
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lamellar silver thiolate coordination polymers with reversibly switchable blue-to-near infrared optical transitions†
Junren
Wang
,
Robert
Graf
 and
Andreas
Riedinger
*
and
Andreas
Riedinger
*
Max Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany. E-mail: riedinger@mpip-mainz.mpg.de
First published on 16th July 2021
Abstract
We discovered that lamellar silver thiolates (Ag-MPA) possess reversible temperature dependent blue-to-near infrared optical transitions. Our analyses revealed that structural flexibility of Ag-MPA is responsible for the transitions. The polymer-like nature of Ag-MPA makes up-scaling the reaction feasible and allows for facile processing for applications ranging from bio-imaging to opto-electronics.
Quantum dots (QDs) are semiconductor crystallites with diameters in the range of a few nanometers and their properties stem largely from quantum confinement effects. This includes a size-dependent optical band-gap.1,2 Ag2S QDs have attracted intense attention in recent years due to their narrow band gap (0.9–1.1 eV at 300 K)3,4 and good biocompatibility.5 Ag2S QDs with tunable photoluminescence feature,6 ultra low cytotoxicity7 and strong emission in the NIR window are promising for biological applications, such as imaging in vivo,8 drug delivery9 and biosensors,10 since they allow for deeper tissue imaging than possible with visible light in addition to lower background photoluminescence and thus lower signal-to-noise ratios.11
Interestingly, in literature, many Ag2S QDs and nanoplatelets were prepared by nearly identical wet-chemical approaches,6,12–15 utilizing AgNO3 and 3-mercaptopropionic acid (MPA) as precursors and ethylene glycol (EG) as solvent with reaction temperatures between 120–145 °C. According to literature, these protocols yield nanomaterials with a variety of polymorphs, sizes, and shapes while at the same time exhibiting nearly identical optical features such as first absorption peaks at ∼800 nm. These results seem to be at odds with the idea of quantum confinement, since the variety in sizes (e.g. QDs with diameters of ∼6.3 nm12) and shapes (e.g. nanoplatelets with thicknesses of 0.35 nm13) should lead to distinctively different optical properties. Equally puzzling are the reports on nearly identical optical properties for α-Ag2S12 and β-Ag2S13,15 nanomaterials, because only the low-temperature monoclinic α-Ag2S (acanthite) phase exhibits semiconducting properties.16 In conclusion, while the optical properties are extremely interesting for a variety of applications, it is unclear to date what kind of material really is generated by syntheses utilizing AgNO3 and MPA precursors in EG.
We investigated the synthesis systematically by changing the molecular structure of the mercapto compounds and the solvent, while keeping the AgNO3 precursors unaltered. We found that the materials formed are predominantly lamellar Ag-MPA coordination polymers. Their optical properties are connected to structural flexibility that can be addressed by temperature. We find that our Ag-MPA coordination polymers display a reversible blue-to-NIR optical transition that is reversible over many cycles. Thus, our study not only reveals that the characteristic optical properties of the materials stemming from the synthesis from AgNO3 and MPA in EG are related to an Ag-MPA coordination polymer with varying acid side chain conformation, rather than quantum confinement effects in Ag2S nanocrystals. It also opens up many new possibilities for applications that can make use of the temperature dependent, fully reversible optical transitions, spanning the blue-to-NIR range. Furthermore, the polymeric nature of Ag-MPA allows for facile scalability to multi-gram syntheses. Due to the polymeric nature of Ag-MPA, we further expect significant advantages in the processing of our Ag-MPA coordination polymers over colloidal quantum dots for solid-state applications.
In a typical synthesis to NIR-photoluminescent materials, AgNO3 is mixed with EG. After degassing under vacuum at room temperature, MPA is added to the reaction flask under argon atmosphere and the temperature is slowly raised to 130 °C. We monitored the products of this reaction from room temperature to heating at 130 °C for 2 h by means of optical spectroscopy and powder X-ray diffraction (PXRD) (see Fig. 1). Four distinct stages were observed as the reaction proceeded.
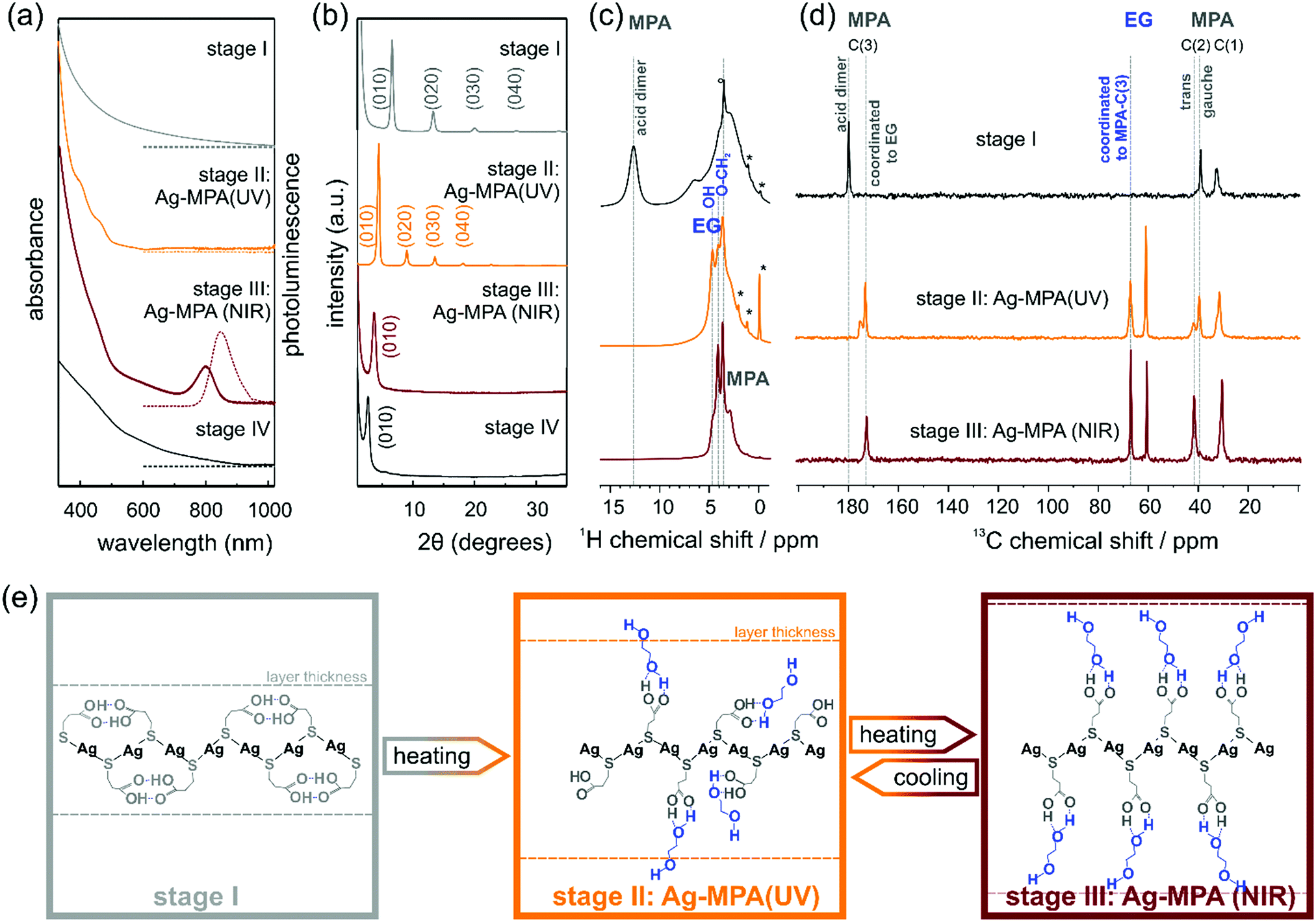 | ||
| Fig. 1 (a) Absorption spectra (solid lines) and photoluminescence spectra (dash lines), (excited by 380 nm); (b) PXRD patterns; (c) 1H MAS NMR spectra (asterisk marked the contaminated with silicon grease and residual solvent); (d) 13C NMR spectra; (e) the scheme of structures at different reaction stages. | ||
After the thiol ligand MPA was injected into the silver nitrate containing EG solution, the colorless solution became cloudy white, this is the first stage until the reactant starting to change to orange ∼130 °C. A first aliquot, a white dispersion, was removed from the flask. This first aliquot did not show any absorption and photoluminescence peaks (see Fig. 1a).
PXRD measurements revealed regular diffraction signals at 6.6°, 13.3° and 20.1° (see Fig. 1b). These diffraction patterns can be interpreted in terms of a crystal structure in which the Ag and S atoms occur in regularly stacked layers with a relatively large interlayer distance.17 Layered Ag thiolate structures are well known in literature.18,19 The intra-layer Ag-thiolate ligands arrangement is usually quasi-hexagonal, with the thiolate terminating both sides of the layers.20–23 Calculated by Bragg's law, the stacking distance of our first aliquot is ∼13.3 Å. The 1H MAS NMR spectrum of the first aliquote shows the expected signals of the aliphatic MPA sites and a sharp peak at ∼13 ppm (see Fig. 1c, first row) assigned to acidic protons, which show a strong auto-correlation signal in 1H double-quantum correlation spectra (see Fig. S1, ESI†) indicating the formation of acid dimers. In the 13C CP-MAS NMR spectrum (Fig. 1d, first row), only the three carbon sites of MPA (COOH ∼180 ppm, CH2 ∼39 ppm, S–CH2 ∼32 ppm) are observed, while the presence of EG in stage I can be safely excluded. The chemical shift value of 39 ppm of the MPA carbon site C(2) indicates that the central carbon site of all MPA molecules in the stage I aliquote is present in gauche conformation, which leads to a bent shape of the molecule. Assuming, that the C–S bond of MPA is oriented perpendicular to the Ag–S layer, thus indicates that the formed acid dimers do not connect MPA molecules of neighboring Ag–S layers, but are intra layer dimers oriented parallel to the Ag–S layer.
When the temperature reached 130 °C, the solution changed to bright orange gradually. Indeed, in the absorption spectra, two absorption shoulders at ∼390 nm and ∼457 nm appeared [see Fig. 1 a; we call this orange material Ag-MPA(UV) from now on]. These two absorption bands are associated with a donor–acceptor charge transfer from sulphur atoms to silver ions24 and metallophilic attraction between silver atoms in Ag-MPA(UV) coordination polymer.25 Therefore, the absorption spectra are determined by the thiolate ligands and the related ligand-to-metal charge transfer (LMCT) and modified by the presence of the Ag–Ag interactions, leading to ligand-to metal–metal charge transfer (LMMCT).26
Similar to aliquot I, the PXRD of aliquot II displayed also a layered structure, but the stacking distance increased to ∼20 Å (see Fig. 1b). In the 1H MAS NMR spectrum of aliquote II, the acid signal at ∼13 ppm of MPA dimers disappears, indicating the opening of the hydrogen bonded MPA dimers during heating, while three distinct signals assigned to EG sites are observed in the range from 3.5–5 ppm. At the same time, the 13C NMR signal of the acid dimers shifts to higher fields and splits into a broader signal at 175 ppm assigned to free acid groups and a narrow signal at 172 ppm of acid sites coordinated to EG. The new methoxy signal of EG in the aliquote II splits as well, into a signal at ∼67 ppm of methoxy sites coordinated to MPA-acid groups and free EG methoxy groups at ∼60.5 ppm. The signal assigned to MPA C(2) splits as well and shows the signal of the trans conformation of MPA at ∼42 ppm. The relative intensities of the two signals gauche![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans is 2:1 indicating a statistical population of the two conformations. Both the presence of MPA in the elongated trans-confirmation as well as the incorporation of EG into the layered structure of Ag-MPA(UV) results in an increasing inter-layer distance compared to the structure of stage I as confirmed by PXRD. The TEM micrographs and AFM images revealed a web-like morphology (see Fig. S2 and S3, ESI†).
:trans is 2:1 indicating a statistical population of the two conformations. Both the presence of MPA in the elongated trans-confirmation as well as the incorporation of EG into the layered structure of Ag-MPA(UV) results in an increasing inter-layer distance compared to the structure of stage I as confirmed by PXRD. The TEM micrographs and AFM images revealed a web-like morphology (see Fig. S2 and S3, ESI†).
When heated to 130 °C for 10 minutes, the color started to become dark and change to brown gradually. In this third stage, the two spectral shoulders in the UV range became weaker and an obvious absorbance peak at ∼800 nm appeared with corresponding photoluminescence at ∼840 nm [see Fig. 1a; we call this material Ag-MPA(NIR) from now on].
The 13C CP-MAS NMR spectra of Ag-MPA(NIR) showed that all MPA molecules have been transferred from lateral space filling gauche conformation to the fully elongated trans conformation and are coordinated to EG by hydrogen bonding as indicated by the single, narrow carbonyl signal at 172 ppm. Therefore, the interlayer distance increases upon the transformation of Ag-MPA(UV) to Ag-MPA(NIR). The 1H MAS NMR spectra show, that the molecular order of coordinated EG molecules increases substantially at this transition, going from a disordered state with broad O–CH2 signals and narrow signals of free OH groups (around ∼4 ppm) to an ordered state with sharper O–CH2 signals and a broad shoulder of hydrogen bonded, less mobile OH groups. The PXRD revealed that the higher order reflections disappeared and the remaining broadened first order peak shifts to ∼3.7°, related to the stacking distance of 23.8 Å. This could indicate that the main structure of the layer is maintained, but the long-range periodicity of the stacked lamellar structure is strongly reduced. We assume that altering of argentophilic interactions causing a lateral contraction of the Ag–S layer is the reason for the change of the optical properties. The lateral contraction of the attached ligand will force the MPA molecules into the entropically unfavorable all-trans state and will lead to an increasing inter-layer distance. It is difficult to imagine that the argentophic lateral contraction of the two-dimensional coordination polymer happen without any local breaking of the Ag–S layer, reducing it's lateral size. The stacking of smaller two-dimensional objects, however, leads to a reduced stacking periodicity as observed by the PXRD.
Importantly, we did not observe any signs of Ag2S diffraction signals, yet the material displayed the typical absorption and emission features often attributed to Ag2S quantum confined nanocrystals.6,12–15 This indicates that the characteristic optical properties (absorbance peak at ∼800 nm) of materials produced by the reaction of AgNO3 and MPA in EG stem from Ag-MPA coordination polymer and not from Ag2S nanocrystals. Interestingly, TEM and AFM analyses revealed that the web-like structure is even more pronounced than for aliquot II, the orange Ag-MPA(UV) (see Fig. S2 and S3, ESI†). We want to emphasize that the obtained Ag-MPA(UV) and Ag-MPA(NIR) materials are all very sensitive to the electron beam, as we observed a gradual increase of particle-like structures with increasing exposure time. This could be the origin of the miss-interpretation of the NIR absorbance feature at ∼800 nm to stem from Ag2S nanocrystals as they could form in situ in the electron microscope. In order to compare with others’ similar work,6,12–15 we also synthesized with different Ag:S ratios (1:2 and 1:20) and reaction temperature (130 °C and 145 °C). Nearly identical NIR optical features are observed in all cases (see Fig. S4, ESI†).
When heating for a longer time (∼50 min), the absorption feature at 800 nm and the NIR photoluminescence feature, together with the web-like structure, disappeared (see Fig. 1a and Fig. S3, ESI†). The PXRD showed a single signal at 2.8° and only traces of monoclinic α-Ag2S around 30–40 °C (see Fig. 1b and Fig. S5, ESI†). Only when heated for more than 2 hours, black precipitation with the clear diffraction pattern of monoclinic Ag2S can be obtained (see Fig. S5, ESI†).
While studying the transition from Ag-MPA(UV) to Ag-MPA(NIR), we noticed that if the heating process was stopped early (∼15 min to ∼25 min heating at 130 °C), the brown Ag-MPA(NIR) sample would change back to orange Ag-MPA(UV) during the cool-down to room temperature. This observation is a second indication that the optical properties are most likely not stemming from Ag2S nanocrystals for which such reversible optical properties are unknown. We followed this transformation optically by absorption spectroscopy during the cool-down from 130 °C (see Fig. 2a). The absorption peak at ∼800 nm decreased and blue shifted during the cooling process and ultimately completely disappeared. When heated to 130 °C again, the optical properties of Ag-MPA(NIR) are fully restored. This heating–cooling process can be repeated at least five times without losing the reversibility (Fig. 2b). Unfortunately, because of thermal fluorescence quenching, the weakening of luminescence intensity when the temperature is increased, the PL spectra for hot Ag-MPA(NIR) can not be measured easily.
 | ||
| Fig. 2 Absorption spectra of (a) Ag-MPA continuously measured during the cooling from 130 °C to room temperature. (b) Ag-MPA measured during after several cycles of heating and cooling; the solid line shows the room temperature absorbance, dash line shows absorbance at 130 °C. (c) Differential scanning calorimetry (DSC) curves of Ag-MPA(UV) powder for five continuous cycles at heating/cooling rates of 5 K min−1. | ||
The reversibility of the transition from Ag-MPA(UV) to Ag-MPA(NIR) and back was confirmed by differential scanning calorimetry (DSC) (Fig. 2c). We cycled the temperature between 25 °C and 150 °C with heating and cooling rates of 5 °C min−1. During the first heating, we measured an endothermic peak at ∼143 °C with the energy of 195.5 mJ and upon cooling an exothermic peak at ∼109 °C with energy of 79.4 mJ (Fig. 2d). This indicates that Ag-MPA(UV) converts to Ag-MPA(NIR) at ∼143 °C and then partially returns to Ag-MPA(UV) at ∼109 °C. This transformation seems to be kinetically limited, since the exothermic peak that most likely is related to the reorganization of the layered Ag-MPA sheets into ordered super structures (see Fig. 1b) does not coincide with the endothermic peak at ∼143 °C of the transition from Ag-MPA(UV) to Ag-MPS(NIR). We speculate that this creates some structural hysteresis that is also the reason the integrated peak area decreased every cycle.
We further used X-ray photoelectron spectroscopy (XPS) to shed some light on the chemical state of S, C and Ag for Ag-MPA(UV) and Ag-MPA(NIR) (see Fig. 3 and Fig. S6 for Ag 3d spectra, Fig. S7, ESI† for survey scans). Purified Ag-MPA(UV) water solution was dropped on two pieces of silicon wafers. Then one of the wafers was heated to 130 °C, to obtain Ag-MPA(NIR). For analysis of the data, the standard spin–orbit splitting of 1.18 eV and a branching ratio of 2:1 (S 2p3/2:S2 p1/2) was used to fit the S 2p3/2,1/2 doublet. S 2p spectra analysis points out three distinct chemical states for both S in Ag-MPA(UV) and Ag-MPA(NIR). The S 2p3/2 signal occurring at ∼162.1 eV is the main contributor of the overall S population, associated with an Ag–S–R structure, which normally showed in silver thiolate.22,27 At lower binding energy the peak at ∼161.2 eV can be associated with S 2p3/2 Ag2S,28 which takes around 27% in the whole content of sulfur. We did not observe these Ag2S traces PXRD. This could be due to an amorphous character or because the Ag2S in the XPS was formed in situ during the high vacuum processing and X-ray exposure during the measurement. Finally, the spin–orbit pair at higher binding energy (S 2p3/2 ∼163.5 eV) is associated with free thiol group.27,28 In the Ag-MPA(UV), there is a small amount of free thiol group (∼6%), most likely stemming from physical absorbed, unbound MPA. In the Ag-MPA(NIR), there is obvious more free thiol group (∼13%).29 This could imply that during the heating of Ag-MPA(UV) to obtain the Ag-MPA(NIR), MPA is partially released from Ag-MPA(UV). The standard spin–orbit splitting of 6 eV and a branching ratio of 3:2 (Ag 3d5/2:Ag 3d3/2) was used to fit the Ag 3d5/2,3/2 doublet. For the main signal, Ag 3d5/2 is centered at 368.2 eV, but the peak separation of Ag, Ag–S–R, Ag2S is too small to be appreciated.29
 | ||
| Fig. 3 High-resolution XP-spectra of Ag-MPA (UV) and Ag-MPA (NIR) (a) S 2p, solid lines represent the signals stemming from S 2p3/2, dash lines represent the signals stemming from S 2p1/2; (b) C 1. | ||
The C 1s spectra exhibit the emission bands at 284.8 eV, 286.4 eV and 288.9 eV. In MPA, there are three types of carbon atoms. The signal at ∼288.9 eV can be attributed to the carbon atom in the COOH group.30 The difference in binding energy for the carbon atoms in the position of S–C–C and C–C–C is too small to be appreciated.31 There is a strong peak in the C 1s spectra at 286.3 eV attributed to C–OH,32 which stemmed from EG. The ratio of C–OH:COOH in Ag-MPA(UV) is 2.4:1 and in Ag-MPA(NIR) is 2.6:1, which reflects a ratio of EG:MPA only slightly larger than 1 (there are two C–OH in EG and one COOH in MPA, the theoretical ratio for EG/MPA would be 2:1). This fits the NMR result that a layer of EG molecules interconnects to the MPA layer through hydrogen bonds, and both Ag-MPA(UV) with MPA in mixed conformation and Ag-MPA (NIR) with MPA in pure trans conformation have almost the same chemical states of Ag, S, and C.
In order to evaluate their importance on the appearance of the NIR feature in Ag thiolate coordination polymers, we assessed a broad range of different ligands and solvents (see Fig. 4 and Fig. S8, ESI†). Our experiments on the various ligands reveal that for the successful synthesis of Ag thiolates with NIR optical properties, the proper coordination between Ag+ and thiol groups is crucial. We show that the group terminating each Ag thiolate layer can be substituted, covering a broad range from CH3 to COOH. We also find that the lengths of the ligand is not of great importance as Ag-12-mercapotundecanoic acid shows nearly identical optical properties to Ag-3-mercaptopropionic acid. However, the solubility of the coordination polymers can be adjusted by choice of the ligands lengths and end group. Moreover, quantum yield (QY) decreases with the decrease of solubility. The QY of Ag-MPA(NIR) is 5.38%, Ag-3-mercapto-1-propanol is 5.35%, Ag-methyl 3-mercaptopropionate is 2.47%, Ag-12-mercaptododecanoic acid is 1.02% and Ag-1-hexanethiol is 0.53% (see Table S1, ESI†).
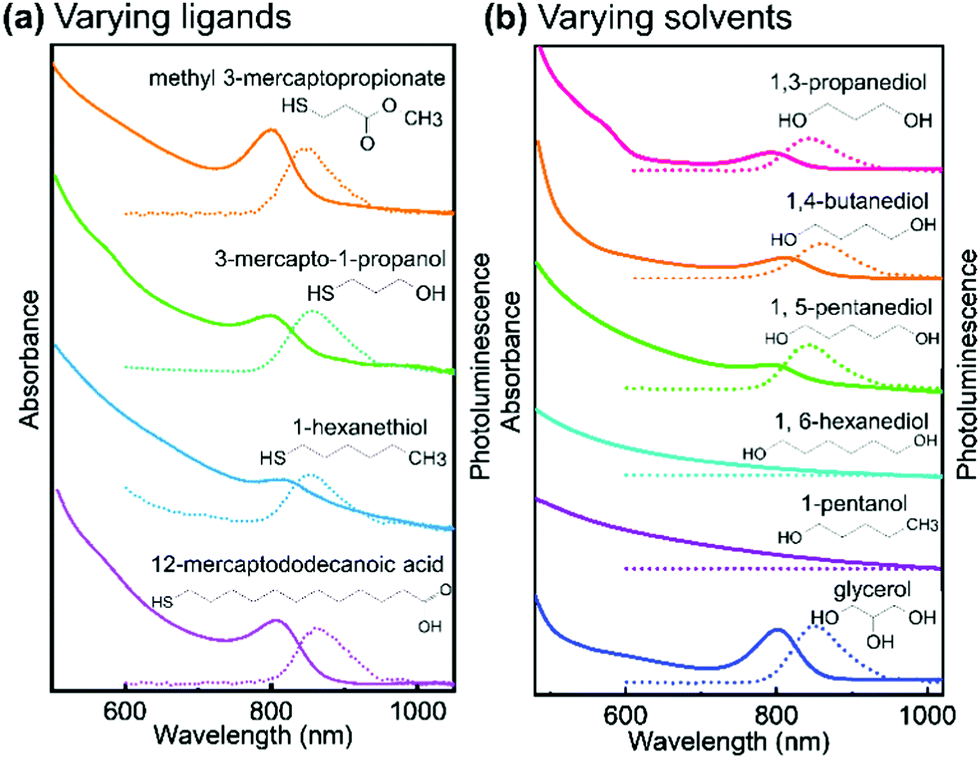 | ||
| Fig. 4 Absorption spectra and photoluminescence spectra of (a) silver thiolate synthesized with different thiol ligands in ethylene glycol, (excited by 380 nm); (b) Ag-MPA synthesized in different solvent, absorption spectra shows by solid lines and photoluminescence spectra shows by dash lines. The PRXD patterns of all samples are shown in Fig. S8 (ESI†). | ||
As our NMR and XPS data demonstrate, EG is not only solvent but also part of the structure in Ag-MPA(UV) and Ag-MPA(NIR). When we replaced EG with 1,3-propanediol, 1,4-butanediol, 1,5-pentanediol, silver thiolate products with similar absorbance and photoluminescence feature still can be obtained. However, the reaction temperature had to be increased from 130 °C to a maximum 155 °C. The higher temperatures could be necessary due to the lower mobility with increasing molecular weights. With 1,6-hexanediol the reaction did not yield a NIR photoluminescent material anymore.
When we compared solvents with one more (glycerol) or one less hydroxyl group (1-pentanol) than EG, we find that we require at least two hydroxyl groups to obtain NIR photoluminescent silver thiolates. However, an additional hydroxyl group seems to be not problematic for the formation of NIR photoluminescent coordination polymers. This can be related to the number of functional groups needed for “cross-linking” the layers. With two or more OH groups in the solvent molecule, the Ag-MPA layers can be easily inter-connected and stabilized.
Since the reaction towards silver thiolate coordination polymers appears to be polymerization-like, we tried to upscale the synthesis by increasing the precursor concentration (see Fig. S9, ESI†) without notable differences in the properties of the materials. By increasing the concentration 15 times we could synthesize 0.36 g Ag-MPA(NIR) from 25 mL of EG. Further increases in the concentration leads to in situ gel formation. At the same time we were able to convert aqueous solutions of Ag-MPA(UV) and Ag-MPA(NIR) into 40 nm sized Ag-MPA(UV) and Ag-MPA(NIR) colloids after emulsification in toluene and evaporation of water (see Fig. S10, ESI†). The versatile processing of Ag-MPA will allow for versatile integration in various applications ranging from bio-imaging to optoelectronics.
Conclusions
In conclusion, we have analyzed the reaction process of AgNO3 and thiolate ligands in EG and other hydroxylated solvents during heating. We found that thiol-bearing molecules act as a ligand for silver in the formation of lamellar silver thiolate coordination polymers. Their end group and overall length of the ligand were not limiting the reaction towards NIR photoluminescent silver thiolates. We further found that EG is not only the solvent but intercalates in the silver thiolate layers at later stages in the synthesis. Similar materials were obtained with solvents that bear at least two hydroxyl groups (diols and glycerol) and are not too high molecular weight (such as 1,6-hexanediol compared to EG). We find that the obtained silver thiolate coordination polymers display a reversible blue-to-NIR optical transition, which are linked to structural transformations as evidenced by PXRD, solid state NMR and DSC. Furthermore, our XPS data support the solid state NMR results that the chemical state in Ag-MPA(UV) and Ag-MPA(NIR) are very similar. Thus we conclude that the (temperature-dependent) optical properties of Ag-MPA coordination polymers are modulated by metallophilic interactions between silver atoms in the layered silver thiolates and stabilized in the reversible range by conformational changes of the ligands. The nearly identical absorption feature at 800 nm and the reversibility (under suitable conditions) suggest that syntheses working with AgNO3 and MPA in ethylene glycol yield NIR photoluminescent coordination polymers rather than the Ag2S nanocrystals that several works of literature linked to the very characteristic optical properties. Therefore, our findings resolve this issue due to the now clarified chemical identity of the NIR photoluminescent material, which opens new avenues for applications. Since the Ag-MPA material forms and can be processed like a polymer material, new possibilities arise for applications ranging from bio-imaging to optoelectronics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Michael Steiert for PXRD measurements, Petra Räder for DSC measurement, Leon Prädel for XPS measurement, and Prof. Katharina Landfester for the fruitful discussions. Open Access funding provided by the Max Planck Society.Notes and references
- S. Baskoutas and A. F. Terzis, J. Appl. Phys., 2006, 99, 013708 CrossRef.
- X. Dai, Y. Deng, X. Peng and Y. Jin, Adv. Mater., 2017, 29, 1607022 CrossRef PubMed.
- P. Junod, H. Hediger, B. Kilchör and J. Wullschleger, Philos. Mag., 1977, 36, 941–958 CrossRef CAS.
- P. Junod, PhD thesis, ETH Zurich, 1959.
- Y. Zhang, G. Hong, Y. Zhang, G. Chen, F. Li, H. Dai and Q. Wang, ACS Nano, 2012, 6, 3695–3702 CrossRef CAS PubMed.
- R. Tang, J. Xue, B. Xu, D. Shen, G. P. Sudlow and S. Achilefu, ACS Nano, 2015, 9, 220–230 CrossRef CAS PubMed.
- R. Gui, A. Wan, X. Liu, W. Yuan and H. Jin, Nanoscale, 2014, 6, 5467–5473 RSC.
- G. Chen, F. Tian, Y. Zhang, Y. Zhang, C. Li and Q. Wang, Adv. Funct. Mater., 2014, 24, 2481–2488 CrossRef CAS.
- L. Tan, A. Wan and H. Li, ACS Appl. Mater. Interfaces, 2013, 5, 11163–11171 CrossRef CAS PubMed.
- X. Zhang, M. Liu, H. Liu and S. Zhang, Biosens. Bioelectron., 2014, 56, 307–312 CrossRef CAS PubMed.
- D. Aydemir, M. Hashemkhani, H. Y. Acar and N. N. Ulusu, Chem. Biol. Drug Des., 2019, 94, 2094–2102 CrossRef CAS PubMed.
- P. Jiang, C. N. Zhu, Z. L. Zhang, Z. Q. Tian and D. W. Pang, Biomaterials, 2012, 33, 5130–5135 CrossRef CAS PubMed.
- L. Kubie, L. A. King, M. E. Kern, J. R. Murphy, S. Kattel, Q. Yang, J. T. Stecher, W. D. Rice and B. A. Parkinson, ACS Nano, 2017, 11, 8471–8477 CrossRef CAS PubMed.
- L. Kubie, M. S. Martinez, E. M. Miller, L. M. Wheeler and M. C. Beard, J. Am. Chem. Soc., 2019, 141, 12121–12127 CrossRef CAS PubMed.
- W. Yu, J. Yin, Y. Li, B. Lai, T. Jiang, Y. Li, H. Liu, J. Liu, C. Zhao, S. C. Singh, J. Chen, B. Lin, H. Idriss and C. Guo, ACS Appl. Energy Mater., 2019, 2, 2751–2759 CrossRef CAS.
- S. Sadovnikov and A. Gusev, J. Mater. Chem. A, 2017, 5, 17676–17704 RSC.
- Z. Ye, P. Lito, M. Y. Efremov, J.-M. Zuo and L. H. Allen, Dalton Trans., 2016, 45, 18954–18966 RSC.
- I. G. Dance, K. J. Fisher, R. H. Banda and M. L. Scudder, Inorg. Chem., 1991, 30, 183–187 CrossRef CAS.
- J. Duan, J. Ma, B. Wu, Q. Li, J. Fang and D. Chen, J. Mater. Chem. C, 2014, 2, 2375–2386 RSC.
- G.-H. Gwak, M.-K. Kim, W.-J. Lee, D.-G. Jeung, J. K. Park, S.-M. Paek and J.-M. Oh, Inorg. Chem., 2019, 59, 2163–2170 CrossRef PubMed.
- O. Veselska, C. Dessal, S. Melizi, N. Guillou, D. Podbevšek, G. Ledoux, E. Elkaim, A. Fateeva and A. Demessence, Inorg. Chem., 2019, 58, 99–105 CrossRef CAS PubMed.
- Y. Li, X. Jiang, Z. Fu, Q. Huang, G.-E. Wang, W.-H. Deng, C. Wang, Z. Li, W. Yin and B. Chen, Nat. Commun., 2020, 11, 1–9 Search PubMed.
- J. Beers, A. Parikh, S. Gillmor, K. Beardmore, R. Cutts and B. Swanson, J. Young Investig., 1998 Search PubMed , https://www.jyi.org/1998-december/1998/12/28/stepwise-assembly-of-silvern-alkanethiolates-an-example-of-hierarchical-or-cooperative-self-assembly/.
- S. D. Khizhnyak, P. V. Komarov, M. M. Ovchinnikov, L. V. Zherenkova and P. M. Pakhomov, Soft Matter, 2017, 13, 5168–5184 RSC.
- P. Casuso, P. Carrasco, I. Loinaz, H. J. Grande and I. Odriozola, Org. Biomol. Chem., 2010, 8, 5455–5458 RSC.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- K. Heister, M. Zharnikov, M. Grunze and L. Johansson, J. Phys. Chem. B, 2001, 105, 4058–4061 CrossRef CAS.
- V. K. Kaushik, J. Electron Spectrosc. Relat. Phenom., 1991, 56, 273–277 CrossRef CAS.
- C. Battocchio, C. Meneghini, I. Fratoddi, I. Venditti, M. V. Russo, G. Aquilanti, C. Maurizio, F. Bondino, R. Matassa and M. Rossi, J. Phys. Chem. C, 2012, 116, 19571–19578 CrossRef CAS.
- M. K. Rabchinskii, A. T. Dideikin, D. A. Kirilenko, M. V. Baidakova, V. V. Shnitov, F. Roth, S. V. Konyakhin, N. A. Besedina, S. I. Pavlov and R. A. Kuricyn, Sci. Rep., 2018, 8, 1–11 CAS.
- P. M. Dietrich, T. Horlacher, P.-L. Girard-Lauriault, T. Gross, A. Lippitz, H. Min, T. Wirth, R. Castelli, P. Seeberger and W. E. Unger, J. Carbohydr. Chem., 2011, 30, 361–372 CrossRef CAS.
- Y. Chen, H. Wang, B. Dang, Y. Xiong, Q. Yao, C. Wang, Q. Sun and C. Jin, Sci. Rep., 2017, 7, 1–9 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc01983j |
| This journal is © The Royal Society of Chemistry 2021 |