
The electronic structure of transition metal oxides for oxygen evolution reaction
Hongxia
Wang
a,
Kelvin H. L.
Zhang
 *a,
Jan P.
Hofmann
b,
Victor A.
de la Peña O'Shea
c and
Freddy E.
Oropeza
*c
*a,
Jan P.
Hofmann
b,
Victor A.
de la Peña O'Shea
c and
Freddy E.
Oropeza
*c
aState Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: kelvinzhang@xmu.edu.cn
bSurface Science Laboratory, Department of Materials and Earth Sciences, Technical University of Darmstadt, Otto-Berndt-Strasse 3, 64287 Darmstadt, Germany
cPhotoactivated Processes Unit, IMDEA Energy, Ramón de la Sagra, 3, 28935, Móstoles, Madrid, Spain. E-mail: freddy.oropeza@imdea.org
First published on 27th July 2021
Abstract
Electrolysis of water to produce hydrogen and oxygen is a promising pathway for the storage of renewable energy in form of chemical fuels. The efficiency of the overall process is usually limited by the sluggish kinetics of the oxygen evolution reaction (OER) due to a complex four-electron/proton transfer mechanism. Therefore, the most crucial step for water electrolysis to become a widespread industrial process is to develop efficient electrocatalysts capable of driving the OER at a low overpotential. In this article, we present a review of the recent understanding of low-cost transition metal oxide-based catalysts for electrochemical OER. Our main focus is on the fundamental understanding of the electronic structure of these materials and its relationship with the catalytic activity and the reaction mechanism. We detail the electronic structure descriptors of the OER performance that have provided guidelines for the development of advanced electrocatalysts. Strategies to enhance the activity based on such electronic structure guidelines, including doping, strain and defect engineering are also discussed. Finally, we summarize the existing challenges and present perspectives for the future development of the field.
 Hongxia Wang | Hongxia Wang received her master degree in chemistry and chemical engineering from Chongqing University in 2020. She is currently a PhD candidate at college of Chemistry and Chemical Engineering, Xiamen University under the supervision of Prof. Kelvin H. L. Zhang. Her research interest focusses on the synthesis of clean and efficient materials and their applications in electrochemical energy storage and conversion. |
 Kelvin H. L. Zhang | Kelvin H. L. Zhang is a professor at college of Chemistry and Chemical Engineering, Xiamen University. His current research interest focus on the synthesis and properties of complex metal oxide thin films and heterostructures relevant to application in oxide electronics and electrocatalysts. He obtained his PhD degree in chemistry of Oxford University in 2012, followed by working as a postdoc at Pacific Northwest National Laboratory and as a Herchel Smith Fellow at University of Cambridge from 2015 to 2017. |
 Jan P. Hofmann | Jan Philipp Hofmann is full professor of Surface Science at Technical University of Darmstadt, Germany since 2020. He received his chemistry diploma and doctoral degree from Justus-Liebig University (JLU) in Giessen, Germany in 2005 and 2009, respectively. After a 1 year postdoctoral stay at JLU Giessen with Prof. Herbert Over, in 2011, he moved to the Netherlands to become DFG postdoc fellow in the Debye Institute of Nanomaterials Science (group of Prof. Bert M. Weckhuysen) at Utrecht University. From 2013–2020, he has been assistant professor of Inorganic Materials Chemistry and Solar Fuels Catalysis at Eindhoven University of Technology, NL. His research focuses on the energetics, chemistry, and dynamics of interfacial processes in energy materials and devices. |
 Victor A. de la Peña O'Shea | Victor A. de la Peña O'Shea received his PhD in Physical Chemistry (2003) from the Universidad Autonoma de Madrid and the Catalysis and Petrochemistry Institute (Spain). After his PhD, he spent three years at Universidad de Barcelona, where he got the M.Sc. in Physics. In 2008, he moved to IMDEA Energy, where he is the head of the photoactivated processes unit. He has co-authored over 120 publications, several patents, and books. He has coordinated more than 25 research projects, including an ERC-CoG. His research group is focused in the design and development of novel multifunctional materials and reactors applied to sustainable energy and environmental technologies. |
 Freddy E. Oropeza | Freddy E Oropeza is a Marie Skłodowska-Curie fellow at IMDEA Energy. His research interests focus on the understanding and control of the structural and electronic properties that define the functionality of metal oxide thin films and interfaces for applications in electronics and (photo)electrochemical technologies. He obtained his PhD in Chemistry from the University of Oxford in 2012, followed by working as postdoc at the Ruhr University Bochum (2012–2013), Imperial College London (2013–2016) and the Eindhoven University of technology (2016–2019). |
1. Introduction
Since the 19th century, non-renewable energy resources such as coal, oil, and natural gas have been utilized as the main energy supply to sustain human activities.1 However, the excessive consumption of fossil fuels caused by the rapidly increasing world population and expanding industrialization as well as concomitant environmental problems have motivated our society to explore a green and sustainable energy system for long-term development.2–5 In this regard, countries around the world have made gigantic efforts to raise the share of renewable and carbon-free sources of energy, such as solar, wind, and tidal energy.6 Unfortunately, these energy sources have a general intermittent availability and heavily depend on the season and weather, which result in a low energy delivery efficiency and restrict their widespread application. Thus, the storage of renewable energy in the form of fuels and useful chemicals is a highly valuable approach to introduce such sources of renewable energy in the mainstream of our current energy economy. In this regard, hydrogen has been considered as a promising energy carrier because it has zero-carbon combustion products and the highest gravimetric energy density.7,8 Hydrogen not only can be used as energy carrier itself but it is also an important industrial feedstock for methanol production and ammonia synthesis among other processes. Since technologies for the conversion of many sources of renewable energy (e.g., solar irradiance and wind) into electrical power are well-developed these days, the electrochemistry provides means of storing the surplus of solar- or wind-driven electricity into fuels and valuable chemicals with a low carbon footprint.9 In a joint effort, scientists, policymakers and politicians around the world have carried extensive research and investment on projects for the large-scale wind power or photovoltaic hydrogen production, which combining the utilization of renewable energy with the hydrogen generation through water electrolysis.10 Currently, the global hydrogen production reaches more than 110 Mt per year.11 However, the H2 production through water electrolysis only takes 4% of whole hydrogen supply,12 because the high cost to drive the electrolytic water splitting (>US$4 per kg of H2) compared with the hydrogen production cost through the reforming of fossil fuels (US$1.3–1.5 per kg of H2).11 The cost of industrial water electrolysis mainly arises from the large electric energy input required, which is closely related with the composition of applied catalysts and their catalytic performance.13 Thus, developing cheap catalysts with high activities is significant to reduce the production cost with better economic benefits (<US$2 per kg of H2) and broaden the commercial and industrial application of electrochemical water splitting.Since Troostwijk and Diemann firstly observed the phenomenon of water electrolysis in 1789,14 it has been extensively investigated by researchers over the past years. It is now well-stablished that water electrolysis involves two fundamental half reactions: hydrogen evolution reaction (HER) at the cathode, and the oxygen evolution reaction (OER) at the anode. Driving the electrochemical water splitting thermodynamically requires to overcome the energy barrier of 237.2 kJ mol−1 at standard conditions, which corresponds to an applied voltage of 1.23 V in an electrochemical cell.15,16 However, in practice, the actual applied cell voltage is much larger than the theoretical value due to the overpotentials required to drive both the HER and the OER.17 As a result, even in well-developed and optimised water electrolysis systems, such as proton-exchange membrane water electrolysers for electrolysis in acidic condition (Fig. 1), a voltage of 1.7–2.2 V is required to deliver a current density of >1.6 A cm−2.18 The situation is similar in anion-exchange membrane electrolysers for electrolysis in alkaline media (Fig. 1) in which a voltage of 1.7–2.4 V is usually required to deliver a current density of <1.0 A cm−2.19 Most of the overpotential loss comes from the OER that involves a multi-step transfer process of electrons and protons with a complex reaction mechanism, resulting in sluggish kinetics and larger activation energy barriers than those associated to the HER process.20 In this scenario, developing more active catalysts toward the OER has become the most important factor to improve the overall efficiency of water electrolysis for industrial applications.
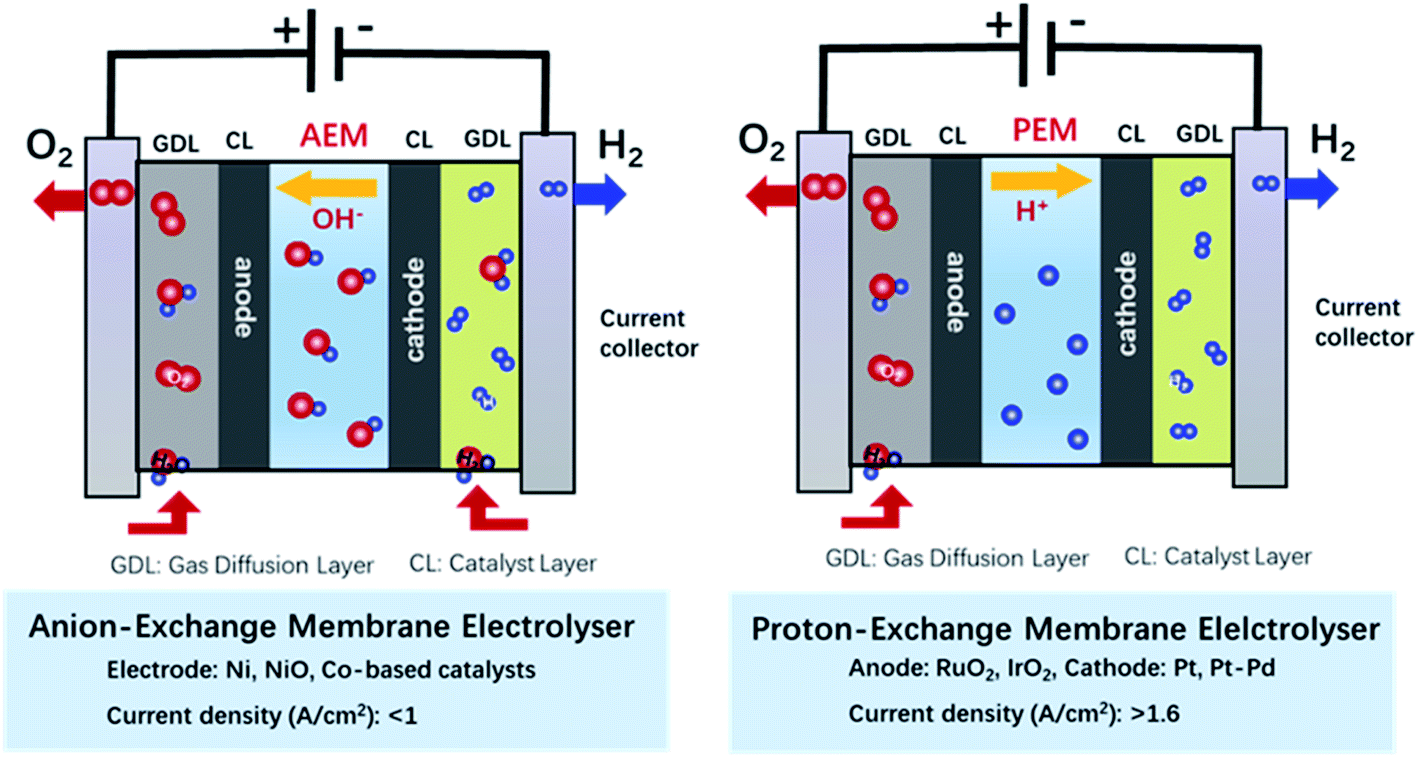 | ||
| Fig. 1 Schematics of the electrochemical water splitting in anion-exchange membrane electrolyser and proton-exchange membrane electrolyser. | ||
After decades of efforts, the worldwide research for OER catalysts have made significant progress in promoting the catalytic activities. Numerous novel materials have emerged as advanced OER catalysts such as metal chalcogenide,21–23 oxides,24,25 nitrids26,27 and molecular/polymeric systems.28,29 Ru- and Ir-based oxides are OER catalysts of high industrial relevance because their high activity in acidic media and implementation in proton-exchange membrane electrolysers.30,31 However, both RuO2 and IrO2 undergo further oxidation and subsequent dissolution in acidic electrolytes under high applied potentials,32,33 leading to unstable performance during water electrolysis. Furthermore, the high price of Ru and Ir also inhibit their large-scale application. On the other hand, one of the major advantages of alkaline water electrolysis over the acidic electrolysis is the replacement of conventional noble metal electrocatalysts with low-cost transition metal (TM) catalysts. Additionally, an alkaline media provides higher stability for the OER metal oxide catalysts.19,34 Nevertheless, the anion-exchange membrane electrolysis is still a developing technology, and the current density and voltage efficiency achieved are lower than those in the electrolysis in acidic media. Therefore, considering the cost and activity for the practical utilization, extensive research efforts must be focused on investigating earth-abundant transition metal oxide (TMO) electrocatalysts including perovskite family,35,36 spinel family,37–39 layer-structured hydroxides40–42 and other types of oxides.43,44 The OER performance of reported outstanding TMOs have exhibited remarkable activities and relatively high electrochemical stability, especially in alkaline electrolytes.45–47
In order to develop a competitive low-cost anion-exchange membrane electrolysis technology for commercially viable hydrogen production, further research and improvements are required, specifically regarding the reaction mechanisms on the surface of the catalysts and the development of guidelines for the preparation of advanced OER electrodes. Earlier in 1955, Rüetschi and Delahay reported an approximate linear relationship between the rate of the OER on metal oxides and the M–OH binding strength.48 They made the pioneering attempt to link the OER activity with a descriptor. Furthermore, in the 1970s, Matsumoto et al.49 firstly studied the correlation between the OER catalytic activity of perovskite oxides and their band structure. They proposed that the perovskite-type oxide formed σ* and π* band, and the rate of electron transfer would increase with the rise of the σ* bandwidth. Later, Bockris and Otagawa systematically investigated the OER activities on perovskite oxides and they connected the OER activity with the number of electrons occupied in the antibonding orbitals.50 Based on the band structure theory and molecular orbital theory, a few typical descriptors have been recently proposed to link the activity with electronic structure and thus theoretically predict the catalytic activity of novel catalysts. The most notable electronic descriptors of the OER activity include the number of electrons in the eg orbitals of metal cations,51,52 the degree of TM 3d–O 2p hybridization,53 the energy position of the O 2p band center,54 charge-transfer energy,35 bulk thermochemistry55 among others. For instance, Shao-Horn and co-workers systematically studied the relation between OER activities and the number of eg electrons for perovskite oxides, conducting a conclusion that the optimal catalytic performance could be achieved when the number of electrons filled in eg orbital was 1.2.51 Later, Xu et al.56 reported that eg orbital occupancy correlates well with the OER activity of spinel type oxides. Zhou et al.57 and Xie et al.58 report that the activity can be optimized by adjusting the spin state of Co 3d orbital electron in the LaCoO3 system. Moreover, Grimaud et al.59 also use the position of O 2p-band centre relative to the Fermi level as a descriptor of the OER catalytic activities. The investigations on the correlation of the OER catalytic activity with electronic structure have provided meaningful results. A comprehensive understanding of these descriptors could help us rationally design TM oxide catalysts with improved performance for the OER and push forward for further developments in this field.
In this review, we provide an overview of the recent progress of the researches on OER catalysts and the generally proposed reaction mechanisms along with the emphasis on the correlation between oxide electronic structures and OER activity. We firstly introduce the most common TM oxide electrocatalysts studied for alkaline water electrolysis along with some recent reports of outstanding catalytic performances, and the main reaction mechanisms that have been proposed for the OER on TM oxides. Then, the electronic structure descriptors in TM oxides for the OER electrochemical activity are discussed, followed by a review on material science strategies to tailor the electronic structure parameters for the design of highly active electrocatalysts. Finally, we briefly conclude the review and discuss the challenges and opportunities for the future development of TM oxides. The in-depth comprehension of the correlation between the OER activity and electronic structure of TM oxide catalysts can be instructive to explore future generation catalysts, and connects the research fields of heterogeneous catalysis with the solid-state chemistry and physics.
2. TM oxide electrocatalysts for alkaline OER
A major advantage of the anion-exchange membrane water electrolysis over the proton-exchange membrane electrolysis is the replacement of conventional noble metal electrocatalysts, such as the traditional Ir- and Ru-based oxides, with low-cost TM oxides. Additionally, the alkaline media provides higher stability to the OER metal oxide catalysts. Since the TMs in the oxide have significant contribution to the valence states of the material, the physicochemical properties are strongly susceptible to the type and chemistry (e.g., oxidation state, coordination and spin state) of the TM. This property provides TM oxides with large flexibility for the design of electrocatalysts for the OER. As a result, many metal oxides have been suggested as promising OER catalysts in alkaline media over the last decade.20,60Table 1 provides a comparison of the performance of various TM oxides electrocatalysts for the OER recently reported in the literature, which provides context to the current state of the art. Although TM oxides in a wide range of compositions are currently under investigation, most can be grouped into complex perovskite oxides, spinel oxides or layered (oxy)hydroxides.| Electrocatalysts | Electrolyte | Catalyst loading amount | Scan rate mV s−1 | Overpotential at specific current density | Tafel slope mV dec−1 | Reference |
|---|---|---|---|---|---|---|
| Defective MnCo2O4 | 0.1 M KOH | ∼0.40 mg cm−2 | 10 | 400 mV at 10 mA cm−2 | 87 | 79 |
| NiMoO4 | 1 M KOH | 0.282 mg cm−2 | 5 | 340 mV at 10 mA cm−2 | 45.6 | 80 |
| NiFeMo oxide | 0.1 M KOH | 0.25 mg cm−2 | 0.5 | 280 mV at 10 mA cm−2 | 49 | 81 |
| NiCo2O4 | 1 M KOH | — | — | 320 at 10 mA cm−2 | 30 | 82 |
| NiCo2O4 | 1 M KOH | 0.25 mg cm−2 | 10 | — | 47 | 68 |
| NiFe2O4/CoFe2O4 | 1 M KOH | 5 mg cm−2 | 5 | 240 mV at 10 mA cm−2 | 42 | 83 |
| Ni0.67Fe0.33 LDH/C | 1 M KOH | 0.28 mg cm−2 | — | 210 mV at 10 mA cm−2 | 35.1 | 84 |
| Ni–Fe hydroxide | 1 M KOH | 32 μg cm−2 | 5 | 270 mV at 80 mA cm−2 | 28 | 85 |
| NiCo2.7(OH)x | 1 M KOH | 0.14 mg cm−2 | 5 | 350 mV at 10 mA cm−2 | 65 | 86 |
| NiVIr-LDH | 1 M KOH | — | — | 180 mV at 10 mA cm−2 | 28 | 46 |
| ZnxCo3−xO4 nanoarray | 1 M KOH | ≈1 mg cm−2 | 0.5 | 320 mV at 10 mA cm−2 | 51 | 87 |
| Spindle-like ZnCo2O4 | 1 M KOH (pH = ∼14) | 0.24 mg cm−2 | 5 | 389 mV at 10 mA cm−2 | 59.5 | 88 |
| ZnCo2O4 | 0.1 M KOH | 51 μg cm−2 | — | 420 mV at 10 mA cm−2 | 38.5 | 37 |
| Cu0.7Co2.3O4 | 1 M KOH | 100 μg cm−2 | 20 | 367 mV at 10 mA cm−2 | 59.6 | 89 |
| SnCoFe hydroxide | 1 M KOH | — | 2 | 270 mV at 10 mA cm−2 | 42.3 | 90 |
| CoO/Co3O4 | 1 M KOH | 0.019 mg cm−2 | 5 | 260 at 10 mA cm−2 | 54 | 47 |
| CoO–MoO2 | 1 M KOH | 0.5 mg cm−2 | 5 | 312 mV at 10 mA cm−2 | 69 | 91 |
| Co2Mo3O8@NC | 1 M KOH | 0.14 mg cm−2 | 5 | 331 mV at 10 mA cm−2 | 87.5 | 92 |
| CoVOx | 1 M KOH | 0.5 mg cm−2 | 10 | 330 mV at 10 mA cm−2 | 46 | 93 |
| CoFe2O4 | 0.1 M KOH | 0.15 mg cm−2 | 10 | 412 mV at 10 mA cm−2 | 53 | 94 |
| Co–MoOx | 1 M KOH | 0.28 mg cm−2 | 340 mV at 10 mA cm−2 | 49 | 95 | |
| CoOx(Ce) | 1 M KOH | 1.1 mg cm−2 | 261 mV at 20 mA cm−2 | 65.67 | 96 | |
| LaMn7O12 | 0.1 M KOH | 0.25 mg cm−2 | 10 | — | ∼100 | 97 |
| LaSr1.4Ni0.6MnO | 1 M KOH | 0.25 mg cm−2 | 10 | 367 mV at 1 mA cm−2 | 86 | 98 |
| LaCoO3-80 nm | 0.1 M KOH | 0.25 mg cm−2 | — | 490 mV at 10 mA cm−2 | 69 | 57 |
| La(Co0.71Ni0.25)0.96O3−δ | 0.1 M KOH | 0.7 mg cm−2 | 10 | 324 mV at 10 mA cm−2 | — | 99 |
| La0.2Sr0.8FeO3 | 0.1 M KOH | 0.25 mg cm−2 | 10 | 318 mV at 50 μA cm−2 | 58 | 63 |
| La0.2Sr0.8FeO3 | 0.1 M KOH | — | 10 | 430 mV 34.6 μA cm−2 | 71 | 100 |
| La0.5Sr1.5Ni0.7Fe0.3O4.04 | 0.1 M KOH | 0.015 mg cm−2 | — | 360 mV at 10 mA cm−2 | — | 101 |
| La0.5Ba0.25Sr0.25CoO2.9−δF0.1 | 1 M KOH | 0.157 mg cm−2 | — | ∼518 mV at 100 mA cm−2 | 113 | 45 |
| LiCo0.8Fe0.2O2 | 0.1 M KOH | 0.232 mg cm−2 | 5 | 340 mV at 10 mA cm−2 | 50 | 102 |
| Li0.5Ni0.5O | 1 M KOH | 0.3 mg cm−2 | 10 | ∼ | 56.6 | 103 |
| SrCo1−xRuxO3−δ | 0.1 M KOH | 0.23 mg cm−2 | 5 | 360 mV at 10 mA cm−2 | 113 | 104 |
| SrCo0.95P0.05O3−δ | 0.1 M KOH | 0.232 mg cm−2 | 10 | 480 mV at 10 mA cm−2 | 84 | 105 |
| BaNi0.83O2.5 | 0.1 M KOH | 0.295 mg cm−2 | 10 | ∼460 mV at 10 mA cm−2 | — | 106 |
| Ba4Sr4(Co0.8Fe0.2)4O15 | 0.1 M KOH | 0.232 mg cm−2 | 10 | 340 mV at 10 mA cm−2 | 47 | 107 |
| BaCo0.7Fe0.2Sn0.1O3−δ | 0.1 M KOH | 0.232 mg cm−2 | 10 | ∼420 mV at 10 mA cm−2 | 69 | 108 |
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ | 0.1 M KOH | 0.202 mg cm−2 | — | 360 mV at 10 mA cm−2 | 55 | 109 |
| Nd1.5Ba1.5CoFeMnO9−δ | 0.1 M KOH | 0.209 mg cm−2 | 10 | 359 mV at 10 mA cm−2 | 81 | 110 |
| Fe2O3 | 0.5 M H2SO4 | — | 5 | 650 mV at 10 mA cm−2 | 56 | 111 |
| Cu1.5Mn1.5O4:10F | 0.5 M H2SO4 | 1 mg cm−2 | 5 | 320 mV at 9.15 mA cm−2 | 60 | 112 |
| Co3O4 | 0.5 M H2SO4 | 10 | 370 mV at 10 mA cm−2 | 70 | 113 | |
| Co0.05Fe0.95Oy | 0.05 M H2SO4 (pH = 0.3) | — | — | 650 mV at 10 mA cm−2 | 110 | 114 |
| Ti–MnO2 | 0.05 M H2SO4 (pH = 0.3) | — | — | >670 mV at 10 mA cm−2 | 170 | 115 |
| MnOx | In phosphate buffer pH = 7 | — | 20 | — | ∼70 | 116 |
| Mn0.67Sb0.33Oz | 1 M H2SO4 (pH = 0.13) | — | 10 | 580 mV at 50 mA cm−2 | 75 | 117 |
| Ni0.2Mn0.8Sb1.6Oy | 1 M H2SO4 | — | — | ∼60 | 118 | |
| NiFePbOx | 0.1 M KPi + 1 M KNO3 (pH = 2.5) | — | — | >700 mV at 1 mA cm−2 | 90 | 119 |
| CaCu3Ru4O12 | 0.5 M H2SO4 | 0.122 mg cm−2 | — | — | 40 | 120 |
2.1 Perovskite TM oxides
Perovskite TM oxides are attractive electrocatalysts for promoting the OER because of their tuneable electronic structure and high activity.61 Perovskite oxides have a general ABO3 structure, with rare-earth and/or alkaline large cations at the A site that have a 12-fold coordination with oxygen anions, and TMs at the B site in the centre of a corner-sharing octahedra of oxygen anions, see Fig. 2a. The chemistry of the TM in the B site can be widely modulated by substitution at A site, e.g., La3+ for Sr2+, which provides means to tailor the OER catalytic properties of the oxides.51,61 Well-known examples of this approach are the systems SrxLa1−xFeO3 and SrxLa1−xCoO3 with 0 ≤ x ≤ 1, in which the substitution of La3+ for Sr2+ induces the oxidation of the TM at the B site, leading to improvements of the catalytic activity for the OER.54,62,63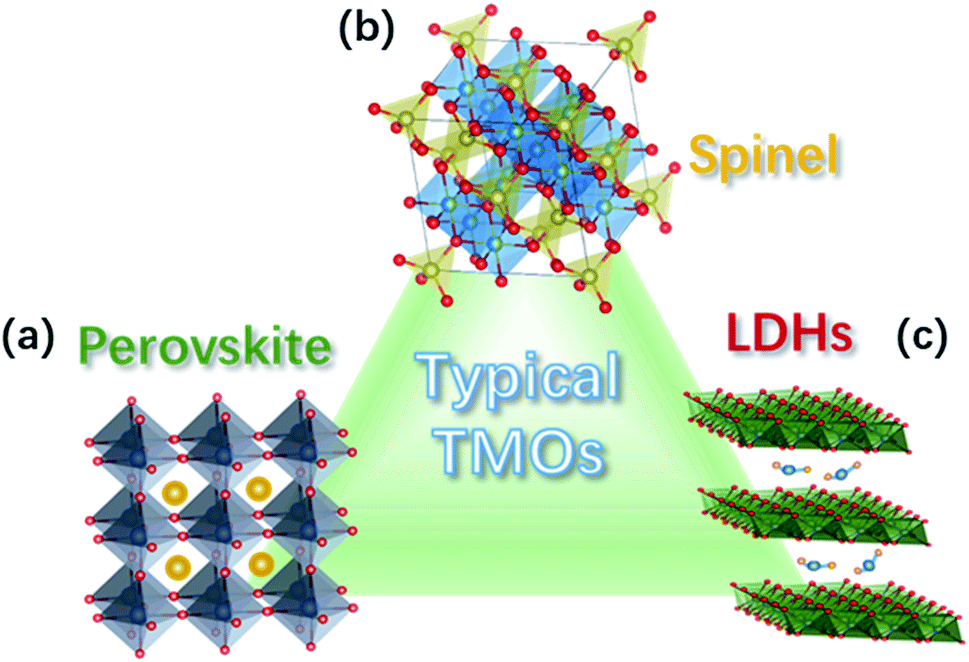 | ||
| Fig. 2 Crystal structure of common TM oxides for OER: (a) perovskite, (b) spinel, and (c) LDH structure. | ||
2.2 Spinel oxides
Spinel oxides have also been shown to drive the OER with excellent catalytic performances.64 These oxides have the general chemical formula AB2O4 and a crystalline structure in which TM cations in tetrahedral (cation A) and octahedral sites (cation B) coexists, as depicted in Fig. 2b. Such arrangement provides multiple sites for accommodating different transition-metal cations and in a wide range of valence states, allowing therefore the preparation of a wide variety of oxides.64,65 The possibility for TM cations in both the tetrahedral and octahedral sites to be involved in the OER may bring ambiguities as to which site is responsible for the OER catalysis. However, theoretical and experimental studies have specifically addressed this issue, revealing that the octahedral sites have a superior OER catalytic activity, therefore, those are generally considered the active catalytic sites for the OER.37,66 The chemistry of the TM in the octahedral site can be tuned by the synthesis conditions and the composition, which provides means to tailor the catalytic activity for the OER.67,68 Ni in NixCo3−xO4 can be prepared in the 0 ≤ x ≤ 1 range,68 whereas MnxCo3−xO4 can be obtained as a single phase in the whole 0 ≤ x ≤ 3 range.67 In both cases, the reorganization of the cation site and their oxidation states leads to substantial modulation of the catalytic activity of the OER.2.3 TM oxyhydroxides
TM oxyhydroxides formed on the surface of TM have been long recognised as highly active catalytic species for OER in alkaline medium.69 Ni oxyhydroxide have received extensive attention because of its particularly high catalytic activity;70 however, the observed high performance have been shown to arise from the presence of Fe impurities.71,72 In a detailed study, L. Trotochaud et al. found that the incorporation of Fe impurities in Ni-based OER electrodes leads to the formation of a mixed Ni–Fe layered double hydroxides (NiFe-LDH) with a fougèrite/pyroaurite mineral structure (Fig. 2c), which is responsible of the high catalytic activity.73 In this structure, both Ni and Fe cations reside in octahedral local coordination; however, it was also shown that the long-range order is not important for the high activity of NiFe-LDH. Optimised synthesis of NiFe-LDH electrodes have led to overpotentials as low as 130 mV to achieve a current density of 10 mA cm−2,74 which has motivated a large number of studies on this type of OER catalysts, in recent years.75 The possibility of intercalating the LDH structure increases the flexibility for material design. For instance, the catalytic activity for the OER of NiFe LDHs can be tailored by the intercalation of anions, and it has been shown that the activity of the intercalated NiFe-LDH varies with the redox potential of the anion: low redox potential of hypophosphites favours the catalysis of the OER.76 Similar to complex oxides, the substitutional doping of NiFe LDHs is also used for tuning the catalytic activity. For example, V-doping has led to an outstanding catalytic performance due to enhanced conductivity, facile electron transfer, and abundant active sites.77 Dionigi and co-workers provided direct evidence that, under applied anodic potentials, NiFe and other Fe-containing LDHs oxidise from as-prepared α-phases to activated γ-phases. The OER-active γ-phases are characterised by about 8% contraction of the lattice spacing and switching of the intercalated ions.783. OER mechanisms for TM oxide electrocatalysts
Early attempts to describe the OER with parameters that could explain and predict the catalytic activity of TM oxides were based on the bond strength between surface of metal oxides and the adsorbates. Based on kinetic studies, Bockris and Otagawa the concluded that the OER on a series of perovskite oxides, containing metal cations from the first transition row, consisted in the electrochemical adsorption of OH−, followed by the rate-determining electrochemical desorption of ˙OH radical, which reacts with water to form O2.50,121 The study of absorbates bonding strengths as descriptors of the OER have been successfully developed with the aid of ab initio density functional theory (DFT) calculations, resulting in the elucidation of a well-stablished adsorbate evolution mechanism (AEM) depicted below in eqn (1)–(4).122–125| H2O + * → OH* + e− + H+ | (1) |
| OH* → O* + e− + H+ | (2) |
| O* + H2O → HOO* + e− + H+ | (3) |
| HOO* → * + O2 + e− + H+ | (4) |
Rossmeisl and co-workers investigated trends in the electrocatalytic OER on basis of a database of O*, HO* and HOO* adsorption energies on oxide surfaces, finding that there is a linear scaling relationship between the adsorption energies of the three species.123 In particular, the adsorption energies of HO* and HOO* are closely related because both species bind with the catalyst surface through an oxygen atom with a single bond so that the difference in adsorption energy for these two species (ΔG0HOO* − ΔG0HO*) is constant for any catalyst surface (see Fig. 3a).126 Thus, the variation in the OER overpotential between different oxide surfaces is determined by the O* adsorption energy. For catalysts that bind oxygen adsorbates strongly to its surface, the formation of the O–O bond in the HOO* adsorbate will be slow, making step 3 (eqn (3)) the rate-determining step (RDS). On the other hand, for catalysts that form labile bonds with oxygen adsorbates the deprotonation of the HO* species will be slow, making step 2 (eqn (2)) the RDS. Another consequence of the fixed adsorption energy difference between adsorbates HO* and HOO* is that the ηOER can be described in terms of the formation free energies of O* and HO*, ΔG0O* and ΔG0HO*, respectively. Plotting ηOERvs. (ΔG0O* − ΔG0HO*) leads to a general volcano-type relationship independent of the catalysts, as shown in Fig. 3b and c. Therefore, in order to rationalised the OER activity, attention must be focused on the oxygen binding strength on the surface of catalysts following the Sabatier principle: the most active catalyst binds oxygen intermediate species to the surface neither too strongly nor too weakly. The most active catalysts are in the centre of this plot and bind these key reaction intermediates neither too strongly nor weakly.
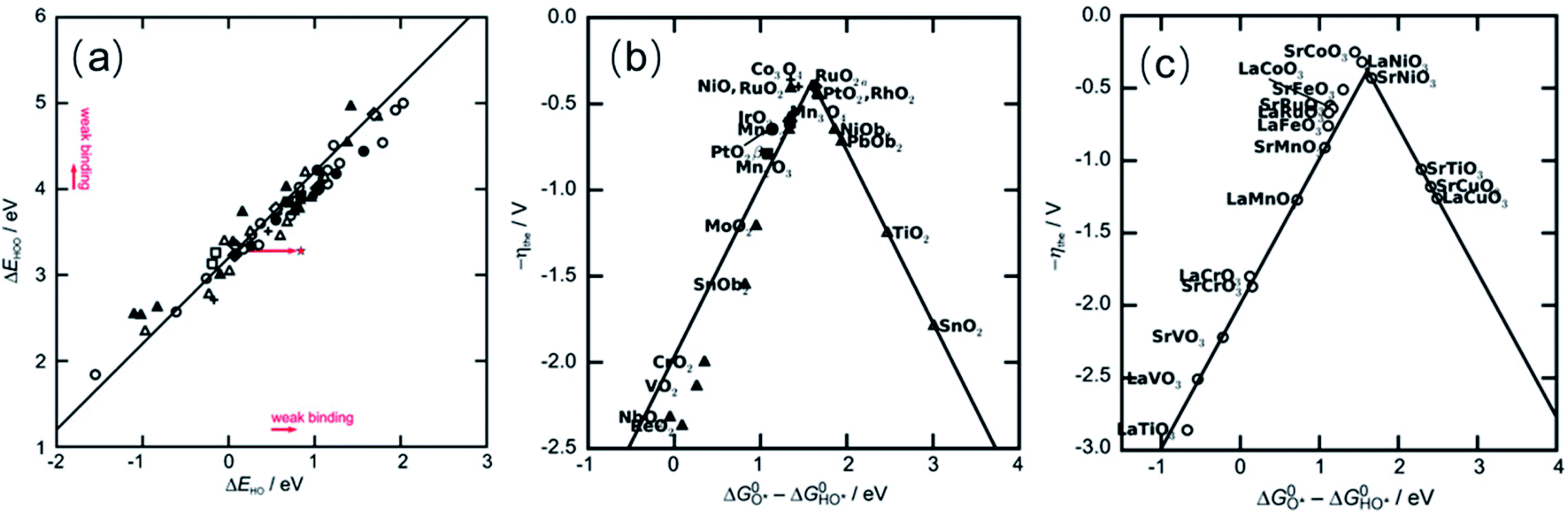 | ||
| Fig. 3 (a) Adsorption energy of HOO* plotted against the adsorption energy of HO* on perovskite, rutile, anatase, MnxOy, Co3O4, and NiO oxides. (b) Trends for the OER activity on rutile, anatase, MnxOy, Co3O4, and NiO oxides. (c) Trends for the OER activity on perovskite. Reproduced with permission.123 Copyright 2011, Wiley-VCH. | ||
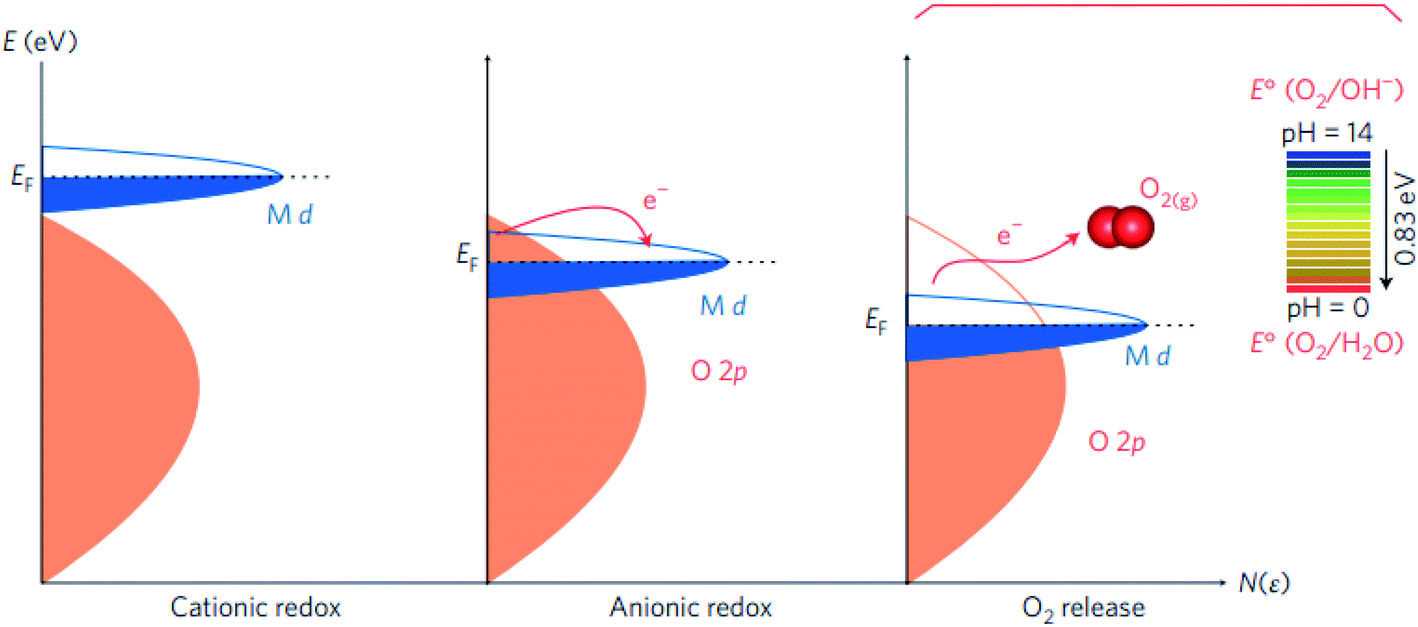
For TM oxides with high covalency in the TM–O bonds, the adsorption energy of oxygen adsorbates generally decreases and the direct O–O bonding with the reversible formation of oxygen vacancy become favourable. In this scenario, a second OER mechanism, the so-called lattice oxygen mechanism (LOM), has been described.126,127 Different from the AEM, the LOM proceeds on two neighbouring metal sites, and consists of 4 steps: (1) two HO* on the metal sites undergo deprotonation, resulting in two neighbouring TM–O− species, (2) these two oxo species directly couple to form the O–O bond, (3) O2 is released leaving behind two oxygen vacancies, (4) reoccupation of these vacancies with OH−. Fig. 4 shows a graphic comparison of the AEM and the LOM. Since HOO* is not an intermediate in the LOM catalytic cycle, catalysts that drive the OER via this mechanism deviate from the trends derived from the AEM which are based on the scaling relation between HO* and HOO*. Over the past few years, the LOM has been confirmed by a series of observations that demonstrate the importance of the bulk oxide electronic structure in the catalytic OER,128,129 and direct evidences of involvement of lattice oxygen in the OER.130–132
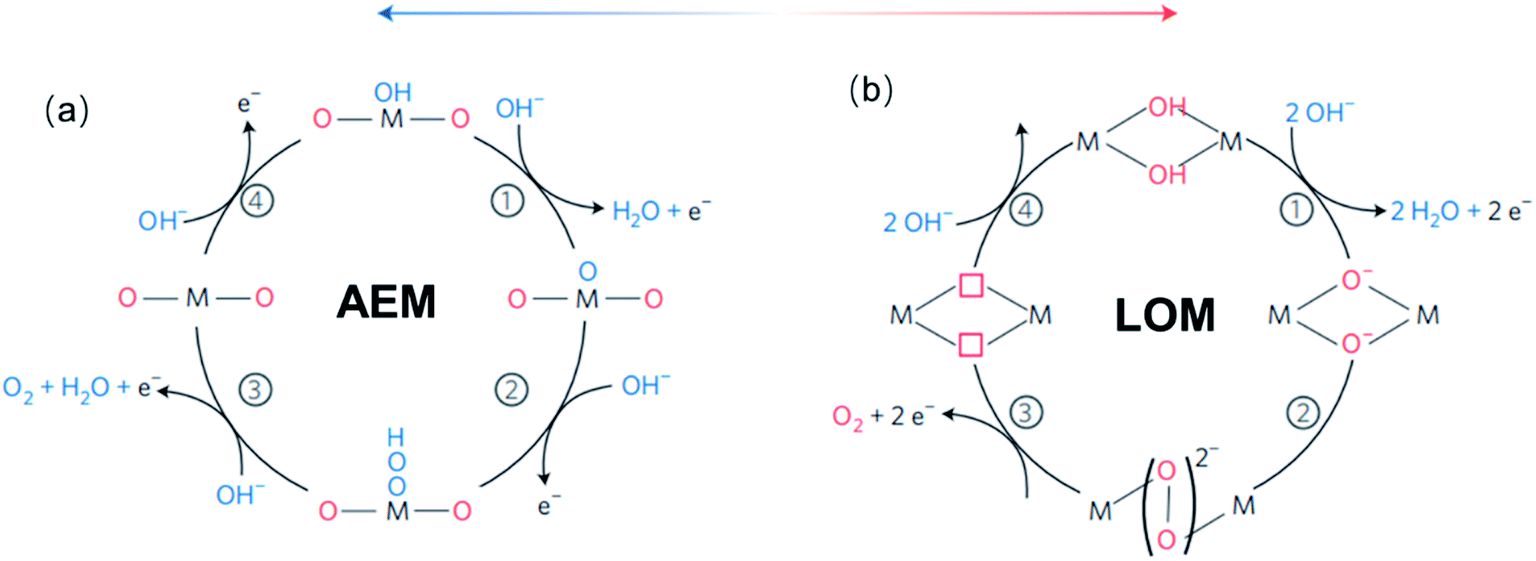 | ||
| Fig. 4 (a) The classical four-steps OER mechanism of proton-coupled electron transfer (AEM), (b) the idealized OER mechanism based on the direct pairing of two lattice oxygens (LOM) in the anionic redox process. Reproduced with permission.127 Copyright 2016, Macmillan Publishers Limited. | ||
4. Electronic structure parameters in TM oxides for OER electrochemical activity
From the reaction mechanisms (AEM and LOM) described in the previous section, it is clear that the OER process is driven by (i) the binding strength of the intermediate oxygen adsorbates (O* and OH*), which is important when the AEM operates, and (ii) the lattice TM–O covalency, which dictates when the LOM becomes active. Additionally, electrons transfer between the electrolyte and the catalyst's surface will always be involved in the OER, and therefore (iii) the Schottky barrier for such electrons transfer,133 is also an important factor the overall OER process. All of these three factors are derived from the electronic structures of the TM oxides, which provides means to directly correlate measurable and controllable electronic properties with the catalytic activity. On the one hand, the electronic configuration of the TM cations drives the covalency of the TM–oxygen bond in the bulk structure, which is ultimately related to the surface TM binding energies with adsorbate oxide species. A coordination chemistry approach is useful for describing these bonds.61 The local structure of most TM metal oxides catalytically active for the OER, e.g., perovskite, rutile, spinel and LDH, feature a TM bonded to six O atoms in an octahedral (TM)O6 coordination. In such coordination, the TM 3d and oxygen 2p atomic orbitals hybridize to form σ orbitals (O 2p with dz2 and dx2–y2 orbitals of the TM) and π orbitals (O 2p orbitals with dxy, dxz and dyz orbitals of the TM), as depicted in Fig. 5a. The σ and π antibonding (σ*and π*) orbitals are named after their symmetry eg and t2g, respectively, and they are the orbital of interest because their filling determines the covalency of the TM–oxygen bonds. On the other hand, the Schottky barrier for an electron transfer between adsorbates (O* and OH*) and TM oxide catalysts is defined by the energy difference between the unoccupied TM 3d–O 2p band and the redox potential of the electrolyte.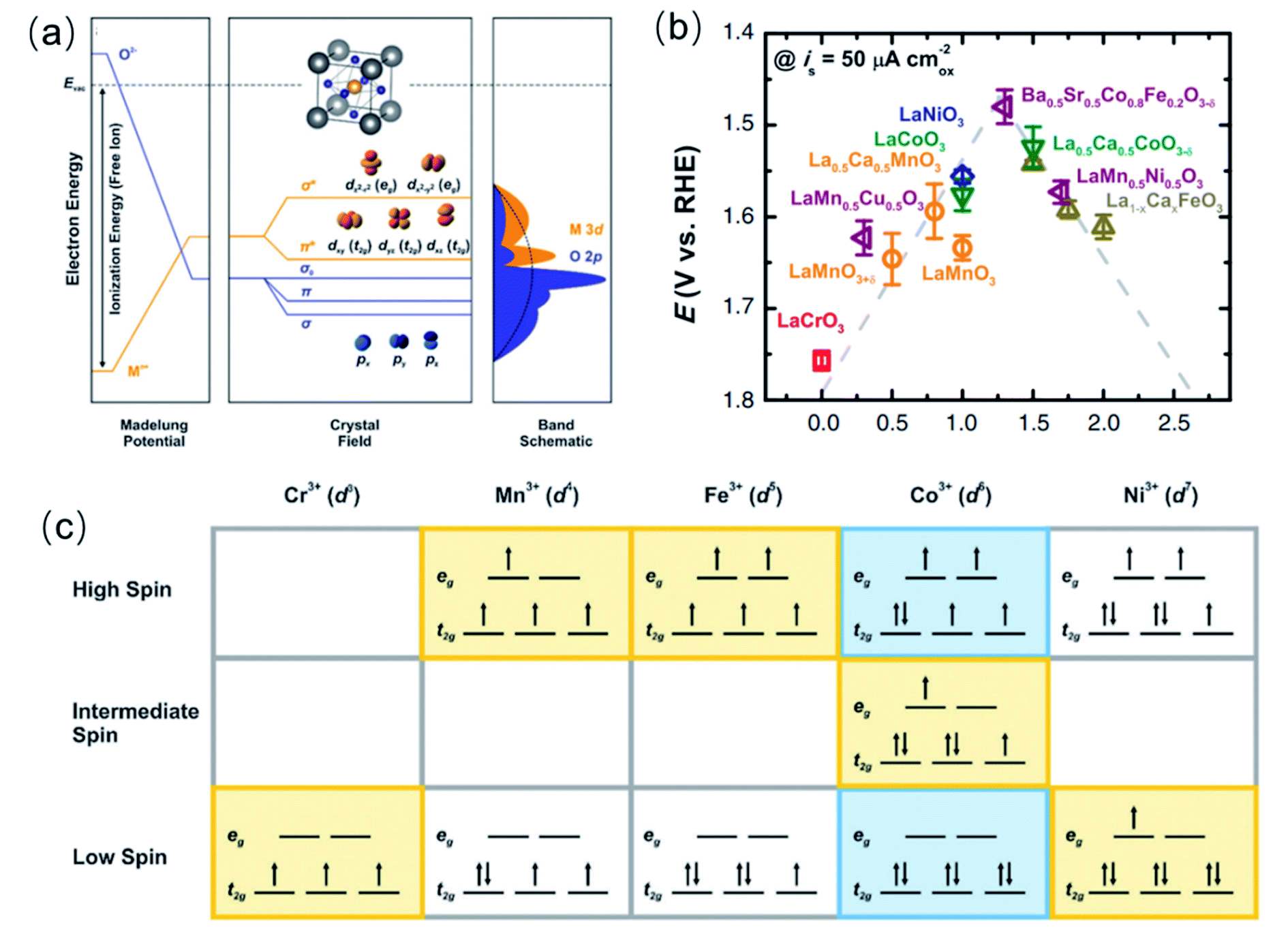 | ||
| Fig. 5 (a) Schematic band structure of perovskite oxides (unit cell inset). (b) Elation between OER activity and eg occupancy of transition metal cations (B in ABO3). (b) Is reproduced with permission.51 Copyright 2011, American Association for the Advancement of Science. (c) eg orbital occupancy of some transition metal cations with different spin states. (a and c) Are reproduced with permission.25 Copyright 2015, Royal Society of Chemistry. | ||
On basis of those factors, several electronic structure descriptors for the OER activity have been recently proposed, addressing the binding strength of adsorbed OER intermediates, the covalency of the TM–O bond, and the energetics for electrons transfer at the interface. The most successful descriptors of the binding strength of adsorbates is the number of electrons in the eg-orbital of the transition metal cations.51 As for the TM-covalency, the degree of TM 3d–O 2p hybridization,128,129 the energy position of the O 2p band centre,59 and the charge-transfer energy, have been used as electronic descriptors.35 Whereas, the Schottky barrier for electrons transfer at the interface has been discussed in terms of the band alignment at the catalysts/electrolyte interface, using parameters like the work function and the electron affinity of the oxide relative to the electrolyte.35,63,68In the following, we will provide the fundamental basis of these electronic descriptor, and how they are used in order to rationalise OER activity and mechanism. In the following Section 5, we also discuss the strategies for modulating the electronic structure parameter in order to optimise the activity for the OER.
4.1 Descriptors for the binding strength of the intermediate oxygen adsorbates: the number of electrons in the eg-orbitals of the TM cations
The filling of the eg-orbitals as a descriptor of the OER catalytic activity of perovskite oxides was first proposed by Shao-Horn and co-workers.51 Since the eg orbitals of surface transition metal ions participate in σ-bonding with adsorbates, its occupancy influences the binding energy of oxygen-related intermediate species on the TM, and therefore the OER activity. They reported that a near-unity occupancy of the eg orbitals of surface transition metal ions and high covalency in bonding to oxygen can enhance the intrinsic OER activity of perovskite transition metal oxides in alkaline solution. Specifically, they found that an occupancy of 1.2 in Ba0.5Sr0.5Co0.8Fe0.2O3−δ was optimum within a group of perovskite oxides with number of eg electrons varying from 0 to 2 (Fig. 5b). The occupancy of eg-orbitals is more relevant than that of t2g orbitals because the σ-bonding eg orbital has a stronger overlap with the oxygen-based adsorbate than does the π-bonding t2g orbital, and therefore can more directly promote electron transfer between surface cation and adsorbed reaction intermediates.51 More recently, it was found that the eg occupancy of the cation in the octahedral site of spinel oxides also works as a descriptor of the OER catalytic activity of these materials,67 with a similar near unity optimum occupancy.In general, the eg orbital occupation of TM oxides of the fist transition series can be modified by tuning the oxidation state and spin state of the TM cation. Although the total number of 3d electrons is only determined by the oxidation state, e.g., Ni3+ is a 3d7 cation and Fe3+ is a 3d5 cation, the spin configuration changes the eg-orbital occupancy. As shown in Fig. 5c, Ni3+ can adopt a low spin configuration t62ge1g or a high spin configuration t52ge2g, respectively providing 1 or 2 eg electrons, whereas Fe3+ can have 2 eg electrons in the high spin configuration t32ge2g or none in the low spin configuration t52ge0g. Therefore, characterisation techniques for the determination of the actual oxidation state and spin configuration are very important in order to rationalise the catalytic activity for the OER based on the eg occupancy. X-ray photoelectron spectroscopy (XPS) and near-edge X-ray absorption fine structure (NEXAFS) are generally used to explore the oxidation state of elements in a compound,134–136 and most studies of OER catalysts include such spectroscopic characterization. Although the characterisation of the spin configuration is less extended, some authors have specifically studied the magnetic properties and spin configurations of TM oxide OER catalysts using field-cooling magnetization measurements,58,137 and superconducting quantum design (SQUID) magnetometer.138 From these measurements, an effective magnetic moment μeff can be calculated, which provides means to determine the average spin configuration. The study of magnetic properties has been particularly extensive in perovskite cobaltite because whether the spin state of Co3+ ions at room temperature is a mixture of low spin (t62ge0g) and high spin (t42ge2g) states or an intermediate-spin (t52ge1g) state (see Fig. 5c) has been controversial in the past decades. However, using either interpretation of the spin state in perovskite cobaltite, there is a general agreement that an average eg occupancy close to the unity is beneficial for the OER activity. Advanced interpretations of the photoelectron spectra and the X-ray adsorption spectra also provide the magnetic properties;139 however well-defined samples are usually required.
4.2 Descriptors of the TM–O covalency: degree of TM 3d–O 2p hybridisation
Aiming at a more comprehensive description of the catalytic OER on complex perovskites, Shao-Horn and co-workers introduced a quantitative measure of the TM–O covalency as an additional descriptor for oxides with an eg-occupancy of 1.51 They estimated the TM 3d–O 2p hybridisation degree (H) from the background-corrected spectral intensity associated with the O 1s → TM 3d–O 2p transition within the O K-edge X-ray absorption (Abs) normalised by the nominal number TM 3d empty levels in eg (heg) and t2g (ht2g) symmetries, using the expression: H = Abs/(heg + ¼ht2g).51,140H is a measure of the covalency because the O K-edge XAS probes the transition from O 1s to unoccupied states with partial O 2p character hybridised with the valence states of the metal cations, due to dipole selection rules. Fig. 6a shows the potentials to reach 50 μA cm−2 current density as a function of the H for LaMnO3, LaCoO3 and LaNiO3. The linear decrease of the overpotentials with the increase of the TM–O covalency led the authors to conclude that a greater covalency between the TM and the oxygen, where the active redox couple of the TM cation has a larger O 2p character, promotes the charge transfer between surface cations and adsorbates, therefore accelerating the rate-determining steps in the OER.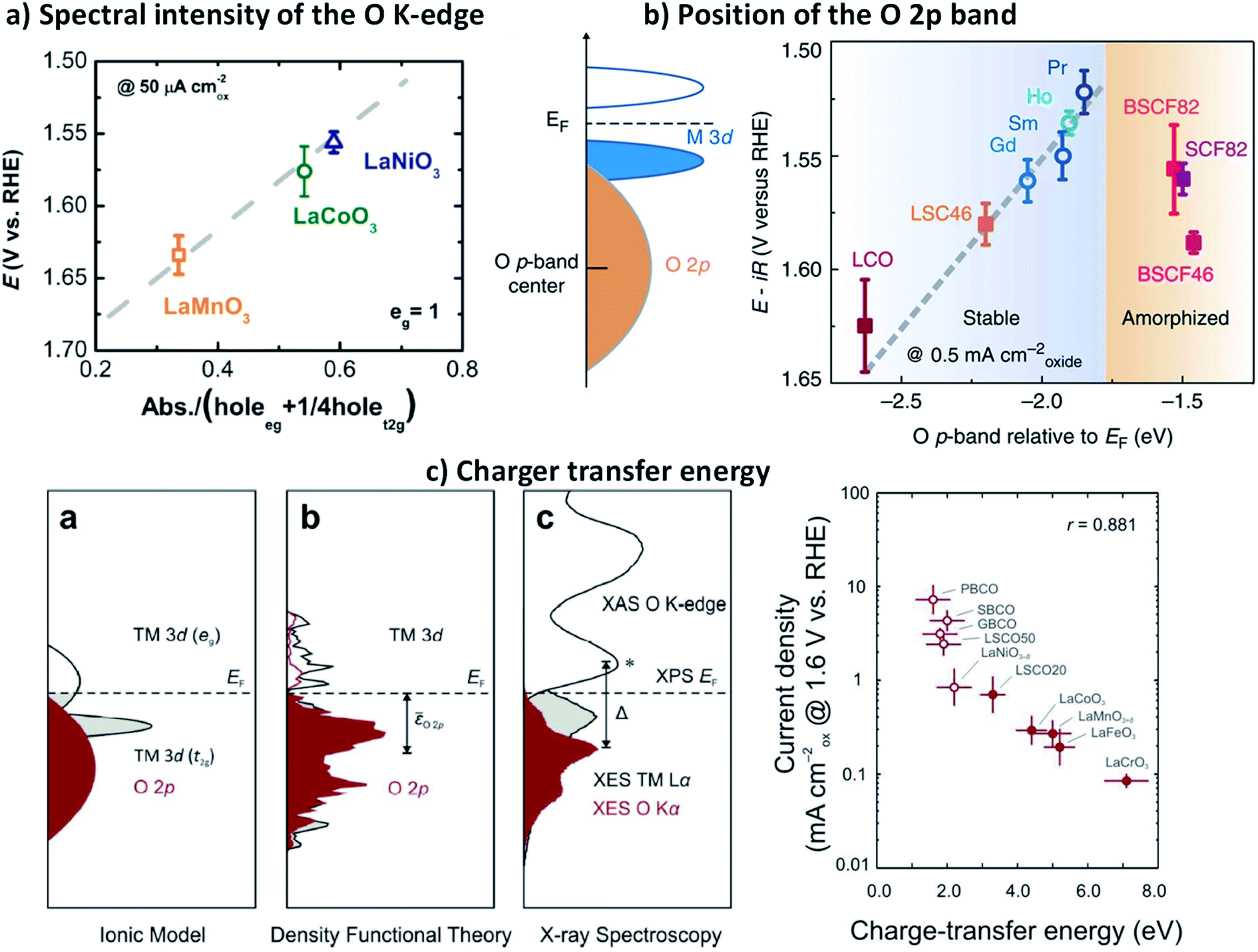 | ||
| Fig. 6 (a) Shows the potentials to reach 50 μA cm−2 current density as a function of the H for LaMnO3, LaCoO3 and LaNiO3. Reproduced with permission.51 Copyright 2011, American Association for the Advancement of Science. (b) Schematic representation of the O p-band for transition metal oxides and evolution of the iR-corrected potential at 0.5 mA cm−2 oxide versus the O p-band centre relative to EF (eV). Reproduced with permission.60 Copyright 2013, Macmillan Publishers Limited. (c) Representations of the transition metal and oxygen partial density of states based on the ionic model, density functional theory and X-ray spectroscopy (XES, XAS, and XPS) of LaCoO3, and correlation between charge transfer energy (Δ) and the OER activity of 10 metal oxides, 5 semi-metallic (open dots) and 5 semiconducting (closed dots). Reproduced with permission.134 Copyright 2017, Royal Society of Chemistry. | ||
Suntivich and co-workers estimated empirical trends of the TM 3d–O 2p hybridisation in complex perovskite using the pre-edge feature of the O K-edge X-ray absorption spectrum.141 The authors found that the hybridisation increases with the number of 3d electrons, which is consistent with the expected electronegativity trend, and also with the oxidation state of the TM cation. It was recognised that a higher oxidation state of the TM in the oxide reduces the energy difference between the TM 3d and the O 2p, leading to an increased covalency.141,142
In 2013, Shao-Horn and co-workers demonstrated that the energy level of the O 2p band relative to the Fermi level (EF) could be also used as a descriptor for the TM–O covalency and activity of complex perovskites catalysts for the OER, as well as their stability.59Fig. 6b shows how the shift of the O 2p band centre closer to the EF in a series of perovskite cobaltite, correlates with the decrease of the overpotential needed to reach a 0.5 mA cm−2. However, they found a shift limit from which the materials undergo amorphization during the OER, which leads to instability and activity losses. These observations suggested the participation of lattice oxygen in the reaction, which has been later confirmed for a series of perovskite cobaltite.130 Although the level of the O 2p band relative to the EF provides satisfying description of the OER on highly conducting oxides, is less reliable to describe OER kinetics on semiconductors and insulating oxides because the Fermi level may lie in forbidden energies and can be poorly defined for this type of materials.
More recently, Hong and co-workers made use of a combination of X-ray emission, X-ray absorption, and X-ray photoelectron spectroscopies to examine the electronic structure of a series of complex perovskite oxides LaBO3 (B = Cr, Mn, Fe, Co and Ni).143 These studies allowed the direct determination of the energy difference between the TM 3d and the O 2p, the so-called charger transfer energy (Δ), which can be directly related with the TM 3d–O 2p hybridisation degree, and therefore with the covalency in the TM–O bonds. The group of authors later reported that reducing Δ greatly enhance the OER activity of oxides (Fig. 6c), and the trend across 10 oxides was statistically stronger than using the O 2p-band centre as the descriptor for both semi-metallic and semiconducting oxides.35 They showed that the charge-transfer energy, determined from the experimental elucidation of the band positions, can be used to evaluate trends in the electronic and chemical interactions at the oxide/electrolyte interface relevant to the OER, specifically the electron-transfer kinetics (electrochemical steps) and hydroxide affinity (chemical steps). As a result, the analysis of the charge-transfer energy of catalytic metal oxides can explain not only OER activities trends but also changes in the mechanisms of the OER from electron-transfer-limited to proton-electron-coupled, to proton-transfer-limited reactions.
 | ||
| Fig. 7 Schematic representation of the energy level versus density of states N(ε), showing the respective motion of the metal d band with respect to the oxygen p band in going from cationic to anionic redox processes and then O2 release. Reproduced with permission.127 Copyright 2016, Macmillan Publishers Limited. | ||
In situ 18O isotope labelling mass spectrometry has been also employed to provide evidences of the LOM in oxides other than covalent perovskites. Amin and co-workers used isotope labelling together with differential electrochemical mass spectrometry to study the OER mechanism on spinel Co3O4.147,148 Their work showed that the alkaline OER in H218O-containing electrolyte generated an amount of 18O16O evolved increases from cycle to cycle before reaching a steady-state value, showing that a surface layer of Co3O4 was taking part in OER via an oxygen exchange mechanism. They found that total number of oxygen atoms of the oxide participating in OER was 0.1 to 0.2% of the total oxide loading, corresponding to about 10–30% of the surface atoms, which represented the catalytically active sites.147
Although the LOM is associated with highly active covalent oxides, the OER mechanism for NiFe oxyhydroxides, the most active OER catalyst in alkaline OER, has been shown not to proceed via lattice oxygen exchange.149,150 Roy and co-workers used a model system of mass-selected NiFe nanoparticles and isotope labelling experiments to show that oxygen evolution in 1 M KOH does not proceed via lattice exchange on very highly active electrodes.150 The lack of lattice oxygen participation in the OER on NiFe oxyhydroxides was further confirmed by Lee and co-workers in a mechanistic study based on 18O-labeling experiments in combination with in situ Raman.151 However, in the same study, they also found that lattice oxygen does participate in the OER on Fe-free Ni and NiCo oxyhydroxides catalysts, specifically in the formation of a NiOO− species that was identified as an OER intermediate. Based on DFT calculation, Huang and co-workers proposed that the OER proceeds via the LOM pathway on the metal oxyhydroxides only if two neighbouring oxidized oxygens can hybridize their oxygen holes without sacrificing metal–oxygen hybridization significantly, finding that the LOM is highly favoured in the OER on Zn0.2Co0.8OOH.152
4.3 Descriptor of the Schottky barrier for e− transfer: band alignment at the catalysts/electrolyte interface
As described in above in Section 4.2, the band energy position in metal oxides dictates the charge transfer energy, which is a measure of the covalency of the TM–O bond. Additionally, the band positions relative to the redox potential of the OER provides information on the electronic and chemical interaction at the catalysts/electrolyte interface relevant to the OER.35 On the one hand, the energy difference between the unoccupied TM 3d–O 2p band and the OER redox potential defines the electron affinity of the oxide relative to the electrolyte, and it represents a Schottky barrier for electron transfer, as schematically shown in Fig. 8a. On the other hand, the energy difference between the potential of the electrolyte and the Fermi level is a measure of the hydroxide affinity of the catalyst (see Fig. 8a). In a comparative study of several oxides, Hong and co-workers found that oxides with a larger charge-transfer energy have a higher Schottky barrier for electron transfer at the oxide-electrolyte interface (Fig. 8b).35 As the charge transfer energy decreases in the series of oxide catalysts, the barrier for electron transfer associated with OER decreases. Additionally, upon decreasing the charge transfer energy, the electrical conductivity of the oxides tends to increase, which brings about an extra benefit of a low resistance to the current flow during the OER.153 However, the intrinsic OER active is more influenced by the presence of low-energy TM 3d orbital states that facilitates the interface electron transfer associated with the OER process. Yun and co-workers recently found that semiconducting LaCoO3 has an intrinsic OER activity more than one order of magnitude higher than that of metallic LaNiO3, in contrast to previous reports.154 The group of authors showed that misleading interpretations could be avoided by using very thin films of the catalyst on conducting interlayer as samples.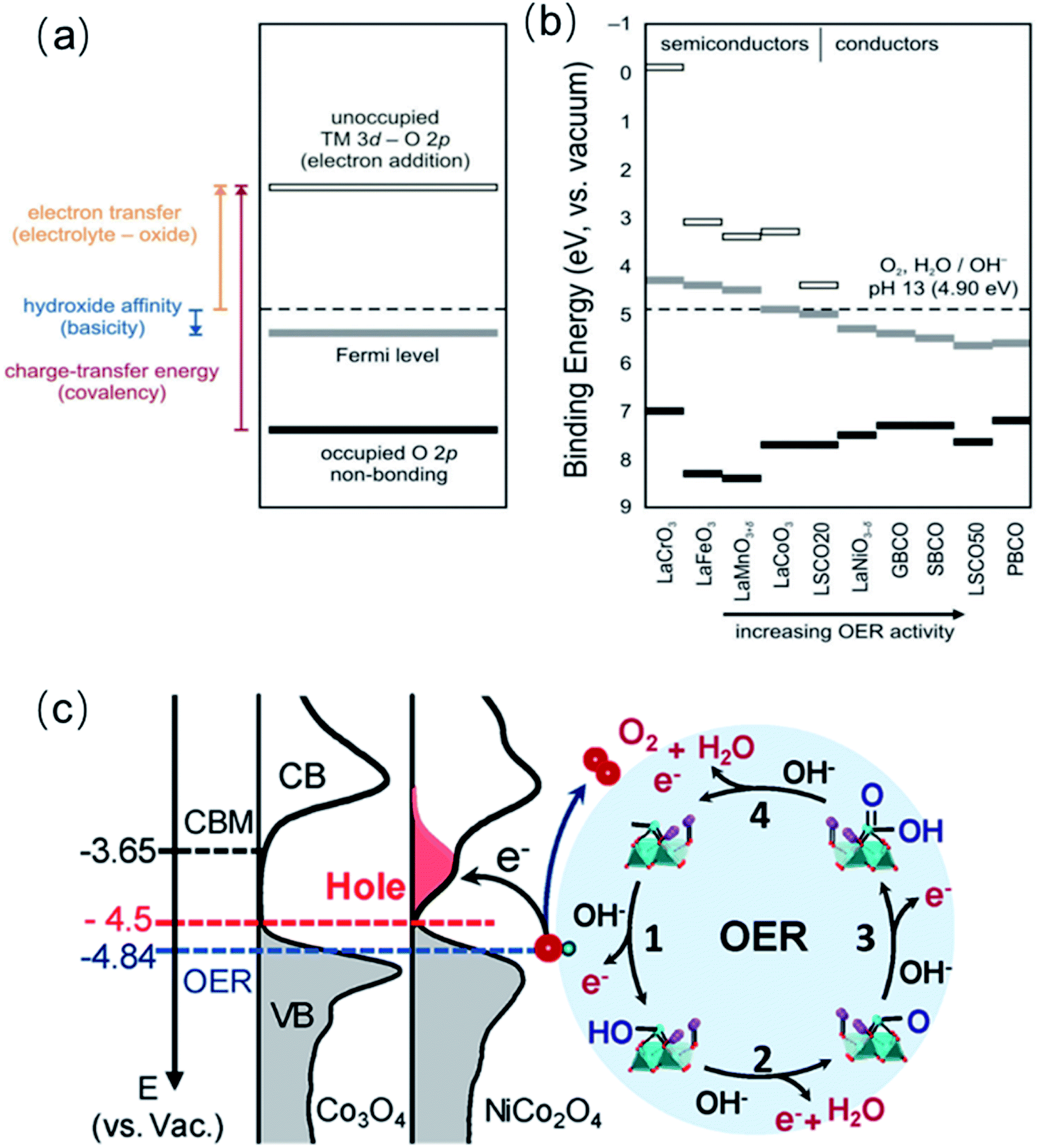 | ||
| Fig. 8 (a) Schematic representation of electron transfer energy at the electrolyte-oxide interface, hydroxide affinity of the oxide, and charge-transfer energy (covalency). (b) Estimation of electron transfer energy, hydroxide affinity, and charge-transfer energy from X-ray spectroscopic data under rigid band assumptions for La0.8Sr0.2CoO3−δ (LSCO20), La0.5Sr0.5CoO3−δ (LSCO50), GdBaCo2O5+δ (GBCO), SmBaCo2O5+δ (SBCO), PrBaCo2O5+δ (PBCO), Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF). Reproduced with permission.35 Copyright 2017, Royal Society of Chemistry. (c) Left panel: the measured occupied and unoccupied density of states (DOS) near the EF for Co3O4 and NiCo2O4; the energy level is relative to vacuum level (vs. Vac.). Right panel: schematics of the four-step reaction pathway of OER. Reproduced with permission.68 Copyright 2019, American Chemical Society. | ||
We have been recently working on a systematic approach to study the relationship between the OER catalytic activity and the electronic structure of complex perovskite, double perovskite and spinel systems such as SrxLa1−xFeO3, La2−xSrxNiMnO6 and NixCo3−xO4.63,68,98 For all three systems, increasing x in the range 0 ≤ x ≤ 1 triggers a shift of the Fermi level (EF) closer to the valence band maximum (VBM) and induces the formation of hole states (unoccupied) below the conduction band minimum (CBM), near the EF. We argue that filled electronic states (i.e., VBM) near the EF improve the orbital overlap with key intermediates, favouring their formation (Fig. 8d). At the same time, unoccupied states close to the EF increase the electron affinity of the oxide semiconductors, thereby facilitating charge transfer at the interface associated with water oxidation. We found linear correlations between the OER activity and the downshift of the EF, as well as with the hole state density, which shows that band energy level descriptors can be very powerful to described the OER activity of TM oxides.
5. Material engineering strategies for the OER tailoring based on electronic parameters
As described above, in Section 4, many efforts have been devoted to discovering the correlation between the electronic structure and OER catalytic activity of TM oxide catalysts. Several electronic structure descriptors of important parameters for the OER catalysis, such as the adsorption energy of adsorbates (described by the eg occupancy), the covalency (described by O 2p energy level and charge transfer energy) and the interface energetics (ionisation potential, EF and electron affinity), have been developed. In this section, we will focus on strategies to modify those electronic parameters that determine the OER catalytic properties of TM oxides through intentional doping, strain and defect engineering.155,1565.1 Doping
Doping is a common and efficient strategy to tuning the electronic structure of TM oxide catalysts, due to the high flexibility in compositions and crystal structures. Introducing or substituting foreign metal ions with different radius size, valence state and electron-donating ability may enable charge transfer occurred through shared oxygen ions or change the lattice parameters and thus the degree of overlap between atomic orbitals, finally altering the electronic, optical and magnetic properties of the oxides as well as their catalytic activity.Various cases of metal doping have been reported to successfully improve the catalytic activities compared with the pristine metal oxide catalysts due to advantageous modification of electronic structure.157 Interestingly, among these cases, the incorporation of Fe can significantly enhance the OER catalytic activities of Co158 or Ni (oxy)-hydroxides.73 The catalytic activity of NixFe1−xOOH even reached 1000-fold higher than the pristine NiOOH.20 Nowadays, layer-structured NixFe1−xOOH electrocatalysts show the best OER catalytic activity in alkaline condition. However, although the superior performance of NixFe1−xOOH has been well accepted, there still remains a confusion how iron dopants influence the OER activity. In the 1980s, Corrigan71 found the crucial effect of Fe impurities on the improvement of the catalytic activity, which has been later verified by Trotochaud and co-workers73 in comparative experiments using an increasing Fe concentration in the KOH solution for the electrochemical tests. The group of authors argued that the incorporation of Fe promoted the electronic conductivity of NiOOH. As illustrated in Fig. 9a, the results of electrochemical investigation revealed that the Ni redox peaks of NixFe1−xOOH catalysts shift toward the anodic direction with increasing x, suggesting the changes happened on the electronic structure.44,159,160 Similar findings were also obtained from electrochemical testing of Fe-doped CoOOH;158 it seemed that Fe incorporation had a similar influence on Ni and Co hosts. To further uncover the effect of Fe in NixFe1−xOOH, Friebel and co-workers used operando X-ray absorption spectroscopy (XAS) to reveal that Fe3+ in NixFe1−xOOH occupies octahedral sites with unusually short Fe–O bond distances induced by edge-sharing with surrounding [NiO6] octahedra and they also identified that Fe cations were the active sites instead of Ni cations by computational results.40 In addition, Ahn and Bard utilized surface interrogation scanning electrochemical microscopy (SI-SECM) technology to directly investigate the OER kinetics of Ni4+ and Fe4+ in Ni(OH)2, FeOOH, and NixFe1−xOOH and they claimed that the OER kinetics were much more fast on iron sites. These results seem to indicate that Fe dopants are the active sites.161 In order to more directly identify the existence of Fe species in NixFe1−xOOH, Mössbauer spectroscopy, has been applied in the investigation of NixFe1−xOOH electrocatalysts.162 For example, Jamie and co-workers reported the first direct evidence for the formation of Fe4+ in NiFe oxide through operando Mössbauer spectroscopy.163 However, they proposed that the detected Fe4+ species did not directly serve as the active site in the OER process, although their existence had great effect on the improvement of OER activity for NiFe oxide electrocatalyst. Using in situ differential electrochemical mass spectrometry (DEMS) and XAS, Görlin et al.164 found that Ni ions remained in the Ni2+ state with the iron incorporation which might be responsible for the enhanced faradaic efficiency, while the Ni ions in pure NiOOH would be oxidized from Ni2+ to Ni3+ or Ni4+ during OER process, leading to a lower faradaic efficiency (Fig. 9b and c). However, later, Nocera et al. proposed that the incorporation of Fe could promote the formation of Ni4+via the Lewis acid effect, causing the observed improved OER performance.165 More recently, Xu and co-workers explained the effect of iron of NixFe1−xOOH on the OER process from a perspective of the local spin configurations of surface iron and nickel.166 As shown in Fig. 9d, the electronic configuration of surface Fe3+ cations under the stable high spin state have 5 unpaired electrons (1 per d orbital), while the low spin state of surface Ni3+ only possesses one single electron at eg orbitals; a spin channel around the Fe sites can be obviously observed in Fig. 9e. During the OER process, the possible orbital interactions between Fe3+/Ni3+ and the *OH, *O, and *OOH species are shown in Fig. 9f. The group of authors found that the different d-orbital occupations of Fe3+ and Ni3+ could result in distinct reaction kinetics, and that iron sites in NixFe1−xOOH could promote the OER process through both the reactant adsorption and product desorption. Actually, other metal cations such as La, Y, Cd, Ce, Cr, and Zn etc., also have been reported to be dopants incorporated in layered metal hydroxides, but their improvement on the OER performance has been less pronounced compared to Fe.167–170
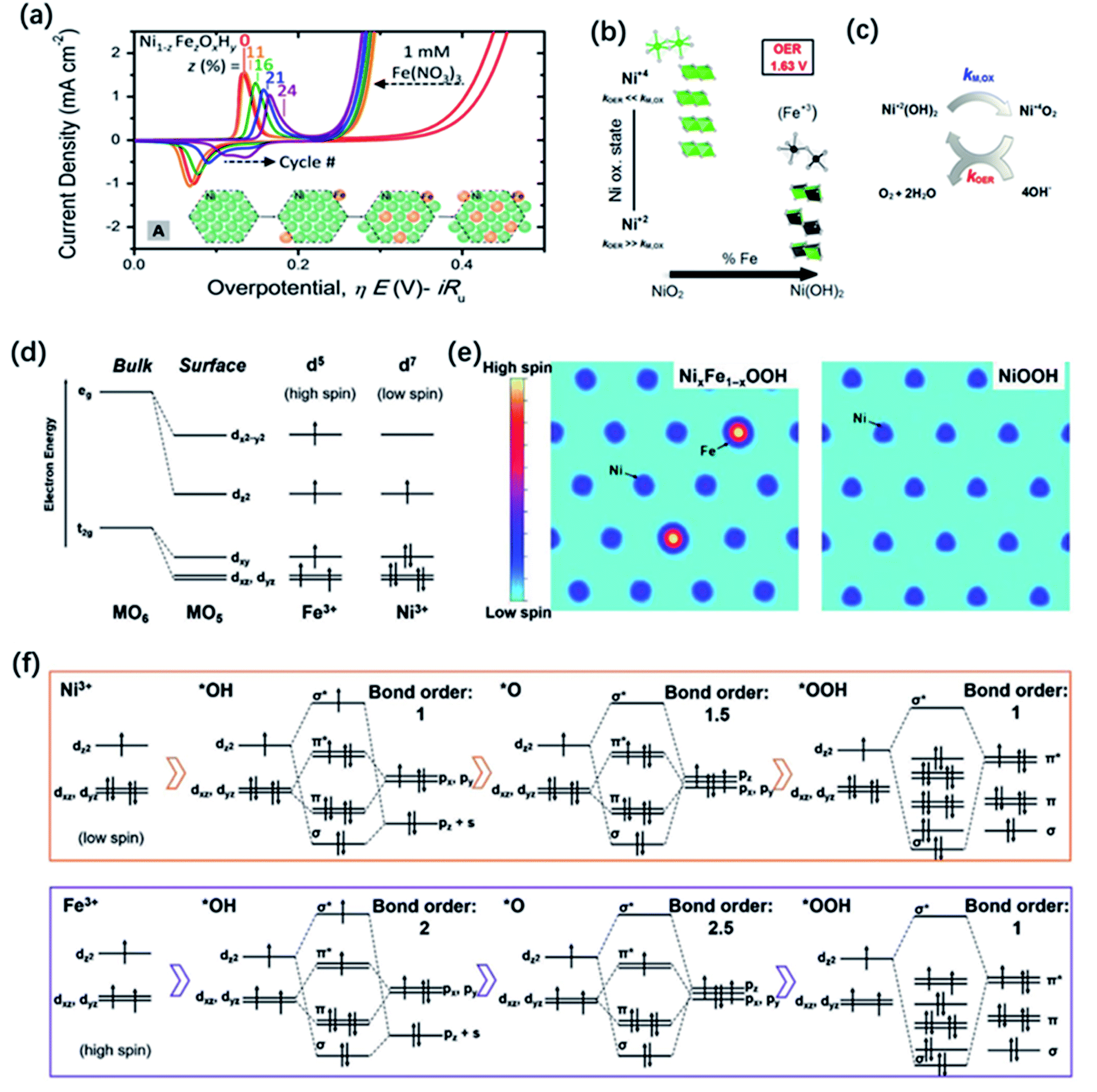 | ||
| Fig. 9 (a) Cyclic voltammetry of NiOxHy with different amount of Fe incorporation. The inset depicts a possible schematic of Fe incorporation into a NiOxHy platelet as a function of cycling in Fe saturated solution. Reproduced with permission.160 Copyright 2017, American Chemical Society. (b) XAS-derived structural motifs prevalent during OER catalysis at high and intermediate Ni-content. (c) Simplified scheme of the electrochemical water splitting cycle with metal oxidation rate constant, kM,OX, and the catalytic OER rate constant, kOER. Reproduced with permission.164 Copyright 2016, American Chemical Society. (d) d-Electron configurations of iron and nickel cations at the surface. (e) The spin channel of Fe sites and Ni sites in NixFe1−xOOH and NiOOH. (f) The orbital interactions between cations and the OER intermediates. Reproduced with permission.166 Copyright 2020, Wiley-VCH. | ||
A typical group of metal oxides featured by the highly compositional and structural flexibility with the unit formula of ABO3, where A usually is the larger ions such as rare-earth metal cations and alkali-earth metal and B is the smaller ions such transition metal cations, is the perovskite-type metal oxides. The ABO3 structure of perovskite can greatly adapt cation incorporation through partially substituting the A or B cation or both of them with other metals giving the 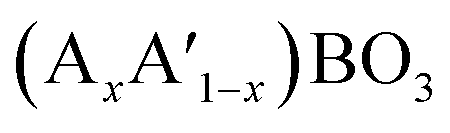 ,
, 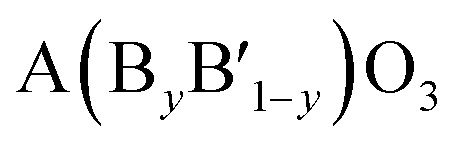 ,
, 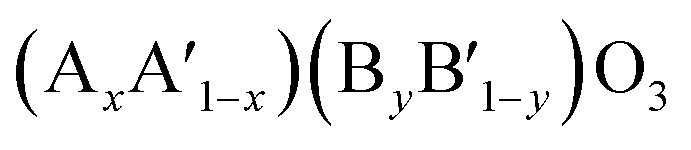 or even more complex compositions. In the periodic table, over 90% of the metal elements could be potentially used to construct perovskite oxides, which provides a broad diversity and possibility for doping.171 Depending on the adjustment of doping, perovskite oxides not only offer controllable electronic structure for modifying the OER activities but they also provide excellent models for establishing correlations between the activity and the electronic properties of surface and/or bulk properties via stablishing electronic descriptors. For example, Xie and co-workers have reported that the Yb doped CaMnO3 electrocatalyst showed a significant enhancement of the OER performance compared with pure CaMnO3, which could be ascribed to optimized electron occupation numbers of eg orbital by doping.172 Zhao and co-workers also found that the co-doping of Sr and Fe into PrBaCo2O5+σ perovskites led to a favourable eg electron filling, which could improve the intrinsic OER activity.109 Mefford and co-workers synthesized a series of La1−xSrxCoO3−δ perovskites with the different amount of Sr dopants. They claimed that the covalency of the Co–O bond can be enhanced through Sr incorporation and thus influence the OER activities.36 Similarly, Xu and co-workers introduced Fe dopants into LaCoO3 for modifying its OER activity,138 and they showed by means of DFT calculation that the increased performance relates to an enlarged covalency of Co–O with 10% incorporation of Fe in LaCoO3. On the other hand, Liu and co-workers analysed the effect of Sr doping in the LaNiO3 on the improvement of OER activity, and they rationalised the observed results based on the analysis of energy levels derived from X-ray photoemission spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) in combination with DFT calculation.173 As is shown in Fig. 10a–c, the results indicated that an up-shift energy of the O 2p band relative to the Fermi level occurred with rising x in La1−xSrxNiO3, which strengthened the degree of Ni 3d–O 2p hybridization and reduced the charge transfer energy, which resulted in a significant enhancement the OER activity. IN a similar analysis approach, Zhang and co-workers recently tuned the electronic structure of double perovskite La2NiMnO6 by Sr doping, which effectively led to higher OER activities of La2−xSrxNiMnO6.98 Their investigation on electronic structures discovered that the Sr doping induced Ni3+ states, resulting in the upshifted valence band centre and enhanced the hybridization of O 2p with Ni 3d and Mn 3d orbitals (Fig. 10d and e). Besides, the increased hole states originated from Ni3+ states decreased the energy barrier of the electron transfer from 0.44 to 0.12 eV. They claimed that the enhanced hybridization of O 2p with Ni and Mn 3d orbitals and the increased hole states should be responsible for the better OER activities.
or even more complex compositions. In the periodic table, over 90% of the metal elements could be potentially used to construct perovskite oxides, which provides a broad diversity and possibility for doping.171 Depending on the adjustment of doping, perovskite oxides not only offer controllable electronic structure for modifying the OER activities but they also provide excellent models for establishing correlations between the activity and the electronic properties of surface and/or bulk properties via stablishing electronic descriptors. For example, Xie and co-workers have reported that the Yb doped CaMnO3 electrocatalyst showed a significant enhancement of the OER performance compared with pure CaMnO3, which could be ascribed to optimized electron occupation numbers of eg orbital by doping.172 Zhao and co-workers also found that the co-doping of Sr and Fe into PrBaCo2O5+σ perovskites led to a favourable eg electron filling, which could improve the intrinsic OER activity.109 Mefford and co-workers synthesized a series of La1−xSrxCoO3−δ perovskites with the different amount of Sr dopants. They claimed that the covalency of the Co–O bond can be enhanced through Sr incorporation and thus influence the OER activities.36 Similarly, Xu and co-workers introduced Fe dopants into LaCoO3 for modifying its OER activity,138 and they showed by means of DFT calculation that the increased performance relates to an enlarged covalency of Co–O with 10% incorporation of Fe in LaCoO3. On the other hand, Liu and co-workers analysed the effect of Sr doping in the LaNiO3 on the improvement of OER activity, and they rationalised the observed results based on the analysis of energy levels derived from X-ray photoemission spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) in combination with DFT calculation.173 As is shown in Fig. 10a–c, the results indicated that an up-shift energy of the O 2p band relative to the Fermi level occurred with rising x in La1−xSrxNiO3, which strengthened the degree of Ni 3d–O 2p hybridization and reduced the charge transfer energy, which resulted in a significant enhancement the OER activity. IN a similar analysis approach, Zhang and co-workers recently tuned the electronic structure of double perovskite La2NiMnO6 by Sr doping, which effectively led to higher OER activities of La2−xSrxNiMnO6.98 Their investigation on electronic structures discovered that the Sr doping induced Ni3+ states, resulting in the upshifted valence band centre and enhanced the hybridization of O 2p with Ni 3d and Mn 3d orbitals (Fig. 10d and e). Besides, the increased hole states originated from Ni3+ states decreased the energy barrier of the electron transfer from 0.44 to 0.12 eV. They claimed that the enhanced hybridization of O 2p with Ni and Mn 3d orbitals and the increased hole states should be responsible for the better OER activities.
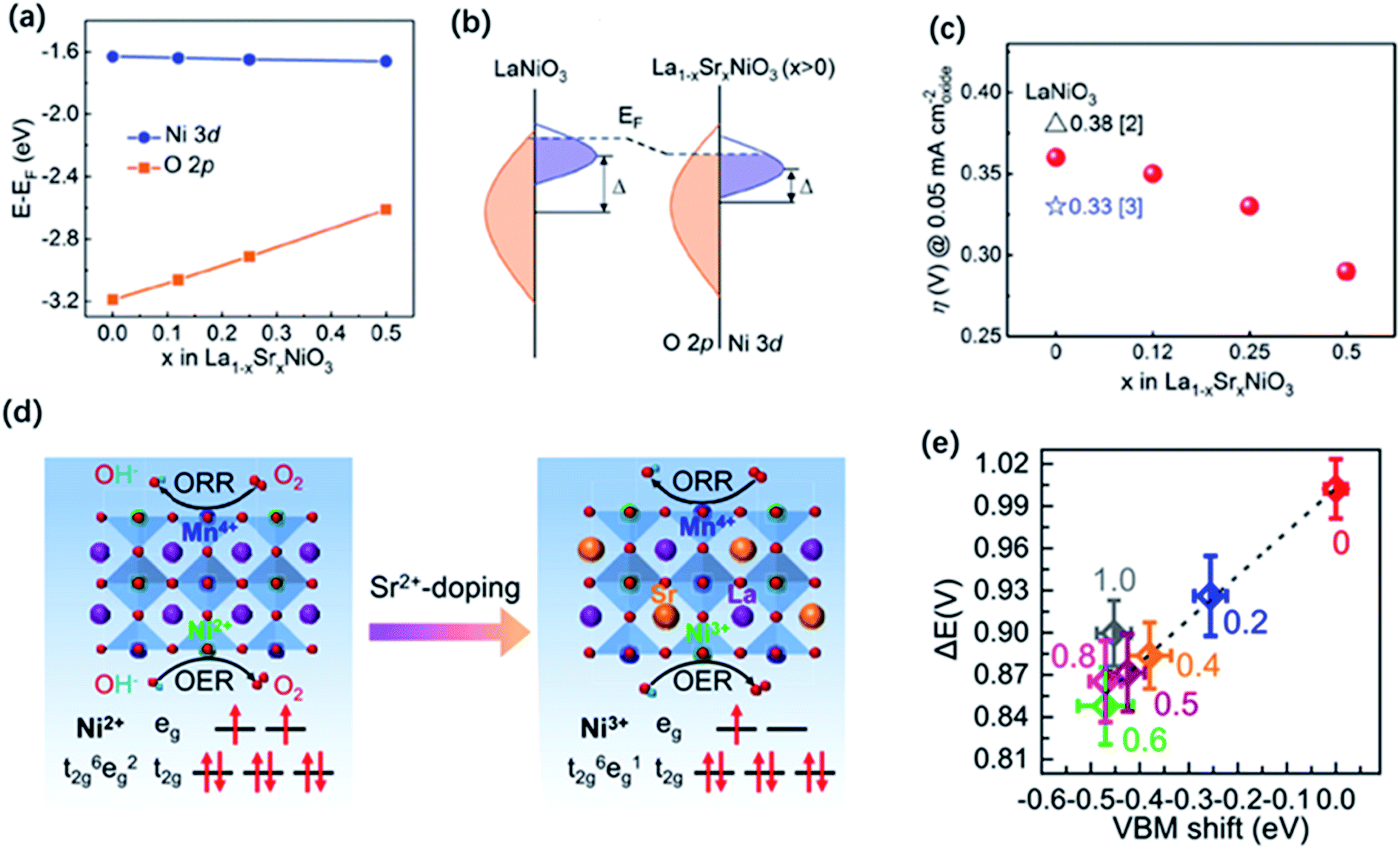 | ||
| Fig. 10 (a) Average onsite energies of O 2p and Ni 3d orbitals with EF as the reference. (b) Schematic energy band diagram for LaNiO3 and La1−xSrxNiO3 (x > 0), where Δ is the charge transfer energy. (c) Overpotentials (η) for LSNO films as required to obtain 0.05 mA cm−2 oxide in CV measurements. Reproduced with permission.173 Copyright 2019, Wiley-VCH. (d) Double perovskite crystal structure of LNMO and LSNMO, and the corresponding electron configuration of Ni2+ and Ni3+ in the oxygen octahedral crystal field. (e) ΔE values as a function of VBM shift. Reproduced with permission.98 Copyright 2021, American Chemical Society. | ||
Another group of metal oxides characterized by controllable composition and electron structure is the spinel family. In the typical structure of spinel oxides, there exist two geometric sites including tetrahedral (Td) sites which are normally half occupied by metal cations and octahedral (Oh) sites of which only one-eighth are filled by metal cations. The unoccupied geometric sites enable spinel-type oxides to largely accommodate the migration of metal ions. Otherwise, two geometric TM cations possess different coordination environment and they would compete for overlapping with the neighbouring oxygen because four neighbouring metal cations (three are in octahedral sites and the other one is in the tetrahedral sites) shared one common oxygen atom. Thus, the electronic properties of the metal cations in both sites and the oxygen anions closely correlate with each other. Doping or substituting other metal ions into Oh or Td sites becomes an effective approach to tuning the electronic structure of spinel oxides and influences their OER performance. For example, Zn, Ni, Cu, and Fe dopants introduced into Co3O4 spinel have been widely reported and their effects on the electronic structure and catalytic properties have been deeply investigated.37,170,174,175 Driess and co-workers found that the OER activity of ZnCo2O4 electrocatalyst was slightly better than Co3O4 in alkaline electrolyte, which could be ascribed to more accessible Co3+ in octahedral sites of ZnCo2O4 surface owing to the preferential loss of Zn ions under reaction conditions.37 Tan and co-workers synthesized different metal incorporated spinel catalysts MCo2O4 (M = Ni, Zn, Mn, etc.) and they uncovered that ZnCo2O4 with a normal spinel structure where all the Co cations were in octahedral sites exhibited the better OER activity.176 The degree of M–O covalency, as measured by the hybridization of M d and O 2p states, is also applied to connect the effect of doping with OER catalytic properties in spinel. Xu co-workers conducted an investigation on linking the Co–O covalency with the OER activity of Fe incorporated ZnFexCo2−xO4 spinel catalyst.129 The results showed that 10–30 at% Fe doping was beneficial to promote the injection/extraction of electrons from oxygen and lead to an enhanced covalency between Co 3d and O 2p, which is responsible for the improvement of the OER performance (Fig. 11a and b). Later, the same group reported that the OER activity followed the trend of Co3O4 > ZnCo2O4 > AlCo2O4, indicating that the octahedral CoO6 block dominated the OER activity, and explained the higher catalytic activities of the octahedral metal cations than the tetrahedral ones by the concept of covalency.177 They also observed a greatly improved OER performance by the doping of Li and Fe into ZnCo2O4, obtaining a highly active Li0.5Zn0.5Fe0.125Co1.875O4 catalysts. They proposed that the enhanced OER performance was attributed to the charge shift from the oxygen to metal cations in octahedral sites via Li doping and enlarged covalency of CoOh–O bond via Fe doping (Fig. 11c and d). Additionally, Zhang and co-workers introduced Ni into Co3O4 and the conducted a systematic investigation on the influence of Ni dopants on the electronic structures of NixCo3−xO4.68 Using XPS and XAS, they found that the incorporation of Ni in the trivalent oxidation state induces the generation of an unoccupied hole state, which could decrease the energy barriers of electron transfer for a faster kinetics. Results also showed that there is a shift of the VBM by 0.27 eV closer to the EF and enhanced the hybridization of O 2p with Ni 3d and Co 3d orbitals, which promoted the adsorption of OH intermediates on the surface of NixCo3−xO4 for a better OER activity than Co3O4 (Fig. 11e and f).
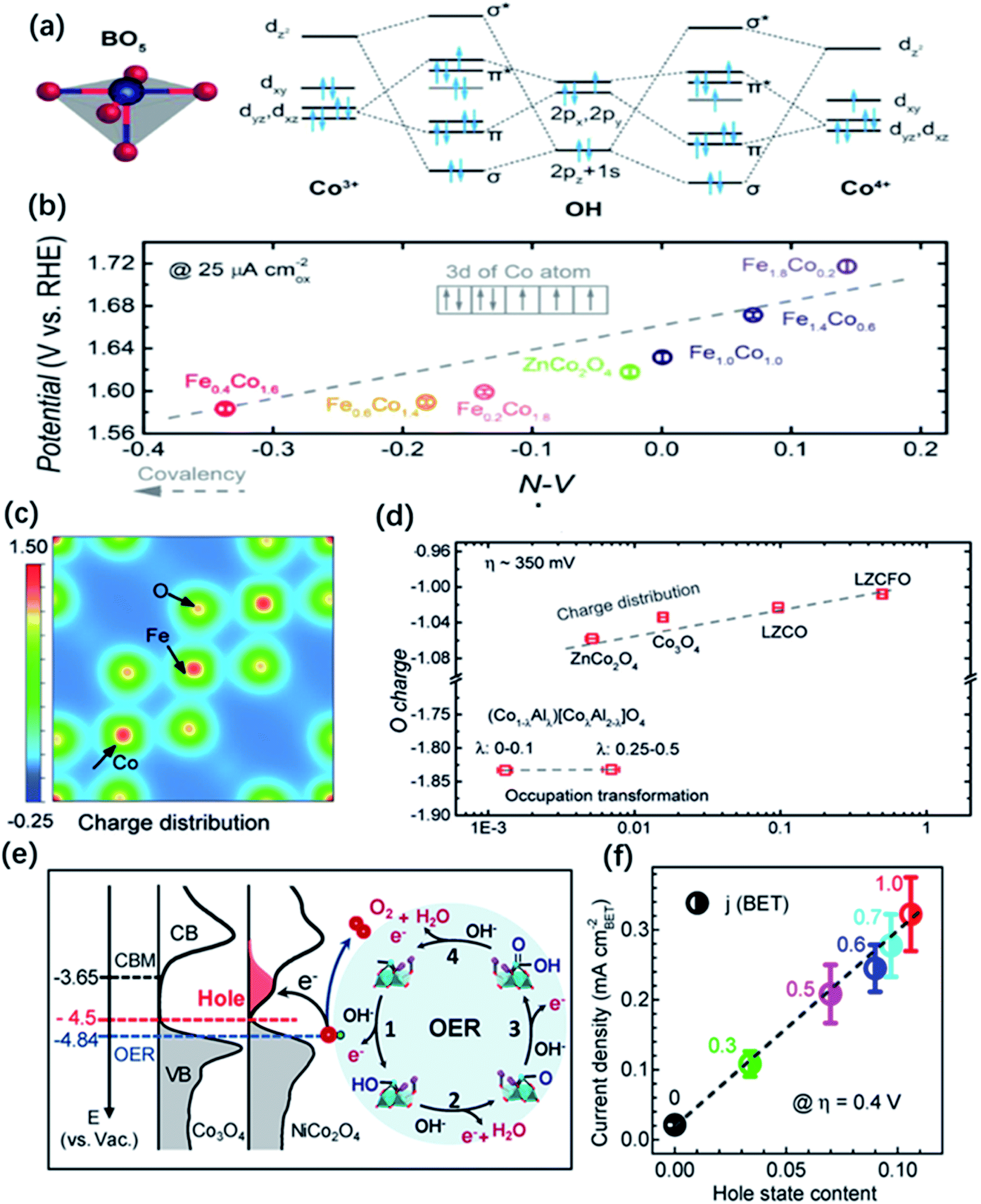 | ||
| Fig. 11 (a) Molecular orbitals diagrams for the Co–OH (Co3+, Co4+) bonding at the surface of spinel oxides. (b) The OER activity, evaluated by the potentials at a current density of 25 μA cm−2, as a function of the N–V parameter. Reproduced with permission.129 Copyright 2018, Wiley-VCH. (c) Illustration of change density on [001] direction of LZCFO. (d) OER activity (specific current at an overpotential of ∼350 mV) as a function of computed oxygen charges. Reproduced with permission.177 Copyright 2019, Wiley-VCH. (e) Left panel: the measured occupied and unoccupied density of states (DOS) near the EF for Co3O4 and NiCo2O4; the energy level is relative to vacuum level (vs. Vac.). Right panel: schematics of the four-step reaction pathway of OER. (f) Correlation of specific OER activity (current density at η = 0.4 V) with the amount of hole state extracted from O K edge XAS spectra. Reproduced with permission.68 Copyright 2019, American Chemical Society. | ||
In addition to the examples mentioned above, other studies on tuning the electronic structure and improving the OER activity of metal oxides by doping have also been reported. For instance, Zhang and co-workers modified the electronic properties of NiO through Li doping (LixNi1−xO) to improve the performance toward OER.103 According to their results, the significantly improved OER activities of LixNi1−xO could be ascribed to the generation of hole states at 1.1 eV above the Fermi level and the enlarged hybridization degree of O 2p–Ni 3d by Li doping which in turn resulted in the optimal adsorption strength of OH intermediates. Patzke et al. improved the OER activity of α-Mn2O3 through Mo doping.178 According to their findings, Mn2O3 with 2.64% Mo dopants exhibited the best OER activity. They claimed that the doping of Mo into the α-Mn2O3 lattice along with the structural distortion had great influence on improving the OER activity of pure α-Mn2O3. Previous research showed that the energy of NiIV/NiIII redox pair located at the top of the O-2p bands resulted in a strong bonding strength between the adsorbates on the surface and NiIV/NiIII redox cation, which was an important premise for high OER activity. However, stoichiometric LiNiO2 with well-ordered Li+ and NiIII is difficult to obtained due to the generation of NiII. Based on these, Gupta and co-workers successfully prepared Al doped LiNiO2 catalysts (Li1−xNi1+xO2) with better ordered lattice structure via solution combustion method at low temperature.179 They proposed that Al dopants could stabilize NiIII with higher concentration for the better OER activity.
5.2 Strain engineering
The compositional flexibility of metal oxides could provide convenience to modulate the electronic structure for a better OER activity, present advanced thin-film deposition techniques also offer the great opportunities to tune the properties of functional metal oxides through strain engineering. The physical and chemical properties of thin film and nanostructured metal oxides may largely differ from their bulk or powder form, which results in extra degrees of freedom for the modification by strain engineering. The modulation of electronic structure through strain effects can change a number of electronic structure parameters such as the metal d-band and oxygen 2p-band centres and eg occupancy etc. In the spinel and perovskite oxides, strain can lead to the distortion of metal–oxygen octahedra from the ideal cubic symmetry. The degeneracy of eg orbitals can be changed through enlarging the electron filling in the dx2–y2 orbital caused by tensile strain and in the dz2 orbital caused by compressive strain. Owing to the alteration in orbital overlap, the metal d-band and oxygen 2p-band centers as well as the binding strength of the adsorbed intermediates can be changed. For example, Petrie and co-workers observed the improvement of the OER activity on compressively strained LaNiO3 thin film in comparison with their unstrained state.180 They proposed that compressive strain could cause orbital asymmetry and induce splitting of the eg orbitals which result in the higher occupancy of the out-of-plane dz2 orbitals. The extra occupancy of the dz2 orbitals reduces the binding strength between the metal cation and adsorbate intermediates, resulting in better activity for OER and ORR.Recently, much attention has focused on the strain engineering of LaCoO3 perovskite to modify its electronic structure, due to unique nature that the electron configuration of Co3+ that can thermally transit from low spin (LS: t62ge0g) to higher spin state with the eg orbital configuration of eg∼1.0 at room temperature. By decreasing the particle size of LaCoO3 to 80 nm, Zeng et al. observed the spin-state transition of Co ions from low-spin to high-spin states and the increase of eg filling from unity to close to the favourable electron number of 1.2. Thus, LaCoO3 catalysts with the particle size of 80 nm displayed a better OER activity than that of other larger sized samples and the bulk.57 Similarly, through strain engineering, Xie and co-workers prepared three kinds of epitaxially strained thin films of LaCoO3 perovskite with (100), (110), and (111) lattice orientation, respectively.58 The LaCoO3 films with different lattice orientations exhibited different distortion degree of the CoO6 octahedron, causing a spin-state transition occurred on cobalt from a low spin state (LS t62ge0g) to an intermediate spin state (IS t52ge1g). As a result, LaCoO3 films with (100) orientation showed the best OER performance duo to its favourable eg electron number of 0.87 and higher electrical conductivity (Fig. 12a and b). Modulating the spin state of cobalt by strain control was also reported by Hsu and co-workers.181 They found that enlarging the lattice constant on the Co3O4 surface could change the spin state of surface Co3+ cations from t62g to the high spin state of t42ge2g, which remarkably enhanced the overlap of the eg orbital of cobalt with the oxygen adsorbates, and thereby improving the OER performance (Fig. 12c and d).181
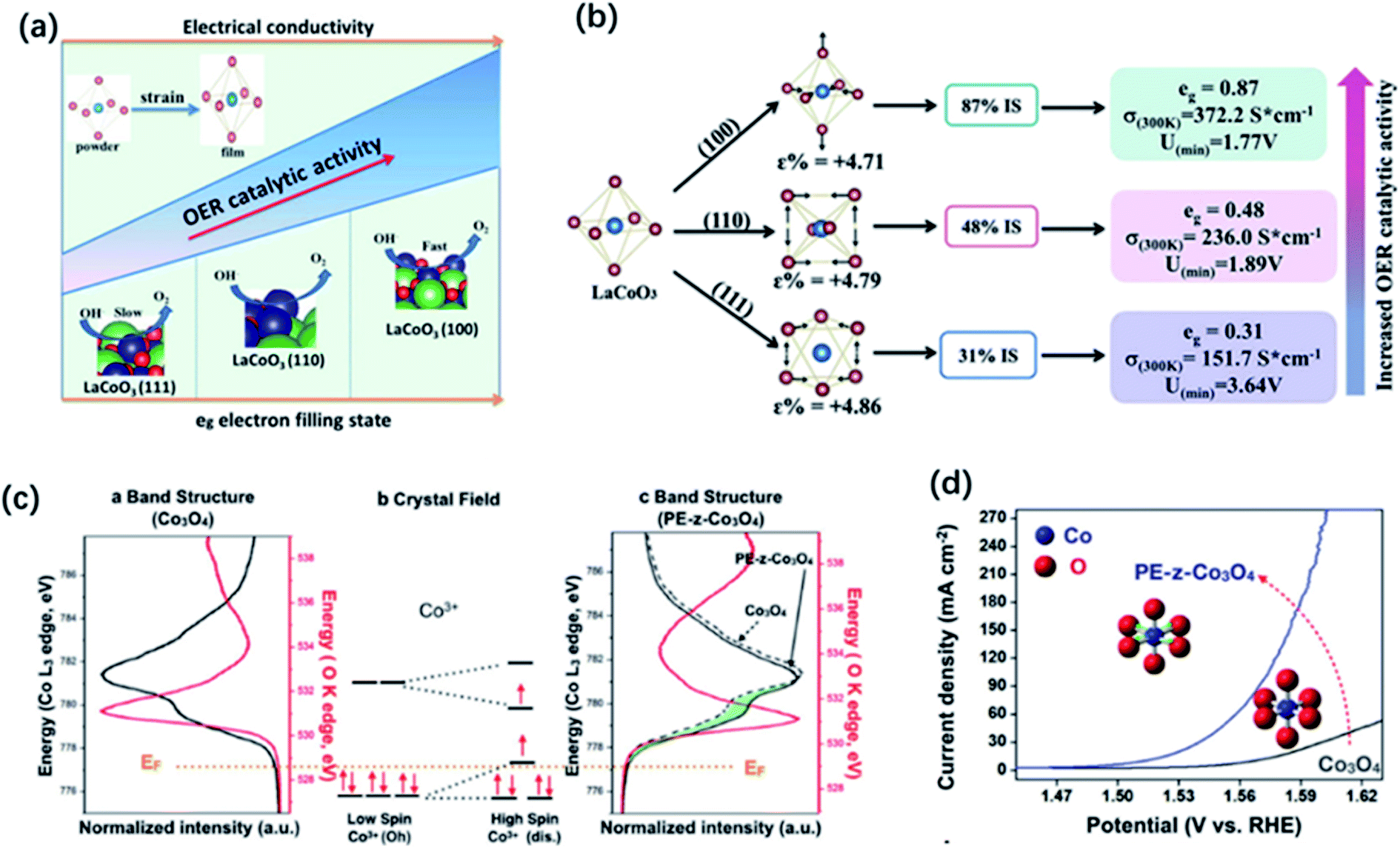 | ||
| Fig. 12 (a) Spin-state regulation by lattice-oriented control of LaCoO3 epitaxial films. (b) The relationship between OER activity and spin configuration (free energy, conductivity as well as eg electrons filling status) of differently oriented LaCoO3 films. Reproduced with permission.58 Copyright 2017, Elsevier Inc. (c) Illustration of Co 3d and O 2p bonding orbitals based on Co (K edge) and O (L edge) spectra and (b) crystal field of cobalt ions with different spin states of Co3+. (d) Linear sweep voltammetry (LSV) curves of Co3O4 and PE-z-Co3O4. Reproduced with permission.181 Copyright 2018, Wiley-VCH. | ||
Strain could also enhance the formation of oxygen vacancy to modify the OER activity of metal oxide catalysts. For SrCoO3−σ thin films, tensile strain would upshift the O 2p-band relative to the Fermi level and it is beneficial for the formation of oxygen vacancy manifesting as the reduction of a small amount of Co4+ to Co3+. Lee et al. reported that the oxygen deficiencies of SrCoO3−δ thin films can be tailored from δ ≤ 0.1 to δ ∼ 0.25 through epitaxial strain engineering.182 The changes in the oxygen content happened on the modest amounts of tensile strain over 1%. The electrochemical measurement showed the improved OER activities of tensile-stained SrCoO3−δ thin films with an increased degree of strain. The authors ascribed the enhancement of OER activity to the existence of oxygen vacancies or the increase of Co3+ caused by strain. Otherwise, Du et al. also confirmed the effect of strain on oxygen vacancy formation.183 They grew the tensile strained NdNiO3 thin films on SrTiO3 substrates and found that enhancing tensile strain would generate a higher content of oxygen vacancies in NdNiO3. They claimed that when the effect of oxygen vacancies could be negligible, the tensile strain was not good for the OER process while compressive strain was beneficial for that due to the strain-induced orbital splitting for a higher filling of the d3z2–r2 orbital and resultant weaker Ni–O binding strength. Nevertheless, when the tensile strain was large enough to enhance the formation of oxygen vacancies, an improvement of the OER performance could be seen. The improved performance was associated with an eg orbitals occupancy slightly larger than 1 due to the partial reduction of Ni3+ to Ni2+ caused by the oxygen vacancy formation.
5.3 Defect engineering
The structural and compositional flexibility of metal oxides also involve high tolerance in element defects, including metal cations and oxygen vacancies.184,185 Defects can greatly influence the electron and orbital distribution of metal oxides. Thereby, defect engineering is an effective strategy to modify the electronic structure and catalytic properties of metal oxide electrocatalysts. For example, Wang et al. produced Sn vacancies on the surface of SnCoFe perovskite hydroxide (SnCo0.9Fe0.1(OH)6) by energetic Ar plasma treatment.90 The generated Sn vacancies reduced the coordination numbers of the neighbouring active sites, which promoted reactant adsorption and charge transfer. And the Sn vacancies also promoted exposure of active Co and Fe sites, tuned conductivity and adsorption energy of the intermediates, thus significantly improving the OER performance. Zhu and co-workers reported the introduction of A-site cation defects in La1−xFeO3−δ, which lead to the formation of highly oxidative oxygen species (O22−/O−) and Fe4+ species.156 They proposed that the O22−/O− species might be the active sites for OER and Fe4+ species with an optimal eg orbital filling (t32ge1g) also promoted the OER processes.Actually, generating oxygen vacancies is easier compared to metal cation vacancies owing to the low formation energy of oxygen vacancies and they usually have a direct influence on the M–O bonds.186 Therefore, many studies focused on the introduction of oxygen vacancies in metal oxides to tune their electronic structure and improve the OER performance. Wang et al. reported that the introduced oxygen vacancies in Zn-doped CoOOH would cause the transformation of the regular CoO6 octahedron to an elongated or compressed CoO6 octahedron (Jahn–Teller distortion), which enhance the lift degeneracy of the t2g and eg orbitals.185 The electron configuration of Co3+ (t62ge0g) transferred to a more optimized eg state filling (t52ge1g), which could facilitate bonding with intermediates due to the stronger overlap of σ-bonding eg with O-2p than π-bonding t2g orbital and finally promote the OER activity. Kim and co-workers prepared the oxygen deficient perovskite Ca2Mn2O5 with a higher OER mass activity of 30.1 A g−1 at 1.70 V (vs. RHE) in comparison to the perovskite CaMnO3.187 They claimed that the unit cells of Ca2Mn2O5 were elongated in b- and c-directions owing to the existence of oxygen vacancies and the electronic configuration of Mn4+ (3d3) with t32g was optimized to t32ge1g of Mn3+ (3d4), which both were favorable for the adsorption of OH intermediates, thereby exhibiting higher OER activity. Moreover, Mefford et al. found that the increase of Sr substitution in La1−xSrxCoO3−δ resulted in more oxygen vacancies, stronger covalency of the Co–O bond, which led to better OER performance.36
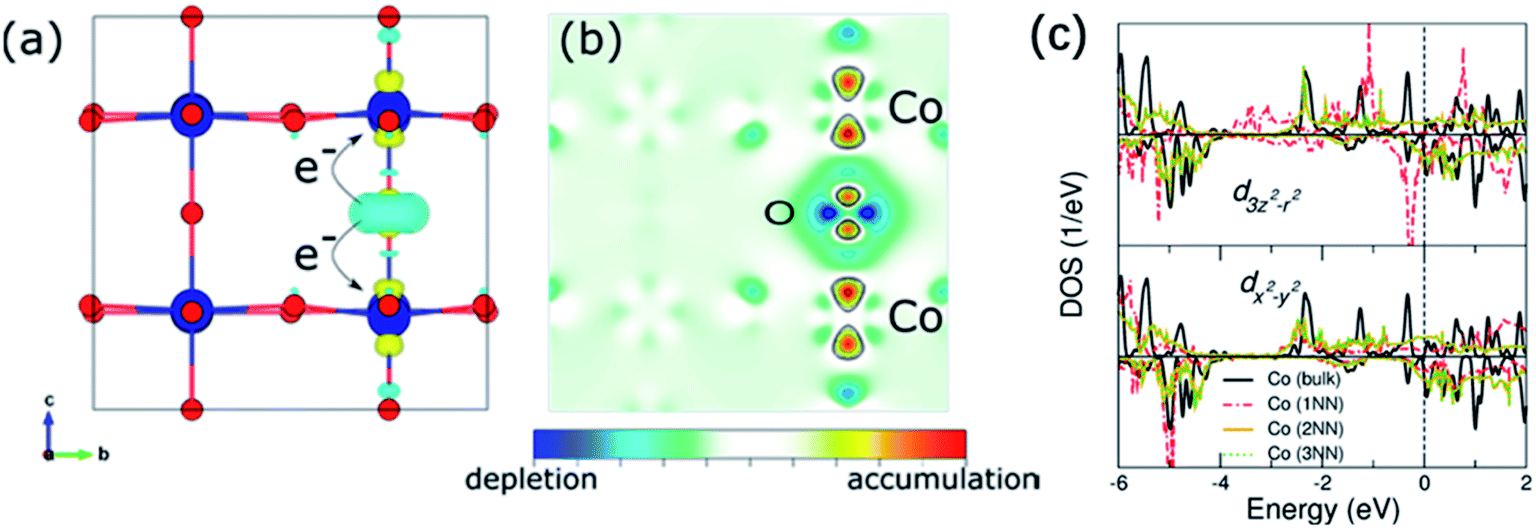
Zhu and co-workers found that controlling oxygen defects in PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) could obviously promote the OER kinetics.188 Using surface-sensitive characterization and DFT calculations, they proved that excessive oxygen vacancies enhanced OH− affinity and lowered the theoretical formation energy of O* intermediates on the surface, thereby significantly improving the OER activity. But they also proposed that excessive oxygen vacancies may lead to larger energy band gap and lower O 2p band position relative to the Fermi level of PrBa0.5Sr0.5Co1.5Fe0.5O5+δ, which may be not beneficial for OER process. Through DFT calculations, Tahini and co-workers found that the introduction of oxygen vacancies in SrCoO3 would lead to a local distortion of the Co–O bonds and donate its electrons to neighbouring Co cations.189 The reduced Co sites on the surface and Co–O bonds in the bulk lead to a reduction in the overlap between the Co 3d and O 2p orbitals, thereby resulting in the decrease of the electron transfer between the catalyst and the reactant intermediates (Fig. 13a–c). In addition, Miao et al.190 and Liu et al.191 also found that OER activities of PrBaCo2O6−δ and La0.7Sr0.3CoO3−δ, respectively, decreased after introducing oxygen vacancies. Thus, optimizing OER performance by properly introducing oxygen vacancies is very important.
 | ||
| Fig. 13 (a) and (b) Differential charge density upon the creation of oxygen vacancies and a slice along the (100) plane. (c) d3z2–r2 and dx2–y2 contributions of the Co ions in a pristine bulk system and the Co ions surrounding oxygen vacancies. Reproduced with permission.189 Copyright 2016, American Chemical Society. | ||
6. Summary and perspectives
Improving the kinetics of the OER with low-cost TM oxide catalysts is one of the big challenges of the scientific and engineering communities owing to the important role of the electrochemical water splitting in the introduction of renewable sources in any future energy scenario. In this article, we reviewed studies of the electronic structure of the TM oxide catalysts for the OER and the design of materials with high catalytic performances on basis of such electronic properties. The identification of electronic structure parameters associated with (i) the adsorbate binding strength, (ii) the covalency of the TM–O bonds, and (iii) the interface energetics can provide rational bases to explain the influence of the composition on the OER catalytic activity and guidelines for the formulation of new catalyst with improved activity. A key observation in the current literature is that highly covalent oxides tend to have higher catalytic activities, triggering a mechanism in which the lattice oxygen has been shown to participate. This has brought the spotlight on the activation of lattice oxygen as a way to improve the OER catalytic activity of TM oxides.127,130,148,151,152Elucidating the plausible involvement of lattice oxygen in highly active oxide catalysts requires the development of novel research approaches to address two important scientific issues: (1) the complex reaction mechanisms and (2) the stability of the TM oxide catalysts. This has motivated the development of in situ and operando characterisation technique such as isotope labelling combined with differential electrochemical mass spectrometry, in situ IR and Raman spectroscopies aiming to capture chemical information of reaction intermediates in mechanistic studies.147 The study of the chemical state and the structure of the catalysts during the OER process has also been studied with operando XAS.192 Additional in situ characterisation studies with techniques such as ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) to study the chemistry and energetics of the solid–liquid interface,193,194 may bring new opportunities to obtain a more realistic understanding of the relationship between electronic structure, adsorbate binding strength, and the OER catalytic activity.
The involvement of lattice oxygen in the OER of highly active catalyst leads to surface reconstructions and may also decrease the stability of the catalysts, especially at higher overpotentials. Therefore, the stability of the catalyst remains an important scientific issue in this field. Studying surface reconstruction phenomena could benefit from the use of well define surfaces of model samples. This experimental approach has been recently used in the description of mechanisms that connect the lattice oxygen activation, metal dissolution, and amorphization in TM OER electrocatalysts.195 Compared with ill-defined porous electrodes commonly used in OER studies, single crystal thin films permit a high degree of control over crystal facets, defects, and compositions, which could be used as model catalysts to gain a definitive structure–property relationships at a molecular level, and the study of surface reconstructions in a controlled way.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. H. L. Zhang is grateful for funding support by the National Natural Science Foundation of China (Grant No. 21872116). K. H. L. Zhang and Jan P. Hofmann also acknowledge the Sino-German Mobility Program (Grant No. M-0377). F. E. Oropeza and V. A. de la Peña ÓShea are greatful to the finatial support from the EU (ERC CoG HyMAP 648319) and Spanish MINECO (PID2019-106315RB-I00). V. A. de la Peña ÓShea also wishes to thank Comunidad de Madrid and European Structural Funds for their financial support for FotoArt-CM project (S2018/NMT-4367) and Fundación Ramón Areces Art-leaf project.References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- A. Morozan, H. Johnson, C. Roiron, G. Genay, D. Aldakov, A. Ghedjatti, C. T. Nguyen, P. D. Tran, S. Kinge and V. Artero, ACS Catal., 2020, 10, 14336–14348 CrossRef CAS.
- C. Yang, G. Rousse, K. Louise Svane, P. E. Pearce, A. M. Abakumov, M. Deschamps, G. Cibin, A. V. Chadwick, D. A. Dalla Corte, H. Anton Hansen, T. Vegge, J. M. Tarascon and A. Grimaud, Nat. Commun., 2020, 11, 1378 CrossRef CAS PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS.
- H. Wang, W. Fu, X. Yang, Z. Huang, J. Li, H. Zhang and Y. Wang, J. Mater. Chem. A, 2020, 8, 6926–6956 RSC.
- N. L. Panwar, S. C. Kaushik and S. Kothari, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524 CrossRef.
- F. Yu, H. Zhou, Y. Huang, J. Sun, F. Qin, J. Bao, W. A. Goddard III, S. Chen and Z. Ren, Nat. Commun., 2018, 9, 2551 CrossRef PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J. E. Trancik, Nature, 2014, 507, 300–302 CrossRef PubMed.
- M. F. Lagadec and A. Grimaud, Nat. Mater., 2020, 19, 1140–1150 CrossRef CAS.
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- M. R. Shaner, H. A. Atwater, N. S. Lewis and E. W. McFarland, Energy Environ. Sci., 2016, 9, 2354–2371 RSC.
- S. Trasatti, J. Electroanal. Chem., 1999, 476, 90–91 CrossRef CAS.
- C. Tang, N. Cheng, Z. Pu, W. Xing and X. Sun, Angew. Chem., 2015, 54, 9351–9355 CrossRef CAS PubMed.
- L. Zhang, Q. Fan, K. Li, S. Zhang and X. Ma, Sustainable Energy Fuels, 2020, 4, 5417–5432 RSC.
- B. Zhang, L. Wang, Z. Cao, S. M. Kozlov, F. P. García de Arquer, C. T. Dinh, J. Li, Z. Wang, X. Zheng, L. Zhang, Y. Wen, O. Voznyy, R. Comin, P. De Luna, T. Regier, W. Bi, E. E. Alp, C.-W. Pao, L. Zheng, Y. Hu, Y. Ji, Y. Li, Y. Zhang, L. Cavallo, H. Peng and E. H. Sargent, Nat. Catal., 2020, 3, 985–992 CrossRef CAS.
- P. Shirvanian and F. van Berkel, Electrochem. Commun., 2020, 114, 106704 CrossRef CAS.
- I. Vincent and D. Bessarabov, Renewable Sustainable Energy Rev., 2018, 81, 1690–1704 CrossRef CAS.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS.
- L. L. Feng, G. Yu, Y. Wu, G. D. Li, H. Li, Y. Sun, T. Asefa, W. Chen and X. Zou, J. Am. Chem. Soc., 2015, 137, 14023–14026 CrossRef CAS PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- J. Wang, H. X. Zhong, Z. L. Wang, F. L. Meng and X. B. Zhang, ACS Nano, 2016, 10, 2342–2348 CrossRef CAS PubMed.
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763–772 CrossRef CAS.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- P. Chen, K. Xu, Z. Fang, Y. Tong, J. Wu, X. Lu, X. Peng, H. Ding, C. Wu and Y. Xie, Angew. Chem., 2015, 54, 14710–14714 CrossRef CAS.
- M. Jiang, Y. Li, Z. Lu, X. Sun and X. Duan, Inorg. Chem. Front., 2016, 3, 630–634 RSC.
- C. Zhang, C. Zhang, Y. Xie, J. W. Su, X. He, J. D. Demaree, M. H. Griep, J. L. Atwood and J. Lin, Chemistry, 2019, 25, 4036–4039 CrossRef CAS PubMed.
- T. N. Pham Truong, H. Randriamahazaka and J. Ghilane, ACS Catal., 2018, 8, 869–875 CrossRef.
- Z. Shi, X. Wang, J. Ge, C. Liu and W. Xing, Nanoscale, 2020, 12, 13249–13275 RSC.
- J. Yu, Q. He, G. Yang, W. Zhou, Z. Shao and M. Ni, ACS Catal., 2019, 9, 9973–10011 CrossRef.
- R. Kötz, H. J. Lewerenz and S. Stucki, J. Electrochem. Soc., 2019, 130, 825–829 CrossRef.
- R. Kötz, H. Neff and S. Stucki, J. Electrochem. Soc., 2019, 131, 72–77 CrossRef.
- J. Luo, D. A. Vermaas, D. Bi, A. Hagfeldt, W. A. Smith and M. Grätzel, Adv. Energy Mater., 2016, 6, 1600100 CrossRef.
- W. T. Hong, K. A. Stoerzinger, Y.-L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. Yang and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2190–2200 RSC.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- P. W. Menezes, A. Indra, A. Bergmann, P. Chernev, C. Walter, H. Dau, P. Strasser and M. Driess, J. Mater. Chem. A, 2016, 4, 10014–10022 RSC.
- Z. Liu, G. Wang, X. Zhu, Y. Wang, Y. Zou, S. Zang and S. Wang, Angew. Chem., 2020, 59, 4736–4742 CrossRef CAS.
- B. Cui, H. Lin, J.-B. Li, X. Li, J. Yang and J. Tao, Adv. Funct. Mater., 2008, 18, 1440–1447 CrossRef CAS.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Norskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- C. Dong, X. Yuan, X. Wang, X. Liu, W. Dong, R. Wang, Y. Duan and F. Huang, J. Mater. Chem. A, 2016, 4, 11292–11298 RSC.
- Z. Lu, W. Xu, W. Zhu, Q. Yang, X. Lei, J. Liu, Y. Li, X. Sun and X. Duan, Chem. Commun., 2014, 50, 6479–6482 RSC.
- K. Fominykh, G. C. Tok, P. Zeller, H. Hajiyani, T. Miller, M. Döblinger, R. Pentcheva, T. Bein and D. Fattakhova-Rohlfing, Adv. Funct. Mater., 2017, 27, 1605121 CrossRef.
- R. D. Smith, M. S. Prevot, R. D. Fagan, S. Trudel and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 11580–11586 CrossRef CAS.
- P. Zhang and J. Gong, Chem, 2018, 4, 2739–2741 CAS.
- D. Wang, Q. Li, C. Han, Q. Lu, Z. Xing and X. Yang, Nat. Commun., 2019, 10, 3899 CrossRef PubMed.
- D. He, X. Song, W. Li, C. Tang, J. Liu, Z. Ke, C. Jiang and X. Xiao, Angew. Chem., 2020, 59, 6929–6935 CrossRef CAS.
- P. Rüetschi and P. Delahay, J. Chem. Phys., 1955, 23, 556–560 CrossRef.
- Y. Matsumoto, S. Yamada, T. Nishida and E. Sato, J. Electrochem. Soc., 2019, 127, 2360–2364 CrossRef.
- J. O. M. Bockris and T. Otagawa, J. Electrochem. Soc., 1984, 131, 290–302 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS.
- M. Jiang, J. Li, Y. Zhao, L. Pan, Q. Cao, D. Wang and Y. Du, ACS Appl. Mater. Interfaces, 2018, 10, 19746–19754 CrossRef CAS PubMed.
- H. Li, S. Sun, S. Xi, Y. Chen, T. Wang, Y. Du, M. Sherburne, J. W. Ager, A. C. Fisher and Z. J. Xu, Chem. Mater., 2018, 30, 6839–6848 CrossRef CAS.
- X. Cheng, E. Fabbri, M. Nachtegaal, I. E. Castelli, M. El Kazzi, R. Haumont, N. Marzari and T. J. Schmidt, Chem. Mater., 2015, 27, 7662–7672 CrossRef CAS.
- F. Calle-Vallejo, O. A. Díaz-Morales, M. J. Kolb and M. T. M. Koper, ACS Catal., 2015, 5, 869–873 CrossRef CAS.
- C. Wei, Z. Feng, G. G. Scherer, J. Barber, Y. Shao-Horn and Z. J. Xu, Adv. Mater., 2017, 29, 1606800 CrossRef PubMed.
- S. Zhou, X. Miao, X. Zhao, C. Ma, Y. Qiu, Z. Hu, J. Zhao, L. Shi and J. Zeng, Nat. Commun., 2016, 7, 11510 CrossRef CAS.
- Y. Tong, Y. Guo, P. Chen, H. Liu, M. Zhang, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Chem, 2017, 3, 812–821 CAS.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 CrossRef.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS PubMed.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- Z. Shen, Y. Zhuang, W. Li, X. Huang, F. E. Oropeza, E. J. M. Hensen, J. P. Hofmann, M. Cui, A. Tadich, D. Qi, J. Cheng, J. Li and K. H. L. Zhang, J. Mater. Chem. A, 2020, 8, 4407–4415 RSC.
- Q. Zhao, Z. Yan, C. Chen and J. Chen, Chem. Rev., 2017, 117, 10121–10211 CrossRef CAS PubMed.
- C. Castillo-Blas, N. López-Salas, M. C. Gutiérrez, I. Puente-Orench, E. Gutiérrez-Puebla, M. L. Ferrer, M. Á. Monge and F. Gándara, J. Am. Chem. Soc., 2019, 141, 1766–1774 CrossRef CAS PubMed.
- Y. Xu, F. Zhang, T. Sheng, T. Ye, D. Yi, Y. Yang, S. Liu, X. Wang and J. Yao, J. Mater. Chem. A, 2019, 7, 23191–23198 RSC.
- C. Wei, Z. Feng, G. G. Scherer, J. Barber, Y. Shao-Horn and Z. J. Xu, Adv. Mater., 2017, 29, 1606800 CrossRef.
- M. Cui, X. Ding, X. Huang, Z. Shen, T.-L. Lee, F. E. Oropeza, J. P. Hofmann, E. J. M. Hensen and K. H. L. Zhang, Chem. Mater., 2019, 31, 7618–7625 CrossRef CAS.
- M. H. Miles, J. Electroanal. Chem. Interfacial Electrochem., 1975, 60, 89–96 CrossRef CAS.
- M. P. B. M. E. G. Lyons, Int. J. Electrochem. Sci., 2008, 3, 1386–1424 CAS.
- D. A. Corrigan, J. Electrochem. Soc., 1987, 134, 377–384 CrossRef CAS.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- Q. Xiang, F. Li, W. Chen, Y. Ma, Y. Wu, X. Gu, Y. Qin, P. Tao, C. Song, W. Shang, H. Zhu, T. Deng and J. Wu, ACS Energy Lett., 2018, 3, 2357–2365 CrossRef CAS.
- J. Mohammed-Ibrahim, J. Power Sources, 2020, 448, 227375 CrossRef CAS.
- D. Zhou, Z. Cai, Y. Bi, W. Tian, M. Luo, Q. Zhang, Q. Zhang, Q. Xie, J. Wang, Y. Li, Y. Kuang, X. Duan, M. Bajdich, S. Siahrostami and X. Sun, Nano Res., 2018, 11, 1358–1368 CrossRef CAS.
- P. Li, X. Duan, Y. Kuang, Y. Li, G. Zhang, W. Liu and X. Sun, Adv. Energy Mater., 2018, 8, 1703341 CrossRef.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. d. Araujo, M. Gliech, D. Teschner, J. Zhu, W.-X. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed.
- K. Zeng, W. Li, Y. Zhou, Z. Sun, C. Lu, J. Yan, J.-H. Choi and R. Yang, Chem. Eng. J., 2021, 421, 127831 CrossRef CAS.
- H. Li, Y. Chen, S. Xi, J. Wang, S. Sun, Y. Sun, Y. Du and Z. J. Xu, Chem. Mater., 2018, 30, 4313–4320 CrossRef CAS.
- Y. Duan, Z. Y. Yu, S. J. Hu, X. S. Zheng, C. T. Zhang, H. H. Ding, B. C. Hu, Q. Q. Fu, Z. L. Yu, X. Zheng, J. F. Zhu, M. R. Gao and S. H. Yu, Angew. Chem., 2019, 58, 15772–15777 CrossRef CAS.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- T. Saravanakumar, S. Sathiya Bama, T. Selvaraju and S. J. Sardhar Basha, Energy Fuels, 2021, 35, 5372–5382 CrossRef CAS.
- S. Yin, W. Tu, Y. Sheng, Y. Du, M. Kraft, A. Borgna and R. Xu, Adv. Mater., 2018, 30, 1705106 CrossRef.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS.
- J. Nai, H. Yin, T. You, L. Zheng, J. Zhang, P. Wang, Z. Jin, Y. Tian, J. Liu, Z. Tang and L. Guo, Adv. Energy Mater., 2015, 5, 1401880 CrossRef.
- X. Liu, Z. Chang, L. Luo, T. Xu, X. Lei, J. Liu and X. Sun, Chem. Mater., 2014, 26, 1889–1895 CrossRef CAS.
- J. Zhang, D. Zhang, Y. Yang, J. Ma, S. Cui, Y. Li and B. Yuan, RSC Adv., 2016, 6, 92699–92704 RSC.
- X. Wu and K. Scott, J. Mater. Chem., 2011, 21, 12344–12351 RSC.
- D. Chen, M. Qiao, Y. R. Lu, L. Hao, D. Liu, C. L. Dong, Y. Li and S. Wang, Angew. Chem., Int. Ed., 2018, 57, 8691–8696 CrossRef CAS PubMed.
- F. Lyu, Y. Bai, Z. Li, W. Xu, Q. Wang, J. Mao, L. Wang, X. Zhang and Y. Yin, Adv. Funct. Mater., 2017, 27, 1702324 CrossRef.
- T. Ouyang, X. T. Wang, X. Q. Mai, A. N. Chen, Z. Y. Tang and Z. Q. Liu, Angew. Chem., 2020, 59, 11948–11957 CrossRef CAS PubMed.
- C. Jiang, J. Yang, T. Zhao, L. Xiong, Z.-X. Guo, Y. Ren, H. Qi, A. Wang and J. Tang, Appl. Catal., B, 2021, 282, 119571 CrossRef CAS.
- C. Si, Y. Zhang, C. Zhang, H. Gao, W. Ma, L. Lv and Z. Zhang, Electrochim. Acta, 2017, 245, 829–838 CrossRef CAS.
- C. Guo, X. Sun, X. Kuang, L. Gao, M. Zhao, L. Qu, Y. Zhang, D. Wu, X. Ren and Q. Wei, J. Mater. Chem. A, 2019, 7, 1005–1012 RSC.
- S. Xu, C. Lv, T. He, Z. Huang and C. Zhang, J. Mater. Chem. A, 2019, 7, 7526–7532 RSC.
- I. Yamada, H. Fujii, A. Takamatsu, H. Ikeno, K. Wada, H. Tsukasaki, S. Kawaguchi, S. Mori and S. Yagi, Adv. Mater., 2017, 29, 1603004 CrossRef PubMed.
- M. Qu, X. Ding, Z. Shen, M. Cui, F. E. Oropeza, G. Gorni, V. A. de la Peña O'Shea, W. Li, D.-C. Qi and K. H. L. Zhang, Chem. Mater., 2021, 33, 2062–2071 CrossRef CAS.
- A. Vignesh, M. Prabu and S. Shanmugam, ACS Appl. Mater. Interfaces, 2016, 8, 6019–6031 CrossRef CAS PubMed.
- G. Fu, W. Li, J.-Y. Zhang, M. Li, C. Li, N. Li, Q. He, S. Xi, D. Qi, J. L. MacManus-Driscoll, J. Cheng and K. H. Zhang, Small, 2021, 17, 2006930 CrossRef CAS.
- R. P. Forslund, W. G. Hardin, X. Rong, A. M. Abakumov, D. Filimonov, C. T. Alexander, J. T. Mefford, H. Iyer, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2018, 9, 3150 CrossRef PubMed.
- Y. Zhu, W. Zhou, Y. Chen, J. Yu, M. Liu and Z. Shao, Adv. Mater., 2015, 27, 7150–7155 CrossRef CAS PubMed.
- G. Fu, X. Wen, S. Xi, Z. Chen, W. Li, J.-Y. Zhang, A. Tadich, R. Wu, D.-C. Qi, Y. Du, J. Cheng and K. H. L. Zhang, Chem. Mater., 2018, 31, 419–428 CrossRef.
- X. Zhang, L. An, J. Yin, P. Xi, Z. Zheng and Y. Du, Sci. Rep., 2017, 7, 43590 CrossRef.
- Y. Zhu, W. Zhou, J. Sunarso, Y. Zhong and Z. Shao, Adv. Funct. Mater., 2016, 26, 5862–5872 CrossRef CAS.
- J. G. Lee, J. Hwang, H. J. Hwang, O. S. Jeon, J. Jang, O. Kwon, Y. Lee, B. Han and Y. G. Shul, J. Am. Chem. Soc., 2016, 138, 3541–3547 CrossRef CAS PubMed.
- Y. Zhu, H. A. Tahini, Z. Hu, Z. G. Chen, W. Zhou, A. C. Komarek, Q. Lin, H. J. Lin, C. T. Chen, Y. Zhong, M. T. Fernandez-Diaz, S. C. Smith, H. Wang, M. Liu and Z. Shao, Adv. Mater., 2020, 32, e1905025 CrossRef PubMed.
- X. Xu, C. Su, W. Zhou, Y. Zhu, Y. Chen and Z. Shao, Adv. Sci., 2016, 3, 1500187 CrossRef.
- B. Zhao, L. Zhang, D. Zhen, S. Yoo, Y. Ding, D. Chen, Y. Chen, Q. Zhang, B. Doyle, X. Xiong and M. Liu, Nat. Commun., 2017, 8, 14586 CrossRef CAS PubMed.
- N. I. Kim, Y. J. Sa, T. S. Yoo, S. R. Choi, R. A. Afzal, T. Choi, Y. S. Seo, K. S. Lee, J. Y. Hwang, W. S. Choi, S. H. Joo and J. Y. Park, Sci. Adv., 2018, 4, eaap9360 CrossRef PubMed.
- W. L. Kwong, C. C. Lee, A. Shchukarev, E. Björn and J. Messinger, J. Catal., 2018, 365, 29–35 CrossRef CAS.
- P. P. Patel, M. K. Datta, O. I. Velikokhatnyi, R. Kuruba, K. Damodaran, P. Jampani, B. Gattu, P. M. Shanthi, S. S. Damle and P. N. Kumta, Sci. Rep., 2016, 6, 28367 CrossRef CAS PubMed.
- X. Yang, H. Li, A.-Y. Lu, S. Min, Z. Idriss, M. N. Hedhili, K.-W. Huang, H. Idriss and L.-J. Li, Nano Energy, 2016, 25, 42–50 CrossRef CAS.
- W. L. Kwong, C. C. Lee, A. Shchukarev and J. Messinger, Chem. Commun., 2019, 55, 5017–5020 RSC.
- R. Frydendal, E. A. Paoli, I. Chorkendorff, J. Rossmeisl and I. E. L. Stephens, Adv. Energy Mater., 2015, 5, 1500991 CrossRef.
- M. Huynh, C. Shi, S. J. Billinge and D. G. Nocera, J. Am. Chem. Soc., 2015, 137, 14887–14904 CrossRef CAS.
- L. Zhou, A. Shinde, J. H. Montoya, A. Singh, S. Gul, J. Yano, Y. Ye, E. J. Crumlin, M. H. Richter, J. K. Cooper, H. S. Stein, J. A. Haber, K. A. Persson and J. M. Gregoire, ACS Catal., 2018, 8, 10938–10948 CrossRef CAS.
- I. A. Moreno-Hernandez, C. A. MacFarland, C. G. Read, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Energy Environ. Sci., 2017, 10, 2103–2108 RSC.
- N. Li, T. P. Keane, S. S. Veroneau, R. G. Hadt, D. Hayes, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 16187–16192 CrossRef CAS.
- X. Miao, L. Zhang, L. Wu, Z. Hu, L. Shi and S. Zhou, Nat. Commun., 2019, 10, 3809 CrossRef PubMed.
- J. O. M. Bockris and T. Otagawa, J. Phys. Chem., 1983, 87, 2960–2971 CrossRef CAS.
- M. García-Mota, M. Bajdich, V. Viswanathan, A. Vojvodic, A. T. Bell and J. K. Nørskov, J. Phys. Chem. C, 2012, 116, 21077–21082 CrossRef.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- V. Tripkovic, H. A. Hansen, J. M. Garcia-Lastra and T. Vegge, J. Phys. Chem. C, 2018, 122, 1135–1147 CrossRef CAS.
- J. Song, C. Wei, Z. F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J. M. Tarascon, Nat. Mater., 2016, 15, 121–126 CrossRef CAS PubMed.
- S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami, H. Fujii, H. Chen, N. Umezawa, H. Abe, N. Nishiyama and S. Mori, Nat. Commun., 2015, 6, 8249 CrossRef PubMed.
- Y. Zhou, S. Sun, J. Song, S. Xi, B. Chen, Y. Du, A. C. Fisher, F. Cheng, X. Wang, H. Zhang and Z. J. Xu, Adv. Mater., 2018, 30, e1802912 CrossRef PubMed.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- M. G. Mavros, T. Tsuchimochi, T. Kowalczyk, A. McIsaac, L. P. Wang and T. V. Voorhis, Inorg. Chem., 2014, 53, 6386–6397 CrossRef CAS PubMed.
- M. Risch, A. Grimaud, K. J. May, K. A. Stoerzinger, T. J. Chen, A. N. Mansour and Y. Shao-Horn, J. Phys. Chem. C, 2013, 117, 8628–8635 CrossRef CAS.
- H. Gerischer, J. Phys. Chem., 1991, 95, 1356–1359 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, B. R. Hart, A. P. Grosvenor, N. S. McIntryre, L. W. Lau and R. S. Smart, J. Phys.: Conf. Ser., 2008, 100, 012025 CrossRef.
- C. J. Powell and M. P. Seah, J. Vac. Sci. Technol., A, 1990, 8, 735–763 CrossRef CAS.
- J. G. Chen, B. Frühberger and M. L. Colaianni, J. Vac. Sci. Technol., A, 1996, 14, 1668–1673 CrossRef CAS.
- S. Zhou, X. Miao, X. Zhao, C. Ma, Y. Qiu, Z. Hu, J. Zhao, L. Shi and J. Zeng, Nat. Commun., 2016, 7, 11510 CrossRef CAS PubMed.
- Y. Duan, S. Sun, S. Xi, X. Ren, Y. Zhou, G. Zhang, H. Yang, Y. Du and Z. J. Xu, Chem. Mater., 2017, 29, 10534–10541 CrossRef CAS.
- F. M. F. de Groot, H. Elnaggar, F. Frati, R.-p. Wang, M. U. Delgado-Jaime, M. van Veenendaal, J. Fernandez-Rodriguez, M. W. Haverkort, R. J. Green, G. van der Laan, Y. Kvashnin, A. Hariki, H. Ikeno, H. Ramanantoanina, C. Daul, B. Delley, M. Odelius, M. Lundberg, O. Kuhn, S. I. Bokarev, E. Shirley, J. Vinson, K. Gilmore, M. Stener, G. Fronzoni, P. Decleva, P. Kruger, M. Retegan, Y. Joly, C. Vorwerk, C. Draxl, J. Rehr and A. Tanaka, J. Electron Spectrosc. Relat. Phenom., 2021, 249, 147061 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- J. Suntivich, W. T. Hong, Y.-L. Lee, J. M. Rondinelli, W. Yang, J. B. Goodenough, B. Dabrowski, J. W. Freeland and Y. Shao-Horn, J. Phys. Chem. C, 2014, 118, 1856–1863 CrossRef CAS.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, J. A. Bradley, S. R. Daly, S. A. Kozimor, W. W. Lukens, R. L. Martin, D. Nordlund, G. T. Seidler, D. K. Shuh, D. Sokaras, T. Tyliszczak, G. L. Wagner, T. C. Weng and P. Yang, J. Am. Chem. Soc., 2013, 135, 1864–1871 CrossRef CAS.
- W. T. Hong, K. A. Stoerzinger, B. Moritz, T. P. Devereaux, W. Yang and Y. Shao-Horn, J. Phys. Chem. C, 2015, 119, 2063–2072 CrossRef CAS.
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich, Y.-L. Lee, A. Grimaud and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 3264–3270 CrossRef CAS.
- J. S. Yoo, Y. Liu, X. Rong and A. M. Kolpak, J. Phys. Chem. Lett., 2018, 9, 1473–1479 CrossRef CAS PubMed.
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, ACS Catal., 2018, 8, 4628–4636 CrossRef CAS.
- H. M. A. Amin and H. Baltruschat, Phys. Chem. Chem. Phys., 2017, 19, 25527–25536 RSC.
- H. M. A. Amin, P. Königshoven, M. Hegemann and H. Baltruschat, Anal. Chem., 2019, 91, 12653–12660 CrossRef CAS.
- S. W. Boettcher, Nat. Catal., 2018, 1, 814–815 CrossRef CAS.
- C. Roy, B. Sebok, S. B. Scott, E. M. Fiordaliso, J. E. Sørensen, A. Bodin, D. B. Trimarco, C. D. Damsgaard, P. C. K. Vesborg, O. Hansen, I. E. L. Stephens, J. Kibsgaard and I. Chorkendorff, Nat. Catal., 2018, 1, 820–829 CrossRef CAS.
- S. Lee, K. Banjac, M. Lingenfelder and X. Hu, Angew. Chem., 2019, 58, 10295–10299 CrossRef CAS.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- J. Zaanen, G. A. Sawatzky and J. W. Allen, Phys. Rev. Lett., 1985, 55, 418–421 CrossRef CAS PubMed.
- T. G. Yun, Y. Heo, H. Bin Bae and S.-Y. Chung, Nat. Commun., 2021, 12, 824 CrossRef CAS PubMed.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, Angew. Chem., 2015, 54, 8722–8727 CrossRef CAS.
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Chem. Mater., 2016, 28, 1691–1697 CrossRef CAS.
- B. J. Kim, E. Fabbri, D. F. Abbott, X. Cheng, A. H. Clark, M. Nachtegaal, M. Borlaf, I. E. Castelli, T. Graule and T. J. Schmidt, J. Am. Chem. Soc., 2019, 141, 5231–5240 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS.
- M. B. Stevens, C. D. M. Trang, L. J. Enman, J. Deng and S. W. Boettcher, J. Am. Chem. Soc., 2017, 139, 11361–11364 CrossRef CAS.
- H. S. Ahn and A. J. Bard, J. Am. Chem. Soc., 2016, 138, 313–318 CrossRef CAS PubMed.
- U. I. Kramm, L. Ni and S. Wagner, Adv. Mater., 2019, 31, e1805623 CrossRef.
- J. Y. Chen, L. Dang, H. Liang, W. Bi, J. B. Gerken, S. Jin, E. E. Alp and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090–15093 CrossRef CAS PubMed.
- M. Görlin, P. Chernev, J. Ferreira de Araujo, T. Reier, S. Dresp, B. Paul, R. Krahnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef.
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486–1491 CrossRef CAS.
- Y. Sun, S. Sun, H. Yang, S. Xi, J. Gracia and Z. J. Xu, Adv. Mater., 2020, 32, 2003297 CrossRef CAS PubMed.
- X. Zou, A. Goswami and T. Asefa, J. Am. Chem. Soc., 2013, 135, 17242–17245 CrossRef CAS.
- L. J. Enman, M. S. Burke, A. S. Batchellor and S. W. Boettcher, ACS Catal., 2016, 6, 2416–2423 CrossRef CAS.
- J. A. Haber, C. Xiang, D. Guevarra, S. Jung, J. Jin and J. M. Gregoire, ChemElectroChem, 2014, 1, 524–528 CrossRef.
- J. S. Kim, B. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702774 CrossRef.
- W.-J. Yin, B. Weng, J. Ge, Q. Sun, Z. Li and Y. Yan, Energy Environ. Sci., 2019, 12, 442–462 RSC.
- Y. Guo, Y. Tong, P. Chen, K. Xu, J. Zhao, Y. Lin, W. Chu, Z. Peng, C. Wu and Y. Xie, Adv. Mater., 2015, 27, 5989–5994 CrossRef CAS.
- J. Liu, E. Jia, L. Wang, K. A. Stoerzinger, H. Zhou, C. S. Tang, X. Yin, X. He, E. Bousquet, M. E. Bowden, A. T. S. Wee, S. A. Chambers and Y. Du, Adv. Sci., 2019, 6, 1901073 CrossRef CAS.
- B. Chi, H. Lin and J. Li, Int. J. Hydrogen Energy, 2008, 33, 4763–4768 CrossRef CAS.
- T. Grewe, X. Deng and H. Tüysüz, Chem. Mater., 2014, 26, 3162–3168 CrossRef CAS.
- Y. Tan, C. Wu, H. Lin, J. Li, B. Chi, J. Pu and L. Jian, Electrochim. Acta, 2014, 121, 183–187 CrossRef CAS.
- S. Sun, Y. Sun, Y. Zhou, S. Xi, X. Ren, B. Huang, H. Liao, L. P. Wang, Y. Du and Z. J. Xu, Angew. Chem., 2019, 58, 6042–6047 CrossRef CAS PubMed.
- S. E. Balaghi, C. A. Triana and G. R. Patzke, ACS Catal., 2020, 10, 2074–2087 CrossRef CAS.
- A. Gupta, W. D. Chemelewski, C. Buddie Mullins and J. B. Goodenough, Adv. Mater., 2015, 27, 6063–6067 CrossRef CAS.
- J. R. Petrie, V. R. Cooper, J. W. Freeland, T. L. Meyer, Z. Zhang, D. A. Lutterman and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 2488–2491 CrossRef CAS.
- S.-H. Hsu, S.-F. Hung, H.-Y. Wang, F.-X. Xiao, L. Zhang, H. Yang, H. M. Chen, J.-M. Lee and B. Liu, Small Methods, 2018, 2, 1800001 CrossRef.
- J. R. Petrie, H. Jeen, S. C. Barron, T. L. Meyer and H. N. Lee, J. Am. Chem. Soc., 2016, 138, 7252–7255 CrossRef CAS PubMed.
- L. Wang, K. A. Stoerzinger, L. Chang, X. Yin, Y. Li, C. S. Tang, E. Jia, M. E. Bowden, Z. Yang, A. Abdelsamie, L. You, R. Guo, J. Chen, A. Rusydi, J. Wang, S. A. Chambers and Y. Du, ACS Appl. Mater. Interfaces, 2019, 11, 12941–12947 CrossRef CAS PubMed.
- J. Du, T. Zhang, F. Cheng, W. Chu, Z. Wu and J. Chen, Inorg. Chem., 2014, 53, 9106–9114 CrossRef CAS PubMed.
- J. Wang, J. Liu, B. Zhang, F. Cheng, Y. Ruan, X. Ji, K. Xu, C. Chen, L. Miao and J. Jiang, Nano Energy, 2018, 53, 144–151 CrossRef CAS.
- K. Zhu, F. Shi, X. Zhu and W. Yang, Nano Energy, 2020, 73, 104761 CrossRef CAS.
- J. Kim, X. Yin, K. C. Tsao, S. Fang and H. Yang, J. Am. Chem. Soc., 2014, 136, 14646–14649 CrossRef CAS.
- Y. Zhu, L. Zhang, B. Zhao, H. Chen, X. Liu, R. Zhao, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Funct. Mater., 2019, 29, 1901783 CrossRef.
- H. A. Tahini, X. Tan, U. Schwingenschlögl and S. C. Smith, ACS Catal., 2016, 6, 5565–5570 CrossRef CAS.
- X. Miao, L. Wu, Y. Lin, X. Yuan, J. Zhao, W. Yan, S. Zhou and L. Shi, Chem. Commun., 2019, 55, 1442–1445 RSC.
- X. Liu, L. Zhang, Y. Zheng, Z. Guo, Y. Zhu, H. Chen, F. Li, P. Liu, B. Yu, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Sci., 2019, 6, 1801898 CrossRef.
- S. Song, J. Zhou, X. Su, Y. Wang, J. Li, L. Zhang, G. Xiao, C. Guan, R. Liu, S. Chen, H.-J. Lin, S. Zhang and J.-Q. Wang, Energy Environ. Sci., 2018, 11, 2945–2953 RSC.
- S. Axnanda, E. J. Crumlin, B. Mao, S. Rani, R. Chang, P. G. Karlsson, M. O. M. Edwards, M. Lundqvist, R. Moberg, P. Ross, Z. Hussain and Z. Liu, Sci. Rep., 2015, 5, 9788 CrossRef CAS PubMed.
- Z. Novotny, D. Aegerter, N. Comini, B. Tobler, L. Artiglia, U. Maier, T. Moehl, E. Fabbri, T. Huthwelker, T. J. Schmidt, M. Ammann, J. A. van Bokhoven, J. Raabe and J. Osterwalder, Rev. Sci. Instrum., 2020, 91, 023103 CrossRef CAS.
- G. Wan, J. W. Freeland, J. Kloppenburg, G. Petretto, J. N. Nelson, D.-Y. Kuo, C.-J. Sun, J. Wen, J. Trey Diulus, G. S. Herman, Y. Dong, R. Kou, J. Sun, S. Chen, K. M. Shen, D. G. Schlom, G.-M. Rignanese, G. Hautier, D. D. Fong, Z. Feng, H. Zhou and J. Suntivich, Sci. Adv., 2021, 7, eabc7323 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |