
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inversion of chirality in GTM-4 enantio-enriched zeolite driven by a minor change of the structure-directing agent†
Ramón
de la Serna
,
Itziar
Arnaiz
,
Carlos
Márquez-Álvarez
 ,
Joaquín
Pérez-Pariente
and
Luis
Gómez-Hortigüela
*
,
Joaquín
Pérez-Pariente
and
Luis
Gómez-Hortigüela
*
Instituto de Catálisis y Petroleoquímica, ICP-CSIC. C/Marie Curie 2, Madrid 28049, Spain. E-mail: lhortiguela@icp.csic.es
First published on 2nd November 2022
Abstract
A surprising inversion of chirality of the -ITV zeolite framework is observed when the ethyl group of the enantiopure N,N-ethyl-methyl-pseudoephedrinium organic structure-directing agent is replaced by a benzyl or 2-methylbenzyl group, keeping the same molecular absolute configuration. Interestingly, chiral zeolite materials obtained with these new benzyl-containing cations reach unprecedentedly high enantiomeric excesses up to 55%.
Chirality is a fundamental property inherent to life.1 The development of easily-recyclable chiral heterogeneous solid catalysts able to asymmetrically process chiral compounds towards desired enantiomers of interest still remains as a great challenge in modern chemistry. One of the most interesting inorganic solids that could drive the realization of chiral enantioselective solids is provided by zeolites with chiral framework structures.2–8 Zeolites are crystalline microporous materials based on periodic silicate frameworks comprising regular pores and cavities of molecular dimensions.9,10 The characteristic shape-selectivity and confinement effects associated to their particular microporous framework structures can be also extended to the asymmetric nature of chiral compounds, being potentially able to discriminate between enantiomers as long as chiral frameworks are available. Of the more than 250 different zeolite frameworks discovered so far,11 a few of them are chiral,3,12–16 crystallizing in a chiral space group with two enantiomorphic polymorphs, and usually containing helicoidal channels.
The challenge in developing chiral zeolites for asymmetric catalysis is not only to promote the crystallization of a chiral framework, but to do so in an enantioselective fashion so that one of the enantiomorphic polymorphs is preferentially (or ideally exclusively) crystallized. Due to the symmetric nature of the basic building units of zeolites (the TO4 tetrahedral units), chirality must be induced from some external component during crystallization. The most straightforward strategy is through the use of chiral organic cations as structure-directing agents (SDAs), in an attempt to transfer their chirality from the molecular component to the inorganic framework.17–20 These organic SDAs drive the crystallization pathway towards a particular framework topology by imprinting their molecular geometric properties (size and shape) into the zeolite framework.21 Despite great efforts applied through the years,2,22 only recently has this strategy met success with the development of two enantiomerically-enriched chiral zeolites with enantioselective catalytic properties, based on the STW23 and -ITV24 chiral frameworks. Davis and coworkers first achieved this goal through the use of a computationally-designed complex SDA cation whose asymmetric molecular structure promoted the growth of the helicoidal channels of the STW framework in one or the other handedness as a function of the molecular absolute configuration of the SDA.4,24,25 Later on, in our group we used a simple organic cation derived from the chiral pool, (1S,2S)-N,N-ethyl-methyl-pseudoephedrinium (or its (1R,2R) enantiomer) (EMPS, see Scheme 1), to drive the crystallization of GTM-3, a new chiral extra-large pore zeolite with the -ITV framework structure that was enantio-enriched in one or the other enantiomorphic polymorph (P4132 or P4332) by using the different enantiomers of the organic cation.24 The discovery of GTM-3 involved a significant breakthrough in asymmetric heterogeneous catalysis since it combined three crucial features for chiral solid catalysts: (i) the presence of active sites; (ii) the occurrence of extra-large pores (with 30-ring windows and pores up to ∼2 nm) that enabled processing very large substrates, and (iii) the enantio-enriched chiral nature of the framework, which qualified to process molecules in an enantioselective fashion, leading to unprecedented enantiomeric excesses of up to 30%.24 Furthermore, the accessibility of the chiral precursor from which the chiral SDA of GTM-3 is synthesized, (1S,2S)- or (1R,2R)-pseudoephedrine, which comes from the chiral pool, comprises a great operational advantage for potential industrial implementation.
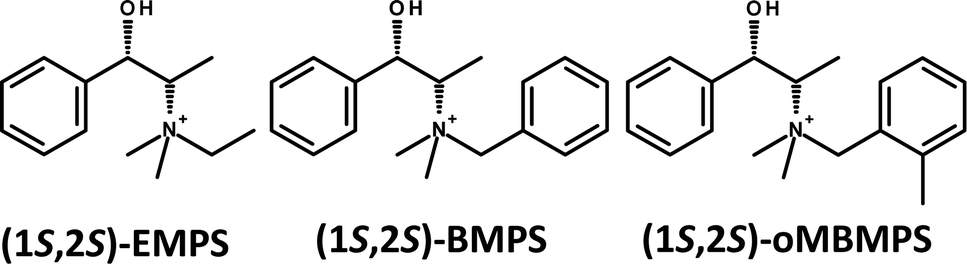 | ||
| Scheme 1 Molecular structure of the (1S,2S)-enantiomers of N,N-ethyl-methyl-pseudoephedrinium (EMPS), N,N-benzyl-methyl-pseudoephedrinium (BMPS), and N,N-(2-methylbenzyl)-methyl-pseudoephedrinium (oMBMPS). | ||
Due to the relevance of the discovery of GTM-3, we have systematically explored the effect of slightly modifying the molecular structure of the organic cation used as SDA (EMPS) in order to understand the relationship between the molecular structure of the chiral cation and the -ITV framework. On the course of these investigations, we found an unexpected observation: when the ethyl group attached to the N atom of the cation was replaced by a benzyl (or a 2-methylbenzyl) group (see Scheme 1, BMPS and oMBMPS), a new chiral zeolite material (that we called GTM-4) with the same -ITV framework was produced, even though the molecular structure of this new organic cation was rather different. Here we report the synthesis, characterization and catalytic activity of GTM-4.
GTM-4 was prepared by hydrothermal method from gels with similar molar compositions as those of GTM-3; Table S1 (ESI†) shows the synthesis results upon a systematic variation of the gel composition and crystallization conditions; temperatures of 100 °C or lower are required in order to avoid the thermal decomposition of the organic cations, while potentially improving the enantio-differentiation of the SDA⋯ITV diastereomeric pairs. A gel composition of 0.25ROH:0.75SiO2:0.25GeO2:0.25HF:3.7H2O (where ROH refers to BMPS hydroxide), with a Si/Ge ratio of 3 (Gef = 0.25), led to the crystallization of GTM-4 (Fig. S2, ESI†), (-ITV), but only at the lower temperature of 80 °C (entry 5 in Table S1, ESI†); higher temperatures (100 °C) provoked the co-crystallization of BEC, the C polymorph of zeolite Beta, together with -ITV (entry 6); BEC is a commonly observed product in the presence of Ge and bulky SDAs. An increase of the Ge fraction to 0.33 (Si/Ge = 2) (entries 1–4) also led to the crystallization of the -ITV framework, but in this case together with minor amounts of GeO2, and also of BEC at high temperature (entries 3,4). These results indicate that the -ITV framework structure is favored at 80 °C crystallization temperature since BEC competes at 100 °C. A decrease of the Ge fraction to 0.17 (Si/Ge = 5, entries 7–10) prevented the crystallization of GTM-4, and only BEC products were observed at both temperatures, while a further decrease to 0.09 (entries 11–14) led mostly to amorphous materials. An increase of the water molar content from 3.7 to 5.0 (entries 15–26) in general led to similar trends in the phase selectivity, but with a lower efficiency for the crystallization of GTM-4.
In an attempt to improve the crystallinity of GTM-4, we introduced slight variations in the molecular structure of the SDA, and observed a more efficient crystallization of the -ITV framework when the benzyl-group of BMPS contained an additional methyl group in ortho-position (see Scheme 1, oMBMPS) (methyl groups in other positions led to worse structure-directing efficiencies). Table S2 (ESI†) shows the compared phase selectivity for BMPS and oMBMPS as a function of the synthesis conditions, where the main difference observed is that the incorporation of the ortho-methyl group prevents the competitive crystallization of the BEC phase at high temperature; Fig. S2 (ESI†) shows the XRD patterns of GTM-4 materials obtained with (1S,2S)-BMPS and (1S,2S)-oMBMPS.
The incorporation of the chiral organic cations in GTM-4 was characterized by thermogravimetric analysis (Fig. S3, ESI†) and 13C CP MAS NMR. 13C NMR spectra (Fig. S4, ESI†) confirmed the integrity of both BMPS and oMBMPS cations confined within the -ITV framework, showing the same bands as those of the original pristine cations, though some resonances slightly shifted as a consequence of the confinement. The SDA content determined by TGA for GTM-4 materials, between ∼15–17 SDAs per unit cell (these packing values might be slightly different because of the presence of some amorphous material), was smaller than that for GTM-3 (∼18–20 SDAs per unit cell), as expected due to the larger size of benzyl-containing cations. 19F MAS NMR showed the incorporation of F− within the D4Rs in both materials, with Ge associated in pairs and/or close clusters (Fig. S5, ESI†).26 N2 adsorption/desorption isotherm of GTM-4(oMBMPS) (Fig. S6, ESI†) gave a micropore volume of 0.20 cm3 g−1 and a surface area (BET) of 692 m2 g−1, similar to those of the original ITQ-3712 and GTM-3,24 though somewhat smaller (particularly the micropore volume) due to the presence of some amorphous material in GTM-4.
The next fundamental step was to analyze the catalytic activity of GTM-4 in asymmetric reactions in order to find if the new material displayed enantio-discrimination properties driven by the chiral -ITV zeolite framework that imposes an asymmetric catalytic environment. Epoxide ring-opening reactions are known to be catalyzed by the weak Lewis acid sites provided by Ge in tetrahedral positions in zeolite frameworks.27 We used a similar chiral catalytic reaction test as in our previous work,24 the ring-opening of trans-stilbene oxide, in this case with 1-butanol instead of 1-hexanol since this gave slightly better results (Scheme 2). As explained in our previous work, this reaction yields two types of products, with inversion of configuration (‘unlike’ products, with (R,S) and (S,R) configurations), and with retention of configuration (‘like’ products, with (R,R) and (S,S) configurations). Enantiomeric excess results (%) are displayed in Table 1 for GTM-4 (prepared with BMPS or oMBMPS) and for GTM-3 (prepared with EMPS); as expected, EE's similar in magnitude but with opposite sign are obtained for catalysts prepared with the two enantiomers of the corresponding SDA, ensuring the control of the chirality of the -ITV framework with the absolute configuration of the SDA. GTM-3 gave EE's for the ‘unlike’ products of +42.6 (with GTM-3 prepared with RR-EMPS) and −38.3% (with GTM-3 prepared with SS-EMPS). When we analyzed the asymmetric catalytic activity of GTM-4 materials, a great surprise was observed: astonishingly, the EE's obtained in the reaction catalyzed by GTM-4 were reversed with respect to those of GTM-3 (Table 1). GTM-4 prepared with BMPS gave EE's for the ‘unlike’ products of −45.0 (GTM-4/RR-BMPS) and +44.0% (GTM-4/SS-EMPS), with chiralities opposite to those of GTM-3. Interestingly, these values notably increased for GTM-4 obtained with oMBMPS, yielding EE's for the ‘unlike’ products of −53.9 and +54.8% for GTM-4 prepared with RR- and SS-oMBMPS, respectively. These EE's values have never been reached before with this type of chiral catalysts, and involve a great breakthrough towards a real application of these heterogeneous chiral catalysts. As for GTM-3, the EE's found for the ‘like’ products were lower, around 26%.
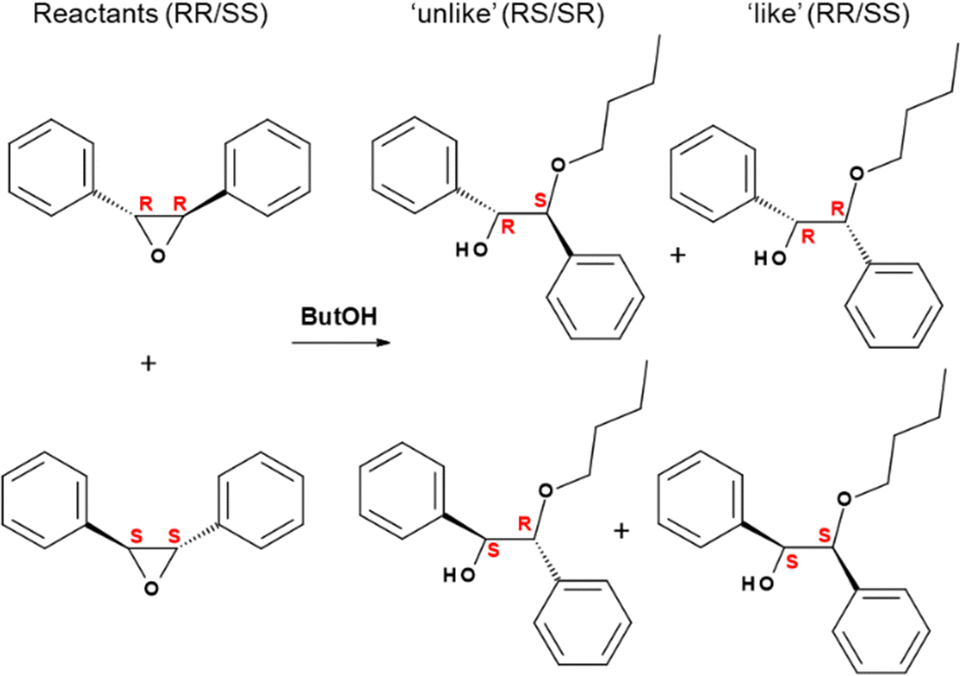 | ||
| Scheme 2 Ring-opening of chiral trans-stilbene oxide with 1-butanol, giving inversion unlike-products (R,S + S,R) or retention like-products (R,R + S,S). Species in columns are enantiomers. | ||
| Catalyst | SDA | EE (%) ‘unlike’ | EE (%) ‘like’ | ||
|---|---|---|---|---|---|
| RR | SS | RR | SS | ||
| GTM-4 | BMPS | −45.0 | +44.0 | +21.6 | −26.3 |
| GTM-4 | oMBMPS | −53.9 | +54.8 | +26.5 | −26.5 |
| GTM-3 | EMPS | +42.6 | −38.3 | −25.9 | +31.6 |
Such discovery of the opposed handedness of GTM-3 and GTM-4 materials prepared from the same chiral precursor comprises a crucial consequence since, thanks to this finding, the -ITV chiral zeolite can be prepared in its two enantiomorphic crystalline polymorphs (with P4132 or P4332 space groups) from the same enantiomer of the chiral precursor available from the chiral pool, (1S,2S)-pseudoephedrine, through GTM-3 (red arrow) or GTM-4 (green arrow) routes (Fig. 1-left). Interestingly, (1S,2S)-pseudoephedrine displays pharmacological properties with adrenergic agonist action, and is used in medicine as a nasal/sinus decongestant, among other uses. Although it occurs naturally as an alkaloid in certain plant species, in particular as a constituent of extracts from the Ephedra species, this process is not very efficient, and hence pseudoephedrine produced for commercial use is derived from yeast fermentation of dextrose in the presence of benzaldehyde in a well-established biotechnological process. The key implication of this synthetic industrial process is the much reduced cost of the (1S,2S)-enantiomer of pseudoephedrine compared with the (1R,2R)-enantiomer. Therefore, the optimized preparation of the starting chiral precursor, (1S,2S)-pseudoephedrine, from which SDAs efficient for the crystallization of both enantiomorphic crystalline forms of the -ITV zeolite framework can be built by attachment of ethyl or benzyl groups, involves a highly advantageous synthetic process for the most expensive component in the synthesis of zeolite catalysts, the SDA, most of all when chiral usually-expensive SDAs are involved, like in the present case.
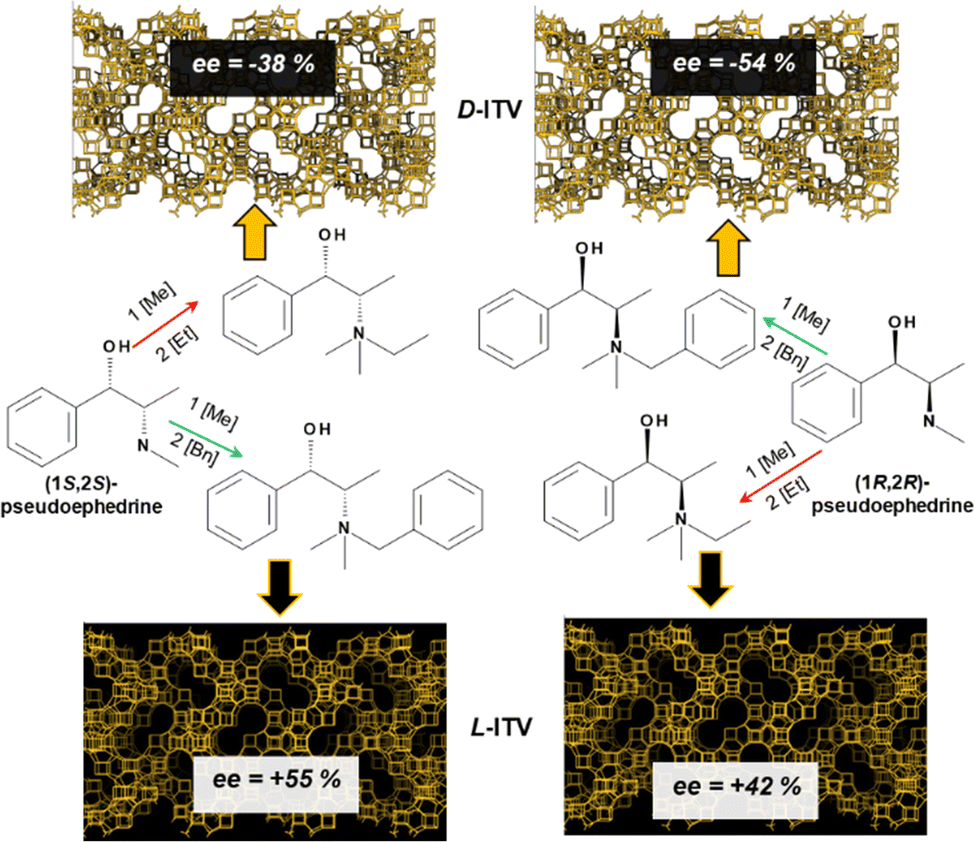 | ||
| Fig. 1 Scheme of the synthetic route to the two enantiomorphic crystalline polymorphs of the -ITV framework (P4132 and P4332 space groups) by using the two derivatives of the enantiomers of pseudoephedrine through GTM-3 (by attachment of an ethyl group, red-arrows routes) or GTM-4 (by attachment of a benzyl group, green-arrows routes). D/L notation is arbitrary and does not mean an actual absolute configuration of the zeolite. | ||
Finally, SEM showed that GTM-4 crystals are smaller than those of GTM-3 (Fig. S7, ESI†), and therefore a higher non-enantioselective reactivity of the external surface (if any) would be expected for GTM-4, what would lead to a decrease of the enantioselectivity. Thus, the notably higher enantioselectivity associated to GTM-4, especially when prepared with oMBMPS, might point to an enhanced enantio-enrichment in one of the two chiral -ITV enantiomorphic polymorphs achieved by these benzyl-containing cations, possibly due to a higher energy difference between the respective SDA⋯ITV chiral diastereomeric pairs. If this is the case, this would involve that GTM-3 and GTM-4 materials are partially enantio-enriched, but are not enantio-pure, and consequently further research in the synthesis of these materials by slight modifications of the SDAs might further improve the enantio-enrichment of the -ITV framework, and hence lead to even stronger enantioselective properties. Attempts to determine the absolute configuration of GTM-3 and GTM-4 materials have so far failed due to their beam-sensitiveness and nanometric crystal size.
In conclusion, the replacement of an ethyl-substituent in chiral N,N-methyl-ethyl-pseudoephedrinium by a benzyl- or 2-methylbenzyl substituent drives the crystallization of the -ITV chiral zeolite framework with a surprising inversion of chirality in the enantio-enrichment in one of the chiral polymorphs, leading to the new GTM-4 materials. Interestingly, these new chiral zeolites display notably improved enantioselective properties during catalytic processes, reaching unprecedented enantiomeric excess values of up to 55% in the ring-opening of trans-stilbene oxide with 1-butanol, representing a major breakthrough in chiral heterogeneous catalysis with zeolites. Such higher enantioselectivity of GTM-4 materials obtained with 2-methylbenzyl-SDAs might be associated to a higher enantio-enrichment of the -ITV framework in the respective chiral polymorphs (P4132 or P4332).
The discovery of the two alternative routes to enantio-enriched -ITV framework through GTM-3 or GTM-4 materials has an important implication for its potential application since the two enantiomorphic chiral polymorphs can be prepared from the same chiral precursor with a given absolute configuration, (1S,2S)-pseudoephedrine, whose synthesis is industrially-optimized as is used as medicinal drug. The ability to tune the chirality of the zeolite solids is essential not only for asymmetric catalysis, but also in other fields where chirality plays a crucial role like bioengineering and biomedicine.28
Conceptualization: L. G. H. Supervision: J. P. P. and L. G. H. Funding acquisition: L. G. H. and J. P. P. Investigation: C. M. A., R. S. and I. A. Formal analysis: all authors. Writing – original draft: L. G. H. Writing – review and editing: all authors.
This work is part of the project PID2019-107968RB-I00, funded by MCIN/AEI/10.13039/501100011033, Spain. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI). RS acknowledges MCIN for predoctoral grant (PRE2020-095946), and IA acknowledges SECAT for a summer internship.
Conflicts of interest
There are no conflicts to declare.References
- W. J. Lough and I. W. Wainer, Chirality in natural and applied science, Blackwell, 2002, ISBN 9780849324345 Search PubMed.
- M. E. Davis, Top. Catal., 2003, 25, 3–7 CrossRef.
- J. Yu and R. J. Xu, Mat. Chem., 2008, 18, 4021–4030 RSC.
- M. E. Davis, ACS Catal., 2018, 8, 10082–10088 CrossRef.
- T. S. van Erp, T. P. Caremans, D. Dubbeldam, A. Martin-Calvo, S. Calero and J. A. Martens, Angew. Chem., Int. Ed., 2010, 49, 3010–3013 CrossRef PubMed.
- J. M. Castillo, T. J. H. Vlugt, D. Dubbeldam, S. Hamad and S. Calero, J. Phys. Chem. C, 2010, 114, 22207–22213 CrossRef.
- R. E. Morris and X. H. Bu, Nat. Chem., 2010, 2, 353–361 CrossRef PubMed.
- D. Dubbledam, T. J. H. Vlugt and S. Calero, Molec. Sim, 2014, 40, 585–598 CrossRef.
- J. Cejka, A. Corma and S. I. Zones, Zeolites and catalysis: synthesis reactions and applications, Wiley-VCH Verlag GmbH & Co. KGaA, 2010 Search PubMed.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef PubMed.
- Atlas of zeolite framework types. https://www.iza-structure.org/databases/ Accessed on 20th of July 2022.
- J. Sun, C. Bonneau, Á. Cantín, A. Corma, M. J. Díaz-Cabañas, M. Moliner, D. Zhang, M. Li and X. Zou, Nature, 2009, 458, 1154–1157 CrossRef PubMed.
- M. M. J. Treacy and J. M. Newsam, Nature, 1988, 332, 249–251 CrossRef.
- N. Rajić, N. Zabukovec Logar and V. Kaučič, Zeolites, 1995, 15, 672–678 CrossRef.
- A. Rojas and M. A. Camblor, Angew. Chem., Int. Ed., 2012, 51, 3854–3856 CrossRef PubMed.
- L. Tang, L. Shi, C. Bonneau, J. Sun, H. Yue, A. Ojuva, B.-L. Lee, M. Kritikos, R. G. Bell, Z. Bacsik, J. Mink and X. Zou, Nat. Mater., 2008, 7, 381–385 CrossRef PubMed.
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756–768 CrossRef.
- Y. Kubota, M. M. Helmkamp, S. I. Zones and M. E. Davis, Microporous Mater., 1996, 6, 213–229 CrossRef.
- L. Gómez-Hortigüela and M. A. Camblor, Introduction to the zeolite structure-directing phenomenon by organic species: general aspects, in Insights into the chemistry of organic structure-directing agents in the synthesis of zeolitic materials. Struct. Bond., ed. L. Gómez-Hortigüela, Springer, Cham, 2017, vol. 175 Search PubMed.
- M. Moliner, F. Rey and A. Corma, Angew. Chem., Int. Ed., 2013, 52, 13880–13889 CrossRef PubMed.
- D. W. Lewis, C. M. Freeman and C. R. A. Catlow, J. Phys. Chem., 1995, 99, 11194–11202 CrossRef.
- L. Gómez-Hortigüela and B. Bernardo-Maestro, Chiral organic structure-directing agents, in Insights into the chemistry of organic structure-directing agents in the synthesis of zeolitic materials. Struct. Bond., ed. L. Gómez-Hortigüela, Springer, Cham, 2017, vol 175 Search PubMed.
- S. K. Brand, J. E. Schmidt, M. W. Deem, F. Daeyaert, Y. Ma, O. Terasaki, M. Orazov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5101–5106 CrossRef PubMed.
- R. de la Serna, D. Nieto, R. Sainz, B. Bernardo-Maestro, A. Mayoral, C. Márquez-Álvarez, J. Pérez-Pariente and L. Gómez-Hortigüela, J. Am. Chem. Soc., 2022, 144, 8249–8256 CrossRef PubMed.
- J. H. Kang, L. B. McCusker, M. W. Deem, C. Baerlocher and M. E. Davis, Chem. Mater., 2021, 33, 1752–1759 CrossRef.
- R. T. Rigo, S. R. G. Balestra, S. Hamad, R. Bueno-Pérez, A. R. Ruiz-Salvador, S. Calero and M. A. Camblor, J. Mater. Chem. A, 2018, 6, 15110 RSC.
- X. Liu, W. Mao, J. Jiang, X. Lu, M. Peng, H. Xu, L. Han, S. Che and P. Wu, Chem. – Eur. J., 2019, 25, 4520–4529 CrossRef PubMed.
- M. Zhang, G. Qinga and T. Sun, Chem. Soc. Rev., 2012, 41, 1972 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc04958a |
| This journal is © The Royal Society of Chemistry 2022 |