
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgress in the development of solid-state electrolytes for reversible room-temperature sodium–sulfur batteries
S. K.
Vineeth
abc,
Mike
Tebyetekerwa
 c,
Hanwen
Liu
c,
Chhail Bihari
Soni
b,
Sungjemmenla
b,
X. S.
Zhao
*c and
Vipin
Kumar
*ab
c,
Hanwen
Liu
c,
Chhail Bihari
Soni
b,
Sungjemmenla
b,
X. S.
Zhao
*c and
Vipin
Kumar
*ab
aUniversity of Queensland – IIT Delhi Academy of Research (UQIDAR), Indian Institute of Technology Delhi, Hauz Khas, New Delhi 110016, India. E-mail: vkumar@dese.iitid.ac.in
bDepartment of Energy Science and Engineering, Indian Institute of Technology Delhi, Hauz Khas, New Delhi 110016, India
cSchool of Chemical Engineering, The University of Queensland, St Lucia, Brisbane 4072, Australia. E-mail: george.zhao@uq.edu.au
First published on 25th July 2022
Abstract
Proliferation in population with booming demand for viable energy storage solutions led to the exploration of storage technology beyond lithium-ion batteries. Sodium–sulfur batteries are potential candidates for post-lithium-ion energy storage courtesy of their high theoretical specific capacity and energy with lower material cost and abundance. However, their general consideration is significantly slowed down by the safety concerns, sodium polysulfide dissolution, and subsequent shuttling in liquid electrolytes, which negatively lower the electrochemical performance and shorten the cycle life. Room-temperature solid-state sodium–sulfur batteries with high electrochemical performances and enhanced safety are excellent analogs based on leakage-free modified electrolytes. However, developments in solid-state electrolytes are in their infancy, with issues such as lower ionic conduction, interfacial instability, and lower capacity retention. This review summarizes developments in room-temperature solid-state sodium–sulfur batteries, focusing on various methods to improve ionic conduction while ensuring interfacial stability and enhancing the overall electrochemical properties. From a combined physico-electro-chemical approach, bifurcation based on the electrolyte material, classified as an inorganic and organic solid polymer electrolyte, has been discussed with its merits and demerits. Finally, several perspectives and insightful conclusions are discussed, citing the crucial challenges that need optimization and rectification.
1. Introduction
The booming energy demand with a rising population has led to an investigation into various new energy sources. The alarming situation due to fossil-fuel-based energy depletion and its negative environmental impacts could be mitigated using alternative energy sources such as solar, hydro, and wind energies.1 However, as these are intermittent, there is an excellent demand for storing the produced energy.2 Thus, energy storage technologies became a core area of research.3,4 In particular considering energy storage for electric vehicles (EVs), hybrid electric vehicles (HEVs), and portable electronics, electrochemical energy storage technology has a profound role.5,61.1 Importance of lithium-ion batteries and their issues
Among the electrochemical energy storage technologies, rechargeable high-density lithium-ion batteries (LIBs) have been the most investigated and deep routed-commercialized technology.7,8 Lithium (Li) with multiple advantages such as high theoretical capacity (∼3800 mA h g−1), a low density of 0.59 g cm−3, and an electrochemical potential of −3.04 V vs. standard hydrogen electrode (SHE) is one of the best anodes in battery technology providing high-energy and high voltage.9,10 Moreover, Li+ has a small ionic radius of 0.76 Å, enabling its better diffusion in solids.11 Continuous development in electrode and electrolyte materials with optimization in packaging and processing led to revolutionizing LIBs in battery technology.12 From conceptualizing the rocking chair model by Michel Armand in the 1970s, the introduction of intercalation electrodes, to developing all-solid-state LIBs, LIB technology has seen innumerable changes.13 Although in LIBs, various transition metal interacted positive electrodes (e.g., LiCoO2 (LCO), LiMn2O4 (LMO), LiFePO4 (LFP), etc.) have been investigated, the principle is a one-electron transfer mechanism.14 Also, the intercalation/de-intercalation process in LIBs is accompanied by a unit change in the oxidation state of transition metals.15 Continuous research progress in intercalation cathodes and graphitic anodes has successfully attained maximum specific capacity.16,17 On the other hand, LIBs possess limitations such as scarcity of precursors availability mainly cobalt, research on alternative materials have regained momentum for enhancing performance and energy density of approximately 15% per year.18,19The cost of recycling components of LIBs remains a challenge for the battery industry. As the current commercial LIBs consist of harmful Li salts containing organic electrolytes, transition metals, their oxides, and other compounds, improper disposal of LIBs results in serious environmental consequences. Hence recycling of these needs to be taken seriously. While the last few decades have witnessed considerable developments in LIB technology to meet the demands of ever-growing energy storage requirements, ‘post lithium era’ storage technologies have been explored.20 Considering alternatives for Li chemistries, other single-valent and multi-valent materials such as sodium (Na), potassium (K), magnesium (Mg), calcium (Ca), aluminum (Al), and zinc (Zn) have been explored.21,22 Metal–sulfur and metal–air/oxygen technologies have also been investigated as promising.23–28 In post lithium battery technology, there are fewer chances for a single battery technology or battery technology monopoly as each system has its advantages and limitations.
1.2 Battery storage for the post LIB era: a focus on Na–S batteries
Among various systems, sodium-based technologies are potential candidates due to their natural abundance and comparable properties with lithium.29–31 Sodium is plentifully available, widely distributed geographically, and the fourth most abundant element on earth.32 The elemental abundance of sodium accounts for nearly 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 670 ppm, while it is 22 for lithium in the upper continental crust.33 Compared to Li, Earth's crust contains 28400 mg kg−1 of sodium, and in water, it is 11000 mg L−1, whereas lithium content in Earth's crust and water is 20 mg kg−1 and 0.18 mg L−1, respectively.34,35 Sodium is the second lightest metal after lithium and has a redox potential of −2.7 V vs. SHE with an ionic radius of 1.02 Å.36,37 Although lithium's specific discharge capacity value is much larger (3800 mA h g−1) than sodium with 1165 mA h g−1, the cost of sodium accounts for nearly 4% of Li, which makes it an attractive candidate.38 Based on the U.S. Geological Survey report, published in 2019, the raw material price of sodium carbonate was approximately 149 U.S. dollars per metric ton, whereas, for lithium carbonate, it was 170000.39 Compared to LIBs, the Na–S system provides higher theoretical energy density, as sulfur possesses a theoretical specific capacity of 1672 mA h g−1 in a two-electron reaction. Besides, when sodium is coupled with sulfur, with an elemental abundance of 953 ppm in the upper continental crust and is ranked as the 17th most abundant element on earth,33 its cost is further reduced with enhancements in electrochemical properties.40,41 Overall, the factors above reduce the cost per kW h for sodium–sulfur (Na–S) batteries compared with LIBs and lithium–sulfur (Li–S) batteries. Fig. 1 shows a comparative analysis of the electrochemical properties of various energy storage devices such as lead-acid (Pb-acid) batteries, nickel–metal hydride (Ni–MH) batteries, electric double-layer capacitors (EDLC), and Li-ion, zinc–air (Zn–air), and Na–S batteries. A comparison is represented as a Ragone plot (specific energy vs. specific power), suggesting a bright future for the Na–S technology (Fig. 1b). Elemental abundance and the raw material price are shown in Fig. 1c and d. Table 1 shows a detailed comparison of the performance of various battery technologies such as high temperature and room temperature Na–S batteries, and Li–S and sodium-ion batteries, based on the literature reported.
670 ppm, while it is 22 for lithium in the upper continental crust.33 Compared to Li, Earth's crust contains 28400 mg kg−1 of sodium, and in water, it is 11000 mg L−1, whereas lithium content in Earth's crust and water is 20 mg kg−1 and 0.18 mg L−1, respectively.34,35 Sodium is the second lightest metal after lithium and has a redox potential of −2.7 V vs. SHE with an ionic radius of 1.02 Å.36,37 Although lithium's specific discharge capacity value is much larger (3800 mA h g−1) than sodium with 1165 mA h g−1, the cost of sodium accounts for nearly 4% of Li, which makes it an attractive candidate.38 Based on the U.S. Geological Survey report, published in 2019, the raw material price of sodium carbonate was approximately 149 U.S. dollars per metric ton, whereas, for lithium carbonate, it was 170000.39 Compared to LIBs, the Na–S system provides higher theoretical energy density, as sulfur possesses a theoretical specific capacity of 1672 mA h g−1 in a two-electron reaction. Besides, when sodium is coupled with sulfur, with an elemental abundance of 953 ppm in the upper continental crust and is ranked as the 17th most abundant element on earth,33 its cost is further reduced with enhancements in electrochemical properties.40,41 Overall, the factors above reduce the cost per kW h for sodium–sulfur (Na–S) batteries compared with LIBs and lithium–sulfur (Li–S) batteries. Fig. 1 shows a comparative analysis of the electrochemical properties of various energy storage devices such as lead-acid (Pb-acid) batteries, nickel–metal hydride (Ni–MH) batteries, electric double-layer capacitors (EDLC), and Li-ion, zinc–air (Zn–air), and Na–S batteries. A comparison is represented as a Ragone plot (specific energy vs. specific power), suggesting a bright future for the Na–S technology (Fig. 1b). Elemental abundance and the raw material price are shown in Fig. 1c and d. Table 1 shows a detailed comparison of the performance of various battery technologies such as high temperature and room temperature Na–S batteries, and Li–S and sodium-ion batteries, based on the literature reported.
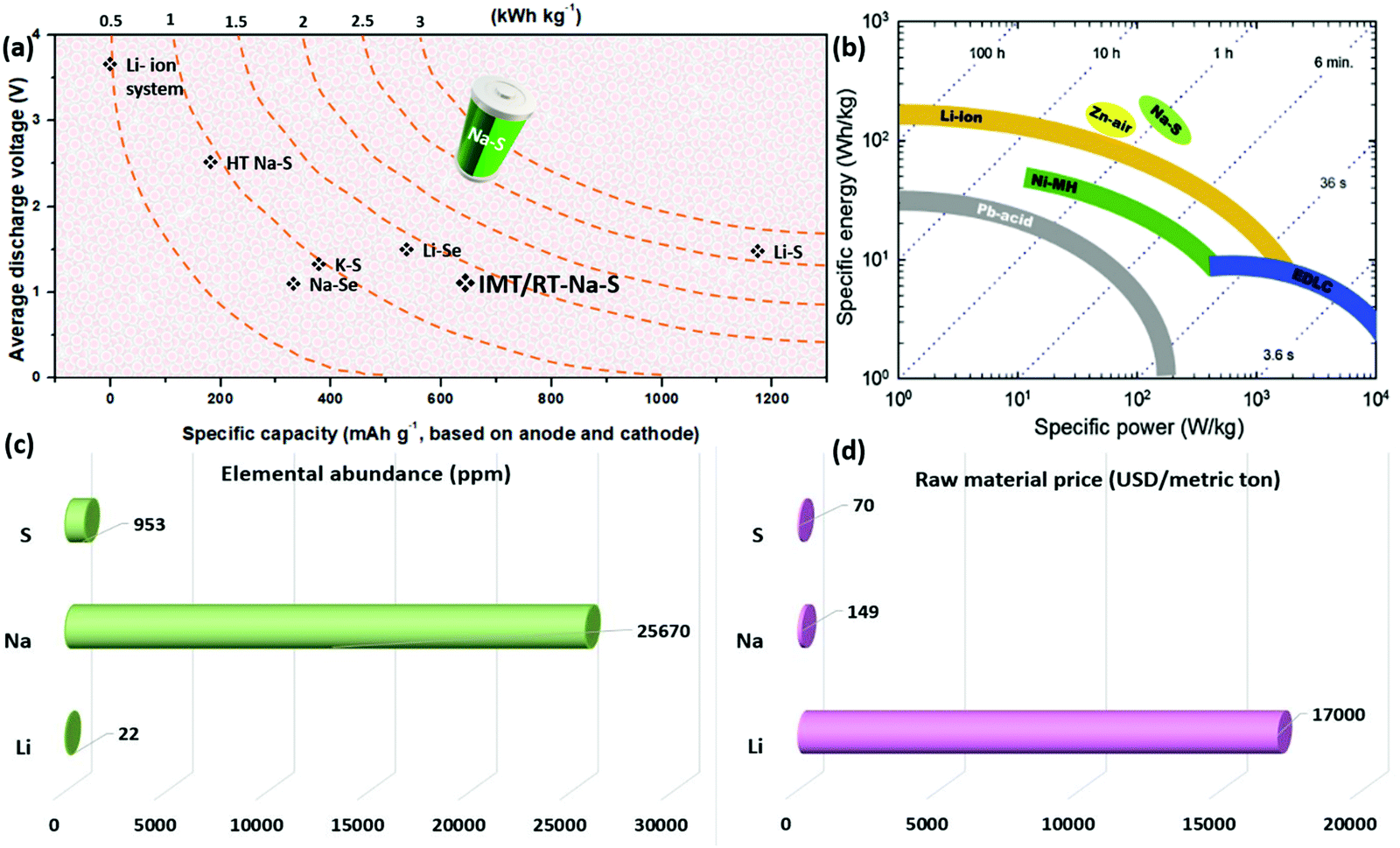 | ||
| Fig. 1 (a) Comparative theoretical electrochemical performance of various alkali-metal rechargeable batteries, (b) Ragone plot showing a comparative analysis of the specific energy and specific power of various energy storage technologies,42 (c) elemental abundance (in ppm) for sulfur, sodium, and lithium in the upper continental crust, and (d) raw material price (USD per metric ton) for elemental sulfur, sodium (carbonate) and lithium (carbonate). | ||
| Battery system | Battery architecture | Ionic conduction, reported temperature (S cm−1, °C) | Electrochemical stability window | Cycles | Coulombic efficiency (%) | ||
|---|---|---|---|---|---|---|---|
| Electrolyte material | Cathode material | Anode material | |||||
| High temperature-Na–S | Beta-alumina, as a solid-state electrolyte50 | Molten sulfur impregnated in a carbon matrix | Molten sodium | 0.1 to 1, 350–300 °C | 2.08 and 1.78 V | 4500 | >98 |
| RT-Na–S with liquid electrolyte | 1 M NaCF3SO3 in diglyme51 | Sulfur-infused microporous carbon | Sodium metal with an interphase containing NaOH and NaNH2 | — | 0.6–2.6 V vs. Na/Na+ | 500 | ∼100 |
| 1 M NaClO4 in EC/DMC/PC (1:1:1)52 |
Macro–microporous carbon–sulfur composite | Sodium metal | 0.6–2.6 V | 500 | ∼100 | ||
| 1 M NaCF3SO3 in diglyme48 | Sulfur infused microporous carbon | Sodium–tin alloy | 0.22 × 10−3, 25 °C | 0.6–2.6 V | 500 | 99.7 | |
| Solid state RT-Na–S | Na3PS453 | Na2S:P2S5 ordered mesoporous carbon (CMK-3) at 30:40:30 |
Na–Sn alloy in AB as a composite | 1.43 × 10−4, 25 °C, 3.45 × 10−4, 60 °C | — | 50 | ∼100 |
| Na3.1Zr1.95Mg0.05Si2PO1254 | Sulfur–carbon–PEO cathode | Sodium metal | 3.5 × 10−3, 25 °C | 4.5 V | 100 | ∼100 (except the first cycle) | |
| PEO–NaCF3SO3 (weight ratio as 9:1)55 |
Elemental sulfur:carbon:PEO (weight percent ratio as 70:20:10) |
Sodium metal | 3.38 × 10−4, 90 °C | — | 10 | — | |
| PEO–NaFSI–TiO256 | Sulfur-carbonized PAN composite | Sodium metal | 4.89 × 10−4, 60 °C | 4.31 V | 100 | ∼100 | |
| PEO–NaCF3SO3–MIL-53(Al)57 | 6.87 × 10−5, 60 °C and 6.52 × 10−4, 100 °C | — | 50 | ∼100 | |||
| Li–S system | 1 M LiTFSI in DME/DOL (1:1)58 |
S-CNT cathode | Lithium metal | — | — | 500 | ∼100 |
| LiTFSI micro fibrillated cellulose-laden polymer electrolyte59 | Sulfur-activated carbon composite | Lithium metal | 1.2 × 10−3, 20 °C | — | 75 | >99 | |
| Modified starch host containing LiTFSI salt60 | Macro-structural sulfur–carbon composite | Lithium metal | 3.39 × 10−4, 25 °C | Up to 4.80 V in 25 °C | 2000 | ∼100 | |
| Sodium-ion | 1 M NaPF6 in EC/PC with 1 wt% FEC61 | Sodium vanadium pyrophosphate | Sodium vanadium pyrophosphate | — | — | 1000 | — |
| 1 M NaCF3SO3 in diglyme and 1 M NaClO4 in PC with 5 vol% FEC62 | No cathode, study was on half-cells with sodium as a counter and reference electrode. Layered bismuth selenide nanosheet–carbon binder composite as an anode | — | 0.05–2.5 V | 500 | ∼100 | ||
| 1 M NaClO4 in EC/DMC (1:1)63 |
Hard carbon as an anode/working electrode and sodium metal as a counter electrode | — | 4.9 V | 100 | ∼100 | ||
In terms of various practical applications, the amount of sulfur loading plays a vital role in achieving a high energy density battery system. For instance, Pope and co-workers analyzed Li–S cathode chemistries and reported “energy density as a function of sulfur loading”.43 Accordingly, in order to achieve a higher specific energy density of ∼400 W h kg−1, a minimum of 2 mg cm−2 is required in a Li–S battery system.44 As a result, the main key component to enhance the specific density is tailoring the high content of sulfur inside the matrix. However, literature studies reported to date on a RT-Na–S battery reveal sulfur loadings in various ranges from <1 mg cm−2 to >5 mg cm−2.45 Notwithstanding the importance of increasing sulfur loading to achieve a high-energy battery system that plays a vital role, the system, however, suffers from certain formidable challenges with an increase in sulfur loadings, viz. the polysulfide dissolution effect.46 As a result, in-depth studies with proper tailoring of sulfur hosts need to be engineered for improving the sulfur loadings without compromising the performance of the battery chemistry. As initial developments in any battery systems depend on the material development, analysis, and electrochemical cell fabrication (usually coin cells), the amount of electrolyte needs to be at the optimum level, generally it ranges from 50–100 μl.47 For instance Kumar and co-workers incorporated 80 μl liquid electrolyte of 1 M sodium trifluoromethanesulfonate in diglyme.48 Considering the thickness of sodium metal as an anode in a coin cell assembly, the value ranges from 0.2 to 0.5 mm, by slicing with a sharp blade/knife inside an inert gas filled glove box.49
1.3 Purpose of review
Considering the availability of raw materials and their respective costs, and equating with the electrochemical performance, it can be unequivocally stated that the RT-Na–S battery technology seems promising.64 As the performance of a battery depends on the material properties of electrodes and electrolytes, the selection of these materials is of prime importance.65,66 In addition to the electrolytes' ion transporting properties, the electrode–electrolyte interface's stability (for anode–electrolyte it is referred to as a SEI, and for cathode–electrolyte it is referred to as a CEI) has a direct relation to the electrochemical properties of a battery.67,68 Stable interfaces provide uniform deposition of sodium, which mitigates dendritic growth over the anode.69 The stability of the SEI is related to the chemical nature of electrolytes; the chemical composition of products formed at the interface decides the electrochemical performance and life of a cell.22,70It has been reported that Na–S batteries with organic solvent-free solid-state electrolytes, having a wide electrochemical stability window, can effectively prevent diffusion and shuttling of sodium polysulfide to the anode while inhibiting the dendrite growth.71,72 In liquid electrolytes (LEs), the discharged products get dissolved and migrate to the negative electrode, decreasing the capacity.73 The process is known as the shuttling effect.74–76 Furthermore, the LE with a narrow electrochemical window is prone to produce an unstable SEI, as the electrolyte oxidizes at the anode, thereby reducing the capacity and Coulombic efficiency of the cell.77–79 As solid electrolytes are free from organic liquids, the bottleneck issue of polysulfide dissolution and the shuttling effect is mitigated; this enhances solid-state batteries’ Coulombic efficiency and maximizes capacity retention.80
However, low ionic conductivity, especially at room temperature, higher interfacial resistance, and poor contact with the electrode are the main issues faced by solid electrolytes.81 Hence this review paper provides an overview of Na–S batteries operated at RT, focusing on the solid electrolytes. A detailed summary of solid-state electrolytes bifurcated as inorganic and organic with electrochemical and battery properties has been systematically highlighted. Various challenges associated with solid electrolytes and approaches to rectify the limitations have been covered. Finally, to accelerate the developments in solid-state RT-Na–S batteries, an outlook section focusing on the advancements and opportunities in the future developments and creation of safer RT-Na–S batteries has been outlined.
2. History and evolution of electrolytes in Na–S
An electrolyte is a central part of a battery that provides an internal connection by promoting ionic mobility while balancing the flow of electrons in the outer circuit. The electrolyte also functions by stabilizing the electrochemistry of the oxidizing cathode and reducing anode.82 The initial investigations of Na–S batteries were focused on high-temperature batteries operating at temperatures above 300 °C. In 1966, Kummer and co-workers developed high-temperature Na–S (HT Na–S) batteries for EV application at the Ford Motor Company, containing ceramic electrolyte: β′′-Al2O3 solid electrolyte (BASE).45,83 In 1965, the Dow chemical company introduced electrolytes based on thin hollow glass capillary tubes.84 They consisted of multiple glass fibers of high surface area and length of 11.5 cm, filled with sodium. However, the tendency of glass fibers to break and the difficulties in the construction of glass fibers made a barrier for broad acceptance. Goodenough and co-workers reported a sodium ion (Na+) conducting inorganic solid electrolyte (ISE), NAtrium Superionic CONductors (NASICON), in 1976, which showed equivalent ionic conduction to BASE at 300 °C.85 In 1979, Gupta and co-workers investigated failure analysis of β′′-alumina electrolyte through post mortem of the failed cells.86 A correlation between the crack patterns and the shape of electrodes was observed. Non-uniform current distribution resulted in high current density, which initiated the crack.Later in the 1980s, and 1990s application-based developments were reported for HT Na–S and intermediate Na–S batteries and their usage in aerospace applications succeeded.87 In 1986, Ansell reviewed the stability of the β′′-alumina electrolyte and factors affecting Na+ conduction referring to Na–S batteries.88 Dendritic growth and development of electronic conductivity account for the primary failure in β′′-alumina electrolytes. From the mid-1980s, a joint expedition by NGK Insulators, Ltd, and the Tokyo Electric Power Company (TEPCO) developed HT-Na–S batteries with BASE and started mass production in 2003.89 The ionic conduction depends on the orientation of crystals in BASE, resistivity, and grain boundary resistance. In 2009, a commercial level 34 MW high temperature Na–S battery system was planted in Japan for stabilizing a 51 MW wind farm.90 Furthermore, Tewari et al. and Rodrigues et al. also reported the usage of Na–S batteries for load leveling of wind energy.91,92 A Na–S system has high energy efficiency and a theoretical energy of ∼760 W h kg−1 with a stable cycle life.93 In conventional HT-Na–S batteries, the operational temperature is maintained above 300 °C.90 The architecture of such HT-Na–S batteries consisted of a sodium anode, BASE, and a sulfur cathode.94
The BASE avoids self-discharge as it is an insulator to electrons but ionically an active conductor.95 The high discharge capacity of sulfur (1675 mA h g−1) with abundant availability accounts for its usage as a sustainable cathode material.96–98 The working involves the transportation of Na+ to sulfur when one electron is withdrawn. Na+ migrates to the cathode, and the electron thus drives an electric current through an electric load. During the cell operation formation of sodium polysulfides occurs; sodium polysulfide is a complex compound that consists of multiple sulfur atoms attached to molten sodium.99 Such cells operated above 300 °C contain all active materials in the molten stage, which lowers the interfacial resistance and increases the Na+ conduction. But as the temperature goes up, the reactivity of metallic sodium also increases, and a minor fracture on BASE can lead to explosions. Also, the corrosive nature of the polysulfide melts with the cathode current collector intensifies the problem.100 The cell temperature increases due to the liberation of high enthalpy (∼−420 kJ mol−1) during the electrochemical reaction.101 Thus, a low Coulombic efficiency, lacking safety with higher operational cost, and the requirement of routine maintenance limit the widespread application of the HT-Na–S battery.
An intermediate temperature Na–S (IMT-Na–S) battery operating at 120–300 °C, possessing similarity in a sulfur redox reaction to HT-Na–S, was introduced at the beginning of the 1980s by Abraham and co-workers.102,103 Followed by the development of IMT-Na–S battery, further developments were reported by US National Aeronautics and Space Administration (NASA).102,103 The IMT-Na–S battery utilizes sodium polysulfides (Na2Sn) dissolved in non-aqueous catholyte, mainly organic solvents with a higher boiling point. The main agenda of introducing the IMT-Na–S battery was to reduce the operating temperature of the HT-Na–S system, thereby reducing the maintenance cost and enhancing safety. Polyethylene oxide (PEO) as a polymer electrolyte for the IMT-Na–S battery, operated at 90 °C, was developed by Park and co-workers in 2006.55 The ionic mobility and transportation of ions between the electrodes are critical factors in enhancing the electrochemical properties of any battery technology. The liquid electrolytes thus have a role in improving the electrochemical properties as they have high ionic conduction. A LE for sodium metal batteries consists of solvated sodium salt in an organic solvent medium. Unlike in HT-Na–S and IMT-Na–S batteries, RT-Na–S ensures higher theoretical energy density (1274 W h kg−1) due to the transformation of sulfur to sodium sulfide (Na2S).104
In 2007 Wang and co-workers reported initial work on LEs for RT-Na–S batteries, with carbonate-based electrolytes containing sodium perchlorate (NaClO4) in ethylene carbonate (EC) and dimethyl carbonate (DMC).105 In 2013, Hwang and co-workers reported RT-Na–S batteries with 0.8 M NaClO4 in EC and diethyl carbonate (DEC).106 Xin and co-workers communicated the applicability of LEs based on 1 M NaClO4 in EC and propylene carbonate (PC) in a 1:1 weight ratio for RT-Na–S batteries.107 Later ether-based solvents such as glyme-based systems including cyclic ether 1,3-dioxolane (DOL), dimethoxyethane (DME), tetraethylene glycol dimethyl ether (TEGDME), and tetraglyme were also reported for RT-Na–S batteries.35,71,75,108 In 2011, Ryu and co-workers reported RT-Na–S cells with sodium trifluoromethanesulfonate (NaCF3SO3) in TEGDME with a high ionic conduction of 1.14 × 10−3 S cm−1.109 Most liquid electrolytes are flammable organic solvents and volatile and are the vital culprits in postmortem fire analysis involving Na–S batteries.110
The advantage of liquid electrolytes is that they demonstrate high ionic conduction (10−2 S cm−1). However, they fail to resist dendrite formation resulting in a lower Coulombic efficiency concerning longer charge–discharge cycles.111 Furthermore, liquid electrolytes result in serious safety issues such as flammability and electrolyte leakage.112 Hence, to rectify the problems of LEs, gel and solid-state electrolytes were introduced as substitutes.113–115 Still, dendrite growth can be effectively suppressed with solid-state electrolytes contributing to better battery cyclability, operation at higher voltages, and compact packing of batteries.116 It can also mitigate other issues associated with us, such as the parasitic reaction between the electrolyte and anode, internal short-circuiting, fire hazards, and thermal runaway.117 In 2016, Wei and co-workers reported room-temperature ionic liquid (IL) tethered silicon dioxide (SiO2) nanoparticles incorporated in 1 M NaClO4 in the EC-PC electrolyte for RT-Na–S batteries, which showed stabilizing effects on the cell.118
Park and co-workers first proposed using polymer-based electrolyte for RT-Na–S batteries in 2007, and the work was the first report on RT-Na–S batteries.119 The team fabricated a solid state RT-Na–S battery with polymer gel electrolyte polyvinylidene fluoride-hexafluoropropene (PVDF-HFP). The work opened new avenues in developing Na–S batteries, which ensured decrement in operational temperature with safer battery operation, paving the way for further exploration of RT-Na–S. Hence after the first report on HT-Na–S batteries in 1966, for the development of IMT-Na–S and RT-Na–S batteries in 2006, progress in the electrolyte has shifted from LEs to gel electrolytes and solid-state electrolytes, considering safety as a primary requirement (Fig. 2).120 Solid-state batteries containing all solid-state components are essential replacements for such a system. Recently, Ma and co-workers reported a solid-state RT-Na–S battery with perfluorinated sulfonic resin powder in the sodium-form (PFSA-Na), which functions as a separator and electrolyte, showing an ionic conductivity of 1.4 × 10−4 S cm−1.121 In another work, Ma and co-workers activated the PFSA-Na membrane with carbonate solvents before fabrication of RT-Na–S cells, which provided conduction paths for Na+.122
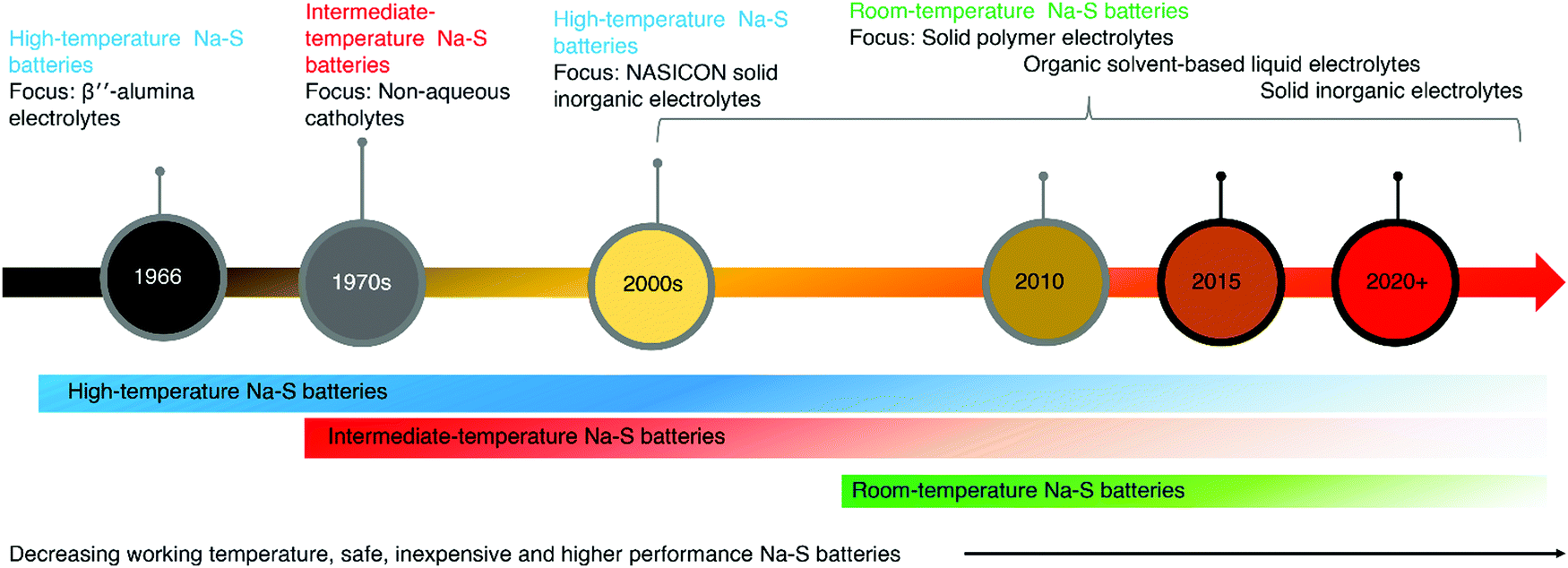 | ||
| Fig. 2 Evolution in high-temperature to room temperature Na–S batteries depicting the corresponding change in electrolytes. | ||
3. Classification of the solid-state electrolytes for RT Na–S batteries
Solid-state electrolytes utilized in RT-Na–S batteries can be broadly categorized based on the materials used, for example, the inorganic and organic solid electrolytes. In contrary to liquid-based systems where a series of reactions occur during discharge reactions, the intermediate reactions (i.e., solid–liquid transition in the range of 2.6–2.2 V, and liquid–liquid transition in the range of 2.2–1.65 V) are effectively eliminated in the presence of a solid electrolyte. As a result of which the solid–solid transition (i.e., S8 + 16Na+ + 16e− → 8Na2S) mainly occurs in the range of 1.65–0.6 V, during discharge reactions. Table 2 summarizes various studies reported on solid-state electrolytes for the RT-Na–S system.| Electrolyte type | Electrolyte formulation | Cathode material | Anode material | Ionic conduction, reported temperature (S cm−1, °C) | Electrochemical stability window (V) | Cycles | Coulombic efficiency (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Sulfide based inorganic solid electrolyte | Na3PS4 (Na2S:P2S5 as 75:25) |
Activated carbon–elemental sulfur composite | Na–Sn alloy | 1.3 × 10−4, 25 °C | — | — | — | 123 |
| Na3PS4 (Na2S:P2S5 as 75:25) |
AB–Na3PS4–Na2S nano composite | Na–Sn alloy in AB as a composite | 1.09 × 10−4, 28 °C | — | — | 124 | ||
| Na3PS4 glass-ceramic electrolyte | Composite of sulfur, AB, and Na3PS4 (25:25:50 weight ratio) |
Na–Sn alloy in AB as a composite | — | — | 25 | ∼100 | 125 | |
| Na3PS4 | Composite of sulfur, Ketjen black, and P2S5 | Na–Sn alloy in AB as a composite | — | — | 25 | — | 126 | |
| Na3PS4 | Na2S:P2S5 ordered mesoporous carbon (CMK-3) at 30:40:30 |
Na–Sn alloy in AB as a composite | 1.43 × 10−4, 25 °C, 3.45 × 10−4, 60 °C | — | 50 | ∼100 | 53 | |
| Na3SbS4 | Active sulfur:Na3SbS4:super P in 1.2:2.0:0.4 mole ratio |
Metallic sodium | 1.14 × 10−3, 25 °C | — | 100 | 98.5 | 127 | |
| Na3PS4 | Selenium (Se) doped S-PAN composite | Na–Sn alloy (in 3:1 mole ratio) |
6.9 × 10−4 | — | 50 | — | 128 | |
| NASICON electrolyte | Na3.1Zr1.95Mg0.05Si2PO12 | Sulfur–carbon–PEO cathode | Sodium metal | 3.5 × 10−3, 25 °C | 4.5 V | 100 | ∼100% (except first cycle) | 54 |
| Polymer coated Na3Zr2Si2PO12 | Carbon nanofiber (CNF)–sulfur composite | Sodium metal | — | — | 100 | — | 72 | |
| Na3.4Zr1.9Al0.1Si2.4P0.6 O12 | CNT–elemental sulfur composite | Sodium metal | 4.43 × 10−3, 50 °C | — | 480 | ∼100% | 129 | |
| Solid polymer electrolyte | PVDF–tetraglyme–NaCF3SO3 (weight ratio as 3:6:1) |
Elemental sulfur:carbon:PEO (weight percent ratio as 70:20:10) |
Sodium metal | 5.1 × 10−4, 25 °C | — | 20 | — | 119 |
| PEO–NaCF3SO3 (weight ratio as 9:1) |
Elemental sulfur:carbon:PEO (weight percent ratio as 70:20:10) |
Sodium metal | 3.38 × 10−4, 90 °C | — | 10 | — | 55 | |
| PEO–NaFSI–TiO2 | Sulfur-carbonized PAN composite | Sodium metal | 4.89 × 10−4, 60 °C | 4.31 V | 100 | ∼100% | 56 | |
| PEO–NaCF3SO3–MIL-53(Al) | 6.87 × 10−5, 60 °C and 6.52 × 10−4, 100 °C | — | 50 | ∼100% | 57 |
4. Inorganic solid electrolytes
Ionic mobility in solid-state electrolytes was introduced by Faraday in 1838, as he discovered the possibility of ionic transport in lead(II) fluoride (PbF2) and silver(II) sulfide (Ag2S).130 As per the understanding of ion diffusion in solids, ionic mobility is described by the ion-hopping mechanism.131,132 As the structural framework of solid materials has interconnected networks, individual ions hop from neighboring lattices.133 Ion-diffusion can be possible following other mechanisms such as direct-hopping (where a cation diffuses between interstitial sites) and the knock-off mechanism (knocking off of adjacent ions in the lattice by an interstitial ion).68 Inorganic solid electrolytes (ISEs) consist of a symmetrical structure with mobile ions (Fig. 3). ISEs can be further classified as sulfide type and oxide-based electrolyte systems.134 Point defects in the skeleton facilitate mobile ions to shift from different sites, thus enabling ionic transportation. ISEs suffer from mechanical brittleness and grain boundaries, limiting their solid-state battery application. Moreover, the materials design, composition, and morphology have a profound role in ionic conduction.135 Overall, the ionic conduction in ISEs depends on the availability of defects/vacancies, the presence of mobile ions, and their energy.136 ISE follows Arrhenius model ionic conduction concerning temperature; this is mainly attributed to the relatively fixed orientations of molecules, which occupy equilibrium positions in a solid.137,138 In the Arrhenius model for ionic conduction in solids, data are generated by varying the temperature and temperature-dependent ionic conduction, which are further equated.139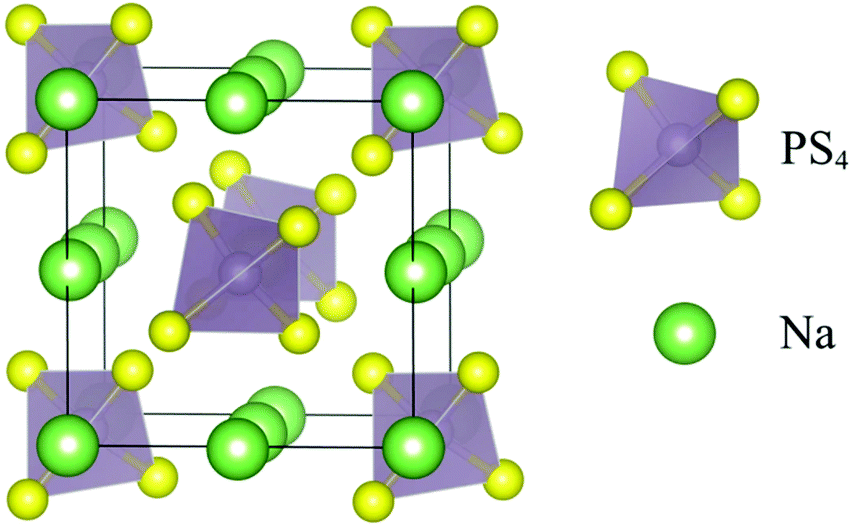 | ||
| Fig. 3 Schematic depicting the crystal structure of pristine t-Na3PS4.146 | ||
4.1 Sulfide-based ISEs
Sulfide-based ISEs constitute a class of solid electrolytes with better intrinsic ionic conduction (∼0.2 mS cm−1), making them ideal electrolyte candidates for high-power batteries.140 The sulfide-based ISE gained acceptance as it can be synthesized at lower temperatures. Moreover, while considering the high ionic conductivity of oxide-based solid electrolytes (in the range of mS cm−1), a major limiting factor is the requirement to reduce grain boundary resistance under extreme conditions, such as a high temperature of 1200 °C, which limits its wide acceptance. However, sulfide electrolytes have the advantage of ductility, with lower grain boundary resistance, allowing ionic conduction under ambient conditions.141 The main reason for the ionic conduction is due to the structural feature, with sulfur having high ionic radius, lower electronegativity, and weak ionic binding, facilitating channels for ion mobility. As solid electrolytes possess a poor electrode–electrolyte contact, sulfide-based ISEs form a better contact by even cold-pressing, contributing to a lower cost for commercial production. Sulfide electrolytes can be subdivided into glass and glass-ceramic electrolytes. Preparation of glassy electrolyte involves cooling below glass transition temperature (Tg) after heating crystalline material above its melting temperature. Preparation methods for the sulfide electrolytes include a mechanochemical method, where the precursors undergo mechanical ball milling and heat treatment at relatively low temperatures.142,143 Famprikis and co-workers established a relation between mechanochemical synthesis and ionic conduction of sulfide-based ISEs through a theory-guided experimental approach.144 With the reduction of crystallite and particle size, ball milling induces micro and macrostrains in the crystal, thereby creating sodium defects enhancing ion transportation. Moreover, the work reported that applying high pressure (∼1 GPa) can also impart enhancement in ionic conduction.The most explored material for sulfide-based ISE is trisodium; sulfanylidene(trisulfido)-lambda5-phosphane (Na3PS4), which has the advantage of scalable synthesis.140 Depending on the synthesis conditions, Na3PS4 crystallizes in either of the two structural polymorphs, tetragonal Na3PS4 (t-Na3PS4) and cubic-Na3PS4 (c-Na3PS4).145,146 In 1992, Jansen and co-workers reported tetragonal modification of Na3PS4 synthesis via solid-state synthesis.147 In 2012, Hayashi and co-workers reported a glass-ceramic electrolyte containing c-Na3PS4, which showed an ionic conduction of 2 × 10−4 S cm−1 at ambient temperature, prepared by reducing glass-ceramic grain boundaries.148 An increase in ionic conduction can also be engineered by partial substitution of P by Si.149 The increase in ionic conduction is mainly due to the structural changes in c-Na3PS4. Ab initio molecular dynamics simulations showed that introducing Si into c-Na3PS4 resulted in sodium disorder, which positively contributed to enhancing the ionic conduction.150,151
Mechanical processes such as ball-milling also affect the electrochemical properties of sulfide-based ISEs. Macroscopic conductivity is significantly enhanced by the reduction in grain boundary resistance.152 Takeuchi and co-workers found that introducing sodium vacancies and expanding them by mechanical milling, sintering, quenching, and annealing can enhance ionic conduction in t-Na3PS4.153 These processes expand the 3D lattice and facilitate 3D diffusion pathways for the sodium ion. Nguyen and co-workers identified that ionic conductivity reaches a maximum at the formation stage during the synthesis process of the Na3PS4 electrolyte by ball-milling. Further milling in the saturation stage has an insignificant effect on ionic conduction.140 The investigators reported a maximum ionic conduction of 1.7 × 10−4 S cm−1 when ball-milled at 550 rpm for 1 h. This high value of ionic conduction of Na3PS4 at ambient temperature attracted researchers over oxide type solid-state electrolytes such as single and poly crystalline β-alumina, NASICON, etc.146
Another strategy to enhance ionic conduction in sulfide electrolytes is by anionic or cationic substitution through doping or defect substitution.150,154 Huang and co-workers conducted ab initio molecular dynamics simulations to investigate the effect of halogen doping in t-Na3PS4 and reported that the highest ionic conduction could be possible in bromine doped t-Na3PS4 (2.37 mS cm−1).155 Halogen doping creates Na+ vacancies with 3D Na+ diffusion paths, which act as pathways for the mobility of cations. As the interactions between the bromine atom and Na+ are relatively weaker compared to other halogens such as fluorine, chlorine, and iodine atoms, weaker bonding corresponds to lowering of the activation energy of Na+; hence this accounts for the enhancement in ionic conduction in bromine doped t-Na3PS4. A similar theoretical investigation by Klerk and co-workers using density functional theory (DFT) molecular dynamics (MD)-simulations reiterated the effect of halogen doping in c-Na3PS4 and t-Na3PS4.156 Introducing 2% vacancies achieved an ionic conductivity of 0.2 S cm−1. Furthermore, the theory predicted that doping with bromine atoms shows the highest bulk conductivity in halogen doped samples.
The ionic conductivity and electrochemical properties of c-Na3PS4 profoundly affect the purity of precursors, their crystal structure, and synthesis methods.157 Sulfide solid electrolytes with c-Na3PS4, synthesized using sodium sulfide (Na2S) and P2S5 as a precursor, showed an ionic conductivity of 2.6 × 10−6 S cm−1 at room temperature as a suitable inorganic electrolyte for all-solid-state sodium batteries.158 Using 99.1% pure crystalline cubic Na2S with heat treatment and incorporated at a mole ratio of 75:25 (Na2S:P2S5), it was found that it showed an ionic conductivity of 4.2 × 10−4 S cm−1.157 Despite the high ionic conductivity of sulfide-based electrolytes (∼10−4 S cm−1), it has been identified that parasitic reactions at the sodium metal anode–electrolyte interface lower the electrochemical properties, affecting its practical use in solid-state sodium metal batteries. Wu and co-workers investigated the nature of reactions affecting cycling performance and longevity of cells with sulfide-based ISEs and a metallic sodium anode through experimental and computational techniques.159 For Na3SbS4 and Na3PS4 electrolytes, the composition of the interface was similar to compounds Na2S and Na3S, as observed from the computational predictions and XPS analysis. In contrast, a chlorine doped Na3PS4 electrolyte interface consisted of NaCl as an additional compound and Na2S and Na3S. The presence of NaCl mitigated the decomposition of Na3PS4, thereby stabilizing the interface. Hence the investigators underline the importance of modifying sulfide-based ISEs by chlorine doping, thereby enhancing the cyclability and producing solid-state sodium metal batteries with long life.
Another work by Hu and co-workers addressed the issue of stabilizing an anode–electrolyte interface in sulfide-based ISEs by an electron-blocking polymer-based interlayer approach.160 The interlayer consisted of cellulose–PEO, which insulates electronic flow and prevents electrolyte decomposition while conducting ions. Investigators demonstrated the improvement of cyclic stability by fabricating a symmetric cell consisting of Na3PS4 electrolyte between two cellulose−PEO interlayers. At 60 °C and 0.1 mA cm−2 as current density, the system showed stable sodium striping-plating for 800 h (800 cycles). Overall, by using the interlayer, effective interfacial stabilization between the metallic sodium anode and Na3SbS4 solid electrolyte arose, thereby enhancing the electrochemical properties in a solid-state sodium metal battery. Sulfide-based ISEs have been used in fabricating RT-Na–S cells as well. In 2014, Nagata and co-workers successfully employed Na3PS4 as a solid electrolyte for all-solid-state Na–S batteries operating at 25 °C.123 Na3PS4 was prepared by the mechanochemical method and the reagent ratio was optimized as 75:25 (Na2S:P2S5), which showed an ionic conductivity of 0.13 mS cm−1 at 25 °C. The cell architecture consisted of a sodium–tin (15:4) alloy anode with a cathode–electrolyte mixture of activated carbon elemental sulfur mixed with the Na3PS4 electrolyte. The electrochemical properties of the cell were measured by sandwiching layered pellets of the anode in contact with the Na3PS4 solid electrolyte and cathode. At 25 °C and a constant current density of 0.13 mA cm−2, the cell delivered a high capacity of 1522 mA h g−1.
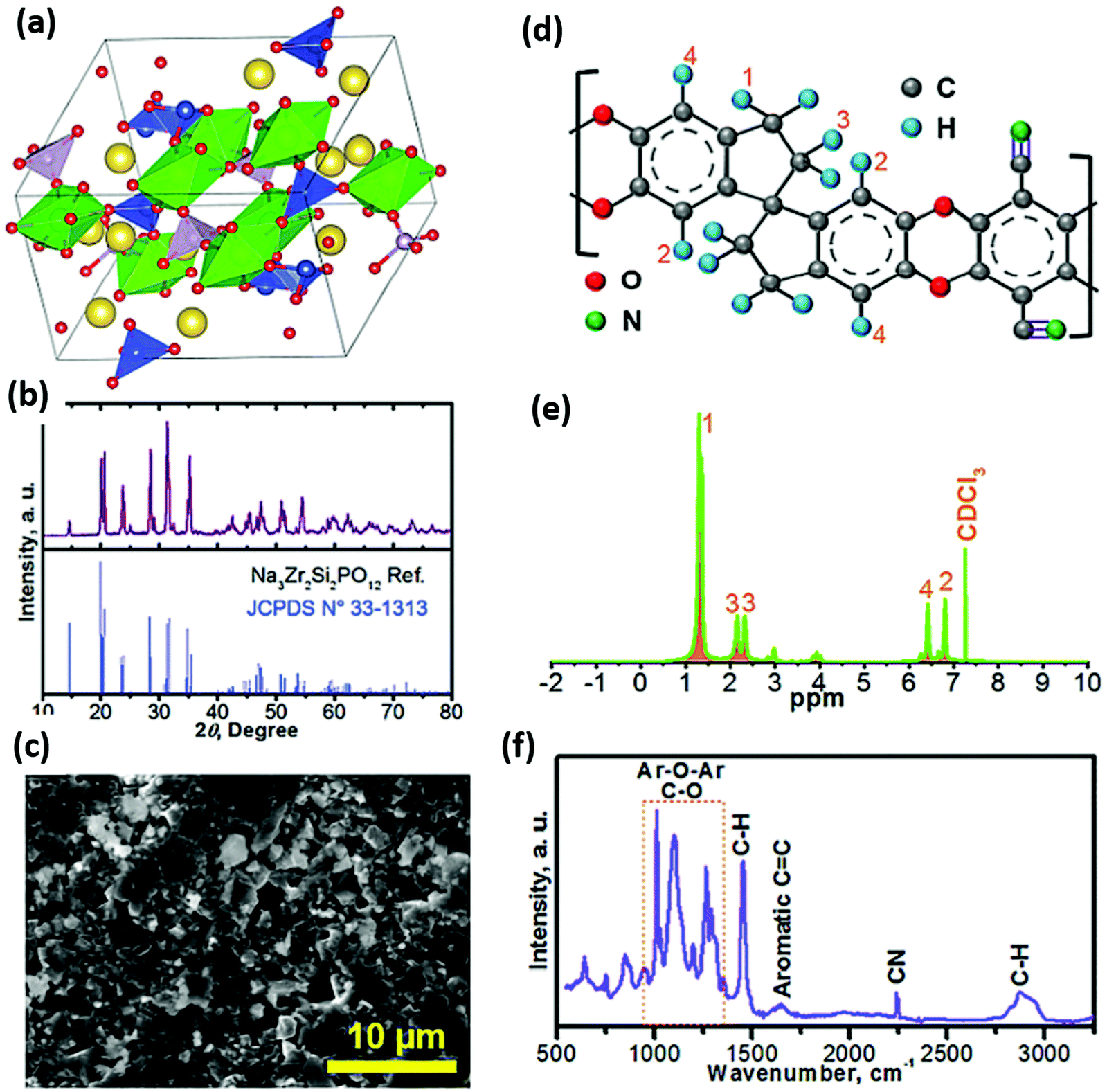
As mechanical milling processes showed a reduction in the interfacial resistance of sulfide-based ISEs,152 Yue and co-workers employed a mechanical milling process followed by annealing in the Na3PS4 electrolyte for solid-state Na–S batteries.124 The cell consisted of a carbon nanocomposite cathode prepared using Na3PS4–Na2S, where Na3PS4 acted as both an active material at catholyte and solid electrolyte. For measuring the ionic-conductivity of the solid electrolyte, electrochemical impedance spectroscopy (EIS) was employed by the ion-blocking method, sandwiching the Na3PS4 electrolyte between platinum electrodes and showing a value of 1.09 × 10−4 S cm−1 at 28 °C. Observations from SEM and elemental mapping of the carbon nanocomposite cathode (shown in Fig. 4) confirmed the formation of a uniform elemental distribution of sodium, phosphorus, and carbon. Furthermore, transmission electron microscopy (TEM) of the nanocomposite cathode (Fig. 4d) showed homogeneity in the distribution of Na3PS4 and Na2S. The carbon content decreased the interfacial resistance of electrolyte–cathode, enhancing the electrochemical properties. In addition to improving energy density and two-fold increment in capacity, nanosized Na2S also worked as an active material. The nano dimension of active material contributed to the reduction of the diffusion path for Na+ and electronic conduction, thereby increasing the active material utilization. Moreover, at a current density of 50 mA g−1, it delivered 869.2 mA h g−1 reversible capacity with good cycling and rate capabilities at 60 °C.
 | ||
| Fig. 4 Morphological analysis and elemental mapping of the Na3PS4 solid electrolyte and nanocomposite cathode. (a and b) Scanning electron microscopy (SEM) of Na3PS4 and Na2S, (c) SEM and elemental mapping analysis, and (d) transmission electron microscopy (TEM) of the Na3PS4–Na2S–carbon nanocomposite cathode.124 | ||
From the literature, it can be observed that HT-Na–S batteries utilized active sulfur material more effectively than in the case of RT-Na–S system with liquid electrolytes. Polysulfide dissolution and its shuttling effect are the leading cause of capacity decay and cycling stability in a liquid electrolyte-based RT-Na–S battery.46,161–163 Tanibata and co-workers devised a strategy to mitigate the capacity decay in a sulfide-based RT-Na–S system.125 The investigators utilized the Na3PS4 glass-ceramic solid electrolyte and fabricated an all-solid-state RT-Na–S battery. As the solid electrolyte mitigated the dissolution of polysulfides, the cell delivered a reversible capacity of 1112 mA h (g of S)−1 at room temperature with a Coulombic efficiency of ∼100% after 25 cycles at different current densities. Compared to a commercially available high-temperature Na–S battery with sintered β′′-solid alumina electrolyte, the cell uses two times more sulfur. OCP measured by galvanostatic intermittent titration technique (GITT) showed curves with plateaus at 2.1 and 1.5 V versus Na+/Na, which were similar to the high-temperature cell with β′′-alumina electrolyte.
Enhancement in the electrochemical properties can be engineered by improving the chemical stability of Na3PS4 based electrolytes. Tanibata and co-workers fabricated an all-solid-state Na–S battery consisting of a composite sulfur cathode with Ketjen black and P2S5 and investigated the effect of adding P2S5.126 Investigators used P2S5 and Na3PS4, which served as electrolyte parts to the composite sulfur cathode, and compared the electrochemical properties. From various analyses, it was observed that a higher mixing degree of crystallites of the P2S5 cathode and Na3PS4 electrolyte contributed to the enhancement in capacity. Moreover, comparative performance on all-solid-state Na–S cell configuration with P2S5 containing electrode showed a high first discharge capacity at a current density of 0.13 mA cm−2, and the value was ∼1240 mA h g−1-sulfur, which is more than twice the value shown by the electrode with Na3PS4 (∼500 mA h g−1-sulfur). However, phosphorus in Na3PS4 reacted with oxygen and moisture, thereby producing hydrogen sulfide gas, and decomposition occurred due to hydrolysis. Wan and co-workers developed phosphorus-free inorganic solid sulfide-based electrolyte sodium thioantimoniate (Na3SbS4) for the RT-Na–S battery.127 All-solid-state Na–S batteries consisted of a nano-scaled electronic/ionic network in the sulfur cathodes for a fast flow of ions and electrons in all the cathode directions. The investigators prepared a composite of active sulfur material in Na3SbS4, Super P acting as an electronically conducting carbon and electrolyte precursor Na3SbS4 as the cathode by a mechanochemical process. Morphological analysis showed the formation of triple-phase contacts among sulfur, carbon, and Na3SbS4, indicating stable interfacial contacts and reduced stress/strain in the cathode material. Moreover, at an ultrahigh cathode loading of 6.34 and 12.74 mg cm−2, the batteries delivered reversible discharge specific capacities of 742.9 and 465.6 mA h g−1 at 100 mA g−1.
To develop the Na3PS4 electrolyte with reduced interfacial resistance and to eliminate the residential stress in all-inorganic solid-state Na–S batteries, the cast-annealing process was employed.53 The Na3PS4 electrolyte consists of Na2S, which lowers the interfacial resistance while enhancing the mechanical properties of the electrolyte. The cell was fabricated with Na2S–Na3PS4 in a mesoporous carbon matrix forming a nanocomposite as the cathode by melting-casting, followed by a stress-release annealing-precipitation process. The cathode composition consisted of Na2S, P2S5, and ordered mesoporous carbon (CMK-3) in the ratio 30:40:30. Material and morphological characterization studies evidenced that Na3PS4 and CMK-3 were reduced through the cast-annealing process, thereby improving the interfacial contacts without producing residential stress. Electrochemical properties were enhanced in the solid-state Na–S battery fabricated with Na2S–Na3PS4 and nanocomposite carbon. The cathode delivered a high reversible capacity of ∼650 mA h g−1 for 50 cycles at a relatively low temperature of 60 °C. Moreover, the nanocomposite cathodes also showed a high Coulombic efficiency (∼100%) without any shuttle reaction, which indicated the reduction of interfacial resistances.
To create a better electrode–solid sulfide-based electrolyte interface, incorporating a few drops of IL at the interface has shown promising results in lithium metal batteries.164,165 ILs have been employed as a wetting agent, which can ensure the enhanced interface between metallic Li anode and electrolyte.166 Following these works, Tao An and co-workers reported the same approach in a solid-state RT-Na–S battery.128 Incorporating IL, N-butyl-N-methylpyrrolidinium bis(fluorosulfonyl)imide (Pyr14FSI), at the sodium–tin alloy anode–Na3PS4 electrolyte interface, the investigators reported enhanced stability in a solid-state RT-Na–S battery. Electrochemical characterizations showed that the addition of drops of Pyr14FSI to the interface of anode and electrolyte supported the formation of an in situ SEI layer. IL penetrated the pores of cold-pressed anode and facilitated Na+ conduction. Moreover, from XPS analysis, the in situ generated SEI over alloy anode consisted majorly of sodium fluoride (NaF). Also, a symmetric cell with NaSn and Na3PS4 electrolytes showed lower impedance than for cells with Na as electrodes. Galvanostatic cycling curves for the symmetric cell with IL at NaSn and Na3PS4 showed better cycling stability and with an overpotential of 0.55 V after 900 h. From the impedance response (concerning time and cycles), IL incorporated cell showed the lowest value (450 Ω after 400 cycles) and prolonged stable cycling performance. The results for various battery stability tests are shown in Fig. 5. Solid-state RT-Na–S battery with a cathode of Se doped sulfurized-poly acrylonitrile (S-PAN) that contributed to enhanced electronic and ionic conduction, had improved cycling performance and high reversible capacities at different current rates. Cells with an electrolyte cathode mixture, Na3PS4, and Se doped S-PAN cathode showed 50 regular cycles at 0.3 A g−1 and displayed high initial capacity, with a value of 708.5 mA h g−1.
 | ||
| Fig. 5 Comparative battery stability tests with and without ionic liquid (IL) incorporated interface. (a and b) Impedance analysis (time-based) of with and without IL. (c) Galvanostatic cycling curves (d) impedance analysis (cycle-based) of with and without IL.128 | ||
4.2 Oxide-based ISEs
Oxide-based ISEs consist of NASICON electrolyte and β-alumina solid electrolytes. These materials possess lower toxicity and better chemical stability with the atmosphere than the sulfide-based ISE. Hence, it enhances solid-state sodium batteries' performance, cyclic stability, and power density.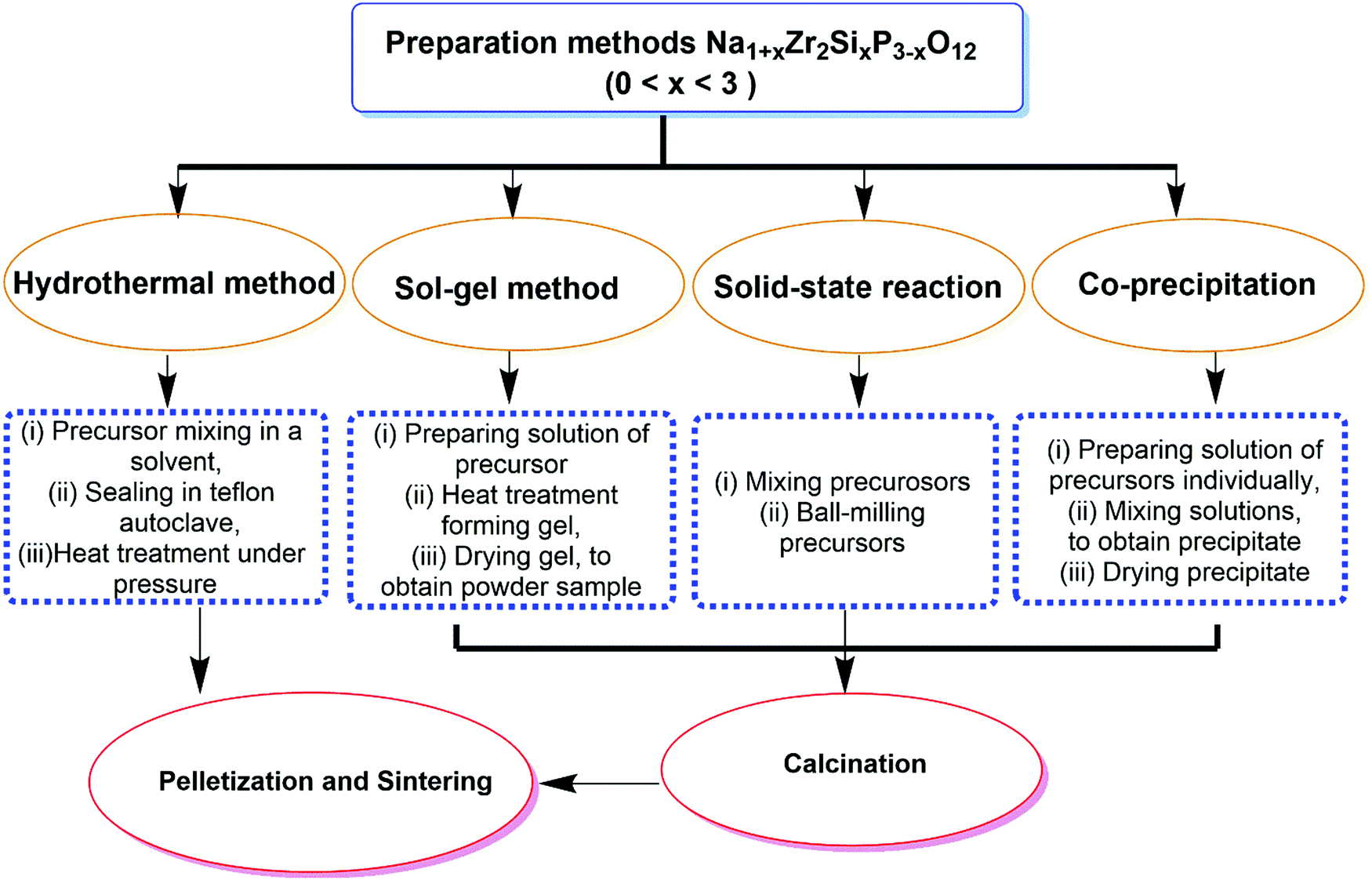 | ||
| Fig. 6 Flow chart depicting various preparation methods for Na1+xZr2SixP3−xO12, where the value of x is given by 0 ≤ x ≤ 3 (NZSPx). | ||
NASICON electrolyte provides a significant advantage that the compositional variations can be engineered.177 Partial or complete replacement of A, M, or X (in AxM2(XO4)3) with other ions alters the ionic conduction.171,178 Saito and co-workers reported that the ionic radius of substitutional ions affects the ionic conduction in NASICON materials.179 NASICON with general structure as Na1.5M0.5Zr1.5P3O12, showed dependency between the ionic radius of trivalent cation (M3+) and its overall ionic conduction. Partial substitution of Zr4+ with M3+ influenced the lattice parameters. Ionic conduction showed proportionate increment with lattice parameters. Increase in the mobile ion content and widening of the bottleneck for ion migration cause enhancement in ion mobility. However, it was identified that the ionic conduction mechanism varies at the grain boundary region and the bulk in ceramic materials. Frequency dispersion analysis showed that the main two factors which influences ionic conduction are the mobile ion concentration and hopping rate. In line with the above observation, Guin and co-workers identified that bulk ionic conductivity depends on the lattice parameters, and change in lattice size by substitution affects the Na+ mobility.180,181
In the development of oxide-based ISEs, there are many bottleneck issues: poor room temperature ionic conduction, instability at the electrode–electrolyte interface, and poor mixed ionic and electronic conduction at the electrodes. Song and co-workers worked on these issues, replaced the liquid electrolyte with a novel NASICON structure, and reported high Na+ conduction of 3.5 × 10−3 S cm−1.54 Preparation of Na3.1Zr1.95Mg0.05Si2PO12 involved a solid-state reaction and mechanochemical process. Prevention of sodium loss was ensured by cold sintering of calcinated product at 1260 °C. A coin cell, operated at a cut-off voltage ranging between 0.8–3.6 V, showed an initial capacity of 527 mA h g−1. Moreover, this value was higher than the HT-Na–S cell with β-alumina electrolyte and RT Na–S cell with LE and sulfur–carbon composite cathode.108,182 However, the cell showed a sharp decline in capacity during the first 10 cycles, indicating polysulfide formation. Investigators also compared a solid-state system with a cell consisting of LE, EC PC 1 M NaClO4. Fig. 7 shows a comparative electrochemical performance of both solid states and LE Na–S cells at 1C and 5C rates. Although solid cells showed slightly lower initial capacity than liquid, the solid cell was observed to have capacity retention of 88% after 100 cycles, showing that the solid-state cell is more stable than the LE cell with 36% capacity retention. At a 5C rate, the solid-state cell showed a slight increment in capacity till 60 cycles and was almost maintained till 100 cycles and showed a value of 60 mA h g−1. In contrast, the capacity faded slightly from a high initial capacity in LE cells.
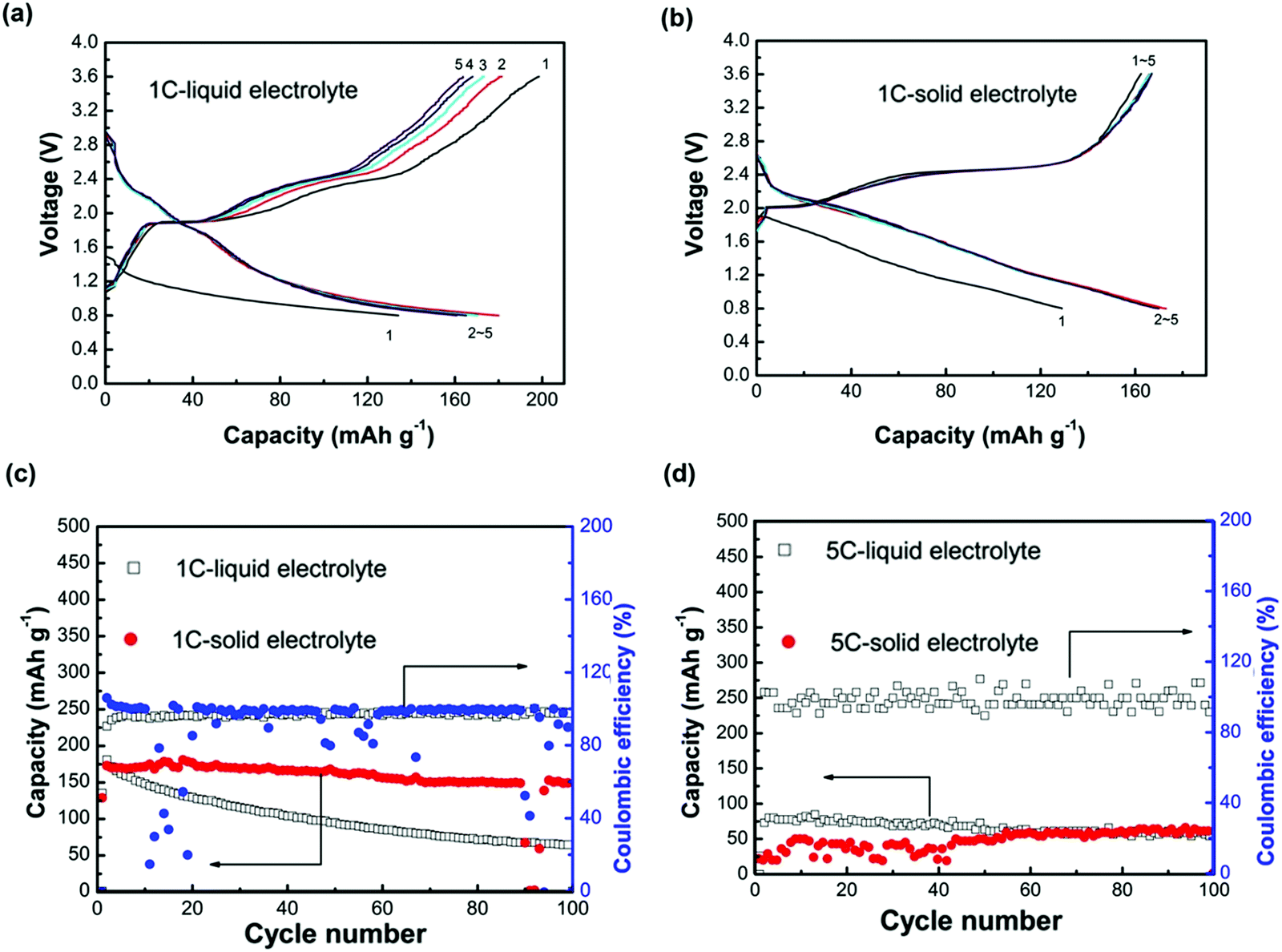 | ||
| Fig. 7 (a and b) Comparative voltage profile is showing charging and discharging of solid-state and LE cells operated at 1C rate and room temperature, (c and d) graphs showing a comparison of cycling performance of the solid-state cell and LE cell operated at 1C rate and 5C rate respectively.54 | ||
A hybrid electrolyte approach was investigated by Manthiram and co-workers, to mitigate the issues associated with the shuttling of sodium polysulfides, improving the electrolyte–electrode interface, and suppressing sodium dendrite formation in RT-Na–S battery.72 Although the NASICON solid-electrolyte membrane (with chemical formula as Na3Zr2Si2PO12), prevents the polysulfide shuttling effect, the investigators addressed the issue of the poor ionic interface between the Na–metal anode and the NASICON membrane. It was found that the issue can be mitigated by coating the NASICON membrane with an intrinsic nano-porous polymer coating. The polymer was synthesized by potassium carbonate (K2CO3) initiated polymerization between 5,5′,6,6′-tetrahydroxy-3,3,3′,3′-tetramethyl-1,1′-spirobisindane and tetrafluoroterephthalonitrile. The chemical structure of the polymer was analyzed through proton-nuclear magnetic resonance (1H-NMR) and Fourier transform infrared spectroscopy (FTIR) (Fig. 8). The Na3Zr2Si2PO12 pellet was then coated with an ultra-thin polymer layer using a tetrahydrofuran dip-coating method. The interface of the polymer–separator was wetted with a few drops of 1 M NaClO4 in the TEGDME electrolyte to provide an ionic conduction path, and the ionic conductivity was found to be 4.1 × 10−5 S cm−1. Although the LE-absorbed membrane's ionic conductivity was lower than a GPE, it showed minor deformation under stress. Moreover, the elastic properties of the polymer-coated membrane enhanced the ionic interface between the ceramic NASICON electrolyte and metallic sodium anode. Thus the cell showed stable discharge capacity throughout 100 cycles. Overall, the hybrid-electrolyte approach facilitated a better electrode–electrolyte interface without the issue of polysulfide migration, thereby enhancing the RT-Na–S battery's cycling performance.
 | ||
| Fig. 8 Various characterization methods of the Na3Zr2Si2PO12 solid electrolyte and the intrinsic nano-porous polymer (a) schematic representation of the structure of NASICON, (b) overlaid XRD of Na3Zr2Si2PO12 membrane and Na3Zr2Si2PO12 reference XRD pattern, (c) SEM image showing the dense structure of Na3Zr2Si2PO12, (d and e) chemical structure of the polymer and corresponding 1H-NMR spectrum, (f) FTIR spectrum with bands showing various stretching/bending vibrations for the polymer.72 | ||
To mitigate the issue of interfacial resistance between solid-state electrolytes and sodium metal anode, Lu and co-workers investigated aluminum doping over a monolithic solid electrolyte, producing a Na3.4Zr1.9Al0.1Si2.4P0.6 O12 (NZSP) structure.129 Polarizing power quantification showed that Al3+ ions possess higher polarity than Zr4+ ions. Furthermore, bond-valence energy landscape (BVEL) maps showed the Na+ ion pathway and diffusion along with the matrix. Aluminum doping facilitated enhancement in ionic conduction by increasing volume fraction for ionic mobility, and the electrolyte showed ionic conduction at 50 °C as 4.43 × 10−3 S cm−1. The schematic representation of depositing Ni(NO3)2 solution and CNT deposition through chemical vapor deposition (CVD), with the final architecture of solid-state RT Na–S cell, is depicted in Fig. 9. To modify the electrode, both the sides of the solid electrolyte were wetted with LE containing 1 M NaPF6, triethylene glycol, 5% fluoroethylene carbonate (FEC), and methoxymethane. EIS analysis for symmetric and Na–S cells showed that the electrolyte resistance is lower than other reported solid electrolytes with values as 248 and 1000 Ω. In addition, the symmetric cell showed 40 mV as polarization after 1600 h of cycling, and the Coulombic efficiency of the full cell was 100%. Furthermore, the cell showed a specific discharge above 300 mA h g−1 even after 480 cycles.
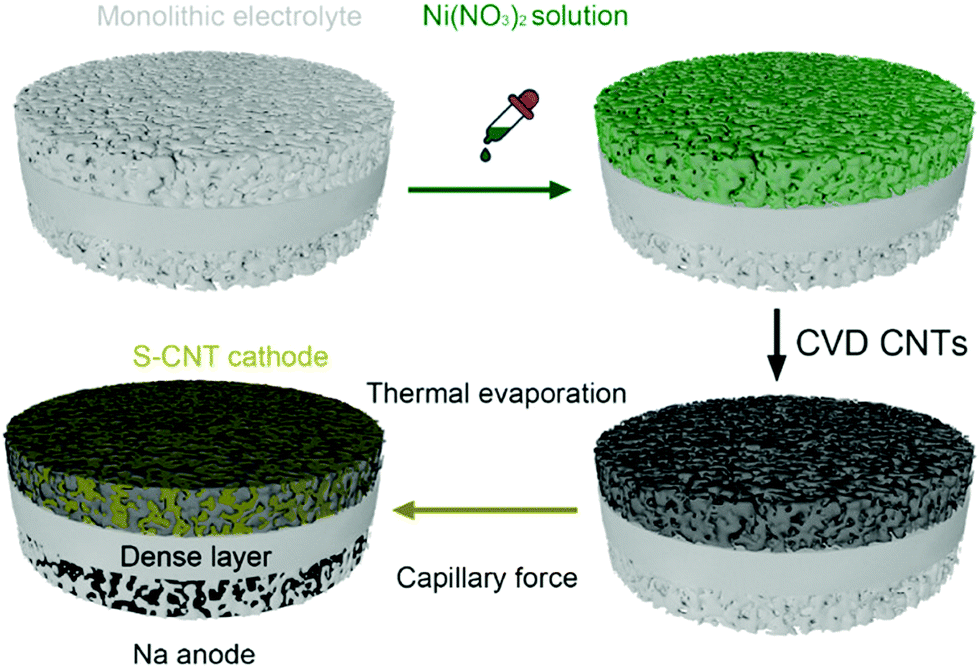 | ||
| Fig. 9 Schematic representation of depositing nickel nitrate (Ni(NO3)2) solution and chemical vapor deposited CNT over monolithic electrolyte with the cell architecture of a solid-state RT Na–S battery.129 | ||
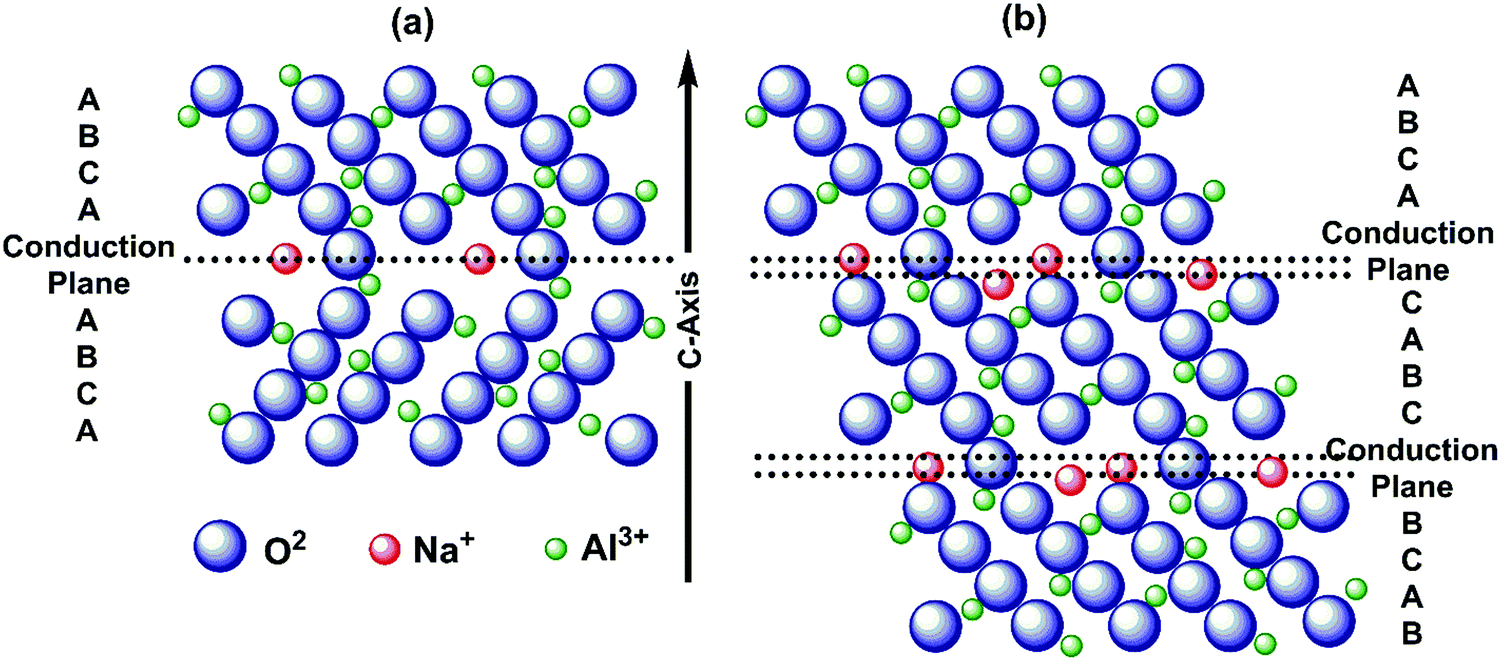 | ||
| Fig. 10 Schematic representation showing the sequence of stacking and sodium ion conduction plane for (a) β-alumina and (b) β′′-alumina. | ||
Ionic conductivity increases with temperatures for β′′-alumina electrolytes, and hence it was used for high-temperature Na–S batteries, operated above 300 °C.186 Single crystal β′′-alumina is reported to have better ionic conductivity than commercial poly crystalline β′′-alumina. Moreover, by doping with metal ions thereby by replacing aluminum, microstructure modifications and by optimizing the ratio of β′′-alumina and β-alumina, ionic conductivity and mechanical properties can be further enhanced.187 An optimum ratio of β′′-alumina and β-alumina enhances not only ionic conductivity but also enhances mechanical strength and moisture sensitivity. Incorporation of stabilizers such as SiO2, magnesium oxide (MgO), manganese dioxide (MnO2), zirconium dioxide (ZrO2), etc. suppresses the formation of β-alumina and promotes a high proportion of β′′-alumina, enhancing ionic conductivity.188,189 Sodium-β/β′′-alumina is often synthesized through sol–gel method, co-precipitation, mechano-chemical and microwave-assisted methods, solid-state reactions, spark plasma sintering, molecular beam epitaxy.190,191 Sintering is common for enhancing ionic conductivity for β′′-alumina solid electrolytes.89 However, loss of active sulfur materials due to vaporization at high temperatures occurs (>1500 °C). Moreover, pellets' low formability and high grain boundary resistance by powder-compression lower ionic conductivity.
Kim and co-workers successfully confined sodium polysulfides in the cathode by the usage of a β′′-alumina solid electrolyte separator in a LE-based RT Na–S battery that delivered long cycling performance.192 The cell was assembled with metallic sodium as the anode, porous activated carbon and sulfur composite cathode, 1 M solution of NaCF3SO3 in the optimum amount of TEGDME as the LE. A porous polypropylene film (Celgard) functioned as a reference separator for a comparative study. The battery with a solid electrolyte separator showed a first discharge capacity of 855 mA h g−1, while the porous separator showed the first capacity of 350 mA h g−1. Surface morphology analysis after 104 cycles showed the absence of cracks or brown-colored polysulfides at the cathode while maintaining a clean surface, indicating the suitability of the β′′-alumina solid electrolyte separator. Furthermore, as the solid electrolyte prevented the migration of dissolved sodium polysulfides to the sodium anode while allowing only Na+ to pass, the cell showed high cyclic stability and a Coulombic efficiency of ∼100%.
5. Solid polymer electrolytes
Polymers are long-chain macromolecular species that are covalently bonded, and have the advantage of solution processability, suitability for bulk manufacturing, and can be engineered depending on the applications.193,194 In addition to the ion-transport properties of polymer electrolytes, these organic molecules possess electrode–electrolyte intimacy, light weight, low-temperature manufacturing compared to inorganic electrolytes.195–199 While ISEs ensure the stable operation of metal batteries, their ionic conductivity and inability to maintain adequate ionic interphases with the metal anode remain significant challenges.124,200,201 In addition, the physical thickness of the electrolyte membrane (which limits ionic transport) plays a crucial role in determining the battery performance.202,203 Polymer electrolytes can be classified as GPEs, SPEs, and composite polymer electrolytes (CPEs), represented in Fig. 11 based on their state of existence, presence of LEs, additives added, etc.204–206 Although ionic conduction is higher for GPEs than in SPEs, the presence of organic solvents in GPE raises severe safety concerns.207 | ||
| Fig. 11 Classification of polymer electrolyte into solid polymer electrolyte, gel polymer electrolyte, and composite polymer electrolyte. | ||
Amongst various solid polymer electrolytes (SPEs), PEO has been one of the most reported SPEs.208–210 Oligoether units with ether-oxygen linkages facilitate effortless segmental motion and inter atomic separation, contributing to better ionic conduction.117,211–213 Besides PEO, which often existed in the ring configuration and has better diffusive properties, other linear-chain polymers such as PVDF and its copolymers, polyvinyl alcohol (PVA) and polyvinylpyrrolidone (PVP) have also been investigated for Na+ conduction.183,214,215 Parker and Wright unveiled the ability of PEO to highly solvate concentrations of alkali metal ions (e.g., Na and K), making PEO a popular choice as a polymer host in SPEs.216 West et al. and Hasmi et al. studied the interface stabilities of PEO–NaClO4 and PEO–NaPF6 in all-solid-state cells at 80 °C, with particular emphasis given to the compatibility between the polymers and metallic Na, and their corresponding interface stabilities.217,218 Corrosive layers were formed over the metal surface and found to be crucial in defining their ionic properties. More importantly, the ratio of Na+ to EO (ethylene oxide of PEO) (typically 0.050–0.080) was significantly affected by the choice of anion: a small anion like PF6−, binding firmly to polymer chains and exhibiting low ionic conductivity. In addition, salt crystallization often affects ionic transport and the uniformity of Na deposition.219 For instance, the larger size of the trifluoromethanesulfonimide anion (TFSI−) and its high degree of delocalization results in PEO–sodium trifluoromethanesulfonimide (NaTFSI) achieving better Na deposition than in the PEO–NaTFSI system.
For the first time, Park and co-workers reported the usage of PEO as an electrolyte all-solid Na–S battery operated at 90 °C.55 The electrolyte film was prepared by solution casting of a mixture of PEO and NaCF3SO3 in acetonitrile at a 9:1 weight ratio. The sulfur cathode was also prepared in acetonitrile by mixing 70 wt% elemental sulfur powder, 20 wt% carbon and 10 wt% PEO, ball milling and then heating at 60 °C. XRD analysis showed an orthorhombic sulfur phase and a crystalline PEO phase only, which confirmed the absence of changes in crystal structure during the fabrication of the sulfur electrode film. DSC analysis showed the melting point of PEO at 68 °C, and for the sulfur electrode, three endothermic peaks corresponding to the existence of PEO and sulfur. The SEM image of the sulfur electrode showed uniformity in mixing the constituents by bright and dark areas. From impedance spectra analysis, the ionic conductivity of SPE at 90 °C was 3.38 × 10−4 S cm−1. Moreover, at 90 °C the cell showed a first discharge capacity of 505 mA h g−1 sulfur and two potential plateaus, at 2.28 V and 1.73 V corresponding to the reduction steps of sulfur by sodium. Still, the discharge capacity decreased during repeated charge–discharge cycling, and after 10 cycles, the value remained at 166 mA h g−1 sulfur.
SPEs have inherent issues like electrode–electrolyte interfacial resistance, lower ionic conduction (compared to LEs), and more significant polarization and capacity decay issues. To rectify these issues Zhou and co-workers employed a quasi-solid state electrolyte/GPE with high ionic conduction of 3.85 × 10−3 S cm−1 (at 25 °C) for RT-Na–S battery application.220 The polymer host for the GPE consisted of pentaerythritol tetraacrylate (PETEA)-tris[2-(acryloyloxy)ethyl]isocyanurate (THEICTA)-based copolymer to which 1 M NaTFSI in PC/FEC solvent was added. The radical polymerization process was initiated by the irradiation of ultraviolet rays to the monomers, and the degree of conversion was observed in the FTIR spectra (Fig. 12). Theoretical study using first principles calculations showed binding energy between sodium polysulfide (Na2S6) with the functional groups of PETEA and THEICTA monomer (Fig. 12). The results evidenced the immobilization of polysulfides rather than dissolution in the carbonate solvents in the matrix. Elemental analysis of sodium metal after cycling also showed a similar result, indicating the mitigation of polysulfide shuttling. Galvanostatic cycling measurement on a symmetric Na/Na cell with GPE showed stability up to 300 cycles, indicating the uniform sodium deposition without short circuit. The RT Na–S cell with quasi-solid state electrolyte sustained a reversible capacity (736 mA h g−1 after 100 cycles) which was twice the value for the cell with LE. Moreover, FESEM analysis for the sodium anode coupled with quasi-solid state electrolyte, after 100 cycles, showed smooth morphology and fewer dendrites compared to the LE counterpart.
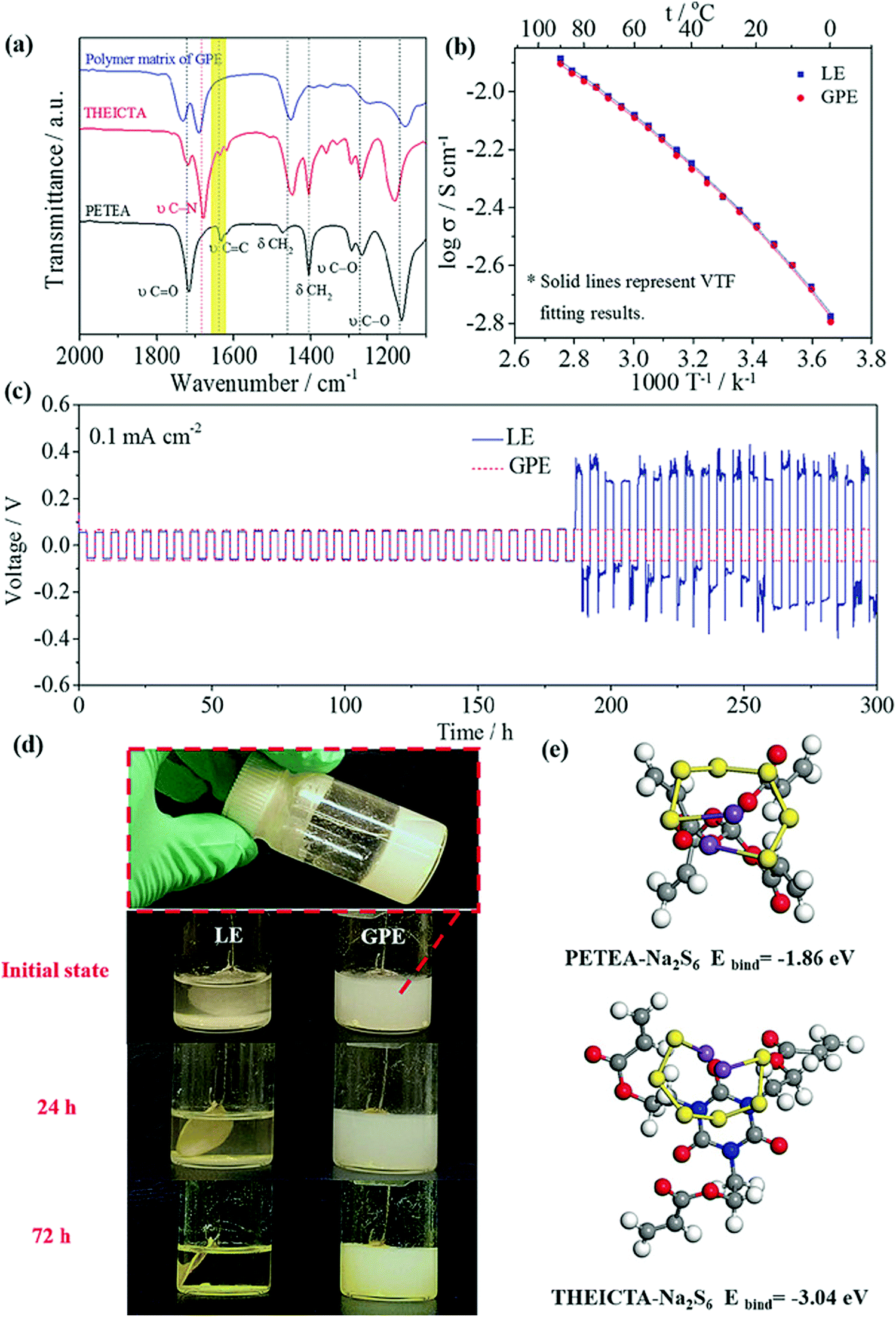 | ||
| Fig. 12 Characterization and electrochemical performance of quasi-solid state electrolyte/GPE. (a) Overlaid FTIR spectra of polymer matrix and monomers showing the conversion of polymer from monomer, (b) comparison of temperature-dependent ionic conduction of GPE and its liquid counterpart, (c) Galvanostatic cycling measurement (at a current density of 0.1 mA cm−2) on a symmetric Na/Na cell with GPE and LE, (d) photographs showing the formation of sodium polysulfides and their dissolution in LE and its comparison with GPE at various times, and (e) binding energy calculation and the molecule bonded with Na2S6, from first principles calculations.220 | ||
5.1 Ion-transport in polymer electrolyte
Ionic conduction in SPE, at various temperatures can be understood using various models such as the Vogel–Tammann–Fulcher (VTF) model, the Arrhenius model, etc.221 The sodium salt incorporated polymer hosts are highly compatible, thus contributing positively towards ionic conduction and mechanical stability. As the microstructure of a polymer consists of crystalline and amorphous regions (Fig. 13a), ionic conduction is facilitated when the Na+ in the sodium salt interacts with the polar groups of the host polymer.222 The interactions between Na+ and polar groups act against the polymer interchain bonds, thereby modifying the crystalline or amorphous characteristics.223 As most polymers are semicrystalline, considering their microstructure, enhancing their amorphous nature affects ionic conduction.223,224 Various approaches have been introduced to reduce the crystalline nature of polymer electrolytes, namely, incorporating plasticizers, preparing polymer blends, filler incorporation, etc.225–227 As crystalline regions of host polymer get destroyed, ionic conduction paths are created, efficiently contributing to ionic transportation.228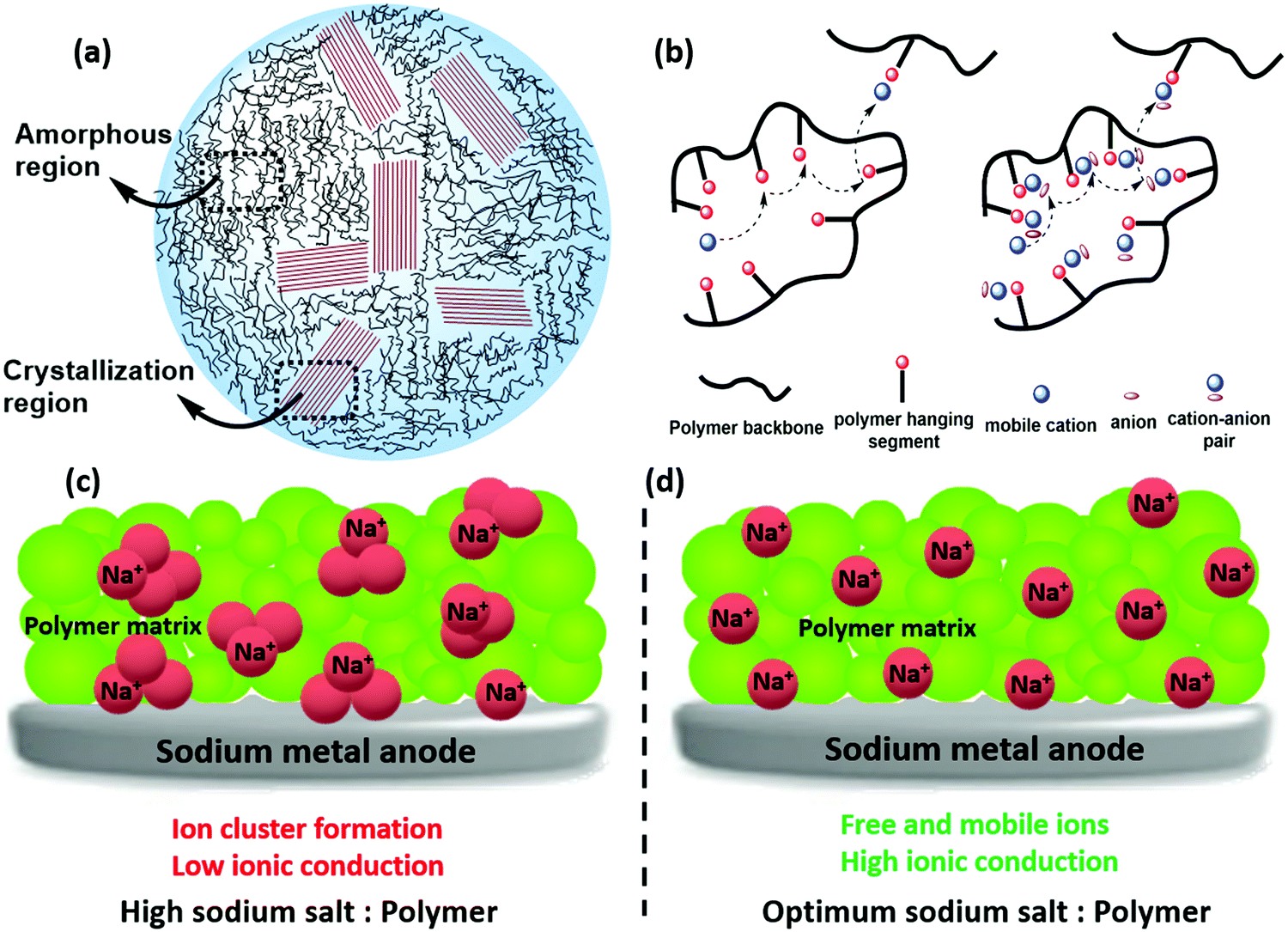 | ||
| Fig. 13 Schematic representation showing the mechanism of ionic conduction in solid polymer electrolyte (SPE). (a) Microstructure of a polymer (b) ionic transport at lower salt concentration, (c and d) schematic representation of the effect of sodium salt concentration in the solid polymer matrix. A high sodium salt concentration causes ion clusters that lower ionic mobility, while the optimum level of sodium salt helps in better ion dispersion producing high ionic conduction. | ||
Moreover, ion transport is determined at lower salt concentrations by the polymer chain segmental motion.229 At high salt concentration, ions, as side chains of polymer, are shielded by salt; hence salt ions self-diffuse to the host polymer matrix. This is mainly due to the lower coupling between ion motion and segmental motion of the polymer.230Tg related to the segmental motion also influences the ionic transport of an SPE, and incorporation of salt modulates Tg.231,232 A simple representation of the same is shown in Fig. 13.
In contrast, ion transport in SPEs is dependent mainly on optimizing the salt-to-polymer ratio, which does not vary linearly with electrolyte salt concentration. Specifically, conductivity increases with the salt concentration as the more significant number of ions decreases the average hopping distance. Beyond a certain threshold, however, further increase of the salt results in the formation of ion clusters, which inadvertently reduces the concentration of available charge carriers.183,233 The schematic representation of this phenomenon is demonstrated in Fig. 13. Arya and co-workers reported an investigation where the ratio of polymer and sodium salt were varied experimentally and considered its effect on ionic conduction.234 Polymer–salt complexes and cationic coordination with the functional group of polymer molecules were evidenced from FTIR analysis, which indicated interactions such as ion–ion and polymer–ion. A two peak percolation model/mechanism based on the correlation between the results of FTIR, impedance study, and transport parameters suggested feasibility of interaction sites in the polymer chains. Ionic transport was achieved by the temporary formation of co-ordination sites as ions move through the polymer matrix. At higher salt–polymer ratio, where the segmental motions are restricted, ion-triplet formation was dominant over ion dissociation, thereby reducing the free charge carriers and hence ionic conduction decreases. Ion cluster/aggregation of ions impedes the ionic conduction, and unveiling the mechanism through a combined approach of theory guided experimental work can provide better insight.235 Webb and co-workers employed a computational and experimental approach to elucidate ionic transport in polymer electrolyte.236 Theoretical predictions showed that the ionic diffusion was based on the density and proximity of solvation shells. Ionic transportation is accelerated in an interconnected solvation shell, whereas a deceleration of ionic conduction was evidenced due to the presence of isolated cluster of sites. Hence the feasibility of solvation sites, its interconnectivity and number of chains involved in co-ordination are the factors affecting ionic conduction.
5.2 Functional additives and fillers
As a polymer matrix inherently has poor mechanical, thermal, chemical, electrical, or electrochemical properties, functional additives are incorporated in the matrix for enhancing the aforementioned properties.231 These are materials that impart a characteristic property to the polymer matrix.193,237 Moreover, since SPEs have inherently lower ionic conduction values than ISEs, fillers that contribute to ionic conduction are usually added.238 Filler incorporation at optimum level decreases crystallinity of the polymer, thereby enhancing the polymer chain mobility.239–241 An increase in mobility contributes to better ionic conductivity and enhancement in thermal and mechanical properties of SPE matrix.208 Chandrasekaran and co-workers added low molecular weight polyethylene glycol (PEG) (4000 g mol−1) to PEO–sodium chlorate (NaClO3) as a means to repair the interfacial layer over the Na surface and reported an improvement in ionic conductivity.242 Another alternative approach is the design of cross-linked network SPEs. Zheng and co-workers designed a hybrid SPE by cross-linking amine-terminated PEG with octakis(3-glycidyloxypropyldimethylsiloxy) octasilsesquioxane (octa-POSS) containing NaClO4.243 The migration of the SEI into the SPE was reported for the first time, attributed to Na metal's mechanical softness and reactivity. Symmetric Na cells demonstrated stable function at 80 °C for over 3550 hours at a current density of 0.5 mA cm−2. The biggest challenge in dealing with a Na metal anode compared to Li is its low melting point close to the operating temperature of many SPEs. This poses a potential risk of cell failure or explosion in extreme cases.As fillers reduce the crystallinity of polymer electrolytes, thereby facilitating ion conduction paths and enhancing a cell's electrochemical performance, researchers found that by blending different polymers, the same can be achieved.244 A hybrid approach of blending and nanofiller incorporation also profoundly affects the electrochemical properties, including ionic mobility.245 Saroja and co-workers fabricated RT-Na–S cells using the hybrid approach, where PVDF-HFP and poly(butyl methacrylate) (PBMA) were blended, with the addition of white graphene (boron nitride), which functioned as a Na+ selective membrane.246 PVDF-HFP/PBMA blended with white graphene showed the highest ionic conductivity as 1.134 × 10−3 S cm−1, and the pristine blend showed an ionic conductivity of 1.04 × 10−4 S cm−1 (both at room temperature). In addition to the selective Na+ transport, the membrane also acted as a shielding layer between cathode and separator, which prevented polysulfide shuttling. The presence of boron and nitrogen atoms in the white graphene restricted the polysulfides from shuttling. Electrochemical properties of the Na–S cell fabricated with PVDF-HFP/PBMA-white graphene membrane showed capacity retention of about 83.1% after 500 charge–discharge cycles. They showed a reduction in self-discharge value, which indicated its ability to resist polysulfide shuttling. Furthermore, the cell delivered an enhanced specific capacity of about 49% compared to the cell without the shielding layer.
Zhu and co-workers found that incorporation of 1% titanium dioxide (TiO2) nanoparticles to PEO matrix containing sodium bis(fluorosulfonyl)-imide (NaFSI) salt showed 4.89 × 10−4 S cm−1 as ionic conductivity at 60 °C for an all-solid-state Na–S battery (shown in Fig. 14).56 SPE was prepared by a solution casting method where, TiO2 nanoparticles were added to the SPE matrix at 1%, 2%, 5%, and 10%, followed by hot-pressing to a thickness of 100 μm. Increasing the content of TiO2 nanoparticles showed agglomeration with porosity in the matrix. The incorporation of TiO2 nanoparticles contributed to lowering the crystallinity of PEO; the same was reflected in the differential scanning calorimetry (DSC) results, showing a decrease in crystalline melting temperature of the SPE (Fig. 14). Thermogravimetric analysis (TGA) showed that the SPE is thermally stable up to 300 °C. Compared to pristine PEO-NaFSI electrolyte with 1.6 × 10−4 S cm−1 as ionic conductivity, SPE with 1% TiO2 nanoparticles showed a high value as 4.89 × 10−4 S cm−1 measured at 60 °C. Although XRD and DSC analysis showed that increasing the amount of TiO2 nanoparticles decreases the crystalline nature of the SPE, which can positively enhance ionic conduction, the agglomeration impedes ionic migration (Fig. 14). Electrochemical analysis was performed between 2.5 and 6 V. At a scan rate of 1 mV s−1, results showed that SPE was stable up to 4.31 V (vs. Na+/Na)), indicating its applicability in the RT Na–S battery. Moreover, the system showed a high Na+ transport number (tNa+) of 0.394, with a low SPE-anode interfacial resistance before and after polarization as 24.98 and 27.31 Ω; these results showed the applicability for an all-solid-state Na–S battery. Furthermore, an all-solid-state Na–S cell at 60 °C delivered ∼100% Coulombic efficiency and showed a stable discharge capacity.
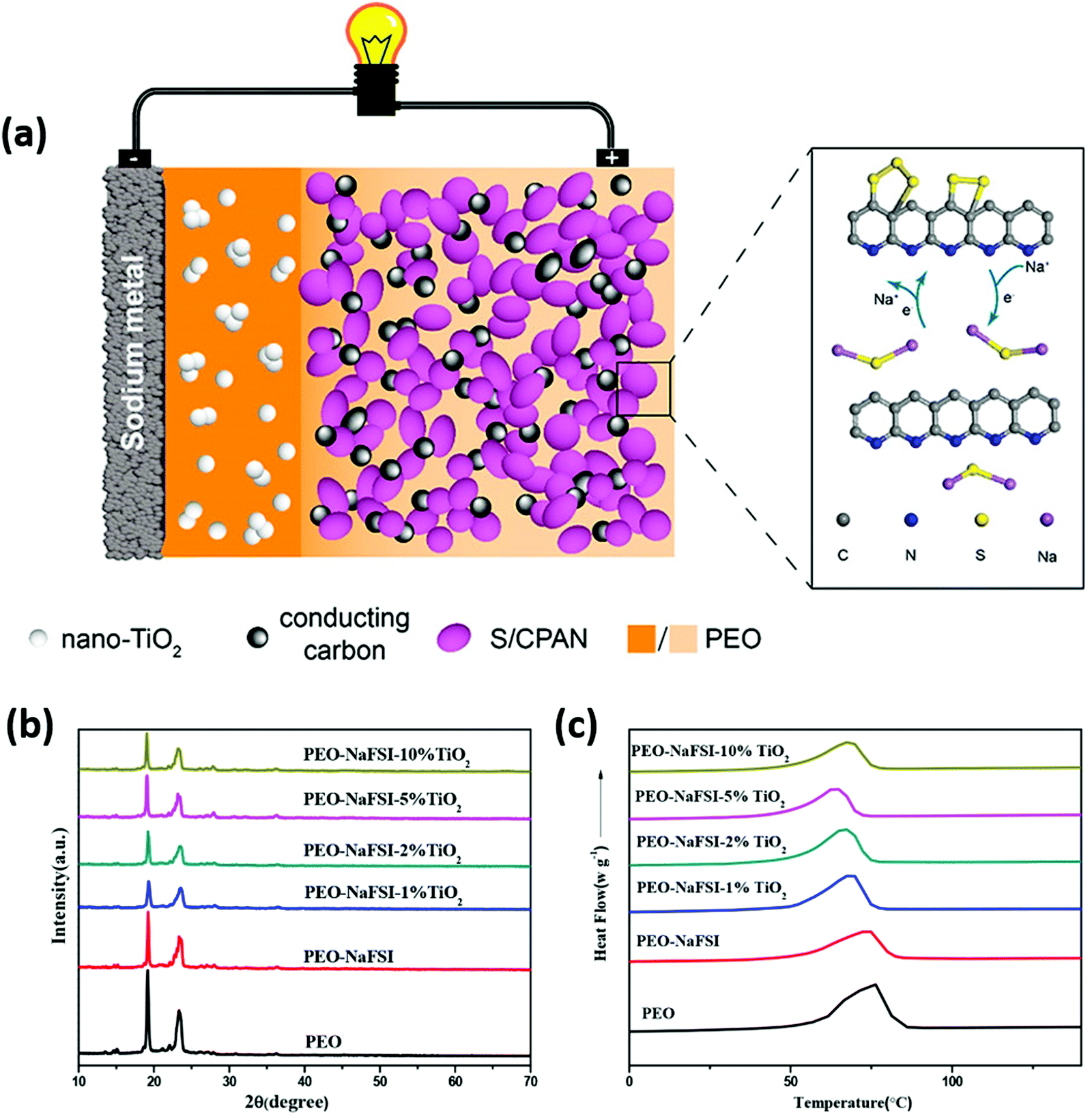 | ||
| Fig. 14 (a) Schematic representation of an all-solid-state Na–S battery with various materials used for fabricating cathode (sulfurized-poly acrylonitrile (S-PAN)), electrolyte and anode, (b) overlaid XRD patterns of PEO, PEO-NaFSI, and PEO-NaFSI with TiO2 at various concentrations (c) overlaid DSC results of various PEO electrolyte membranes.56 | ||
Ge and co-workers investigated the applicability of an aluminum-based MOF (MIL-53(Al)) as a filler for a solid PEO-based electrolyte and fabricated the all-solid-state RT-Na–S.57 The addition of the MOF improved the segmental motion of PEO and increased the dissociation of sodium salts. The work included a comparative study between PEO-NaCF3SO3 and MIL-53(Al) containing a PEO-NaCF3SO3 electrolyte. A uniform dispersion without agglomeration and lumps was observed from SEM analysis of the MOF-incorporated electrolyte. With an increase of NaCF3SO3, the number of free Na+ ions increased due to the enhanced dissociative ability of PEO to NaCF3SO3 and finally got saturated, which led to insufficient dissociation, hence decreasing the ionic conductivity. High ionic conductivity of 6.87 × 10−5 S cm−1 at 60 °C and 6.52 × 10−4 S cm−1 at 100 °C was reached when the EO:Na mole ratio was at 20 with MIL-53(Al) content as 3.24 wt%. By incorporating MIL-53(Al), crystalline parts of PEO were suppressed, ensuring segmental motion and increasing ionic conductivity (Fig. 15).
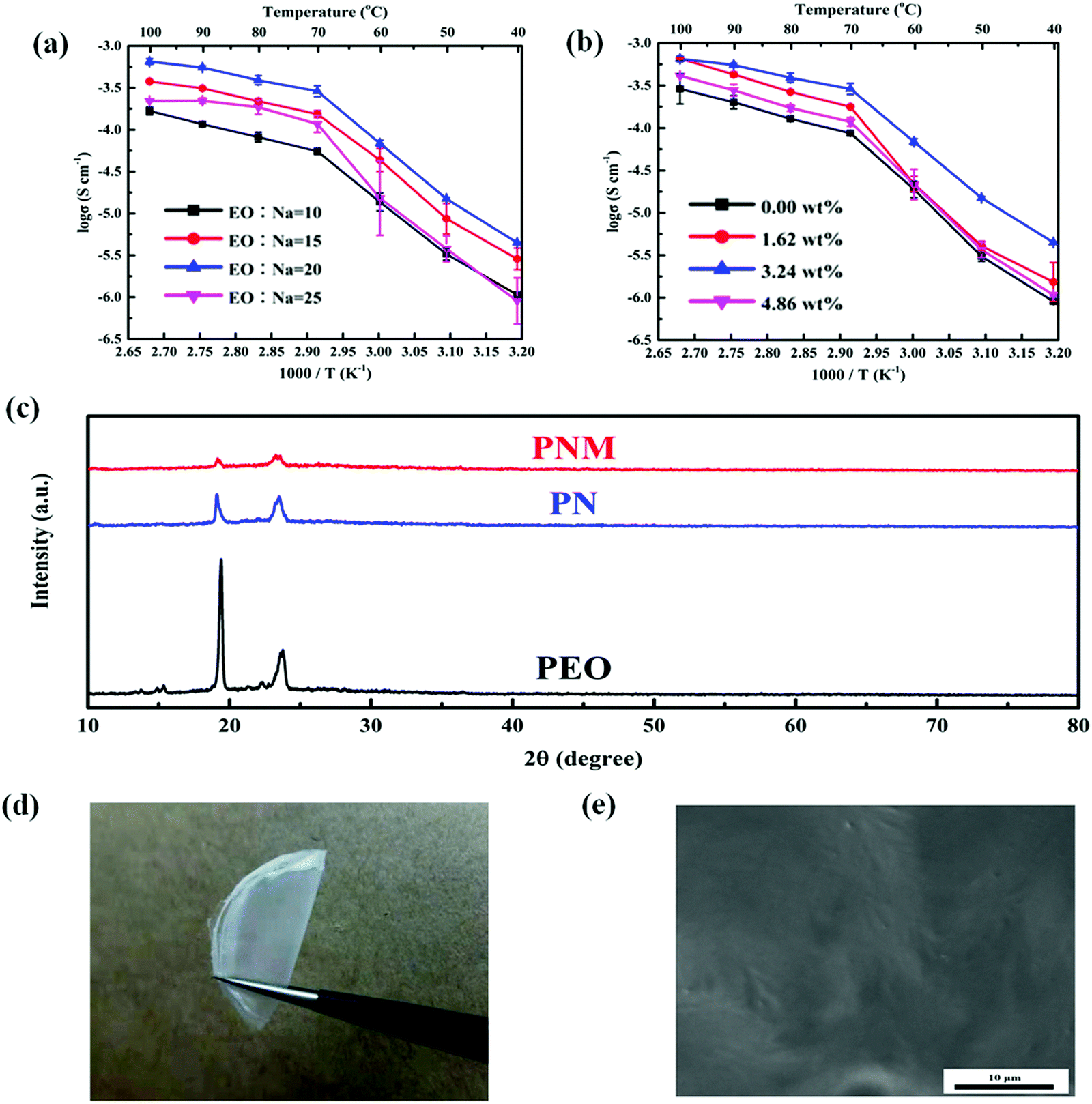 | ||
| Fig. 15 Comparison of various properties of SPE with and without filler (a) ionic conduction of MIL-53(Al)-PEO-NaCF3SO3 (PNM) electrolyte with varying ratios of EO and Na, (b) ionic conduction of PEO-NaCF3SO3 at varying content of MIL-53(Al) filler, (c) overlaid XRD of various electrolytes used in the study (d) photograph showing MIL-53(Al)-PEO-NaCF3SO3 (PNM) electrolyte, and (e) SEM image of the PNM electrolyte depicting uniformity of dispersion of filler in the polymer matrix.57 | ||
Furthermore, the Na+ transference number (t+) of PNM showed a higher value due to Lewis acidity of MIL-53(Al), which attracted CF3SO3− ions while Na+ was repelled. PNM and PN show an electrochemical stability window up to 4.0 V at 60 °C, with thermal stability up to 175 °C. It was also observed that PNM showed the least bulk resistance for all the cycles compared to PN, indicating the mitigation of electrolyte/electrode interfacial resistance by adding MIL 53(Al). The rate performance of the cell assembled with PNM and PN electrolytes at 60 °C shows that the MOF contributed to capacity exertion at the high rates. Also, the full battery configuration displayed a high Coulombic efficiency (100.63 ± 1.09%) and superior capacity retention with more extended cyclic stability.
6. Outlook and future perspectives
Research in RT-Na–S batteries has been a hot topic since it was first reported. Although HT-Na–S batteries are already a commercialized technology, issues related to high-temperature operation and safety concerns limited their widespread acceptance. This led to the development of intermediate temperature Na–S batteries operated at 150 °C, and further research in lowering the operational temperature led to the development of a RT-Na–S battery. Various approaches have been investigated to enhance the electrochemical properties of RT-Na–S cells. As ionic mobility is superior for LEs, RT-Na–S batteries with various LE systems were reported. Despite the easiness of ion transportation, the LE-based Na–S battery showed capacity decay with cycles and unstable solid-electrolyte interface formation. Dissolution of sodium polysulfides and their shuttling from the cathode to the anode were some of the significant drawbacks that reduced the performance of LE-based RT-Na–S batteries. Furthermore, electrolyte leakage, usage of flammable and volatile organic solvents, providing a threat to the environment and human safety, extend the drawbacks of LE-based RT-Na–S batteries.With proliferation in the human population and booming demand for EVs, quest for high energy density batteries have been at its peak. Furthermore, technological advancement accelerated the growth of portable electronic sector. Hence, the aforementioned factors led to the revolution of battery technology. Coupled with the requirement of high performance, researchers identified the undeniable importance and need for safe battery technology and safer battery operations. As the electrolyte part has been identified as that which decreases safety aspects, researchers explored approaches to rectify the problem. With organic solvents being volatile, toxic, carcinogenic and possessing a lower flash point, the probability of thermal runaway is very high. Although many developments have been made in the past decades in incorporating flame retardant additives to mitigate the issues of fire hazards, the inherent nature of organic solvents remains the same. Another approach developed was to replace the liquid electrolyte with a solid counterpart, such as solid state electrolytes. Hence, the development of all-solid-state-RT-Na–S batteries has gained momentum, exclusively identified as promising safe battery chemistry as it is free from flammable and organic solvent-based electrolytes. Solid-state RT-Na–S batteries have shown enhancement in cycling stability, and Coulombic and energy efficiency by the elimination of polysulfide formation and its subsequent cycling. Furthermore, stable plating and stripping of sodium enhance the stability and cycling of the cell. Solid electrolytes that have inorganic and organic backbones have been found to be an effective solution, which promotes ionic conduction and has been found to bypass the issues related to electrolyte leaking, shuttling of polysulfides, capacity decay with cycles, etc. Although considering safety aspects, solid-state electrolytes provide a better option than LEs, the research on solid electrolytes for RT-Na–S is still in its infancy.
The bottleneck challenges related to using sold state electrolytes for RT-Na–S batteries are the resistance at the electrode–electrolyte interface, sluggish Na+ mobility and solvation-desolvation kinetics, and the fading of capacity with cycle number and interfacial instability. Ideally, the solid-state electrolyte should possess the requirements, (i) high ionic conductivity (in the range of 10−3 S cm−1) with a wide temperature range, (ii) wide electrochemical stability window (1.5–4.5 V vs. Na/Na+), (iii) stability against electrodes and chemicals, (iv) mechanically stable to suppress dendritic growth, (v) resistive to polysulfide shuttling and dissolution, (vi) environmental benignancy and cost-effective, and (vii) thermal and mechanical properties with ease of processing. Researchers have been exclusively working on mitigating the limitations of solid-state electrolytes through multi-faced strategies. Solid electrolytes possess poor interfacial properties, engineering the interface and microstructure of ISEs by mechanical milling and thermal processes such as annealing and sintering, thereby reducing mechanical brittleness and grain boundaries. The creation of ion transport pathways via tailoring the lattice parameters such as lattice size by the influence of dopants and substitutional ions has been shown to enhance ionic mobility in the electrolytes and reduce interfacial resistance. Furthermore, it should be noted that the purity of various precursors and optimizing the parameters for the synthesis of ISEs also play a crucial role in the electrochemical properties. Activating interfaces by adding a few drops of LE can also reduce the interfacial resistance and promote the formation of stable SEI. Another effective approach is the preparation of a composite cathode electrolyte by mechanical milling of cathode active material and ISE, thereby a compact cathode–electrolyte interface can be engineered (Fig. 16).
 | ||
| Fig. 16 Schematic illustration showing the trend in solid-state electrolyte engineering, (a) synthesis of composite cathode containing inorganic solid electrolyte by mechanical milling process and its assembly, (b) ion selective membrane containing functional filler in a polymer host and the operando analysis. | ||
Although polymer electrolytes have been revolutionized in LIB chemistry, the same is not observed in RT-Na–S systems. Polymer electrolytes can be tailor-made by modifying the microstructure and chemical structure with ease in processing at lower temperatures compared to ISEs. Advanced fabrication processes, such as electrospinning, phase inversion, and drop-casting create micro-nano porous interconnected network structures, which preferentially allow ionic transport by creating pathways. Moreover, exploring functional fillers that can reduce the crystalline nature, lower cation coordination, and improve the segmental motion enhances the electrochemical properties with substantial increment in mechanical and chemical properties. Optimized amount of sodium salt incorporation mitigates the formation of ion clusters. LE incorporated GPEs, and quasi-solid-state electrolytes are a hybrid approach wherein the system behaves similar to the electrochemical properties of LE. Still, it provides mechanical, thermal, and chemical properties of solid electrolytes, which have been successful in RT-Na–S systems. CPEs, composite polymer electrolytes, and a hybrid have also shown promising effects in Na–S batteries. Functional groups in the filler materials can also trap polysulfides and hence mitigate the shuttling (Fig. 16). Such filler incorporated polymer as membrane can also function as a separator, applicable for solid-state batteries and LE-based batteries.
It is explicitly essential in understanding the interface of electrodes and electrolytes. Hence, real-time monitoring is essential to unveil the complexities and reactions at the interface. Development of in situ or operando characterization techniques can be useful. Such techniques provide better insights into interfacial chemistry, thereby divulging the information on the suitability of electrolytes with their ability to form a stable SEI. As the battery's overall performance depends on the electrode and electrolyte materials and their compatibility, a judicious selection of these materials has prime importance. In addition to the electrochemical properties of a battery, such as energy-power densities, cyclic stability, and Coulombic efficiency, availability of cost-effective raw materials, its safety concerns and environmental benignity also have a significant role. With continuous development in all-solid-state-RT-Na–S technology, efforts have been made to rectify the challenges above, thus, providing a vast scope for a viable technology for future energy storage applications which is sustainable and safe.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Vipin Kumar acknowledges the Science and Engineering Research Board (SERB) for the financial support under the Start-up Research Grant (SRG2021/000101-G). S. K. V., C. B. S., and Sungjemmenla acknowledge the scholarship awarded by the University of Queensland-IIT Delhi Academy of Research (UQIDAR) and the Indian Institute of Technology Delhi (IIT Delhi). XSZ acknowledges the Australian Research Council for financial support under the Laureate Fellowship Program (FL170100101).References
- M. S. Javed, T. Ma, J. Jurasz and M. Y. Amin, Renewable Energy, 2020, 148, 176–192 CrossRef.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- S. Maddukuri, D. Malka, M. S. Chae, Y. Elias, S. Luski and D. Aurbach, Electrochim. Acta, 2020, 354, 136771 CrossRef CAS.
- S. Sharma, A. K. Panwar and M. M. Tripathi, J. Traffic Transp. Eng., 2020, 7, 340–361 Search PubMed.
- Y. Liang, C. Zhao, H. Yuan, Y. Chen, W. Zhang, J. Huang, D. Yu, Y. Liu, M. Titirici, Y. Chueh, H. Yu and Q. Zhang, InfoMat, 2019, 1, 6–32 CrossRef CAS.
- T. Placke, R. Kloepsch, S. Dühnen and M. Winter, J. Solid State Electrochem., 2017, 21, 1939–1964 CrossRef CAS.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- J. Xie and Y.-C. Lu, Nat. Commun., 2020, 11, 2499 CrossRef CAS PubMed.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- J. Mei, T. Liao, G. A. Ayoko, J. Bell and Z. Sun, Prog. Mater. Sci., 2019, 103, 596–677 CrossRef CAS.
- A. Manthiram, ACS Cent. Sci., 2017, 3, 1063–1069 CrossRef CAS PubMed.
- A. Mauger, C. M. Julien, J. B. Goodenough and K. Zaghib, J. Electrochem. Soc., 2020, 167, 070507 CrossRef CAS.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- S.-K. Jung, I. Hwang, D. Chang, K.-Y. Park, S. J. Kim, W. M. Seong, D. Eum, J. Park, B. Kim, J. Kim, J. H. Heo and K. Kang, Chem. Rev., 2020, 120, 6684–6737 CrossRef CAS PubMed.
- F. Wu and G. Yushin, Energy Environ. Sci., 2017, 10, 435–459 RSC.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- J.-M. Tarascon, Philos. Trans. R. Soc. A, 2010, 368, 3227–3241 CrossRef PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 RSC.
- Y. E. Durmus, H. Zhang, F. Baakes, G. Desmaizieres, H. Hayun, L. Yang, M. Kolek, V. Küpers, J. Janek, D. Mandler, S. Passerini and Y. Ein-Eli, Adv. Energy Mater., 2020, 10, 2000089 CrossRef CAS.
- X. Yu and A. Manthiram, Adv. Energy Sustainable Res., 2021, 2, 2000102 CrossRef.
- C. B. Soni, Sungjemmenla, S. K. Vineeth and V. Kumar, Energy Storage, 2022, 4, e264 CrossRef CAS.
- X. Zhang, Metal–Air Batteries Fundamentals and Applications, John Wiley & Sons, Weinheim, Germany, 2018 Search PubMed.
- Y. Pan, S. Li, M. Yin and J. Li, Energy Technol., 2019, 7, 1900164 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- M. A. Rahman, X. Wang and C. Wen, J. Electrochem. Soc., 2013, 160, A1759–A1771 CrossRef CAS.
- Sungjemmenla, C. B. Soni and V. Kumar, Nanoscale Adv., 2021, 3, 1569–1581 RSC.
- P. Hartmann, C. L. Bender, M. Vračar, A. K. Dürr, A. Garsuch, J. Janek and P. Adelhelm, Nat. Mater., 2013, 12, 228–232 CrossRef CAS PubMed.
- B. L. Ellis and L. F. Nazar, Curr. Opin. Solid State Mater. Sci., 2012, 16, 168–177 CrossRef CAS.
- C. Delmas, Adv. Energy Mater., 2018, 8, 1703137 CrossRef.
- F. Duffner, N. Kronemeyer, J. Tübke, J. Leker, M. Winter and R. Schmuch, Nat. Energy, 2021, 6, 123–134 CrossRef CAS.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- K. Hans Wedepohl, Geochim. Cosmochim. Acta, 1995, 59, 1217–1232 CrossRef.
- I. Kim, C. H. Kim, S. H. Choi, J. P. Ahn, J. H. Ahn, K. W. Kim, E. J. Cairns and H. J. Ahn, J. Power Sources, 2016, 307, 31–37 CrossRef CAS.
- P. Adelhelm, P. Hartmann, C. L. Bender, M. Busche, C. Eufinger and J. Janek, Beilstein J. Nanotechnol., 2015, 6, 1016–1055 CrossRef CAS PubMed.
- L. Li, Y. Zheng, S. Zhang, J. Yang, Z. Shao and Z. Guo, Energy Environ. Sci., 2018, 11, 2310–2340 RSC.
- J. Yang, H. Gao and X. Kang, in Advanced Nanomaterials for Electrochemical-Based Energy Conversion and Storage, ed. F. Ran and S. Chen, Elsevier, 2020, pp. 69–114 Search PubMed.
- K. M. Abraham, ACS Energy Lett., 2020, 5, 3544–3547 CrossRef CAS.
- Mineral Commodity Summaries, 2019.
- Z. Wen, J. Cao, Z. Gu, X. Xu, F. Zhang and Z. Lin, Solid State Ionics, 2008, 179, 1697–1701 CrossRef CAS.
- J. G. Wagenfeld, K. Al-Ali, S. Almheiri, A. F. Slavens and N. Calvet, Waste Manage., 2019, 95, 78–89 CrossRef PubMed.
- H. D. Yoo, E. Markevich, G. Salitra, D. Sharon and D. Aurbach, Mater. Today, 2014, 17, 110–121 CrossRef CAS.
- M. A. Pope and I. A. Aksay, Adv. Energy Mater., 2015, 5, 1500124 CrossRef.
- Y. Liu, S. Liu, G.-R. Li, T.-Y. Yan and X.-P. Gao, Adv. Sci., 2020, 7, 1903693 CrossRef CAS PubMed.
- Y. Wang, D. Zhou, V. Palomares, D. Shanmukaraj, B. Sun, X. Tang, C. Wang, M. Armand, T. Rojo and G. Wang, Energy Environ. Sci., 2020, 13, 3848–3879 RSC.
- Sungjemmenla, C. B. Soni, S. K. Vineeth and V. Kumar, Mater. Adv., 2021, 2, 4165–4189 RSC.
- M. S. Syali, D. Kumar, K. Mishra and D. K. Kanchan, Energy Storage Mater., 2020, 31, 352–372 CrossRef.
- V. Kumar, A. Y. S. Eng, Y. Wang, D.-T. Nguyen, M.-F. Ng and Z. W. Seh, Energy Storage Mater., 2020, 29, 1–8 CrossRef.
- J. Wang, Y. Ni, J. Liu, Y. Lu, K. Zhang, Z. Niu and J. Chen, ACS Cent. Sci., 2020, 6, 1955–1963 CrossRef CAS PubMed.
- G. Nikiforidis, M. C. M. van de Sanden and M. N. Tsampas, RSC Adv., 2019, 9, 5649–5673 RSC.
- V. Kumar, Y. Wang, A. Y. S. Eng, M.-F. Ng and Z. W. Seh, Cell Rep. Phys. Sci., 2020, 1, 100044 CrossRef.
- Y. Liu, X. Li, Y. Sun, R. Yang, Y. Lee and J.-H. Ahn, J. Alloys Compd., 2021, 853, 157316 CrossRef CAS.
- X. Fan, J. Yue, F. Han, J. Chen, T. Deng, X. Zhou, S. Hou and C. Wang, ACS Nano, 2018, 12, 3360–3368 CrossRef CAS PubMed.
- S. Song, H. M. Duong, A. M. Korsunsky, N. Hu and L. Lu, Sci. Rep., 2016, 6, 32330 CrossRef CAS PubMed.
- C. Park, H. Ryu, K. Kim, J.-H. Ahn, J. Lee and H. Ahn, J. Power Sources, 2007, 165, 450–454 CrossRef CAS.
- T. Zhu, X. Dong, Y. Liu, Y. Wang, C. Wang and Y. Xia, ACS Appl. Energy Mater., 2019, 2, 5263–5271 CrossRef CAS.
- Z. Ge, J. Li and J. Liu, Ionics, 2020, 26, 1787–1795 CrossRef CAS.
- X. Zhang, G. Li, Y. Zhang, D. Luo, A. Yu, X. Wang and Z. Chen, Nano Energy, 2021, 86, 106094 CrossRef CAS.
- J. R. Nair, F. Bella, N. Angulakshmi, A. M. Stephan and C. Gerbaldi, Energy Storage Mater., 2016, 3, 69–76 CrossRef.
- Y. Lin, J. Li, K. Liu, Y. Liu, J. Liu and X. Wang, Green Chem., 2016, 18, 3796–3803 RSC.
- J. Li, R. Wang, W. Zhao, X. Hou, E. Paillard, D. Ning, C. Li, J. Wang, Y. Xiao, M. Winter and J. Li, J. Power Sources, 2021, 507, 230183 CrossRef CAS.
- M. A. Ud Din, S. Irfan, S. Jamil, S. U. Dar, Q. U. Khan, M. S. Saleem and N. Cheng, J. Alloys Compd., 2022, 901, 163572 CrossRef CAS.
- R. Arunkumar, A. P. Vijaya Kumar Saroja and R. Sundara, ACS Appl. Mater. Interfaces, 2019, 11, 3889–3896 CrossRef CAS PubMed.
- S. Zhang, Y. Yao and Y. Yu, ACS Energy Lett., 2021, 6, 529–536 CrossRef CAS.
- D. Kumar, D. K. Kanchan, S. Kumar and K. Mishra, Mater. Sci. Energy Technol., 2019, 2, 117–129 Search PubMed.
- Y. X. Ren, H. R. Jiang, T. S. Zhao, L. Zeng and C. Xiong, J. Power Sources, 2018, 396, 304–313 CrossRef CAS.
- A. Kotronia, H. D. Asfaw, C.-W. Tai, M. Hahlin, D. Brandell and K. Edström, ACS Appl. Mater. Interfaces, 2021, 13, 3867–3880 CrossRef CAS PubMed.
- S. K. Vineeth, C. B. Soni, Y. Sun, V. Kumar and Z. W. Seh, Trends Chem., 2022, 4, 48–59 CrossRef CAS.
- C. B. Soni, V. Kumar and Z. W. Seh, Batter. Supercaps, 2022, 5, e202100207 CAS.
- M. Schellenberger, R. Golnak, W. G. Quevedo Garzon, S. Risse and R. Seidel, Mater. Today Adv., 2022, 14, 100215 CrossRef CAS.
- Y.-X. Wang, B. Zhang, W. Lai, Y. Xu, S.-L. Chou, H.-K. Liu and S.-X. Dou, Adv. Energy Mater., 2017, 7, 1602829 CrossRef.
- X. Yu and A. Manthiram, Matter, 2019, 1, 439–451 CrossRef.
- M. K. Aslam, T. Hussain, H. Tabassum, Z. Wei, W. Tang, S. Li, S. J. Bao, X. S. Zhao and M. Xu, Chem. Eng. J., 2022, 429, 132389 CrossRef CAS.
- S. Wenzel, H. Metelmann, C. Raiß, A. K. Dürr, J. Janek and P. Adelhelm, J. Power Sources, 2013, 243, 758–765 CrossRef CAS.
- I. Bauer, M. Kohl, H. Althues and S. Kaskel, Chem. Commun., 2014, 50, 3208 RSC.
- M. S. Nahian, R. Jayan, T. Kaewmaraya, T. Hussain and M. M. Islam, ACS Appl. Mater. Interfaces, 2022, 14, 10298–10307 CrossRef CAS PubMed.
- A. Y. S. Eng, V. Kumar, Y. Zhang, J. Luo, W. Wang, Y. Sun, W. Li and Z. W. Seh, Adv. Energy Mater., 2021, 11, 2003493 CrossRef CAS.
- P. Peljo and H. H. Girault, Energy Environ. Sci., 2018, 11, 2306–2309 RSC.
- Q. Pan, D. Gong and Y. Tang, Energy Storage Mater., 2020, 31, 328–343 CrossRef.
- F. Wu, K. Zhang, Y. Liu, H. Gao, Y. Bai, X. Wang and C. Wu, Energy Storage Mater., 2020, 33, 26–54 CrossRef.
- G. Cui, Matter, 2020, 2, 805–815 CrossRef.
- E. Lizundia and D. Kundu, Adv. Funct. Mater., 2021, 31, 2005646 CrossRef CAS.
- J. T. Kummer and W. Neill, US pat., 3413150, 1968 Search PubMed.
- J.-N. Anand, 14th Intersociety Energy Conversion Engineering Conference, 1979, vol. 1, pp. 698–702 Search PubMed.
- J. B. Goodenough, H. Y.-P. Hong and J. A. Kafalas, Mater. Res. Bull., 1976, 11, 203–220 CrossRef CAS.
- N. K. Gupta and G. J. Tennenhouse, J. Electrochem. Soc., 1979, 126, 1451–1455 CrossRef CAS.
- R. Chang and R. Minck, J. Power Sources, 1990, 29, 555–563 CrossRef CAS.
- R. O. Ansell, J. Mater. Sci., 1986, 21, 365–379 CrossRef CAS.
- T. Oshima, M. Kajita and A. Okuno, Int. J. Appl. Ceram. Technol., 2005, 1, 269–276 CrossRef.
- N. Kawakami, Y. Iijima, M. Fukuhara, M. Bando, Y. Sakanaka, K. Ogawa and T. Matsuda, 2010 IEEE International Symposium on Industrial Electronics, IEEE, 2010, pp. 2371–2376 Search PubMed.
- S. Tewari and N. Mohan, IEEE Trans. Power Syst., 2013, 28, 532–541 Search PubMed.
- E. M. G. Rodrigues, G. J. Osório, R. Godina, A. W. Bizuayehu, J. M. Lujano-Rojas, J. C. O. Matias and J. P. S. Catalão, Energy, 2015, 90, 1606–1617 CrossRef CAS.
- X. Xu, D. Zhou, X. Qin, K. Lin, F. Kang, B. Li, D. Shanmukaraj, T. Rojo, M. Armand and G. Wang, Nat. Commun., 2018, 9, 3870 CrossRef PubMed.
- X. Lu, G. Xia, J. P. Lemmon and Z. Yang, J. Power Sources, 2010, 195, 2431–2442 CrossRef CAS.
- K. B. Hueso, M. Armand and T. Rojo, Energy Environ. Sci., 2013, 6, 734 RSC.
- R. Singhal, S.-H. Chung, A. Manthiram and V. Kalra, J. Mater. Chem. A, 2015, 3, 4530–4538 RSC.
- R. X. Chu, J. Lin, C. Q. Wu, J. Zheng, Y. L. Chen, J. Zhang, R. H. Han, Y. Zhang and H. Guo, Nanoscale, 2017, 9, 9129–9138 RSC.
- J. Zhu, J. Zou, H. Cheng, Y. Gu and Z. Lu, Green Energy Environ., 2019, 4, 345–359 CrossRef.
- Y. Wang, W.-H. Lai, S.-L. Chou, H.-K. Liu and S.-X. Dou, Adv. Mater., 2020, 32, 1903952 CrossRef CAS PubMed.
- M. Liu and L. C. De Jonghe, J. Electrochem. Soc., 1988, 135, 741–749 CrossRef CAS.
- M. Andriollo, R. Benato, S. Dambone Sessa, N. Di Pietro, N. Hirai, Y. Nakanishi and E. Senatore, J. Energy Storage, 2016, 5, 146–155 CrossRef.
- K. M. Abraham, R. D. Rauh and S. B. Brummer, Electrochim. Acta, 1978, 23, 501–507 CrossRef CAS.
- W. L. Fielder and J. Singer, NASA Tech. Pap., 1978, 1–40 Search PubMed.
- H. Liu, W. Lai, Y. Lei, H. Yang, N. Wang, S. Chou, H. K. Liu, S. X. Dou and Y. Wang, Adv. Energy Mater., 2022, 2103304 CrossRef CAS.
- J. Wang, J. Yang, Y. Nuli and R. Holze, Electrochem. Commun., 2007, 9, 31–34 CrossRef CAS.
- T. H. Hwang, D. S. Jung, J.-S. Kim, B. G. Kim and J. W. Choi, Nano Lett., 2013, 13, 4532–4538 CrossRef CAS PubMed.
- S. Xin, Y. Yin, Y. Guo and L. Wan, Adv. Mater., 2014, 26, 1261–1265 CrossRef CAS PubMed.
- X. Yu and A. Manthiram, J. Phys. Chem. C, 2014, 118, 22952–22959 CrossRef CAS.
- H. Ryu, T. Kim, K. Kim, J. H. Ahn, T. Nam, G. Wang and H. J. Ahn, J. Power Sources, 2011, 196, 5186–5190 CrossRef CAS.
- T. Li, J. Xu, C. Wang, W. Wu, D. Su and G. Wang, J. Alloys Compd., 2019, 792, 797–817 CrossRef CAS.
- Y. Wang and W.-H. Zhong, ChemElectroChem, 2015, 2, 22–36 CrossRef CAS.
- D. Lu, Y. Shao, T. Lozano, W. D. Bennett, G. L. Graff, B. Polzin, J. Zhang, M. H. Engelhard, N. T. Saenz, W. A. Henderson, P. Bhattacharya, J. Liu and J. Xiao, Adv. Energy Mater., 2015, 5, 1400993 CrossRef.
- N. S. Jishnu, S. K. Vineeth, A. Das, N. T. M. Balakrishnan, A. P. Thomas, M. J. Jabeen Fatima, J.-H. Ahn and R. Prasanth, in Electrospinning for Advanced Energy Storage Applications, ed. N. T. M. Balakrishnan and R. Prasanth, Springer Singapore, Singapore, 1st edn, 2021, pp. 201–234 Search PubMed.
- L. Wang, Z. Wang, Y. Sun, X. Liang and H. Xiang, J. Membr. Sci., 2019, 572, 512–519 CrossRef CAS.
- X. Fu, D. Yu, J. Zhou, S. Li, X. Gao, Y. Han, P. Qi, X. Feng and B. Wang, CrystEngComm, 2016, 18, 4236–4258 RSC.
- Q. Zhang, K. Liu, F. Ding and X. Liu, Nano Res., 2017, 10, 4139–4174 CrossRef.
- Y. Zhang, W. Lu, L. Cong, J. Liu, L. Sun, A. Mauger, C. M. Julien, H. Xie and J. Liu, J. Power Sources, 2019, 420, 63–72 CrossRef CAS.
- S. Wei, S. Xu, A. Agrawral, S. Choudhury, Y. Lu, Z. Tu, L. Ma and L. A. Archer, Nat. Commun., 2016, 7, 11722 CrossRef CAS PubMed.
- C.-W. Park, J.-H. Ahn, H.-S. Ryu, K.-W. Kim and H.-J. Ahn, Electrochem. Solid-State Lett., 2006, 9, A123 CrossRef CAS.
- Q. Li, J. Chen, L. Fan, X. Kong and Y. Lu, Green Energy Environ., 2016, 1, 18–42 CrossRef.
- Q. Ma, G. Du, B. Guo, W. Tang, Y. Li, M. Xu and C. Li, Chem. Eng. J., 2020, 388, 124210 CrossRef CAS.
- Q. Ma, G. Du, W. Zhong, W. Du, S. J. Bao, M. Xu and C. Li, J. Colloid Interface Sci., 2020, 578, 710–716 CrossRef CAS PubMed.
- H. Nagata and Y. Chikusa, Chem. Lett., 2014, 43, 1333–1334 CrossRef CAS.
- J. Yue, F. Han, X. Fan, X. Zhu, Z. Ma, J. Yang and C. Wang, ACS Nano, 2017, 11, 4885–4891 CrossRef CAS PubMed.
- N. Tanibata, M. Deguchi, A. Hayashi and M. Tatsumisago, Chem. Mater., 2017, 29, 5232–5238 CrossRef CAS.
- N. Tanibata, H. Tsukasaki, M. Deguchi, S. Mori, A. Hayashi and M. Tatsumisago, Solid State Ionics, 2017, 311, 6–13 CrossRef CAS.
- H. Wan, W. Weng, F. Han, L. Cai, C. Wang and X. Yao, Nano Today, 2020, 33, 100860 CrossRef CAS.
- T. An, H. Jia, L. Peng and J. Xie, ACS Appl. Mater. Interfaces, 2020, 12, 20563–20569 CrossRef CAS PubMed.
- L. Lu, Y. Lu, J. A. Alonso, C. A. López, M. T. Fernández-Díaz, B. Zou and C. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 42927–42934 CrossRef CAS PubMed.
- M. Faraday, Philos. Trans. R. Soc. London, 1839, 129, 1–12 CrossRef.
- X. He, Y. Zhu and Y. Mo, Nat. Commun., 2017, 8, 15893 CrossRef CAS PubMed.
- F. Sun, C. Wang, M. Osenberg, K. Dong, S. Zhang, C. Yang, Y. Wang, A. Hilger, J. Zhang, S. Dong, H. Markötter, I. Manke and G. Cui, Adv. Energy Mater., 2022, 12, 2103714 CrossRef CAS.
- H. Yildirim, A. Kinaci, M. K. Y. Chan and J. P. Greeley, ACS Appl. Mater. Interfaces, 2015, 7, 18985–18996 CrossRef CAS PubMed.
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Chem. Rev., 2019, 119, 5416–5460 CrossRef CAS PubMed.
- S. Ohno, A. Banik, G. F. Dewald, M. A. Kraft, T. Krauskopf, N. Minafra, P. Till, M. Weiss and W. G. Zeier, Prog. Energy, 2020, 2, 022001 CrossRef.
- Z. Chen, H. Zhang, H. Xu, S. Dong, M. Jiang, Z. Li and G. Cui, Chem. Eng. J., 2022, 433, 133589 CrossRef CAS.
- B. Santhoshkumar, D. L. R. Khanna, M. B. Choudhary, P. Lokeswara Rao, K. V. Ramanathan, A. K. Bera, S. M. Yusuf and B. Pahari, Chem. Phys. Lett., 2021, 776, 138706 CrossRef CAS.
- M. Petrowsky and R. Frech, J. Phys. Chem. B, 2010, 114, 8600–8605 CrossRef CAS PubMed.
- R. B. Nuernberg, Ionics, 2020, 26, 2405–2412 CrossRef CAS.
- H. Nguyen, A. Banerjee, X. Wang, D. Tan, E. A. Wu, J.-M. Doux, R. Stephens, G. Verbist and Y. S. Meng, J. Power Sources, 2019, 435, 126623 CrossRef CAS.
- Z. Zhang, Y. Shao, B. Lotsch, Y.-S. Hu, H. Li, J. Janek, L. F. Nazar, C.-W. Nan, J. Maier, M. Armand and L. Chen, Energy Environ. Sci., 2018, 11, 1945–1976 RSC.
- S. S. Berbano, I. Seo, C. M. Bischoff, K. E. Schuller and S. W. Martin, J. Non-Cryst. Solids, 2012, 358, 93–98 CrossRef CAS.
- K. Noi, A. Hayashi and M. Tatsumisago, J. Power Sources, 2014, 269, 260–265 CrossRef CAS.
- T. Famprikis, Ö. U. Kudu, J. A. Dawson, P. Canepa, F. Fauth, E. Suard, M. Zbiri, D. Dambournet, O. J. Borkiewicz, H. Bouyanfif, S. P. Emge, S. Cretu, J.-N. Chotard, C. P. Grey, W. G. Zeier, M. S. Islam and C. Masquelier, J. Am. Chem. Soc., 2020, 142, 18422–18436 CrossRef CAS PubMed.
- T. Krauskopf, S. P. Culver and W. G. Zeier, Inorg. Chem., 2018, 57, 4739–4744 CrossRef CAS PubMed.
- I. H. Chu, C. S. Kompella, H. Nguyen, Z. Zhu, S. Hy, Z. Deng, Y. S. Meng and S. P. Ong, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- M. Jansen and U. Henseler, J. Solid State Chem., 1992, 99, 110–119 CrossRef CAS.
- A. Hayashi, K. Noi, A. Sakuda and M. Tatsumisago, Nat. Commun., 2012, 3, 856 CrossRef PubMed.
- N. Tanibata, K. Noi, A. Hayashi and M. Tatsumisago, RSC Adv., 2014, 4, 17120–17123 RSC.
- Z. Zhu, I.-H. Chu, Z. Deng and S. P. Ong, Chem. Mater., 2015, 27, 8318–8325 CrossRef CAS.
- R. P. Rao, H. Chen, L. L. Wong and S. Adams, J. Mater. Chem. A, 2017, 5, 3377–3388 RSC.
- J. A. Dawson, P. Canepa, M. J. Clarke, T. Famprikis, D. Ghosh and M. S. Islam, Chem. Mater., 2019, 31, 5296–5304 CrossRef CAS.
- S. Takeuchi, K. Suzuki, M. Hirayama and R. Kanno, J. Solid State Chem., 2018, 265, 353–358 CrossRef CAS.
- N. Tanibata, K. Noi, A. Hayashi, N. Kitamura, Y. Idemoto and M. Tatsumisago, ChemElectroChem, 2014, 1, 1130–1132 CrossRef CAS.
- H. Huang, H.-H. Wu, X. Wang, B. Huang and T.-Y. Zhang, Phys. Chem. Chem. Phys., 2018, 20, 20525–20533 RSC.
- N. J. J. de Klerk and M. Wagemaker, Chem. Mater., 2016, 28, 3122–3130 CrossRef CAS.
- A. Hayashi, K. Noi, N. Tanibata, M. Nagao and M. Tatsumisago, J. Power Sources, 2014, 258, 420–423 CrossRef CAS.
- S. Yubuchi, A. Hayashi and M. Tatsumisago, Chem. Lett., 2015, 44, 884–886 CrossRef CAS.
- E. A. Wu, C. S. Kompella, Z. Zhu, J. Z. Lee, S. C. Lee, I.-H. Chu, H. Nguyen, S. P. Ong, A. Banerjee and Y. S. Meng, ACS Appl. Mater. Interfaces, 2018, 10, 10076–10086 CrossRef CAS PubMed.
- P. Hu, Y. Zhang, X. Chi, K. Kumar Rao, F. Hao, H. Dong, F. Guo, Y. Ren, L. C. Grabow and Y. Yao, ACS Appl. Mater. Interfaces, 2019, 11, 9672–9678 CrossRef CAS PubMed.
- Sungjemmenla, C. B. Soni, S. K. Vineeth and V. Kumar, Adv. Energy Sustain. Res., 2022, 3, 2100157 CrossRef.
- D.-J. Lee, J.-W. Park, I. Hasa, Y.-K. Sun, B. Scrosati and J. Hassoun, J. Mater. Chem. A, 2013, 1, 5256 RSC.
- T. Yang, B. Guo, W. Du, M. K. Aslam, M. Tao, W. Zhong, Y. Chen, S. Bao, X. Zhang and M. Xu, Adv. Sci., 2019, 6, 1901557 CrossRef CAS PubMed.
- N.-W. Li, Y.-X. Yin, J.-Y. Li, C.-H. Zhang and Y.-G. Guo, Adv. Sci., 2017, 4, 1600400 CrossRef PubMed.
- E. Umeshbabu, B. Zheng, J. Zhu, H. Wang, Y. Li and Y. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 18436–18447 CrossRef CAS PubMed.
- B. Zheng, J. Zhu, H. Wang, M. Feng, E. Umeshbabu, Y. Li, Q.-H. Wu and Y. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 25473–25482 CrossRef CAS PubMed.
- J. A. S. Oh, L. He, A. Plewa, M. Morita, Y. Zhao, T. Sakamoto, X. Song, W. Zhai, K. Zeng and L. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 40125–40133 CrossRef CAS PubMed.
- B. Ouyang, J. Wang, T. He, C. J. Bartel, H. Huo, Y. Wang, V. Lacivita, H. Kim and G. Ceder, Nat. Commun., 2021, 12, 1–11 CrossRef PubMed.
- S. Roy and P. P. Kumar, Solid State Ionics, 2013, 253, 217–222 CrossRef CAS.
- M. Guin and F. Tietz, J. Power Sources, 2015, 273, 1056–1064 CrossRef CAS.
- N. Anantharamulu, K. Koteswara Rao, G. Rambabu, B. Vijaya Kumar, V. Radha and M. Vithal, J. Mater. Sci., 2011, 46, 2821–2837 CrossRef CAS.
- R. Rajagopalan, Z. Zhang, Y. Tang, C. Jia, X. Ji and H. Wang, Energy Storage Mater., 2021, 34, 171–193 CrossRef.
- A. M. Nieto-Muñoz, J. F. Ortiz-Mosquera and A. C. M. Rodrigues, Electrochim. Acta, 2019, 319, 922–932 CrossRef.
- W. Zhou, Y. Li, S. Xin and J. B. Goodenough, ACS Cent. Sci., 2017, 3, 52–57 CrossRef CAS PubMed.
- O. Bohnke, S. Ronchetti and D. Mazza, Solid State Ionics, 1999, 122, 127–136 CrossRef CAS.
- Y. B. Rao, K. K. Bharathi and L. N. Patro, Solid State Ionics, 2021, 366–367, 115671 CrossRef CAS.
- O. Nakamura, Y. Saito, M. Kodama and Y. Yamamoto, Solid State Ionics, 1996, 89, 159–164 CrossRef CAS.
- A. Tsuji, H. Takahashi and T. Oi, J. Mater. Chem., 2003, 13, 542–549 RSC.
- Y. Saito, K. Ado, T. Asai, H. Kageyama and O. Nakamura, Solid State Ionics, 1992, 58, 327–331 CrossRef CAS.
- M. Guin, F. Tietz and O. Guillon, Solid State Ionics, 2016, 293, 18–26 CrossRef CAS.
- Z. Khakpour, Electrochim. Acta, 2016, 196, 337–347 CrossRef CAS.
- X. Lu, G. Li, J. Y. Kim, D. Mei, J. P. Lemmon, V. L. Sprenkle and J. Liu, Nat. Commun., 2014, 5, 4578 CrossRef CAS PubMed.
- K. Vignarooban, R. Kushagra, A. Elango, P. Badami, B.-E. Mellander, X. Xu, T. G. Tucker, C. Nam and A. M. Kannan, Int. J. Hydrogen Energy, 2016, 41, 2829–2846 CrossRef CAS.
- W. Hou, X. Guo, X. Shen, K. Amine, H. Yu and J. Lu, Nano Energy, 2018, 52, 279–291 CrossRef CAS.
- X. Lu, J. P. Lemmon, V. Sprenkle and Z. Yang, JOM, 2010, 62, 31–36 CrossRef CAS.
- J. T. Kummer, Prog. Solid State Chem., 1972, 7, 141–175 CrossRef CAS.
- X. Wei, Y. Cao, L. Lu, H. Yang and X. Shen, J. Alloys Compd., 2011, 509, 6222–6226 CrossRef CAS.
- S.-T. Lee, D.-H. Lee, J.-S. Kim and S.-K. Lim, Met. Mater. Int., 2017, 23, 246–253 CrossRef CAS.
- Z. Wen, Z. Gu, X. Xu, J. Cao, F. Zhang and Z. Lin, J. Power Sources, 2008, 184, 641–645 CrossRef CAS.
- J. Lin, Z. Wen, X. Wang, S. Song and Y. Liu, J. Solid State Electrochem., 2010, 14, 1821–1827 CrossRef CAS.
- A. Mali and A. Petric, J. Eur. Ceram. Soc., 2012, 32, 1229–1234 CrossRef CAS.
- I. Kim, J.-Y. Park, C. H. Kim, J.-W. Park, J.-P. Ahn, J.-H. Ahn, K.-W. Kim and H.-J. Ahn, J. Power Sources, 2016, 301, 332–337 CrossRef CAS.
- R. V. Gadhave, V. S. K, P. A. Mahanwar and P. T. Gadekar, J. Adhes. Sci. Technol., 2021, 35, 1072–1086 CrossRef CAS.
- S. K. Vineeth, R. V. Gadhave and P. T. Gadekar, Indian Chem. Eng., 2022, 64, 197–207 CrossRef CAS.
- M. I. Diana, P. C. Selvin, S. Selvasekarapandian and M. V. Krishna, J. Solid State Electrochem., 2021, 25, 2009–2020 CrossRef CAS.
- D.-H. Lim, M. Agostini, J.-H. Ahn and A. Matic, Energy Technol., 2018, 6, 1214–1219 CrossRef CAS.
- J. Zhang, H. Wen, L. Yue, J. Chai, J. Ma, P. Hu, G. Ding, Q. Wang, Z. Liu, G. Cui and L. Chen, Small, 2017, 13, 1601530 CrossRef PubMed.
- X. Yu, L. Wang, J. Ma, X. Sun, X. Zhou and G. Cui, Adv. Energy Mater., 2020, 10, 1–9 Search PubMed.
- Y. Wang, J. Ju, S. Dong, Y. Yan, F. Jiang, L. Cui, Q. Wang, X. Han and G. Cui, Adv. Funct. Mater., 2021, 31, 2101523 CrossRef CAS.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- Y. Yan, J. Ju, S. Dong, Y. Wang, L. Huang, L. Cui, F. Jiang, Q. Wang, Y. Zhang and G. Cui, Adv. Sci., 2021, 8, 2003887 CrossRef CAS PubMed.
- Y. Wang, S. Song, C. Xu, N. Hu, J. Molenda and L. Lu, Nano Mater. Sci., 2019, 1, 91–100 CrossRef.
- H. Verma, K. Mishra and D. K. Rai, J. Solid State Electrochem., 2020, 24, 521–532 CrossRef CAS.
- S. Janakiraman, A. Surendran, R. Biswal, S. Ghosh, S. Anandhan and A. Venimadhav, Mater. Res. Express, 2019, 6, 086318 CrossRef CAS.
- C. Xu, Y. Yang, H. Wang, B. Xu, Y. Li, R. Tan, X. Duan, D. Wu, M. Zhuo and J. Ma, Chem. – Asian J., 2020, 15, 3584–3598 CrossRef CAS PubMed.
- J. Yang, M. Zhang, Z. Chen, X. Du, S. Huang, B. Tang, T. Dong, H. Wu, Z. Yu, J. Zhang and G. Cui, Nano Res., 2019, 12, 2230–2237 CrossRef CAS.
- D. Kumar, Solid State Ionics, 2018, 318, 65–70 CrossRef CAS.
- W. Wang and P. Alexandridis, Polymers, 2016, 8, 387 CrossRef PubMed.
- J. Feng, L. Wang, Y. Chen, P. Wang, H. Zhang and X. He, Nano Convergence, 2021, 8, 2 CrossRef CAS PubMed.
- J. Yang, H. Zhang, Q. Zhou, H. Qu, T. Dong, M. Zhang, B. Tang, J. Zhang and G. Cui, ACS Appl. Mater. Interfaces, 2019, 11, 17109–17127 CrossRef CAS PubMed.
- B. Jinisha, K. M. Anilkumar, M. Manoj, V. S. Pradeep and S. Jayalekshmi, Electrochim. Acta, 2017, 235, 210–222 CrossRef CAS.
- R. Prasanth, N. Shubha, H. H. Hng and M. Srinivasan, J. Power Sources, 2014, 245, 283–291 CrossRef CAS.
- V. K. Singh, S. K. Singh, H. Gupta, Shalu, L. Balo, A. K. Tripathi, Y. L. Verma and R. K. Singh, J. Solid State Electrochem., 2018, 22, 1909–1919 CrossRef CAS.
- F. Gebert, J. Knott, R. Gorkin, S.-L. Chou and S.-X. Dou, Energy Storage Mater., 2021, 36, 10–30 CrossRef.
- D. Kumar and D. K. Kanchan, J. Energy Storage, 2019, 22, 44–49 CrossRef.
- D. E. Fenton, J. M. Parker and P. V. Wright, Polymer, 1973, 14, 589 CrossRef CAS.
- S. A. Hashmi and S. Chandra, Mater. Sci. Eng., B, 1995, 34, 18–26 CrossRef.
- L. Qiao, X. Judez, T. Rojo, M. Armand and H. Zhang, J. Electrochem. Soc., 2020, 167, 070534 CrossRef CAS.
- Z. Li, P. Liu, K. Zhu, Z. Zhang, Y. Si, Y. Wang and L. Jiao, Energy Fuels, 2021, 35, 9063–9079 CrossRef CAS.
- D. Zhou, Y. Chen, B. Li, H. Fan, F. Cheng, D. Shanmukaraj, T. Rojo, M. Armand and G. Wang, Angew. Chem., Int. Ed., 2018, 57, 10168–10172 CrossRef CAS PubMed.
- Y. Mallaiah, V. R. Jeedi, R. Swarnalatha, A. Raju, S. N. Reddy and A. S. Chary, J. Phys. Chem. Solids, 2021, 155, 110096 CrossRef CAS.
- S. B. Aziz, T. J. Woo, M. F. Z. Kadir and H. M. Ahmed, J. Sci. Adv. Mater. Devices, 2018, 3, 1–17 CrossRef.
- M. Patel, K. G. Chandrappa and A. J. Bhattacharyya, Solid State Ionics, 2010, 181, 844–848 CrossRef CAS.
- R. Prasanth, V. Aravindan and M. Srinivasan, J. Power Sources, 2012, 202, 299–307 CrossRef CAS.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 RSC.
- R. Prasanth, N. Shubha, H. H. Hng and M. Srinivasan, Eur. Polym. J., 2013, 49, 307–318 CrossRef CAS.
- N. Shubha, R. Prasanth, H. H. Hoon and M. Srinivasan, Mater. Res. Bull., 2013, 48, 526–537 CrossRef CAS.
- D. Kumar and S. A. Hashmi, Solid State Ionics, 2010, 181, 416–423 CrossRef CAS.
- C. A. Vincent, Electrochim. Acta, 1995, 40, 2035–2040 CrossRef CAS.
- M. Kumar and N. Srivastava, Ionics, 2015, 21, 1301–1310 CrossRef CAS.
- B. Jinisha, K. M. Anilkumar, M. Manoj, A. Abhilash, V. S. Pradeep and S. Jayalekshmi, Ionics, 2018, 24, 1675–1683 CrossRef CAS.
- J. W. Fergus, Solid State Ionics, 2012, 227, 102–112 CrossRef CAS.
- D. Kumar, K. Gohel, D. K. Kanchan and K. Mishra, J. Mater. Sci.: Mater. Electron., 2020, 31, 13249–13260 CrossRef CAS.
- A. Arya and A. L. Sharma, J. Solid State Electrochem., 2018, 22, 2725–2745 CrossRef CAS.
- A. Y. S. Eng, C. B. Soni, Y. Lum, E. Khoo, Z. Yao, S. K. Vineeth, V. Kumar, J. Lu, C. S. Johnson, C. Wolverton and Z. W. Seh, Sci. Adv., 2022, 8 DOI:10.1126/sciadv.abm2422.
- M. A. Webb, Y. Jung, D. M. Pesko, B. M. Savoie, U. Yamamoto, G. W. Coates, N. P. Balsara, Z.-G. Wang and T. F. Miller, ACS Cent. Sci., 2015, 1, 198–205 CrossRef CAS PubMed.
- R. V. Gadhave, S. K. Vineeth, P. V. Dhawale and P. T. Gadekar, Des. Monomers Polym., 2020, 23, 188–196 CrossRef CAS PubMed.
- H. Verma, K. Mishra and D. K. Rai, Mater. Today Proc., 2020, 28, 346–349 CrossRef CAS.
- L. Zhou, N. Wu, Q. Cao, B. Jing, X. Wang, Q. Wang and H. Kuang, Solid State Ionics, 2013, 249–250, 93–97 CrossRef CAS.
- N. Wu, Q. Cao, X. Wang, S. Li, X. Li and H. Deng, J. Power Sources, 2011, 196, 9751–9756 CrossRef CAS.
- D. Kumar, M. Suleman and S. A. Hashmi, Solid State Ionics, 2011, 202, 45–53 CrossRef CAS.
- R. Sathiyamoorthi, R. Chandrasekaran, S. Selladurai and T. Vasudevan, Ionics (Kiel)., 2003, 9, 404–410 CrossRef CAS.
- Y. Zheng, Q. Pan, M. Clites, B. W. Byles, E. Pomerantseva and C. Y. Li, Adv. Energy Mater., 2018, 8, 1801885 CrossRef.
- P. Raghavan, X. Zhao, C. Shin, D.-H. Baek, J.-W. Choi, J. Manuel, M.-Y. Heo, J.-H. Ahn and C. Nah, J. Power Sources, 2010, 195, 6088–6094 CrossRef CAS.
- P. Raghavan, X. Zhao, J.-K. Kim, J. Manuel, G. S. Chauhan, J.-H. Ahn and C. Nah, Electrochim. Acta, 2008, 54, 228–234 CrossRef CAS.
- A. P. Vijaya Kumar Saroja, A. Rajamani, K. Muthusamy and R. Sundara, Adv. Mater. Interfaces, 2019, 6, 1901497 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |