
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamics of the alkyne → copper(I) interaction and its use in a heteroleptic four-component catalytic rotor†
Suchismita
Saha
a,
Sohom
Kundu
a,
Pronay Kumar
Biswas
a,
Michael
Bolte
 b and
Michael
Schmittel
*a
b and
Michael
Schmittel
*a
aCenter of Micro and Nanochemistry and (Bio)Technology, Organische Chemie I, Universität Siegen, Adolf-Reichwein-Str. 2, Siegen D-57068, Germany. E-mail: schmittel@chemie.uni-siegen.de; Tel: +49(0) 2717404356
bInstitut für Organische Chemie und Chemische Biologie, Johann Wolfgang Goethe-Universität, Max-von-Laue Strasse 7, Frankfurt am Main D-60438, Germany
First published on 7th November 2022
Abstract
The HETPYNE (HETeroleptic Phenanthroline and alkYNE metal) and DABCO·(zinc porphyrin)2 interactions were used to assemble the four-component nanorotor ROT-1 that exhibited a highly dynamic alkyne → copper(I) dissociation (k298 = 240 kHz) at 298 K. Quantitative click reaction transformed ROT-1 into the new rotor ROT-2 (k298 = 77 kHz) with a triazole → copper(I) linkage thus opening perspectives for bioorthogonal click strategies to biohybrid machinery.
Inspired by nanomechanical motions1 in biological machines,2–4 scientists have developed an enormous interest in the development of artificial molecular devices.5–8 Among them, molecular motors,9–11 rotors,12 shuttles,13–15 tweezers,16–18 turnstiles,19 muscles,20 elevators,21 pumps,10 walkers22etc.5–8 are well studied. Though numerous examples of artificial covalent molecular devices are known in the literature,5–8 evolution toward multicomponent artificial machineries still represents a major challenge due to the limited amount of dynamic orthogonality in hetero-assemblies.23,24
For designing artificial multicomponent rotors, orthogonal dynamic interactions are a key requirement.25 To the best of our knowledge, all literature known dynamic interactions that have been used to construct artificial multicomponent rotors are derived from H-bonding or N,O-donor19 metal interactions.26–30 Clearly, development of any new dynamic interaction will open further opportunities. Here, we demonstrate for the first time a supramolecular assembly and a rotor built on the dynamic alkyne → copper(I) interaction.31,32 Specifically, we designed a four-component supramolecular assembly and nanorotor based on the heteroleptic Cu+-phenanthroline alkyne (HETPYNE: HETeroleptic Phenanthroline and alkYNE metal) complexation (Fig. 1). Addition of stoichiometric quantities of azide to the rotor afforded the new class of a Cu+-triazole rotor through an in situ copper(I) catalysed click reaction.
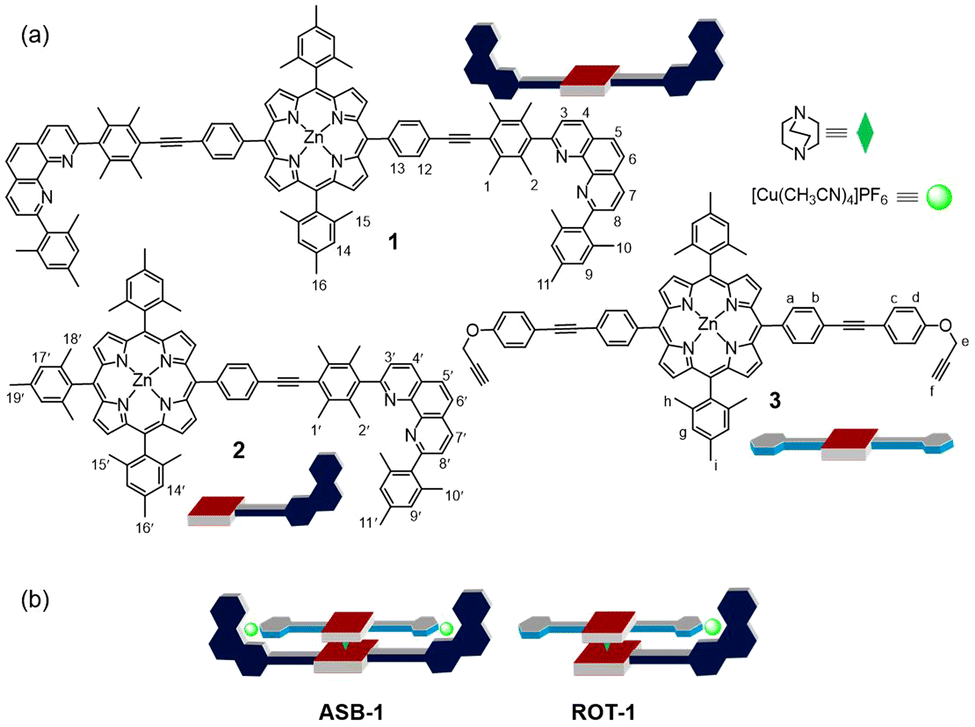 | ||
| Fig. 1 (a) Chemical structure and cartoon representation of the ligands 1, 2, 3 and DABCO. (b) Cartoon representation of the four-component assembly ASB-1 and nanorotor ROT-1. | ||
For our study, we decided to use the phenanthroline-appended zinc(II) porphyrin ligands 1 or 2 as stator. Bulky aryl groups33 at the 2,9-position of the phenanthroline phenAr2 are essential to avoid the unwanted formation of the corresponding homoleptic Cu+ complexes.34–36 In order to design rotator 3, we performed a few model experiments to evaluate the binding of a terminal ethynyl group to [Cu(phenAr2)]+. Mixing of 4, 5 and [Cu(CH3CN)4]PF6 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio (2.5 mM each) in CD2Cl2 accomplished quantitative formation of C1 = [Cu(4)(5)]+ (Fig. 2a). In the 1H NMR, a downfield shift of all phenanthroline protons indicated binding of 5 to [Cu(4)]+, for instance, proton 4′′-H shifted from 8.67 to 8.74 ppm and 5′′-H from 8.14 to 8.20 ppm (Fig. 2b). In contrast, protons d′-H (from 6.77 to 6.60 ppm) and e′-H (from 4.68 to 4.09 ppm) of the ethynyl ligand 5 shifted upfield upon its complexation to [Cu(4)]+, due to the shielding of these protons by the π-ring current of the mesityl groups. On the other hand, despite being in the shielding region of a strong π-electron cloud, the downfield shift of proton f′-H (from 5 to C1: 2.58 to 2.68 ppm) validated the ethynyl binding to the Cu+ center. Single crystal X-ray analysis of C1 revealed a triclinic crystal system with the space group P
:1:1 ratio (2.5 mM each) in CD2Cl2 accomplished quantitative formation of C1 = [Cu(4)(5)]+ (Fig. 2a). In the 1H NMR, a downfield shift of all phenanthroline protons indicated binding of 5 to [Cu(4)]+, for instance, proton 4′′-H shifted from 8.67 to 8.74 ppm and 5′′-H from 8.14 to 8.20 ppm (Fig. 2b). In contrast, protons d′-H (from 6.77 to 6.60 ppm) and e′-H (from 4.68 to 4.09 ppm) of the ethynyl ligand 5 shifted upfield upon its complexation to [Cu(4)]+, due to the shielding of these protons by the π-ring current of the mesityl groups. On the other hand, despite being in the shielding region of a strong π-electron cloud, the downfield shift of proton f′-H (from 5 to C1: 2.58 to 2.68 ppm) validated the ethynyl binding to the Cu+ center. Single crystal X-ray analysis of C1 revealed a triclinic crystal system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (ESI,† Fig. S27). Importantly, it clearly demonstrated the side-on binding of Cu+ to both ethynyl carbons whereas there was no binding visible between oxygen and Cu+ center (Fig. 2c). The solid state structure disclosed the bond lengths of Cu(1)–C(41), Cu(1)–C(42), Cu(1)–N(11) and Cu(1)–N(1) to be 1.958(5) Å, 1.969(4) Å, 2.002(3) Å and 2.013(3) Å, respectively. The angle between the planes defined by N(1)–Cu(1)–N(11) and C(41)–Cu(1)–C(42) was determined as 16°. This geometry around the copper(I) center is not very common. From an NMR titration, the binding constant of 5 to [Cu(4)]+ was determined as log K = 2.81 ± 0.16 (ESI,† Fig. S26). We propose to denote the heteroleptic complexation motif between a [Cu(phenAr2)]+ and an alkyne as HETPYNE interaction (vide supra).
(ESI,† Fig. S27). Importantly, it clearly demonstrated the side-on binding of Cu+ to both ethynyl carbons whereas there was no binding visible between oxygen and Cu+ center (Fig. 2c). The solid state structure disclosed the bond lengths of Cu(1)–C(41), Cu(1)–C(42), Cu(1)–N(11) and Cu(1)–N(1) to be 1.958(5) Å, 1.969(4) Å, 2.002(3) Å and 2.013(3) Å, respectively. The angle between the planes defined by N(1)–Cu(1)–N(11) and C(41)–Cu(1)–C(42) was determined as 16°. This geometry around the copper(I) center is not very common. From an NMR titration, the binding constant of 5 to [Cu(4)]+ was determined as log K = 2.81 ± 0.16 (ESI,† Fig. S26). We propose to denote the heteroleptic complexation motif between a [Cu(phenAr2)]+ and an alkyne as HETPYNE interaction (vide supra).
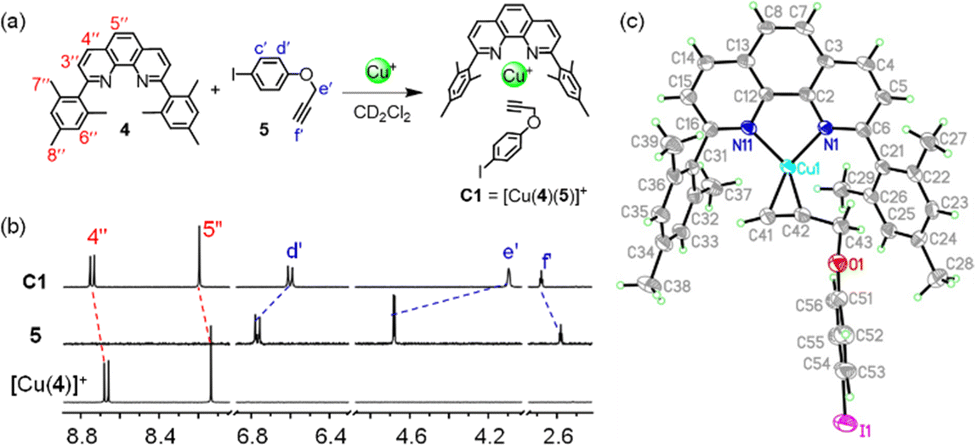 | ||
| Fig. 2 (a) Formation of model complex C1. (b) Partial 1H NMR (400 MHz, 298 K) of [Cu(4)]+, 5 and C1 in CD2Cl2 (2.5 mM). (c) X-ray crystal structure of complex C1. Carbons are shown in light grey; H, light green; N, blue; O, red; Cu+, cyan and I, violet. | ||
After establishing the HETPYNE motif, the zinc(II) porphyrin 3 with two ethynyl terminals was designed. To synthesize ligand 3, we first reacted 5,15-di(4-iodophenyl)-10,20-dimesityl zinc(II) porphyrin and 4-ethynylphenol under Sonogashira coupling conditions providing the corresponding diphenol. In the final step, a Williamson ether synthesis between the phenol-substituted zinc(II) porphyrin and propargyl bromide in presence of base furnished ligand 3 in 85% yield. Protons e-H of 3 appear in the 1H NMR well separated from other proton signals and should serve as good indicator of any binding.
As expected from the model studies, the four-component self-assembly ASB-1 was quantitatively afforded by mixing DABCO, ligands 1 & 3, and [Cu(CH3CN)4]PF6 in a 1:1:1:2 ratio in CD2Cl2 (Fig. 3a). Two characteristic multiplets for the CH2-units of DABCO in the negative region of the 1H NMR indicated quantitative formation of the hetero-sandwich complex (Fig. 3b).37 Significant changes at all phenanthroline protons in the 1H NMR upon moving from [Cu2(1)]2+ to ASB-1 supported the binding of 3 to the copper(I)-loaded phenanthroline stations (Fig. 3c and d). Downfield shift of proton f-H from 2.64 to 3.10 ppm in ASB-1 attested the terminal ethynyl binding of 3 at the Cu+ center of 1 (Fig. 3d). Drastic upfield shifts of proton signal e-H from 4.79 to 3.60 ppm and of d-H from 7.05 to 6.77 ppm along with a downfield shift of proton signal c-H from 7.64 to 7.75 ppm validated the formation of the HETPYNE complex. Furthermore, a single peak in the ESI-MS at m/z = 1489.1 confirmed formation of the hetero-assembly (ESI,† Fig. S23) and a single diffusion trace in the 1H-DOSY NMR representing structure ASB-1 excluded the presence of other undesired assemblies (ESI,† Fig. S20).
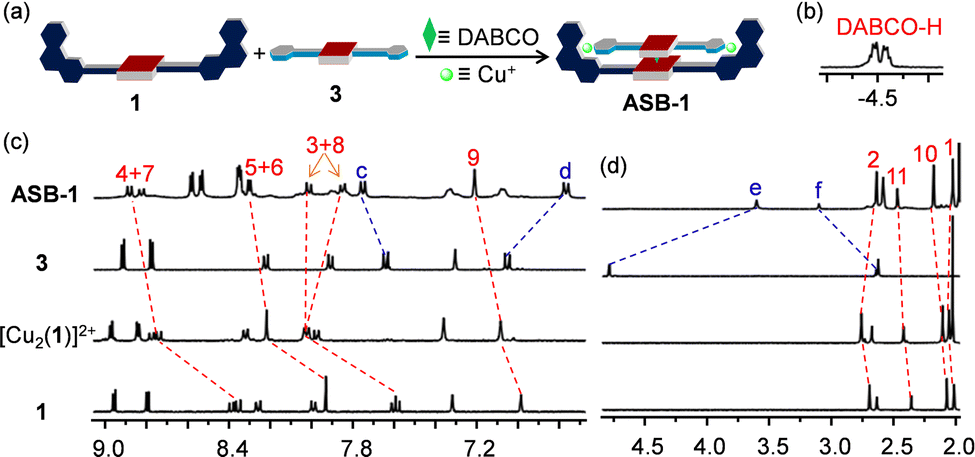 | ||
| Fig. 3 (a) Cartoon representation of the four-component self-assembly leading to the formation of ASB-1. (b) DABCO-H signal of ASB-1 in 1H NMR (CD2Cl2, 400 MHz, 298 K). Partial 1H NMR (CD2Cl2, 400 MHz, 298 K) of 1, [Cu2(1)]2+, 3 and ASB-1 showing the (c) aromatic and (d) aliphatic region. | ||
The clean formation of the heteroleptic sandwich complex encouraged us to test the HETPYNE motif as a dynamic interaction in a multicomponent rotor. To assemble the rotor, we selected zinc(II) porphyrin 2 containing just one phenanthroline station as stator and ligand 3 as rotator. Dissolving the ligands 2, 3, DABCO and [Cu(CH3CN)4]PF6 in a 1:1:1:1 ratio in CD2Cl2 quantitatively furnished rotor ROT-1 irrespective of the sequence of addition (Fig. 4a). As in ASB-1, two broad signals in the negative region corresponding to DABCO and significant shifts of all phenanthroline protons in the 1H NMR validated formation of the heteroassembly (Fig. 4b–d). Upfield shifts of rotator proton signals e-H from 4.79 to 4.19 ppm along with downfield shift of f-H from 2.64 to 2.88 ppm authenticated the rotor structure (Fig. 4c and d). Its formation was further confirmed by DOSY NMR and ESI-MS data (ESI,† Fig. S21 and S24).
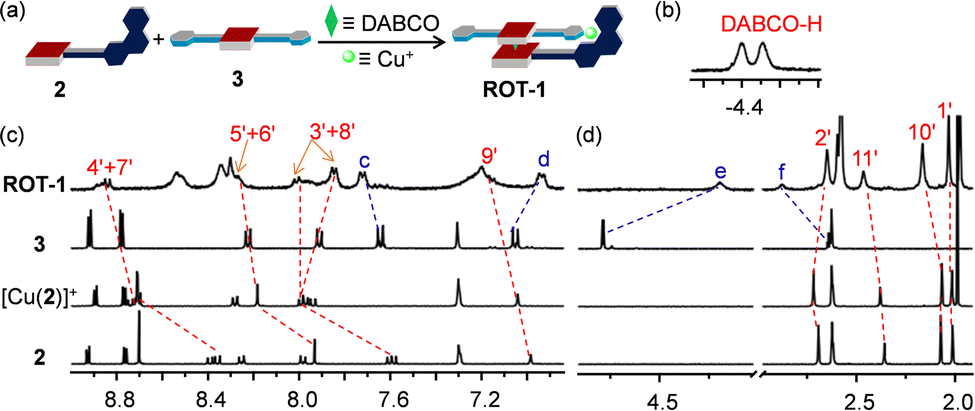 | ||
| Fig. 4 (a) Cartoon representation of the self-assembly of rotor ROT-1 from four components. (b) (CH2)DABCO signal of ROT-1 in the 1H NMR (CD2Cl2, 400 MHz). Partial 1H NMR (CD2Cl2, 400 MHz, 298 K) of 2, [Cu(2)]+, 3 and ROT-1 in the (c) aromatic and (d) aliphatic region. | ||
A single set of 1H NMR signals for protons c-H, d-H, e-H and f-H of ROT-1 suggested fast rotation of the rotor on the NMR time scale (Fig. 4c and d). Comparison of the 1H NMR spectra of the free rotator 3, ROT-1 and ASB-1 showed that the proton signals d-H, e-H and f-H of rotor ROT-1 appeared approximately in the averaged position of those of free 3 and ASB-1 (Fig. 5a). Variable temperature (VT) 1H NMR of ROT-1 was thus performed to evaluate its dynamic behavior. Upon lowering the temperature, the sharp singlet at 4.19 ppm corresponding to proton e-H broadened and split into two singlets in a 1:1 ratio at −75 °C with a coalescence temperature around −50 °C (Fig. 5b). The upfield signal at 3.50 ppm was assigned to the HETPYNE-complexed proton e-H and the downfield signal at 4.71 ppm is ascribed to proton e-H at the uncomplexed arm. The rotational frequency of the rotor at different temperatures was evaluated using winDNMR-based spectral simulations.38 The activation data for the rotation was derived from the Eyring plot (Table 1 and ESI,† Fig. S18). The rotational frequency turned out to be 240 kHz at 25 °C and ΔG‡298 = 42.5 kJ mol−1.
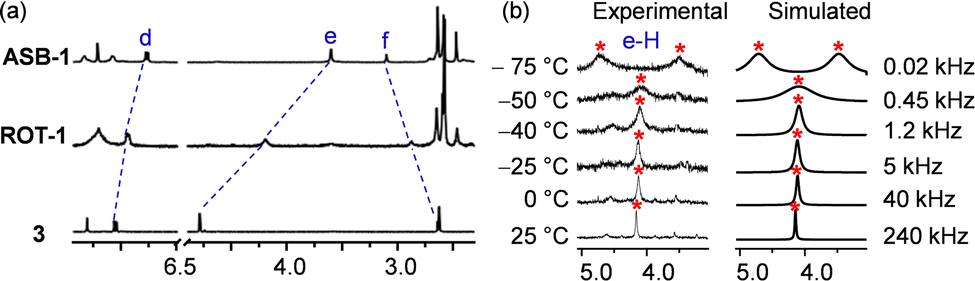 | ||
| Fig. 5 (a) Partial 1H NMR (CD2Cl2, 400 MHz, 298 K) of 3, ROT-1 and ASB-1. (b) VT-1H NMR (CD2Cl2, 600 MHz) of ROT-1 exhibiting the splitting of proton signal e-H into a 1:1 set and the corresponding rotational frequency at different temperatures. | ||
| Rotor | k 298/kHz | ΔH‡/kJ mol−1 | ΔS‡/J K−1 mol−1 | ΔG‡298/kJ mol−1 |
|---|---|---|---|---|
| a The higher ΔH‡ for ROT-2 than ROT-1 reflects the stronger binding constant of a triazole to [Cu(4)]+ (see triazole 6 in ESI, Fig. S27). As often seen in enthalpy–entropy compensation, strong binding leads to higher positive activation entropy. | ||||
| ROT-1 | 240 | 44.0 ± 0.2 | 5.0 ± 0.7 | 42.5 |
| ROT-2 | 77 | 50.1 ± 0.4 | 16.7 ± 0.6 | 45.2 |
After the clean formation of rotor ROT-1, our next target was the in situ rotor-to-rotor transformation. The presence of a copper(I) ion and terminal alkynes in the rotor suggested a conversion of ROT-1 to a triazole rotor through an in situ click reaction. For this purpose, 2.0 equiv. of benzyl azide was added to ROT-1 in CD2Cl2 (Fig. 6a). To accelerate the reaction, 1 μL of Et3N was added. After 24 h of heating at 40 °C, the solvent was evaporated to remove NEt3 and the residue was redissolved in CD2Cl2. 1H NMR showed quantitative formation of ROT-2 and a disappearance of the proton signal f-H (Fig. 6b). Upon moving from ROT-1 to ROT-2, characteristic shifts for all phenanthroline protons were observed. The downfield shift of proton signal e-H (from 4.19 to 4.81 ppm), upfield shifts of proton signals d-H (from 6.93 to 6.78 ppm) and c-H (from 7.72 ppm to 7.61 ppm) along with the appearance of a new singlet at 5.52 ppm (j-H) corroborated the formation of ROT-2. The broad signal of the DABCO protons at −4.39 ppm confirmed the intactness of the assembly (ESI,† Fig. S15). ROT-2 was further characterized by ESI-MS and DOSY NMR data (ESI,† Fig. S25 and S22).
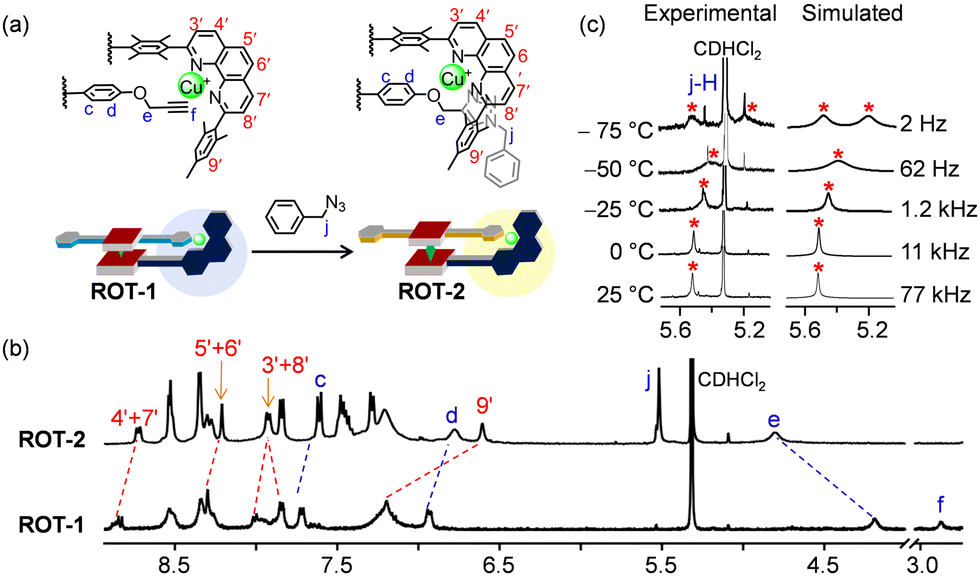 | ||
| Fig. 6 (a) In situ transformation of ROT-1 to ROT-2 upon addition of 2.0 equiv. of benzyl azide. (b) Partial 1H NMR (CD2Cl2, 400 MHz, 298 K) of ROT-1 and ROT-2. (c) VT-1H NMR (CD2Cl2, 600 MHz) of ROT-2 showing splitting of the proton signal j-H (1:1 ratio) and the corresponding rotational frequency at different temperatures. | ||
A single set of 1H NMR signals for protons c-H, d-H, e-H and j-H of ROT-2 indicated a fast rotation on the NMR time scale. Upon performing the VT 1H NMR the proton signal for j-H split into two singlets in 1:1 ratio at −75 °C (Fig. 6c). Rotational frequencies at different temperature along with activation parameters were calculated (Fig. 6c and Table 1). The facile transformation of the self-catalyzing rotor ROT-1 to rotor ROT-2 opens interesting perspectives to generate biohybrid materials via bioorthogonal click reactions.39
In conclusion, we have synthesized a four-component heterosandwich complex and a four-component rotor based on the dynamic [Cu(phenAr2)(alkyne)]+ motif. Though alkyne → copper(I) interactions are well known in the literature,40 for the first time its high dynamics has been determined and used to assemble a high-speed multicomponent rotor. The utility of this dynamic orthogonal motif in supramolecular rotors opens new venues for molecular machines. Furthermore, a successful quantitative transformation of the Cu+-alkyne rotor to a new Cu+-triazole rotor was achieved through in situ click reaction. It is expected that thermal self-catalyzing rotors will find their way into diverse applications, e.g., in catalysis,35,41 biohybrid materials via bioorthogonal functionalization42 and elsewhere.43
We are indebted to the Deutsche Forschungsgemeinschaft for continued support under Schm 647/22-1 (No 491092614). We thank Dr Paululat for measuring the VT-1H NMR.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Dutta and B. Jana, Chem. Commun., 2021, 57, 272–283 RSC.
- J. E. Walker, Biochem. Soc. Trans., 2013, 41, 1–16 Search PubMed.
- R. D. Vale and R. A. Milligan, Science, 2000, 288, 88–95 Search PubMed.
- H. Noji, R. Yasuda, M. Yoshida and K. Kinosita, Jr, Nature, 1997, 386, 299–302 CrossRef CAS PubMed.
- A. Goswami, S. Saha, P. Biswas and M. Schmittel, Chem. Rev., 2020, 120, 125–199 CrossRef CAS PubMed.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 Search PubMed.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Chem. Rev., 2015, 115, 7729–7793 CrossRef CAS PubMed.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- Y. Feng, M. Ovalle, J. S. W. Seale, C. K. Lee, D. J. Kim, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 5569–5591 Search PubMed.
- M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200–268 Search PubMed.
- B. Lin, I. Karki, P. J. Pellechia and K. D. Shimizu, Chem. Commun., 2022, 58, 5869–5872 Search PubMed.
- T. Kumpulainen, M. R. Panman, B. H. Bakker, M. Hilbers, S. Woutersen and A. M. Brouwer, J. Am. Chem. Soc., 2019, 141, 19118–19129 Search PubMed.
- H. V. Schröder, F. Stein, J. M. Wollschläger, S. Sobottka, M. Gaedke, B. Sarkar and C. A. Schalley, Angew. Chem., Int. Ed., 2019, 58, 3496–3500 CrossRef PubMed.
- J. E. M. Lewis, R. J. Bordoli, M. Denis, C. J. Fletcher, M. Galli, E. A. Neal, E. M. Rochette and S. M. Goldup, Chem. Sci., 2016, 7, 3154–3161 RSC.
- B. Doistau, L. Benda, J.-L. Cantin, L.-M. Chamoreau, E. Ruiz, V. Marvaud, B. Hasenknopf and G. Vives, J. Am. Chem. Soc., 2017, 139, 9213–9220 CrossRef CAS PubMed.
- H. Yoon, J. M. Lim, H.-C. Gee, C.-H. Lee, Y.-H. Jeong, D. Kim and W.-D. Jang, J. Am. Chem. Soc., 2014, 136, 1672–1679 CrossRef CAS PubMed.
- A. Petitjean, R. G. Khoury, N. Kyritsakas and J.-M. Lehn, J. Am. Chem. Soc., 2004, 126, 6637–6647 Search PubMed.
- B. Godde, A. Jouaiti, A. Fluck, N. Kyritsakas, M. Mauro and M. W. Hosseini, Dalton Trans., 2017, 46, 14897–14906 Search PubMed.
- M. C. Jimenez, C. O. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284–3287 Search PubMed.
- J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 Search PubMed.
- M. von Delius, E. M. Geertsema and D. A. Leigh, Nat. Chem., 2010, 2, 96–101 Search PubMed.
- M. Schmittel, Chem. Commun., 2015, 51, 14956–14968 RSC.
- M. Schmittel, Isr. J. Chem., 2018, 59, 197–208 CrossRef.
- P. K. Biswas, S. Saha, Y. Nanaji, A. Rana and M. Schmittel, Inorg. Chem., 2017, 56, 6662–6670 Search PubMed.
- B. E. Dial, P. J. Pellechia, M. D. Smith and K. D. Shimizu, J. Am. Chem. Soc., 2012, 134, 3675–3678 CrossRef CAS PubMed.
- S. Saha, P. K. Biswas and M. Schmittel, Inorg. Chem., 2019, 58, 3466–3472 CrossRef CAS PubMed.
- P. K. Biswas, A. Goswami, S. Saha and M. Schmittel, Chem. – Eur. J., 2020, 26, 14095–14099 Search PubMed.
- I. Paul, A. Goswami, N. Mittal and M. Schmittel, Angew. Chem., Int. Ed., 2018, 57, 354–358 Search PubMed.
- S. Hiraoka, M. Shiro and M. Shionoya, J. Am. Chem. Soc., 2004, 126, 1214–1218 Search PubMed.
- D. Parasar, T. T. Ponduru, A. Noonikara-Poyil, N. B. Jayaratna and H. V. R. Dias, Dalton Trans., 2019, 48, 15782–15794 Search PubMed.
- H. V. R. Dias, J. A. Flores, J. Wu and P. Kroll, J. Am. Chem. Soc., 2009, 131, 11249–11255 Search PubMed.
- S. K. Samanta and M. Schmittel, J. Am. Chem. Soc., 2013, 135, 18794–18797 Search PubMed.
- B. Ralahy, U. Hahn, E. Wasielewski and J.-F. Nierengarten, Eur. J. Inorg. Chem., 2021, 2625–2635 Search PubMed.
- S. Saha, A. Ghosh, T. Paululat and M. Schmittel, Dalton Trans., 2020, 49, 8693–8700 RSC.
- S. De, S. Pramanik and M. Schmittel, Angew. Chem., Int. Ed., 2014, 53, 14255–14259 CrossRef CAS PubMed.
- S. K. Samanta, D. Samanta, J. W. Bats and M. Schmittel, J. Org. Chem., 2011, 76, 7466–7473 Search PubMed.
- H. J. Reich, NMR Spectrum Calculations: WinDNMR, Version 7.1.13, Department of Chemistry, University of Wisconsin, 2008 Search PubMed.
- C. G. Parker and M. R. Pratt, Cell, 2020, 180, 605–632 Search PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 Search PubMed.
- A. Goswami, T. Paululat and M. Schmittel, J. Am. Chem. Soc., 2019, 141, 15656–15663 CrossRef CAS PubMed.
- https://www.nobelprize.org/uploads/2022/10/popular-chemistryprize2022.pdf .
- K. T. Fam, L. Saladin, A. S. Klymchenko and M. Collot, Chem. Commun., 2021, 57, 4807–4810 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterizations, spectral data, NMR titration, and VT-1H-NMR kinetics. CCDC 2199510. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc04497h |
| This journal is © The Royal Society of Chemistry 2022 |