
Covalent drugs targeting histidine – an unexploited opportunity?
Jianwei
Che
ab and
Lyn H.
Jones
 *ab
*ab
aCenter for Protein Degradation, Dana-Farber Cancer Institute, 360 Longwood Avenue, Boston, MA, USA
bDepartment of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA, USA. E-mail: lyn_jones@dfci.harvard.edu
First published on 28th September 2022
Abstract
Covalent drugs and chemical probes often possess pharmacological advantages over reversible binding ligands, such as enhanced potency and pharmacodynamic duration. The highly nucleophilic cysteine thiol is commonly targeted using acrylamide electrophiles, but the amino acid is rarely present in protein binding sites. Sulfonyl exchange chemistry has expanded the covalent drug discovery toolkit by enabling the rational design of irreversible inhibitors targeting tyrosine, lysine, serine and threonine. Probes containing the sulfonyl fluoride warhead have also been shown to serendipitously label histidine residues in proteins. Histidine targeting is an attractive prospect because the residue is frequently proximal to protein small molecule ligands and the imidazole side chain possesses desirable nucleophilicity. We recently reported the design of cereblon molecular glues to site-selectively modify a histidine in the thalidomide binding site using sulfonyl exchange chemistry. We believe that histidine targeting holds great promise for future covalent drug development and this Opinion highlights these opportunities.
Introduction: beyond cysteine targeting
Covalent drugs and chemical probes often possess enhanced potency, selectivity, and pharmacodynamic duration compared to reversible binding small molecules.1–3 The rational design of covalent compounds has historically focussed on targeting cysteine residues in protein active sites due to its high intrinsic nucleophilicity, and several drugs have been approved that operate via this mechanism of inhibition.4 However, cysteine has a very low occurrence in protein binding sites, and the high intrinsic nucleophilicity of the side chain can hamper the development of highly selective probes and drugs.5,6 For example, the clinical candidate ritlecitinib was designed to engage a rare cysteine in the ATP-site of JAK3, rendering selectivity against the related JAK enzymes (and broader kinome). However, the inhibitor was shown to react with other kinases possessing the corresponding cysteine that determines its phenotypic effects.7,8 Additionally, targeted cysteine residues are often not functionally critical, and this may facilitate the rapid emergence of mutational resilience in cancer.9,10 Therefore, a need exists to target nucleophilic residues beyond cysteine.5,11 Recently, in several different studies we have demonstrated the utility of a sulfonyl exchange chemical biology platform to enable the rational design of covalent small molecules that site-selectively engage tyrosine, lysine, and serine in protein binding pockets.12–17 Nevertheless, opportunities remain to further expand the synthetic chemical biology toolkit to facilitate the targeting of additional nucleophilic amino acid side chains.Histidine is a targetable residue in proteins
Based on an analysis of 1350 measured pKa values for ionizable protein residues, 82% of the 253 histidine values fall within 6 and 8, with a mean of 6.45 (Fig. 1).18 We also noticed from this analysis that <1 in 5 protein histidines possess a pKa > 7.4 and most of these are proximal to acidic residues, which is expected to increase the basicity of the side chain. Thus, histidine residues in proteins are usually unprotonated, which facilitates nucleophilic chemical reactions of the imidazole ring and targeting by electrophiles. This contrasts with the considerably higher pKa of lysine, which is usually fully protonated in proteins at physiological pH (Fig. 1).18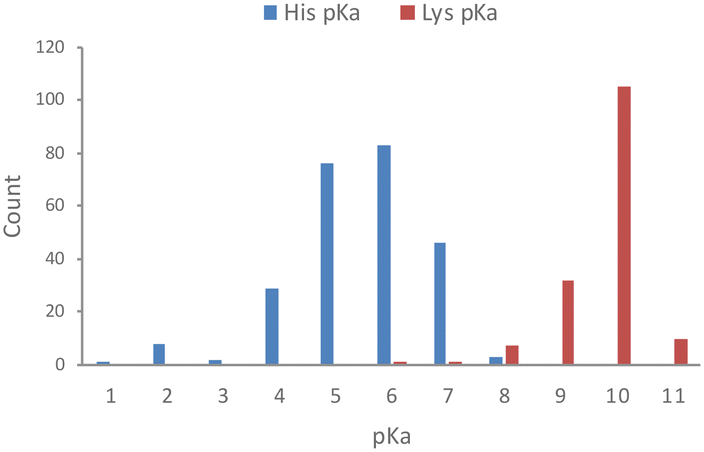 | ||
| Fig. 1 Measured pKa values for histidine and lysine residues in proteins (data taken from the pKa database PKAD).18 | ||
Histidine is the most common catalytic residue in enzyme sites, and it appears 8.2 times more often than expected based on its natural frequency.19 One reason for this is that the pKa range of histidine often translates to the residue acting either as a proton acceptor or donor in its catalytic mechanisms. Additionally, histidine is far more frequently represented in DNA/RNA binding sites than cysteine for example20 and it regularly acts as a catalytic nucleophile for phosphates.19
Importantly, histidine has a low surface frequency compared to other amino acids6,18 and has one of the highest preferences to be within 4.5 Å of drugs and drug-like compounds in binding sites, as determined by an analysis of protein structures in the PDB.21 By surveying the entire PDB databank (as of July 2022) we confirmed that histidine is indeed enriched in small molecule binding sites (2-fold over its expected natural abundance). These findings suggest that histidine could be a natural choice for the development of covalent chemical probes and drugs, but this opportunity appears to have been mostly unexplored and underexploited in the published literature.
