50 mg L−1), the degradation ratio slightly declined, only 90.29 ± 0.26% despite prolonging incubation for another 2 h, which demonstrated that a few RhB molecules were not completely degraded. The Langmuir–Hinshelwood model was adopted to quantitatively analyze pseudo-first-order kinetics: ln(C0/Ct) = kt.50 The ratio constant k could be calculated from the plot of -ln(C0/Ct) versus t within 60 min and the k values were 0.1481, 0.1322, 0.06233, 0.04568, 0.03915, and 0.0238 min−1, corresponding to RhB concentrations of 10, 20, 30, 40, 50, and 60 mg L−1. Regression coefficient (R2) values of the fitted lines were 0.8958, 0.9605, 0.9784, 0.9161, 0.9209, and 0.9435, respectively (Fig. 4c). The results showed that the overall trend of degradation efficiency declined as the RhB concentration increased.
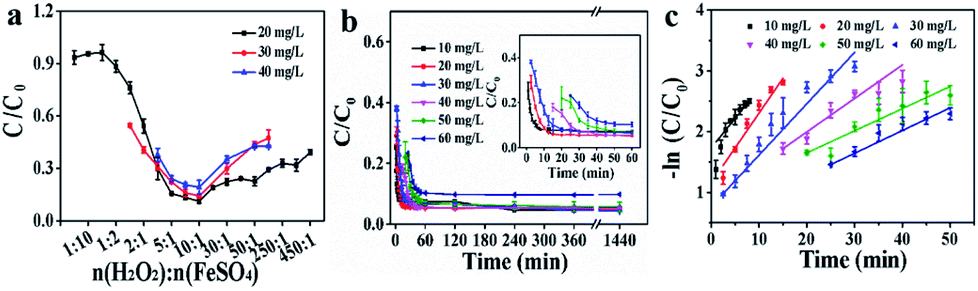 | ||
| Fig. 4 (a) The effect of the ratio of H2O2 to FeSO4 on RhB degradation; (b) the effect of substrate RhB concentrations on degradation; (c) the −ln(C/C0) vs. time curves of RhB degradation. | ||
3.6 Fine chemical treatment and the possible degradation mechanism
Since RhB was decomposed, flocculent precipitation occurred. The mixture solution was separated through centrifugation. The amounts of Fe and Cu in either the filtrates or the aggregates were quantified by ICP-MS. The Fe ratio was denoted as the amounts of metal in the filtrates to those in the aggregates. The Cu ratio was calculated by the same method. The results showed that the Fe ratio decreased as the RhB concentration increased (Fig. 5a), suggesting that the Fenton reaction needed more Fe2+ to achieve RhB degradation, leading to the abundant formation of iron sludge. Yet the Cu ratios slightly increased (Fig. 5a). Perhaps CuCDs could participate in the Fenton reaction to yield more copper ions and fewer unreactive CuCDs were adsorbed onto the surface of the sludge, resulting in an increased Cu ratio. Inspired by the above results, we further assessed whether the addition of H2O2 and RhB alone to the filtrate could continue degrading RhB. It was noticed that the RhB degradation ratios remained above 85% in the Fe2+ + H2O2 + CuCD system for five successive experiments (Fig. 5b), which was superior to the RhB degradation rates in the Fe2+ + H2O2 system, implying that the RhB filtrate was able to be reused to degrade RhB as long as replenishing H2O2. However, the RhB solutions with various concentrations from 10 to 60 mg L−1 were decomposed in the case of Fe2+ + H2O2 + CuCDs. The results showed that the values of chemical oxygen demand (COD) and ammonia nitrogen tended to be high with increasing substrate RhB concentrations, suggesting that more RhB molecules were not decomposed with increasing RhB concentrations. When the substrate RhB concentration was at 40 mg L−1, the values of COD and ammonia nitrogen were 97.93 ± 11.11 mg L−1 and 12.36 ± 2.76 mg L−1, respectively (Fig. 5c).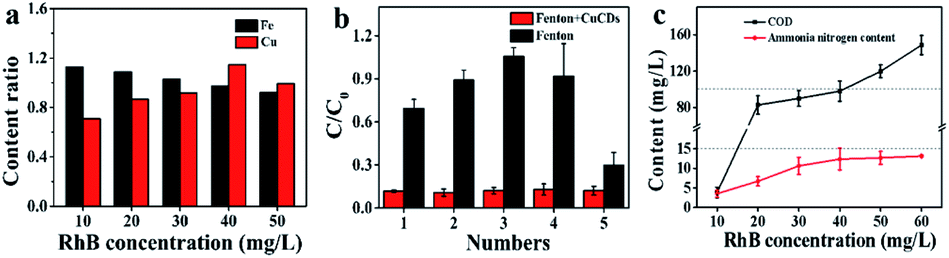 | ||
| Fig. 5 (a) The liquid-to-solid ratio of Fe and Cu contents under different substrate RhB concentrations; (b) the recycles of H2O2 and RhB on degradation; (c) COD and ammonia nitrogen content of the degraded solution. | ||
With the help of LC-MS, more detailed information about intermediates was obtained. The RhB degradation filtrates were measured as depicted in Fig. S4a-f.† The major mass peaks appeared at m/z 487, 293, 197, 160 and 109, which could be assigned to C26H27Cl2N2O3−, C19H19N2O−, C13H12NO−, C6H5FeNO− and C6H6NO−, respectively. As the RhB concentration increased, the intensities of these two peaks at 487 and 293 increased, indicating that the RhB degradation underwent a de-ethylation process. Based on the results of LC-MS, a possible RhB degradation pathway was proposed (Fig. 6). The strong oxidation of ROS caused the cleavage of the chromophore aromatic ring and the de-ethylation process from the aromatic ring with a hypsochromic shift. Then, RhB was converted into low-toxicity even non-toxic compounds.
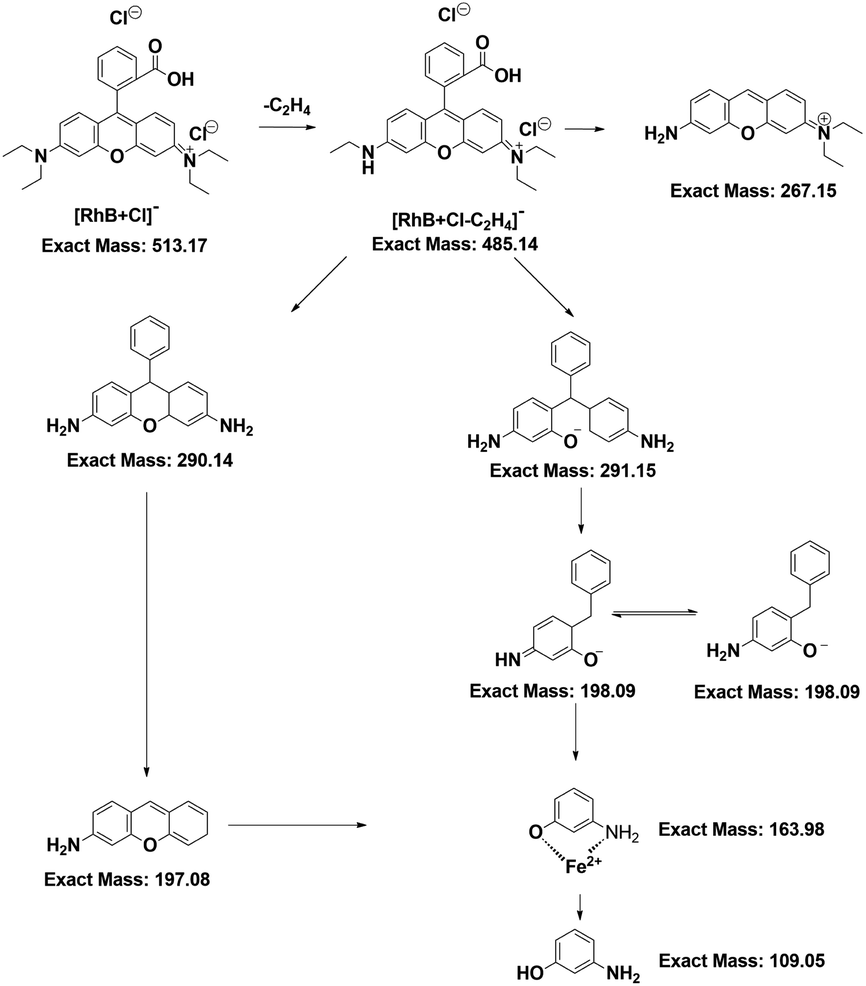 | ||
| Fig. 6 RhB degradation pathway in the case of Fe2+ + H2O2 + CuCDs. | ||
3.7 Toxicity of RhB degradation filtrates in vitro and in vivo
The toxicity of the RhB degradation filtrates was investigated after centrifugal separation using the MTT assay. NaCl, KCl, Na2HPO4 and KH2PO4 were dissolved in the RhB degradation filtrates and the mixture was incubated with 4T1 or 293T cells for 24 h. As depicted in Fig. 7a, the degradation filtrate corresponding to a substrate RhB concentration of 10 mg L−1 showed low cytotoxicity toward cells (4T1 and 293T) and the cell viabilities were 78.06 ± 2.64% and 84.19 ± 3.95%, respectively. The decrease in cell viability showed that the cell viability depended on the substrate RhB concentration. Additionally, the effect of the RhB degradation filtrates on the secondary structure of BSA was investigated. However, no obvious secondary structure difference in BSA was observed at 209 nm and 222 nm (Fig. 7b), suggesting that the RhB degradation filtrates had weak effects on the α-helix structure of BSA. A hemolysis assay was performed to further evaluate the blood biocompatibility in vitro. The hemolysis percentages were 1.41 ± 0.12%, 1.37 ± 0.05%, 1.63 ± 0.03%, 2.75 ± 0.29%, and 2.88 ± 0.23%, corresponding to the substrate RhB concentrations in the range of 10–50 mg L−1 (Fig. 7c), indicating that the RhB degradation filtrates had negligible hemolytic activity.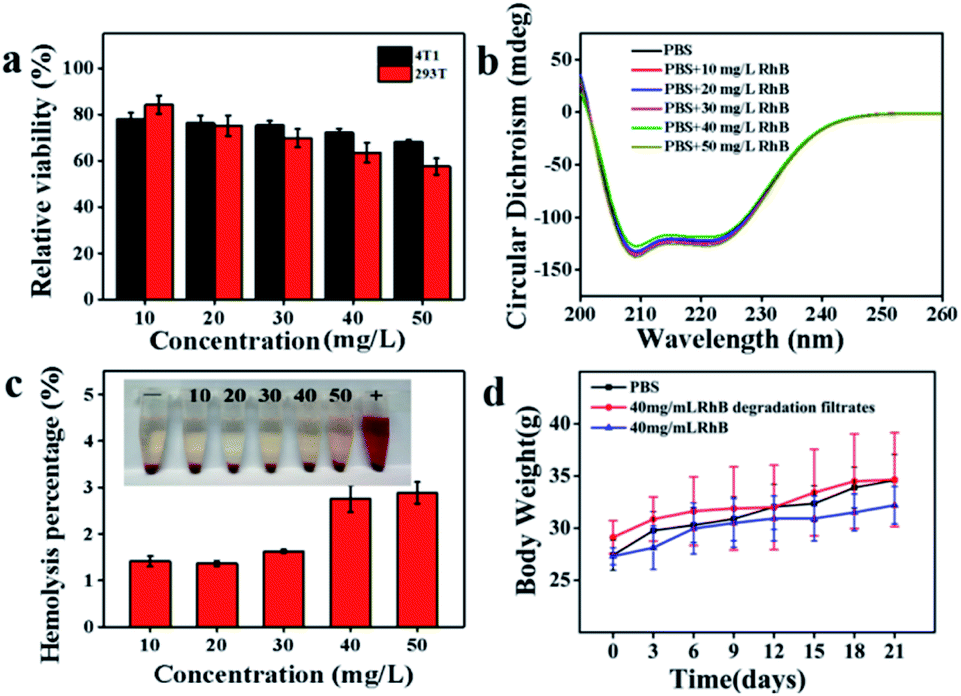 | ||
| Fig. 7 (a) The mixture incubated with 4T1 and 293T cells in the different concentrations of RhB degradation filtrates; (b) the effects of the RhB degradation filtrates on the secondary structure of BSA; (c) hemolysis assay in the different RhB degradation filtrates; (d) the effect of different concentrations of RhB on body weight. | ||
Hematology, blood biochemistry and histopathology examinations were performed to evaluate the biocompatibility of RhB degradation filtrates in vivo. The Kunming mice were implanted with the RhB degradation buffer solution for treatment. The body weights of mice increased during 21 days treatment (Fig. 7d). White blood cells (WBCs), red blood cells (RBCs), hemoglobin (HGB), hematocrit (HCT), and mean corpuscular hemoglobin (MCH) of the RhB solution groups exhibited obvious statistical differences with the control groups (P < 0.01, Table 1).
| Blood routine index | Control group | RhB degradation filtrate | P* | RhB solution | P* |
|---|---|---|---|---|---|
| a *P, compared with the control groups. | |||||
| WBC (109 L−1) | 2.53 ± 0.79 | 2.16 ± 0.73 | 0.394 | 2.28 ± 0.64 | 0.002 |
| RBC (1012 L−1) | 8.97 ± 1.03 | 8.15 ± 0.47 | 0.070 | 7.15 ± 0.54 | 0.001 |
| HGB (g L−1) | 118.83 ± 13.35 | 146.16 ± 25.93 | 0.083 | 103.67 ± 2.50 | 0.009 |
| HCT (%) | 39.38 ± 3.67 | 35.66 ± 2.61 | 0.048 | 33.80 ± 2.57 | 0.006 |
| MCV (fL) | 43.95 ± 1.32 | 43.80 ± 1.86 | 0.883 | 42.48 ± 1.93 | 0.163 |
| MCH (pg) | 13.13 ± 0.43 | 13.36 ± 0.56 | 0.480 | 14.88 ± 0.88 | 0.000 |
| MCHC (g L−1) | 310.00 ± 4.86 | 306.83 ± 5.95 | 0.333 | 306.00 ± 5.58 | 0.226 |
| PLT (109 L−1) | 731.5 ± 104.1 | 722.8 ± 120.4 | 0.879 | 715.00 ± 53.12 | 0.772 |
| RDW (%) | 15.68 ± 0.56 | 15.55 ± 0.93 | 0.813 | 16.30 ± 1.25 | 0.283 |
| MPV (fL) | 5.11 ± 0.17 | 5.50 ± 0.39 | 0.092 | 5.18 ± 0.47 | 0.758 |
| RDW (%) | 15.68 ± 0.56 | 15.55 ± 0.93 | 0.211 | 16.30 ± 1.25 | 0.437 |
| PCT (%) | 0.45 ± 0.05 | 0.40 ± 0.08 | 0.136 | 0.44 ± 0.06 | 0.713 |
Compared to the control groups, most of the hematological results of RhB degradation filtrates exhibited no obvious statistical differences, except HCT (P = 0.048). Additionally, the blood chemical assay was performed. Alanine aminotransferase (ALT), aspartate aminotransferase (AST), albumin (ALB), alkaline phosphatase (ALP), triglycerides (TG) and total cholesterol (CHO) were the major biomarkers of liver function. The blood urea nitrogen (BUN) and creatinine (CREA) were related to kidney function. Also no obvious statistical differences were observed in RhB degradation filtrate groups. However, the ALT and AST of the RhB solution groups were greatly higher than those of the control groups (P < 0.01), which was associated with impaired liver function (Table 2). The pathological changes of major organs were compared with those of the control groups using the H&E staining assay (Fig. 8). The RhB solution groups showed high toxicity toward major organs. Hepatocytes with moderate degeneration were widely seen accompanied by a small amount of liver nuclear pyknosis. Occasional necrosis and nucleolysis were detected in liver cells. More aggregates of inflammatory cell infiltrates in liver lobules, lungs and kidneys were also seen. Yet the RhB degradation filtrate groups did not exhibit obvious pathological changes, compared to the controls. The toxicity of RhB degradation filtrates was assessed in vitro and in vivo, demonstrating that RhB degradation filtrates exhibited lower toxicity compared with those of RhB injection alone.
| Markers | Control group | RhB degradation filtrate | P* | RhB solution | P* |
|---|---|---|---|---|---|
| a *P, compared with the control groups. b U L−1. c mmol L−1. d μmol L−1. | |||||
| ALTb | 45.95 ± 9.73 | 58.52 ± 11.33 | 0.053 | 91.39 ± 9.99 | 0.000 |
| ASTb | 142.16 ± 16.16 | 149.91 ± 21.34 | 0.620 | 195.89 ± 37.31 | 0.003 |
| ALBb | 37.65 ± 4.57 | 41.08 ± 3.98 | 0.120 | 36.01 ± 1.43 | 0.442 |
| ALPb | 250.86 ± 78.84 | 234.66 ± 52.76 | 0.672 | 252.31 ± 60.32 | 0.970 |
| TGc | 0.96 ± 0.19 | 1.13 ± 0.23 | 0.313 | 1.11 ± 0.34 | 0.349 |
| CHOc | 2.19 ± 0.39 | 2.24 ± 0.50 | 0.856 | 2.29 ± 0.55 | 0.709 |
| BUNd | 6.11 ± 0.88 | 7.49 ± 1.58 | 0.077 | 6.70 ± 1.21 | 0.426 |
| CREAd | 25.24 ± 9.27 | 27.57 ± 3.57 | 0.226 | 14.71 ± 11.09 | 0.051 |
| UAd | 291.44 ± 97.01 | 324.36 ± 84.22 | 0.502 | 313.48 ± 64.09 | 0.652 |
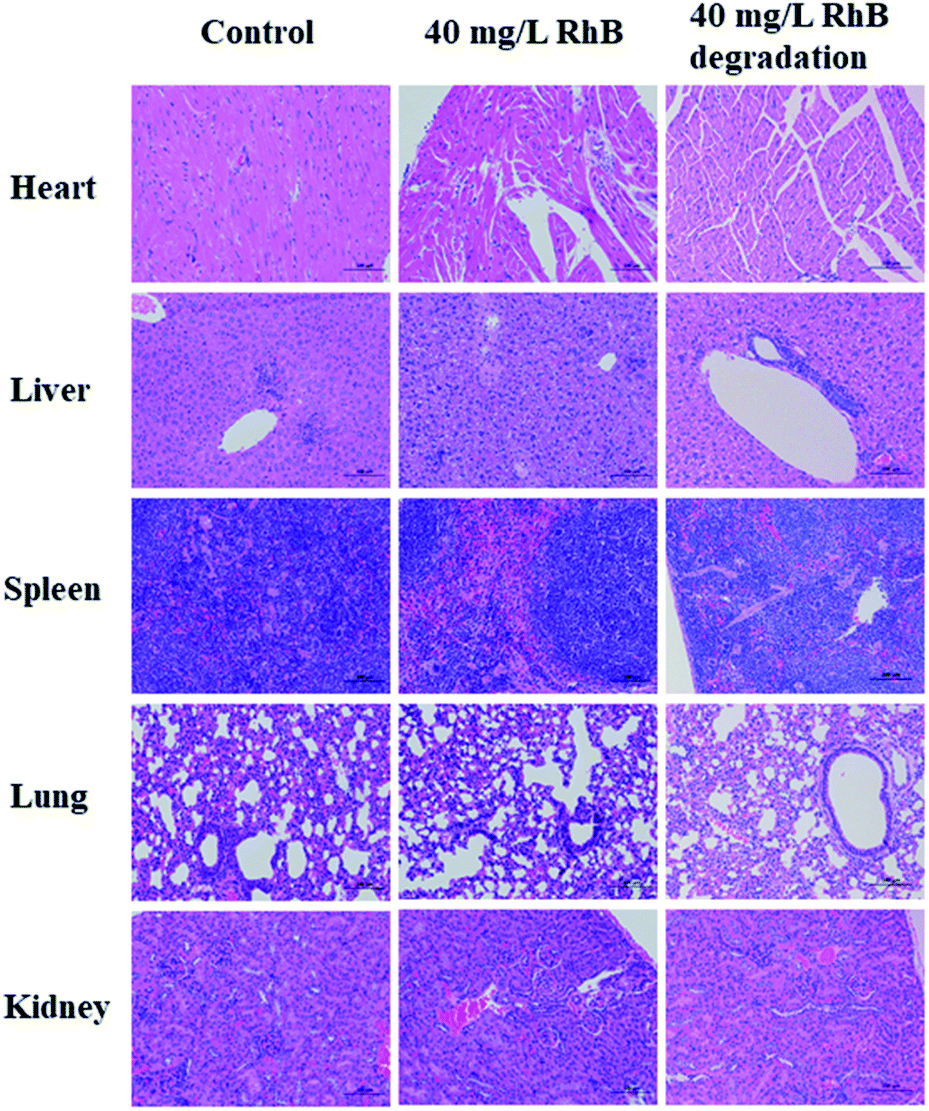 | ||
| Fig. 8 H&E stained sections of the heart, liver, spleen, lung and kidney after 21 days treatment with the RhB degradation filtrate (substrate RhB concentration = 40 mg L−1) or RhB solution (40 mg L−1). | ||
4. Conclusions
In this work, CuCDs were successfully prepared by a facile hydrothermal synthesis strategy. It was found that the addition of CuCDs helped the Fenton reaction accelerate RhB degradation in neutral solution. The catalytic activity of CuCDs was influenced by temperature, pH, concentration of CuCDs, metallic ions and electric charges. It was worth mentioning that electron transfer on the surface of CuCDs was found to be crucial in RhB degradation upon the Fenton reaction process as well as more ˙OH and 1O2 generation promoted by CuCDs. Moreover, the composition and microstructures of CuCDs had contributions to RhB degradation. The RhB degradation pathway was proposed on the basis of the results of LC-MS. Unlike other RhB degradation mechanisms, the strong oxidation of ROS caused the cleavage of the chromophore aromatic ring and the de-ethylation process in this work. In addition, the toxicity of RhB degradation filtrates was assessed in vitro and in vivo, demonstrating that the highly toxic RhB solution could convert to low-toxicity degradation products through the CuCD accelerated Fenton reaction. Therefore, CuCDs would be a promising Fenton catalyst to enhance the RhB degradation efficiency.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81960558) and the Science Foundation of Guangxi Academy of Sciences (2018YJJ908).References
- Y. Liang, G. Huang, Q. Zhang, Y. Yang, J. Zhou and J. Cai, J. Mol. Liq., 2021, 330, 115580 CrossRef CAS.
- D. Joshy, S. Chakko, Y. A. Ismail and P. Periyat, Nanoscale Adv., 2021, 3(23), 6704–6718 RSC.
- S. Mishra, L. Cheng and A. Maiti, J. Environ. Chem. Eng., 2021, 9(1), 104901 CrossRef CAS.
- S. Nayak, G. Swain and K. Parida, ACS Appl. Mater. Interfaces, 2019, 11(23), 20923–20942 CrossRef CAS PubMed.
- S. Steplin Paul Selvin, A. Ganesh Kumar, L. Sarala, R. Rajaram, A. Sathiyan, J. Princy Merlin and I. Sharmila Lydia, ACS Sustainable Chem. Eng., 2017, 6(1), 258–267 CrossRef.
- X. Li, J. Liu, A. I. Rykov, H. Han, C. Jin, X. Liu and J. Wang, Appl. Catal., B, 2015, 179, 196–205 CrossRef CAS.
- M. Cheng, G. Zeng, D. Huang, C. Lai, P. Xu, C. Zhang and Y. Liu, Chem. Eng. J., 2016, 284, 582–598 CrossRef CAS.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- X. Lai, X. A. Ning, J. Chen, Y. Li, Y. Zhang and Y. Yuan, J. Hazard. Mater., 2020, 398, 122826 CrossRef CAS PubMed.
- M. A. Tony and S. A. Mansour, Nanoscale Adv., 2019, 1(4), 1362–1371 RSC.
- X. Huang, X. Hou, F. Jia, F. Song, J. Zhao and L. Zhang, ACS Appl. Mater. Interfaces, 2017, 9(10), 8751–8758 CrossRef CAS PubMed.
- W. Meng, Y. Wang, Y. Zhang, C. Liu, Z. Wang, Z. Song, B. Xu, F. Qi and A. Ikhlaq, J. Taiwan Inst. Chem. Eng., 2020, 111, 162–169 CrossRef CAS.
- Y. Leng, W. Guo, X. Shi, Y. Li and L. Xing, Ind. Eng. Chem. Res., 2013, 52(38), 13607–13612 CrossRef CAS.
- Y. Cui, Z. Zhou, Y. Gao, L. Lei, J. Cao, R. Wu, L. Liang and Z. Huang, J. Clean. Prod., 2021, 289, 125807 CrossRef CAS.
- S. Ni, T. Zhou, H. Zhang, Y. Cao and P. Yang, ACS Appl. Nano Mater., 2018, 1(9), 5128–5141 CrossRef CAS.
- G. Huang, X. Chen, C. Wang, H. Zheng, Z. Huang, D. Chen and H. Xie, RSC Adv., 2017, 7(75), 47840–47847 RSC.
- Q. Jiang, L. Liu, Q. Li, Y. Cao, D. Chen, Q. Du, X. Yang, D. Huang, R. Pei, X. Chen and G. Huang, J. Nanobiotechnol., 2021, 19(1), 64 CrossRef CAS PubMed.
- A. Sharma and J. Das, J. Nanobiotechnol., 2019, 17(1), 92–116 CrossRef PubMed.
- J. Zhang, X. Liu, J. Zhou, X. Huang, D. Xie, J. Ni and C. Ni, Nanoscale Adv., 2019, 1(6), 2151–2156 RSC.
- M. Pan, Z. Xu, Q. Jiang, J. Feng, J. Sun, F. Wang and X. Liu, Nanoscale Adv., 2019, 1(2), 765–771 RSC.
- R. Wang, K. Q. Lu, Z. R. Tang and Y. J. Xu, J. Mater. Chem. A, 2017, 5(8), 3717–3734 RSC.
- V. Arul, T. N. Edison, Y. R. Lee and M. G. Sethuraman, J. Photochem. Photobiol., B, 2017, 168, 142–148 CrossRef CAS PubMed.
- X. Xu, Z. Chen, Q. Li, D. Meng, H. Jiang, Y. Zhou, S. Feng and Y. Yang, Microchem. J., 2021, 160, 105708 CrossRef CAS.
- Y. Duan, Y. Huang, S. Chen, W. Zuo and B. Shi, ACS Omega, 2019, 4(6), 9911–9917 CrossRef CAS PubMed.
- L. Lin, Y. Luo, P. Tsai, J. Wang and X. Chen, Trac. Trends Anal. Chem., 2018, 103, 87–101 CrossRef CAS.
- Z. X. Liu, B. B. Chen, M. L. Liu, H. Y. Zou and C. Z. Huang, Green Chem., 2017, 19(6), 1494–1498 RSC.
- W. Wu, L. Zhan, W. Fan, J. Song, X. Li, Z. Li, R. Wang, J. Zhang, J. Zheng, M. Wu and H. Zeng, Angew. Chem., Int. Ed., 2015, 54(22), 6540–6544 CrossRef CAS PubMed.
- C. Han, X. Zhang, F. Wang, Q. Yu, F. Chen, D. Shen, Z. Yang, T. Wang, M. Jiang, T. Deng and C. Yu, Carbon, 2021, 183, 789–808 CrossRef CAS.
- W. Qing, K. Chen, Y. Yang, Y. Wang and X. Liu, Microchem. J., 2020, 152, 104262 CrossRef CAS.
- P. P. Zhu, Z. Cheng, L. L. Du, Q. Chen and K. J. Tan, Langmuir, 2018, 34(34), 9982–9989 CrossRef CAS PubMed.
- X. Ren, J. Liu, J. Ren, F. Tang and X. Meng, Nanoscale, 2015, 7(46), 19641–19646 RSC.
- J. Du, Y. Zhao, J. Chen, P. Zhang, L. Gao, M. Wang, C. Cao, W. Wen and C. Zhu, RSC Adv., 2017, 7(54), 33929–33936 RSC.
- D. Yang, Q. Li, S. K. Tammina, Z. Gao and Y. Yang, Sens. Actuators B Chem., 2020, 319, 128273 CrossRef CAS.
- Z. Chai, C. Li, Y. Zhu, X. Song, M. Chen, Y. Yang, D. Chen, X. Liang and J. Wu, Int. J. Bio. Macromol., 2020, 165, 506–516 CrossRef CAS PubMed.
- X. Song, Z. Chai, Y. Zhu, C. Li and X. Liang, Carbohydr. Polym., 2019, 219, 316–327 CrossRef CAS PubMed.
- J. Guo, W. Lu, H. Zhang, Y. Meng, F. Du, S. Shuang and C. Dong, Sens. Actuators B Chem., 2021, 330, 129360 CrossRef CAS.
- N. Wang, W. Ma, Y. Du, Z. Ren, B. Han, L. Zhang, B. Sun, P. Xu and X. Han, ACS Appl. Mater. Interfaces, 2019, 11(1), 1174–1184 CrossRef CAS PubMed.
- V. Lakshmi Prasanna and R. Vijayaraghavan, J. Phys. Chem. C, 2017, 121(34), 18557–18563 CrossRef CAS.
- S. Pandiyan, L. Arumugam, S. P. Srirengan, R. Pitchan, P. Sevugan, K. Kannan, G. Pitchan, T. A. Hegde and V. Gandhirajan, ACS Omega, 2020, 5(47), 30363–30372 CrossRef CAS PubMed.
- J. Xiao, J. Lai, R. Li, X. Fang, D. Zhang, P. Tsiakaras and Y. Wang, Ind. Eng. Chem. Res., 2020, 59(27), 12431–12440 CrossRef CAS.
- Z. Liang, M. Kang, G. F. Payne, X. Wang and R. Sun, ACS Appl. Mater. Interfaces, 2016, 8(27), 17478–17488 CrossRef CAS PubMed.
- D. F. Eaton, Pure Appl. Chem., 1988, 60(7), 1107–1114 CrossRef CAS.
- B. Sun, H. Li, X. Li, X. Liu, C. Zhang, H. Xu and X. S. Zhao, Ind. Eng. Chem. Res., 2018, 57(42), 14011–14021 CrossRef CAS.
- H. Yin, D. Gao, Y. Qiu, G. Yi, J. Li, Y. Dong, K. Zhang, Z. Xia and Q. Fu, Nanoscale Adv., 2020, 2(4), 1483–1492 RSC.
- N. Dhenadhayalan, K. C. Lin and T. A. Saleh, Small, 2020, 16(1), 1905767 CrossRef CAS PubMed.
- B. Palanivel and A. Mani, ACS Omega, 2020, 5(31), 19747–19759 CrossRef CAS PubMed.
- W. Yu, T. Liu, M. Zhang, Z. Wang, J. Ye, C. X. Li, W. Liu, R. Li, J. Feng and X. Z. Zhang, ACS Nano, 2019, 13(2), 1784–1794 CAS.
- S. Sahar, A. Zeb, C. Ling, A. Raja, G. Wang, N. Ullah, X. M. Lin and A. W. Xu, ACS Nano, 2020, 14(3), 3017–3031 CrossRef CAS PubMed.
- Y. J. Chung, B. I. Lee, J. W. Ko and C. B. Park, Adv. Healthc. Mater., 2016, 5(13), 1560–1565 CrossRef CAS PubMed.
- B. Wang, G. Zhang, Z. Sun, S. Zheng and R. L. Frost, J. Environ. Chem. Eng., 2015, 3(3), 1444–1451 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2na00269h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |