
Synergistic complexation of phenol functionalized polymer induced in situ microfiber formation for 3D printing of marine-based hydrogels†
Hafez
Jafari
 *a,
Christine
Delporte
b,
Katrien V.
Bernaerts
c,
Houman
Alimoradi
d,
Lei
Nie
e,
Daria
Podstawczyk
f,
Kam Chiu
Tam
*g and
Amin
Shavandi
*a
*a,
Christine
Delporte
b,
Katrien V.
Bernaerts
c,
Houman
Alimoradi
d,
Lei
Nie
e,
Daria
Podstawczyk
f,
Kam Chiu
Tam
*g and
Amin
Shavandi
*a
aUniversité libre de Bruxelles (ULB), École polytechnique de Bruxelles – BioMatter unit, Avenue F.D. Roosevelt, 50 – CP 165/61, 1050 Brussels, Belgium. E-mail: Hafez.Jafari@ulb.be; amin.shavandi@ulb.be; Tel: +326503681
bLaboratory of Pathophysiological and Nutritional Biochemistry, Faculty of Medicine, Université libre de Bruxelles (ULB), Route de Lennik, 808 – CP611, 1070 Brussels, Belgium
cMaastricht University, Aachen-Maastricht Institute for Biobased Materials (AMIBM), Brightlands Chemelot campus, Urmonderbaan 22, 6167 RD Geleen, the Netherlands
dSchool of Biomedical Sciences, University of Otago, Dunedin, New Zealand
eCollege of Life Sciences, Xinyang Normal University (XYNU), Xinyang 464000, China
fDepartment of Process Engineering and Technology of Polymer and Carbon Materials, Faculty of Chemistry, Wroclaw University of Science and Technology, Norwida 4/6, Wroclaw, 50–373 Poland
gDepartment of Chemical Engineering, Waterloo Institute for Nanotechnology, University of Waterloo, 200 University Avenue West, Waterloo, Ontario N2L 3G1, Canada. E-mail: mkctam@uwaterloo.ca
First published on 5th January 2022
Abstract
The design of 3D printable bio-based hydrogels with enhanced mechanical properties and minimal chemical modification can open new opportunities in the field of biomedical applications. A facile and safe approach is proposed to prepare mechanically reinforced chitosan-based hydrogels via a phenolated polyelectrolyte complex (PHEC) and enzyme-mediated crosslinking. PHEC was formed between phenolated chitosan and alginate, leading to the formation of in situ phenol-functionalized microfibers that exhibited excellent 3D printability. The synergistic complexation enhanced the loss modulus (60 times), toughness, flexibility, and moldability of hydrogel as well as dynamic viscosity (20 times) of the hydrogel precursor compared to individual phenolated chitosan and alginate hydrogels. This complexation endowed the material with excellent printability without sacrificing the hydrogel's elasticity. This study proposes a strategy to design tough and 3D printable marine-based hydrogels based on the synergistic complexation of a phenolated polyelectrolyte complex and enzyme-mediated crosslinking.
Introduction
In recent years, there has been increasing interest in developing bioprintable hydrogels for tissue engineering and wound healing applications.1 However, biopolymer-based hydrogels generally lack the required resilience and toughness for biomedical applications, such as tissue regeneration, wound healing, and wearable sensors.2 The toughness and viscoelasticity of hydrogels can be improved by incorporating high aspect ratio fillers such as microfibres and nanowhiskers.3 Another way of improving the mechanical properties of biobased hydrogels is combining additional covalent or physical interactions or synthetic polymers to form a tough and flexible hydrogel by increasing the energy dissipation of hydrogels.4,5 Among the biopolymers, polysaccharides such as chitin, chitosan, alginate, hyaluronic acid, and dextran have been widely used to develop bio-based hydrogels due to their biocompatibility and biodegradability.6However, natural polysaccharides have poor mechanical strength, low elasticity, easy breakability, and brittleness which hamper the biomedical application of natural hydrogels. Chitosan is extensively used in hydrogel development for biomaterials engineering with inherent biological properties such as antioxidant, antibacterial, and anti-inflammatory activity.7–9 However, in addition to low solubility, the poly-β-(1,4)-D-glucosamine structure endows the chitosan hydrogel with high rigidity and an unsatisfactory energy dissipation mechanism resulting in brittle hydrogels.10–12
The introduction of reversible interactions as secondary crosslinks to chitosan hydrogels can resolve the limitation caused by the polysaccharides’ rigidity. For example, Xu et al. developed a series of chitosan/vanillin hydrogels originating from reversible Schiff base reactions between the aldehyde group of vanillin and the amino group of chitosan.13 However, the Schiff-base linkage can be hydrolyzed under acidic conditions, which hampers its applicability.14 Besides, Zhou et al. developed a tough and self-healable chitosan/polyacrylic acid hydrogel via the conjugation of quaternary ammonium groups onto the chitosan backbone.15 However, the time-consuming grafting process of the quaternized amino groups with extensive use of chemicals such as sodium bicarbonate, sodium borohydride, and methanol as well as the low biocompatibility of polyacrylic acid hindered the biomedical application of the hydrogel.10
Furthermore, several methods have been reported on the 3D printability of chitosan-based hydrogels. Wu et al. reported a solvent evaporation method for 3D printing of chitosan pre-solution in acidic media.16 Zhou et al. printed chitosan pre-solution (alkali solution) using a high-temperature method.17 However, the long fabrication time and the use of acidic or alkaline media to dissolve the chitosan pre-gel solution in these methods render it unsuitable for direct cell encapsulation and biomedical application. Alternatively, Liu et al. reported a 3D printed chitosan-based hydrogel using photocrosslinking of phenol functionalized chitosan and dibenzaldehyde-terminated telechelic poly(ethylene glycol) (DF-PEG) pre-gel crosslinked by dynamic imine bonds.18 However, using synthetic polymers and extensive chemical modification to prepare DF-PEG does not involve green chemistry and hinders biomedical applications. Hence, more green approaches should be considered to develop chitosan-based hydrogels for 3D printing without using toxic chemicals, solvents, and extensive modification for clinical applications such as wound healing, tissue engineering, drug delivery, and biosensor applications.
To address these limitations, we have developed a tough and self-healable chitosan-based hydrogel via a green and sustainable approach by taking advantage of chitosan's inherent cationic nature, enabling chitosan to form a dynamic reversible electrostatic interaction with an anionic polymer. Hence, a dual crosslinked hydrogel via the synergy of horseradish peroxidase (HRP) mediated crosslinking and the phenolated polyelectrolyte complexation (PHEC) between the cationic chitosan and anionic alginate was developed. We hypothesize that the brittleness and rigid structure of the hydrogel resulting from the primary enzymatic crosslinking can be addressed by incorporating weak and reversible electrostatic interactions as well as in situ microfiber formation using PHEC, which is responsible for the toughness and flexibility of the hydrogel as a sacrificial bond for energy dissipation.19–21 The in situ microfiber formation is expected to synergistically reinforce the hydrogel mechanical stability and increase the gel viscosity endowing the hydrogel with excellent 3D printability.
Results
The design rationale of the hydrogel
To prepare the phenolated polyelectrolyte complex (PHEC), phenolic compounds were conjugated to the chitosan and alginate backbone by conjugation with 3-(4-hydroxyphenyl) propionic acid (HPA) and tyramine, respectively, via carbodiimide coupling chemistry (Fig. S1 and 2†). Moreover, phenolic compounds are required for hydrogel development via enzyme-mediated crosslinking using horseradish peroxidase (HRP).22 The formation of chitosan-phenol (Ch-Ph) and alginate-tyramine (Alg-Ty) was confirmed by 1HNMR and UV-Vis spectra of the purified samples (Fig. S1 and 2†). In 1H NMR, the presence of new peaks at around 7 ppm belonging to aromatic protons confirmed the presence of aromatic protons in the structure of the polymers.23 Similarly, chitosan and alginate have no absorption at 275 nm; however, adding a phenolic group to the structure gives Ch-Ph and Alg-Ty a maximum absorbance at that wavelength.24We then developed a series of hydrogels based on Ch-Ph solution, Alg-Ty solution, and the PHEC suspension. To this end, hydrogel precursors containing different concentrations of Ch-Ph or Alg-Ty (Table S1†) were mixed with HRP and H2O2. HRP is activated by H2O2, which catalyzes the oxidation of the phenolic groups in the chitosan or alginate chains, generating two phenoxy radicals in one catalytic cycle. The phenoxy radicals can then react with each other through a radical coupling reaction resulting in C–C and C–O bonds leading to the formation of single crosslinked chitosan and alginate hydrogel via covalent bonding.22
For the double crosslinked chitosan alginate hydrogel (DCCA), the Alg-Ty solution was added dropwise to Ch-Ph with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 under vigorous agitation and multidirectional mixing to obtain a viscous PHEC suspension. Given the difference in the charge density between positively charged amino groups of chitosan and carboxyl groups of alginate, the electrostatic interaction simultaneously occurred upon mixing the two solutions, leading to the formation of a polyion complex followed by a physically crosslinked weak hydrogel.25 Vigorous shear agitation of the weak hydrogel led to the breakage of the hydrogel and in situ microfiber formation (Fig. 1a), resulting in a heterogenous PHEC suspension with significantly higher viscosity (20 times) compared to the Ch-Ph and Alg-Ty solutions (Fig. 2a). Generally, polyelectrolyte complex between chitosan and alginate led to the formation of microfibers at acidic and neutral pH, and microparticle under alkaline conditions (pH 8.5) and the fiber surface charge undergoes a transition from positive to the negative surface by increasing the pH from acidic (pH 3.5) to alkaline conditions (pH 8.5).26 Indeed, instantaneous complexation led to a microfibrous colloidal suspension with hierarchical morphology with micron-scale fibers branching into thin fibers at natural pH (Fig. 1a).
:1 under vigorous agitation and multidirectional mixing to obtain a viscous PHEC suspension. Given the difference in the charge density between positively charged amino groups of chitosan and carboxyl groups of alginate, the electrostatic interaction simultaneously occurred upon mixing the two solutions, leading to the formation of a polyion complex followed by a physically crosslinked weak hydrogel.25 Vigorous shear agitation of the weak hydrogel led to the breakage of the hydrogel and in situ microfiber formation (Fig. 1a), resulting in a heterogenous PHEC suspension with significantly higher viscosity (20 times) compared to the Ch-Ph and Alg-Ty solutions (Fig. 2a). Generally, polyelectrolyte complex between chitosan and alginate led to the formation of microfibers at acidic and neutral pH, and microparticle under alkaline conditions (pH 8.5) and the fiber surface charge undergoes a transition from positive to the negative surface by increasing the pH from acidic (pH 3.5) to alkaline conditions (pH 8.5).26 Indeed, instantaneous complexation led to a microfibrous colloidal suspension with hierarchical morphology with micron-scale fibers branching into thin fibers at natural pH (Fig. 1a).
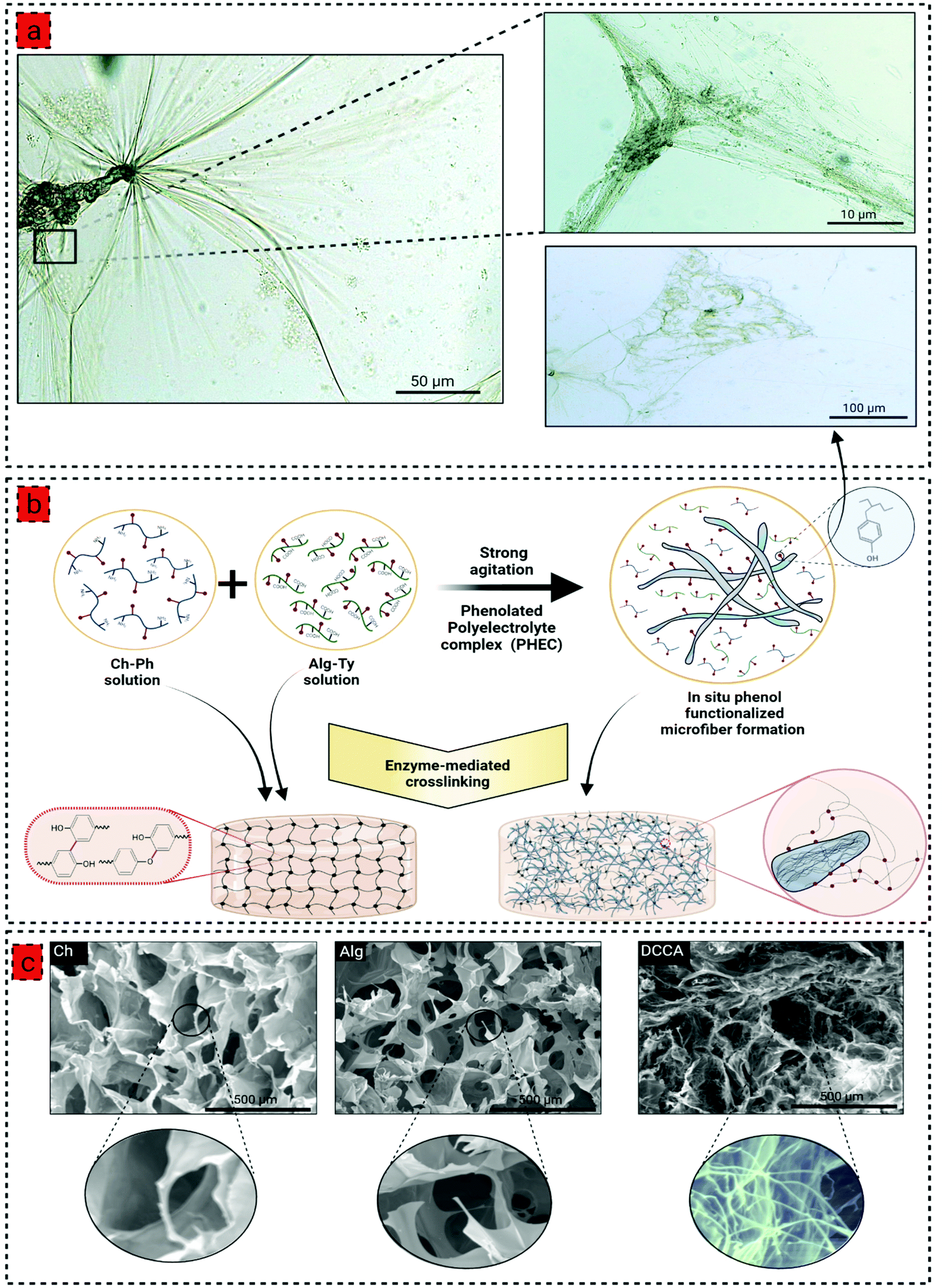 | ||
| Fig. 1 (a) Optical microscopy images of in situ phenol functionalized microfiber formation by phenolated polyelectrolyte complex (PHEC), showing a hierarchical morphology of microfibers, (b) schematic illustration of phenol functionalized microfiber formation via the PHEC formation between the Ch-Ph and Alg-Ty solution, and subsequent hydrogel formation by enzyme-mediated crosslinking using horseradish peroxidase (HRP) and hydrogen peroxide (H2O2), (c) the scanning electron microscopy (SEM) images of Ch, Alg, and DCCA hydrogels illustrating a compact and heterogenous microstructure of DCCA hydrogel compared to Ch and Alg hydrogels due to formation of in situ phenol functionalized microfibers induced by PHEC; the figure was created with BioRender.com. | ||
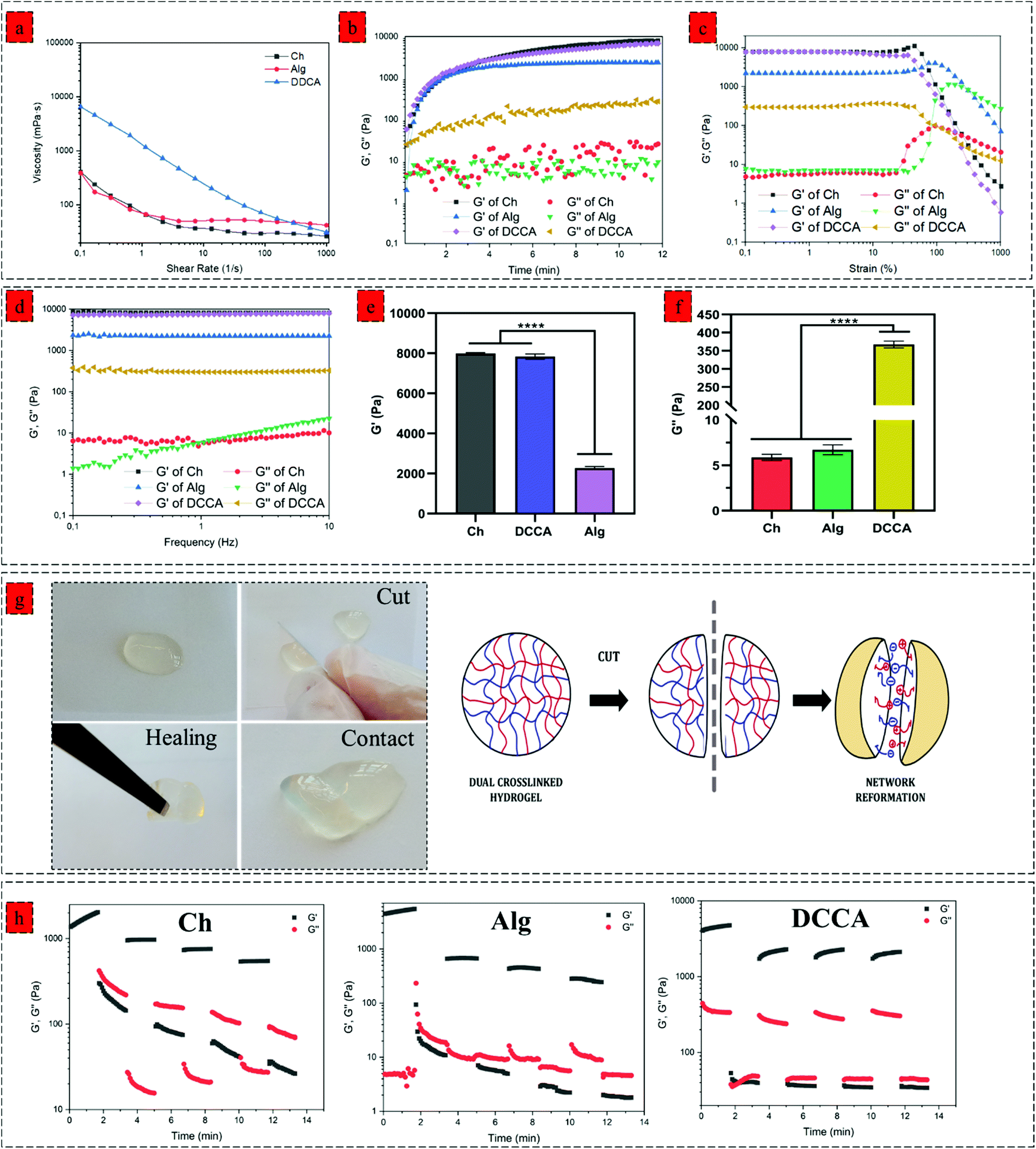 | ||
| Fig. 2 Rheological properties of the hydrogels. (a) Shear-rate dependent variations gel precursors viscosity over the shear rate of 0.1–1000 1 s−1 at 37 °C, (b) gelling kinetics of the Ch, Alg and DCCA hydrogel investigated by a time sweep test at a constant strain of 0.1% and frequency of 1 Hz at 37 °C, (c) storage modulus (G′) and loss modulus (G′′)–strain dependence of Ch, Alg and DCCA hydrogels at a constant frequency of 1 Hz at 37 °C, (d) G′ and G′′–frequency dependence Ch, Alg and DCCA hydrogels at a constant strain of 1% at 37 °C, (e) mean value of hydrogels G′ at linear viscoelastic region (LVR), (f) mean value of hydrogels G′′ at linear viscoelastic region (LVR), (g) macroscopical observation of self-healing behaviour DCCA hydrogel and the self-healing mechanism of DCCA hydrogels based on the dynamic electrostatic interactions, (h) self-healing capability of Ch, Alg and DCCA hydrogels evaluated by 4 cycle step-strain test with 100 s time interval for each step (strain = 1%/300% /1%…). Data were analyzed using a one-way ANOVA test. ****p < 0.0001. | ||
Interestingly, we hypothesized that the surface of the in situ microfibers are functionalized with phenol groups, letting the microfibers to participate in the enzyme-mediated crosslinking with each other and the phenolated chitosan and alginate (Fig. 1b). Then, the PEC suspension with different concentrations of Ch-Ph and Alg-Ty (Table S1†) was used for the hydrogel formation via the enzyme-mediated crosslinking using HRP and H2O2. The gelation time was determined using the vial tilting method; all hydrogels exhibited a concentration-dependent gelation time (Fig. S4b†) with a controllable sol–gel transition from approximately 2 min to a few seconds, indicating that the polymer (phenol group) concentration significantly affects the gelation time due to availability of higher phenolic groups for the enzyme-mediated crosslinking.27 Although the in situ microfiber formation increased the viscosity of the gel precursors, the gelation times of hydrogels were not affected by this phenomenon (Table S1† and Fig. 4b). These results suggested that the PHEC formation did not influence the velocity of the HRP-catalyzed crosslinking reaction and phenol group mobility.28 Hence, the electrostatic interaction did not interfere with the HRP mediated cross-linking. Finally, Ch1.5 (named Ch), Alg1.5 (named Alg), and a dual crosslinked Ch1.5–Alg1.5 hydrogel (named DCCA) presenting the shortest gelation time were chosen for further characterization to examine the effect of PHEC on the mechanical properties of the hydrogels.
Physiochemical characterization
Before investigating the PHEC effect on the viscoelastic properties, PHEC formation was investigated by FTIR (Fig. S5†) and XRD (Fig. S6†) on freeze-dried Ch, Alg, and DCCA hydrogels. Both Ch and Alg hydrogels demonstrated typical FTIR bands of the polymers (Fig. S5†). The DCCA hydrogel showed a similar spectrum as the Ch and Alg; however, the amide I peak of Ch had disappeared and a new sharp peak showed overlapping with carboxylate groups of alginate at 1567 cm−1 which was shifted from 1557 cm−1 in the Alg spectra. Moreover, the carboxylate stretch peak of Alg shifted from 1397 to 1406 after the PHEC formation. These shifts in the assigned bands of alginate carboxylic groups and chitosan amino groups could account for the electrostatic formation.25 Furthermore, the XRD pattern of the freeze-dried hydrogels, chitosan, and alginate powder revealed that phenol modification of chitosan and alginate reduced the intensity of semicrystalline peaks of chitosan and alginate (at around 12° and 20°).This phenomenon could be due to the reduction in the inter-and intramolecular hydrogen bonds of chitosan that reduced the crystallinity, in agreement with the water solubility of chitosan after conjugation.29 Besides, the peaks intensity was further reduced in the DCCA hydrogel, indicating that PHEC formation further decreased the crystallinity due to the formation of electrostatic interactions resulting in the breakage of intermolecular hydrogen bonding.29
The morphological investigation of the hydrogel showed a typical porous microstructure for Ch and Alg hydrogels with an average pore size of 258 ± 53 μm 297 ± 13 μm (Fig. 1c). However, due to the in situ microfiber formation, the DCCA hydrogel exhibited a denser microstructure with a hierarchical morphology containing large pores filled with a fibrous structure distributed throughout the hydrogel. The microfibers possessed an average diameter of 1.3 ± 0.4 μm (Fig. 1c), which could reinforce the mechanical properties of the hydrogel.30 The microstructure investigation demonstrated that the fiber-like structure is preserved, indicating the stability of in situ microfibers upon enzyme-mediated crosslinking even after the freeze-drying process.
We investigated the effect of PEC on the swelling rate and degradation behaviour of the hydrogels. PHEC formation reduced the swelling ratio of Alg hydrogel from 311 ± 15% to 47 ± 9% (Fig. S7†). Besides, it prolonged the degradation compared to the Ch and Alg hydrogel in lysozyme solution (Fig. S8†) due to the formation of a dense network with a high crosslinking density, which results in a smaller pore size filled with microfibers. The results showed that the PHEC formation could significantly affect the hydrogel's swelling ratio and degradation behaviour.
Viscoelastic properties of hydrogels
We next investigated the effect of complexation on the viscoelastic properties of the hydrogel. We characterized the dynamic viscosity and shear thinning properties of the hydrogel precursors (Fig. 2a). DCCA hydrogel precursor exhibited 20 times higher dynamic viscosity (6500 mPa S) compared to the Ch and Alg hydrogel precursors indicating the significant effect of in situ microfibers on the viscosity of the hydrogel precursors. More importantly, the shear-thinning behaviour of the DCCA hydrogel precursors was enhanced due to the dynamic nature of non-covalent electrostatic interaction, which dissociated under the applied shear and subsequent network recovery following the shear removal.31,32We monitored the gelation kinetics via a time sweep test (Fig. 2b). The Ch and Alg hydrogel showed a gelation point at around 40 s. However, the gelation point was not observed for the DCCA hydrogel due to its higher G′ than the G′′ at the beginning, showing a solid-like behaviour expected due to the PHEC formation. However, after initializing the HRP-mediated crosslinking, the G′ was further increased up to 12 min, similar to the Ch and Alg hydrogel indicating the solidification of the hydrogels. The G′ of the DDCA approached 7.1 kPa, which is 50 times higher than previously reported studies on chitosan and alginate polyelectrolyte complex hydrogel.25,33 These data suggest that the enzymatic crosslinking significantly improved the stiffness of the hydrogel. Furthermore, an amplitude sweep test (constant frequency of 0.1 Hz) (Fig. 2c) and a frequency sweep (0.1 to 10 Hz, at 1% strain) (Fig. 2d) were performed to evaluate the viscoelastic properties of the hydrogels. All hydrogels exhibited strain-independent G′ and a stable structure up to 200% strain showing a wide range of linear viscoelastic region (LVR) and confirming the elasticity and stability of the hydrogels.34,35
The mean values of G′ and G′′ of hydrogels at the linear viscoelastic region (LVR) are shown in Fig. 2e and f, respectively. The Ch hydrogel showed the highest G′ value (7.9 kPa), while the Alg hydrogel exhibited the lowest (2.3 kPa) value. Interestingly, the DCCA hydrogel revealed a high G′ (7.1 kPa) without any significant difference from the Ch hydrogel indicating the significant role of microfibers in the stiffness of the hydrogel. Ch and Alg hydrogels showed a G′′ value of around 7 Pa, while the DCCA hydrogel exhibited a significantly higher G′′ value of around 370 Pa (Fig. 2f). Although G′ of DCCA was close to the Ch hydrogel, its G′′ was 30 times higher than that of Ch and Alg, indicating significantly higher energy dissipation during the deformation of the DCCA hydrogel. In our previous study, the addition of silk fibroin with lower G′ compared to that of chitosan significantly lowered the G′ of the hydrogel (3 times).36 However, in this study, the in situ microfibers could maintain the hydrogel elasticity in addition to increasing the G′′ of the DCCA.
The hydrogels’ loss factor (tanδ) was measured from the frequency test at 0.1 Hz, indicating how a material can absorb and dissipate energy in response to deformation.37 Tanδ for the Ch and Alg hydrogels were around 0.001, indicating that the Ch and Alg hydrogels were highly elastic and brittle, while DCCA hydrogel exhibited a tanδ of 0.06, showing that the DCCA hydrogel is much more viscous than the Ch, and Alg hydrogels which can dissipate energy during the deformation.38
This phenomenon is due to the synergy of covalent enzymatic crosslinking of a brittle irreversible bond and dynamic non-covalent electrostatic interaction as a weak and recoverable bond.39,40 The uniformly distributed microfibers limit the amount of stress accumulation in the structure and therefore act as sacrificial bonds that enable energy dissipation and improve the resilience of the hydrogel.39,40 Hence, the DCCA hydrogels revealed high flexibility and toughness without sacrificing the stiffness of the hydrogel thanks to the PHEC and phenol functionalized microfibers without using any synthetic polymer.
Self-healing and compression properties of hydrogels
We investigated the effect of complexation on the self-healing property of the hydrogel via macroscopic observations and rheological investigations. The hydrogel was cut into two pieces, and then the two pieces of hydrogels were contacted to examine their self-healing property. After a few minutes, the hydrogel was lifted using a tweezer (Fig. 2g). The two pieces stayed together, showing that new bonding had occurred and healed the fractured network. This self-healing capability is due to the interpolymer complex driven by the electrostatic interaction between the negatively charged alginate's carboxyl group and the positively charged chitosan's amino group. The dynamic interaction between these functional groups regenerated upon sealing the gap, showing the reversibility of these interactions.41Moreover, a rheological investigation of self-healing properties of Ch, Alg, and DCCA hydrogels was performed (Fig. 2h) using a 4 cycle step–strain test. At each cycle, a 300% strain was applied for 100 s to break the gel. The recovery cycle of 100 s at a strain of 1% was used to allow the reformation of the network. Both Ch and Alg hydrogels showed a significant reduction in G′ (10 times) after the first cycle, indicating that the applied strain damaged the hydrogel network without any recovery due to the presence of only non-dynamic covalent bonds. The DCCA hydrogel showed a complete sol–gel transition during the first interval of 300% strain. Interestingly, after the first cycle, the G′ of DCCA was recovered to 2.2 kPa (60% of initial G′) and showed an increasing trend with time, indicating that the DCCA hydrogel could recover the G′. The G′ of Ch and Alg decreased continuously after each cycle. The G′ values at each cycle were constant, showing the inability of the hydrogel to recover the broken bonds, while DCCA hydrogel could recover 60% of the G′.
Furthermore, it is worth mentioning that the ascending trend of the G′ value of DCCA during the relaxation cycle indicates that the hydrogel probably needs more time to recover the higher amount of the broken bonds. Indeed, the covalent phenolic network of the DCCA hydrogel could preserve the structure from the nonrecoverable damages, while the dynamic chain entanglement and electrostatic interactions broke quickly and acted as sacrificial bonds to dissipate energy resulting in the self-healing ability after the deformation.10 Furthermore, the loss modulus of DCCA was 30 times higher than for the other hydrogels confirming the previous results regarding the high loss factor representing the toughness and flexible structure of the DCCA hydrogel.
Further examination of the toughness and deformation resistance of the hydrogels was performed using a compression test (Fig. 3) by recording the force until the breakage of the hydrogel using a constant pressure (1 mm min−1). The compressive strain–stress curve demonstrates the brittleness of the Ch hydrogel compared to Alg and DCCA hydrogels, indicating the brittle and stiff structure of the Ch hydrogel (Fig. 3a). DCCA hydrogel showed the highest fracture strain (55%) compared to the Ch (41%) and Alg (48%) hydrogels. Moreover, an investigation of the compressive modulus of the hydrogels (Fig. 3b) revealed that the DCCA hydrogel exhibited a compressive modulus of 1.37 ± 0.05 kPa without any significant difference compared to Ch hydrogel with the highest modulus (1.51 ± 0.07 kPa). In comparison, Alg hydrogel showed a compressive modulus of 0.43 ± 0.06 kPa, which is in line with the rheological results.
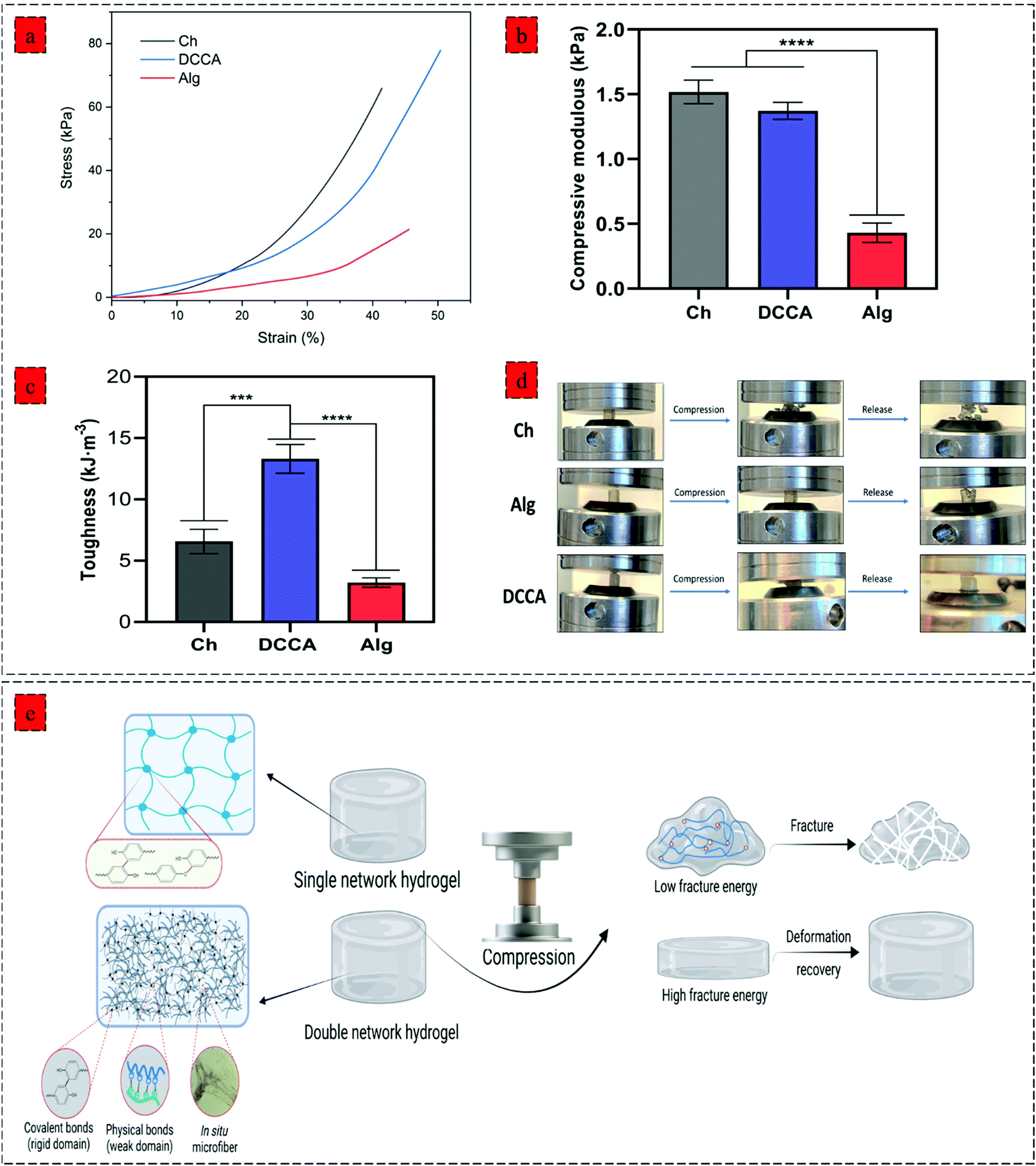 | ||
| Fig. 3 Mechanical properties of hydrogels. (a) compression stress–strain curves of hydrogels, (b) compressive modulus of hydrogels calculating from the stress–strain curves (strain 10–15%), (c) the corresponding dissipated energy of hydrogels Data were analyzed using a one-way ANOVA test. ***p < 0.0005; ****p < 0.0001. (d) photographs of Ch, Alg, and DCCA hydrogels showing their compressibility and elasticity under compression, (e) schematic illustration of hydrogel deformation recovery after the compression showing destruction of Ch and Alg hydrogel due to their single covalent network and high deformation recovery capability of DCCA hydrogel due to the synergistic reinforcement effect of PHEC by inducting electrostatic interaction and in situ microfibers; the figure was created with BioRender.com. | ||
Furthermore, the toughness of the hydrogels indicating their capacity to absorb mechanical energy was measured by integrating the compressive stress–strain curve12 (Fig. 3c). The DCCA hydrogel showed a significantly higher toughness (13.3 ± 0.9 kJ m−3) compared to Ch (6.5 ± 0.8 kJ m−3) and Alg (3.2 ± 0.3 kJ m−3) hydrogels. Our results showed that the synergistic reinforcement in the DCCA hydrogel could increase the hydrogel's toughness and deformability without sacrificing the hydrogel's stiffness. Unlike Ch and Alg hydrogel, the DCCA hydrogel could recover the deformation after the compression, while both Ch and Alg hydrogels were completely broken without any recovery (Fig. 3d and 4a). This phenomenon is due to the synergistic complexation induced by PHEC via microfiber formation, which could increase the hydrogel's stiffness and significantly improve the energy dissipation leading to delay in the network fracture42 (Fig. 3e). Hence, the results showed that a straightforward mixing of Ch and Alg hydrogel could significantly improve the toughness and flexibility without requiring additional synthetic polymers.
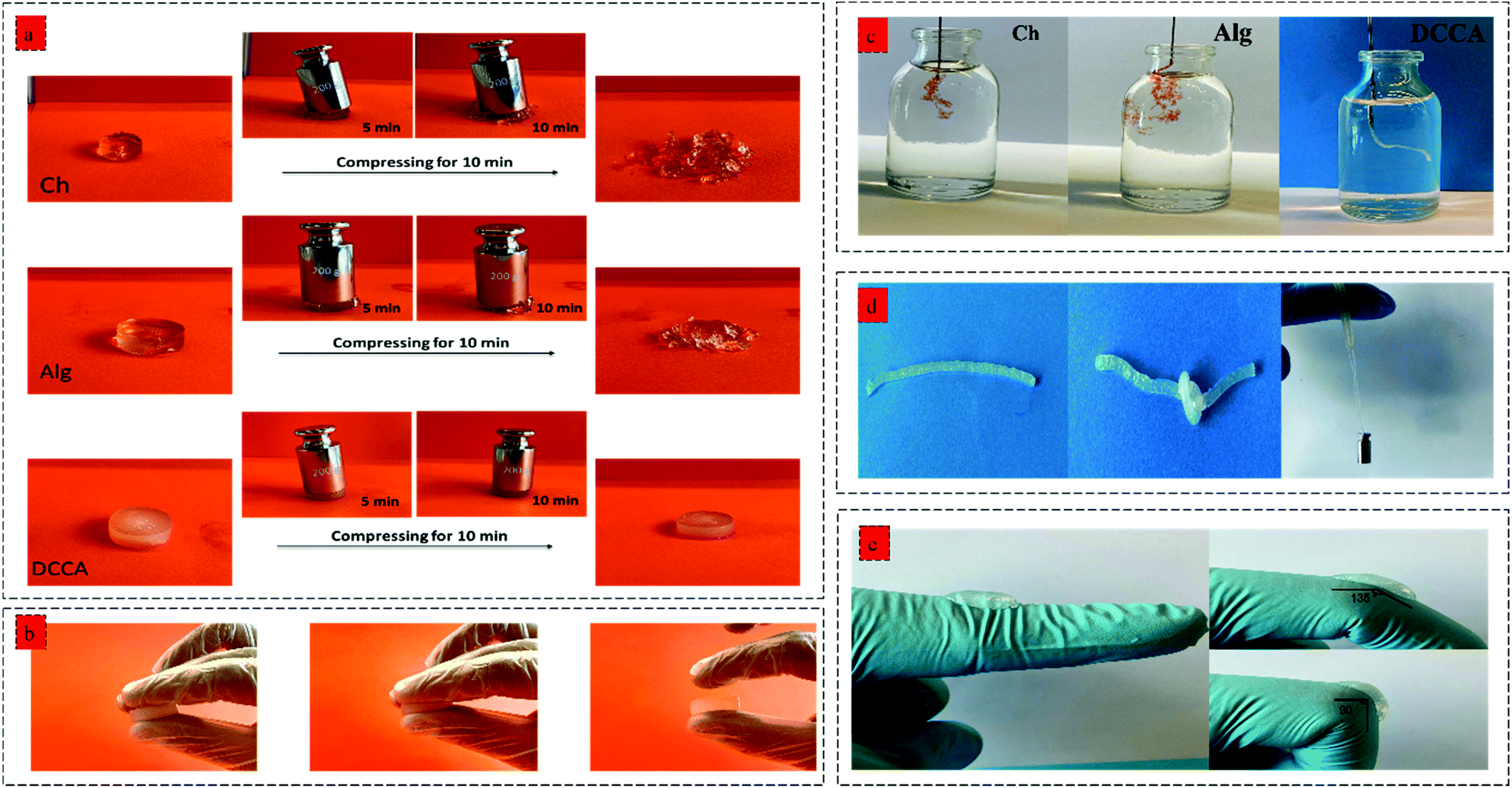 | ||
| Fig. 4 (a) Deformation capability of Ch, Alg and DCCA hydrogels after being pressed by the 200 g weight for 10 min, and (b) deformation resistance of DCCA hydrogel by pressing between fingers, (c) injectability of Ch, Alg, and DCCA evaluation using a syringe with 30 G needles, (d) foldability and knotting ability of DCCA hydrogel and high mechanical flexibility could sustain a 50 g weight, (e) the DCCA hydrogel adherence to a knuckle which was repeatedly bent from 180° to 90° in the flexibility test 50 times to examine the flexibility and adhesion performance. | ||
The hydrogels’ injectability was investigated by loading the gel precursors into a syringe and evaluating the injection capability by extruding the gel precursors through a 30 G needle into PBS (Fig. 4b). Both Ch and Alg hydrogels did not show a successful injection into the PBS buffer, and the gel was broken after the injection without any recovery due to the rigid and brittle structure of the hydrogels. However, the DCCA hydrogel could be extruded through the needle due to the presence of weak electrostatic interactions endowing the hydrogel with self-recovery capability.43 Indeed, due to the reversible nature of these weak physical interactions, the formed network can be broken and reformed when the shear caused by the injection through the needle was removed.44
Moreover, the DCCA hydrogel could exhibit good flexibility by knotting the hydrogel and could sustain a 50 g weight for more than 30 min indicating high mechanical strength and flexibility of the hydrogel (Fig. 4d), which was not observed in the Ch and Alg hydrogel due to their brittle and non-reversible covalent bonds. Besides, the adhesive performance of the DCCA hydrogel was tested by bending the hydrogel to a knuckle of more than 50 elongation/recovery cycles (Fig. 4e). During the elongation/recovery cycles, the DCCA hydrogel could maintain its structure while the hydrogel's adhesiveness prevented it from falling off due to the presence of electrostatic interaction between the polymers.45
Our results demonstrated that DCCA hydrogel has high mechanical strength, toughness, flexibility, and moldability as an injectable hydrogel for clinical applications with a minimally invasive approach for regenerating irregular-shaped defects, particularly wound healing applications. As a biopolymer-based hydrogel without using any synthetic polymer or harmful crosslinking agent, the approach used in this study could open new insights into the design and development of bio-based hydrogels for biomedical applications.
Biological properties
We evaluated the hydrogels’ biological activity to examine the effect of PHEC on the antioxidant, antibacterial, and cell proliferation capacity of the hydrogel. The DPPH scavenging evaluation (Fig. 5a) revealed a scavenging activity of 51.3 ± 3.5% for Ch, 37.9 ± 2.6% for Alg and 44.4 ± 6.4% for DCCA, demonstrating that the PHEC formation did not have any significant effect (P < 0.05) on the inherent antioxidant activity of chitosan.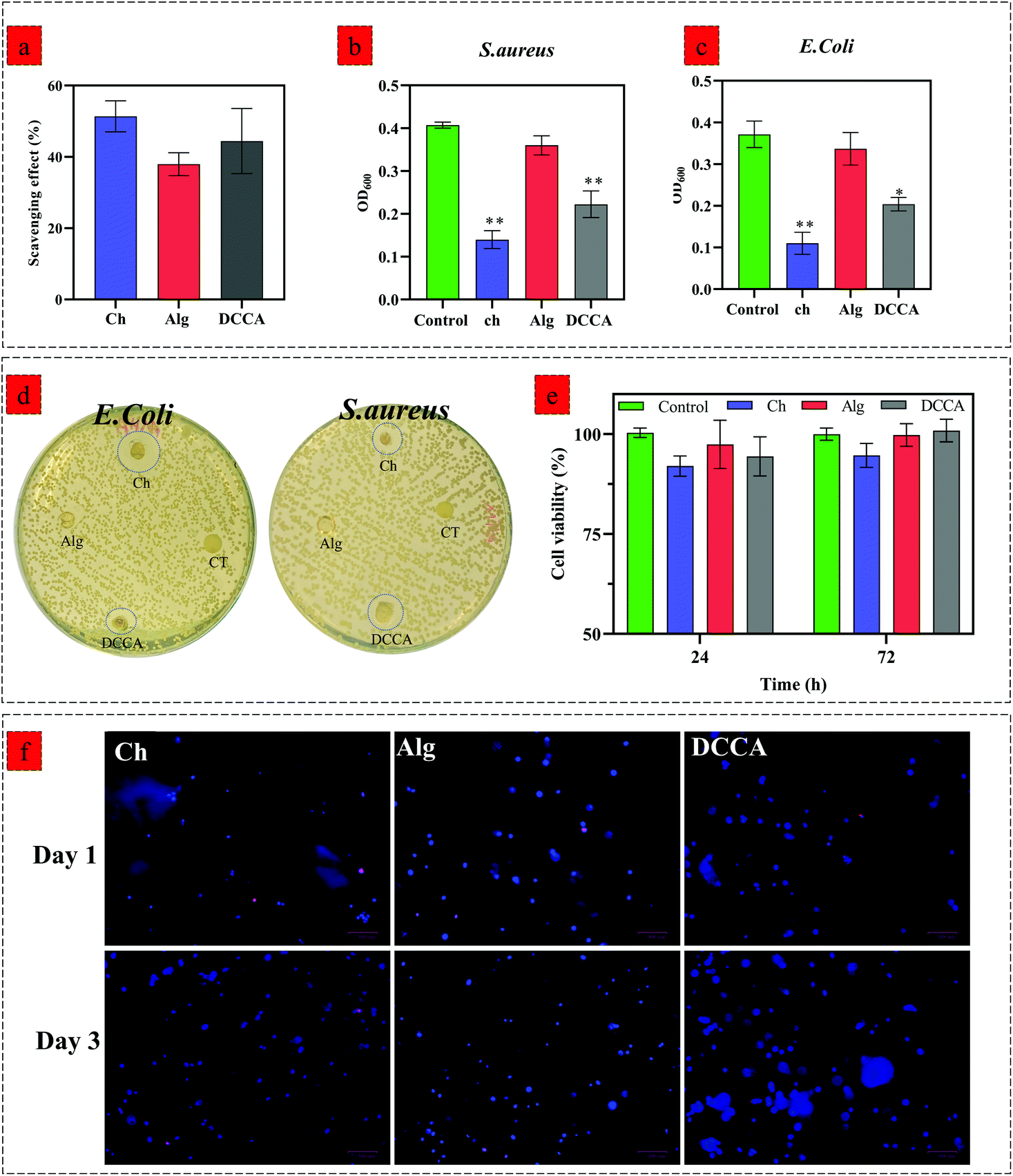 | ||
| Fig. 5 (a) DDPH scavenging effect of Ch, Alg and DCCA hydrogels, results are expressed as % scavenging activity and are the mean ± SD of three independent experiments, (b and c) bacterial growth of E. coli (b) and S. aureus (c) treaded by Ch, Alg, and DCCA hydrogels after 24 h incubation at 37 °C, results are expressed as optical density (OD600) and are the mean ± SD of three independent experiments, Data were analyzed using a one-way ANOVA test. *p < 0.05, **p < 0.005, (b) Zone inhibition investigation of E. coli and S. aureus treated by Ch, Alg, and DCCA hydrogels, (e) cell viability of 3T3-L1 cells seeded on the Ch, Alg, and DCCA hydrogels for 24 and 72 h. Results are expressed as % cell viability and are the mean ± SD of three independent experiments. Data were analyzed using a one-way ANOVA test. *p < 0.05 as compared to the control (cell culture media), (f) fluorescent microscopic images of cell-laden Ch, Alg, and DCCA hydrogels. The 3T3-L1 fibroblast was encapsulated into cell-laden hydrogels and was stained via Hoescht and ethidium bromide (dead cells) after 1 and 3 days. | ||
Furthermore, the antibacterial activity of Ch, Alg and DCCA hydrogels was investigated by an inhibition test against S. aureus (Fig. 5b) and E. coli (Fig. 5c) growth via measuring the optical density (OD) of a bacterial suspension at 600 nm and the disk diffusion test (Fig. 5d). The Alg hydrogel did not show any inhibition activity against both Gram-negative and Gram-positive bacteria. The OD of the bacterial suspension treated with Alg hydrogel did not show any significant difference compared to the control groups demonstrating no antibacterial activity of the Alg hydrogel.46 However, the Ch hydrogel exhibited growth inhibition against both Gram-negative and Gram-positive bacteria with a mean OD of 0.11 ± 0.02 and 0.14 ± 0.01 against E. coli and S. aureus, respectively.
The results showed that the Ch hydrogel demonstrated a higher antibacterial activity against E. coli probably due to its cationic nature enabling chitosan to have more antibacterial activity against Gram-negative bacteria.47 DCCA had lower antibacterial activity than the Ch hydrogel; however, the OD of the bacterial suspension treated with DCCA hydrogels is significantly lower than that of the control groups against E. coli (0.2 ± 0.004) and S. aureus (0.22 ± 0.02). Similar to the inhibition growth test, the disk diffusion test, carried out to further investigate the antibacterial activity of the hydrogels, showed no inhibition zone for the Alg hydrogels. However, an inhibition zone was observed for Ch and DCCA hydrogels against both strains of bacteria, which indicated the bactericidal activities of the hydrogels. On the other hand, due to the use of similar H2O2 concentrations for all the hydrogels and because alginate has no antibacterial activity, it was confirmed that H2O2 did not have any effect on the antibacterial activity of Ch and the DCCA hydrogel. Hence, these results revealed that the DCCA hydrogel has antibacterial activity due to the presence of chitosan with inherent microbiocidal properties. Indeed, two antibacterial activity mechanisms can be considered for chitosan-based material. Positively charged chitosan would interact with the negatively charged microbial cell wall.48 For E. coli (Gram-negative), the bacteria possess an outer covering of phospholipids and lipopolysaccharides, leading to a negatively charged surface. The interaction between chitosan and the microbial cell wall leads to leakage of proteinaceous and other intracellular constituents. The second factor is the bonding of chitosan with the DNA of bacteria, resulting in the prevention of transcription and translation by DNA.48,49
We further investigated the effect of Ch, Alg, and DCCA hydrogels on the viability of 3T3-L1 fibroblasts by a direct contact test using an MTS assay after 24 and 72 h of culture (Fig. 5e). No significant effect on the cell viability of 3T3-L1 was observed for the hydrogels compared to the control (cell culture media) (p < 0.05). To further investigate the cell encapsulation capability of hydrogels, 3D encapsulation of 3T3-L1 fibroblast cells into Ch, Alg, and DCCA hydrogels was performed, and the cell nucleus spreading distribution as well as cell viability was evaluated via Hoescht/ethidium bromide staining (Fig. 5f). A uniform spreading distribution of 3T3-L1 fibroblast cells encapsulated into hydrogels was observed after 1 and 3 days of culture, showing the capability of the hydrogels for homogenous cell spreading and proliferation with a low amount of dead cells (stained with ethidium bromide) compared to the live cells (stained by Hoescht). The results indicate that the PHEC formation in the DCCA hydrogel did not adversely affect biological activities. This hydrogel with toughness and flexibility is a promising candidate for 3D cell encapsulation for various biomedical applications such as cell and gene delivery, regenerative medicine, and wound healing.
3D printing
By using a synergistic complexation approach, the properties of the mixture can be precisely controlled to facilitate 3D printing via extrusion, which has traditionally been difficult with hydrogel precursors. For printing of the DCCA hydrogel, we investigated the effect of a higher concentration (2–4%) of PHEC suspension on the printability of the hydrogel. Viscoelastic properties of PHEC suspension (2, 2.5, 3, 3.5, and 4%) were determined, and the results demonstrated that the PHEC suspension with a concentration of 2% had an inadequate viscosity (10 Pa s−1) at a shear rate of 0.1 1 s−1 (Fig. S9a†) and was still in the sol-phase (G′ = 50 Pa, and G′′ = 80 Pa) at 1% strain according to the amplitude test (Fig. S9b†) which is not suitable for 3D printing.50 However, by increasing the PHEC concentration to 2.5%, the suspension exhibited a solid-like behaviour (G′ = 122 Pa, and G′′ = 70 Pa) (Fig. S10b†) with a higher viscosity (17 Pa s) (Fig. S10a†). Indeed, increasing the PHEC concentration led to a higher amount of in situ microfibers with self-assembly features, resulting in the formation of instantaneous solid-like hydrogel upon the addition of phenolated alginate to phenolated chitosan solution (Video S1,† PHEC 3%). Finally, the PHEC suspension with 4% concentration was chosen for 3D printing. It is worth mentioning that no Ch-Ph, Alg-Ty, nor non-phenolated polyelectrolyte complex (PEC) can be used alone as a printing ink at the same concentration of PHEC (4%). As mentioned above, the dynamic viscosity of Ch-Ph and Alg-Ty was not high enough to be printed. On the other hand, we evaluated the viscosity of PEC (Fig. 6b) to examine the effect of phenol modification on the viscoelasticity of the hydrogel precursor. PEC possessed a viscosity of 41 Pa s−1, which was twice lower than the viscosity of PHEC (110 Pa s−1), indicating the significant effect of chitosan and alginate phenol modification in increasing the viscosity of the gel precursor (Video S2†). This phenomenon can be explained by the formation of hydrophobic interactions between the side groups (phenyl groups) of chitosan and alginate, leading to a significant increase in the viscosity of phenolated PEC compared to its non-phenolated counterpart.51 Furthermore, the amplitude sweep test revealed (Fig. 6c) the insufficient stiffness of PEC as standalone ink due to its low G′ (50 Pa) compared to the G′′ (43 Pa) at the strain of 0.1%. However, PHEC possessed a solid-like network with 24 times higher G′ (1200 Pa) compared to the non-phenolated one due to the synergy of electrostatic and hydrophobic interaction at low strain (<1%) and by increasing the strain beyond the yield point, sol-like behaviour was observed resulting in the extruding ability of the PHEC suspension.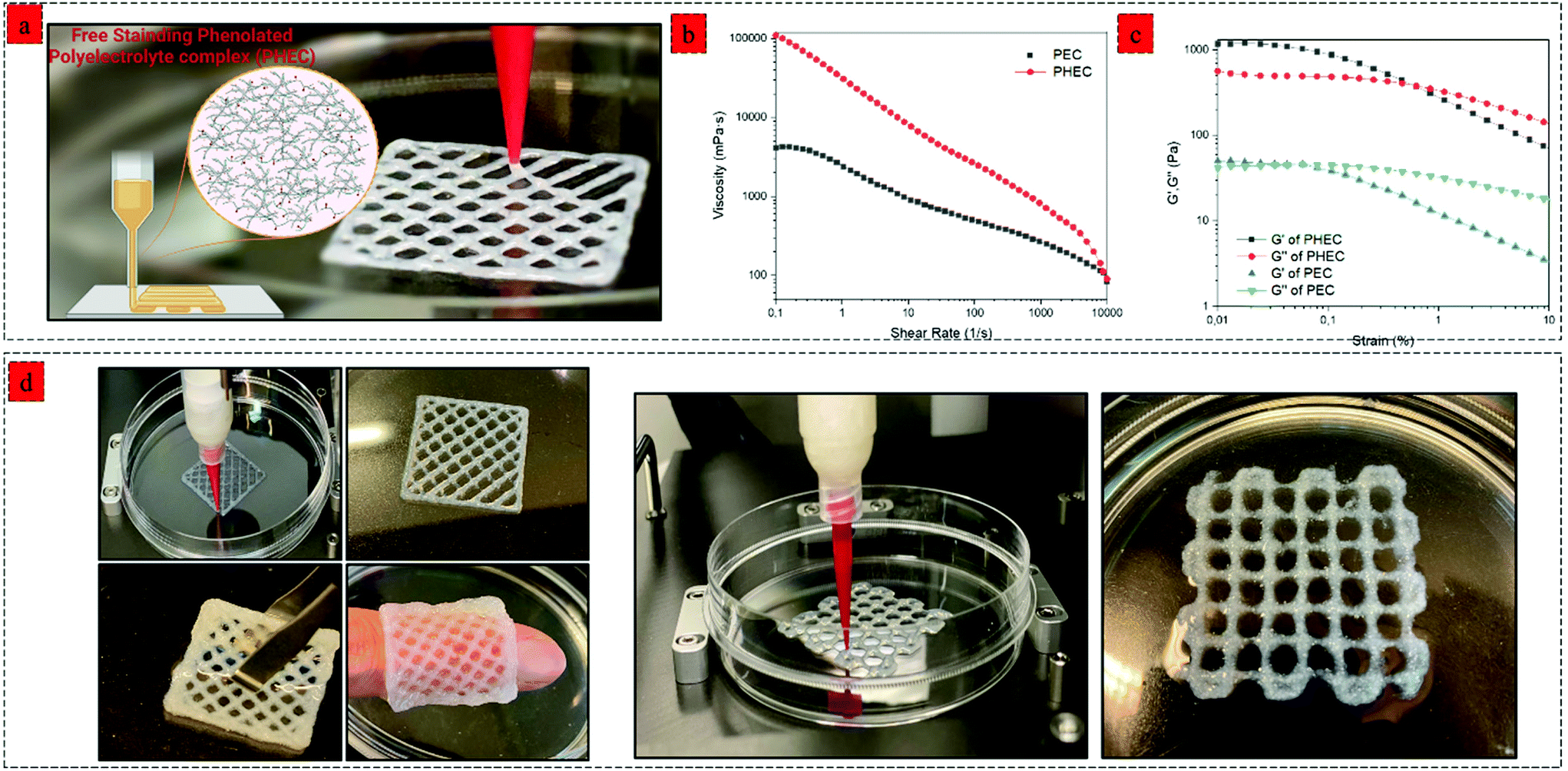 | ||
| Fig. 6 3D printing with phenolated polyelectrolyte complex (PHEC) hydrogels. (a) Schematic illustration and image of 3D printing of PHEC, the figure was created with BioRender.com. (b) shear-rate dependent dynamic viscosity of PHEC and non-phenolated over the shear rate of 0.1–1000 1 s−1, (c) storage modulus (G′) and loss modulus (G′′)–strain dependence of phenolated PEC and non-phenolated PEC at a constant frequency of 1 Hz at 37 °C, (d) Images of extruded PHEC (4%) that has been layered in a 3D structure. | ||
The 3D printing process of the phenolated PEC hydrogel was performed using a bioplotter pneumatic dispensing system (BioScaffolder 3.1, GeSiM, Germany) (Fig. 6a). PHEC suspension was extruded via an 18 G plastic needle (diameter: 250 μm) at 140 kPa with a speed of 11 mm s−1, and the gel retained its shape neatly after extrusion due to its reversible non-dynamic bonds (Video S3†). Several examples of 3D-printed hydrogels are shown in Fig. 6d. The extrusion rate of the hydrogel was not slowed, even after adding more than 7 layers of the hydrogel. For further solidification, the 3D printed hydrogels containing HRP (1 U mL−1) were soaked in diluted H2O2 solution (5 mM) for 5 min to increase the hydrogels’ stiffness through mild and nontoxic enzymatic crosslinking of phenol groups presented on the microfibers surface, chitosan, and alginate chain which enables for 3D encapsulation of cells and bioactive agent.52
Discussion
We introduced a new class of printable hydrogel reinforced by the synergistic effect of phenolated polyelectrolyte complex (PHEC) between phenolated chitosan and alginate chains. The complexation not only enables the 3D printability of the hydrogel but also significantly improves the toughness, moldability, flexibility, and self-healing capacity of the hydrogel. Indeed, PHEC self-induced reinforcement by in situ microfiber formation between positively charged phenolated chitosan and negatively charged phenolated chitosan led to a significant increase in the suspension's viscosity. On the other hand, increasing the concentration of PHEC beyond 2.5% (Ch-Ph and Alg-Ty 2.5%) endowed the PHEC suspension with adequate viscosity and solid-like behaviour necessary for 3D printing without the addition of any other compound and free of extensive chemical modification. This phenolated polyelectrolyte complex was prepared using chitosan and alginate, and this principle can be applied to a wide range of combinations of phenolated cationic and anionic biopolymers indicating the broad applicability of this method. While various 3D printing methods such as ionic crosslinking or photo-crosslinking of alginate and chitosan-based hydrogels have been reported previously,18,53–55 there are several advantages in using the proposed method, such as high flexibility, toughness, and foldability of the hydrogel due to the in situ microfiber formation and weak physical interaction leading to increase in the energy dissipation. Besides, the PHEC suspension shows tunable concentration-dependent viscoelastic properties from sol to solid-like behaviour due to the rapid physical solidification (Video S1†) between the microfibers via hydrophobic, electrostatic, and van der Waals interactions. The dynamic nature of these reactions allows for easy extrusion and injection as a crucial requirement of hydrogels for 3D printing applications. The PHEC approach can be an efficient solution for common hydrogel inks with low yield stress to improve the extrusion capability.Our novel 3D printable PHEC hydrogels exhibited superior properties compared to the recent 3D printable biobased hydrogel. For example, a recent study showed that incorporating alginate soft dendritic colloids into an alginate solution could endow the hydrogel with 3D printability.56 However, due to the low stiffness and long solidification time (60 min), the hydrogel formation required additional ionic crosslinking, hampering its biomedical applicability. Alternatively, another recent study reported a 3D printable polyelectrolyte complex between alginate and ε-polylysine. However, a high concentration of alginate (40%) and ε-polylysine (∼30%) was required to reach an adequate viscosity, and additional ionic crosslinking was needed for further solidification.57
In contrast, our new bioink showed adequate physical stability required for 3D printability at a much lower concentration (2.5% polymers) due to the synergistic effect of PHEC. Furthermore, in contrast to typical chitosan and alginate 3D printed hydrogels, our 3D printed hydrogel possesses greater flexibility and foldability, thanks to the synergistic reinforcement by in situ microfiber formation (Video S4†). Moreover, the phenol modification can endow the 3D printed hydrogel with photo-crosslinking capability using visible light as a safe and biocompatible method, further providing a suitable substrate for 3D cell encapsulation and bioink development.18 Hence, this work employed natural marine resources to fabricate 3D printable hydrogels using green chemistry technology. Indeed, we showed that the phenolated polyelectrolyte complex as a new concept could be formed between phenolated alginate and chitosan close to neutral pH without the use of acidic media compared to the previously described polyelectrolyte complex between chitosan and alginate.26,58 Moreover, further modification, such as oxidation of phenolated alginate, can be performed to increase the hydrogel's self-healing efficacy by introducing reversible imine bonds. Furthermore, tuneable viscoelasticity of the double crosslinked hydrogel can be optimized to promote fundamental cell processes and behaviours dependent on the matrix's viscoelasticity.59 We believe that the proposed method described in this study offers a green approach for the 3D printing of chitosan-based hydrogels for biomedical applications, such as biomaterials,60 biosensors,61 food stuffs,62 and personalized medicine.63
Materials and methods
Materials
Chitosan with a deacetylation degree ≥75%, sodium alginate (9005-38-3), 1-ethyl-3-(3dimethylaminopropyl)carbodiimide (EDC) (98%), N-hydroxysuccinimide (NHS) (98%), horseradish peroxidase (HRP) (Type VI, essentially salt-free, lyophilized powder, ≥250 units per mg solid), sodium chloride, hydrogen peroxide (H2O2) (30%), 4 morpholineethanesulfonic acid, 2-(N-morpholino)ethanesulfonic acid (MES), Hoechst (H33342) and ethidium homodimer I (EH1) (E1903) were purchased from Sigma Aldrich (St Louis, MO, USA). 3-(4-Hydroxyphenyl) propionic acid (>98%) and tyramine hydrochloride (>98%) were obtained from Carbosynth (Compton, United Kingdom).Fabrication and characterization of phenolated chitosan and alginate
Chitosan and alginate were conjugated with phenol groups via carbodiimide coupling chemistry using 3-(4-hydroxyphenyl) propionic acid (HPA) and tyramine hydrochloride, respectively.64,65 Briefly, 1 g (6 mmol) of 3-(4-hydroxyphenyl) propionic acid (HPA) was dissolved in 20 mL of aqueous ethanol (50% v/v), then 0.5 g (3 mmol) of EDC was added to the solution, followed by the addition of 0.3 g of NHS (2.6 mmol) and allowed to stir at 25 °C for 30 min. The resulting solution was added dropwise to 100 mL of 1 wt% chitosan (solubilized in diluted HCl solution) at the pH of 4.75. The pH of the solution was maintained at 4.75 by the dropwise addition of 1 M HCl. After 24 h of stirring at 25 °C, the solution was dialyzed against 4.5 L of NaCl solution (0.6%) at pH 4.5 for 3 days with changing the dialysis solution every 8 h. Then the sample was dialyzed against distilled water for another 4 h, and the resulting dialysate was freeze-dried for 48 h and kept in a moisture-free desiccator before use.For the alginate modification, briefly, 0.5 g (3 mmol) of EDC and 0.3 g (2.6 mol) of NHS were added to a solution of 1 g of sodium alginate dissolved in 100 ml of MES (2-(N-morpholino)ethanesulfonic acid) buffer (50 mM), and the pH was adjusted to 6 by 1 M NaOH. Then, 0.7 g (4 mmol) of tyramine hydrochloride dissolved in 20 ml of MES (50 mM) was added dropwise to the alginate solution and continuously stirred for 24 h at 25 °C. The solution was dialyzed against distilled water for three days with change of water every 8 h. The final product was freeze-dried and kept in a moisture-free desiccator prior to use. The modification of phenolic groups on the chitosan and alginate backbones was investigated using 1H NMR and ultraviolet-visible spectroscopy.
Preparation of hydrogel
A series of hydrogels based on chitosan, alginate, and mixtures was prepared in 1 mL vials at 37 °C.66 To prepare chitosan or alginate hydrogels, different concentrations of Ch-Ph and Alg-Ty (0.5, 1, and 1.5% w/v) (Table S1†) were dissolved in PBS (pH 7.4). The gelation was performed using two vial methods; one vial containing 90 μL of the polymers (Ch-Ph or Alg-Ty) and 10 μL of HRP (0.1 mg mL−1), while the other containing 90 μL of the polymers and 10 μL of H2O2 was then mixed and gently stirred to homogenize the solution and allow subsequent gel formation. The gelation time was recorded by the tube inversion method. The final concentration of HRP and H2O2 was 1 U mL−1 and 1 mM, respectively.To prepare the double crosslinked chitosan-alginate hydrogel (DCCA), different combinations (Table S1†) of Ch-Ph and Alg-Ty were prepared by dissolving Ch-Ph in deionized water at pH 6.5 and Alg-Ty in deionized water at pH 8 to make sure that the amino groups of chitosan and carboxylic acid groups of alginate are in the form of protonated, and deprotonated, respectively. Then, the Alg-Ty solution was dropwise added to the Ch-Ph solution with a volume ratio of 1:1, and the mixture was vigorously stirred with a magnetic stirring to allow homogenization of the PHEC suspension. After the homogenization, the hydrogel formation and gelation time measurement were performed similarly on the individual chitosan and alginate hydrogels.
Characterization
The infrared spectra of the freeze-dried hydrogels were recorded using an FT/IR-6600 FT-IR spectrometer (JASCO, Japan) with a resolution of 4 cm−1 and 64 scans per spectrum. High-resolution X-ray diffraction (XRD) analysis was performed using an X-ray diffractometer (XRD; Bruker ecoD8 advance). The microporous structure of the hydrogel was investigated using a scanning electron microscope (HITACHI, SU-70, Japan). The average pore size and the microfiber diameter were analyzed using the Image J software (version 1.53k, National Institutes of Health, Bethesda, MD, USA). The hydrogels’ swelling ratio and degradation rate were evaluated by the gravimetric method in PBS and lysozyme solution at 37 °C, respectively. Details are described in the ESI notes 1–2.†Rheological and compression characterization of hydrogels
The rheological properties were investigated in a rheometer (Modular Compact Rheometer MCR 302, Anton Paar, Austria) coupled with a parallel plate (25 mm, and 0.5 mm gap size) with the temperature maintained at 37 °C for all experiments. The hydrogel precursors’ dynamic viscosity was measured via the flowability test over a range of shear rates (0.1–10000 1 s−1). Hydrogel gelation kinetics was investigated by a time sweep oscillatory test at a constant frequency of 1 Hz and a strain of 0.1% (LVR). The frequency sweep test was carried out over the frequency range between 0.1 to 10 Hz at a constant strain of 1%. Amplitude sweep ranging from 0.01 to 1000% strain was performed at a constant frequency of 1 Hz to determine the linear viscoelastic region (LVR).
The compression properties of the hydrogels were evaluated using a Zwick/Roell Z020 universal testing machine (Zwick GmbH, Ulm, Germany) with 2 K N cell load. Cylindrical hydrogels with a diameter and height of 6 mm were prepared in flat-bottomed vials. The compression test was performed with a 1 mm min−1 crosshead speed, and the stress–strain curves were recorded. The compressive modulus was calculated from the slope of the linear stress–strain curves, and the toughness was measured by the integration of stress–strain curves using OriginPro (9.6.5.169).
Antioxidant and antibacterial activity
DPPH‡ radical scavenging assay was performed to investigate the antioxidant activity of hydrogels according to a previously reported method.66 The antibacterial activity of the hydrogels against Gram-negative bacteria E. coli and Gram-positive bacteria S. aureus were investigated using the growth inhibition assay67 and disk diffusion test.68 Details are described in the ESI notes 3 and 4.†3D Cell encapsulation and biocompatibility evaluation
The cytocompatibility of hydrogels was investigated using CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS, Promega) and live/dead staining using Hoechst 33342/PI double-staining assay. 3T3-L1 cells were seeded on the hydrogels at a density of 5 × 103 cells per well and incubated for three days, and the media was replaced every day. The cytotoxicity of the hydrogels was evaluated on days 1, and 3 using an MTS assay. For 3D cell encapsulation, sterile gel precursors containing 3T3-L1 cell suspension with a cell density of 5 × 105 cells per mL were used to form the hydrogels in a Millicell EZ SLIDE 8-well glass (Merck, Kenilworth, NJ, USA) followed by 30 min incubation at 37 °C prior to culture. After 1 and 3 days of culture, the encapsulated cell was stained using Hoechst/ethidium homodimer I (EH1) staining. Details are described in the ESI notes 5.†Statistical analysis
All experiments were performed in triplicate, and the results are expressed as means ± standard deviations (SD). Statistical analyses were performed using GraphPad Prism 8 (GraphPad Software Inc.) using either one-way ANOVA or two-way ANOVA followed by Bonferroni's post hoc test. P-Values < 0.05 were considered statistically significant. Wherever significance has been proved it is indicated by *p < 0.05; **p < 0.005; ***p < 0.0005; ****p < 0.0001.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
H. J and A. S. acknowledge funding from Innoviris Brussels, Belgium (https://innoviris.brussels) under the project 2019 – BRIDGE – 4: RE4BRU. The content is solely the responsibility of the authors and does not necessarily represent the official views of the above-mentioned funding agency. The authors would like to thank Dr Véronique Fontaine for her assistance with the antibacterial test.References
- Q. V. Nguyen, J. H. Park and D. S. Lee, Eur. Polym. J., 2015, 72, 602–619 CrossRef CAS.
- J.-Y. Sun, X. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- Y. Huang, D. R. King, T. L. Sun, T. Nonoyama, T. Kurokawa, T. Nakajima and J. P. Gong, Adv. Funct. Mater., 2017, 27, 1605350 CrossRef.
- D. Zhao, J. Huang, Y. Zhong, K. Li, L. Zhang and J. Cai, Adv. Funct. Mater., 2016, 26, 6279–6287 CrossRef CAS.
- S. Xia, S. Song and G. Gao, Chem. Eng. J., 2018, 354, 817–824 CrossRef CAS.
- T. Zhu, J. Mao, Y. Cheng, H. Liu, L. Lv, M. Ge, S. Li, J. Huang, Z. Chen and H. Li, Adv. Mater. Interfaces, 2019, 6, 1900761 CrossRef.
- I.-Y. Kim, S.-J. Seo, H.-S. Moon, M.-K. Yoo, I.-Y. Park, B.-C. Kim and C.-S. Cho, Biotechnol. Adv., 2008, 26, 1–21 CrossRef CAS PubMed.
- F. Croisier and C. Jérôme, Eur. Polym. J., 2013, 49, 780–792 CrossRef CAS.
- M. Rajabi, M. McConnell, J. Cabral and M. A. Ali, Carbohydr. Polym., 2021, 117768 CrossRef CAS PubMed.
- C. Cui, C. Shao, L. Meng and J. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 39228–39237 CrossRef CAS PubMed.
- J. Liu, B. Yang, M. Li, J. Li and Y. Wan, Carbohydr. Polym., 2020, 227, 115335 CrossRef CAS PubMed.
- H. Y. Jung, P. Le Thi, K.-H. HwangBo, J. W. Bae and K. D. Park, Carbohydr. Polym., 2021, 261, 117810 CrossRef CAS PubMed.
- C. Xu, W. Zhan, X. Tang, F. Mo, L. Fu and B. Lin, Polym. Test., 2018, 66, 155–163 CrossRef CAS.
- M. C. Pellá, M. K. Lima-Tenório, E. T. Tenório-Neto, M. R. Guilherme, E. C. Muniz and A. F. Rubira, Carbohydr. Polym., 2018, 196, 233–245 CrossRef PubMed.
- J. You, S. Xie, J. Cao, H. Ge, M. Xu, L. Zhang and J. Zhou, Macromolecules, 2016, 49, 1049–1059 CrossRef CAS.
- Q. Wu, M. Maire, S. Lerouge, D. Therriault and M. C. Heuzey, Adv. Biosyst., 2017, 1, 1700058 CrossRef.
- L. Zhou, H. Ramezani, M. Sun, M. Xie, J. Nie, S. Lv, J. Cai, J. Fu and Y. He, Biomater. Sci., 2020, 8, 5020–5028 RSC.
- Y. Liu, C.-W. Wong, S.-W. Chang and S.-h. Hsu, Acta Biomater., 2021, 122, 211–219 CrossRef CAS PubMed.
- J. Berger, M. Reist, J. M. Mayer, O. Felt and R. Gurny, Eur. J. Pharm. Biopharm., 2004, 57, 35–52 CrossRef CAS PubMed.
- W. Pu, F. Jiang, P. Chen and B. Wei, Soft Matter, 2017, 13, 5645–5648 RSC.
- H. Fan, J. Wang, Z. Tao, J. Huang, P. Rao, T. Kurokawa and J. P. Gong, Nat. Commun., 2019, 10, 1–8 CrossRef CAS PubMed.
- S. Sakai and M. Nakahata, Chem. – Asian J., 2017, 12, 3098–3109 CrossRef CAS PubMed.
- M. H. Kim, J. N. Lee, J. Lee, H. Lee and W. H. Park, ACS Biomater. Sci. Eng., 2020, 6, 3103–3113 CrossRef CAS PubMed.
- Y. Zhong, J. Wang, Z. Yuan, Y. Wang, Z. Xi, L. Li, Z. Liu and X. Guo, Colloids Surf., B, 2019, 179, 462–469 CrossRef CAS PubMed.
- D. Komoto, T. Furuike and H. Tamura, Int. J. Biol. Macromol., 2019, 126, 54–59 CrossRef CAS PubMed.
- G. K. Wasupalli and D. Verma, Int. J. Biol. Macromol., 2018, 114, 10–17 CrossRef CAS PubMed.
- R. Jin, C. Lin and A. Cao, Polym. Chem., 2014, 5, 391–398 RSC.
- T. T. H. Thi, Y. Lee, S. B. Ryu, H.-J. Sung and K. D. Park, RSC Adv., 2017, 7, 34053–34062 RSC.
- Y. Wang, M. Xie, G. Ma, Y. Fang, W. Yang, N. Ma, D. Fang, Q. Hu and F. Pei, Carbohydr. Polym., 2019, 225, 115238 CrossRef CAS PubMed.
- M. de Ruijter, A. Hrynevich, J. N. Haigh, G. Hochleitner, M. Castilho, J. Groll, J. Malda and P. D. Dalton, Small, 2018, 14, 1702773 CrossRef PubMed.
- S. Uman, A. Dhand and J. A. Burdick, J. Appl. Polym. Sci., 2020, 137, 48668 CrossRef CAS.
- M. Truong and L. Walker, Langmuir, 2002, 18, 2024–2031 CrossRef CAS.
- X. Lv, Y. Liu, S. Song, C. Tong, X. Shi, Y. Zhao, J. Zhang and M. Hou, Carbohydr. Polym., 2019, 205, 312–321 CrossRef CAS PubMed.
- R. Jin, C. Hiemstra, Z. Zhong and J. Feijen, Biomaterials, 2007, 28, 2791–2800 CrossRef CAS PubMed.
- S. V. Gohil, S. B. Brittain, H.-M. Kan, H. Drissi, D. W. Rowe and L. S. Nair, J. Mater. Chem. B, 2015, 3, 5511–5522 RSC.
- H. Jafari, A. Dadashzadeh, S. Moghassemi, P. Zahedi, C. A. Amorim and A. Shavandi, Gels, 2021, 7, 138 CrossRef CAS PubMed.
- O. Yuksel, M. Sandberg, I. Baran, N. Ersoy, J. H. Hattel and R. Akkerman, Composites, Part B, 2021, 207, 108543 CrossRef CAS.
- O. Okay and W. Oppermann, Macromolecules, 2007, 40, 3378–3387 CrossRef CAS.
- M. Sun, J. Qiu, C. Lu, S. Jin, G. Zhang and E. Sakai, Polymers, 2020, 12, 2263 CrossRef CAS PubMed.
- S. Talebian, M. Mehrali, N. Taebnia, C. P. Pennisi, F. B. Kadumudi, J. Foroughi, M. Hasany, M. Nikkhah, M. Akbari and G. Orive, Adv. Sci., 2019, 6, 1801664 CrossRef PubMed.
- N. Barroso, O. Guaresti, L. Pérez-Álvarez, L. Ruiz-Rubio, N. Gabilondo and J. L. Vilas-Vilela, Eur. Polym. J., 2019, 120, 109268 CrossRef CAS.
- W. Diao, L. Wu, X. Ma, Z. Zhuang, S. Li, X. Bu and Y. Fang, J. Appl. Polym. Sci., 2019, 136, 47783 CrossRef.
- S. Potiwiput, H. Tan, G. Yuan, S. Li, T. Zhou, J. Li, Y. Jia, D. Xiong, X. Hu and Z. Ling, Mater. Chem. Phys., 2020, 241, 122354 CrossRef CAS.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- Z. Deng, Y. He, Y. J. Wang, Y. Zhao and L. Chen, Soft Matter, 2020, 16, 6128–6137 RSC.
- X. Zhao, Q. Li, X. Ma, F. Quan, J. Wang and Y. Xia, J. Ind. Eng. Chem., 2015, 24, 188–195 CrossRef CAS.
- R. C. Goy, D. d. Britto and O. B. Assis, Polímeros, 2009, 19, 241–247 CrossRef CAS.
- N. Bhardwaj and S. C. Kundu, Carbohydr. Polym., 2011, 85, 325–333 CrossRef CAS.
- A. Rasool, S. Ata and A. Islam, Carbohydr. Polym., 2019, 203, 423–429 CrossRef CAS PubMed.
- T. Kreller, T. Distler, S. Heid, S. Gerth, R. Detsch and A. Boccaccini, Mater. Des., 2021, 109877 CrossRef CAS.
- A. Aljawish, L. Muniglia, A. Klouj, J. Jasniewski, J. Scher and S. Desobry, Food Hydrocolloids, 2016, 60, 551–558 CrossRef CAS.
- Q. Li, S. Xu, Q. Feng, Q. Dai, L. Yao, Y. Zhang, H. Gao, H. Dong, D. Chen and X. Cao, Bioact. Mater., 2021, 6, 3396–3410 CrossRef CAS PubMed.
- T. M. Valentin, A. K. Landauer, L. C. Morales, E. M. DuBois, S. Shukla, M. Liu, L. H. S. Valentin, C. Franck, P.-Y. Chen and I. Y. Wong, Carbon, 2019, 143, 447–456 CrossRef CAS.
- O. Jeon, Y. B. Lee, T. J. Hinton, A. W. Feinberg and E. Alsberg, Mater. Today Chem., 2019, 12, 61–70 CrossRef CAS PubMed.
- J. H. Teoh, A. Mozhi, V. Sunil, S. M. Tay, J. Fuh and C. H. Wang, Adv. Funct. Mater., 2021, 2105932 CrossRef CAS.
- A. H. Williams, S. Roh, A. R. Jacob, S. D. Stoyanov, L. Hsiao and O. D. Velev, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- Z. Lin, M. Wu, H. He, Q. Liang, C. Hu, Z. Zeng, D. Cheng, G. Wang, D. Chen and H. Pan, Adv. Funct. Mater., 2019, 29, 1808439 CrossRef.
- M. Gierszewska, J. Ostrowska-Czubenko and E. Chrzanowska, Eur. Polym. J., 2018, 101, 282–290 CrossRef CAS.
- O. Chaudhuri, J. Cooper-White, P. A. Janmey, D. J. Mooney and V. B. Shenoy, Nature, 2020, 584, 535–546 CrossRef CAS PubMed.
- C. Yu, J. Schimelman, P. Wang, K. L. Miller, X. Ma, S. You, J. Guan, B. Sun, W. Zhu and S. Chen, Chem. Rev., 2020, 120, 10695–10743 CrossRef CAS PubMed.
- D. Han, C. Farino, C. Yang, T. Scott, D. Browe, W. Choi, J. W. Freeman and H. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 17512–17518 CrossRef CAS PubMed.
- F. Ding, B. Hu, S. Lan and H. Wang, Food Packag. Shelf Life, 2020, 26, 100559 CrossRef.
- C. Tonda-Turo, I. Carmagnola, A. Chiappone, Z. Feng, G. Ciardelli, M. Hakkarainen and M. Sangermano, Bioprinting, 2020, 18, e00082 CrossRef.
- D. Lee, J. P. Park, M.-Y. Koh, P. Kim, J. Lee, M. Shin and H. Lee, Biomater. Sci., 2018, 6, 1040–1047 RSC.
- Y. Liu, S. Sakai and M. Taya, Acta Biomater., 2013, 9, 6616–6623 CrossRef CAS PubMed.
- D. L. Tran, P. Le Thi, T. T. H. Thi and K. D. Park, Prog. Nat. Sci.: Mater. Int., 2020, 30, 661–668 CrossRef CAS.
- G. Tao, R. Cai, Y. Wang, H. Zuo and H. He, Mater. Sci. Eng., C, 2021, 119, 111597 CrossRef CAS PubMed.
- Y. Liang, X. Zhao, T. Hu, Y. Han and B. Guo, J. Colloid Interface Sci., 2019, 556, 514–528 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc04347a |
| ‡ 2,2-Diphenyl-1-picryl-hydrazyl-hydrate. |
| This journal is © The Royal Society of Chemistry 2022 |