Serendipitous histidine labelling
There are some reports of histidine labelling in proteins by small molecules containing thiophosphorodichloridate,22 α-cyanoenone,23 and epoxide electrophiles.24,25 While serving as useful precedence, it is challenging to apply such highly reactive warheads in drug development due to non-specific labelling. Aryl sulfonyl fluoride has been shown to be a privileged electrophilic warhead suitable for the development of numerous covalent chemical probes.12,26 Fahrney and Gold elucidated the mode of inhibition of serine hydrolases by sulfonyl fluoride containing molecules, which occurs via the sulfonylation of the catalytic serine residue.27,28 Bernard Baker was the first to then incorporate the electrophile deliberately into reversible binding ligands to convert them into irreversible inhibitors.29 Later, Roberta Colman developed a nucleoside derivative bearing a sulfonyl fluoride, 5′-p-fluorosulfonyl benzoyladenosine (FSBA) that was used to map nucleophilic residues in several protein binding sites.30,31 At that time, the specific amino acids labelled by these probes were determined after the fact, and subsequent work has shown they have a natural preference to react with tyrosine side chains, followed by lysine.12,16,32–36 Recently, sulfonyl triazole exchange covalent chemistry was developed, and unbiased chemoproteomics demonstrated a preference for this warhead to attack tyrosine residues also.37,38Importantly, FSBA was shown previously to label His427 in the binding site of bovine mitochondrial ATPase at pH 6.5, while at pH 8.0, Tyr368 was preferentially modified, presumably aided by deprotonation of the phenol moiety.39 Additionally, a histidine residue in the ATP site of phosphoribosyl pyrophosphate synthetase was engaged preferentially over two proximal lysine side chains in the same binding pocket.40 These serendipitous findings suggest that sulfonyl exchange chemistry may be suitable for the rational context-dependent targeting of histidine in protein binding sites.
Rational histidine targeting: cereblon covalent modulators
Inspired by the possibility of histidine modification using sulfonyl exchange chemistry we rationally designed the first covalent ligands of cereblon (CRBN), an adapter protein for an E3 ubiquitin ligase complex. Small molecule ligands called immunomodulatory drugs (IMiDs), including thalidomide, lenalidomide, pomalidomide and EM12, bind in a site that modifies the CRBN surface which induces interactions with neosubstrates resulting in their polyubiquitination and proteasomal degradation.41,42 We reasoned that covalent engagement of His353 in the binding site would be feasible due to its proximity to the small molecule IMiDs in these structures, and for the general reasons described above. His353 resides in the so-called ‘sensor loop’ β-hairpin that flips towards the ligand when bound, stabilizing a closed CRBN conformation that induces neosubstrate binding.43 His353 makes interactions with the key structural motif that resides within neosubstrates referred to as the G-loop degron. We hypothesized that labelling of His353 would staple CRBN in a conformation that either inhibits neosubstrate recruitment or subtly modifies the CRBN surface to enable binding of alternative substrate proteins. Using structure-based drug design, a series of sulfonyl fluorides and triazoles, and intrinsically less electrophilic fluorosulfates, were developed that exclusively labelled His353.44 The sulfonyl fluoride EM12-SO2F (Fig. 2a) blocked the IMiD binding site and did not appreciably degrade any protein in the cell lines tested (as shown by quantitative mass spectrometry proteomics). Remarkably, the congeneric fluorosulfate EM12-FS (Fig. 2a), which differs from EM12-SO2F by the addition of a single oxygen atom, degraded one protein significantly called protein N-terminal glutamine amidohydrolase (NTAQ1). Presumably, the sulfonyl fluoride and fluorosulfate warheads engage His353 in a slightly different manner to impact the surface structure of CRBN, resulting in these different phenotypic effects. Docking of the probes into CRBN reveals a likely steric clash between the –SO2– groups and the traditional neosubstrate degron (Fig. 2b), but more work is required to elucidate the structural details of NTAQ1 recruitment. Biochemical studies suggested that modification of His353 is required for binding NTAQ1 causing its subsequent degradation.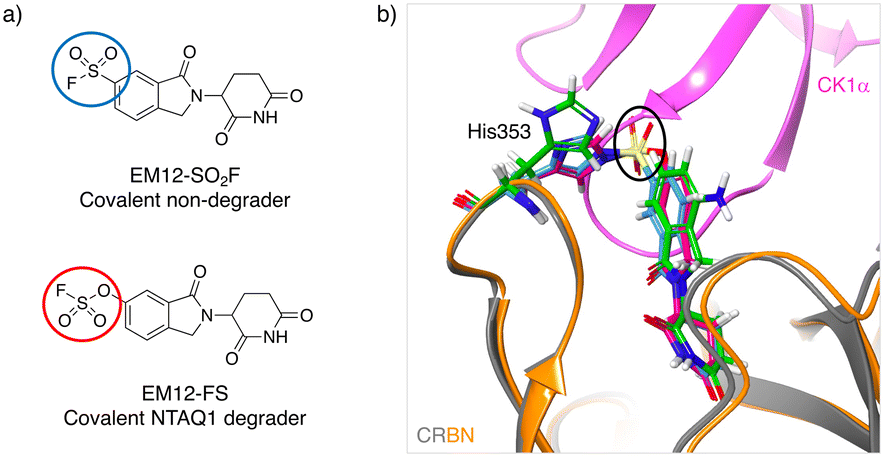 | ||
| Fig. 2 a) Structures of covalent cereblon modulators that modify His353 in the IMiD binding pocket (electrophilic warheads are circled). b) Docking model of EM12-SO2F (blue) and EM12-FS (red) into the crystal structure of the lenalidomide (green)/cereblon (orange ribbon)/CK1α (magenta ribbon) complex (PDB 5FQD).47 The sulfonyl groups (black oval) would appear to clash with the traditional neosubstrate G-loop degron (CK1α in this case). | ||
NTAQ1 is involved in the N-end rule pathway, mediating the hydrolysis of N-terminal glutamine to glutamate, which triggers arginylation to create an N-degron that is recognised by E3 ligases called N-recognins.45 There are no chemical probes for NTAQ1, a protein that is annotated undruggable,46 and EM12-FS and its derivatives may be useful additions to the toolkit to further study the N-end rule pathway.
Another important aspect of this work was that the His adducts appeared to be formed in an irreversible manner (shown by the mass spectrometry and biochemical studies). Presumably in other instances the sulfonylation event may indeed be reversible and, as discussed below, metabolism in plasma would be expected to cleave the N–S bond to return the parent His, although these studies were not performed.
Targeted protein degradation by IMiD derivatives, also referred to as ‘molecular glues’,48 is a very active area of drug discovery research because changes to the IMiD scaffold results in the degradation of diverse neosubstrates, including difficult-to-drug proteins like zinc-finger transcription factors.42
Additionally, IMiDs have been incorporated into heterobifunctional molecules called proteolysis targeting chimeras (PROTACs).49–51 PROTACs chemically link an E3 ligase ligand to a binder for the protein-of-interest (POI). PROTACs thus hijack the ubiquitin machinery to induce degradation of the POI. Target validation and deconvolution experiments are required following the phenotypic assessment of IMiD-based degraders in order to confirm the involvement of CRBN in the mode-of-action. Current methods use genetically engineered cell lines to knock out CRBN, or PROTAC degraders of CRBN itself, which can be a relatively slow process.52–54 EM12-SO2F provides a simple and fast-acting alternative by blocking the function of CRBN in native cells. As a proof-of-principle study, the probe was shown to block the degradation of the IKZF1 neosubstrate by lenalidomide.44
As an aside, the first PROTAC that was developed incorporated ovalicin as the POI ligand which is a natural product with an embedded spiroepoxide electrophile that covalently engages a histidine residue in the binding site of methionine aminopeptidase 2 (MetAP2).55 Our work demonstrates the feasibility of developing a covalent CRBN PROTAC through His353 engagement which may engender heterobifunctional degraders with high catalytic efficiency,56–58 in contrast to degraders designed to covalently bind the POI.
Metabolic stability of sulfonyl exchange warheads
Targeted covalent drugs may possess PKPD advantages over reversible binders, but metabolic stability is still a necessary requirement to achieve the requisite drug levels in vivo that drives the functional pharmacological effects. Although sulfonyl fluorides and triazoles may possess good stability in microsomes and hepatocytes, generally low plasma stability makes them a challenging warhead for incorporation into drugs (Fig. 3). However, as described above, sulfonyl fluorides/triazoles are perfectly suitable for the development of in-cell target engagement and proteomic chemical tools, such as EM12-SO2F. Conversely, the fluorosulfate warhead is a considerably weaker electrophile and as a result possesses high metabolic stability rendering the motif suitable for drug design strategies.14 In the CRBN example described above, EM12-FS has high stability in human plasma, liver microsomes and hepatocytes (Fig. 3). Further optimization of equilibrium binding, and modification of the intrinsic reactivity of the warhead would likely provide the basis for optimizing site-selective histidine labelling whilst retaining metabolic stability.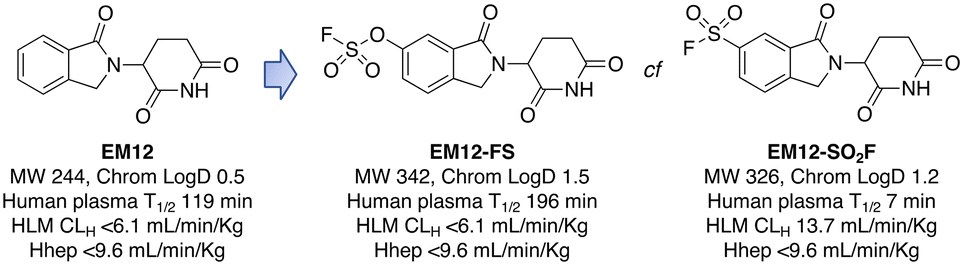 | ||
| Fig. 3 Metabolic stability of the IMiD EM12 (ref. 59) and the covalent cereblon modulators EM12-SO2F (a sulfonyl fluoride) and EM12-FS (a fluorosulfate). | ||
Outlook
Although the intrinsic nucleophilicity of histidine is weaker than cysteine, our work shows that optimal equilibrium binding interactions enable the use of targeting electrophilic warheads such as fluorosulfate with ameliorated reactivity which appears to be suitable for drug development. Considerable advances are being made targeting residues beyond cysteine and we feel that histidine should be the focus of more research. The following points highlight further opportunities and challenges for the field:• Expand the toolkit of histidine targeting electrophiles and further explore the relationships between intrinsic nucleophilicity of the warhead and the propensity for developing a site-selective covalent modulator with the requisite metabolic stability for drug development.
• Create histidine-selective warheads to enable the development of covalent probes and drugs. Currently, there are no electrophiles that are known to intrinsically engage histidines over other amino acids. Although we do not believe this is entirely necessary (because equilibrium binding optimization and warhead placement also drives amino acid side chain labelling specificity) the development of such selective chemistry would clearly provide new opportunities for the field.
• Convert histidine-targeting ligands into clickable occupancy biomarker probes, to aid in-cell target engagement, selectivity, and validation studies.
• Develop promiscuous chemoproteomic probes that map the targetable histidinome, analogous to such methods used for cysteine, lysine, and tyrosine. This has the potential to considerably expand the druggable proteome.
• Specifically, target non-catalytic histidines in kinase ATP sites, to expand the druggable kinome. Based on our analysis of the PDB databank there are 171 human kinase structures (without removing redundant proteins) that possess histidines within 4.5 Å of a bound small molecule. Moreover, based on primary sequence alignment, there are 162 kinases out of 556 that contain a histidine within the ATP binding site (Fig. 4). These residues present opportunities for the development of selective covalent kinase inhibitors and chemical probes.
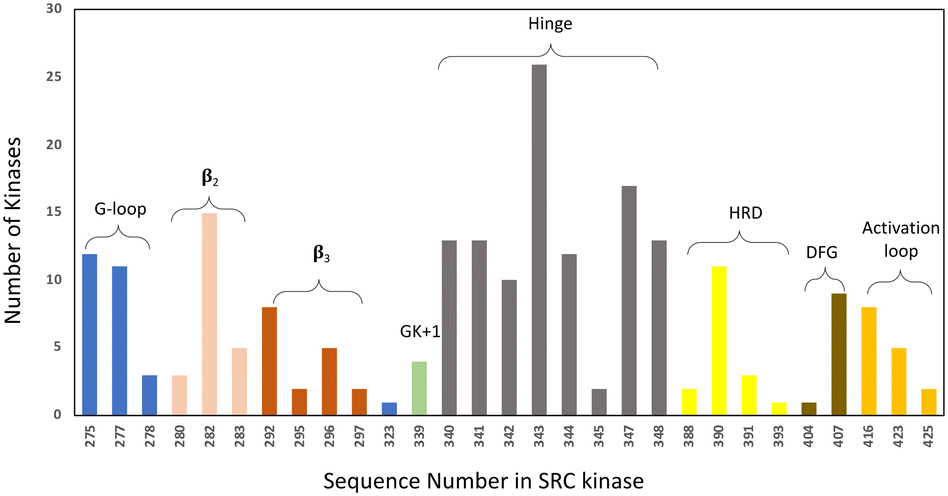 | ||
| Fig. 4 Histidine distribution in the kinase ATP binding site. The x axis is labelled according to the SRC kinase amino acid number. The ATP site is defined by 7 Å proximity to AMP-PNP in the SRC cocrystal structure (PDB 2SRC). The colouring indicates the structural motifs present in common protein kinases (GK = gatekeeper). | ||
• Map the E3 ligase histidinome and develop new molecular degraders, as shown for CRBN, to expand the degradable proteome.
• The frequency of nucleophilic histidines in RNA-binding protein sites highlights the potential of chemogenomic targeting of this family using covalent probes and drugs.
• Better understand the features of the microenvironment of binding pockets that enhance histidine nucleophilicity to aid in the selection of druggable therapeutic targets. This information will also expedite structure-based drug design.
• Apply the knowledge attained from targeting histidine to bioconjugation, expanding the chemistry toolkit beyond cysteine, lysine and tyrosine which dominate the field.
• Explore the susceptibility for histidine to mutate when covalently engaged by an anticancer drug – this may provide an advantage over cysteine targeted drugs where serine mutations rapidly emerge following treatment.
• Study the biological roles of histidine phosphorylation. Since phosphohistidine is labile and transient in many cellular processes, it is difficult to study in a native environment. With selective modification of a given histidine using stable chemical structures, it is possible to interrogate the functional consequences of histidine modification. Labelling of histidine using chemical probes would likely be inhibited by phosphorylation, providing an alternative readout of this relatively understudied post-translational modification.
There is clearly a need to advance covalent drug discovery beyond cysteine labelling and we believe that histidine targeting will play an important future role. Although there are challenges that need to be navigated, we believe histidine-targeted covalent chemical probes and drug candidates will emerge in the coming years.
Author contributions
Both authors contributed to the writing of this manuscript.Conflicts of interest
L. H. J. serves on the scientific advisory boards for, and holds equity in, Interline Therapeutics, Rapafusyn Pharmaceuticals, Ananke Therapeutics and Umbra Therapeutics, consults for Matchpoint Therapeutics, and holds equity in Jnana Therapeutics. The Center for Protein Degradation at DFCI receives research funding from Deerfield. J. C. is a consultant for Soltego, Allorion and Matchpoint Therapeutics, and holds equity in Soltego, Allorion, Matchpoint and M3 Bioinformatics & Technology Inc.Acknowledgements
We thank members of the Center for Protein Degradation for their contributions to the research reviewed here and for useful discussions.References
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673–5724 CrossRef CAS PubMed.
- Q. Liu, Y. Sabnis, Z. Zhao, T. Zhang, S. J. Buhrlage, L. H. Jones and N. S. Gray, Chem. Biol., 2013, 20, 146–159 CrossRef CAS PubMed.
- F. Daryaee, Z. Zhang, K. R. Gogarty, Y. Li, J. Merino, S. L. Fisher and P. J. Tonge, Chem. Sci., 2017, 8, 3434–3443 RSC.
- F. Sutanto, M. Konstantinidou and A. Dömling, RSC Med. Chem., 2020, 11, 876–884 RSC.
- L. H. Jones, Annu. Rep. Med. Chem., 2021, 56, 95–134 CAS.
- N. A. Khazanov and H. A. Carlson, PLoS Comput. Biol., 2013, 9, e1003321 CrossRef PubMed.
- J. B. Telliez, M. E. Dowty, L. Wang, J. Jussif, T. Lin, L. Li, E. Moy, P. Balbo, W. Li, Y. Zhao, K. Crouse, C. Dickinson, P. Symanowicz, M. Hegen, M. E. Banker, F. Vincent, R. Unwalla, S. Liang, A. M. Gilbert, M. F. Brown, M. Hayward, J. Montgomery, X. Yang, J. Bauman, J. I. Trujillo, A. Casimiro-Garcia, F. F. Vajdos, L. Leung, K. F. Geoghegan, A. Quazi, D. Xuan, L. Jones, E. Hett, K. Wright, J. D. Clark and A. Thorarensen, ACS Chem. Biol., 2016, 11, 3442–3451 CrossRef CAS PubMed.
- H. Xu, M. I. Jesson, U. I. Seneviratne, T. H. Lin, M. N. Sharif, L. Xue, C. Nguyen, R. A. Everley, J. I. Trujillo, D. S. Johnson, G. R. Point, A. Thorarensen, I. Kilty and J. B. Telliez, ACS Chem. Biol., 2019, 14, 1235–1242 CrossRef CAS PubMed.
- J. A. Woyach, R. R. Furman, T. M. Liu, H. G. Ozer, M. Zapatka, A. S. Ruppert, L. Xue, D. H. Li, S. M. Steggerda, M. Versele, S. S. Dave, J. Zhang, A. S. Yilmaz, S. M. Jaglowski, K. A. Blum, A. Lozanski, G. Lozanski, D. F. James, J. C. Barrientos, P. Lichter, S. Stilgenbauer, J. J. Buggy, B. Y. Chang, A. J. Johnson and J. C. Byrd, N. Engl. J. Med., 2014, 370, 2286–2294 CrossRef PubMed.
- M. J. Niederst, H. Hu, H. E. Mulvey, E. L. Lockerman, A. R. Garcia, Z. Piotrowska, L. V. Sequist and J. A. Engelman, Clin. Cancer Res., 2015, 21, 3924–3933 CrossRef CAS PubMed.
- L. H. Jones, Angew. Chem., Int. Ed., 2018, 57, 9220–9223 CrossRef CAS PubMed.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680–685 CrossRef CAS PubMed.
- O. O. Fadeyi, L. R. Hoth, C. Choi, X. Feng, A. Gopalsamy, E. C. Hett, R. E. Kyne, R. P. Robinson and L. H. Jones, ACS Chem. Biol., 2017, 12, 2015–2020 CrossRef CAS PubMed.
- S. Liu, L. A. Dakin, L. Xing, J. M. Withka, P. V. Sahasrabudhe, W. Li, M. E. Banker, P. Balbo, S. Shanker, B. A. Chrunyk, Z. Guo, J. M. Chen, J. A. Young, G. Bai, J. T. Starr, S. W. Wright, J. Bussenius, S. Tan, A. Gopalsamy, B. A. Lefker, F. Vincent, L. H. Jones, H. Xu, L. R. Hoth, K. F. Geoghegan, X. Qiu, M. E. Bunnage and A. Thorarensen, Sci. Rep., 2016, 6, 30859 CrossRef CAS PubMed.
- E. C. Hett, H. Xu, K. F. Geoghegan, A. Gopalsamy, R. E. Kyne, C. A. Menard, A. Narayanan, M. D. Parikh, S. Liu, L. Roberts, R. P. Robinson, M. A. Tones and L. H. Jones, ACS Chem. Biol., 2015, 10, 1094–1098 CrossRef CAS PubMed.
- O. Fadeyi, M. D. Parikh, M. Z. Chen, R. E. Kyne, A. P. Taylor, I. O'Doherty, S. E. Kaiser, S. Barbas, S. Niessen, M. Shi, S. L. Weinrich, J. C. Kath, L. H. Jones and R. P. Robinson, ChemBioChem, 2016, 17, 1925–1930 CrossRef CAS PubMed.
- S. Pahari, L. Sun and E. Alexov, Database, 2019, baz024 CAS.
- A. J. M. Ribeiro, J. D. Tyzack, N. Borkakoti, G. L. Holliday and J. M. Thornton, J. Biol. Chem., 2020, 295, 314–324 CrossRef CAS PubMed.
- D. Corcoran, N. Maltbie, S. Sudalairaj, F. N. Baker, J. Hirschfeld and A. Porollo, Front. Bioinform., 2021, 1, 653681 CrossRef PubMed.
- S. Soga, H. Shirai, M. Kobori and N. Hirayama, J. Chem. Inf. Model., 2007, 47, 400–406 CrossRef CAS PubMed.
- S. Jia, D. He and C. J. Chang, J. Am. Chem. Soc., 2019, 141, 7294–7301 CrossRef CAS PubMed.
- C. G. Jakob, A. K. Upadhyay, P. L. Donner, E. Nicholl, S. N. Addo, W. Qiu, C. Ling, S. M. Gopalakrishnan, M. Torrent, S. P. Cepa, J. Shanley, A. R. Shoemaker, C. C. Sun, A. Vasudevan, K. R. Woller, J. B. Shotwell, B. Shaw, Z. Bian and J. E. Hutti, J. Med. Chem., 2018, 61, 6647–6657 CrossRef CAS PubMed.
- E. C. Griffith, Z. Su, S. Niwayama, C. A. Ramsay, Y. H. Chang and J. O. Liu, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15183–15188 CrossRef CAS.
- S. Liu, J. Widom, C. W. Kemp, C. M. Crews and J. Clardy, Science, 1998, 282, 1324–1327 CrossRef CAS PubMed.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- A. M. Gold and D. Fahrney, Biochemistry, 1964, 3, 783–791 CrossRef CAS PubMed.
- A. M. Gold, Biochemistry, 1965, 4, 897–901 CrossRef CAS PubMed.
- B. R. Baker, Annu. Rev. Pharmacol., 1970, 10, 35–50 CrossRef CAS PubMed.
- P. K. Pal, W. J. Wechter and R. F. Colman, Biochemistry, 1975, 14, 707–715 CrossRef CAS.
- R. F. Colman, Annu. Rev. Biochem., 1983, 52, 67–91 CrossRef CAS PubMed.
- X. Hanoulle, J. van Damme, A. Staes, L. Martens, M. Goethals, J. Vandekerckhove and K. Gevaert, J. Proteome Res., 2006, 5, 3438–3445 CrossRef CAS PubMed.
- C. Gu, D. A. Shannon, T. Colby, Z. Wang, M. Shabab, S. Kumari, J. G. Villamor, C. J. McLaughlin, E. Weerapana, M. Kaiser, B. F. Cravatt and R. A. van der Hoorn, Chem. Biol., 2013, 20, 541–548 CrossRef CAS PubMed.
- A. Cuesta and J. Taunton, Annu. Rev. Biochem., 2019, 88, 365–381 CrossRef CAS PubMed.
- M. E. Abbasov, M. E. Kavanagh, T. A. Ichu, M. R. Lazear, Y. Tao, V. M. Crowley, C. W. Am Ende, S. M. Hacker, J. Ho, M. M. Dix, R. Suciu, M. M. Hayward, L. L. Kiessling and B. F. Cravatt, Nat. Chem., 2021, 13, 1081–1092 CrossRef CAS PubMed.
- B. Yang, H. Wu, P. D. Schnier, Y. Liu, J. Liu, N. Wang, W. F. DeGrado and L. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11162–11167 CrossRef CAS.
- A. L. Borne, J. W. Brulet, K. Yuan and K. L. Hsu, RSC Chem. Biol., 2021, 2, 322–337 RSC.
- J. W. Brulet, A. L. Borne, K. Yuan, A. H. Libby and K. L. Hsu, J. Am. Chem. Soc., 2020, 142, 8270–8280 CrossRef CAS.
- D. A. Bullough and W. S. Allison, J. Biol. Chem., 1986, 261, 5722–5730 CrossRef CAS PubMed.
- K. W. Harlow and R. L. Switzer, J. Biol. Chem., 1990, 265, 5487–5493 CrossRef CAS.
- T. Ito, H. Ando, T. Suzuki, T. Ogura, K. Hotta, Y. Imamura, Y. Yamaguchi and H. Handa, Science, 2010, 327, 1345–1350 CrossRef CAS PubMed.
- P. P. Chamberlain and L. G. Hamann, Nat. Chem. Biol., 2019, 15, 937–944 CrossRef CAS.
- E. R. Watson, S. J. Novick, M. E. Matyskiela, P. P. Chamberlain, A. Hernandez de la Pena, J.-Y. Zhu, E. Tran, P. R. Griffin, I. E. Wertz and G. C. Lander, bioRxiv, 2022, preprint, DOI:10.1101/2022.07.02.498551.
- J. T. Cruite, G. P. Dann, J. Che, K. A. Donovan, S. Ferrao, E. S. Fischer, N. S. Gray, F. Huerta, N. R. Kong, H. Liu, J. Marto, R. J. Metivier, R. P. Nowak, B. L. Zerfas and L. H. Jones, RSC Chem. Biol., 2022, 3, 1105–1110 RSC.
- H. Wang, K. I. Piatkov, C. S. Brower and A. Varshavsky, Mol. Cell, 2009, 34, 686–695 CrossRef CAS PubMed.
- C. Mitsopoulos, P. Di Micco, E. V. Fernandez, D. Dolciami, E. Holt, I. L. Mica, E. A. Coker, J. E. Tym, J. Campbell, K. H. Che, B. Ozer, C. Kannas, A. A. Antolin, P. Workman and B. Al-Lazikani, Nucleic Acids Res., 2021, 49, D1074–D1082 CrossRef CAS PubMed.
- G. Petzold, E. S. Fischer and N. H. Thomä, Nature, 2016, 532, 127–130 CrossRef CAS PubMed.
- S. L. Schreiber, Cell, 2021, 184, 3–9 CrossRef CAS PubMed.
- G. M. Burslem and C. M. Crews, Cell, 2020, 181, 102–114 CrossRef CAS PubMed.
- J. Lu, Y. Qian, M. Altieri, H. Dong, J. Wang, K. Raina, J. Hines, J. D. Winkler, A. P. Crew, K. Coleman and C. M. Crews, Chem. Biol., 2015, 22, 755–763 CrossRef CAS PubMed.
- G. E. Winter, D. L. Buckley, J. Paulk, J. M. Roberts, A. Souza, S. Dhe-Paganon and J. E. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS.
- C. E. Powell, G. Du, J. W. Bushman, Z. He, T. Zhang, E. S. Fischer and N. S. Gray, RSC Med. Chem., 2021, 12, 1381–1390 RSC.
- M. Girardini, C. Maniaci, S. J. Hughes, A. Testa and A. Ciulli, Bioorg. Med. Chem., 2019, 27, 2466–2479 CrossRef CAS PubMed.
- A. Lopez-Girona, D. Mendy, T. Ito, K. Miller, A. K. Gandhi, J. Kang, S. Karasawa, G. Carmel, P. Jackson, M. Abbasian, A. Mahmoudi, B. Cathers, E. Rychak, S. Gaidarova, R. Chen, P. H. Schafer, H. Handa, T. O. Daniel, J. F. Evans and R. Chopra, Leukemia, 2012, 26, 2326–2335 CrossRef CAS PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- N. P. Grimster, RSC Med. Chem., 2021, 12, 1452–1458 RSC.
- X. Zhang, V. M. Crowley, T. G. Wucherpfennig, M. M. Dix and B. F. Cravatt, Nat. Chem. Biol., 2019, 15, 737–746 CrossRef CAS PubMed.
- J. N. Spradlin, X. Hu, C. C. Ward, S. M. Brittain, M. D. Jones, L. Ou, M. To, A. Proudfoot, E. Ornelas, M. Woldegiorgis, J. A. Olzmann, D. E. Bussiere, J. R. Thomas, J. A. Tallarico, J. M. McKenna, M. Schirle, T. J. Maimone and D. K. Nomura, Nat. Chem. Biol., 2019, 15, 747–755 CrossRef CAS PubMed.
- N. R. Kong, H. Liu, J. Che and L. H. Jones, ACS Med. Chem. Lett., 2021, 12, 1861–1865 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |