
Competitive gold/nickel transmetalation†
Mitchell J.
Demchuk‡
 a,
Joseph A.
Zurakowski‡
a,
Brady J. H.
Austen
a,
David J.
Nelson
*b and
Marcus W.
Drover
*a
a,
Joseph A.
Zurakowski‡
a,
Brady J. H.
Austen
a,
David J.
Nelson
*b and
Marcus W.
Drover
*a
aDepartment of Chemistry and Biochemistry, The University of Windsor, 401 Sunset Avenue, Windsor, ON N9B 3P4, Canada. E-mail: marcus.drover@uwindsor.ca
bWestCHEM Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, Scotland, UK. E-mail: david.nelson@strath.ac.uk
First published on 26th November 2021
Abstract
Transmetalation is a key method for the construction of element–element bonds. Here, we disclose the reactivity of [NiII(Ar)(I)(diphosphine)] compounds with arylgold(I) transmetalating agents, which is directly relevant to cross-coupling catalysis. Both aryl-for-iodide and unexpected aryl-for-aryl transmetalation are witnessed. Despite the strong driving force expected for Au–I bond formation, aryl scrambling can occur during transmetalation and may complicate the outcomes of attempted catalytic cross-coupling reactions.
The forging of metal–element bonds via transmetalation is a pivotal elementary step in catalytic cross-coupling for access to a variety of functionalized molecules, to the extent that cross-coupling reactions are defined and categorised by their transmetalation event.1 Frequently, this reaction follows substrate oxidative addition and precedes product reductive elimination.1 Many such coupling reactions have emerged, relying on electropositive metals for functional group (R) delivery viz. Kumada (R—MgBr), Suzuki (R—BR2), Negishi, (R—ZnX), Stille (R—SnR3), and Hiyama (R—SiR3), to name a few.1,2 In 2009, Hashmi et al. introduced organogold(I) compounds ([R—Au(Ln)]; Ln = ligand) as competent partners for catalytic cross-coupling using palladium(0).3
Blum and co-workers subsequently reported the room temperature [NiCl2(PCy3)2]-catalysed cross-coupling of [R—Au(PPh3)] complexes with aryl bromides.4 This reaction is proposed to proceed via an open-shell Ni complex wherein [NiII(Cl)2(PCy3)2] first undergoes transmetalation with 2 equivalents of [Ar—Au(PPh3)] to give [NiII(Ar)2(PCy3)2] and 2 equivalents of [Cl—Au(PPh3)], presumably via [NiII(Ar)(Cl)(PCy3)2]; subsequent halide transfer and oxidation by Au(I) gives [NiIII(Ar)2(Cl)(PCy3)2] and Au(0). This NiIII species would thus reductively eliminate Ar—Ar to provide [NiI(Cl)(PCy3)2] and enter the catalytic cycle, undergoing transmetalation, oxidative addition, and reductive elimination, giving a C—C coupled product. To buttress this proposal, the stoichiometric reaction of [NiCl2(PCy3)2] with 10 equivalents of [(4-(MeO)C6H4)—Au(PPh3)] resulted in the homocoupled biaryl product, and a paramagnetic nickel complex that was characterised by EPR spectroscopy (Chart 1A).4
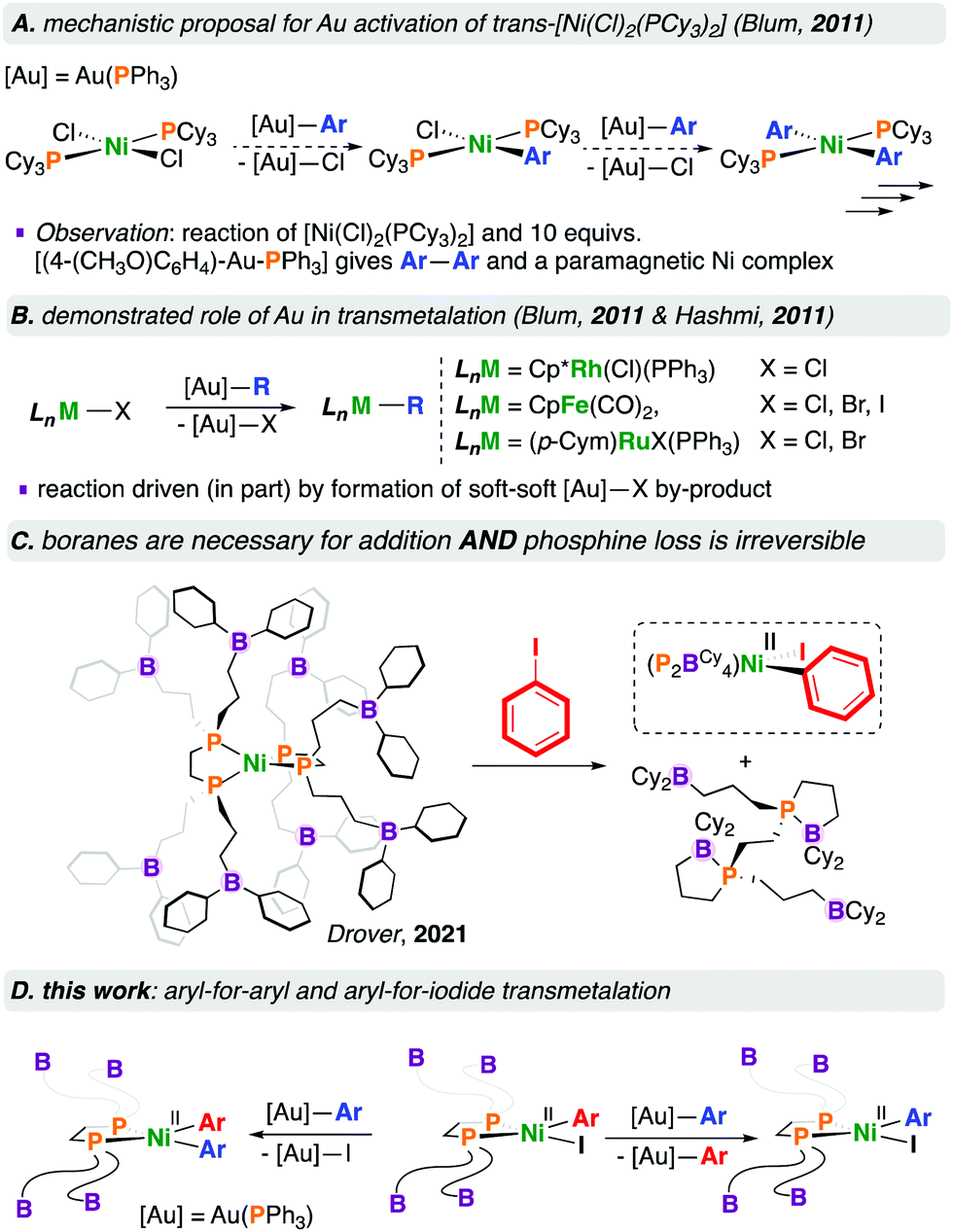 | ||
| Chart 1 Literature precedent and accessing a [Ni(diphosphine)(Ar)(I)] precursor and its reactivity with organogold(I) transmetalating reagents. | ||
Organogold(I) compounds are competent for transmetalation to (e.g.) [CpFe(CO)2(X)] (Cp = C5H5−, X = Cl, Br, I),5 [(p-Cym)Ru(X)2(PPh3)] (p-Cym = p-cymene, X = Br, I),5 and [Cp*Rh(Cl)2(PPh3)] (Cp* = C5Me5−) (Chart 1B).6 For the reaction of [CpFe(CO)2(X)] with [(4-(NO2)C6H4)—Au(PPh3)], the rate of reaction followed the order I > Br > Cl, consistent with the affinity of gold(I) for soft halides in the [X—Au(PPh3)] by-product.5 We refer interested readers to several reviews pertaining to [Au]—based transmetalations.7
We recently reported the fundamental reactivity of [Ni0(P2BCy4)2] (P2BCy4 = 1,2-bis(di(3-dicyclohexylboraneyl)propylphosphino)ethane), which undergoes room temperature iodoarene activation to afford [NiII(P2BCy4)(Ar)(I)] and P2BCy4 (Chart 1C).8 This reaction is noteworthy as the treatment of alkyl/aryl-substituted diphosphine complexes of the form [Ni0(diphosphine)2] with haloarenes generally does not result in C–X bond activation, due to the significant endergonicity associated with forming the [Ni0(κ1-diphosphine)(κ2-diphosphine)] complex required for substrate activation;9 [Ni0(dnppe)2] (dnppe = 1,2-bis(di-n-propylphosphino)ethane) does not undergo oxidative addition under similar conditions.
To extend the reactivity of such P2BCy4-ligated complexes, we elected to study the behaviour of [NiII(P2BCy4)(Ar)(I)] with a transmetalating reagent, as a means to explore the second elementary step in a viable cross-coupling sequence using [Ni0(P2BCy4)2]. Our interests turned to [Au]—based reagents because these reagents are accurately weighed in small quantities, soluble in hydrocarbon solvents, readily tailored, and are unlikely to undergo transfer to the sp2-hybridized boranes present on the P2BCy4 ligand.8 Furthermore, these agents are bench-stable and provide [X—Au(PPh3)] as a by-product, whose formation can be conveniently monitored by 31P NMR spectroscopy.10 In addition to probing the stepwise reactivity of “[Ni0/II(P2BCy4)]”-type compounds, we were equally interested in contributing to the growing area of gold-to-metal transmetalations starting from a bona-fide [NiII(diphosphine)(Ar)(X)] complex, informing studies such as that noted for [NiII(Cl)2(PCy3)2].4
[NiII(P2BCy4)(4-FC6H4)(I)] (2-F), generated from oxidative addition of 4-fluoroiodobenzene at [Ni(P2BCy4)2] (1), was selected as the transmetalation partner, enabling reaction monitoring by 19F NMR spectroscopy.8 Transmetalation was examined using gold(I) aryl compounds with electron-donating or -withdrawing groups at the para-position, [(4-X-C6H4)—Au(PPh3)] (X = H, OCH3, CF3) (Scheme 1). Initially, compound 2-F was combined with 1 equiv. of [(C6H5)—Au(PPh3)], and the reaction was analysed by NMR spectroscopy. Given the literature precedent noted above, we hypothesized that this would represent an ideal pairing, owing to the favourable generation of [I—Au(PPh3)]. However, monitoring the reaction by 31P NMR spectroscopy evidenced formation of [Ni(P2BCy4)(C6H5)(I)]8 (2-H) in 25% yield after 7 mins, resulting from ‘aryl-to-aryl’ transmetalation. The fate of the exchanged “4-FC6H4” unit was deduced by 19F NMR spectroscopy, matching data reported for [(4-FC6H4)—Au(PPh3)].10 In addition to signals for 2-H, a signal at δP = 43 ppm was also noted for [(4-FC6H4)—Au(PPh3)] in the 31P NMR spectrum. The formation of 4-fluorobiphenyl, from reductive elimination via [Ni(P2BCy4)(4-FC6H4)(C6H5)], was observed by 19F NMR spectroscopy, indicating that both Ar-for-Ar and Ar-for-I transmetalation processes operate. The treatment of 2-F with 1 equiv. of [(4-X-C6H4)—Au(PPh3)] (X = OCH3, CF3) gave [Ni(P2BCy4)(4-X-C6H4)(I)] (2-X; X = OCH3 (17%), CF3 (38%)) after 7 minutes.
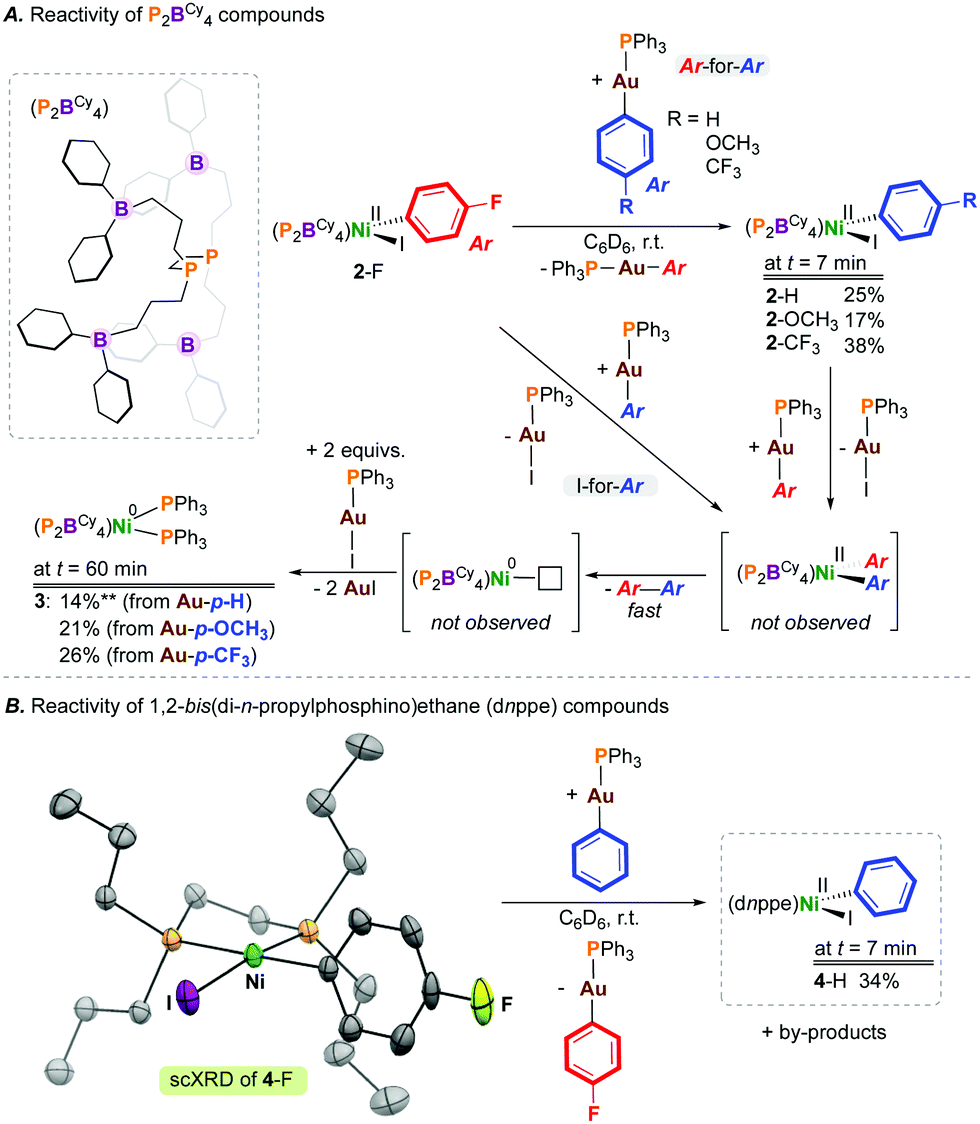 | ||
| Scheme 1 Transmetalation of 2-F and 4-F using [Ar—Au(PPh3)] reagents. Yields taken after 7 min. Inset of B shows the scXRD structure of 4-F (50% occupancy, hydrogens omitted for clarity). ** The maximum theoretical yield of 3 is 50%. dnppe = 1,2-bis(di-n-propylphosphino)ethane. | ||
Compounds 2-X were highly reactive under the conditions studied. Up to 50% of [Ni]total (after ca. 60 mins) in these elementary reactions is [Ni0(P2BCy4)(PPh3)2] (3) δP = 34.8 (t, 2JP,P = 27 Hz; PPh3), 19.7 (t, 2JP,P = 27 Hz; P2BCy4), which forms via PPh3 transfer from Au following C(Ar)—C(Ar) reductive elimination in yields from 14–26%, based on Ni (Scheme 1). The observation of some S = 0 Ni(0) product suggests reductive elimination from Ni(II). The identity of 3 was confirmed by its independent preparation from the reaction of [Ni0(P2BCy4)2] with 2 equiv. of PPh3. This reaction illustrates the lability of the P2BCy4 ligand scaffold; the related species [Ni0(dnppe)2], which is devoid of pendant boranes, does not coordinate PPh3.
To demonstrate the generality of our observations regarding transmetalation between gold and nickel, and to show that this is not a boron effect, [Ni(dnppe)(4-FC6H4)(I)] (4-F) (see ESI†) was prepared, characterized, and exposed to 1 equiv. of [(C6H5)—Au(PPh3)]. Analysis of the reaction mixture after 7 min by NMR spectroscopy showed formation of [Ni(dnppe)(C6H5)(I)] (4-H) in 34% yield, consistent with previous results for P2BCy4.
The relevance of such compounds viz. [Ni(PP2BCy4)2] (1) in cross-coupling was also confirmed with 10 mol% 1 enabling the cross-coupling of 4-fluoroiodobenzene and [(4-FC6H4)—Au(PPh3)], delivering the corresponding biaryl in 86% conversion by 19F NMR spectroscopy.
To probe the thermodynamics associated with transmetalation, density functional theory (DFT)11 and DLPNO-CCSD(T) calculations12 were carried out on model reactions (Scheme 2). In all cases, there was excellent agreement between these methods. The data from DLPNO-CCSD(T) calculations are discussed here, but DFT-derived energies are reported in Scheme 2 and in the ESI.† The outcomes from I-for-Aryl and Ar-for-Ar transmetalation at 4-F are endergonic and energetically neutral, respectively. Somewhat surprisingly, the exchange of iodine for phenyl is endergonic (ΔG° = +2.6 kcal mol−1). The exchange of para-fluorophenyl for phenyl (i.e. formation of 4-H from 4-F) is essentially energetically neutral (ΔG° = −0.1 kcal mol−1) (similar trends are observed with gold reagents having p-F3CC6H4, p-H3COC6H4, and 4-pyridyl substituents (vide infra) – see ESI†). In the context of catalysis, the product of I-for-Ar transmetalation can undergo reductive elimination, driving catalysis forward if the overall reaction is exergonic; however, these experimental and computational results indicate the potential for aryl scrambling during cross-coupling reactions, eroding reaction selectivity and forming undesired homocoupling products.
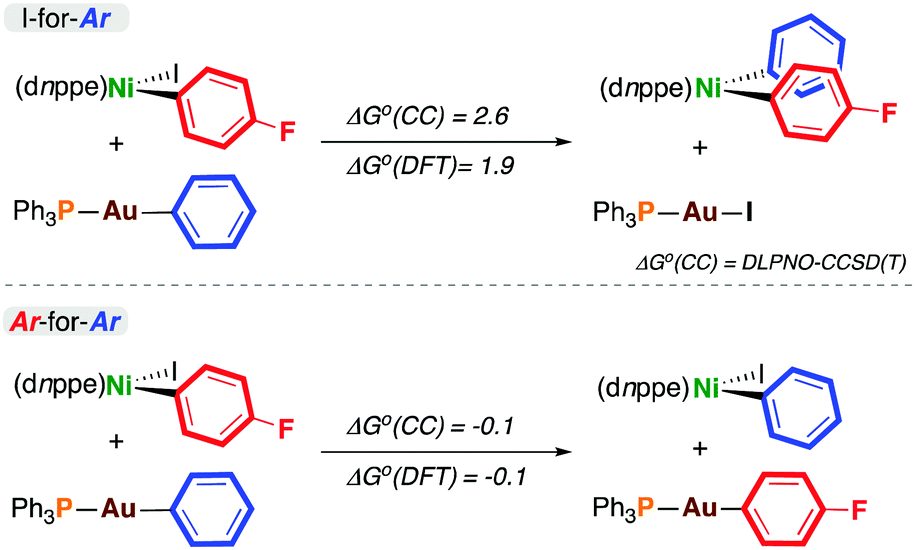 | ||
| Scheme 2 Thermodynamics of transmetalation (kcal mol−1). ΔG°(CC) was calculated using DLPNO-CCSD(T), and ΔG°(DFT) using DFT.11,12 | ||
Previously, we showed that [Ni(P2BCy4)2] (1) coordinates Lewis bases, binding up to eight equiv. of 4-N,N-dimethylaminopyridine (DMAP).13 We wondered whether an appropriate gold(I)-pyridyl fragment (i.e. [Au]—(4-NC5H4)) might instead be used for coordination, with the P2BCy4 ligand serving in a directing capacity, luring transmetalating agents into close proximity with a Ni(II) fragment. A new compound, [(4-NC5H4)—Au(PPh3)] (5) was prepared via the reaction of [Br—Au(PPh3)] with 4-pyridylboronic acid (75% yield) (Scheme 3A).14 The formation of 5 is substantiated by NMR spectroscopy (δP = 43 ppm) and single crystal X-ray diffraction(Scheme 3A). Importantly, 5 boasts an accessible Lewis basic pyridine, allowing for possible interaction with the pendant boraneyl groups of [Ni(P2BCy4)2] (1).
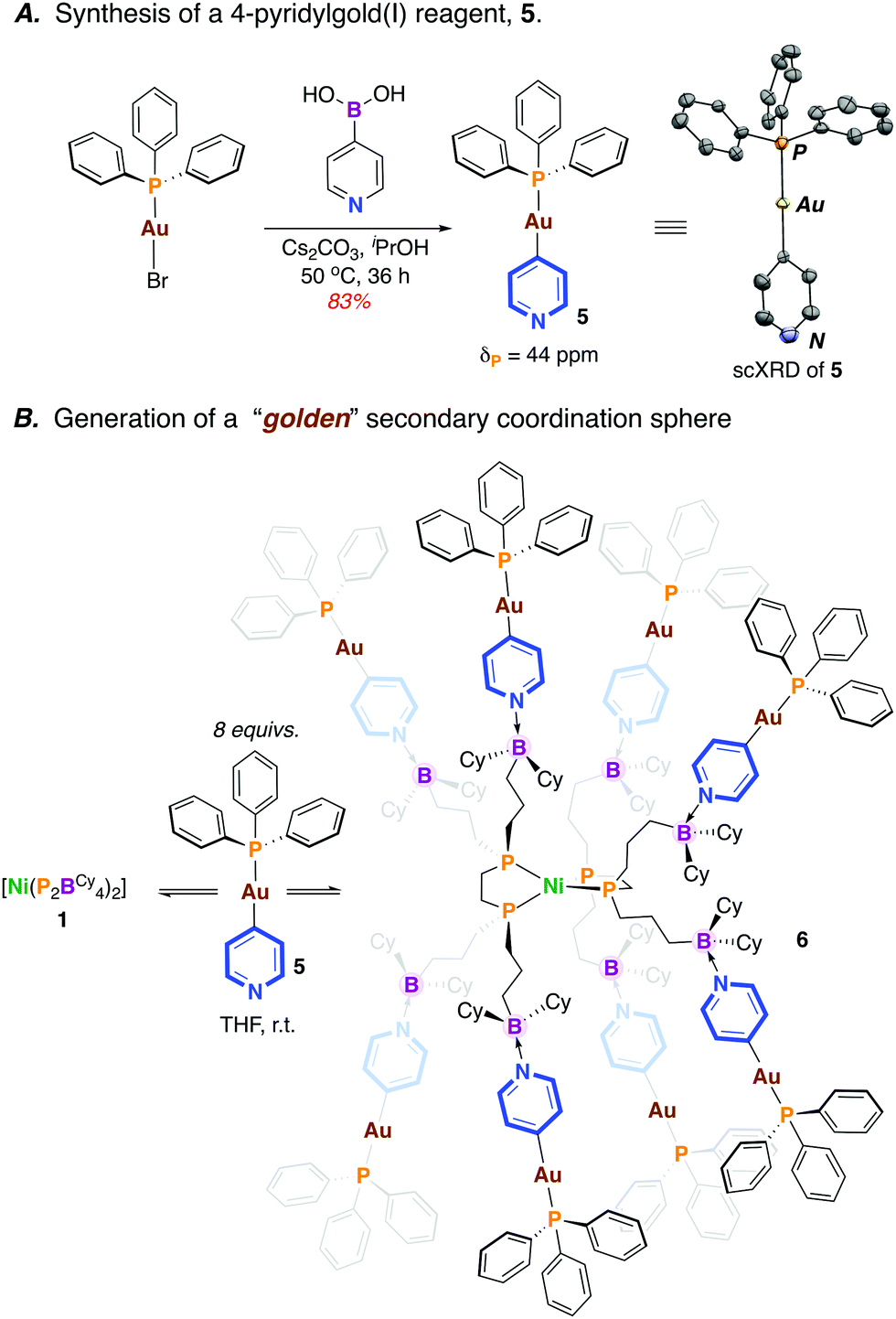 | ||
| Scheme 3 A. Synthesis of a 4-pyridylgold(I) reagent 5. Inset shows the scXRD structure of 5 (50% occupancy, hydrogens omitted for clarity). B. Synthesis of octaaurated compound 6. | ||
The reaction of 1 with 8 equiv. of 5 in THF15 provided an immediate colour change from light yellow to dark brown to give [Ni0(P2BCy4)2{(4—NC5H4)—AuI(PPh3)}8] (6) (Scheme 3B). Analysis by 31P NMR spectroscopy (THF-d8) provided two broad signals (δP = 42, 36 ppm) that are shifted upfield compared to uncoordinated 5 (ΔδP = −1 ppm) and 1 (ΔδP = −3 ppm) (see ESI†).13 Consistent with an array of interacting borane–pyridine groups, the 31P NMR signature for the equivalent [Ni]—P fragments is similar to [Ni0(P2BCy4)2(DMAP)8] (δP = 35 ppm).13 No signal is detected by 11B NMR spectroscopy at 298 K cf. δB = 84 ppm for free [Ni0(P2BCy4)2] and δB = +4 ppm for [Ni0(P2BCy4)2(DMAP)8].13 To probe the possibility of fluxional solution behaviour, a variable temperature (VT) NMR study was undertaken. At 193 K, the 31P NMR spectrum is markedly different; the broad signal associated with [Au]—P groups decoalesces, while the signal for the [Ni]—P groups moves upfield and the baseline broadens (see ESI†). Fluxional behaviour is also witnessed by VT 11B NMR spectroscopy, which at 263 K shows an averaged signal for the sp3-hybridized boraneyl groups at δB = −4 ppm; this sharpens considerably at 193 K. These data demonstrate the proclivity of the pendant boranes of the [Ni0(P2BCy4)2] framework to support a metal-rich secondary coordination sphere, an attractive approach towards accessing multimetallic complexes that are “docked” via Lewis acid/base interactions.
Previously, we showed that reaction of [Ni0(P2BCy4)2(DMAP)8] with PhI resulted in the formation of [NiII(P2BCy4)2(DMAP)8(I)]I via iodine atom abstraction.8 We probed the reactivity of 6 with 4-fluoroiodobenzene but protection of the Ni(0) site by a “golden” secondary coordination sphere prevents oxidative addition. Nonetheless, switching the order of addition and allowing 1 to fully react with 4-fluoroiodobenzene to give [NiII(P2BCy4)(4-FC6H4)(I)] (2-F), followed by addition of 5, provided the pyridyl-linked oligomer, [NiII(P2BCy4)(4-NC5H4)(I)]n8 in 24% yield after 7 mins by 31P NMR spectroscopy, which results from aryl-to-pyridyl transmetalation (Scheme 4).16 Under these conditions 2-Pyr was noted to be more robust than 2-X, producing less 3; 4-(4-fluorophenyl)pyridine was not detected by 19F NMR spectroscopy. Interestingly, the reaction between [NiII(dnppe)(4-FC6H4)(I)] (3-F) and 5 in THF provides a cloudy reaction mixture and [NiII(dnppe)(4-Pyr)(I)] is not observed.
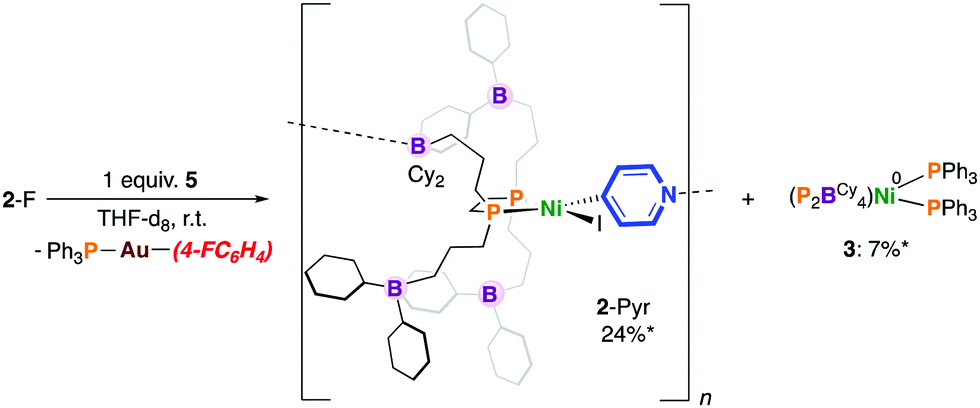 | ||
| Scheme 4 Transmetalation using a Au-based 4-pyridyl reagent. * = Yield after 60 min (the maximum theoretical yield of 3 is 50%). | ||
In sum, we have confirmed that isolated [NiII(diphosphine)(Ar)(I)] compounds undergo aryl-for-aryl in addition to aryl-for-iodide transmetalation with arylgold(I) reagents. Furthermore, we have exploited the boron-rich secondary coordination sphere of [Ni(P2BCy4)2] to host a metal (gold)-rich secondary coordination sphere via pyridine–borane dative interactions, a starting point toward achieving metal–metal cooperativity using such boron-rich ligand scaffolds in an elementary cross-coupling transformation. This study expands our knowledge of Au-to-metal transmetalation and provides insights into the initial steps associated with metal-to-metal functional group transfer relevant to carbon–carbon cross-coupling.
The authors are grateful to the University of Windsor, the Council of Ontario Universities, Compute Canada, and the Natural Sciences and Engineering Research Council of Canada (Discovery Grant, RGPIN-2020-04480, Discovery Launch Supplement, DGECR-2020-00183, and a graduate award (CGS-M) to J. A. Z.) for funding. This work was also made possible by the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET: www.sharcnet.ca) and Compute/Calcul Canada. D. J. N. thanks the Carnegie Trust for the Universities of Scotland for a Research Incentive Grant (RIG008165). Some of the calculations were performed using the Archie-WEST High Performance Computer (http://www.archie-west.ac.uk) at the University of Strathclyde; we thank Mr J. Buzzard, Dr K. Kubiak-Ossowska, and Dr R. Martin for their assistance with this facility.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. F. Hartwig, Organotransition Metal Chemistry From Bonding to Catalysis. Chapter 7, University Science Books, Mill Valley, California. 2010 Search PubMed.
- (a) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS; (b) N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem., 2017, 1, 0025 CrossRef CAS PubMed; (c) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS.
- (a) A. S. K. Hashmi, C. Lothschütz, R. Döpp, M. Rudolph, T. D. Ramamurthi and F. Rominger, Angew. Chem., Int. Ed., 2009, 48, 8243–8246 CrossRef CAS; (b) M. Hansmann, M. Pernpointner, R. Dopp and A. S. K. Hashmi, Chem. – Eur. J., 2013, 19, 15290–15303 CrossRef CAS PubMed; (c) For a related study, see: S. Witzel, M. Hoffmann, M. Rudolph, F. Rominger, A. Dreuw and A. S. K. Hashmi, Adv. Synth. Catal., 2021 DOI:10.1002/adsc.202101113.
- J. J. Hirner and S. A. Blum, Organometallics, 2011, 30, 1299–1302 CrossRef CAS.
- A. S. K. Hashmi and L. Molinari, Organometallics, 2011, 30, 3457–3460 CrossRef CAS.
- Y. Shi and S. A. Blum, Organometallics, 2011, 30, 1776–1779 CrossRef CAS.
- (a) J. J. Hirner, Y. Shi and S. A. Blum, Acc. Chem. Res., 2011, 44, 603–613 CrossRef CAS; (b) L.-P. Piu and G. B. Hammond, Chem. Soc. Rev., 2012, 41, 3129–3139 RSC.
- J. A. Zurakowski, B. J. H. Austen, M. C. Dufour, D. M. Spasyuk, D. J. Nelson and M. W. Drover, Chem. – Eur. J., 2021, 27(64), 16021–16027 CrossRef CAS.
- (a) C. Amatore, G. Broeker, A. Jutand and F. Khalil, J. Am. Chem. Soc., 1997, 119, 5176–5185 CrossRef CAS; (b) P. Fitton and E. A. Rick, J. Organomet. Chem., 1971, 28, 287–291 CrossRef CAS; (c) A. L. Clevenger, R. M. Stolley, N. D. Staudaher, N. Al, A. L. Rheingold, R. T. Vanderlinden and J. Louie, Organometallics, 2018, 37, 3259–3268 CrossRef CAS; (d) G. Yin, I. Kalvet, U. Englert and F. Schoenebeck, J. Am. Chem. Soc., 2015, 137, 4164–4172 CrossRef CAS.
- C. Croix, A. Longeau-Balland, H. Allouchi, M. Giorgi, A. Duchêne and J. Thibonnet, J. Organomet. Chem., 2005, 690, 4835–4843 CrossRef CAS.
- DFT calculations were carried out using Gaussian16 Rev. C.01 (Gaussian 16, Revision C.01, M. J. Frisch, et al., Gaussian, Inc., Wallingford CT, 2016) at the M06/6-311 + G(d,p) + LANL2DZ(d,p) + LANL2DZ(f) + SMD(benzene)//B3LYP-D3/6-31G(d) + LANL2DZ(d,p) + LANL2TZ(f) level of theory. DLPNO-CCD(T) single points were obtained using ORCA 5.0.0 on DFT-optimised geometries using a cc-pVTZ-PP basis set and SK-MCDHF-RSC ECP on Au and I, and a cc-pVTZ basis set on all other atoms. For more details, see the ESI.
- (a) C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, J. Chem. Phys., 2016, 144, 024109 CrossRef; (b) C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed.
- M. W. Drover, M. C. Dufour, L. A. Lesperance-Nantau, R. P. Noriega, K. Levin and R. W. Schurko, Chem. – Eur. J., 2020, 26, 11180–11186 CrossRef CAS.
- For examples of Au-arene synthesis, see: (a) N. Ahlsten, G. J. P. Perry, X. C. Cambeiro, T. C. Boorman and I. Larrosa, Catal. Sci. Technol., 2013, 3, 2892–2897 RSC; (b) N. V. Tzouras, M. Saab, W. Janssens, T. Cauwenbergh, K. Van Hecke, F. Nahra and S. P. Nolan, Chem. – Eur. J., 2020, 26, 5541–5551 CrossRef CAS PubMed; (c) F. J. L. Ingner, Z. X. Giustra, S. Novosedlik, A. Orthaber, P. J. Gates, C. Dyrager and L. T. Pilarski, Green Chem., 2020, 22, 5648–5655 RSC; (d) D. V. Partyka, M. Zeller, A. D. Hunter and T. G. Gray, Inorg. Chem., 2012, 51, 8394–8401 CrossRef CAS.
- This reaction mixture precipitated from benzene or toluene, and required THF as solvent. Notably, both components (1 and 5) are soluble in benzene or toluene, indicating persistent adduct formation.
- In addition, reaction of [NiII(P2BCy4)(4-CH3OC6H4)(I)] (2-OCH3) with 5 provided the pyridyl-linked oligomer, [NiII(P2BCy4)(4-NC5H4)(I)]n in 48% yield.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C{1H}, 31P{1H}, and 11B NMR spectra for all complexes. XYZ coordinates for DFT calculations. CCDC 2109283 and 2109284. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc06064c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |