
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterfaces in metal halide perovskites probed by solid-state NMR spectroscopy
Clayton J.
Dahlman
a,
Dominik J.
Kubicki
bc and
G. N. Manjunatha
Reddy
 *d
*d
aMaterials Department, University of California, Santa Barbara, California 93106, USA
bCavendish Laboratory, Department of Physics, University of Cambridge, JJ Thomson Avenue, CB3 0HE, Cambridge, UK
cDepartment of Chemistry, University of Cambridge, Lensfield Road, CB2 1EW, Cambridge, UK
dUniv. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: gnm.reddy@univ-lille.fr
First published on 24th June 2021
Abstract
Metal halide perovskites (MHPs) are promising light harvesting and emitting materials that have enabled solar energy conversion efficiencies of over 25% in solution-processed single-junction cells, and found applications in flexible electronics, detectors and other display technologies. Research on MHPs has achieved significant fundamental and technological advancements over the last decade, in large part due to improvements in characterization approaches to understand these materials. It has become clear that engineering the interfaces between device layers, and within the MHP layer itself, is crucially important to develop stable and efficient optoelectronic devices. Interfaces in MHP-based devices exhibit varying degrees of order, which manifest heterogeneities in compositions, structures and optoelectronic properties. This review assesses the overall prospects for a range of solid-state (ss)NMR spectroscopy techniques to facilitate structure-based understanding of complex interfaces in MHPs and contact layers. The role of ssNMR in elucidating local compositions and structures, intermolecular connectivity, phase transitions, degradation products and molecular passivation at MHP interfaces is discussed. In addition, an overview of different dynamic processes in MHPs probed by ssNMR is provided. Finally, we discuss perspectives on the development of ssNMR spectroscopy for investigating interfaces in MHPs for various optoelectronics.
 Clayton J. Dahlman | Clayton Dahlman is a postdoctoral researcher in the Materials Department at the University of California, Santa Barbara, working in the research group of Prof. Michael Chabinyc. He received his bachelor's degree from Columbia University in 2011 and a master's degree from the University of California, Berkeley in 2014. He received his PhD from the University of Texas at Austin in 2017 under the supervision of Prof. Delia Milliron. Clayton is interested in the dynamic behavior of solution-processed, colloidal and hybrid organic–inorganic nanomaterials for energy and information technologies. |
 Dominik J. Kubicki | Dominik J. Kubicki is a Marie Skłodowska-Curie Actions Fellow in the Cavendish Laboratory in the group of Sam Stranks and in the Department of Chemistry in the group of Clare Grey at the University of Cambridge (UK). During his PhD at EPFL he was developing MAS DNP strategies to enhance the sensitivity of solid-state NMR. During his subsequent postdoctoral stay with Michael Grätzel, he began applying solid-state NMR to obtain atomic-level structural and dynamic information on lead halide perovskites. He is currently developing new materials for optoelectronics with a particular focus on the structure–property relationships resulting from the local structure and disorder. He received the Young Scientist Award at the 20th International Society of Magnetic Resonance (ISMAR) Conference. |
 G. N. Manjunatha Reddy | G. N. Manjunatha Reddy is an Assistant Professor of Chemistry at the University of Lille, France. He received his PhD from Aix-Marseille University, France. Subsequently he has been a postdoctoral researcher at the University of Warwick (UK), and research associate at the University of California Santa Barbara (USA). In 2016, he has been acquired by the Royal Society of Chemistry as member. He received Marie Skłodowska-Curie Individual Fellowship Award in 2017 on behalf of European Union Framework Programme Horizon-2020. His research is focused on establishing structure–property relationships in energy materials and self-assembled nanomaterials using solid-state NMR spectroscopy and modelling approaches. |
1. Introduction
Metal halide perovskites (MHPs) have garnered considerable interest in the past decade as photoactive materials in low-cost and high-performance optoelectronic devices. Perovskite is originally the name of the mineral CaTiO3, but the term is used to describe isostructural materials with a composition of ABX3, where X is an anion and A and B are various cations.1 In MHPs, A is a monovalent cation (e.g. Cs+, methylammonium, formamidinium), B is a divalent metal (e.g. Pb2+, Sn2+) and X is a halide (I−, Br−, Cl−). Structures with similar octahedral bonding, coordination and chemical composition are also commonly described as MHPs,2 such as layered homologues.3–8 Halide perovskites have been heralded as next-generation materials for photovoltaics (PV),9–12 with power conversion efficiencies (PCE) currently exceeding 25%.13,14 MHPs have also been explored for applications including light-emitting diodes (LEDs),15–23 phosphors for display technology,24,25 lasers,26–30 photodetectors,31–35 ionizing radiation detectors30,36–40 and as quantum emitters.41,42 Significant improvements in device performance have been driven by interfacial engineering, either between layers of a device or within MHP phases.43 However, atomic structures and dynamic interactions at MHP interfaces are notoriously difficult to probe. In this review, we discuss the challenges of studying interfaces in MHP materials and devices and how solid-state nuclear magnetic resonance (ssNMR) spectroscopy can access these regions.MHPs, and particularly lead iodides, stand out from other optoelectronic semiconductors because of their long charge carrier lifetimes and diffusion lengths in spite of the presence of many defects.44,45 The defect tolerance of lead halide perovskites has enabled the manufacture of high-performance devices through a variety of scalable solution-phase processes,46–51 solid-phase mechanochemical syntheses,52,53 and vapor-phase approaches.54 MHPs have relatively low formation energies and exhibit low-temperature phase transformations55 compared to other semiconductors for optoelectronic applications. The substantially ionic character of bonds in MHPs renders them simple to form but also quick to degrade.2,56–58 Weak coulombic attractions between large constituent ions, including small organic molecules such as methylammonium, are easily disrupted by polar solvents or vapors, pressure, temperature and high-flux photoexcitation. The weak bonding between constituent ions also leads to significant lattice disorder, polarizability and ion migration.2,59–61 For this reason, perovskite active layers possess varying degrees of order and crystallinity between and within layers of MHP based devices.44 The desire for probing short-range (<1 nm) to middle-range (1–10 nm) structural order and competing interactions at interfaces is particularly relevant here. A bottleneck challenge remains to develop reliable structure-processing-property relationships at different length scales in MHP devices.
Many experimental techniques used to characterize the structure of MHPs, such as X-ray diffraction, are sensitive to bulk crystalline regions. However, MHPs demonstrate heterogeneity and disorder across many length scales – either in the bulk or at interfaces – that are difficult to probe by diffraction alone.44 Interfacial regions make up the minority of the material and exhibit atomic-scale disorder, but may have an outsized impact on device performance.43 In addition, intrinsic interfaces can form within MHP layers that may dominate optical and electronic properties. For example, low-dimensional Ruddlesden–Popper (RP), Dion–Jacobson (DJ) phases are composed of 2D slabs of MHP sub-phases separated by insulating ‘spacer’ cations.3–8 Characterizing amorphous, disordered and interfacial structures in MHPs remains an experimental challenge. Multi-technique approaches to bridge different length- and timescales, such as diffraction, electron microscopy and ssNMR spectroscopy, are well suited to correlate bulk behaviors with interfacial and atomic-scale structures in MHPs.
SsNMR spectroscopy has emerged as a powerful tool to understand local chemical environments, disorder and dynamic behavior of MHPs. ssNMR techniques have enjoyed decades of development for organic, inorganic and biomolecular materials (e.g. catalysts, energy conversion and storage materials, polymers, glasses, solution-processable semiconductors).62,63 The recent upsurge of interest in MHPs has introduced a new set of compositions, bonding moieties and interfaces for ssNMR spectroscopists to explore. The length-scales that ssNMR experiments can study range from atomic-scale bond lengths to macroscopic diffusion/transport distances. In addition, ssNMR can probe dynamic processes at timescales ranging from picoseconds to hours or days. Sample preparation is also versatile – ssNMR can probe samples with different compositions, morphologies, crystallinity and solid/solution combinations – so realistic synthesis and deposition conditions can be studied. Specifically, wide range of one (1D) and two-dimensional (2D) techniques, surface-enhanced NMR spectroscopy, NMR crystallography, relaxation measurements and analysis, variable temperature NMR, and ex situ and in situ techniques have been employed to study MHPs.64–67 The rapid growth in structure-based understanding of this material class, and steady improvements in NMR instrumentation, have cultivated a rich field of study for MHPs by ssNMR.
This review describes the growing use of ssNMR spectroscopy as a tool to understand interfacial structures in MHPs. Several reviews have recently explored the role of ssNMR in probing local structures and dynamics in micro- and nanocrystalline MHPs.64–67 Here, we focus on how ssNMR techniques can probe the interfaces present in MHPs and on an overarching goal of extending ssNMR capabilities to investigate reactive surfaces and interfaces in device stacks. We begin by categorizing different types of interfaces in MHPs and MHP-based optoelectronic devices. Next, we describe the length and timescales of physical processes that occur in optoelectronic devices and compare these to the scales accessible to ssNMR and other characterization techniques. A practical tutorial on ssNMR methods is provided for both non-experts and NMR spectroscopists to identify opportunities and limitations for relevant techniques. We then highlight recent findings that illustrate state-of-the-art ssNMR spectroscopy approaches to study MHP interfaces in crystalline 3D phases, layered structures and nanocrystals, and contact layers. In addition, an overview of dynamic aspects in MHPs probed by 1D and 2D ssNMR techniques is provided. Finally, we offer an outlook on opportunities for future research on MHP interfaces by ssNMR methods.
2. Interfaces in MHPs and devices
Interfaces in MHPs can be categorized based on the composition of the metal halide phase and its assembly into functional devices or composite materials. Interfaces span different length scales from the molecular level (Å) to device scales (mm). In this review we distinguish between (i) extrinsic interfaces between the halide perovskite layer and different contact layers (e.g., charge-transport layers, electrodes, ambient atmosphere), and (ii) intrinsic interfaces that exist within MHPs (e.g., grain boundaries). Both of these interfaces exhibit varying degrees of structural order and manifest heterogeneities. Many, but not all, of these interfaces have been studied by ssNMR techniques in MHPs, yet each type of interface presents a characterization and design challenge.2.1. Extrinsic interfaces
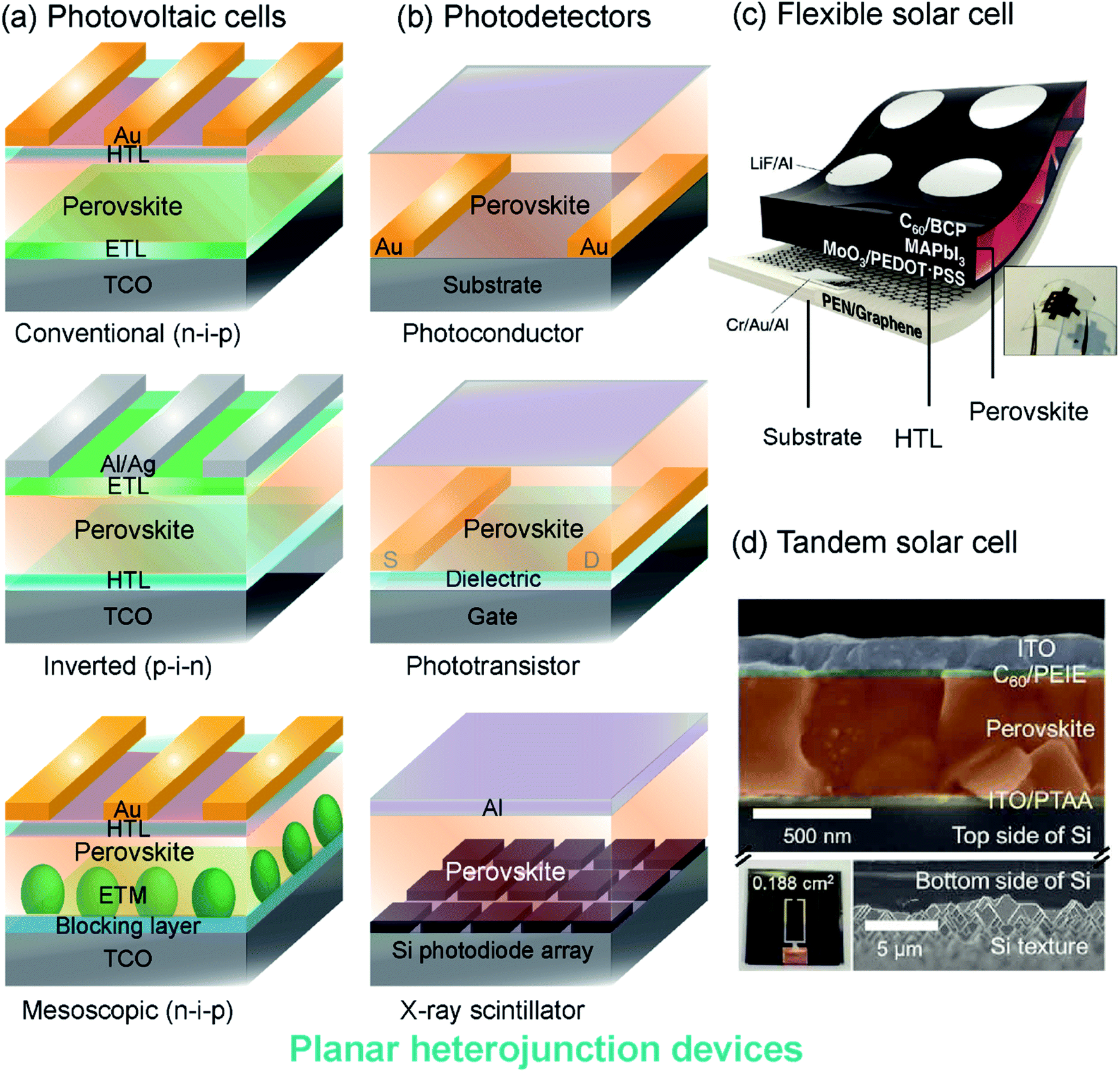 | ||
| Fig. 1 Perovskite device architectures: (a) planar heterojunction solar cells with mesoporous, conventional and inverted structures. (b) Photodetectors including photoconductor, phototransistor and scintillator architectures. HTL: hole transporting layer, ETL: electron transporting layer, TCO: transparent conductive oxide, ETM: electron transporting material, S: source electrode, D: drain electrode. (c) A schematic and photograph of a flexible solar cell with flexible photoactive thin films, contact layers and substrates. Reproduced from ref. 81 with permission from The Royal Society of Chemistry. (d) A colored SEM cross-section of the top-cell (upper) and back side of the bottom cell (lower) of a tandem perovskite-silicon solar cell. The scale bars in the upper and lower panels are different as indicated. Reproduced from ref. 82 with permission from The Royal Society of Chemistry. | ||
Solar cells and LEDs. Photovoltaic cells are composed of a device stack that sandwiches a photoactive MHP layer between transport layers (ETLs and HTLs), as shown in Fig. 1a. Typically, the MHP layer is several hundred nanometers thick and the contact layers are much thinner (Fig. 1d). In a conventional (n-i-p) cell, the ETL (e.g. titanium dioxide, TiO2) contacts the transparent conductive substrate (e.g. tin-doped indium oxide (ITO)-coated glass) and the HTL (e.g. 2,2′,7,7′-tetrakis-9,9′-spirobifluorene, spiro-OMeTAD) contacts gold electrodes. An inverted cell has an opposite arrangement, where the HTL (e.g. poly(3,4-ethylenedioxythiophene):polystyrene sulfonate, PEDOT:PSS) contacts the transparent substrate and the ETL (e.g. phenyl-C61-butyric acid methyl ester, PCBM) contacts silver or aluminum electrodes. Mesoscopic architectures are also common, where the MHP is dispersed within an electron-transporting material scaffold (e.g. mesoporous TiO2). The development of improved organic and inorganic transport materials is an active area of research for MHP solar cells and LEDs.68–75 Each of these interfaces can have properties that differ from the bulk. For instance, MHPs may become sub-stoichiometric at the interface with contact layers, leading to significant changes in electronic transport. Similarly, the structure of ETL and HTL materials may be perturbed when in contact with MHPs, and electrodes (e.g., gold back-contacts) can diffuse through contact layers to the MHP. Interface management is essential to the development of stable high-performing MHP-based devices, but characterizing interfacial regions remains challenging.43
Photodetectors. MHPs have also found use in photodetector applications that do not rely on a photovoltaic architecture. Lateral devices, such as photoconductors and phototransistors, use the MHP layer to detect incident light by transporting photocarriers between a source and drain electrode (Fig. 1b). The geometry of lateral photoconductors and phototransistors may require photocarriers to travel further distances than in vertical photovoltaic cells. Interfacial charging and depletion zones, defect passivation, and structural rearrangements near the gate or substrate interface, are particularly consequential in lateral devices because these regions form electronic transport pathways. Interfacial doping of MHPs through chemical surface treatments has been attempted, but the unique chemistry of MHPs leads to convoluted effects on structure and electronic defect compensation.76–79 Photodetectors for ionizing radiation (e.g., X-rays, γ-rays) often use a scintillator architecture instead of lateral heterostructures. The heavy elements in MHPs can efficiently down-convert high-energy radiation to visible light for detection by optical photodiodes. Scintillators do not rely on electrical contact between transport layers and MHPs, but MHP interfaces may determine device stability and optical characteristics.36,40,80
Flexible devices. Mechanically flexible photoactive and contact layers are required for the development of portable, light weight and wearable technology. Substrates such as polyethylene naphthalate (PEN), polyethylene terephthalate (PET), and flexible glass support mechanically flexible devices that can be fabricated using solution-processing methods.83–85Fig. 1c illustrates a flexible perovskite solar cell with a CH3NH3PbI3 (MAPbI3) absorber layer, organic PEDOT:PSS HTL, and a fullerene (C60) ETL, all deposited on a graphene-coated PEN substrate.81 Large-scale manufacture of flexible MHP devices can be accomplished through roll-to-roll printing,86 but these processing methods impart interfacial strain and mechanical work to device layers. Interfacial strain impacts the crystalline structure and optical properties of MHPs,87–91 but disordered regions at strained interfaces are difficult to probe. Composite materials with MHP particles embedded in a flexible matrix have also been explored for additive manufactured and printable luminescent devices, and may exhibit similar interfacial characterization challenges.92
Tandem cells. Multi-junction (tandem) MHP solar cells, have promising commercial applications because of their high efficiencies, which can break the thermodynamic limit for single-junction cells.93 Record setting MHP-Si tandem cells have PCEs approaching 30%.13 A cross-section of an example MHP-Si tandem cell with PCE > 26% is shown in Fig. 1d.82 As additional layers are added to MHP-based devices, the properties and behaviors of interfacial regions become more important to characterize and control. The structure and behavior of interfaces between each layer of a tandem MHP can be impacted by other layers, so it is crucial to develop tools to probe these regions.
2.2. Intrinsic interfaces
Intrinsic interfaces exist within MHP layers between different grains, domains or substructures. Unlike the extrinsic interfaces described in the prior section, these interfaces can exist independently of device architecture. The tolerance of MHPs to atomic defects allows for a great diversity of interfaces to form, ranging from the unit cell to the device scale. These interfaces may form under equilibrium conditions due to compositional heterogeneity (e.g. layered Ruddlesden–Popper phases), or during non-equilibrium processes such as film growth and transient light exposure. Fig. 2 illustrates several intrinsic interfaces that may form in MHPs. While interfaces may be defined in various ways, here we consider an expansive set of examples that present exciting opportunities for characterization by ssNMR.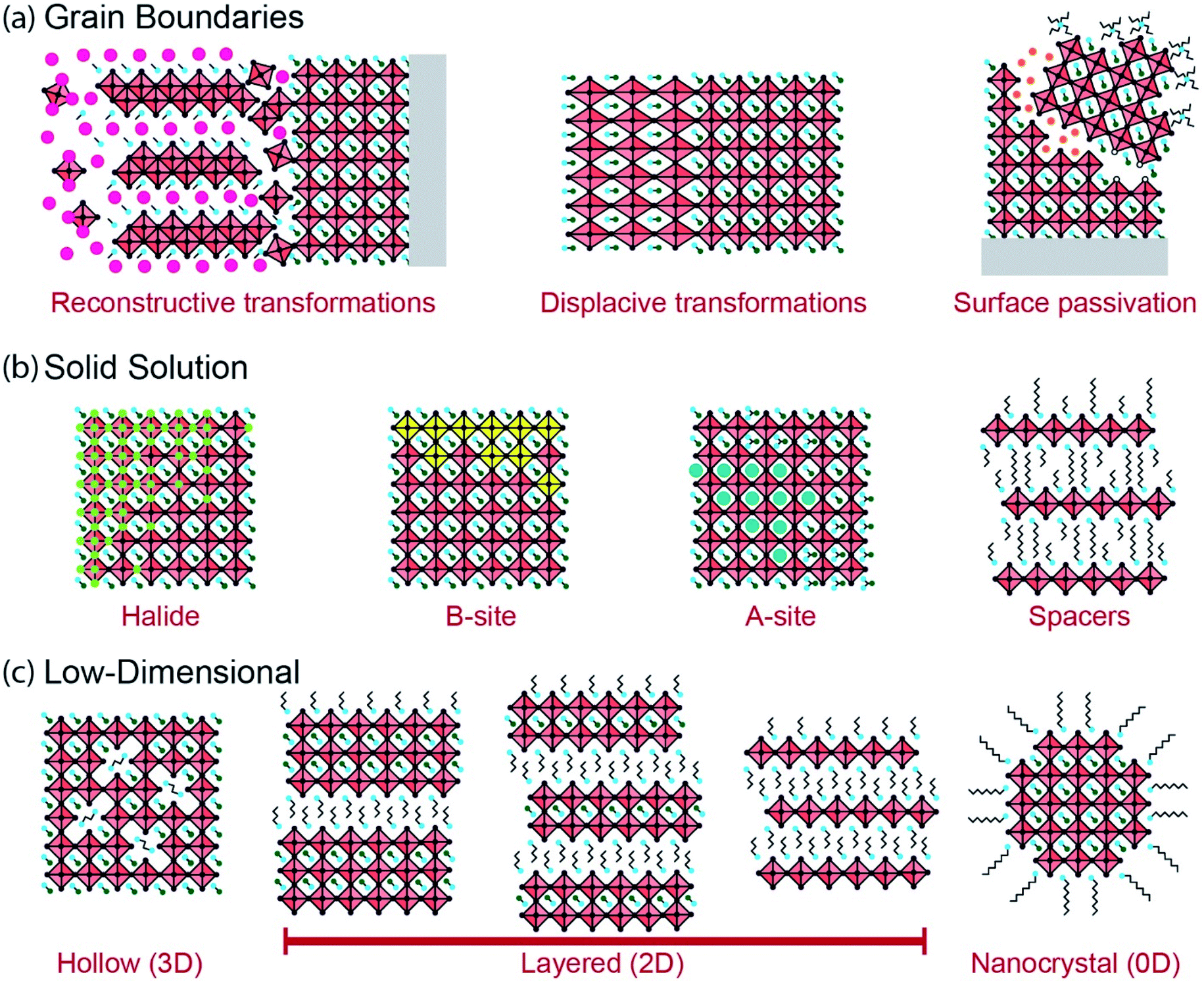 | ||
| Fig. 2 Intrinsic interfaces in MHPs: interfaces can form within MHP layers due to (a) grain boundaries, (b) interfaces caused by segregation and clustering of mixed anions and cations in MHP solid solutions, and (c) the formation of low-dimensional structures separated by octahedral voids, large spacer cations and ligands. | ||
The formation of intrinsic interfaces in MHPs is mediated by the geometry of the halide perovskite crystal structure. The stability of the simplest MHP structure, a cubic ABX3 lattice composed of spherical ions, can be described by the size and bonding character of constituent ions. The ‘tolerance factor’ (TF) introduced by Goldschmidt in 1926,106 establishes stability limits for perovskites based on the relative ionic radii (r) of A, B and X: 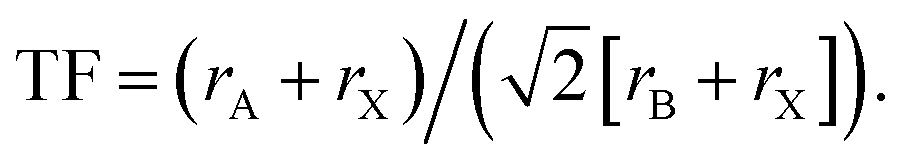 Another geometric ratio, the ‘octahedral factor’ (μ = rB/rX), establishes additional constraints on stability and octahedral distortions.107 Covalency of B–X bonds, lone pair interactions, and the sterics and hydrogen bonding interactions of small molecules in hybrid organic–inorganic MHPs extend this structural model to a great diversity of materials.2,108–110 When these geometric constraints are broken, either due to composition or processing conditions, intrinsic interfaces can form.
Another geometric ratio, the ‘octahedral factor’ (μ = rB/rX), establishes additional constraints on stability and octahedral distortions.107 Covalency of B–X bonds, lone pair interactions, and the sterics and hydrogen bonding interactions of small molecules in hybrid organic–inorganic MHPs extend this structural model to a great diversity of materials.2,108–110 When these geometric constraints are broken, either due to composition or processing conditions, intrinsic interfaces can form.
Reconstructive reactions, either during growth or degradation, inherently occur at grain boundaries. These reactions involve breaking and reforming bonds at an interface. A large body of research has been devoted to tuning grain microstructure in polycrystalline MHPs through processing techniques.9,44,46,47,49,119 Growth often occurs through sequential chemical pathways, forming intermediate gel or solvate phases during solution-processing that eventually form MHPs (Fig. 2a).46,47,120–125 The density and composition of grain boundaries in polycrystalline MHPs affects their stability to ambient exposure, heat or illumination. Additive engineering to alter the chemical environment of these grain boundaries has proven fruitful to prevent interfacial degradation in MHPs.43,103,104,118,126,127 Moreover, some thermal phase transformations in MHPs are reconstructive and may be catalyzed by solvation or hydration at grain boundaries. For instance, CsPbIBr2 transforms between a low-temperature one-dimensional phase and a high-temperature perovskite phase through a reconstructive reaction across a liquid-like interface.128 Reconstructive reactions form complex interfacial regions that may be disordered and short-lived. However, these regions determine how MHPs grow and degrade, and present a powerful opportunity for characterization techniques such as ssNMR that can probe reconstruction of interfaces.
Phase transformations can occur in MHPs without atomic reconstruction, forming interfaces between phases or domains (Fig. 2a). Polymorphism in MHPs is well-documented across different temperatures and pressures.55 MHP phase transformations are often displacive, without any bonds forming or breaking. As a result, the interfaces that form during displacive transformations are more like the bulk chemical environment than during reconstructive transformations. Nonetheless, these boundaries mediate the phase stability of MHPs, and may develop distinct properties. Ferroelastic displacive transformations can be induced by pressure or anisotropic stress. In this case, interfaces may exist either between distinct phases across a ferroelastic transformation (e.g. between cubic and tetragonal phases), or between different orientations of the same phase (i.e. twinning).91,129–135 Subtle transformations can also occur due to ordering of electronic dipoles or spins in MHPs, creating interfaces between ferroic domains, although there is active debate about ferroic properties in MHPs.135–141 The interfaces that form across non-reconstructive phase transformations in MHPs may be difficult to observe by ssNMR techniques, but present an important characterization and engineering challenge nonetheless.
The electronic properties and stability of MHPs can also be tuned in solid solutions of mixed halides. Halides participate in octahedral B–X bonds that form the electronic bands of MHPs. Changing the halide composition in MHPs can tune the band-gap continuously across the entire solar spectrum.149,157,158 However, halide species are mobile in MHPs and can diffuse due to temperature changes, mechanical strain, potential gradients or illumination.59,159 For instance, light illumination of MAPbI1−xBrx causes a reversible phase segregation into iodide-rich and bromide-rich regions.160,161 Sustained illumination can also redistribute halides and point defects in single-halide MHPs such as MAPbI3, leading to photo-brightening (i.e. increased photoluminescence intensity).162,163 Clustering of segregated halides in MHPs, or defect-rich and defect-poor regions, forms concentration gradients or interfaces (Fig. 2b) that may behave as semiconductor heterojunctions.164–170 Electronic traps can also become clustered during halide redistribution, or segregate to interfaces between domains, with significant impacts on photocarrier recombination.171 Halide redistribution is impacted by the A-site cations, and the effect can be minimized in mixed-cation MHPs.150,172 ssNMR is well-suited to characterize halide redistribution because of its sensitivity to the halide coordination environment of B-site cations as well as the ability to probe the local structure of halides.64,67
The pursuit of more environmentally friendly and less-toxic lead-free MHPs has inspired the development of halide perovskites with different B-site cations.173–175 For instance, divalent tin,176–179 manganese180,181 or germanium182 can be substituted for lead in MHPs, with improvements in stability and optoelectronic properties in some cases.183–185 Similar to mixed A-site and X-site MHPs, solid-solutions of mixed B-site perovskites may segregate into distinct domains of different cations (Fig. 2b).180 Multivalent B-site substitutes, such as tin, present an additional challenge because they can oxidize (Sn2+ to Sn4+) and degrade the MHPs.185 Understanding the distribution and stabilization of multivalent B-site cations in MHPs is an important characterization challenge. Aliovalent doping of B-site cations has been explored for several years in the pursuit of n- or p-type MHPs. However, MHPs are generally resistant to deviations from electroneutrality due to the ease of formation and mobility of compensating defects, so doping remains a challenge.78 MHPs can also be composed of heterovalent B-site substitutes including a combination of mono-, di-, tri- and tetravalent cations (e.g., Ag+, Cu+, Bi3+, In3+, Sb3+, Ti4+).183,184 For instance, MHPs with a formula unit of A2B(I)B′(III)X6 form elpasolite phases, more commonly known as double perovskites.186–189 However, photovoltaic device efficiencies of double perovskites have lagged behind single B-site MHPs due to large band-gaps and poor dispersion of electronic bands.184,190 The exploration of new compositions and processing routes for B-site substituted MHPs requires characterization techniques such as ssNMR that are sensitive to the different nuclei and oxidation states present, and that can resolve segregation, domain interfaces and transformations among these cations.
Layered 2D MHP phases can form when large ammonium or diammonium cations are introduced that exceed the tolerance and octahedral factors of perovskites.3–8 The layered phases that form bear intrinsic interfaces between spacer molecule and octahedral metal halide substructures. 2D metal halide phases have been explored for decades as promising anisotropic optoelectronic materials.201–204 These spacers are typically insulating, although there is currently a strong incentive to make them electroactive to improve charge transport in optoelectronic devices.205–211 The insulating layer of spacer molecules induces quantum and dielectric confinement of electronic states in the metal halide octahedra.212–219 Therefore, the bandgap, exciton binding and recombination dynamics can be changed by varying the number of metal halide octahedra, n, within each perovskite layer. For instance, adding butylammonium (BA) to the growth precursors for MAPbI3 forms layered Ruddlesden–Popper phases (BA2MAn−1PbnI3n+1) composed of separated 2D MAPbI3 slabs ranging from n = 1 to about n = 7 octahedra thick in the layer stacking direction, depending on stoichiometry (Fig. 2c).4,5,220–222 Organic ammonium spacer cations containing long-chain alkyl-, aryl-, adamantantyl- and alkylphenyl- and heterocyclic moieties can be readily incorporated to generate layered structures of different compositions.5 The lattice plane along which this layering occurs depends on the composition of the MHP.3–5,7 Research on low-dimensional halide perovskites is currently flourishing because these materials demonstrate tunable quantum confinement, anisotropic optoelectronic properties and improved ambient stability compared to 3D MHPs.3–8,218 The 2D interfaces that form at the unit-cell scale in layered MHPs require local characterization techniques such as ssNMR that can probe the short-range interactions between organic and inorganic species.
The layered phases present the same variety of intrinsic interfaces as the higher-dimensional MHPs. Grain boundaries of layered MHPs can have distinct optoelectronic properties from the bulk and serve as efficient recombination or transport centers.112,223,224 Mixed A-site, halide and B-site solid solutions and heterostructures have also been explored to tune the properties and stability of various layered MHPs.164,225–230 Moreover, layered MHPs synthesized from mixtures of different spacer cations can form solid solutions within the spacer substructure (Fig. 2b).231–234 During polycrystalline film growth, phase segregation between regions with different 2D confinement indices (the n value from the formula unit R2An−1BnX3n+1) can occur. Layered MHPs with a nominal stoichiometry of n greater than 2 typically segregate into regions of higher and lower n-values due to solution-processing kinetics.5,235–241 These polycrystalline materials have optical and transport properties characteristic of bulk heterojunctions.239,242–246 The desirable stability of layered MHPs have inspired efforts to incorporate these phases within 3D MHPs, or as capping and passivation layers, for high-performing photovoltaic devices.5,8 The local structures that form in these mixed low-dimensional MHP films are challenging to characterize and control. Mixtures of layered phases with different n-values can form due to epitaxial intergrowth, nanoscale disorder or nucleation and growth of segregated grains.236,238,240,241,247,248 Low-dimensional MHPs exhibit interfacial disorder and interactions at the molecular, unit cell and grain scales, and offer a compelling opportunity for study by multinuclear ssNMR methods.
Nanocrystals (NCs) of MHP phases can be synthesized with tunable composition, morphology and interfacial chemistry (Fig. 2c). Controlled nucleation and growth of 0D, 1D and 2D MHP nanostructures can be accomplished from liquid or vapor phases, ranging from quantum-confined nanoparticles to polycrystalline microparticles.56,249–254 The structure and properties of MHP NCs are largely defined by their interfaces with the surrounding solution (in colloidal phases) or matrix (in solid composites) because of their large surface-to-volume ratio.126,255–258 There has been tremendous interest in MHP NCs (often called perovskite quantum dots, even for particles larger than the ∼5 nm limit for quantum confinement) for lighting applications because of their near-unity photoluminescence quantum yield, narrow emission and tunable color gamut.15,25,158,257,259,260 The unique combination of these optical properties and solution processability makes them suitable candidates for display technologies. MHP NCs can assemble into microparticles and superlattices in dispersions or upon deposition.42,233,261–269 Perovskite NC films are promising candidates for optoelectronic devices, but their limited ambient stability and charge transport across ligand–NC interfaces remains a challenge.15,51,257,259,270,271 The small size of MHP NCs can also induce strain and modify surface energies, altering the energetics of particular phases (e.g. stabilized cubic CsPbI3 at room temperature).272,273 This in turn may cause local distortions and allow for the existence of metastable structures that would not be observed in the bulk. Finally, the growth processes used to synthesize MHP NCs can introduce interfacial species with binding motifs and compositions that would be difficult or impossible to achieve in microcrystalline MHPs.65,274–276 All of these properties have attracted a great deal of interest from spectroscopists who have used NMR techniques in the liquid and solid state to explore interfacial phenomena in MHP NCs.64,65,251
3. Length and timescales of physical processes
The following section outlines the length and time scales of different physical processes occurring in MHPs. We place these interactions in context with commonly used characterization techniques, with an emphasis on ssNMR techniques, and describe practical limitations for experimentalists. Fig. 3 illustrates the length and timescales of dynamic processes in MHPs along with fundamental limits of common characterization techniques, including ssNMR.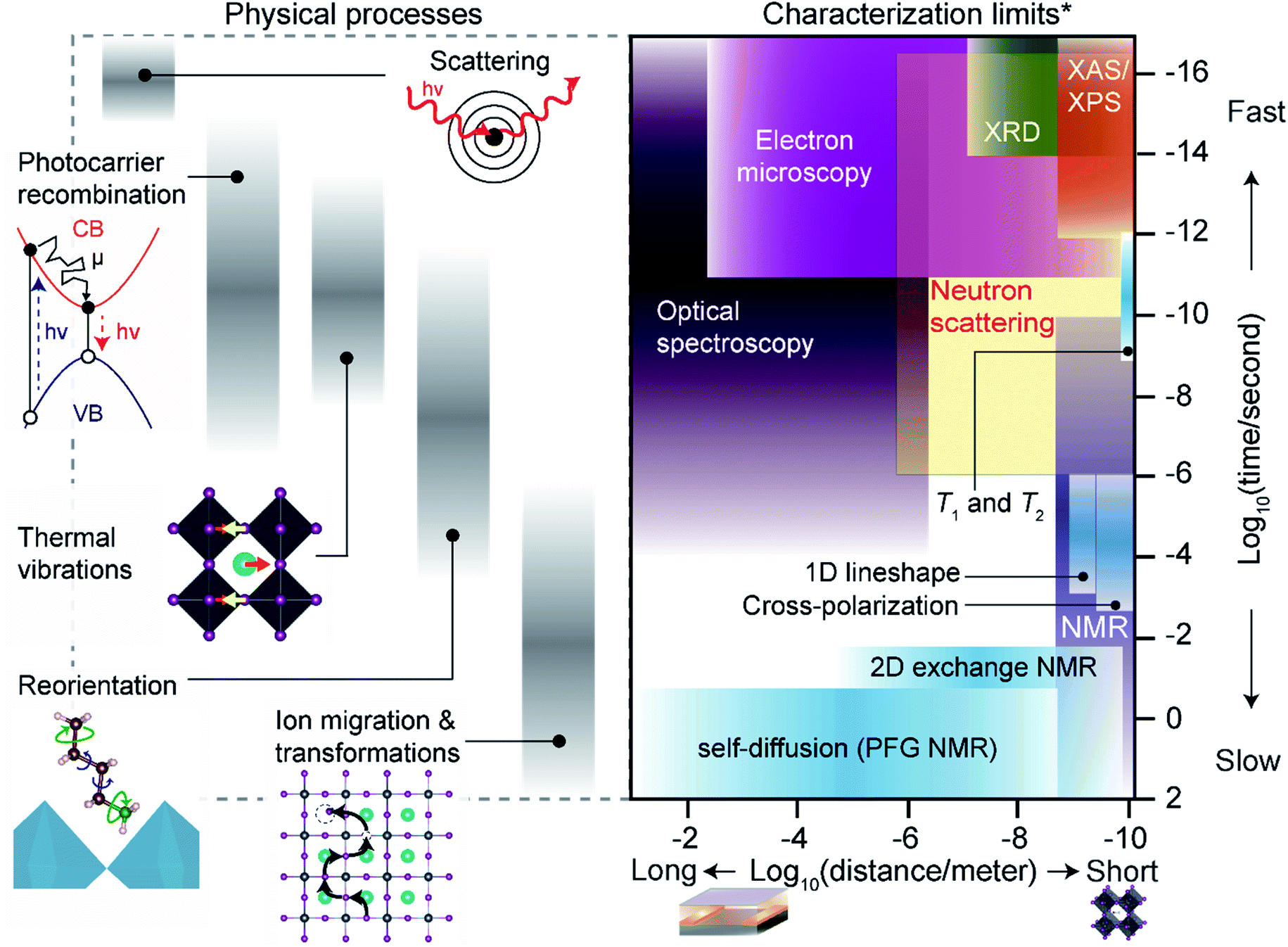 | ||
| Fig. 3 Comparison of length and timescales: the range of timescales of physical processes in MHPs is presented alongside the fundamental time and length scales that several characterization techniques can probe in MHPs. XRD: X-ray diffraction, XAS – X-ray absorption spectroscopy, XPS – X-ray photoelectron spectroscopy, PFG – pulsed-field gradient, CB – conduction band, VB – valence band. *Characterization limits are defined based on their fundamental interactions, not constraints on acquisition times or sample size. | ||
The physical processes that affect MHPs for device applications range from sub-femtoseconds (fs) to multi-year timescales. The fastest interactions probed in MHPs are scattering events between incident photons, electrons or ions and constituent atoms. For instance, X-ray diffraction (XRD) relies on elastic scattering of X-rays with core electrons in MHP atoms. At fs time-scales ion positions can be treated as a rigid ‘snapshot’ during each scattering event (i.e. the adiabatic Born–Oppenheimer approximation).277,278 Ultrafast optical spectroscopy of MHPs (e.g. transient absorbance and fluorescence) approach fs resolution,279 but direct observations of atom positions by X-ray and electron scattering have recently reached ps resolution.280–282 Nonetheless, even non-ultrafast scattering integrates distinct ‘snapshots’ of atomic positions. The photocarriers formed after initial scattering events migrate, scatter and recombine over timescales ranging from fs to μs. Photocarrier dynamics in MHPs have attracted a great deal of experimental and theoretical research,279,283–286 and impact optoelectronic device performance. Fast radiative recombination rates and strong exciton binding are beneficial for light-emitting applications to prevent non-radiative recombination,15 but longer lifetimes and diffusion distances aid in photogenerated charge extraction for photovoltaic devices.286,287 The collective motion of ions (i.e. vibrations, phonons) in MHPs occurs at timescales that overlap with photocarrier dynamics in MHPs (Fig. 3). The fastest ionic vibrations occur in the infrared spectrum (∼fs) for covalent bonds in molecular A-site and spacer cations. Lower-frequency octahedral phonons have important effects on exciton binding, carrier mobility, lifetimes and non-radiative recombination.45,60,288–290
Solid-state NMR techniques can access the timescales of photocarrier dynamics and vibrational modes through analysis of T1 and T2 relaxation times and other techniques (Section 4).66 Molecules in MHPs also reorient at longer timescales (nanoseconds to microseconds) than MHP phonon vibrations or photocarrier lifetimes. Molecular reorientations can introduce local octahedral distortions110,291,292 and transient electronic293,294 or dielectric disorder.295 Techniques including temperature-dependent broadband dielectric spectroscopy,291,296–298 inelastic neutron scattering,292,299–302 and NMR dynamics66,291,303–309 studies can access the frequencies of different reorientation modes, ranging from fast tumbling of small A-site cations such as methylammonium (ps–ns), to slow rotations (μs) of phenylethylammonium spacers in 2D Ruddlesden–Popper phases. When gradients in chemical potential, heat, light and electromagnetic fields are present, ions can diffuse at timescales ranging from nanoseconds to seconds.59,60,310,311 Section 6 describes ssNMR approaches to measure halide diffusion in MHPs, but the technique is also well-suited to probe A-site, B-site, spacer, additive and solvent transport. At the longest timescales of device lifetimes (hours to years), most characterization techniques have adequate time resolution to capture in situ or ex situ transformation kinetics.
The practical acquisition times required for the techniques described above may be much more restrictive than their fundamental limits. For instance, a typical Cu Kα sourced X-ray diffractometer may integrate fs-scale scattering events but require several minutes to obtain useful information. Acquisition times in ssNMR experiments vary tremendously depending on the sample material and interactions probed, but often require hours or days of signal averaging for adequate signal-to-noise ratios. Acquisition time constraints are imposed by equipment, type of nuclei and experiments, and labor costs, as well as the environmental stability of MHPs. For instance, Sn-based MHPs often degrade within minutes of ambient exposure, for which acquisition of multidimensional ssNMR experiments is less straightforward.185,312,313 Acceleration of acquisition times for ssNMR experiments, particularly for mass-limited samples such as thin films, have been pivotal for the characterization of MHPs.314
The length scales of physical interactions in MHP devices span from the unit-cell (Å) scale to many cm. Laboratory scale optoelectronic device stacks tend to be roughly a micron thick (not including the substrate) and ∼cm2 in area. Heterogeneity across this entire span of distances can have important effects on optoelectronic device properties.44Fig. 3 highlights the fundamental length scales that some common techniques can access, although practical spatial resolutions (i.e. the resolvable size of the sample or probe) can be much coarser. The atomic (Å) scale structure of MHPs can be probed by spectroscopic or scattering techniques with short-wavelength particles (photons, neutrons, electrons, ions). X-ray, electron or neutron scattering can resolve the atomic structure of crystalline materials with sub-Å precision. However, the spatial resolution of sample regions that can be probed by these techniques may be much more limited. ssNMR can probe interactions and structure across a particularly wide range of length scales. Homo- and heteronuclear correlation methods can probe distances with atomic-scale resolution,63 and pulsed-field gradient (PFG) diffusion and magnetic resonance imaging (MRI) methods resolve mm scale features and transport distances.315,316 However, the length scales that can be probed by ssNMR techniques are generally limited by the sensitivity of the instrument to particular nuclei or type of NMR experiment. MRI techniques have some ability to localize signal to microscopic scales, but this is highly specialized.317,318 Practical considerations for the spatial resolution of different ssNMR techniques are described in Section 4.
4. NMR spectroscopy tutorial
This section covers some basic aspects, sample preparation and practical considerations of ssNMR spectroscopy for the characterization of MHPs. The essence of ssNMR is elemental specificity and natural abundance of NMR active isotopes (Fig. 4a), which can be separately excited and detected. The MHPs and the contact layers contain several NMR active nuclei: 1H, 7Li, 11B, 13C, 14N, 15N, 19F, 27Al, 33S, 35Cl, 37Cl, 39K, 41K, 49Ti, 55Mn, 63Cu, 65Cu, 67Zn, 73Ge, 79Br, 81Br, 85Rb, 87Rb, 107Ag, 109Ag, 113Cd, 119Sn, 121Sb, 123Sb, 123Te, 125Te, 127I, 133Cs, 207Pb, and 209Bi. A wide variety of NMR interactions such as chemical shift (characterized by chemical shift anisotropy, CSA), dipole–dipole couplings, quadrupolar interactions, knight shifts, hyperfine interactions and paramagnet-induced interactions can be used to characterize local bonding environments and inter- and intramolecular interactions at the sub-nanometer to nanometer length scales. For a detailed description of basic concepts of NMR and different nuclear spin interactions, we refer the reader to textbooks.319–321 Chemical shifts are sensitive to local bonding environments. Dipole–dipole couplings are sensitive to through-space inter- and intramolecular proximities between specific nuclei and the fluctuations in the local structures. Quadrupolar interaction arises from the electric field gradient at the nucleus, which is sensitive to symmetry of the local bonding environments and structural changes in the coordination sphere and molecular fluctuations. The dynamic motion associated with various molecular processes can be investigated at different time scales through ssNMR spectroscopy techniques.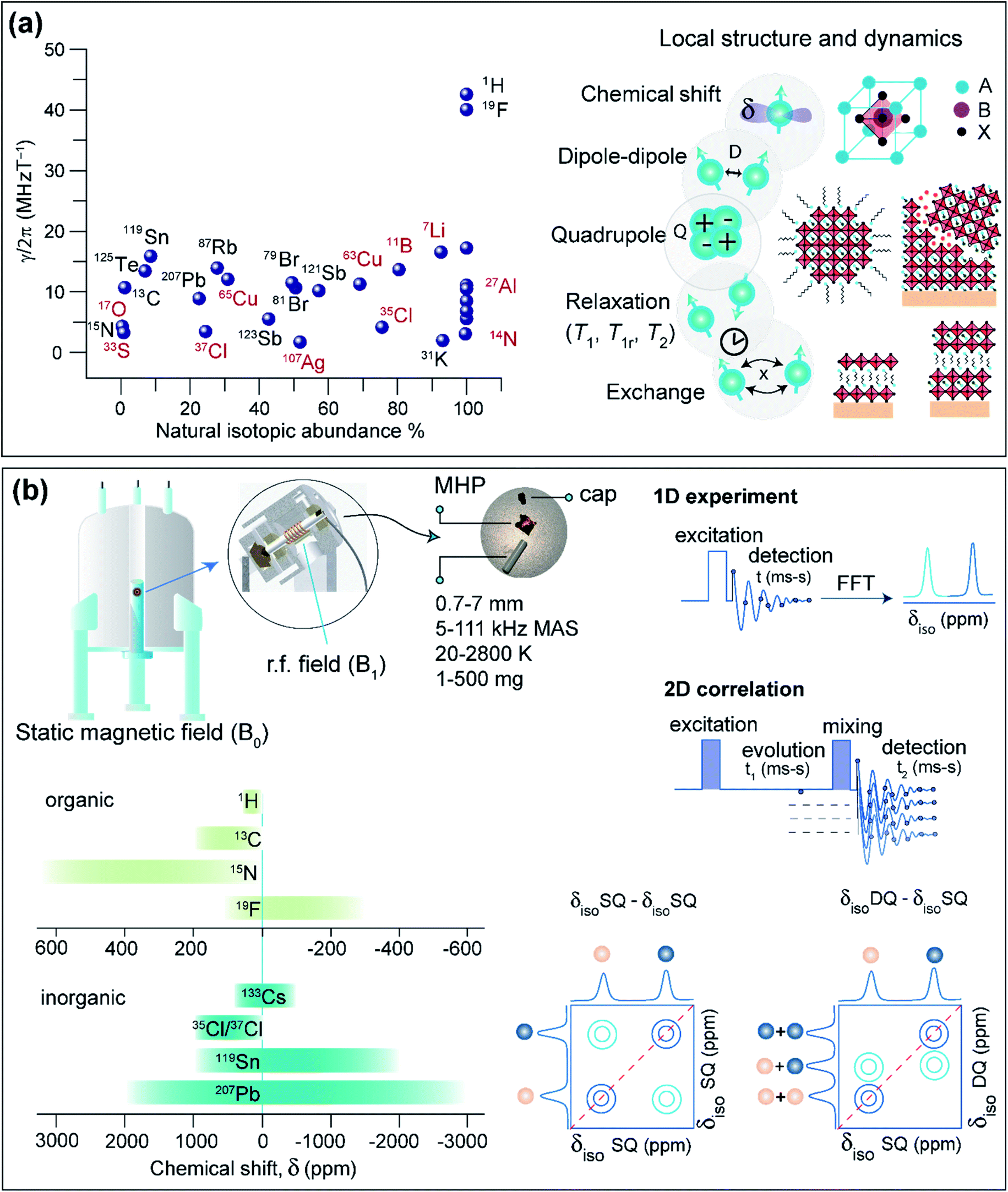 | ||
| Fig. 4 ssNMR spectroscopy of MHPs: (a) NMR nuclei relevant for MHPs characterization plotted as a function of sensitivity and natural abundance, and a schematic of nuclear spin interactions, relaxation and exchange that can be used to elucidate structures and dynamics. (b) schematic of solid-sate NMR setup along with the acquisition of 1D and 2D ssNMR experiments, and chemical shift ranges of organic and inorganic sites in MHPs. FFT – Fast Fourier-Transformation; γ – gyromagnetic ratio; rf – radiofrequency; δiso – isotropic chemical shift; SQ – Single-Quantum; DQ – Double-Quantum. | ||
4.1. Sample preparation, data acquisition and analysis
Solid-state NMR spectra of MHPs, HTMs, ETMs and their blends can be acquired in a variety of forms, such as microcrystalline powders, thin films scraped off from glass substrates, crushed thin films, or slurry-like materials. These samples can be packed into cylindrical zirconia or ceramic sample holders, referred to as rotors (Fig. 4b). A packed rotor is then inserted into a probehead placed in a static magnetic field (B0), and excited with a radio frequency (rf) pulse or a sequence of rf pulses and delays. Most of the solid-state NMR experiments are performed under Magic-Angle Spinning (MAS) conditions, that is by spinning rotors at a set angle 54.74° with respect to the external field (B0). MAS approach partially or even completely averages out the anisotropic interactions that broaden static NMR spectra. Today, probeheads and rotors of varying diameters in the range of 0.7 mm to 7 mm (which can be filled with ∼1 mg to up to 1 g of material), capable of achieving spinning speeds in the range of 5 kHz to over 111 kHz, are available for carrying out MAS NMR experiments. Most of the rotors used in MAS experiments are air-tight such that the moisture sensitive samples can be characterized. Variable Temperature (VT) experiments can be performed in the range of 20–450 K, although dedicated high temperature probes increase this extent up to 2800 K.322–324 High pressure experiments of up to 90 GPa are feasible,325 and a combination of high temperatures and pressures can be achieved.326 In addition, static NMR experiments of bulk crystals and intact thin films may be carried out as functions of moisture exposure and light illumination.327,328In a simple 1D NMR experiment, a single pulse rf excitation is applied to acquire a time domain signal which is referred to as free-induction decay (FID). The fast Fourier transformation (FFT) of the time-domain signal yields a frequency domain spectrum. If the MAS rate is high enough to average out anisotropic interactions, 1D signals corresponding to chemically distinct sites can be resolved (at least for spin-1/2 nuclei in crystalline materials). The isotropic chemical shifts (δiso) are characteristic to the local chemical environments and structures, which can be obtained in most cases by analyzing 1D NMR spectra.
A sequence of rf pulses and delays can be applied to acquire 2D NMR spectra. 2D pulse sequences can be generally classified into excitation, evolution, mixing and detection steps (Fig. 4b). Each step can be engineered to obtain specific information on structure and dynamics. The analysis of 2D correlation spectra allows the local structures and intermolecular interactions to be elucidated in MHPs. 2D correlation experiments involving isotropic chemical shifts (δiso–δiso), δiso and dipolar couplings (δiso–D), δiso and J-couplings (δiso–J) are widely used to characterize materials including MHPs. For example, information about through-bond connectivity or through-space interactions can be obtained by analyzing scalar (J) and dipolar(D)-mediated 2D correlation spectra. The Double-Quantum (DQ) signals can be excited and detected for spatially proximate and dipole–dipole coupled spin pairs (e.g., through-space 1H–1H proximities within 5 Å).329 In a 2D Double-Quantum–Single-Quantum (DQ–SQ) correlation experiment, the DQ signal (δDQ) evolves at the sum of the SQ signals (δSQ) leading to 2D peaks on the diagonal that originate from chemically equivalent sites, or off-diagonal 2D peaks between chemically distinct sites within the same molecule or between neighboring molecules (Fig. 4b). Notably, homonuclear DQ–SQ correlation (1H–1H, 19F–19F, 13C–13C) spectroscopy provides information on the intermolecular interactions and interatomic distances. Of particular interest is the magnetization exchange, also referred to as the spin diffusion (SD) experiment, which allows the through-space proximities between neighboring sites to be probed and analyzed. Spin diffusion experiments mediated through dipolar couplings (2D SQ–SQ correlation, Fig. 4b) extend the NMR length scales beyond a nanometer by enabling the magnetization to propagate between more distant chemical sites through a network of dipolar coupled spins. 2D 1H–1H DQ–SQ and SD experiments are particularly suitable to probe intrinsic and extrinsic interfaces in MHPs, as will be discussed in Section 5. Likewise, 2D heteronuclear correlation experiments (1H–13C, 1H–207Pb and 1H–133Cs) can be carried out to probe the chemical nature and vicinities between A and B sites in MHPs at sub-nm to nm distances, facilitating NMR-based structure elucidation.
4.2. Resolution and sensitivity
NMR spectral resolution plays an important role in the structural elucidation of material solids such as MHPs. Resolution in ssNMR spectra can be attained by carrying out experiments at high magnetic fields, fast MAS or a combination of these two. When a fast-spinning limit is reached, i.e., the MAS frequency is greater than the chemical-shift anisotropy and multitudes of dipole–dipole couplings, high-resolution can be fulfilled. Resolution enhancement can also be attained by decoupling of dipole–dipole couplings between homo- (e.g., 1H–1H)330,331 and heteronuclei (e.g., 13C–1H)332 and/or by Combined Rotation and Multiple Pulse Spectroscopy (CRAMPS)333 which employs moderate MAS rates (often <15 kHz) in conjunction with decoupling sequences. Motional averaging of A-site cations (MA+, FA+), specific functional groups such as –CH3, –NH3 and phenyl groups in spacer cations, or translational mobilities of cations in the voids of BX6 octahedra lead to partially averaged anisotropic interactions, which improves resolution in the ssNMR spectra. As discussed in the previous section, 2D NMR spectroscopy provides improved resolution by spreading the signals into two frequency dimensions. In contrast, ssNMR spectra of B site cations and X site anions face a resolution challenge due to the large chemical shift anisotropy (CSA) and quadrupolar interactions, respectively. CSA scales linearly with B0, whereas quadrupolar interaction is inversely proportional to B0. In this respect, a combination of high B0 and fast MAS are expected to help to enhance the resolution in the NMR spectra of quadrupolar nuclei (11B, 14N, 27Al, 33S, 35Cl, 37Cl, 39K, 41K, 49Ti, 55Mn, 63Cu, 65Cu, 67Zn, 73Ge, 79Br, 81Br, 85Rb, 87Rb, 121Sb, 123Sb, 127I, 133Cs and 209Bi), and the resolution in 207Pb and 119Sn NMR spectra can be improved using fast MAS at relatively low B0.Sensitivity gain in ssNMR can be achieved by acquiring the spectra at higher magnetic fields. Static magnetic fields in the range of 2.4 T to 35.2 T (1H Larmor frequency range from 100 MHz to 1.5 GHz) are available to carryout ssNMR experiments of material solids. Other methods of sensitivity enhancements include Cross-Polarization (CP) MAS NMR experiments. CP-MAS experiments exploit the transfer of polarization from abundant nuclei (e.g., a = 1H, 19F) to dilute nuclei (e.g., b = 13C, 15N) through the network of dipole–dipole interactions, which results in a signal-to-noise (S/N) ratio increased by a factor of up to γa/γb, γ being the corresponding gyromagnetic ratios.
One of the most important techniques in the context of atomic-level studies of surfaces is Dynamic Nuclear Polarization Surface Enhanced NMR Spectroscopy (DNP SENS).334 DNP SENS makes it possible to substantially increase the sensitivity of NMR by transferring the electron spin polarization to nuclei via hyperfine interactions. This leads to a theoretical sensitivity gains of a factor of γe/γn, where γe and γn are the gyromagnetic ratios of the electron and nuclear spins, respectively. Accordingly, enhancements of up to ∼660, ∼2600, and ∼6600 for 1H, 13C, and 15N, respectively, can be attained. The source of the unpaired electrons is typically a stable nitroxide biradical, which is introduced as a dilute solution in a solvent which does not dissolve the material under study (i.e. an antisolvent with respect to the solid material). One successful implementation of this protocol is the incipient wetness impregnation method, in which the amount of radical-containing solution is just enough to coat the particles without making the powder excessively wet. Since the radical resides on the surface of the particles, the DNP effect is most pronounced for the surface sites, which are enhanced preferentially compared to the bulk of the crystallites. The high surface polarization can also be used to increase the sensitivity of the nuclei in the bulk by taking advantage of spin diffusion (SD).335 While in typical organic solids SD relies on the dense network of 1H nuclei, it has also been demonstrated to occur in proton-free solids, considerably extending the applicability of DNP in inorganic materials.336 Taken together, DNP can enhance the sensitivity of species starting from those located directly at the surface to those tens to hundreds of nanometers away from the surface inside the bulk, depending on the material, type of ssNMR experiment and how the experiment is set up. DNP experiments are typically carried out at low temperatures (∼100 K) but protocols have been devised to make it feasible up to room temperature.337
DNP of MHPs has only been moderately successful so far with the reported 1H–207Pb enhancements not exceeding 20 (for nanocrystalline MAPbCl3) and no measurable enhancement for MAPbI3.314,338 This is caused by the short 1H T1 times under the typical DNP experimental conditions, which in turn make 1H–1H SD inefficient and prevent the polarization of deeper sites which are far from the surface. Piveteau et al. have reported DNP SENS of CsPbBr3 nanocrystals in a frozen solution, although in that case the enhancement was not quantified because of the high dilution of the material.338 Enabling high efficiency DNP SENS for MHPs remains an ongoing challenge.
5. Interfaces in MHPs probed by solid-state NMR
Traditional multinuclear NMR approaches can be used for the characterization of local chemical environments in a wide variety of MHPs including 3D phases, solid solutions, defect-engineered phases, 2D layered structures, and surface-passivated MHPs. Despite the strong similarities in molecular topologies, MHPs exhibit structurally diverse interfaces. ssNMR can resolve pertinent questions about the interfacial chemistry of MHPs, including (i) short-range order and disorder in A sites and BX6 octahedra, (ii) the degree of A-cation intermixing and phase separation, (iii) spacer cation packing arrangements in low-dimensional MHPs, (iv) the extent of octahedral tilt and local structural distortions, (v) structural transformations and degradation products, and (vi) slow dynamic processes at organic–inorganic interfaces including reorientational modes of organic cations, cation diffusion and halide migration.The local environments of A and B site cations can be identified and distinguished by analyzing 1H, 13C, 14N, 15N, 133Cs, 207Pb and 119Sn chemical shifts. In addition, through-space proximities of A–A sites, B–B sites, and A–B sites can be probed through the dipolar-based 2D correlation experiments such as, for example, 1H–1H, 133Cs–1H, 207Pb–1H and 207Pb–207Pb 2D correlation NMR spectra, which manifest weak and strong 2D peaks corresponding to long and short distances, respectively. In addition, quadrupolar nuclei (e.g., 35Cl, 79Br, 81Br, 115In, 127I, 209Bi) exhibit large quadrupolar interactions, which make these nuclei amenable to both NMR and NQR, providing information on the local chemical environments of halogen atoms. ssNMR experiments have also been employed to map the A-site cation distribution, interfacial structures, degradation products, point defects and dynamics in MHPs, as discussed below. For a recent summary on ssNMR studies of MHPs, we refer the reader to the reviews by Franssen et al. (structural and dynamical aspects),67 Bernard et al. (dynamics of MA in MAPbX3),306 Moudrakovski (dynamics in MHPs),66 Senocrate et al. (ion conduction),311 Smock et al. (MHP nanocrystal surfaces)65 and Piveteau et al. (ssNMR and NQR of MHPs).64 In the subsequent sections, we demonstrate how ssNMR techniques can be used to probe intrinsic and extrinsic interfacial structure and properties in MHPs.
5.1. Cations, anions and degradation products in 3D MHPs
Pure and mixed-cation/anion MHPs exhibit various organic–inorganic interfaces. In 3D crystalline phases, the local structures and interactions involving BX6 and chemically diverse monovalent cations have been investigated by 1D and 2D ssNMR techniques.152–154,154,339–341 Specifically, analysis of multinuclear (1H, 2H, 13C, 14N, 15N, 39K, 87Rb, and 133Cs) NMR chemical shifts has been shown to be an effective approach to gain insight into the local structural environments of monovalent cations in MHPs.153,154,304,339,342–348 Kubicki et al. studied cation doping in 3D MHPs using 39K, 87Rb and 133Cs NMR spectroscopy, and revealed that the K+ and Rb+ ions do not incorporate into the 3D perovskite structure of MHPs, while Cs+ ions do.153,154 Atomic-level understanding including local order in BX6 octahedra and mapping of cation distributions and interactions between them is expected to help to better understand the optoelectronic properties.In a study by Grüninger et al., 2D 1H–1H DQ-SQ correlation NMR (Fig. 5) was combined with molecular dynamics (MD) simulations using machine-learning force-fields (MLFFs) to unravel the clustering of MA+ and FA+ in mixed composition MA1−xFAxPbI3 (x = 0.25, 0.5 and 0.75) and MA0.15FA0.85PbI2.55Br0.45.151Fig. 5a shows the 1H DQ-SQ signal intensities originating from intramolecular 1H–1H proximities within MA (red circles and red thick line) and FA (blue circle and blue thick line), and intermolecular 1H–1H proximities between MA and FA cations (blue-to-red-gradient thick lines). The 1H DQ intensity buildup curves associated with intermolecular FA–MA proximities have been analyzed using a second moment approach in order to estimate the average 1H–1H dipolar coupling strengths between FA+ and MA+ (Fig. 5b).151,349,350 These results are corroborated by MD simulations which indicate that the intramolecular dipolar couplings within the A cation (MA+, FA+) are averaged through the rapid reorientation at the ssNMR time scales (micro to milliseconds), thus intermolecular 1H–1H dipolar interactions are attributed to the DQ signal intensity build-up. For three different MA1−xFAxPbI3 mixed formulations (x = 0.25, 0.5 and 0.75), the average 1H–1H dipolar couplings and theoretical populations of MA–FA contacts are calculated. The populations of FA–MA contacts are analyzed (see ref. 151 for a detailed procedure) by means of an order parameter S, which takes different values in the range of 0–1 depending on the degree of FA-MA mixing: S = 0 for random cation distribution, 0 < S < 1 for clustering, S = 1 for phase segregation. The clustering tendency and the A-site occupancies (rMA and rFA) have been parametrized as rMA = (1 − x) + Sx and rFA = x + S(1 − x). The calculated populations indicated that partial MA–MA and FA–FA clustering occurs for x = 0.25 and x = 0.5 (S values in the range of 0.2–0.4), whereas majority-FA formulations (x = 0.75) have slightly more random distributions (S values in the range of 0–0.3, Fig. 5c). This work suggests that heterogeneities in the cation distributions lead to local variations in the electrostatic interactions at the organic–inorganic sublattice (Section 2, Fig. 2), which influence the optoelectronic properties.351,352
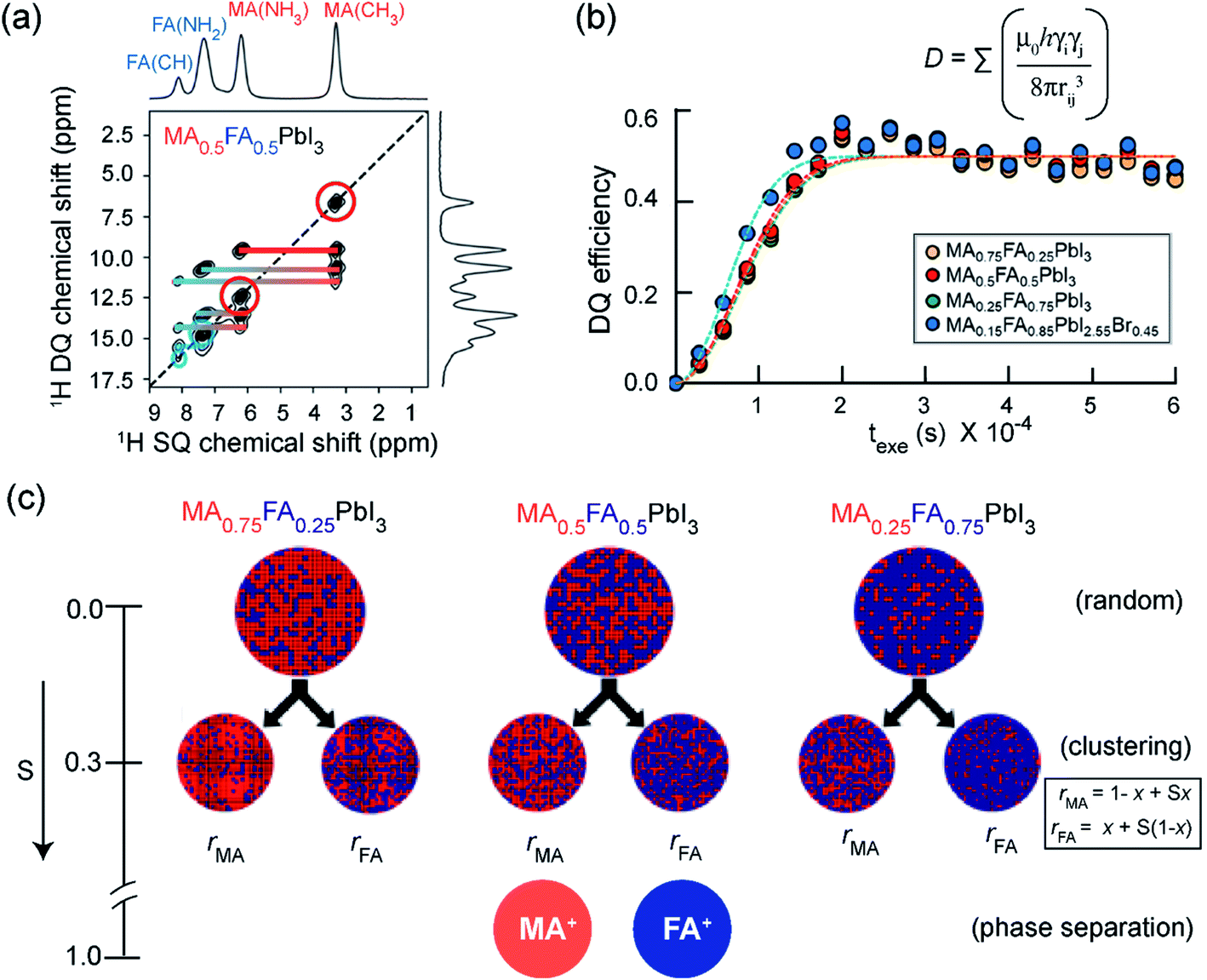 | ||
| Fig. 5 Clustering of cations in mixed A-site MHPs: (a) 2D 1H–1H DQ–SQ correlation spectra of mixed A-site cation and anion MHPs: thick horizontal lines depict intramolecular 1H–1H proximities associated with MA cations (red), FA cations (blue), and intermolecular H–H proximities between MA and FA cations (blue-to-red) in MA0.5FA0.5PbI3. (b) Comparison of 1H DQ build-up curves in MA1−xFAxPbI3 (yellow, red, green) as well as MA0.15FA0.85PbI2.55Br0.45 (blue) formulations. The dashed lines are fits of the DQ build-up curves based on analytical expression (ref. 151, eqn (2)) which allow to calculate the average dipolar couplings. (c) Schematic representation of MA(red)/FA(blue) cation distributions within MA1−xFAxPbI3 (x = 0.25, 0.50, 0.75) perovskites following random statistics (S = 0), MA/FA clustering with S = 0.3 parametrized using rMA and rFA, and phase separation (S = 1), respectively. As the experimental DQ intensity buildup analysis does not provide information about domain sizes of MA-rich and FA-rich regions, arbitrary sized circles were chosen to represent the statistical distribution of cations. Adapted with permission from ref. 151. Copyright 2021 American Chemical Society. | ||
The type of halogen atom forming the BX6 octahedra strongly influences the chemical shifts and lineshapes of B-site cations (Pb2+, Sn2+).152,153,312,314,345,353–357 In this context, 207Pb and 119Sn NMR studies have been increasingly pursued to examine the local environments of BX6 octahedra in lead and tin halide perovskites, respectively. The subtle changes in the organic–inorganic interfaces such as electrostatic interactions, cation dynamics and octahedral tilts and B–X bond distances and phase transitions may have dramatic effects on the 207Pb and 119Sn NMR lineshapes. In lead MHPs, the substantial chemical shift range of 207Pb signals of up to 3000 ppm and peak widths (full-width-at-half-maximum, FWHM in the range of a fraction of kHz to several tens of kHz) provides a powerful way to characterize structural properties such as Pb–X distances, Pb–X–Pb bond angles and octahedral tilts in the PbX6 octahedra.304,314,353–355,358,359 1D NMR often suffices for identifying and distinguishing the local coordination spheres and the impact of A and X site doping on the 207Pb and 119Sn shifts in ABX6 MHPs. In addition, 2D 207Pb exchange spectroscopy has been used to gain insight into the solid-solution behavior of MAPbX3 (X = Cl, Br, I) obtained from mechanochemical synthesis.360
In tin-based MHPs, the 119Sn chemical shifts provide a wealth of structural and dynamics information.312,313,361–364119Sn chemical shifts span a wide range (∼12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm, e.g., δ (Sn metal) = 7500 ppm and δ(MA2SnI6) = −4684 ppm) and typically exhibit chemical shift anisotropy, which is readily averaged out in iodide- and bromide-based tin halide perovskite compositions due to halide hopping. Knight shifts, resulting from the interaction of nuclear spins with itinerant electrons, have also been reported.312,363,364 It has been shown that the 119Sn chemical shift is a sensitive local probe of the A-site cation (Cs, MA, FA) composition.312 In addition, ssNMR has been shown to be particularly useful for probing B-site cation mixing in MHPs by specifically probing the local structure of each of the contributing cations. Cadmium doping in hybrid and all-inorganic MHPs has been investigated by 113Cd ssNMR spectroscopy. Cd2+ ions can be incorporated into PbI2 and CsPbBr3, whereas Cd2+ ions phase segregate into non-perovskite halocadmates in MHPs based on MA and FA.365
000 ppm, e.g., δ (Sn metal) = 7500 ppm and δ(MA2SnI6) = −4684 ppm) and typically exhibit chemical shift anisotropy, which is readily averaged out in iodide- and bromide-based tin halide perovskite compositions due to halide hopping. Knight shifts, resulting from the interaction of nuclear spins with itinerant electrons, have also been reported.312,363,364 It has been shown that the 119Sn chemical shift is a sensitive local probe of the A-site cation (Cs, MA, FA) composition.312 In addition, ssNMR has been shown to be particularly useful for probing B-site cation mixing in MHPs by specifically probing the local structure of each of the contributing cations. Cadmium doping in hybrid and all-inorganic MHPs has been investigated by 113Cd ssNMR spectroscopy. Cd2+ ions can be incorporated into PbI2 and CsPbBr3, whereas Cd2+ ions phase segregate into non-perovskite halocadmates in MHPs based on MA and FA.365
Another important feature of ssNMR is the ability to study and distinguish B site cations in double perovskites. In a study by Michaelis and co-workers, halide double perovskites (HDPs) with the general formula (A(I)2B′(III)B′′(I)X6) have been investigated by ssNMR, XRD and DFT modelling. A series of Bi3+/In3+ mixed-cation Cs2Bi1−xInxAgCl6 solid solutions has been characterized by 115In and 209Bi ssNMR spectroscopy (Fig. 6).366 The impact of short to medium-range octahedral symmetry in the first B′(III) coordination sphere of Cs2Bi1−xInxAgCl6 compounds on the 209Bi and 115In quadrupolar lineshapes is analyzed and compared with the calculated quadrupolar coupling constants (CQ) of 115In and 209Bi sites in the supercells (Fig. 6a). In particular, when the symmetry starts to breakdown in (BiCl6){AgCl6}6[BiCl6]12 – that is the central (BiCl6)3− unit surrounded by six (AgCl6)3− and twelve [BiCl6]3− octahedra – upon doping with one or more [InCl6]3− in the first coordination sphere, the central 209Bi sites experience different local chemical environments, leading to significant changes in the calculated 209Bi CQ values (Fig. 6a, top). For example, a replacement of four and eight [BiCl6]3− sites (blue dots) by [InCl6]3− sites (red dots) in the first coordination sphere causes change in the CQ values of central 209Bi sites up to 35.8 and 45.9 MHz. In contrast, the replacement of all twelve [BiCl6]3− octahedra by [InCl6]3− results in only subtle changes in the CQ values (0.4 MHz) because of the recreation of the symmetry. Similar trends are observed in the CQ values of 115In (25.7 MHz for nearly equal Bi/In populations) when the symmetry in (InCl6){AgCl6}6[InCl6]12 is interrupted upon doping with [BiCl6]3− sites. Specifically, high-field (21.14 T) ssNMR enabled the local bonding environments of 115In and 209Bi sites in mixed-cationic HDPs to be resolved (Fig. 6b and c), whereby 115In and 209Bi signals corresponding to different octahedral sites and symmetries in the first coordination sphere are identified and distinguished. This study showed that there is a high degree of B′(III)/B′′(I) cation ordering and cation mixing in Cs2Bi1−xInxAgCl6 HDPs. The high degree of Bi/In alloying allows a delicate tuning of the band-gap and light emitting properties of HDPs. The combined high field ssNMR, XRD, PL and computational modelling has been used to establish structure-based understanding of broad-band white-light emission properties.
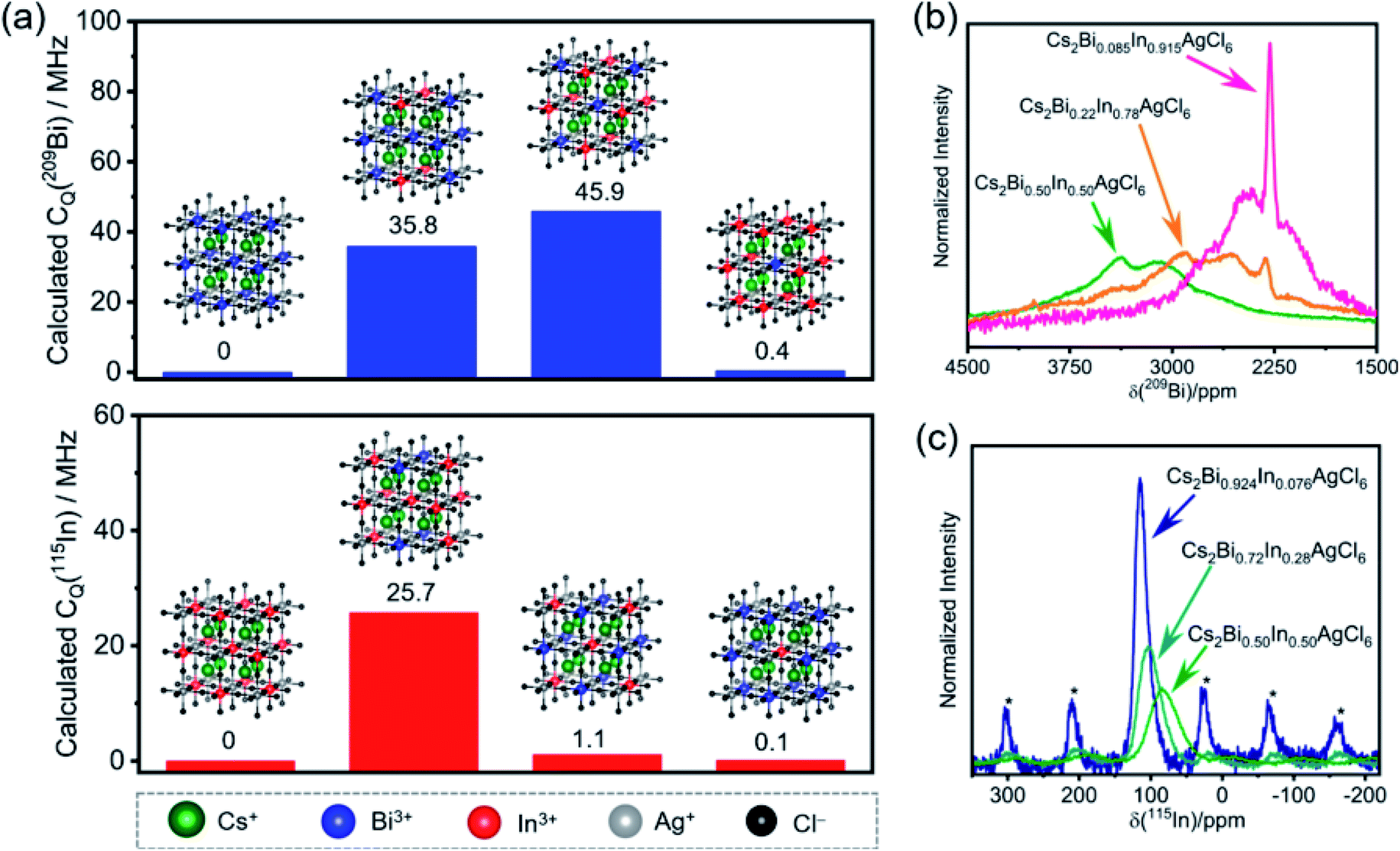 | ||
| Fig. 6 Local chemical environments of B-site cations in halide double perovskites: (a) calculated CQ (quadrupolar coupling constants) of the central 209Bi and 115In sites depending on the nearest neighbors in the first B′(III) coordination sphere (insight, blue and red dots) in the superlattice. Experimental (b) 209Bi and (c) 115In NMR spectra acquired at 21.14 T under static and MAS (18 kHz) conditions, respectively. Asterisks in (c) indicate spinning side bands. Adapted with permission from ref. 366. Copyright 2020 American Chemical Society. | ||
The environmental stability and degradation processes of a wide variety of 3D MHPs have been investigated by multinuclear (1H, 207Pb, 119Sn, 133Cs) ssNMR spectroscopy techniques.312,313,359,367,368 The hydrothermal stability and degradation of MAPbI3 has been characterized through 1D 207Pb NMR spectroscopy, and revealed that pristine MAPbI3 and its degradation products, MAPbI3·H2O, and PbI2, have distinct chemical shifts (Fig. 7a). This analysis shows that MAPbI3 degrades into MAPbI3·H2O upon exposure to moisture, while no (MA)4PbI6·2H2O or PbI2 is formed as a decomposition product upon prolonged exposure to moderate humidity (∼40% RH or below) at ambient temperature. Nevertheless, simultaneous exposure of MAPbI3 to heat and humidity, or liquid water, leads to the irreversible formation of PbI2. These results are corroborated by in situ XRD and electron microscopy techniques, which also show the formation of MAPbI3·H2O and PbI2 upon exposure to moisture at room temperature under controlled humidity (85% RH, relatively humidity).368
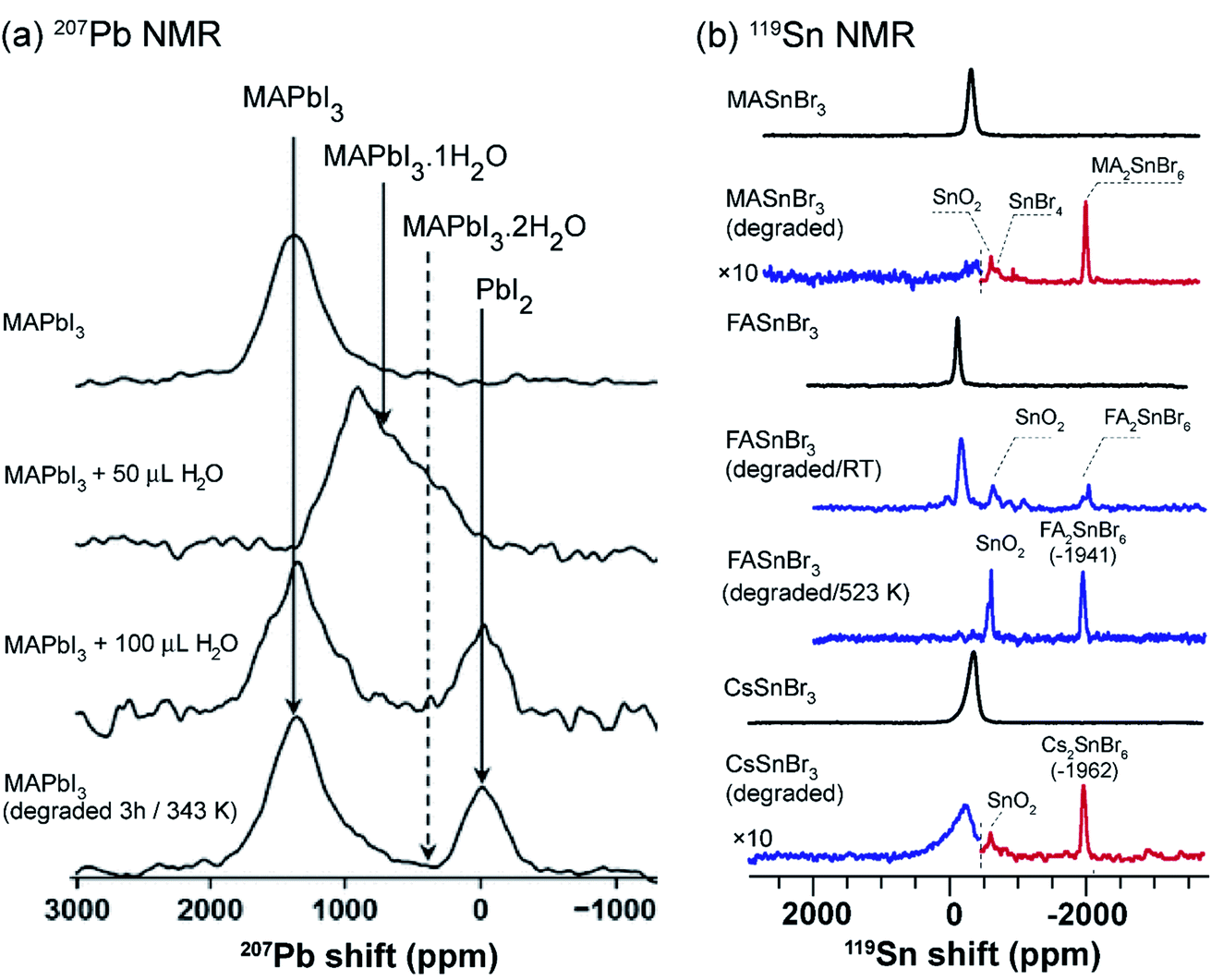 | ||
| Fig. 7 Probing degradation of MHPs through ssNMR: (a) static 1D 207Pb NMR spectra (7.05 T, 294 K) of pristine MAPbI3 and the same material after the addition of 50 μL and 200 μL of H2O, and the water treated MAPbI3 after heated for 3 h at 343 K. The 207Pb signals associated with MAPbI3 and degraded byproducts are shown in solid lines, and the dotted lines correspond to proposed decomposition product (not observed). Adapted with permission from ref. 367. Copyright 2017 American Chemical Society. (b) 119Sn MAS NMR spectra (4.7 T, 12 kHz MAS, 298 K), from top-to-bottom: fresh MASnBr3, aged MASnBr3 (1 h at 523 K in air); and fresh FASnBr3, aged FASnBr3 (5 days at RT in air), aged FASnBr3 (0.5 h at 523 K in air); and fresh CsSnBr3, aged CsSnBr3 (0.5 h at 623 K in air). The dashed vertical lines represent the combined spectra acquired at different transmitter offsets (blue and red). Adapted with permission from ref. 312. Copyright 2020 American Chemical Society. | ||
Ex situ 119Sn NMR has been used to probe the degradation products in tin based MHPs (Fig. 7b).312 For example, (hydro)thermal degradation of MASnBr3 (exposed to air for 1 h at 523 K) leads to the formation of MA2SnBr6, SnO2, SnBr4, and dilute concentrations of ionic species as a result of the reaction between tin(II) and decomposition products of the organic cation. By comparison, FASnBr3 exposed to air for 5 days at RT yields signals corresponding to SnO2 and FA2SnBr6, while still containing signals corresponding to undegraded FASnBr3 (∼45% of the pristine material). However, exposure at 523 K leads to a complete degradation of the perovskite phase. In the case of CsSnBr3, exposure to air for 0.5 h at 623 K leads to the formation of SnO2 and Cs2SnBr6. Similar trends have been observed for ternary tin(II) iodides (halostannates(II)), which degrade into different byproducts and traces of metallic β-Sn. There is a high degree of complementarity between the MHP degradation processes studied by different analytical techniques such as XRD, SEM, ssNMR and thermogravimetric analysis (TGA). While the crystalline inorganic degradation products are amenable to the XRD and SEM techniques, MHPs often degrade into disordered phases which are better identified by ssNMR techniques. For example, the degradation of tin-based perovskites into SnO2 and SnX4 has been studied by TGA.369 However, 119Sn NMR study refines this picture by identifying other intermediate products corresponding to tin(IV) halostannates, A2SnX6.312
Much interest has been directed towards ssNMR study of halides (19F, 35Cl, 79Br), which provides a suitable complementary method to explore halogen sites in MHPs. For instance, 19F NMR has been used to investigate local structures and halogen bonding in MHPs doped with fluorine-containing organic species.370,371 However, the scenario becomes complicated with NMR investigations of MHPs involving halides such as 35Cl, 79Br and 127I, due to the very large quadrupole interaction associated with these nuclei, which leads to relatively broad spectra for non-symmetric local environments. In the case of bulk and nanocrystalline CsPbX3 (X = Cl, Br, I), local halide sites have been characterized by 35Cl and 79Br ssNMR, and compared with 79Br and 127I NQR results.64,372 NQR is readily applicable to static samples with minimum restrictions on the material type, and is thus expected to help in the analysis of halide perovskites and the associated optoelectronic devices.
5.2. B-site deficient perovskites
The incorporation of large A-site cations in MHPs that exceed perovskite tolerance factors can produce crystalline phases with improved environmental stability and optoelectronic properties. Lead-deficient perovskites with periodically localized octahedral vacancies have recently been synthesized by adding hydroxyethylammonium (HEA+) or thioethylammonium (TEA+) to MAPbI3, FAPbI3 or FASnI3 precursors during growth.191–193 Analysis of 1D 207Pb NMR lineshapes and intensities provides an insight into the local environments of Pb2+ cations in 3D-like lead-deficient MAPI3 (d-MAPbI3). Specifically, 207Pb NMR signal intensities are relatively lower in d-MAPbI3 than in pristine MAPbI3,191 which corroborates the reduced lead content in Pb-deficient perovskites. The different distributions of large cations in the 3D lattice and their local chemical environments have been characterized by 13C and 1H ssNMR.191,192 While the reduced 207Pb signal intensity in d-MAPI3 indicates the presence of defects, 13C NMR spectral analysis of (MA)1−2.48x(HEA)3.48x[Pb1−xI3−x] (x = 0.1, 0.13, 0.16, 0.19 and 0.2) compounds showed that the 13C signal intensities of HEA+ increase as the x value increases, indicating the progressive incorporation of HEA cations into the perovskite framework.B-site deficient MHP phases can also be formed with randomly distributed pairs of large cations and octahedral voids (Fig. 8a).194 These phases are commonly described as hollow perovskites (Section 2). Although single crystals suitable for X-ray diffraction studies can be grown,194–198,200 the local environments of the large cations in hollow perovskites are exceedingly challenging to obtain. Spanopoulos et al. used 1D and 2D 1H MAS NMR to elucidate the local structures of organic cations in ethylene diammonium (en) incorporated FAPbBr3 hollow perovskites.373 The 1H signals corresponding to FA (7.5 and 8.1 ppm) and en (4.1 and 7.5 ppm) cations are distinguished when sufficient en is incorporated. The lineshape associated with the 1H signal at ∼4.1 ppm can be deconvoluted into broad and narrow features, which are attributable to en cations in the perovskite framework and phase segregated regions, respectively. This result demonstrates that en cations are not homogeneously distributed in hollow (en)FAPbBr3, at least at stoichiometries of 33% or higher. A quantitative 1H NMR analysis of 12%, 33% and 44% en FAPbBr3 reveals that up to ∼29% en can be incorporated into the perovskite framework (Fig. 8b), while en loadings greater than 30% leads to the formation an additional type of en defect in the crystal structure. In the case of 33% en and 41% en FAPbBr3, there is ∼5% and ∼12% aggregation of the total en cations, as depicted by the broad signals in the deconvoluted 1H NMR spectra (Fig. 8b, inset). This result is consistent with the changes in the bulk optoelectronic properties and 3D lattice expansion determined XRD as function of %en incorporation into FAPbI3, although diffraction and optoelectronic measurements alone would not have revealed this phase segregation of en.
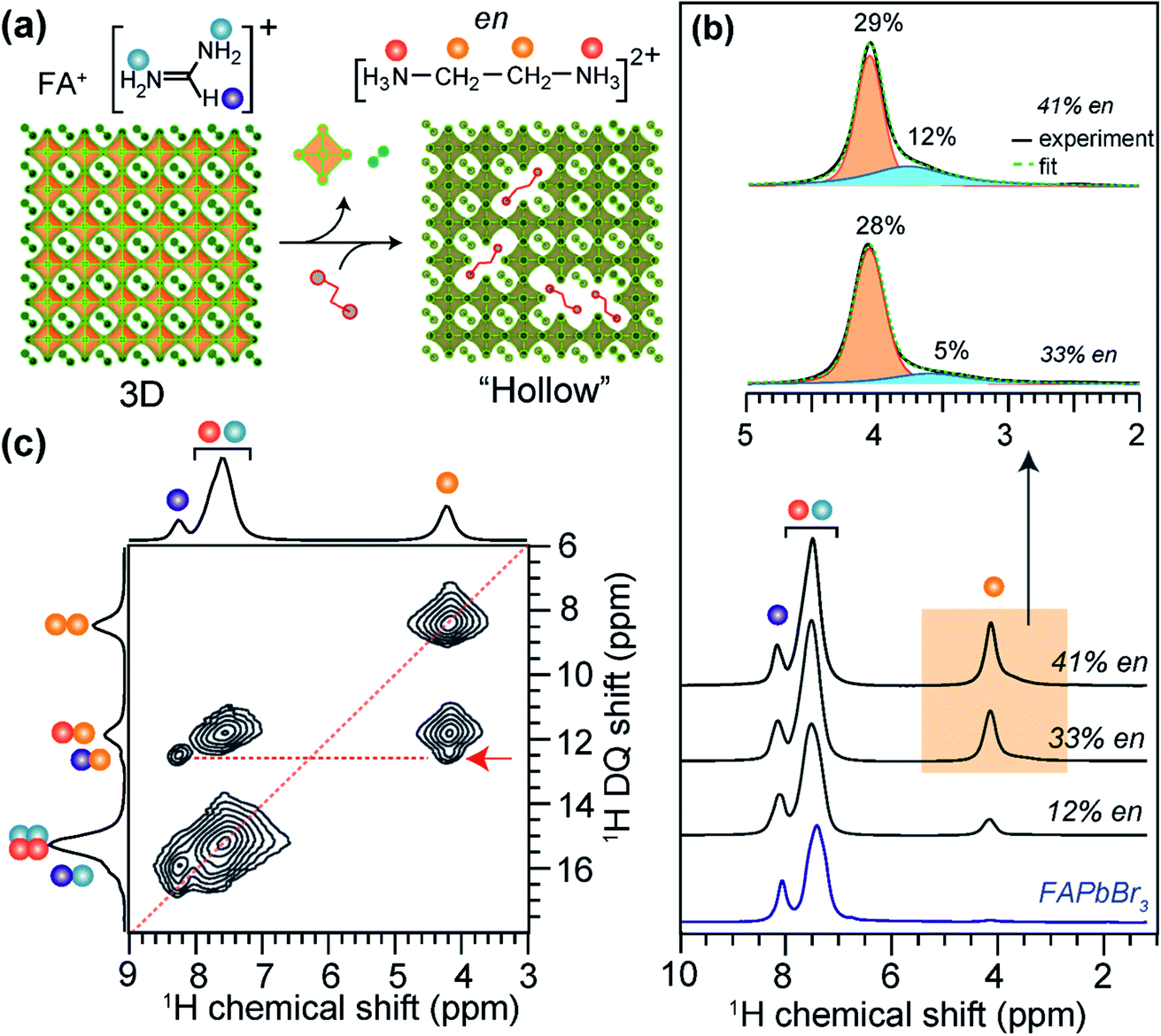 | ||
| Fig. 8 Large cation incorporation in hollow FAPbBr3: (a) schematic of the formation of hollow perovskites, (b) solid-state 1H MAS NMR spectra of FAPbBr3 and (en)FAPbBr3, (en content = 12%, 33% and 41% of available sites) hollow perovskites together with the assignment of signals corresponding to FA+ and en protons depicted in color dots as shown in the figure inset. (c) 2D 1H–1H DQ–SQ correlation NMR spectra of 41% en/FAPbBr3 exhibit the cross peaks (red arrow) indicating the sub-nm molecular proximity between FA+ and en. Adapted with permission from ref. 373. Copyright 2021 American Chemical Society. | ||
Insight into local chemical environment of organic cations in 41% en FAPbBr3 were obtained by analyzing 2D 1H–1H DQ–SQ correlation spectrum (Fig. 8c). The on-diagonal DQ signals associated with –CH2– groups of en (8.2 ppm) and ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– site of FA+ (15.6 ppm) are distinguishable, and a broad feature at ∼15 ppm is attributed to overlapped contributions from the –NH3+ groups in en and FA+. The resolved off-diagonal peak at 12.2 ppm (red arrow) indicates the intermolecular through-space 1H⋯1H proximity (<0.5 Å) between –CH2– groups of en and CH– group of FA+, confirming the incorporation of en cations into the perovskite framework. Such structural details obtained from ssNMR analysis are highly complementary to X-ray diffraction and pair distribution function (PDF) analysis of hollow perovskites.
CH– site of FA+ (15.6 ppm) are distinguishable, and a broad feature at ∼15 ppm is attributed to overlapped contributions from the –NH3+ groups in en and FA+. The resolved off-diagonal peak at 12.2 ppm (red arrow) indicates the intermolecular through-space 1H⋯1H proximity (<0.5 Å) between –CH2– groups of en and CH– group of FA+, confirming the incorporation of en cations into the perovskite framework. Such structural details obtained from ssNMR analysis are highly complementary to X-ray diffraction and pair distribution function (PDF) analysis of hollow perovskites.
5.3. 2D layered perovskites
There is a growing interest in the use of layered 2D perovskites as a strategy to develop MHP-based optoelectronic devices, owing to their outstanding stability and structural diversity. The emerging field of 2D perovskite-based optoelectronics has inspired research to develop a systematic understanding of the impact of organic spacer cations on the structure and optoelectronic properties of layered phases. ssNMR spectroscopy is a powerful characterization tool to understand the local molecular structures of spacer cations, and their interactions with A sites and X sites within the perovskite slabs.232,309,374,375 One notable example is the use of 1H–207Pb heteronuclear correlation NMR to gain insights into the octahedral structures of 2D BA2MAn−1PbnI3n+1 (n = 1 to n = 4, a value determining the thickness of the lead halide octahedral layer) Ruddlesden–Popper phases with butylammonium (BA) spacers (Fig. 9).374 These investigations by Lee et al. enabled layer-by-layer identification of 2D halide perovskites by n value, whereby the surface (“outer”) and bulk (“inner”) Pb2+ sites in the octahedral slabs could be distinguished and resolved.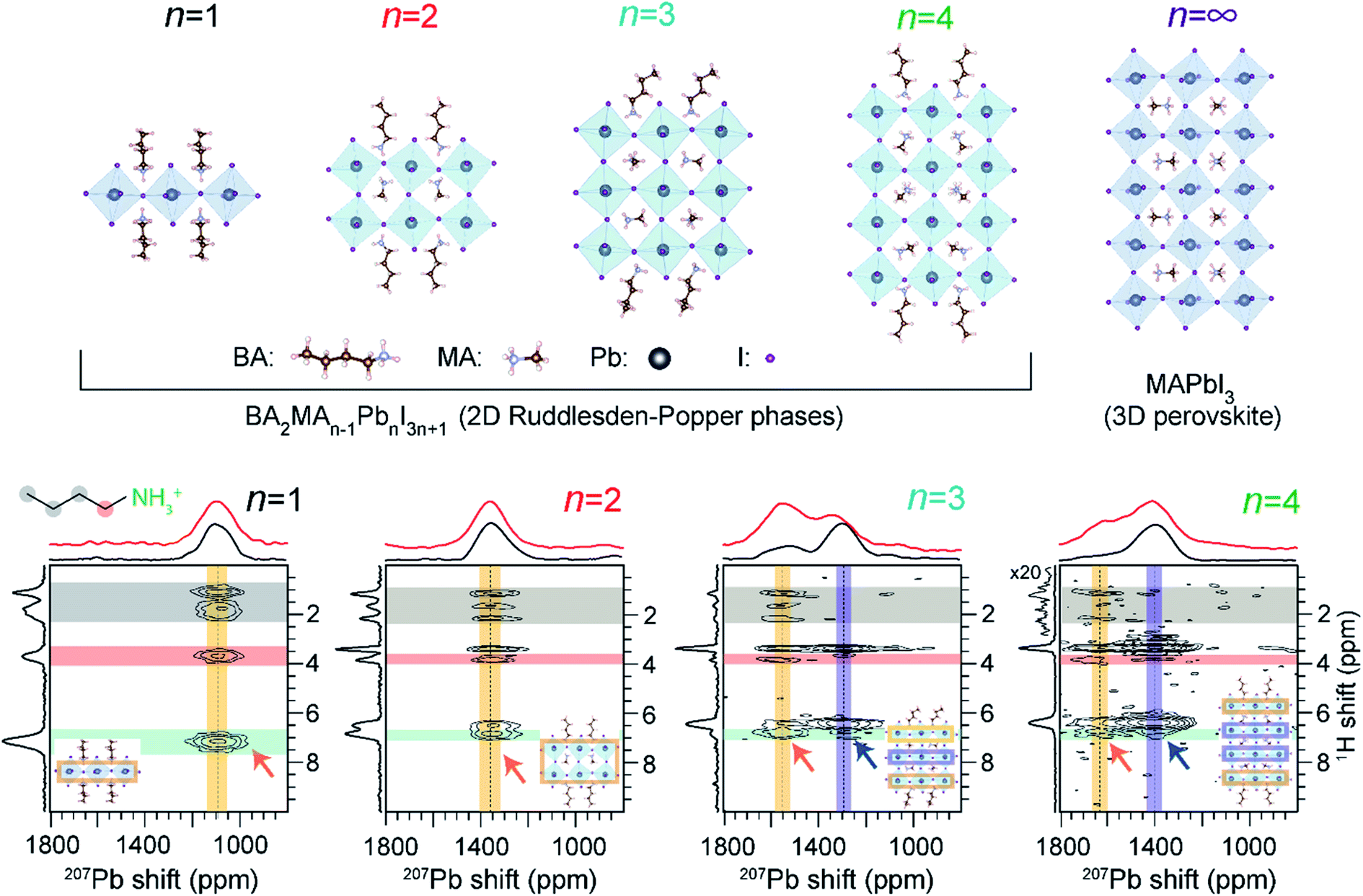 | ||
| Fig. 9 Layer-by-layer identification of 2D RP phases: 2D 1H-detected 207Pb–1H correlation NMR spectra of RP BA2MAn−1PbnI3n+1 materials acquired under fast (62.5 kHz) MAS with CP contact time of 4 ms. The top horizontal spectra compare the skyline 1D projections (black) and the 1D 207Pb direct polarization spectra (red). In each 2D spectrum, the vertical yellow and blue bands indicate the 207Pb signals corresponding to outer and inner Pb2+ sites, respectively, as depicted in the insets. Adapted with permission from ref. 374. Copyright 2021 American Chemical Society. | ||
The octahedral tilts associated with 3D-like “inner” PbX6 sites differ substantially from the surface-like “outer” PbX6 sites, which manifests in different isotropic 207Pb chemical shifts. A correlation between 207Pb NMR shifts and mean Pb–I bond lengths has been established. The intermolecular interactions between PbX6 and spacer cations are characterized by 2D 207Pb–1H correlation spectroscopy for different layered MHPs of varying n values (Fig. 9). For n = 1 and n = 2 RP phases, 2D correlation peaks occur between BA(–NH3) and MA (–NH3) protons (orange arrows) and Pb at a single 207Pb resonance. Thus, only a single Pb chemical environment is present in n = 1, 2 phases. This result also confirms that through-space correlations can be resolved between outer Pb nuclei and remote spacer protons, which may be up to 6 Å away from the octahedral slab. In the case of n = 3 and n = 4 RP phases, two distinct 207Pb–1H correlation peaks are observed, indicating through-space proximity between MA/BA protons and the “outer” Pb sites (depicted in orange arrows) and the peaks attributed to “inner” Pb sites (depicted in blue arrows). This ssNMR study confirmed that octahedral distortions in n = 3 and 4 RP phases lead to two distinct bonding environments for Pb in these materials, which are reflected in different 207Pb signals.
SsNMR spectroscopy has been employed to probe the molecular arrangement of spacer molecules in layered MHPs. The packing arrangement of different spacer cations in n = 1 lead iodide RP phases, including butylammonium {C4}, octylammonium {C8}, dodecylammonium {C12} and aromatic phenylethylammonium {PEA} cations, have been analyzed by multinuclear (1H, 13C and 15N) ssNMR spectroscopy.309 In these 2D MHPs, the 1H chemical shift (7.2 ppm) associated with the spacer cation NH3+ groups in the n = 1 RP phases is shifted to a higher frequency than the 1H chemical shift (6.3 ppm) of NH3+ groups in 3D MAPbI3, indicating stronger affinity interactions (e.g. H-bonding) between RP spacer cations and lead iodide octahedra. In addition, 2D 1H DQ–SQ spectra (Fig. 10) show signals corresponding to an interleaved arrangement of long alkyl spacer cations within the spacer region of {C4}2PbI4, {C8}2PbI4 and {C12}2PbI4. Specifically, the off-diagonal 2D peaks that are characteristic to the side chain interdigitation (red arrows) are observed in {C4}2PbI4, {C8}2PbI4. In the case of {C4}2PbI4, the 1H DQ signals at 1.8 + 3.7 = 5.5 ppm, 3.7 + 7.2 = 10.9 ppm, and at 1.1 + 7.2 = 8.3 ppm indicate the through-space proximities in CH2↔CH2, CH2↔NH3 and CH3↔NH3 pairs within the same spacer or between adjacent C4 chains. The closest CH3↔NH3 distance between adjacent molecules in the refined crystal structure of {C4}2PbI4 is ≈5 Å, similar to the end-to-end distance between NH3 and CH3 groups in a single {C4} molecule, indicating that the CH3↔NH3 off-diagonal peak (red arrow) is expected to show signal from both inter and intramolecular 1H–1H proximities.
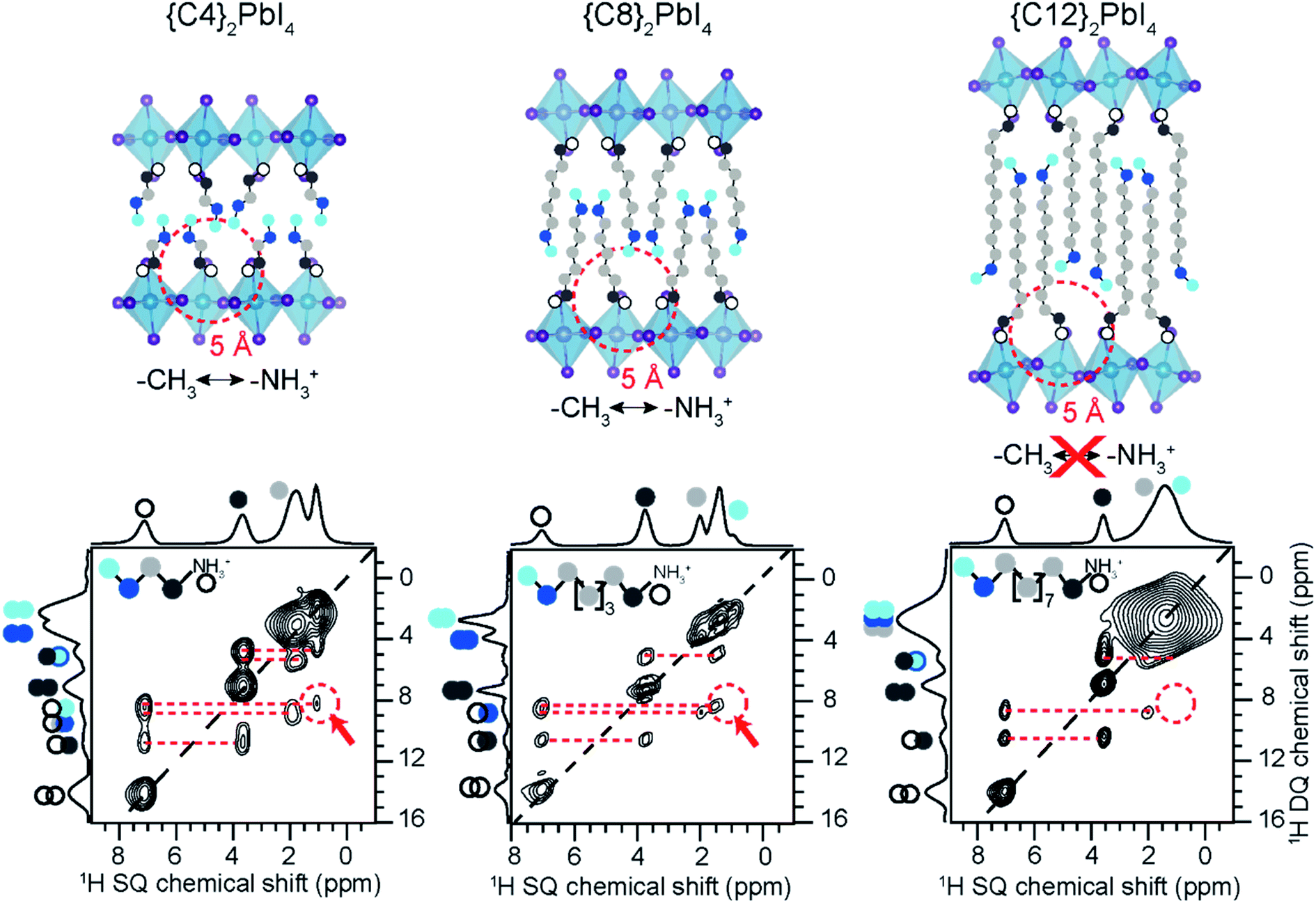 | ||
| Fig. 10 Spacer cation packing interactions in 2D RP phases: 2D 1H–1H DQ–SQ NMR correlation spectra acquired at 18.8 T and at 50 kHz MAS of {C4}2PbI4, {C8}2PbI4 and {C12}2PbI4. The corresponding skyline projections are shown along the top 1H SQ horizontal and left 1H DQ vertical axes, respectively. Correlated signal intensity originates from dipolar-coupled 1H–1H pairs and are depicted in colored circles. Adapted with permission from ref. 309. Copyright 2021 American Chemical Society. | ||
The longer spacer cations are too large to produce an off-diagonal peak corresponding to an intramolecular H–H proximity between the endgroups. The intrachain distance from CH3 to NH3 is ≈10 Å for {C8}2PbI4 and ≈15 Å for {C12}2PbI4 in the extended trans orientation (Fig. 10, top), which is too far for through-space correlations to show well-resolved features. The 2D 1H DQ–SQ NMR spectra of both {C8}2PbI4 and {C12}2PbI4 show intramolecular off-diagonal features between NH3 and nearby CH2 groups (dark and light grey dots), but no intermolecular correlations with terminal CH3 or CH2 groups (light blue, dark blue). However, the DQ features between CH2↔CH3 (dark grey to light blue) for long {C8} and {C12} spacers can only be caused by intermolecular correlations between the interleaved spacers. The nearest intermolecular CH2↔CH3 (dark grey to light blue) distance is 4.7 Å in the published crystal structures (Fig. 10, top), which is consistent with the DQ–SQ NMR analysis. Single crystal XRD analysis of light atoms in RP structures is often impractical, and 2D 1H DQ–SQ NMR data provides an alternative means to characterize the molecular arrangement of spacers in these phases.
Protocols that combine ssNMR spectroscopy, X-ray scattering and modelling approaches (i.e., NMR crystallography) have been employed to characterize the structures and intermolecular interactions in MHPs and other perovskite materials.232,376 Development of such integrated methods are important to understand the chemical nature of mixed spacer cations in layered MHPs at the atomic scale, which is a challenge for conventional diffraction-based techniques or ssNMR spectroscopy alone. Hope et al. applied an NMR crystallography approach to elucidate the supramolecular structure of the mixed cation organic spacers in layered hybrid perovskites.232 By using this approach, 3D structures of layered hybrid perovskites consisting of 2-phenylethylammonium (PEA+) and 2-(perfluorophenyl)ethylammonium (FEA+) moieties have been proposed. This study revealed that the spacer cations in a mixed arene–fluoroarene model system most closely resemble those of phase-segregated PEA+/FEA+ compositions, rather than a solid solution (Fig. 11). Nonetheless, it was found that there are local interfacial contacts between PEA+ and FEA+ in the spacer layer.
 | ||
| Fig. 11 Nanoscale phase segregation of organic cations in layered MHPs: (a) 1D 1H→13C (left) and 19F→13C (middle) CP-MAS, and 19F MAS (right) NMR spectra of the spacer cations together with the chemical structures of PEA+ and FEA+ cations. The top panels (filled circles) correspond to the spectra of layered MHPs and the lower half (open circles) correspond to the neat spacers and their blends (PF = 1:1, PEA+:FEA+), and the dashed boxes depict the 13C signals associated with PEA+ enhanced by intermolecular 19F→13C CP transfer from the FEA+. (b) Trial structures for DFT calculations and NMR crystallography of (spacer)2PbI4 2D layered perovskites consisting of twisted and parallel arrangements of spacer cations with interlayer spacing (d1) and top view of the spacer layer indicating the square lattice and octahedral tilting (φ). Bottom panel depicts schematic of different possible orientation of PEA+ (P) and FEA+ (F) moieties (1–5) on the two opposing lattices representing the spacer bilayer within the layered perovskite. (c) Correlation between the calculated aromatic 13C (top) and 19F (bottom) chemical shifts plotted against the experimental (PF)2PbI4 chemical shifts for the structures shown in (b). Adapted with permission from ref. 232. Copyright 2021 American Chemical Society. | ||
To understand how the spacer cation chemical environment changes upon formation of layered 2D perovskites, a suite of 1D 1H→13C, 19F→13C CP-MAS and 19F MAS spectra of layered 2D perovskites (Fig. 11a, filled circles), precursor salts and their blends (Fig. 11a, open circles) were analyzed and compared. The 13C and 19F signals associated with 2D perovskites (Fig. 11a, filled circles) are displaced and change intensities compared to the iodide salts (Fig. 11a, open circles). To establish a correlation between the 13C and 19F chemical shifts and the atomic-level structures, NMR chemical shielding calculations were carried out for a range of modelled structures.
Trial structures of n = 1 RP phases with pure PEA and FEA spacer cations were created based on previously published structures, and modelled using Molecular Dynamics (MD) simulations.232 For pure cation RP phases of (PEA)2PbI4 and (FEA)2PbI4 (Fig. 11b, top), structural models to imitate the nanoscale phase segregation of twisted or parallel packing arrangements were considered. For trial structures of pure (PEA)2PbI4 and (FEA)2PbI4 phases (Fig. 11b, top), the NMR shieldings were calculated using DFT (Density Functional Theory). In the case of (PEA)2PbI4, the DFT-calculated 13C chemical shifts of twisted structure agree better with the experimental 13C shifts than the parallel structure, which is consistent with the previously reported single crystal structure. In contrast, for (FEA)2PbI4, the DFT-calculated 13C and 19F chemical shifts of parallel structure are in better agreement with the experimental 13C and 19F chemical shifts. In the case of mixed-spacer (PF)2PbI4 (PF = PEA:FEA) formulation, putative structures with different packing arrangements, such as layered, checkerboard and striped (Fig. 11b, bottom), were examined.
A comparison between DFT calculated chemical shifts and the experimental 13C and 19F chemical shifts (Fig. 11c) for the trial structures of (PF)2PbI4 and the phase-segregated (PEA)2PbI4 and (FEA)2PbI4 structures reveal that segregated models are in better agreement. It is noteworthy that the hydrogen, carbon and fluorine atoms close to the heavy Pb and I atoms may require fully relativistic treatment to obtain a good correlation between DFT and experimental results, though it has been shown that these relativistic effects do not significantly influence the agreement between experimental and calculated 1H and 13C chemical shifts.377 The root-mean-square errors (RMSEs) for the 13C and 19F chemical shifts for the segregated models are less than the expected RMSE in DFT calculated chemical shifts for 13C (2.32 ppm) and 19F (1.98 ppm). It has been suggested that the layered hybrid perovskite structure comprises of segregated regions of PEA+ and FEA+ spacers with nanoscale domain sizes. This is further corroborated by comparing the 13C signals of PEA+ sites in the 19F→13C CP MAS spectra (Fig. 11a, middle column, dashed rectangles), which are enhanced by the 19F sites from FEA+ in both (PF)2PbI4 and physical mixture (PEA)2PbI4/(FEA)2PbI4, hence the two spacers in the (PF)2PbI4 phase are in close proximity despite the nanoscale domain segregation. Overall, this study demonstrates the potential of NMR crystallography to elucidate structures and organic–organic interfaces in low-dimensional MHPs.
5.4. Molecularly passivated MHPs
Electronic defects at the interface between the perovskite film and the transport layers, or between grains within the MHP layer, can be effectively passivated with ammonium salts to augment the stability and performance of MHPs.118 A wide verity of ammonium salts have been employed to passivate MHPs. ssNMR has been used to study how a few of these salts, including choline, ethylammonium (EA), imidazolium, guanidinium (G) and tetrapropylammonium iodide (TPA), can passivate defects in MHPs.375,378–382 One important question presented by passivated MHPs is how the passivating agents incorporate within the device. Do the passivating salts mix microscopically with MHP grains, form a layer at the surface of the MHP film, or segregate away from the MHP layer? Passivating salts are often chemically similar to A-site cations in MHPs, so NMR signals arising from passivating agents and MHP constituent ions can be difficult to deconvolute. Structure elucidation of passivating overlayers is also challenging because of their small thickness and dilute concentrations in MHP films and devices. Nevertheless, 2D ssNMR spectroscopy has been applied to investigate the local proximities and interactions between MHP surfaces and passivating agents, and to elucidate the microscopic mechanisms of surface passivation.375,378,379,381,382 Unlike the 2D 1H–1H DQ–SQ correlation experiment that probes through-space 1H–1H proximities within 5 Å, the 2D 1H–1H SD experiment extends the length scale from sub-nanometer to several nanometers, and is thus well-suited to gain insight into intermolecular interactions and passivation mechanism in surface-modified MHPs.Interfaces between MHPs and passivating cations in ethylammonium iodide (EAI)-treated (FA:Cs:MA)Pb(I:Br)3 films have been probed by ssNMR spectroscopy. In a study by Alharbi et al., 2D 1H–1H spin diffusion spectrum of an EAI-treated FA0.93Cs0.07PbI3 thin film has been analyzed to shed light on the through-space atomic-level contact between FA+ and EA+ in the passivated material.379 The off-diagonal 2D peaks between ethylammonium (EA+, 3.0–4.3 ppm) and FA+ (∼7.5 ppm) cations (Fig. 12a, dashed orange lines), indicate close through-space proximity between these two cations (i.e. between the passivating agent and MHP phase). This result implies that EA+ passivating salts are in intimate contact with A-site cations in FA0.93Cs0.07PbI3 films.
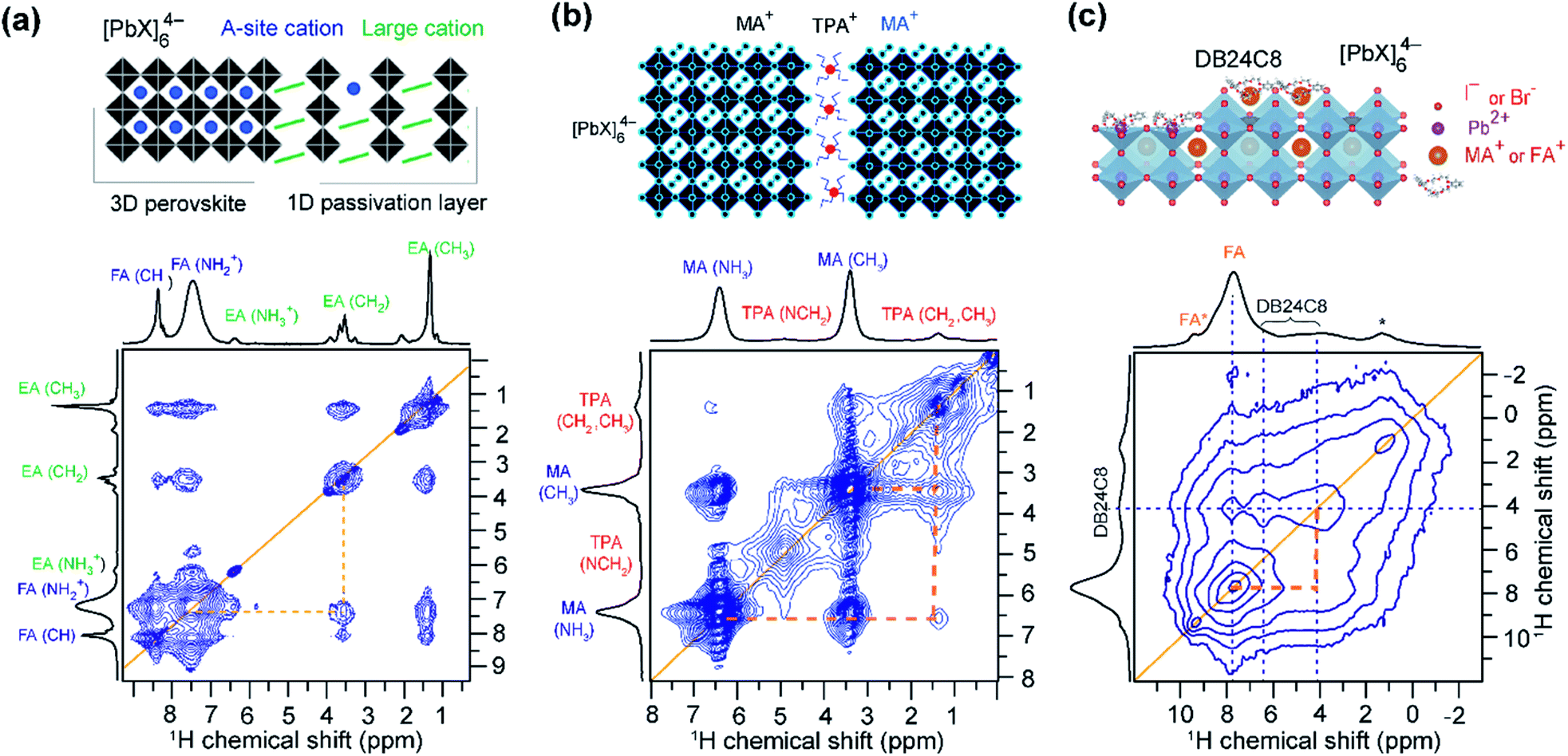 | ||
| Fig. 12 Elucidating interfacial structures in molecularly passivated MHPs: (a) 2D 1H–1H spin-diffusion spectrum (21.1 T, 20 kHz MAS, mixing time = 50 ms) evidencing atomic-level proximity between FA+ and EA+ in the FA0.9EA0.1PbI3, thin film treated with 5 mg ml−1 EAI (FA/EA yellow dotted lines). Adapted from ref. 379. (b) 2D 1H–1H spin-diffusion NMR spectrum of TPAxMA1−xPbI3 (x = 0.04) acquired at 18.8 T (35 kHz MAS, mixing time = 100 ms) shows cross-peaks corresponding to intra-molecular 1H–1H dipolar interactions between MA+ and TPA+ cations (yellow dotted lines). Adapted with permission from ref. 381. Copyright 2020, John Wiley and Sons. (c) 2D 1H–1H spin-diffusion NMR spectrum of crown ether (DB24C8) modulated FAPbI3 (11.7 T, 40 kHz MAS, mixing time = 10 ms) indicate the close proximity between DB24C8 and FA+ proton sites depicted in yellow dashed lines. The asterisk (*) indicates polypropylene from the ball-milling jar. The top panels depict the schematic illustration of proposed passivation mechanisms and potential interaction sites. Adapted with permission from ref. 382. Copyright 2020 American Chemical Society. | ||
Defect passivation using a Lewis-acid (tetrapropylammonium, TPA) has been employed to enhance the stability of MAPbI3-based single junction solar cells.381 Krishna et al. applied 2D ssNMR techniques to understand the molecular passivation mechanism in TPAxMA1−xPbI3 (x = 0.04) thin films. Isotropic chemical shifts of NCH2 (4.8 ppm) and CH2 (1.3 ppm) groups of TPA+ are distinguishable from the 1H signals of CH3 (3.3 ppm) and NH3 (6.3 ppm) groups of MA+. The close proximity between passivating TPA+ and MAPbI3 is confirmed by the analysis of 2D 1H–1H spin diffusion spectrum. The off-diagonal peaks corresponding to the intramolecular proximities between MA+ and TPA+ cations (Fig. 12b, dashed orange lines) were resolved, indicating that the TPA+ cations reside close to MAPbI3 A-site cations.
In a more recent study by Su et al., a combined ssNMR and DFT modelling has been used to understand the passivation mechanism of surface uncoordinated lead sites by a crown ether (DB24C8).382 The close proximity between MHP layers and the passivating agent (DB24C8) were elucidated by 2D 1H–207Pb and 1H–1H correlation NMR. These results are corroborated by DFT modelling of DB24C8 passivated perovskite structures. In particular, the analysis of a 2D 1H–1H spin diffusion spectrum revealed through-space proximities between the 1H sites in crown ether (∼4 ppm) and A-site FA+ cations (∼7.8 ppm) at the sub-surface perovskite layers (Fig. 12c, black dashed lines).
5.5. MHP nanoparticles
Halide perovskite nanocrystals (NCs) have gained significant attention as next-generation optoelectronic materials. However, their properties are highly dependent on the interface between the perovskite crystals and organic ligands. The interfacial structure of MHP NCs can be substantially different from the bulk, and lead to changes in phase stability, defect compensation and transport, and surface passivation effects.65,255,256 A variety of non-covalent bonding interactions can occur between MHP surfaces and capping ligands, leading to a corresponding diversity of structural and optoelectronic properties in MHP NCs. It is crucially important to understand how MHP interfacial chemistry changes upon treatment or exchange with different ligands. To this end, some fundamental questions pertaining to the surfaces of precipitated or assembled MHP NCs, such as atomic surface termination, binding interactions, and molecular structure of ligands at the surface can be addressed by ssNMR spectroscopy.64,65,372,383A recent study by Rossini and coworkers illustrates how ssNMR can probe the interfaces of precipitated ligand-capped CsPbBr3 NCs. MHP-ligand proximities and distances between ligands and terminal MHP atoms can be determined by analyzing dipolar couplings between dodecylammonium, oleate, and/or 10-undecenylphosphonate –NH3+ protons and the surface/subsurface Cs and Pb sites.383 ssNMR results are complemented by the simulation of multi-spin dipolar dephasing curves to provide an atomistic picture of NC termination. The results demonstrated that CsPbBr3 NCs are terminated by CsBr, rather than PbBr2, and that alkylammonium ligands substitute Cs+ at some surface sites. These results are corroborated by analyzing 2D 207Pb–1H and 133Cs–1H HETCOR spectra, as well as previous computational models of NC surfaces.258 The binding chemistry of alkylphosphonate ligands on CsPbBr3 nanocrystals has been probed by 31P–1H HETCOR analysis upon different surface and washing treatments.384,385 In addition, 207Pb NMR has been employed to characterize local 207Pb environments in MHP NCs.314,354 These studies revealed that 207Pb spectra of NCs exhibited much broader signals than the analogous 3D MHPs due to structural disorder at the NC interface. Solid-state DNP SENS has also been used to study the core and surface of MHP-inspired Cs2ZrCl6 NCs, including the composition of the surface ligands and exposed atomic sites.386 Such atomic-level insights into the MHP NC interfaces, precipitated solids or dispersed colloids, may enable rational design of new ligands and provide opportunities to develop molecular passivation strategies.
SsNMR studies of colloidal MHP NCs can build on the literature of solution-state NMR studies of dispersed MHP colloids. Colloidal nanocrystals exhibit rich solution phase chemistry, enabled by the broad range of interactions between ligands, solvent molecules and MHPs.274 Solution-phase NMR experiments have been used to determine how ligands interact and exchange at the surface of MHP NCs within a liquid environment.65,251,275,387–391 For example, solution 1H NMR studies have been used to quantify the thermodynamics of ligand binding to CsPbBr3 QDs, and revealed that both oleic acid and oleylamine ligands dynamically interact with CsPbBr3 QD surface.390 Pulse-Field-Gradient (PFG) based 1H diffusion ordered spectroscopy (DOSY) experiments have also been used to gain insight into the self-diffusion of surface bounded oleic-species and free ligands in solution.389 In this respect, high-resolution DOSY experiments are expected to aid the analysis of different diffusion behaviors of ligands at solid-solution interfaces.392,393
6. Dynamics at organic–inorganic interfaces in MHPs
Dynamic motion of molecules and ions at the interface between photoactive and contact layers, or at intrinsic interfaces within MHPs, influence the overall bulk optoelectronic properties in MHP-based devices. Although fast ion migration and charge carrier dynamics in MHPs at fs–ps time scales can be characterized by ultrafast spectroscopy, many relevant photophysical processes extend to slower timescales accessible by NMR (Fig. 3). The wide range of timescales (ns–s) associated with ssNMR spectroscopy has sparked interest in studying different dynamic processes in MHPs with site specificity.66,312 In MHPs, relaxation, lineshape analysis and exchange measured by ssNMR spectroscopy techniques reveal information on different dynamic processes associated with A, B and X sites in MHPs. For example, fast dynamics on the timescale of tens of ps to hundreds of ns can be studied by measuring T1 and T2 values. Dynamic processes and structural transformations that occur at much slower timescales, in the order of milliseconds to minutes (or slower), can be measured by carrying out 2D exchange spectroscopy and time-resolved experiments. The analysis of 2H quadrupole splittings of deuterated cations and 14N spectral lineshapes provides insights into the correlation times and modes of reorientation of A-site organic cations in MHPs. The above experiments are typically carried out as a function of temperature to determine the activation energies and correlation times associated with the cation reorientation, diffusion and ion migration, as well as to evaluate the homogeneous (order, rotational or translational mobility) and inhomogeneous (disorder) contributions to lineshapes, and as discussed in the below sections.The structure and, to a lesser extent, photophysical properties of high-performing MHP absorbers such as MAPbI3 are influenced by the dynamics of A-site reorientations.291,394,395 The different dynamic processes of A-site reorientations in MAPbX3 have been investigated by 1H, 2H, 13C, and 14N, 15N ssNMR spectroscopy,152,291,304,306,343,344 revealing the presence of various degrees of freedom at different temperatures. 14N NMR has been used in conjunction with DFT calculations to understand the symmetry and orientational dynamics of MA+ cations in MAPbI3.306 These trends have been extended to study the dynamic motion of other A-site cations such as MA+, FA+, DMA+ (dimethylammonium) and G+ (guanidinium) or inorganic monovalent cations in MHPs153,154,291,339,340 along with solid solutions of cations such as FA0.67MA0.33PbI3, DMA1−xMAxPbI3 and G0.25MA0.75PbI3.152,345,396 In another example, a broad 2H NMR signal envelope has been observed in a 1D non-perovskite G(d6)PbI3 indicating no rotational dynamics of the C–N bond of G, whereas narrow envelopes were observed for G in G(d6)0.25MA0.75PbI3 confirming the nearly isotropic reorientation of the G moiety in that material. The correlation times associated with the G reorientation are shorter than (18 ± 8) ps, which has been correlated with high charge carrier lifetimes and promising performance of this material in solar cells.345
The dynamic nature of both A-site cations and halides influences the local BX6 octahedral structure. For instance, ion hopping should manifest itself as the presence of exchange between 207Pb environments associated with different halide coordination. 2D 207Pb exchange spectroscopy (EXSY) does indeed confirm that the different Pb environments exchange their halides.355,360 An important practical consideration is the choice of the recycle delay to optimize the sensitivity in 207Pb NMR experiments. There are several mechanisms that drive T1 relaxation of 207Pb nuclei in MHPs, which depend on the experimental condition, structure and composition of the material. We first highlight a mechanism known as the MAS-induced heteronuclear longitudinal relaxation enhancement, which occurs in lead iodides and bromides, and leads to a dramatic reduction of T1 when the sample is spun (Fig. 13a). This behavior is related to the crossing between the energy levels of the spin-1/2 nucleus (207Pb) and the very fast relaxing quadrupolar spin (127I, 79/81Br) during sample rotation.397 The effect is absent in MAPbCl3. 207Pb sensitivity benefits substantially from spinning, which leads to a significant (2 orders of magnitude) decrease in the necessary recycle delay.
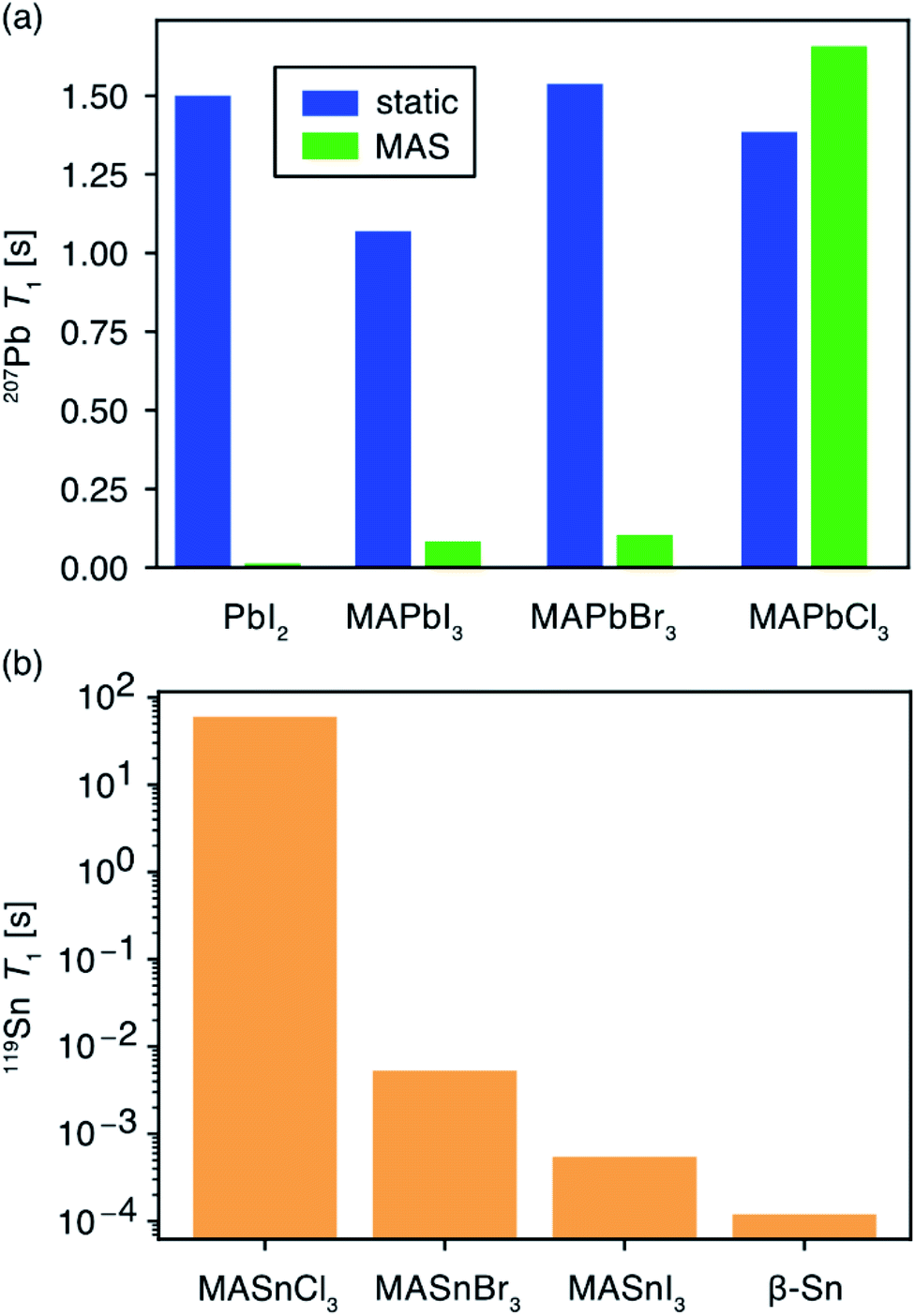 | ||
| Fig. 13 Longitudinal relaxation times in lead- and tin-based MHPs: (a) Comparison of spin-lattice (T1) relaxation rates of 207Pb nuclei in different MHPs. MAS-induced longitudinal relaxation enhancement is a process leading to a substantial shortening of 207Pb T1 when the sample is spun. (b) 119Sn T1 span 6 orders of magnitude and are strongly dependent on the halide composition. | ||
In tin-based MHPs, 119Sn ssNMR has been applied to probe the mixing of halides and local dynamics of [SnX6]4− sites.312,313 Longitudinal relaxation times (T1) of 119Sn sites in tin-based MHPs span 6 orders of magnitudes owing to the presence of different relaxation mechanisms depending on the halide composition. For example, in tin halides, the 119Sn T1 is determined by the strength of the 119Sn–X scalar couplings, which is as follows: 1JSn–I > 1JSn–Br > 1JSn–Cl.398,399 The presence of multiple composition-dependent relaxation mechanisms for 119Sn lead to a large spread of T1 values (Fig. 13b). For example, the dominating relaxation mechanism in MASnBr3 is scalar relaxation caused by the modulation of the 119Sn–79/81Br J-coupling. If this relaxation pathway is not effective, other mechanisms, such as CSA or dipolar, may be at play. In tin metal (β-Sn), the dominant sources of relaxation are the conduction electrons, leading to efficient Korringa relaxation.400 This variability highlights the importance of choosing the recycle delay judiciously, especially if quantitative spectra are required of materials containing multiple species with different relaxation characteristics.
Activation energies (Ea) associated with halide migration in MHPs have been measured by ssNMR spectroscopy and compared with other bulk techniques (Table 1). In MAPbI3, the activation energies of iodine migration are in the range of 0.17–0.44 eV as determined from ssNMR, NQR and electrical measurements. However, in the mixed-halide compositions MAPbI3−xClx, these values are in the range of 0.31–0.55 eV, indicating that in chloride-doped MAPbI3, iodine migration requires higher activation energies. By comparison, in tin-based perovskites the activation energy for bromide hopping in MASnBr3 is 0.37 eV (determined from variable-temperature 119Sn T1 relaxation at multiple magnetic fields), which is consistent with the a.c. and d.c. conductivity measurements as well as DFT calculations. Overall, ssNMR spectroscopy provides a wealth of information on the dynamic motion of organic cations and halides in MHPs.
| MHP | E a (eV) | Technique | Reference |
|---|---|---|---|
| MAPbI3 | 0.17 | ssNMR | 344 |
| 0.17 | 127I NQR | 344 | |
| 0.43 | Impedance | 401 | |
| 0.5 | Thermally stimulated current | 402 | |
| 0.44 | Ab initio/DFT | 403 | |
| 0.33 | Temperature dependent J–V curves | 404 | |
| 0.29 | Transient ion drift | 405 | |
| 0.44 | Ab initio simulations | 406 | |
| MAPbI3−xClx | 0.55 | Impedance and IMVS | 407 |
| 0.45 | Temperature dependent capacitance | 408 | |
| 0.31 | Temperature dependent current | 409 | |
| MASnBr3 | 0.44 | ssNMR (4.7 T) | 312 |
| 0.33 | ssNMR (9.4 T) | ||
| 0.35 | ssNMR (17.6 T) | ||
| 0.37 | ssNMR (average) | ||
| 0.30 | a.c. conductivity | 410 | |
| 0.31 | d.c. conductivity | 411 | |
| MASnI3 | 0.37 (VI), 0.65 (Ii) | DFT | 412 |
| CsPbCl3 | 0.27 | Diffusion/DFT | 413 |
| CsSnBr3 | 0.29 | 119Sn ssNMR (11.75 T) | 313 |
Low-dimensional MHPs exhibit different ion dynamics and reorientational modes than the 3D MHPs. For example, different trends in ion motion have been reported for 2D layered perovskites. Lin et al. employed ion conductivity measurements to characterize ion dynamics and showed that the ion motion is suppressed in low-dimensional perovskites compared to 3D perovskites, both in the dark and under illumination.414,415 Jiang et al. used Scanning Kelvin Probe Microscopy (SKPM) to study ion migration in layered 2D perovskites with varying dimensionality.416 For n-butylammonium lead iodide perovskites BA2PbI4 (n = 1), and methylammonium-incorporated quasi-2D perovskites (BA2MA3Pb4I13) 〈n〉 = 4, different ion motion was observed under the effects of illumination and temperature. Activation energies associated with ion motion in 2D perovskites studied by SKPM technique are compared in Table 2. This study proposes that ion motion in neat BA2PbI4 perovskite films is dominated by paired halide and halide vacancy, whereas in the case of quasi-2D BA2MA3Pb4I13 perovskites it can be viewed as a collective motion of both halide and methylammonium (vacancy) migration. This study indicates that the dimensionality plays a significant role in ion dynamics in layered perovskites.
| Material | E a (eV) | Technique |
|---|---|---|
| BA2PbI4 (n = 1) | 0.61 | SKPM, dark (negative potential) |
| 0.55 | SKPM, dark (positive potential) | |
| 0.37 | SKPM, light (negative potential) | |
| 0.30 | SKPM, light (positive potential) | |
| BA2MA3Pb4I13 (〈n〉 = 4) | 0.64 | SKPM, dark (negative potential) |
| 0.24 | SKPM, dark (positive potential) | |
| 0.39 | SKPM, light (negative potential) | |
| 0.25 | SKPM, light (positive potential) |
There is a tremendous diversity of organic spacer cations and packing interactions in layered and nanocrystalline MHPs, so it is less straightforward to probe molecular dynamics of spacer cations with atomic site specificity. The typical method of analyzing dynamics in MHPs is to perform temperature-dependent relaxation experiments (and/or 2H or 14N lineshape analysis). However, this approach becomes impractical in larger organic spacers with many different local environments and convoluted 1D NMR spectra. Organic spacer cations may also show glassy packing arrangements at elevated temperatures,417 further complicating interpretations of dynamics by NMR. Dynamics can instead be probed at different temperatures using 1D and 2D CP-MAS ssNMR methods. This approach has been used to understand spacer dynamics in some layered Ruddlesden–Popper phases. For instance, temperature-dependent measurements of T1 and line-broadening, using 1D 1H→13C CP-MAS spectra, distinguished activation energies of different reorientation modes of phenylethylammonium spacers in 2D lead iodide RP phases (n = 1) from 110 to 400 K.307,308 However, many MHPs, including PEA2PbI4, undergo phase transitions in the typical temperature range (∼100–400 K) of variable-temperature MAS ssNMR experiments.55,418 Therefore, other approaches that do not rely on large temperature variations can be helpful to understand cation dynamics in MHPs with narrow ranges of phase stability.
At room temperature, rotational and librational motions of spacer cations in RP phases typically occur on much faster timescales than CP transfer (microseconds to milliseconds). However, the CP transfer efficiency is affected by reorientation rates, so CP transfer kinetics can be used to compare molecular dynamics on the scale of μs to ms. In a study by Dahlman et al., the room temperature dynamic motion of different organic spacer cations in layered RP phases has been characterized by CP-MAS ssNMR techniques.309Fig. 14a compares the CP buildup rate for different carbon sites in a butylammonium {C4} RP n = 1 crystal, {C4}2PbI4 (an alternative notation for BA2PbI4). A comparison of 1H→13C CP build-up time constants (shown as TCP = 1/kCP, where k defines the rate at which CP signal intensity builds up) for different carbon sites of linear alkylammonium spacers is shown (Fig. 14b), alongside a complementary measure of site-specific disorder, Uiso, obtained from single-crystal X-ray diffraction refinements. The isotropic displacement parameter (Uiso) from single-crystal XRD measurements can help to visualize site-specific spatial disorder in spacer cations, which can be compared to the CP buildup rates of different carbon sites. A larger Uiso from XRD indicates greater atomic spatial disorder as the average of many X-ray scattering events (∼fs), either due to static disorder or dynamic fluctuations. CP build-up rates (kCP) probe dynamic motion on the scale of μs to ms, which may manifest as spatial disorder during X-ray scattering. Intramolecular trends in Uiso are consistent with CP build-up rates; for {C4}2PbI4 the CH2 groups near to NH3+ sites exhibit faster CP intensity buildup and smaller atomic displacement compared to carbon sites further away. Similar trends between CP time constants and Uiso values are observed for the RP phases with longer spacer cations, {C8}2PbI4 and {C8}2PbI4. The agreement between XRD and ssNMR indicates that Uiso values are determined by dynamic fluctuations that affect CP build-up, rather than static disorder. Thus, ssNMR provides complementary information to other characterization techniques to study dynamics and site-specific disorder, including X-ray diffraction and inelastic neutron scattering.
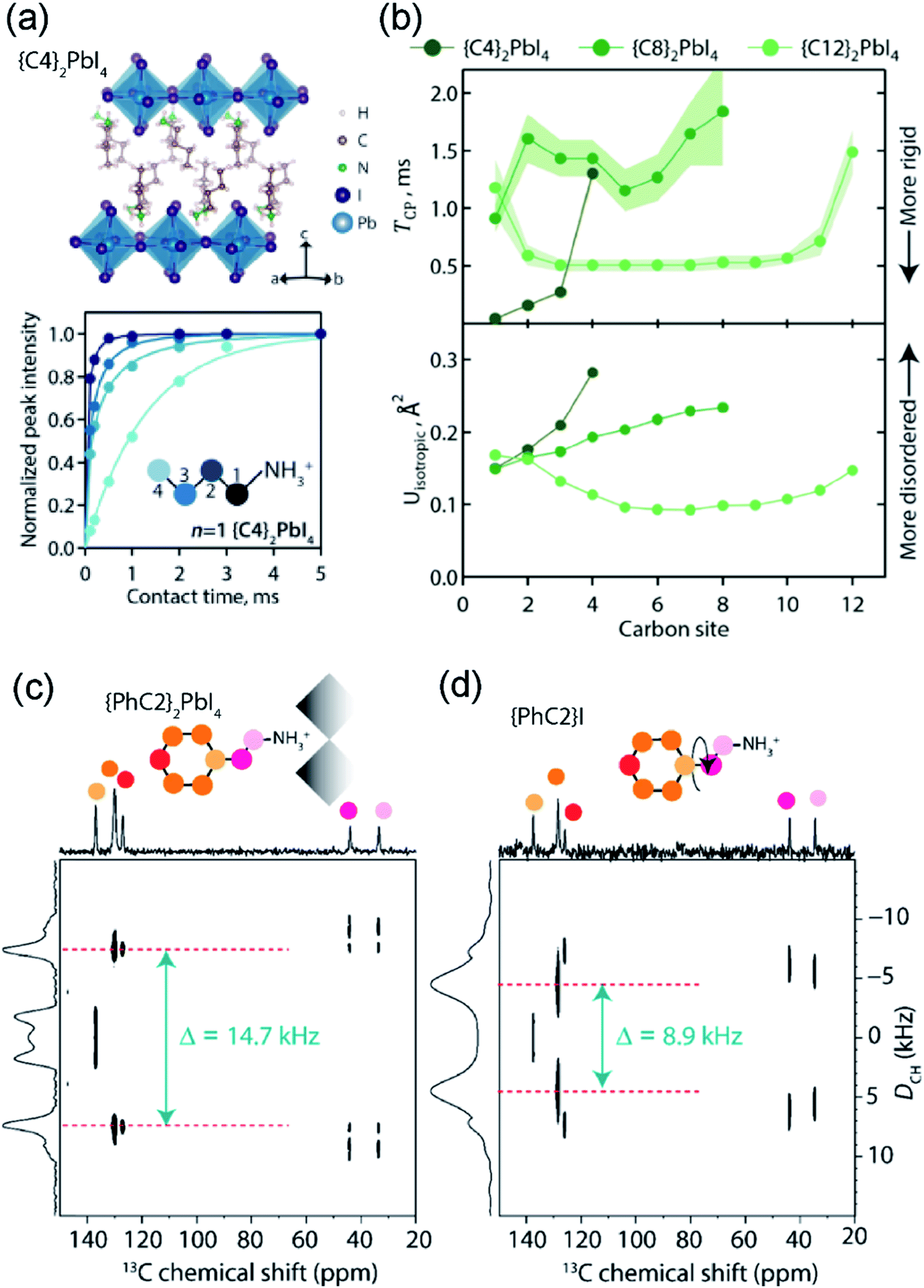 | ||
| Fig. 14 Dynamic motion of spacer cations in layered perovskites: (a) crystal structure of {C4}2PbI4 obtained from previously published XRD studies at 293 K with a projection along 〈110〉 of the orthorhombic Pbca unit cell, and 1H→ 13C CP-MAS NMR build-up curves (dots) for the four distinguishable carbon sites in {C4}2PbI4 spectra acquired at 9.4 T, 8 kHz MAS and at 300 K, accompanied by the monoexponential fitted CP signal intensity buildup (solid lines). (b) Site-specific 13C{1H} CP build-up time constants (top), TCP, for different carbon atoms in alkylammonium spacers in {C4}2PbI4, {C8}2PbI4 and {C12}2PbI4 compared with calculated isotropic displacement parameters (bottom) derived from room-temperature single-crystal XRD measurements and analyses. (c and d) Comparison of the solid-state 2D 13C{1H} CP-VC NMR spectrum acquired at 14.1 T for (a) the {PhC2}2PbI4 (n = 1) RP phase and (b) the {PhC2}I precursor salt. The separation of signals Δ = (DCH/√2) that leads to the estimation of 13C–1H dipole–dipole couplings of (c) 20.8 kHz and (d) 12.6 kHz. Adapted with permission from ref. 309. Copyright 2021 American Chemical Society. | ||
Aromatic spacers such as phenylethylammonium {PhC2} cations exhibit different dynamic motions in RP phases. For example, the phenyl ring rotation in {PhC2} is an important reorientation mode that can affect both the spacer dynamics and dielectric environment of RP phases. Dipolar-mediated 2D 13C{1H} cross-polarization variable contact (CP-VC) experiments can directly probe the room-temperature C2 rotational mode of {PhC2}.309Fig. 14c and d compares 2D CP-VC spectra of {PhC2} within the RP phase and iodide salt, which show that dipolar frequency splittings (inset arrows) for aromatic carbon sites are much higher in the RP phase, {PhC2}2PbI4 (DCH = √2 Δ = 20.8 kHz, close to a theoretically rigid CH bonds), than in the precursor iodide salt, {PhC2}I (DCH = √2 Δ = 12.6 kHz). This analysis indicates that templating by lead iodide octahedra makes phenylethylammonium cations more rigid. Analysis of CP build-up processes provide a useful probe of molecular dynamics at μs to ms timescales in low-dimensional MHPs without requiring temperature variations.
7. Contact layers
In a layer-by-layer device stack (Fig. 1) obtained by solution- and vapor-processing techniques, the MHP film forms an interface with the HTL and ETLs. Energy alignment of the valence and conduction band of the MHP and transport layers (or HOMO and LUMO levels) is essential to the performance of the device. The structural integrity and stability of transport layers is crucially important for device operation.43 The transport layers can also determine the ambient stability of MHP layers. Charge transfer between MHP photoactive layers and transport layers is sensitive to the atomic interface between the two. Significant gain in the MHPs device efficiency and stability can be obtained by tailoring the contact layers, chemically doping ETL and HTLs, and processing techniques.68–70,72–75,419–421 Many studies have attempted to determine the precise molecular structure of this interface. Organic semiconductors are commonly used as HTLs, either in pure form or doped with inorganic salts such as, for example, spiro-OMeTAD (2,20,7,70-tetrakis(N,N-di-p-methoxyphenylamine)9,90-spirobifluorene) doped with hydrophilic LiTFSI (lithium bis(trifluoromethylsulfonyl)-imide) or tert-butylpyridine.99,420 Synthetic modifications of organic HTLs, e.g. fluorinated analogues of spiro-OMeTAD,144 can also improve MHP device stability and performance. In addition, the formation of self-assembled monolayers (SAMs) at the interface between MHP and transport layers can improve the stability and performance of assembled devices.422,423The local structures of organic semiconductors, with and without molecular dopants, have been characterized by ssNMR spectroscopy. ssNMR studies have focused on providing insight into intermolecular interactions in pristine and doped conducting polymers.62,421,424–427 The roles of Li-TFSI doping on spiro-OMeTAD has been elucidated by 7Li MAS NMR spectroscopy.424 A comparison of 7Li NMR spectra showed different displacements of 7Li signals corresponding to neat Li-TFSI, doped Li-TFSI:spiro-OMeTAD, and the oxidized product of Li-TFSI:spiro-OMeTAD (upon exposure to air over 12 h). These findings indicate a doping mechanism that is triggered by consumption of the Li+ ions during device operation, which is troublesome because the concentration of Li+ must be maintained at approximately 20 mol% with respect to spiro-OMeTAD for optimal device performance. ssNMR spectroscopy has also offered insight into the molecular doping of other conjugated polymers that show promise as HTL materials, such as poly-3-hexylthiophene (P3HT) and PBTTT-C14 doped with 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ).425 Cochran et al., used combined 1D 19F and 2D 1H{13C} ssNMR analysis to provide crucial insights into the co-facial arrangement of F4TCNQ and PBTTT-C14, where charge transfer can be 100% efficient in the solid state.428 In a study by Yurash et al., the local structures and binding properties of small molecule Lewis acid dopants such as tris(pentafluorophenyl)borane (BCF) in doped organic semiconductors have also been characterized by 1D 11B, 13C{1H} and 13C{19F} CP-MAS and 2D 1H–19F and 1H–1H correlation ssNMR spectroscopy.62,421
The transport layers can also serve to prevent environmental pollution from MHP devices upon degradation. In a recent work by Lee et al., a HTL material was synthesized that can chelate Pb ions to prevent degradation and avoid lead leakage.429 The lead capturing ability of alkoxy–poly tetraethylene glycol was examined by solution 1H NMR spectroscopy as a function of the concentration of lead iodide in dimethylformamide. The 1H signals of TEG groups were displaced to lower ppm values as the concentration of lead ions increased, which can be used to estimate the Pb binding constants. This example, along with the studies of conjugated polymers described above, illustrate the important role that NMR can play to examine interfaces between MHPs and contact layers for high-performing devices.
8. Outlook
8.1. Materials design and interfacial engineering
The search for improved perovskite absorbers and light emitters is enticing. We envision that the characterization of interfacial structures, both between device layers and within MHP materials, will demand further scrutiny to improve performance in a variety of applications. The remarkable optoelectronic properties of MHPs lend them to applications in high-performance light-emitting devices such as LEDs and lasers, flexible and wearable electronics, and tandem solar cell architectures, and it is likely that we will see these areas rapidly develop in the immediate future.One of the key materials design challenges is to rationally control halide mixing and transport properties of existing and novel MHP phases. This problem has only recently been tackled in depth using electrical measurements,430–432 and ssNMR has the potential to provide unique insights into the atomic-level mechanism of ionic transport in new materials. ssNMR will continue to offer insight into synthesis, processing and device performance puzzles as research into MHPs extends to new materials, stability and performance benchmarks.
MHPs composed of mixed A-site, B-site and halide species will continue to pose characterization challenges that ssNMR spectroscopy is particularly well-suited to address. ssNMR has provided, in some cases, the only direct probe of clustering and local disorder in mixed-phase MHPs. Low-dimensional phases in particular present a compelling case for ssNMR applications, because minority phases, disordered or defective regions, or even glassy and amorphous sub-phases may dominate photophysical properties. The density of interfaces in these materials is also much higher than dense 3D MHP phases. Careful accounting of intrinsic interfacial structures and dynamics in low-dimensional MHPs may yield unexpected insights and engineering solutions. There is a wide range of ssNMR approaches that have not yet been attempted with these materials, and may prove lucrative to better understand hollow, layered, colloidal and quantum-confined MHP phases.
8.2. NMR methodology
Improvements in ssNMR resolution and detection limits are crucially important for the structural elucidation of mass-limited MHPs and contact layers. The vast majority of ssNMR experiments are carried out on bulk MHP materials and HTLs (microcrystalline samples of 1–100 mg). The main challenge of NMR spectroscopy is its low sensitivity, which inhibits the investigation of MHPs in device stacks, particularly when low-sensitivity nuclei are involved. Although sensitivity-enhanced methods have been demonstrated,314 the most versatile and promising method for enhancing sensitivity in NMR – DNP SENS – has yet to be optimized to yield large enhancements comparable to those achievable in other material solids.334 DNP experiments achieve their maximum sensitivity enhancements at cryogenic temperatures of 20–110 K. However, at cryogenic temperatures many MHPs undergo phase transitions and the heterogeneous interfaces manifest much broader linewidths with signals from MHPs and contact layers yielding to severe spectral overlap. DNP NMR protocols suitable for ambient/near-ambient temperatures might be sought to fully explore the potential of ssNMR spectroscopy for the investigation of device stacks. An efficient MAS DNP strategy for MHPs would allow routine investigation of MHP thin films, charge-selective transport layers, and extremely dilute surface passivation agents. Notably, it would also allow 2D correlation experiments between dilute and low-sensitivity nuclei, which are currently out of reach for the conventional NMR techniques.While ssNMR studies of nuclei of high importance to MHP research have been demonstrated (Fig. 4a), there remain pertinent nuclei that are notoriously challenging to detect, owing to their high quadrupole moment and low sensitivity. These include, for example, 17O, 33S, 35Cl, 49Ti and 67Zn. Compounds with these elements are frequency employed in different layers of device stacks. ssNMR of these nuclei will benefit from the application of the highest available magnetic fields,433,434 fast MAS,435 and the development r.f pulse sequences suitable for the direct and indirect detection of NMR nuclei436–438 in conjunction with sensitivity enhanced techniques.439 The hypothetical 2D experiments correlating NMR signals originating from A and X sites (e.g., X= Cl, Br, I), or X sites and contact layers, would be exceedingly challenging to obtain. In addition, 119Sn and 207Pb spectra of MHPs are characterized by very large chemical shift ranges and may feature substantial chemical shifts anisotropies, which make the necessary bandwidth challenging for conventional 1D experiments. The use of low magnetic fields, fast MAS, short high-powered adiabatic pulses (SHAPs) for refocusing440 as well as microcoils to increase the RF strength441 can be viewed as viable strategies to resolve some of these issues.
8.3. Extending NMR length scales
While ssNMR is particularly well suited to address short-range structural and dynamics problems in MHPs, full potential of the technique for the study of MHP-HTL and MHP-ETL contacts and other intrinsic and extrinsic interfaces in MHP-based devices is yet to be explored. For example, there is an increasing interest in mapping ion diffusion at interfaces, understanding degradation processes at MHP/contact layer interfaces, and probing the chemistry of low-dimensional perovskites. Each of these challenges will benefit from the application of ssNMR spectroscopy. ssNMR techniques that allow structure elucidation beyond a nanometer distance are expected to be suitable for the investigation of layers and interfaces in device stacks. In addition, approaches that combine experiment and modelling such as NMR crystallography and modelling of CP or SD build-up curves, are expected to provide unique insight into low-dimensional MHPs, most notably at interfaces. There are many areas where it can provide unique structural insights, and may require additional method development. For example, the assignment of experimental NMR spectra of MHPs386 often requires fully relativistic first principles calculations that include spin orbit coupling. Inspiration can be taken from prior literature combining ssNMR experiments and modelling of material solids.442–445 In addition, the development of PFG-based NMR methods would help in understanding the diffusion properties of small molecules and ions in MHPs, nanocrystals, across grain boundaries and contact layers.8.4. In situ characterization
Solid-state NMR is also expected to play an increasing role in in situ characterization of materials degradation in the presence of light, temperature and moisture. For such applications to be realized, ssNMR probes must be integrated with in situ photo-illumination, moisture and temperature controls. We expect that existing ssNMR protocols can operate in parallel with new developments of in situ and in operando approaches. Kinetics of crystallization, additive engineering and degradation reactions in MHP-based devices can be probed by ssNMR techniques in conjunction with other characterization tools to reach consensus on the environmental stability of MHPs and contact layers. In mixed dimensional MHPs, we envision that in situ ssNMR spectroscopy may be leveraged in conjunction with other complementary techniques to build more complete understanding of layer-by-layer structural evolution, formation kinetics, stability and properties.In situ and in operando studies of MHPs may be uniquely enabled by NQR techniques. NQR spectroscopy can be further developed towards a broader range of MHP compositions and sample preparations, including single crystals, thin films and device stacks. Improved NQR methodology and wider availability of NQR spectrometers will complement insights obtained from ssNMR techniques. The short experiment times accessible with NQR are promising for in situ and operando studies of molecular events occurring on relatively short timescales.
8.5. Data repository
Data mining and machine leaning (ML) techniques are among the most interesting modern tools of materials discovery. Harnessing the power of such protocols for MHP research requires accurate, well-categorized and available experimental data. While databases for crystal structures (Cambridge Structural Database-CSD;446 Inorganic Crystal Structure Database-ICSD),447 and structure prediction and modelling (The Materials Project),448 are well-established, we hope that ssNMR databases may be built and integrated to accelerate MHP research. Data classes for different MHP compositions including spectra with labeled chemical shifts, T1 and T2 relaxation rates, distance, orientation and ambient stability constraints, and even the activation energies of ion migration and molecular reorientations would be tremendously valuable for data-driven MHP discovery. In addition, raw ssNMR data can be deposited in different open-source formats (e.g., ASCII format, .fid and .ser formats generated by TopSpin software) for future data analysis.Author contributions
The manuscript was written through the contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors thank Dr Douglas Fabini for helpful discussion and review of the manuscript. We thank J. Lee, S. K. Lee and Vlad Michaelis for providing us with figures. G. N. M. R gratefully acknowledges the financial support University of Lille, UCCS laboratory, the IR-RMN-THC FR-3050 CNRS France for conducting some of the ssNMR experiments and results discussed in this review. G. N. M. R. gratefully acknowledges the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant no. 795091.References
- H.-R. Wenk and A. Bulakh, Minerals: Their Constitution and Origin, Cambridge University Press, 2nd edn, 2016 Search PubMed.
- W. Li, Z. Wang, F. Deschler, S. Gao, R. H. Friend and A. K. Cheetham, Nat. Rev. Mater., 2017, 2, 1–18 Search PubMed.
- M. D. Smith, B. A. Connor and H. I. Karunadasa, Chem. Rev., 2019, 119, 3104–3139 CrossRef CAS PubMed.
- L. Mao, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 141, 1171–1190 CrossRef PubMed.
- X. Li, J. M. Hoffman and M. G. Kanatzidis, Chem. Rev., 2021, 121, 2230–2291 CrossRef.
- G. Grancini and M. K. Nazeeruddin, Nat. Rev. Mater., 2019, 4, 4 CrossRef.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef PubMed.
- F. Zhang, H. Lu, J. Tong, J. J. Berry, M. C. Beard and K. Zhu, Energy Environ. Sci., 2020, 13, 1154–1186 RSC.
- J. Y. Kim, J.-W. Lee, H. S. Jung, H. Shin and N.-G. Park, Chem. Rev., 2020, 120, 7867–7918 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef.
- M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–647 CrossRef PubMed.
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 CrossRef.
- NREL, Best Research-Cell Efficiency Chart, https://www.nrel.gov/pv/cell-efficiency.html, (accessed April 6, 2021) Search PubMed.
- M. Green, E. Dunlop, J. Hohl-Ebinger, M. Yoshita, N. Kopidakis and X. Hao, Prog. Photovoltaics, 2021, 29, 3–15 Search PubMed.
- X.-K. Liu, W. Xu, S. Bai, Y. Jin, J. Wang, R. H. Friend and F. Gao, Nat. Mater., 2021, 20, 10–21 CrossRef PubMed.
- Z.-K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef PubMed.
- S. D. Stranks and H. J. Snaith, Nat. Nanotechnol., 2015, 10, 391–402 CrossRef.
- Y. Cao, N. Wang, H. Tian, J. Guo, Y. Wei, H. Chen, Y. Miao, W. Zou, K. Pan, Y. He, H. Cao, Y. Ke, M. Xu, Y. Wang, M. Yang, K. Du, Z. Fu, D. Kong, D. Dai, Y. Jin, G. Li, H. Li, Q. Peng, J. Wang and W. Huang, Nature, 2018, 562, 249–253 CrossRef PubMed.
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang, C. Yan, W. Li, X. Liu, Y. Lu, J. Kirman, E. H. Sargent, Q. Xiong and Z. Wei, Nature, 2018, 562, 245–248 CrossRef PubMed.
- T. Chiba, Y. Hayashi, H. Ebe, K. Hoshi, J. Sato, S. Sato, Y.-J. Pu, S. Ohisa and J. Kido, Nat. Photonics, 2018, 12, 681–687 CrossRef CAS.
- B. Zhao, S. Bai, V. Kim, R. Lamboll, R. Shivanna, F. Auras, J. M. Richter, L. Yang, L. Dai, M. Alsari, X.-J. She, L. Liang, J. Zhang, S. Lilliu, P. Gao, H. J. Snaith, J. Wang, N. C. Greenham, R. H. Friend and D. Di, Nat. Photonics, 2018, 12, 783–789 CrossRef CAS.
- W. Xu, Q. Hu, S. Bai, C. Bao, Y. Miao, Z. Yuan, T. Borzda, A. J. Barker, E. Tyukalova, Z. Hu, M. Kawecki, H. Wang, Z. Yan, X. Liu, X. Shi, K. Uvdal, M. Fahlman, W. Zhang, M. Duchamp, J.-M. Liu, A. Petrozza, J. Wang, L.-M. Liu, W. Huang and F. Gao, Nat. Photonics, 2019, 13, 418–424 CrossRef CAS.
- Q. Wang, X. Wang, Z. Yang, N. Zhou, Y. Deng, J. Zhao, X. Xiao, P. Rudd, A. Moran, Y. Yan and J. Huang, Nat. Commun., 2019, 10, 5633 CrossRef CAS.
- Y. Wei, Z. Cheng and J. Lin, Chem. Soc. Rev., 2019, 48, 310–350 RSC.
- Z. He, C. Zhang, Y. Dong and S.-T. Wu, Crystals, 2019, 9, 59 CrossRef.
- H. Dong, C. Zhang, X. Liu, J. Yao and Y. Sheng Zhao, Chem. Soc. Rev., 2020, 49, 951–982 RSC.
- G. Xing, N. Mathews, S. S. Lim, N. Yantara, X. Liu, D. Sabba, M. Grätzel, S. Mhaisalkar and T. C. Sum, Nat. Mater., 2014, 13, 476–480 CrossRef CAS PubMed.
- Y. Jia, R. A. Kerner, A. J. Grede, B. P. Rand and N. C. Giebink, Nat. Photonics, 2017, 11, 784–788 CrossRef CAS.
- T. J. S. Evans, A. Schlaus, Y. Fu, X. Zhong, T. L. Atallah, M. S. Spencer, L. E. Brus, S. Jin and X.-Y. Zhu, Adv. Opt. Mater., 2018, 6, 1700982 CrossRef.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS PubMed.
- H. Wang and D. H. Kim, Chem. Soc. Rev., 2017, 46, 5204–5236 RSC.
- L. Li, S. Ye, J. Qu, F. Zhou, J. Song and G. Shen, Small, 2021, 2005606 CrossRef , early view..
- X. Wang, M. Li, B. Zhang, H. Wang, Y. Zhao and B. Wang, Org. Electron., 2018, 52, 172–183 CrossRef.
- M. Ahmadi, T. Wu and B. Hu, Adv. Mater., 2017, 29, 1605242 CrossRef.
- F. P. García de Arquer, A. Armin, P. Meredith and E. H. Sargent, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- H. Wei and J. Huang, Nat. Commun., 2019, 10, 1066 CrossRef.
- Y. C. Kim, K. H. Kim, D.-Y. Son, D.-N. Jeong, J.-Y. Seo, Y. S. Choi, I. T. Han, S. Y. Lee and N.-G. Park, Nature, 2017, 550, 87–91 CrossRef PubMed.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef PubMed.
- Y. He, L. Matei, H. J. Jung, K. M. McCall, M. Chen, C. C. Stoumpos, Z. Liu, J. A. Peters, D. Y. Chung, B. W. Wessels, M. R. Wasielewski, V. P. Dravid, A. Burger and M. G. Kanatzidis, Nat. Commun., 2018, 9, 1609 CrossRef PubMed.
- Y. Zhou, J. Chen, O. M. Bakr and O. F. Mohammed, ACS Energy Lett., 2021, 6, 739–768 CrossRef.
- H. Utzat, W. Sun, A. E. K. Kaplan, F. Krieg, M. Ginterseder, B. Spokoyny, N. D. Klein, K. E. Shulenberger, C. F. Perkinson, M. V. Kovalenko and M. G. Bawendi, Science, 2019, 363, 1068–1072 CrossRef.
- G. Rainò, M. A. Becker, M. I. Bodnarchuk, R. F. Mahrt, M. V. Kovalenko and T. Stöferle, Nature, 2018, 563, 671–675 CrossRef.
- P. Schulz, D. Cahen and A. Kahn, Chem. Rev., 2019, 119, 3349–3417 CrossRef.
- E. M. Tennyson, T. A. S. Doherty and S. D. Stranks, Nat. Rev. Mater., 2019, 4, 573–587 CrossRef.
- L. M. Herz, ACS Energy Lett., 2017, 2, 1539–1548 CrossRef.
- I. A. Howard, T. Abzieher, I. M. Hossain, H. Eggers, F. Schackmar, S. Ternes, B. S. Richards, U. Lemmer and U. W. Paetzold, Adv. Mater., 2019, 31, 1806702 CrossRef PubMed.
- C. Liu, Y.-B. Cheng and Z. Ge, Chem. Soc. Rev., 2020, 49, 1653–1687 RSC.
- Z. Li, T. R. Klein, D. H. Kim, M. Yang, J. J. Berry, M. F. A. M. van Hest and K. Zhu, Nat. Rev. Mater., 2018, 3, 1–20 CrossRef.
- L. Qiu, S. He, L. K. Ono, S. Liu and Y. Qi, ACS Energy Lett., 2019, 4, 2147–2167 CrossRef.
- F. Huang, M. Li, P. Siffalovic, G. Cao and J. Tian, Energy Environ. Sci., 2019, 12, 518–549 RSC.
- J. Yuan, A. Hazarika, Q. Zhao, X. Ling, T. Moot, W. Ma and J. M. Luther, Joule, 2020, 4, 1160–1185 CrossRef.
- D. Prochowicz, M. Saski, P. Yadav, M. Grätzel and J. Lewiński, Acc. Chem. Res., 2019, 52, 3233–3243 CrossRef PubMed.
- B. A. Rosales, L. Wei and J. Vela, J. Solid State Chem., 2019, 271, 206–215 CrossRef.
- J. A. Raiford, S. T. Oyakhire and S. F. Bent, Energy Environ. Sci., 2020, 13, 1997–2023 RSC.
- A. Alaei, A. Circelli, Y. Yuan, Y. Yang and S. S. Lee, Mater. Adv., 2021, 2, 47–63 RSC.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394 CrossRef CAS.
- Y. Yang and J. You, Nature, 2017, 544, 155–156 CrossRef CAS.
- T. Leijtens, K. Bush, R. Cheacharoen, R. Beal, A. Bowring and M. D. McGehee, J. Mater. Chem. A, 2017, 5, 11483–11500 RSC.
- A. Walsh and S. D. Stranks, ACS Energy Lett., 2018, 3, 1983–1990 CrossRef CAS.
- L. M. Herz, J. Phys. Chem. Lett., 2018, 9, 6853–6863 CrossRef CAS.
- Y. Yuan and J. Huang, Acc. Chem. Res., 2016, 49, 286–293 CrossRef CAS.
- M. Seifrid, G. N. M. Reddy, B. F. Chmelka and G. C. Bazan, Nat. Rev. Mater., 2020, 5, 910–930 CrossRef.
- B. Reif, S. E. Ashbrook, L. Emsley and M. Hong, Nat. Rev. Dis. Primers, 2021, 1, 1–23 CrossRef.
- L. Piveteau, V. Morad and M. V. Kovalenko, J. Am. Chem. Soc., 2020, 142, 19413–19437 CrossRef CAS PubMed.
- S. R. Smock, Y. Chen, A. J. Rossini and R. L. Brutchey, Acc. Chem. Res., 2021, 54, 707–718 CrossRef CAS.
- I. L. Moudrakovski, in Annual Reports on NMR Spectroscopy, Elsevier, 2021, vol. 102 Search PubMed.
- W. M. J. Franssen and A. P. M. Kentgens, Solid State Nucl. Magn. Reson., 2019, 100, 36–44 CrossRef CAS.
- K. Wang, S. Olthof, W. S. Subhani, X. Jiang, Y. Cao, L. Duan, H. Wang, M. Du and S. Liu, Nano Energy, 2020, 68, 104289 CrossRef CAS.
- X. Sun, X. Yu and Z. Li, ACS Appl. Energy Mater., 2020, 3, 10282–10302 CrossRef CAS.
- X. Zhao and M. Wang, Mater. Today Energy, 2018, 7, 208–220 CrossRef.
- G.-W. Kim, H. Choi, M. Kim, J. Lee, S. Y. Son and T. Park, Adv. Energy Mater., 2020, 10, 1903403 CrossRef CAS.
- K. Mahmood, S. Sarwar and M. Taqi Mehran, RSC Adv., 2017, 7, 17044–17062 RSC.
- H. Duc Pham, L. Xianqiang, W. Li, S. Manzhos, A. K. Ko Kyaw and P. Sonar, Energy Environ. Sci., 2019, 12, 1177–1209 RSC.
- A. Krishna and A. C. Grimsdale, J. Mater. Chem. A, 2017, 5, 16446–16466 RSC.
- L. Hu, M. Li, K. Yang, Z. Xiong, B. Yang, M. Wang, X. Tang, Z. Zang, X. Liu, B. Li, Z. Xiao, S. Lu, H. Gong, J. Ouyang and K. Sun, J. Mater. Chem. A, 2018, 6, 16583–16589 RSC.
- Arramel, H. Pan, A. Xie, S. Hou, X. Yin, C. S. Tang, N. T. Hoa, M. D. Birowosuto, H. Wang, C. Dang, A. Rusydi, A. T. S. Wee and J. Wu, Nano Res., 2019, 12, 77–84 CrossRef CAS.
- E. E. Perry, J. G. Labram, N. R. Venkatesan, H. Nakayama and M. L. Chabinyc, Adv. Electron. Mater., 2018, 4, 1800087 CrossRef.
- J. Euvrard, Y. Yan and D. B. Mitzi, Nat. Rev. Mater., 2021, 1–19 Search PubMed.
- N. K. Noel, S. N. Habisreutinger, A. Pellaroque, F. Pulvirenti, B. Wenger, F. Zhang, Y.-H. Lin, O. G. Reid, J. Leisen, Y. Zhang, S. Barlow, S. R. Marder, A. Kahn, H. J. Snaith, C. B. Arnold and B. P. Rand, Energy Environ. Sci., 2019, 12, 3063–3073 RSC.
- Q. Chen, J. Wu, X. Ou, B. Huang, J. Almutlaq, A. A. Zhumekenov, X. Guan, S. Han, L. Liang, Z. Yi, J. Li, X. Xie, Y. Wang, Y. Li, D. Fan, D. B. L. Teh, A. H. All, O. F. Mohammed, O. M. Bakr, T. Wu, M. Bettinelli, H. Yang, W. Huang and X. Liu, Nature, 2018, 561, 88–93 CrossRef.
- J. Yoon, H. Sung, G. Lee, W. Cho, N. Ahn, H. Suk Jung and M. Choi, Energy Environ. Sci., 2017, 10, 337–345 RSC.
- E. Köhnen, M. Jošt, A. B. Morales-Vilches, P. Tockhorn, A. Al-Ashouri, B. Macco, L. Kegelmann, L. Korte, B. Rech, R. Schlatmann, B. Stannowski and S. Albrecht, Sustainable Energy Fuels, 2019, 3, 1995–2005 RSC.
- J. Zhang, W. Zhang, H.-M. Cheng and S. R. P. Silva, Mater. Today, 2020, 39, 66–88 CrossRef.
- H. S. Jung, G. S. Han, N.-G. Park and M. J. Ko, Joule, 2019, 3, 1850–1880 CrossRef.
- D. Yang, R. Yang, S. Priya and S. Liu, Angew. Chem., Int. Ed., 2019, 58, 4466–4483 CrossRef CAS PubMed.
- Y.-S. Jung, K. Hwang, Y.-J. Heo, J.-E. Kim, D. Vak and D.-Y. Kim, Adv. Opt. Mater., 2018, 6, 1701182 CrossRef.
- T. W. Jones, A. Osherov, M. Alsari, M. Sponseller, B. C. Duck, Y.-K. Jung, C. Settens, F. Niroui, R. Brenes, C. V. Stan, Y. Li, M. Abdi-Jalebi, N. Tamura, J. E. Macdonald, M. Burghammer, R. H. Friend, V. Bulović, A. Walsh, G. J. Wilson, S. Lilliu and S. D. Stranks, Energy Environ. Sci., 2019, 12, 596–606 RSC.
- C. Wang, L. Ma, D. Guo, X. Zhao, Z. Zhou, D. Lin, F. Zhang, W. Zhao, J. Zhang and Z. Nie, J. Mater. Chem. C, 2020, 8, 3374–3379 RSC.
- N. Rolston, K. A. Bush, A. D. Printz, A. Gold-Parker, Y. Ding, M. F. Toney, M. D. McGehee and R. H. Dauskardt, Adv. Energy Mater., 2018, 8, 1802139 CrossRef.
- J. Zhao, Y. Deng, H. Wei, X. Zheng, Z. Yu, Y. Shao, J. E. Shield and J. Huang, Sci. Adv., 2017, 3, eaao5616 CrossRef PubMed.
- R. M. Kennard, C. J. Dahlman, R. A. DeCrescent, J. A. Schuller, K. Mukherjee, R. Seshadri and M. L. Chabinyc, Chem. Mater., 2021, 33, 298–309 CrossRef.
- N. Zhou, Y. Bekenstein, C. N. Eisler, D. Zhang, A. M. Schwartzberg, P. Yang, A. P. Alivisatos and J. A. Lewis, Sci. Adv., 2019, 5, eaav8141 CrossRef PubMed.
- H. Li and W. Zhang, Chem. Rev., 2020, 120, 9835–9950 CrossRef PubMed.
- R. Wang, M. Mujahid, Y. Duan, Z.-K. Wang, J. Xue and Y. Yang, Adv. Funct. Mater., 2019, 29, 1808843 CrossRef.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef.
- D. Bryant, N. Aristidou, S. Pont, I. Sanchez-Molina, T. Chotchunangatchaval, S. Wheeler, J. R. Durrant and S. A. Haque, Energy Environ. Sci., 2016, 9, 1655–1660 RSC.
- J. M. Howard, E. M. Tennyson, S. Barik, R. Szostak, E. Waks, M. F. Toney, A. F. Nogueira, B. R. A. Neves and M. S. Leite, J. Phys. Chem. Lett., 2018, 9, 3463–3469 CrossRef.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. S. Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef.
- Z. Hawash, L. K. Ono, S. R. Raga, M. V. Lee and Y. Qi, Chem. Mater., 2015, 27, 562–569 CrossRef.
- J. A. Christians, P. Schulz, J. S. Tinkham, T. H. Schloemer, S. P. Harvey, B. J. Tremolet de Villers, A. Sellinger, J. J. Berry and J. M. Luther, Nat. Energy, 2018, 3, 68–74 CrossRef.
- I. Spanopoulos, W. Ke and M. G. Kanatzidis, Helv. Chim. Acta, 2021, 104, e2000173 CrossRef.
- T. H. Schloemer, J. A. Christians, J. M. Luther and A. Sellinger, Chem. Sci., 2019, 10, 1904–1935 RSC.
- A. N. Singh, S. Kajal, J. Kim, A. Jana, J. Y. Kim and K. S. Kim, Adv. Energy Mater., 2020, 10, 2000768 CrossRef.
- F. Zhang and K. Zhu, Adv. Energy Mater., 2020, 10, 1902579 CrossRef.
- J. A. Christians, S. N. Habisreutinger, J. J. Berry and J. M. Luther, ACS Energy Lett., 2018, 3, 2136–2143 CrossRef.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef.
- C. Li, X. Lu, W. Ding, L. Feng, Y. Gao and Z. Guo, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 702–707 CrossRef PubMed.
- G. Kieslich, S. Sun and A. K. Cheetham, Chem. Sci., 2015, 6, 3430–3433 RSC.
- M. R. Filip and F. Giustino, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5397–5402 CrossRef.
- D. H. Fabini, R. Seshadri and M. G. Kanatzidis, MRS Bull., 2020, 45, 467–477 CrossRef.
- D. W. de Quilettes, S. M. Vorpahl, S. D. Stranks, H. Nagaoka, G. E. Eperon, M. E. Ziffer, H. J. Snaith and D. S. Ginger, Science, 2015, 348, 683–686 CrossRef PubMed.
- J.-C. Blancon, H. Tsai, W. Nie, C. C. Stoumpos, L. Pedesseau, C. Katan, M. Kepenekian, C. M. M. Soe, K. Appavoo, M. Y. Sfeir, S. Tretiak, P. M. Ajayan, M. G. Kanatzidis, J. Even, J. J. Crochet and A. D. Mohite, Science, 2017, 355, 1288–1292 CrossRef CAS.
- D. W. deQuilettes, S. Jariwala, S. Burke, M. E. Ziffer, J. T.-W. Wang, H. J. Snaith and D. S. Ginger, ACS Nano, 2017, 11, 11488–11496 CrossRef CAS.
- O. G. Reid, M. Yang, N. Kopidakis, K. Zhu and G. Rumbles, ACS Energy Lett., 2016, 1, 561–565 CrossRef CAS.
- J. Xing, Q. Wang, Q. Dong, Y. Yuan, Y. Fang and J. Huang, Phys. Chem. Chem. Phys., 2016, 18, 30484–30490 RSC.
- J. S. Yun, J. Seidel, J. Kim, A. M. Soufiani, S. Huang, J. Lau, N. J. Jeon, S. I. Seok, M. A. Green and A. Ho-Baillie, Adv. Energy Mater., 2016, 6, 1600330 CrossRef.
- Y. Shao, Y. Fang, T. Li, Q. Wang, Q. Dong, Y. Deng, Y. Yuan, H. Wei, M. Wang, A. Gruverman, J. Shield and J. Huang, Energy Environ. Sci., 2016, 9, 1752–1759 RSC.
- B. Chen, P. N. Rudd, S. Yang, Y. Yuan and J. Huang, Chem. Soc. Rev., 2019, 48, 3842–3867 RSC.
- X.-D. Wang, W.-G. Li, J.-F. Liao and D.-B. Kuang, Sol. RRL, 2019, 3, 1800294 CrossRef.
- M. Jung, S.-G. Ji, G. Kim and S. Il Seok, Chem. Soc. Rev., 2019, 48, 2011–2038 RSC.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS PubMed.
- R. Munir, A. D. Sheikh, M. Abdelsamie, H. Hu, L. Yu, K. Zhao, T. Kim, O. E. Tall, R. Li, D.-M. Smilgies and A. Amassian, Adv. Mater., 2017, 29, 1604113 CrossRef.
- J. C. Hamill, J. Schwartz and Y.-L. Loo, ACS Energy Lett., 2018, 3, 92–97 CrossRef CAS.
- R. A. Kerner, L. Zhao, Z. Xiao and B. P. Rand, J. Mater. Chem. A, 2016, 4, 8308–8315 RSC.
- A. Dubey, N. Adhikari, S. Mabrouk, F. Wu, K. Chen, S. Yang and Q. Qiao, J. Mater. Chem. A, 2018, 6, 2406–2431 RSC.
- J. Ye, M. M. Byranvand, C. O. Martínez, R. L. Hoye, M. Saliba and L. Polavarapu, Angew. Chem., Int. Ed., 2021, 60, 2–27 CrossRef.
- A. Mahapatra, D. Prochowicz, M. Mahdi Tavakoli, S. Trivedi, P. Kumar and P. Yadav, J. Mater. Chem. A, 2020, 8, 27–54 RSC.
- C. G. Bischak, M. Lai, Z. Fan, D. Lu, P. David, D. Dong, H. Chen, A. S. Etman, T. Lei, J. Sun, M. Grünwald, D. T. Limmer, P. Yang and N. S. Ginsberg, Matter, 2020, 3, 534–545 CrossRef.
- H. T. Pham, T. Duong, K. J. Weber and J. Wong-Leung, ACS Mater. Lett., 2020, 2, 415–424 CrossRef CAS.
- M. U. Rothmann, W. Li, Y. Zhu, U. Bach, L. Spiccia, J. Etheridge and Y.-B. Cheng, Nat. Commun., 2017, 8, 1–8 CrossRef.
- I. M. Hermes, S. A. Bretschneider, V. W. Bergmann, D. Li, A. Klasen, J. Mars, W. Tremel, F. Laquai, H.-J. Butt, M. Mezger, R. Berger, B. J. Rodriguez and S. A. L. Weber, J. Phys. Chem. C, 2016, 120, 5724–5731 CrossRef CAS.
- M. Chabin and F. Gilletta, J. Appl. Crystallogr., 1980, 13, 539–543 CrossRef CAS.
- F. Bertolotti, L. Protesescu, M. V. Kovalenko, S. Yakunin, A. Cervellino, S. J. L. Billinge, M. W. Terban, J. S. Pedersen, N. Masciocchi and A. Guagliardi, ACS Nano, 2017, 11, 3819–3831 CrossRef CAS.
- M. Li, T. Liu, Y. Wang, W. Yang and X. Lü, Matter Radiat. Extremes, 2020, 5, 018201 CrossRef CAS.
- X. Xiao, W. Li, Y. Fang, Y. Liu, Y. Shao, S. Yang, J. Zhao, X. Dai, R. Zia and J. Huang, Nat. Commun., 2020, 11, 2215 CrossRef CAS.
- H. Röhm, T. Leonhard, M. J. Hoffmann and A. Colsmann, Energy Environ. Sci., 2017, 10, 950–955 RSC.
- H. Röhm, T. Leonhard, M. J. Hoffmann and A. Colsmann, Adv. Funct. Mater., 2020, 30, 1908657 CrossRef.
- E. Strelcov, Q. Dong, T. Li, J. Chae, Y. Shao, Y. Deng, A. Gruverman, J. Huang and A. Centrone, Sci. Adv., 2017, 3, e1602165 CrossRef.
- B. Huang, G. Kong, E. N. Esfahani, S. Chen, Q. Li, J. Yu, N. Xu, Y. Zhang, S. Xie, H. Wen, P. Gao, J. Zhao and J. Li, npj Quantum Mater., 2018, 3, 1–8 CrossRef CAS.
- A. Gómez, Q. Wang, A. R. Goñi, M. Campoy-Quiles and A. Abate, Energy Environ. Sci., 2019, 12, 2537–2547 RSC.
- J. N. Wilson, J. M. Frost, S. K. Wallace and A. Walsh, APL Mater., 2019, 7, 010901 CrossRef.
- N. J. Jeon, H. Na, E. H. Jung, T.-Y. Yang, Y. G. Lee, G. Kim, H.-W. Shin, S. Il Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682–689 CrossRef CAS.
- X. Zheng, Y. Hou, C. Bao, J. Yin, F. Yuan, Z. Huang, K. Song, J. Liu, J. Troughton, N. Gasparini, C. Zhou, Y. Lin, D.-J. Xue, B. Chen, A. K. Johnston, N. Wei, M. N. Hedhili, M. Wei, A. Y. Alsalloum, P. Maity, B. Turedi, C. Yang, D. Baran, T. D. Anthopoulos, Y. Han, Z.-H. Lu, O. F. Mohammed, F. Gao, E. H. Sargent and O. M. Bakr, Nat. Energy, 2020, 5, 131–140 CrossRef CAS.
- M. Jeong, I. W. Choi, E. M. Go, Y. Cho, M. Kim, B. Lee, S. Jeong, Y. Jo, H. W. Choi, J. Lee, J.-H. Bae, S. K. Kwak, D. S. Kim and C. Yang, Science, 2020, 369, 1615–1620 CrossRef CAS PubMed.
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- J. J. Yoo, G. Seo, M. R. Chua, T. G. Park, Y. Lu, F. Rotermund, Y.-K. Kim, C. S. Moon, N. J. Jeon, J.-P. Correa-Baena, V. Bulović, S. S. Shin, M. G. Bawendi and J. Seo, Nature, 2021, 590, 587–593 CrossRef CAS.
- H. Lu, A. Krishna, S. M. Zakeeruddin, M. Grätzel and A. Hagfeldt, iScience, 2020, 23, 101359 CrossRef CAS.
- R. E. Beal, N. Z. Hagström, J. Barrier, A. Gold-Parker, R. Prasanna, K. A. Bush, D. Passarello, L. T. Schelhas, K. Brüning, C. J. Tassone, H.-G. Steinrück, M. D. McGehee, M. F. Toney and A. F. Nogueira, Matter, 2020, 2, 207–219 CrossRef.
- H. Grüninger, M. Bokdam, N. Leupold, P. Tinnemans, R. Moos, G. A. De Wijs, F. Panzer and A. P. M. Kentgens, J. Phys. Chem. C, 2021, 125, 1742–1753 CrossRef.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, P. Péchy, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2017, 139, 10055–10061 CrossRef CAS.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2017, 139, 14173–14180 CrossRef CAS.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2018, 140, 7232–7238 CrossRef CAS.
- H. X. Dang, K. Wang, M. Ghasemi, M.-C. Tang, M. De Bastiani, E. Aydin, E. Dauzon, D. Barrit, J. Peng, D.-M. Smilgies, S. De Wolf and A. Amassian, Joule, 2019, 3, 1746–1764 CrossRef CAS.
- J.-P. Correa-Baena, Y. Luo, T. M. Brenner, J. Snaider, S. Sun, X. Li, M. A. Jensen, N. T. P. Hartono, L. Nienhaus, S. Wieghold, J. R. Poindexter, S. Wang, Y. S. Meng, T. Wang, B. Lai, M. V. Holt, Z. Cai, M. G. Bawendi, L. Huang, T. Buonassisi and D. P. Fenning, Science, 2019, 363, 627–631 CrossRef CAS PubMed.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS.
- A. J. Knight and L. M. Herz, Energy Environ. Sci., 2020, 13, 2024–2046 RSC.
- E. T. Hoke, D. J. Slotcavage, E. R. Dohner, A. R. Bowring, H. I. Karunadasa and M. D. McGehee, Chem. Sci., 2015, 6, 613–617 RSC.
- M. C. Brennan, S. Draguta, P. V. Kamat and M. Kuno, ACS Energy Lett., 2018, 3, 204–213 CrossRef CAS.
- D. W. deQuilettes, W. Zhang, V. M. Burlakov, D. J. Graham, T. Leijtens, A. Osherov, V. Bulović, H. J. Snaith, D. S. Ginger and S. D. Stranks, Nat. Commun., 2016, 7, 11683 CrossRef CAS PubMed.
- Z. Andaji-Garmaroudi, M. Anaya, A. J. Pearson and S. D. Stranks, Adv. Energy Mater., 2020, 10, 1903109 CrossRef CAS.
- E. Shi and L. Dou, Acc. Mater. Res., 2020, 1, 213–224 CrossRef CAS.
- R. A. Scheidt and P. V. Kamat, J. Chem. Phys., 2019, 151, 134703 CrossRef PubMed.
- T. Elmelund, R. A. Scheidt, B. Seger and P. V. Kamat, ACS Energy Lett., 2019, 4, 1961–1969 CrossRef CAS.
- R. M. Kennard, C. J. Dahlman, H. Nakayama, R. A. DeCrescent, J. A. Schuller, R. Seshadri, K. Mukherjee and M. L. Chabinyc, ACS Appl. Mater. Interfaces, 2019, 11, 25313–25321 CrossRef CAS.
- Y. Wang, Z. Chen, F. Deschler, X. Sun, T.-M. Lu, E. A. Wertz, J.-M. Hu and J. Shi, ACS Nano, 2017, 11, 3355–3364 CrossRef CAS PubMed.
- L. Dou, M. Lai, C. S. Kley, Y. Yang, C. G. Bischak, D. Zhang, S. W. Eaton, N. S. Ginsberg and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7216–7221 CrossRef CAS.
- N. T. Shewmon, H. Yu, I. Constantinou, E. Klump and F. So, ACS Appl. Mater. Interfaces, 2016, 8, 33273–33279 CrossRef CAS.
- T. A. S. Doherty, A. J. Winchester, S. Macpherson, D. N. Johnstone, V. Pareek, E. M. Tennyson, S. Kosar, F. U. Kosasih, M. Anaya, M. Abdi-Jalebi, Z. Andaji-Garmaroudi, E. L. Wong, J. Madéo, Y.-H. Chiang, J.-S. Park, Y.-K. Jung, C. E. Petoukhoff, G. Divitini, M. K. L. Man, C. Ducati, A. Walsh, P. A. Midgley, K. M. Dani and S. D. Stranks, Nature, 2020, 580, 360–366 CrossRef CAS.
- Y. Liu, Y. Zhang, X. Zhu, J. Feng, I. Spanopoulos, W. Ke, Y. He, X. Ren, Z. Yang, F. Xiao, K. Zhao, M. Kanatzidis and S. Liu, Adv. Mater., 2021, 33, 2006010 CrossRef CAS PubMed.
- Q. Zhang, F. Hao, J. Li, Y. Zhou, Y. Wei and H. Lin, Sci. Technol. Adv. Mater., 2018, 19, 425–442 CrossRef CAS.
- G. Schileo and G. Grancini, J. Mater. Chem. C, 2021, 9, 67–76 RSC.
- W. Ke and M. G. Kanatzidis, Nat. Commun., 2019, 10, 965 CrossRef.
- A. B. F. Vitoreti, S. Agouram, M. Solis de la Fuente, V. Muñoz-Sanjosé, M. A. Schiavon and I. Mora-Seró, J. Phys. Chem. C, 2018, 122, 14222–14231 CrossRef CAS PubMed.
- J. Liu, G. Wang, Z. Song, X. He, K. Luo, Q. Ye, C. Liao and J. Mei, J. Mater. Chem. A, 2017, 5, 9097–9106 RSC.
- B. Zhao, M. Abdi-Jalebi, M. Tabachnyk, H. Glass, V. S. Kamboj, W. Nie, A. J. Pearson, Y. Puttisong, K. C. Gödel, H. E. Beere, D. A. Ritchie, A. D. Mohite, S. E. Dutton, R. H. Friend and A. Sadhanala, Adv. Mater., 2017, 29, 1604744 CrossRef PubMed.
- Z. Yang, A. Rajagopal, S. B. Jo, C.-C. Chueh, S. Williams, C.-C. Huang, J. K. Katahara, H. W. Hillhouse and A. K.-Y. Jen, Nano Lett., 2016, 16, 7739–7747 CrossRef CAS.
- D. Bartesaghi, A. Ray, J. Jiang, R. K. M. Bouwer, S. Tao and T. J. Savenije, APL Mater., 2018, 6, 121106 CrossRef.
- D. Parobek, B. J. Roman, Y. Dong, H. Jin, E. Lee, M. Sheldon and D. H. Son, Nano Lett., 2016, 16, 7376–7380 CrossRef CAS PubMed.
- F. Yang, D. Hirotani, G. Kapil, M. A. Kamarudin, C. H. Ng, Y. Zhang, Q. Shen and S. Hayase, Angew. Chem., Int. Ed., 2018, 57, 12745–12749 CrossRef CAS.
- R. Nie, R. Raj Sumukam, S. Harshavardhan Reddy, M. Banavoth and S. Il Seok, Energy Environ. Sci., 2020, 13, 2363–2385 RSC.
- Z. Xiao, Z. Song and Y. Yan, Adv. Mater., 2019, 31, 1803792 CrossRef CAS PubMed.
- W. Ke, C. C. Stoumpos and M. G. Kanatzidis, Adv. Mater., 2019, 31, 1803230 CrossRef CAS PubMed.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef CAS.
- G. Volonakis, M. R. Filip, A. A. Haghighirad, N. Sakai, B. Wenger, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2016, 7, 1254–1259 CrossRef CAS.
- E. T. McClure, M. R. Ball, W. Windl and P. M. Woodward, Chem. Mater., 2016, 28, 1348–1354 CrossRef CAS.
- M. R. Filip, S. Hillman, A. A. Haghighirad, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2016, 7, 2579–2585 CrossRef CAS.
- A. H. Slavney, L. Leppert, A. Saldivar Valdes, D. Bartesaghi, T. J. Savenije, J. B. Neaton and H. I. Karunadasa, Angew. Chem., Int. Ed., 2018, 57, 12765–12770 CrossRef CAS.
- A. Leblanc, N. Mercier, M. Allain, J. Dittmer, V. Fernandez and T. Pauporté, Angew. Chem., Int. Ed., 2017, 56, 16067–16072 CrossRef CAS.
- A. Leblanc, N. Mercier, M. Allain, J. Dittmer, T. Pauporté, V. Fernandez, F. Boucher, M. Kepenekian and C. Katan, ACS Appl. Mater. Interfaces, 2019, 11, 20743–20751 CrossRef.
- C.-M. Tsai, Y.-P. Lin, M. K. Pola, S. Narra, E. Jokar, Y.-W. Yang and E. W.-G. Diau, ACS Energy Lett., 2018, 3, 2077–2085 CrossRef CAS.
- W. Ke, C. C. Stoumpos, M. Zhu, L. Mao, I. Spanopoulos, J. Liu, O. Y. Kontsevoi, M. Chen, D. Sarma, Y. Zhang, M. R. Wasielewski and M. G. Kanatzidis, Sci. Adv., 2017, 3, e1701293 CrossRef PubMed.
- I. Spanopoulos, W. Ke, C. C. Stoumpos, E. C. Schueller, O. Y. Kontsevoi, R. Seshadri and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 5728–5742 CrossRef CAS PubMed.
- W. Ke, I. Spanopoulos, Q. Tu, I. Hadar, X. Li, G. S. Shekhawat, V. P. Dravid and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 8627–8637 CrossRef CAS.
- M. Worku, Y. Tian, C. Zhou, H. Lin, M. Chaaban, L. Xu, Q. He, D. Beery, Y. Zhou, X. Lin, Y. Su, Y. Xin and B. Ma, Sci. Adv., 2020, 6, eaaz5961 CrossRef CAS.
- A. Senocrate, I. Spanopoulos, N. Zibouche, J. Maier, M. S. Islam and M. G. Kanatzidis, Chem. Mater., 2021, 33, 719–726 CrossRef CAS.
- W. Ke, C. C. Stoumpos, I. Spanopoulos, M. Chen, M. R. Wasielewski and M. G. Kanatzidis, ACS Energy Lett., 2018, 3, 1470–1476 CrossRef CAS.
- J. A. McNulty, A. M. Z. Slawin and P. Lightfoot, Dalton Trans., 2020, 49, 15171–15174 RSC.
- Y. I. Dolzhenko, T. Inabe and Y. Maruyama, Bull. Chem. Soc. Jpn., 1986, 59, 563–567 CrossRef CAS.
- T. Ishihara, J. Takahashi and T. Goto, Solid State Commun., 1989, 69, 933–936 CrossRef CAS.
- J. Calabrese, N. L. Jones, R. L. Harlow, N. Herron, D. L. Thorn and Y. Wang, J. Am. Chem. Soc., 1991, 113, 2328–2330 CrossRef CAS.
- D. B. Mitzi, C. A. Feild, W. T. A. Harrison and A. M. Guloy, Nature, 1994, 369, 467–469 CrossRef CAS.
- D. B. Mitzi, Inorg. Chem., 2000, 39, 6107–6113 CrossRef CAS.
- Y. Gao, E. Shi, S. Deng, S. B. Shiring, J. M. Snaider, C. Liang, B. Yuan, R. Song, S. M. Janke, A. Liebman-Peláez, P. Yoo, M. Zeller, B. W. Boudouris, P. Liao, C. Zhu, V. Blum, Y. Yu, B. M. Savoie, L. Huang and L. Dou, Nat. Chem., 2019, 11, 1151–1157 CrossRef CAS.
- C. Liu, W. Huhn, K.-Z. Du, A. Vazquez-Mayagoitia, D. Dirkes, W. You, Y. Kanai, D. B. Mitzi and V. Blum, Phys. Rev. Lett., 2018, 121, 146401 CrossRef CAS.
- W. A. Dunlap-Shohl, E. Tomas Barraza, A. Barrette, S. Dovletgeldi, G. Findik, D. J. Dirkes, C. Liu, M. K. Jana, V. Blum, W. You, K. Gundogdu, A. D. Stiff-Roberts and D. B. Mitzi, Mater. Horiz., 2019, 6, 1707–1716 RSC.
- J. V. Passarelli, D. J. Fairfield, N. A. Sather, M. P. Hendricks, H. Sai, C. L. Stern and S. I. Stupp, J. Am. Chem. Soc., 2018, 140, 7313–7323 CrossRef CAS PubMed.
- J. Xue, R. Wang, X. Chen, C. Yao, X. Jin, K.-L. Wang, W. Huang, T. Huang, Y. Zhao, Y. Zhai, D. Meng, S. Tan, R. Liu, Z.-K. Wang, C. Zhu, K. Zhu, M. C. Beard, Y. Yan and Y. Yang, Science, 2021, 371, 636–640 CrossRef CAS.
- J. V. Passarelli, C. M. Mauck, S. W. Winslow, C. F. Perkinson, J. C. Bard, H. Sai, K. W. Williams, A. Narayanan, D. J. Fairfield, M. P. Hendricks, W. A. Tisdale and S. I. Stupp, Nat. Chem., 2020, 12, 672–682 CrossRef CAS PubMed.
- T. Ishihara, J. Takahashi and T. Goto, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 11099–11107 CrossRef CAS.
- K. Tanaka, T. Takahashi, T. Ban, T. Kondo, K. Uchida and N. Miura, Solid State Commun., 2003, 127, 619–623 CrossRef CAS.
- L. Pedesseau, D. Sapori, B. Traore, R. Robles, H.-H. Fang, M. A. Loi, H. Tsai, W. Nie, J.-C. Blancon, A. Neukirch, S. Tretiak, A. D. Mohite, C. Katan, J. Even and M. Kepenekian, ACS Nano, 2016, 10, 9776–9786 CrossRef CAS.
- C. Katan, N. Mercier and J. Even, Chem. Rev., 2019, 119, 3140–3192 CrossRef CAS PubMed.
- E. A. Muljarov, S. G. Tikhodeev, N. A. Gippius and T. Ishihara, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 14370–14378 CrossRef CAS PubMed.
- J.-C. Blancon, A. V. Stier, H. Tsai, W. Nie, C. C. Stoumpos, B. Traoré, L. Pedesseau, M. Kepenekian, F. Katsutani, G. T. Noe, J. Kono, S. Tretiak, S. A. Crooker, C. Katan, M. G. Kanatzidis, J. J. Crochet, J. Even and A. D. Mohite, Nat. Commun., 2018, 9, 2254 CrossRef.
- K. Leng, W. Fu, Y. Liu, M. Chhowalla and K. P. Loh, Nat. Rev. Mater., 2020, 5, 482–500 CrossRef CAS.
- R. A. DeCrescent, N. R. Venkatesan, C. J. Dahlman, R. M. Kennard, M. L. Chabinyc and J. A. Schuller, ACS Nano, 2019, 13, 10745–10753 CrossRef CAS PubMed.
- C. M. M. Soe, G. P. Nagabhushana, R. Shivaramaiah, H. Tsai, W. Nie, J.-C. Blancon, F. Melkonyan, D. H. Cao, B. Traoré, L. Pedesseau, M. Kepenekian, C. Katan, J. Even, T. J. Marks, A. Navrotsky, A. D. Mohite, C. C. Stoumpos and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 58–66 CrossRef PubMed.
- L. Mao, R. M. Kennard, B. Traore, W. Ke, C. Katan, J. Even, M. L. Chabinyc, C. C. Stoumpos and M. G. Kanatzidis, Chem, 2019, 5, 2593–2604 CAS.
- C. C. Stoumpos, D. H. Cao, D. J. Clark, J. Young, J. M. Rondinelli, J. I. Jang, J. T. Hupp and M. G. Kanatzidis, Chem. Mater., 2016, 28, 2852–2867 CrossRef CAS.
- E. Shi, S. Deng, B. Yuan, Y. Gao, Akriti, L. Yuan, C. S. Davis, D. Zemlyanov, Y. Yu, L. Huang and L. Dou, ACS Nano, 2019, 13, 1635–1644 CrossRef CAS PubMed.
- K. Wang, C. Wu, Y. Jiang, D. Yang, K. Wang and S. Priya, Sci. Adv., 2019, 5, eaau3241 CrossRef CAS.
- L. Mao, Y. Wu, C. C. Stoumpos, B. Traore, C. Katan, J. Even, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 11956–11963 CrossRef CAS PubMed.
- Y. Fu, X. Jiang, X. Li, B. Traore, I. Spanopoulos, C. Katan, J. Even, M. G. Kanatzidis and E. Harel, J. Am. Chem. Soc., 2020, 142, 4008–4021 CrossRef CAS.
- D. Pan, Y. Fu, N. Spitha, Y. Zhao, C. R. Roy, D. J. Morrow, D. D. Kohler, J. C. Wright and S. Jin, Nat. Nanotechnol., 2021, 16, 159–165 CrossRef CAS.
- E. Shi, B. Yuan, S. B. Shiring, Y. Gao, Akriti, Y. Guo, C. Su, M. Lai, P. Yang, J. Kong, B. M. Savoie, Y. Yu and L. Dou, Nature, 2020, 580, 614–620 CrossRef CAS PubMed.
- J. Li, Q. Yu, Y. He, C. C. Stoumpos, G. Niu, G. G. Trimarchi, H. Guo, G. Dong, D. Wang, L. Wang and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 11085–11090 CrossRef CAS PubMed.
- B. A. Connor, L. Leppert, M. D. Smith, J. B. Neaton and H. I. Karunadasa, J. Am. Chem. Soc., 2018, 140, 5235–5240 CrossRef CAS.
- L. Mao, P. Guo, M. Kepenekian, I. Spanopoulos, Y. He, C. Katan, J. Even, R. D. Schaller, R. Seshadri, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2020, 142, 8342–8351 CrossRef CAS PubMed.
- M. A. Hope, T. Nakamura, P. Ahlawat, A. Mishra, M. Cordova, F. Jahanbakhshi, M. Mladenović, R. Runjhun, L. Merten, A. Hinderhofer, B. I. Carlsen, D. J. Kubicki, R. Gershoni-Poranne, T. Schneeberger, L. C. Carbone, Y. Liu, S. M. Zakeeruddin, J. Lewinski, A. Hagfeldt, F. Schreiber, U. Rothlisberger, M. Grätzel, J. V. Milić and L. Emsley, J. Am. Chem. Soc., 2021, 143, 1529–1538 CrossRef CAS.
- C. J. Dahlman, N. R. Venkatesan, P. T. Corona, R. M. Kennard, L. Mao, N. C. Smith, J. Zhang, R. Seshadri, M. E. Helgeson and M. L. Chabinyc, ACS Nano, 2020, 14, 11294–11308 CrossRef CAS PubMed.
- C. M. M. Soe, C. C. Stoumpos, M. Kepenekian, B. Traoré, H. Tsai, W. Nie, B. Wang, C. Katan, R. Seshadri, A. D. Mohite, J. Even, T. J. Marks and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 16297–16309 CrossRef CAS PubMed.
- J. M. Hoffman, J. Strzalka, N. C. Flanders, I. Hadar, S. A. Cuthriell, Q. Zhang, R. D. Schaller, W. R. Dichtel, L. X. Chen and M. G. Kanatzidis, Adv. Mater., 2020, 32, 2002812 CrossRef CAS PubMed.
- J. Dong, S. Shao, S. Kahmann, A. J. Rommens, D. Hermida-Merino, G. H. ten Brink, M. A. Loi and G. Portale, Adv. Funct. Mater., 2020, 30, 2001294 CrossRef CAS.
- T. Li, A. M. Zeidell, G. Findik, W. A. Dunlap-Shohl, J. Euvrard, K. Gundogdu, O. D. Jurchescu and D. B. Mitzi, Chem. Mater., 2019, 31, 4267–4274 CrossRef CAS.
- C. J. Dahlman, R. A. DeCrescent, N. R. Venkatesan, R. M. Kennard, G. Wu, M. A. Everest, J. A. Schuller and M. L. Chabinyc, Chem. Mater., 2019, 31, 5832–5844 CrossRef CAS.
- A. H. Proppe, R. Quintero-Bermudez, H. Tan, O. Voznyy, S. O. Kelley and E. H. Sargent, J. Am. Chem. Soc., 2018, 140, 2890–2896 CrossRef CAS PubMed.
- X. Zhang, R. Munir, Z. Xu, Y. Liu, H. Tsai, W. Nie, J. Li, T. Niu, D.-M. Smilgies, M. G. Kanatzidis, A. D. Mohite, K. Zhao, A. Amassian and S. Liu, Adv. Mater., 2018, 30, 1707166 CrossRef.
- R. Quintero-Bermudez, A. Gold-Parker, A. H. Proppe, R. Munir, Z. Yang, S. O. Kelley, A. Amassian, M. F. Toney and E. H. Sargent, Nat. Mater., 2018, 17, 900 CrossRef CAS PubMed.
- A. H. Proppe, M. H. Elkins, O. Voznyy, R. D. Pensack, F. Zapata, L. V. Besteiro, L. N. Quan, R. Quintero-Bermudez, P. Todorovic, S. O. Kelley, A. O. Govorov, S. K. Gray, I. Infante, E. H. Sargent and G. D. Scholes, J. Phys. Chem. Lett., 2019, 10, 419–426 CrossRef.
- Y. Lin, Y. Fang, J. Zhao, Y. Shao, S. J. Stuard, M. M. Nahid, H. Ade, Q. Wang, J. E. Shield, N. Zhou, A. M. Moran and J. Huang, Nat. Commun., 2019, 10, 1008 CrossRef PubMed.
- K. Zheng, Y. Chen, Y. Sun, J. Chen, P. Chábera, R. Schaller, M. J. Al-Marri, S. E. Canton, Z. Liang and T. Pullerits, J. Mater. Chem. A, 2018, 6, 6244–6250 RSC.
- N. Liu, P. Liu, H. Ren, H. Xie, N. Zhou, Y. Gao, Y. Li, H. Zhou, Y. Bai and Q. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 3127–3133 CrossRef CAS PubMed.
- M. Yuan, L. N. Quan, R. Comin, G. Walters, R. Sabatini, O. Voznyy, S. Hoogland, Y. Zhao, E. M. Beauregard, P. Kanjanaboos, Z. Lu, D. H. Kim and E. H. Sargent, Nat. Nanotechnol., 2016, 11, 872–877 CrossRef CAS PubMed.
- R. F. Moral, L. G. Bonato, J. C. Germino, W. X. Coelho Oliveira, R. Kamat, J. Xu, C. J. Tassone, S. D. Stranks, M. F. Toney and A. F. Nogueira, Chem. Mater., 2019, 31, 9472–9479 CrossRef CAS.
- N. R. Venkatesan, R. M. Kennard, R. A. DeCrescent, H. Nakayama, C. J. Dahlman, E. E. Perry, J. A. Schuller and M. L. Chabinyc, Chem. Mater., 2018, 30, 8615–8623 CrossRef CAS.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS.
- Y. Zhang, T. D. Siegler, C. J. Thomas, M. K. Abney, T. Shah, A. De Gorostiza, R. M. Greene and B. A. Korgel, Chem. Mater., 2020, 32, 5410–5423 CrossRef CAS.
- Y. Fu, H. Zhu, J. Chen, M. P. Hautzinger, X.-Y. Zhu and S. Jin, Nat. Rev. Mater., 2019, 4, 169 CrossRef CAS.
- E. Shi, Y. Gao, B. P. Finkenauer, Akriti, A. H. Coffey and L. Dou, Chem. Soc. Rev., 2018, 47, 6046–6072 RSC.
- H. Huang, M. I. Bodnarchuk, S. V. Kershaw, M. V. Kovalenko and A. L. Rogach, ACS Energy Lett., 2017, 2, 2071–2083 CrossRef CAS PubMed.
- M. Kazes, T. Udayabhaskararao, S. Dey and D. Oron, Acc. Chem. Res., 2021, 54, 1409–1418 CrossRef CAS PubMed.
- J. Xue, R. Wang and Y. Yang, Nat. Rev. Mater., 2020, 5, 809–827 CrossRef CAS.
- T. Chiba and J. Kido, J. Mater. Chem. C, 2018, 6, 11868–11877 RSC.
- M. I. Bodnarchuk, S. C. Boehme, S. ten Brinck, C. Bernasconi, Y. Shynkarenko, F. Krieg, R. Widmer, B. Aeschlimann, D. Günther, M. V. Kovalenko and I. Infante, ACS Energy Lett., 2019, 4, 63–74 CrossRef CAS.
- F. Yan, S. T. Tan, X. Li and H. V. Demir, Small, 2019, 15, 1902079 CrossRef CAS PubMed.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS PubMed.
- S. Toso, D. Baranov, C. Giannini, S. Marras and L. Manna, ACS Mater. Lett., 2019, 1, 272–276 CrossRef CAS.
- Y. Nagaoka, K. Hills-Kimball, R. Tan, R. Li, Z. Wang and O. Chen, Adv. Mater., 2017, 29, 1606666 CrossRef PubMed.
- Y. Tong, E.-P. Yao, A. Manzi, E. Bladt, K. Wang, M. Döblinger, S. Bals, P. Müller-Buschbaum, A. S. Urban, L. Polavarapu and J. Feldmann, Adv. Mater., 2018, 30, 1801117 CrossRef.
- M. C. Brennan, S. Toso, I. M. Pavlovetc, M. Zhukovskyi, S. Marras, M. Kuno, L. Manna and D. Baranov, ACS Energy Lett., 2020, 5, 1465–1473 CrossRef CAS.
- J. Jagielski, S. F. Solari, L. Jordan, D. Scullion, B. Blülle, Y.-T. Li, F. Krumeich, Y.-C. Chiu, B. Ruhstaller, E. J. G. Santos and C.-J. Shih, Nat. Commun., 2020, 11, 387 CrossRef CAS PubMed.
- Y. Liu, M. Siron, D. Lu, J. Yang, R. dos Reis, F. Cui, M. Gao, M. Lai, J. Lin, Q. Kong, T. Lei, J. Kang, J. Jin, J. Ciston and P. Yang, J. Am. Chem. Soc., 2019, 141, 13028–13032 CrossRef CAS.
- Z. Dang, B. Dhanabalan, A. Castelli, R. Dhall, K. C. Bustillo, D. Marchelli, D. Spirito, U. Petralanda, J. Shamsi, L. Manna, R. Krahne and M. P. Arciniegas, Nano Lett., 2020, 20, 1808–1818 CrossRef CAS PubMed.
- D. Zhang, Y. Yu, Y. Bekenstein, A. B. Wong, A. P. Alivisatos and P. Yang, J. Am. Chem. Soc., 2016, 138, 13155–13158 CrossRef CAS.
- C. J. Thomas, Y. Zhang, A. Guillaussier, K. Bdeir, O. F. Aly, H. G. Kim, J. Noh, L. C. Reimnitz, J. Li, F. L. Deepak, D.-M. Smilgies, D. J. Milliron and B. A. Korgel, Chem. Mater., 2019, 31, 9750–9758 CrossRef CAS.
- X. Zhao, J. D. A. Ng, R. H. Friend and Z.-K. Tan, ACS Photonics, 2018, 5, 3866–3875 CrossRef CAS.
- Y. Wei, Z. Cheng and J. Lin, Chem. Soc. Rev., 2019, 48, 310–350 RSC.
- P. Cottingham and R. L. Brutchey, Chem. Mater., 2018, 30, 6711–6716 CrossRef CAS.
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS PubMed.
- G. Almeida, I. Infante and L. Manna, Science, 2019, 364, 833–834 CrossRef CAS.
- D. P. Nenon, K. Pressler, J. Kang, B. A. Koscher, J. H. Olshansky, W. T. Osowiecki, M. A. Koc, L.-W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2018, 140, 17760–17772 CrossRef CAS.
- M. A. Boles, D. Ling, T. Hyeon and D. V. Talapin, Nat. Mater., 2016, 15, 141–153 CrossRef CAS PubMed.
- M. Ben-Nun, J. Cao and K. R. Wilson, J. Phys. Chem. A, 1997, 101, 8743–8761 CrossRef CAS.
- J. Cao and K. R. Wilson, J. Phys. Chem. A, 1998, 102, 9523–9530 CrossRef CAS.
- C. Li, A. Wang, X. Deng, S. Wang, Y. Yuan, L. Ding and F. Hao, ACS Photonics, 2020, 7, 1893–1907 CrossRef CAS.
- X. Wu, L. Z. Tan, X. Shen, T. Hu, K. Miyata, M. T. Trinh, R. Li, R. Coffee, S. Liu, D. A. Egger, I. Makasyuk, Q. Zheng, A. Fry, J. S. Robinson, M. D. Smith, B. Guzelturk, H. I. Karunadasa, X. Wang, X. Zhu, L. Kronik, A. M. Rappe and A. M. Lindenberg, Sci. Adv., 2017, 3, e1602388 CrossRef.
- E. J. VandenBussche, C. P. Clark, R. J. Holmes and D. J. Flannigan, ACS Omega, 2020, 5, 31867–31871 CrossRef CAS.
- B. Guzelturk, T. Winkler, T. W. J. Van de Goor, M. D. Smith, S. A. Bourelle, S. Feldmann, M. Trigo, S. W. Teitelbaum, H.-G. Steinrück, G. A. de la Pena, R. Alonso-Mori, D. Zhu, T. Sato, H. I. Karunadasa, M. F. Toney, F. Deschler and A. M. Lindenberg, Nat. Mater., 2021, 1–6 Search PubMed.
- L. M. Herz, Annu. Rev. Phys. Chem., 2016, 67, 65–89 CrossRef CAS PubMed.
- N. Mondal, A. De, S. Das, S. Paul and A. Samanta, Nanoscale, 2019, 11, 9796–9818 RSC.
- Z. Li, Y. Chen and C. Burda, J. Phys. Chem. C, 2019, 123, 3255–3269 CrossRef CAS.
- J. Shi, Y. Li, Y. Li, D. Li, Y. Luo, H. Wu and Q. Meng, Joule, 2018, 2, 879–901 CrossRef CAS.
- J.-P. Correa-Baena, M. Saliba, T. Buonassisi, M. Grätzel, A. Abate, W. Tress and A. Hagfeldt, Science, 2017, 358, 739–744 CrossRef CAS.
- D. Meggiolaro, F. Ambrosio, E. Mosconi, A. Mahata and F. D. Angelis, Adv. Energy Mater., 2020, 10, 1902748 CrossRef CAS.
- C. Katan, A. D. Mohite and J. Even, Nat. Mater., 2018, 17, 377–379 CrossRef CAS PubMed.
- P. P. Joshi, S. F. Maehrlein and X. Zhu, Adv. Mater., 2019, 31, 1803054 CrossRef CAS PubMed.
- D. H. Fabini, T. A. Siaw, C. C. Stoumpos, G. Laurita, D. Olds, K. Page, J. G. Hu, M. G. Kanatzidis, S. Han and R. Seshadri, J. Am. Chem. Soc., 2017, 139, 16875–16884 CrossRef CAS PubMed.
- E. M. Mozur, A. E. Maughan, Y. Cheng, A. Huq, N. Jalarvo, L. L. Daemen and J. R. Neilson, Chem. Mater., 2017, 29, 10168–10177 CrossRef CAS.
- J. D. Dow and D. Redfield, Phys. Rev. B: Solid State, 1972, 5, 594–610 CrossRef.
- M. Ledinsky, T. Schönfeldová, J. Holovský, E. Aydin, Z. Hájková, L. Landová, N. Neyková, A. Fejfar and S. De Wolf, J. Phys. Chem. Lett., 2019, 10, 1368–1373 CrossRef CAS.
- A. Raja, L. Waldecker, J. Zipfel, Y. Cho, S. Brem, J. D. Ziegler, M. Kulig, T. Taniguchi, K. Watanabe, E. Malic, T. F. Heinz, T. C. Berkelbach and A. Chernikov, Nat. Nanotechnol., 2019, 14, 832–837 CrossRef CAS.
- A. M. A. Leguy, A. R. Goñi, J. M. Frost, J. Skelton, F. Brivio, X. Rodríguez-Martínez, O. J. Weber, A. Pallipurath, M. Isabel Alonso, M. Campoy-Quiles, M. T. Weller, J. Nelson, A. Walsh and P. R. F. Barnes, Phys. Chem. Chem. Phys., 2016, 18, 27051–27066 RSC.
- A. Poglitsch and D. Weber, J. Chem. Phys., 1987, 87, 6373–6378 CrossRef CAS.
- S. Govinda, B. P. Kore, M. Bokdam, P. Mahale, A. Kumar, S. Pal, B. Bhattacharyya, J. Lahnsteiner, G. Kresse, C. Franchini, A. Pandey and D. D. Sarma, J. Phys. Chem. Lett., 2017, 8, 4113–4121 CrossRef CAS PubMed.
- J. R. Neilson, Neutron News, 2021, 32, 11–12 CrossRef.
- X. Hu, D. Zhang, T. Chen, A. Z. Chen, E. N. Holmgren, Q. Zhang, D. M. Pajerowski, M. Yoon, G. Xu, J. J. Choi and S.-H. Lee, J. Chem. Phys., 2019, 152, 014703 CrossRef.
- T. Chen, B. J. Foley, B. Ipek, M. Tyagi, J. R. D. Copley, C. M. Brown, J. J. Choi and S.-H. Lee, Phys. Chem. Chem. Phys., 2015, 17, 31278–31286 RSC.
- X. Gong, O. Voznyy, A. Jain, W. Liu, R. Sabatini, Z. Piontkowski, G. Walters, G. Bappi, S. Nokhrin, O. Bushuyev, M. Yuan, R. Comin, D. McCamant, S. O. Kelley and E. H. Sargent, Nat. Mater., 2018, 17, 550–556 CrossRef CAS PubMed.
- A. Senocrate, I. Moudrakovski and J. Maier, Phys. Chem. Chem. Phys., 2018, 20, 20043–20055 RSC.
- C. Roiland, G. Trippé-Allard, K. Jemli, B. Alonso, J.-C. Ameline, R. Gautier, T. Bataille, L. L. Pollès, E. Deleporte, J. Even and C. Katan, Phys. Chem. Chem. Phys., 2016, 18, 27133–27142 RSC.
- R. E. Wasylishen, O. Knop and J. B. Macdonald, Solid State Commun., 1985, 56, 581–582 CrossRef CAS.
- G. M. Bernard, R. E. Wasylishen, C. I. Ratcliffe, V. Terskikh, Q. Wu, J. M. Buriak and T. Hauger, J. Phys. Chem. A, 2018, 122, 1560–1573 CrossRef CAS.
- T. Ueda, K. Shimizu, H. Ohki and T. Okuda, Z. Naturforsch. A, 1996, 51, 910–914 CrossRef CAS.
- T. Ueda, M. Omo, K. Shimizu, H. Ohki and T. Okuda, Z. Naturforsch. A, 1997, 52, 502–508 CrossRef CAS.
- C. J. Dahlman, R. M. Kennard, P. Paluch, N. R. Venkatesan, M. L. Chabinyc and G. N. Manjunatha Reddy, Chem. Mater., 2021, 33, 642–656 CrossRef CAS.
- M. Lai, A. Obliger, D. Lu, C. S. Kley, C. G. Bischak, Q. Kong, T. Lei, L. Dou, N. S. Ginsberg, D. T. Limmer and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11929–11934 CrossRef CAS.
- A. Senocrate and J. Maier, J. Am. Chem. Soc., 2019, 141, 8382–8396 CrossRef CAS PubMed.
- D. J. Kubicki, D. Prochowicz, E. Salager, A. Rakhmatullin, C. P. Grey, L. Emsley and S. D. Stranks, J. Am. Chem. Soc., 2020, 142, 7813–7826 CrossRef CAS.
- A. Karmakar, A. Bhattacharya, D. Sarkar, G. M. Bernard, A. Mar and V. K. Michaelis, Chem. Sci., 2021, 12, 3253–3263 RSC.
- M. P. Hanrahan, L. Men, B. A. Rosales, J. Vela and A. J. Rossini, Chem. Mater., 2018, 30, 7005–7015 CrossRef CAS.
- L. Avram and Y. Cohen, Chem. Soc. Rev., 2014, 44, 586–602 RSC.
- A. J. Ilott, N. M. Trease, C. P. Grey and A. Jerschow, Nat. Commun., 2014, 5, 4536 CrossRef CAS.
- S. Chandrashekar, N. M. Trease, H. J. Chang, L.-S. Du, C. P. Grey and A. Jerschow, Nat. Mater., 2012, 11, 311–315 CrossRef CAS PubMed.
- A. J. Ilott, M. Mohammadi, H. J. Chang, C. P. Grey and A. Jerschow, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 10779–10784 CrossRef CAS.
- M. J. Duer, Introduction to Solid-State NMR Spectroscopy, Wiley-Blackwell, 2005 Search PubMed.
- M. H. Levitt, Spin Dynamics: Basics of Nuclear Magnetic Resonance, John Wiley & Sons, 2013 Search PubMed.
- K. Schmidt-Rohr and H. W. Spiess, Multidimensional Solid-State NMR and Polymers, Elsevier, 2012 Search PubMed.
- H. Kirchhain and L. van Wüllen, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114–115, 71–85 CrossRef CAS.
- P. Florian, D. Massiot, B. Poe, I. Farnan and J.-P. Coutures, Solid State Nucl. Magn. Reson., 1995, 5, 233–238 CrossRef CAS.
- M. ConcistrÈ, O. G. Johannessen, E. Carignani, M. Geppi and M. H. Levitt, Acc. Chem. Res., 2013, 46, 1914–1922 CrossRef PubMed.
- T. Meier, S. Khandarkhaeva, S. Petitgirard, T. Körber, A. Lauerer, E. Rössler and L. Dubrovinsky, J. Magn. Reson., 2018, 292, 44–47 CrossRef CAS.
- A. Chamas, L. Qi, H. S. Mehta, J. A. Sears, S. L. Scott, E. D. Walter and D. W. Hoyt, Magn. Reson. Imaging, 2019, 56, 37–44 CrossRef CAS.
- A. Senocrate, I. Moudrakovski, T. Acartürk, R. Merkle, G. Y. Kim, U. Starke, M. Grätzel and J. Maier, J. Phys. Chem. C, 2018, 122, 21803–21806 CrossRef CAS.
- W.-C. Qiao, J. Wu, R. Zhang, W. Ou-Yang, X. Chen, G. Yang, Q. Chen, X. L. Wang, H. F. Wang and Y.-F. Yao, Matter, 2020, 3, 1–13 CrossRef.
- J. Gottwald, D. E. Demco, R. Graf and H. W. Spiess, Chem. Phys. Lett., 1995, 243, 314–323 CrossRef CAS.
- E. Vinogradov, P. K. Madhu and S. Vega, Chem. Phys. Lett., 2002, 354, 193–202 CrossRef CAS.
- A. Lesage, D. Sakellariou, S. Hediger, B. Eléna, P. Charmont, S. Steuernagel and L. Emsley, J. Magn. Reson., 2003, 163, 105–113 CrossRef CAS.
- P. Hodgkinson, Prog. Nucl. Magn. Reson. Spectrosc., 2005, 46, 197–222 CrossRef CAS.
- B. C. Gerstein, R. G. Pembleton, R. C. Wilson and L. M. Ryan, J. Chem. Phys., 1977, 66, 361–362 CrossRef CAS.
- A. J. Rossini, A. Zagdoun, M. Lelli, A. Lesage, C. Copéret and L. Emsley, Acc. Chem. Res., 2013, 46, 1942–1951 CrossRef CAS.
- A. J. Rossini, A. Zagdoun, F. Hegner, M. Schwarzwälder, D. Gajan, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2012, 134, 16899–16908 CrossRef CAS PubMed.
- S. Björgvinsdóttir, B. J. Walder, A. C. Pinon and L. Emsley, J. Am. Chem. Soc., 2018, 140, 7946–7951 CrossRef PubMed.
- M. Lelli, S. R. Chaudhari, D. Gajan, G. Casano, A. J. Rossini, O. Ouari, P. Tordo, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2015, 137, 14558–14561 CrossRef CAS.
- L. Piveteau, T.-C. Ong, A. J. Rossini, L. Emsley, C. Copéret and M. V. Kovalenko, J. Am. Chem. Soc., 2015, 137, 13964–13971 CrossRef CAS PubMed.
- D. Prochowicz, P. Yadav, M. Saliba, D. J. Kubicki, M. M. Tavakoli, S. M. Zakeeruddin, J. Lewiński, L. Emsley and M. Grätzel, Nano Energy, 2018, 49, 523–528 CrossRef CAS.
- E. M. Mozur, M. A. Hope, J. C. Trowbridge, D. M. Halat, L. L. Daemen, A. E. Maughan, T. R. Prisk, C. P. Grey and J. R. Neilson, Chem. Mater., 2020, 32, 6266–6277 CrossRef CAS.
- A. Kanwat, N. Yantara, Y. F. Ng, T. J. N. Hooper, P. J. S. Rana, B. Febriansyah, P. C. Harikesh, T. Salim, P. Vashishtha, S. G. Mhaisalkar and N. Mathews, ACS Energy Lett., 2020, 5, 1804–1813 CrossRef CAS.
- A. J. Lehner, D. H. Fabini, H. A. Evans, C.-A. Hébert, S. R. Smock, J. Hu, H. Wang, J. W. Zwanziger, M. L. Chabinyc and R. Seshadri, Chem. Mater., 2015, 27, 7137–7148 CrossRef CAS.
- T. Baikie, N. S. Barrow, Y. Fang, P. J. Keenan, P. R. Slater, R. O. Piltz, M. Gutmann, S. G. Mhaisalkar and T. J. White, J. Mater. Chem. A, 2015, 3, 9298–9307 RSC.
- A. Senocrate, I. Moudrakovski, G. Y. Kim, T.-Y. Yang, G. Gregori, M. Grätzel and J. Maier, Angew. Chem., Int. Ed., 2017, 56, 7755–7759 CrossRef CAS PubMed.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, M. Saski, P. Yadav, D. Bi, N. Pellet, J. Lewiński, S. M. Zakeeruddin, M. Grätzel and L. Emsley, J. Am. Chem. Soc., 2018, 140, 3345–3351 CrossRef CAS.
- Z.-J. Yong, S.-Q. Guo, J.-P. Ma, J.-Y. Zhang, Z.-Y. Li, Y.-M. Chen, B.-B. Zhang, Y. Zhou, J. Shu, J.-L. Gu, L.-R. Zheng, O. M. Bakr and H.-T. Sun, J. Am. Chem. Soc., 2018, 140, 9942–9951 CrossRef CAS PubMed.
- Y.-F. Chen, Y.-T. Tsai, L. Hirsch and D. M. Bassani, J. Am. Chem. Soc., 2017, 139, 16359–16364 CrossRef CAS.
- W. M. J. Franssen, S. G. D. van Es, R. Dervişoğlu, G. A. de Wijs and A. P. M. Kentgens, J. Phys. Chem. Lett., 2017, 8, 61–66 CrossRef CAS.
- K. Saalwächter, F. Lange, K. Matyjaszewski, C.-F. Huang and R. Graf, J. Magn. Reson., 2011, 212, 204–215 CrossRef.
- K. Saalwächter, ChemPhysChem, 2013, 14, 3000–3014 CrossRef.
- R. Chatterjee, I. M. Pavlovetc, K. Aleshire, G. V. Hartland and M. Kuno, ACS Energy Lett., 2018, 3, 469–475 CrossRef CAS.
- S. Maheshwari, S. Patwardhan, G. C. Schatz, N. Renaud and F. C. Grozema, Phys. Chem. Chem. Phys., 2019, 21, 16564–16572 RSC.
- B. A. Rosales, L. Men, S. D. Cady, M. P. Hanrahan, A. J. Rossini and J. Vela, Chem. Mater., 2016, 28, 6848–6859 CrossRef CAS.
- B. A. Rosales, M. P. Hanrahan, B. W. Boote, A. J. Rossini, E. A. Smith and J. Vela, ACS Energy Lett., 2017, 2, 906–914 CrossRef CAS.
- A. M. Askar, A. Karmakar, G. M. Bernard, M. Ha, V. V. Terskikh, B. D. Wiltshire, S. Patel, J. Fleet, K. Shankar and V. K. Michaelis, J. Phys. Chem. Lett., 2018, 9, 2671–2677 CrossRef CAS PubMed.
- A. Karmakar, A. Bhattacharya, G. M. Bernard, A. Mar and V. K. Michaelis, ACS Mater. Lett., 2021, 3, 261–267 CrossRef CAS.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381–385 CrossRef CAS.
- M. Aebli, L. Piveteau, O. Nazarenko, B. M. Benin, F. Krieg, R. Verel and M. V. Kovalenko, Sci. Rep., 2020, 10, 8229 CrossRef CAS PubMed.
- M. Mączka, M. Ptak, D. L. M. Vasconcelos, L. Giriunas, P. T. C. Freire, M. Bertmer, J. Banys and M. Simenas, J. Phys. Chem. C, 2020, 124, 26999–27008 CrossRef.
- A. Karmakar, A. M. Askar, G. M. Bernard, V. V. Terskikh, M. Ha, S. Patel, K. Shankar and V. K. Michaelis, Chem. Mater., 2018, 30, 2309–2321 CrossRef CAS.
- M. Ozaki, Y. Katsuki, J. Liu, T. Handa, R. Nishikubo, S. Yakumaru, Y. Hashikawa, Y. Murata, T. Saito, Y. Shimakawa, Y. Kanemitsu, A. Saeki and A. Wakamiya, ACS Omega, 2017, 2, 7016–7021 CrossRef CAS PubMed.
- M. Ha, A. Karmakar, G. M. Bernard, E. Basilio, A. Krishnamurthy, A. M. Askar, K. Shankar, S. Kroeker and V. K. Michaelis, J. Phys. Chem. C, 2020, 124, 15015–15027 CrossRef CAS.
- K. Yamada, K. Fujise, S. Hino, Y. Yamane and T. Nakagama, Chem. Lett., 2019, 48, 749–752 CrossRef CAS.
- K. Yamada, Y. Kuranaga, K. Ueda, S. Goto, T. Okuda and Y. Furukawa, Bull. Chem. Soc. Jpn., 1998, 71, 127–134 CrossRef CAS.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, B. J. Walder and L. Emsley, ACS Energy Lett., 2020, 5, 2964–2971 CrossRef CAS.
- A. Karmakar, G. M. Bernard, A. Meldrum, A. O. Oliynyk and V. K. Michaelis, J. Am. Chem. Soc., 2020, 142, 10780–10793 CrossRef CAS.
- A. M. Askar, G. M. Bernard, B. Wiltshire, K. Shankar and V. K. Michaelis, J. Phys. Chem. C, 2017, 121, 1013–1024 CrossRef CAS.
- M. A. A. Kazemi, P. Raval, K. Cherednichekno, J.-N. Chotard, A. Krishna, A. Demortiere, G. N. M. Reddy and F. Sauvage, Small Methods, 2021, 5, 2000834 CrossRef.
- T. Leijtens, R. Prasanna, A. Gold-Parker, M. F. Toney and M. D. McGehee, ACS Energy Lett., 2017, 2, 2159–2165 CrossRef CAS.
- M. A. Ruiz-Preciado, D. J. Kubicki, A. Hofstetter, L. McGovern, M. H. Futscher, A. Ummadisingu, R. Gershoni-Poranne, S. M. Zakeeruddin, B. Ehrler, L. Emsley, J. V. Milić and M. Grätzel, J. Am. Chem. Soc., 2020, 142, 1645–1654 CrossRef CAS PubMed.
- A. Abate, M. Saliba, D. J. Hollman, S. D. Stranks, K. Wojciechowski, R. Avolio, G. Grancini, A. Petrozza and H. J. Snaith, Nano Lett., 2014, 14, 3247–3254 CrossRef CAS.
- L. Piveteau, M. Aebli, N. Yazdani, M. Millen, L. Korosec, F. Krieg, B. M. Benin, V. Morad, C. Piveteau, T. Shiroka, A. Comas-Vives, C. Copéret, A. M. Lindenberg, V. Wood, R. Verel and M. V. Kovalenko, ACS Cent. Sci., 2020, 6, 1138–1149 CrossRef CAS PubMed.
- I. Spanopoulos, I. Hadar, W. Ke, P. Guo, E. M. Mozur, E. Morgan, S. Wang, D. Zheng, S. Padgaonkar, G. N. M. Reddy, E. A. Weiss, M. C. Hersam, R. Seshadri, R. D. Schaller and M. G. Kanatzidis, J. Am. Chem. Soc., 2021, 143, 7069–7080 CrossRef CAS PubMed.
- J. Lee, W. Lee, K. Kang, T. Lee and S. K. Lee, Chem. Mater., 2021, 33, 370–377 CrossRef CAS.
- J. V. Milić, J. Im, D. J. Kubicki, A. Ummadisingu, J. Seo, Y. Li, M. A. Ruiz-Preciado, M. I. Dar, S. M. Zakeeruddin, L. Emsley and M. Grätzel, Adv. Energy Mater., 2019, 9, 1900284 CrossRef.
- K. L. Svane, A. C. Forse, C. P. Grey, G. Kieslich, A. K. Cheetham, A. Walsh and K. T. Butler, J. Phys. Chem. Lett., 2017, 8, 6154–6159 CrossRef CAS.
- C. Quarti, E. Furet and C. Katan, Helv. Chim. Acta, 2021, 104, e2000231 CrossRef CAS.
- D. Bi, X. Li, J. V. Milić, D. J. Kubicki, N. Pellet, J. Luo, T. LaGrange, P. Mettraux, L. Emsley, S. M. Zakeeruddin and M. Grätzel, Nat. Commun., 2018, 9, 4482 CrossRef PubMed.
- E. A. Alharbi, A. Y. Alyamani, D. J. Kubicki, A. R. Uhl, B. J. Walder, A. Q. Alanazi, J. Luo, A. Burgos-Caminal, A. Albadri, H. Albrithen, M. H. Alotaibi, J.-E. Moser, S. M. Zakeeruddin, F. Giordano, L. Emsley and M. Grätzel, Nat. Commun., 2019, 10, 3008 CrossRef.
- X. Zheng, B. Chen, J. Dai, Y. Fang, Y. Bai, Y. Lin, H. Wei, X. C. Zeng and J. Huang, Nat. Energy, 2017, 2, 1–9 Search PubMed.
- A. Krishna, M. A. A. Kazemi, M. Sliwa, G. N. M. Reddy, L. Delevoye, O. Lafon, A. Felten, M. T. Do, S. Gottis and F. Sauvage, Adv. Funct. Mater., 2020, 30, 1909737 CrossRef CAS.
- T.-S. Su, F. T. Eickemeyer, M. A. Hope, F. Jahanbakhshi, M. Mladenović, J. Li, Z. Zhou, A. Mishra, J.-H. Yum, D. Ren, A. Krishna, O. Ouellette, T.-C. Wei, H. Zhou, H.-H. Huang, M. D. Mensi, K. Sivula, S. M. Zakeeruddin, J. V. Milić, A. Hagfeldt, U. Rothlisberger, L. Emsley, H. Zhang and M. Grätzel, J. Am. Chem. Soc., 2020, 142, 19980–19991 CrossRef CAS.
- Y. Chen, S. R. Smock, A. H. Flintgruber, F. A. Perras, R. L. Brutchey and A. J. Rossini, J. Am. Chem. Soc., 2020, 142, 6117–6127 CrossRef CAS.
- A. A. M. Brown, T. J. N. Hooper, S. A. Veldhuis, X. Y. Chin, A. Bruno, P. Vashishtha, J. N. Tey, L. Jiang, B. Damodaran, S. H. Pu, S. G. Mhaisalkar and N. Mathews, Nanoscale, 2019, 11, 12370–12380 RSC.
- J. Shamsi, D. Kubicki, M. Anaya, Y. Liu, K. Ji, K. Frohna, C. P. Grey, R. H. Friend and S. D. Stranks, ACS Energy Lett., 2020, 5, 1900–1907 CrossRef CAS PubMed.
- A. Abfalterer, J. Shamsi, D. J. Kubicki, C. N. Savory, J. Xiao, G. Divitini, W. Li, S. Macpherson, K. Gałkowski, J. L. MacManus-Driscoll, D. O. Scanlon and S. D. Stranks, ACS Mater. Lett., 2020, 2, 1644–1652 CrossRef CAS PubMed.
- R. Grisorio, E. Fanizza, I. Allegretta, D. Altamura, M. Striccoli, R. Terzano, C. Giannini, V. Vergaro, G. Ciccarella, N. Margiotta and G. P. Suranna, Nanoscale, 2020, 12, 623–637 RSC.
- R. Grisorio, M. E. D. Clemente, E. Fanizza, I. Allegretta, D. Altamura, M. Striccoli, R. Terzano, C. Giannini, M. Irimia-Vladu and G. Paolo Suranna, Nanoscale, 2019, 11, 986–999 RSC.
- G. Almeida, O. J. Ashton, L. Goldoni, D. Maggioni, U. Petralanda, N. Mishra, Q. A. Akkerman, I. Infante, H. J. Snaith and L. Manna, J. Am. Chem. Soc., 2018, 140, 14878–14886 CrossRef PubMed.
- S. R. Smock, T. J. Williams and R. L. Brutchey, Angew. Chem., Int. Ed., 2018, 57, 11711–11715 CrossRef CAS PubMed.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- M. Reddy G. N., R. Ballesteros-Garrido, J. Lacour and S. Caldarelli, Angew. Chem., Int. Ed., 2013, 52, 3255–3258 CrossRef CAS PubMed.
- G. N. M. Reddy, M. Yemloul and S. Caldarelli, Magn. Reson. Chem., 2017, 55, 492–497 CrossRef PubMed.
- J. Gong, M. Yang, X. Ma, R. D. Schaller, G. Liu, L. Kong, Y. Yang, M. C. Beard, M. Lesslie, Y. Dai, B. Huang, K. Zhu and T. Xu, J. Phys. Chem. Lett., 2016, 7, 2879–2887 CrossRef CAS PubMed.
- C. Motta, F. El-Mellouhi, S. Kais, N. Tabet, F. Alharbi and S. Sanvito, Nat. Commun., 2015, 6, 7026 CrossRef CAS PubMed.
- W. M. J. Franssen, C. M. M. van Heumen and A. P. M. Kentgens, Inorg. Chem., 2020, 59, 3730–3739 CrossRef CAS PubMed.
- A. A. Shmyreva, M. Safdari, I. Furó and S. V. Dvinskikh, J. Chem. Phys., 2016, 144, 224201 CrossRef PubMed.
- R. R. Sharp, J. Chem. Phys., 1972, 57, 5321–5330 CrossRef CAS.
- R. R. Sharp, J. Chem. Phys., 1974, 60, 1149–1157 CrossRef CAS.
- J. Korringa, Physica, 1950, 16, 601–610 CrossRef CAS.
- T.-Y. Yang, G. Gregori, N. Pellet, M. Grätzel and J. Maier, Angew. Chem., Int. Ed., 2015, 54, 7905–7910 CrossRef CAS PubMed.
- A. Baumann, S. Väth, P. Rieder, M. C. Heiber, K. Tvingstedt and V. Dyakonov, J. Phys. Chem. Lett., 2015, 6, 2350–2354 CrossRef CAS PubMed.
- J. Haruyama, K. Sodeyama, L. Han and Y. Tateyama, J. Am. Chem. Soc., 2015, 137, 10048–10051 CrossRef CAS PubMed.
- S. Meloni, T. Moehl, W. Tress, M. Franckevičius, M. Saliba, Y. H. Lee, P. Gao, M. K. Nazeeruddin, S. M. Zakeeruddin, U. Rothlisberger and M. Graetzel, Nat. Commun., 2016, 7, 10334 CrossRef CAS PubMed.
- M. H. Futscher, J. Min Lee, L. McGovern, L. A. Muscarella, T. Wang, M. Irfan Haider, A. Fakharuddin, L. Schmidt-Mende and B. Ehrler, Mater. Horiz., 2019, 6, 1497–1503 RSC.
- D. W. Ferdani, S. R. Pering, D. Ghosh, P. Kubiak, A. B. Walker, S. E. Lewis, A. L. Johnson, P. J. Baker, M. Saiful Islam and P. J. Cameron, Energy Environ. Sci., 2019, 12, 2264–2272 RSC.
- A. Pockett, G. E. Eperon, N. Sakai, H. J. Snaith, L. M. Peter and P. J. Cameron, Phys. Chem. Chem. Phys., 2017, 19, 5959–5970 RSC.
- O. Almora, I. Zarazua, E. Mas-Marza, I. Mora-Sero, J. Bisquert and G. Garcia-Belmonte, J. Phys. Chem. Lett., 2015, 6, 1645–1652 CrossRef CAS PubMed.
- C. Li, S. Tscheuschner, F. Paulus, P. E. Hopkinson, J. Kießling, A. Köhler, Y. Vaynzof and S. Huettner, Adv. Mater., 2016, 28, 2446–2454 CrossRef CAS PubMed.
- N. Onoda-Yamamuro, T. Matsuo and H. Suga, J. Chem. Thermodyn., 1991, 23, 987–999 CrossRef CAS.
- K. Yamada, H. Kawaguchi, T. Matsui, T. Okuda and S. Ichiba, Bull. Chem. Soc. Jpn., 1990, 63, 2521–2525 CrossRef CAS.
- T. Shi, H.-S. Zhang, W. Meng, Q. Teng, M. Liu, X. Yang, Y. Yan, H.-L. Yip and Y.-J. Zhao, J. Mater. Chem. A, 2017, 5, 15124–15129 RSC.
- J. Mizusaki, K. Arai and K. Fueki, Solid State Ionics, 1983, 11, 203–211 CrossRef CAS.
- Y. Lin, Y. Bai, Y. Fang, Q. Wang, Y. Deng and J. Huang, ACS Energy Lett., 2017, 2, 1571–1572 CrossRef CAS.
- X. Xiao, J. Dai, Y. Fang, J. Zhao, X. Zheng, S. Tang, P. N. Rudd, X. C. Zeng and J. Huang, ACS Energy Lett., 2018, 3, 684–688 CrossRef CAS.
- F. Jiang, J. Pothoof, F. Muckel, R. Giridharagopal, J. Wang and D. S. Ginger, ACS Energy Lett., 2021, 6, 100–108 CrossRef CAS.
- N. S. Dahod, W. Paritmongkol, A. Stollmann, C. Settens, S.-L. Zheng and W. A. Tisdale, J. Phys. Chem. Lett., 2019, 10, 2924–2930 CrossRef CAS.
- W. Paritmongkol, N. S. Dahod, A. Stollmann, N. Mao, C. Settens, S.-L. Zheng and W. A. Tisdale, Chem. Mater., 2019, 31, 5592–5607 CrossRef CAS.
- A. Gheno, S. Vedraine, B. Ratier and J. Bouclé, Metals, 2016, 6, 21 CrossRef.
- Y. Saygili, H.-S. Kim, B. Yang, J. Suo, A. B. Muñoz-Garcia, M. Pavone and A. Hagfeldt, ACS Energy Lett., 2020, 5, 1271–1277 CrossRef CAS.
- B. Yurash, D. Leifert, G. N. M. Reddy, D. X. Cao, S. Biberger, V. V. Brus, M. Seifrid, P. J. Santiago, A. Köhler, B. F. Chmelka, G. C. Bazan and T.-Q. Nguyen, Chem. Mater., 2019, 31, 6715–6725 CrossRef CAS.
- E. Yalcin, M. Can, C. Rodriguez-Seco, E. Aktas, R. Pudi, W. Cambarau, S. Demic and E. Palomares, Energy Environ. Sci., 2019, 12, 230–237 RSC.
- C. M. Wolff, L. Canil, C. Rehermann, N. Ngoc Linh, F. Zu, M. Ralaiarisoa, P. Caprioglio, L. Fiedler, M. Stolterfoht, S. Kogikoski, I. Bald, N. Koch, E. L. Unger, T. Dittrich, A. Abate and D. Neher, ACS Nano, 2020, 14, 1445–1456 CrossRef CAS PubMed.
- A. Abate, T. Leijtens, S. Pathak, J. Teuscher, R. Avolio, M. E. Errico, J. Kirkpatrik, J. M. Ball, P. Docampo, I. McPherson and H. J. Snaith, Phys. Chem. Chem. Phys., 2013, 15, 2572–2579 RSC.
- J. Hynynen, D. Kiefer, L. Yu, R. Kroon, R. Munir, A. Amassian, M. Kemerink and C. Müller, Macromolecules, 2017, 50, 8140–8148 CrossRef CAS PubMed.
- A. Karki, G.-J. A. H. Wetzelaer, G. N. M. Reddy, V. Nádaždy, M. Seifrid, F. Schauer, G. C. Bazan, B. F. Chmelka, P. W. M. Blom and T.-Q. Nguyen, Adv. Funct. Mater., 2019, 29, 1901109 CrossRef.
- M. T. Seifrid, G. N. M. Reddy, C. Zhou, B. F. Chmelka and G. C. Bazan, J. Am. Chem. Soc., 2019, 141, 5078–5082 CrossRef CAS.
- J. E. Cochran, M. J. N. Junk, A. M. Glaudell, P. L. Miller, J. S. Cowart, M. F. Toney, C. J. Hawker, B. F. Chmelka and M. L. Chabinyc, Macromolecules, 2014, 47, 6836–6846 CrossRef CAS.
- J. Lee, G.-W. Kim, M. Kim, S. A. Park and T. Park, Adv. Energy Mater., 2020, 10, 1902662 CrossRef CAS.
- L. McGovern, M. H. Futscher, L. A. Muscarella and B. Ehrler, J. Phys. Chem. Lett., 2020, 11, 7127–7132 CrossRef CAS.
- M. H. Futscher, M. K. Gangishetty, D. N. Congreve and B. Ehrler, ACS Appl. Electron. Mater., 2020, 2, 1522–1528 CrossRef CAS.
- M. H. Futscher, M. K. Gangishetty, D. N. Congreve and B. Ehrler, J. Chem. Phys., 2020, 152, 044202 CrossRef CAS PubMed.
- Z. Gan, I. Hung, X. Wang, J. Paulino, G. Wu, I. M. Litvak, P. L. Gor'kov, W. W. Brey, P. Lendi, J. L. Schiano, M. D. Bird, I. R. Dixon, J. Toth, G. S. Boebinger and T. A. Cross, J. Magn. Reson., 2017, 284, 125–136 CrossRef CAS PubMed.
- V. Martins, J. Xu, X. Wang, K. Chen, I. Hung, Z. Gan, C. Gervais, C. Bonhomme, S. Jiang, A. Zheng, B. E. G. Lucier and Y. Huang, J. Am. Chem. Soc., 2020, 142, 14877–14889 CrossRef CAS.
- A. Samoson, J. Magn. Reson., 2019, 306, 167–172 CrossRef CAS PubMed.
- F. A. Perras, A. Venkatesh, M. P. Hanrahan, T. W. Goh, W. Huang, A. J. Rossini and M. Pruski, J. Magn. Reson., 2017, 276, 95–102 CrossRef CAS.
- Y. Hong, G. N. Manjunatha Reddy and Y. Nishiyama, Solid State Nucl. Magn. Reson., 2020, 106, 101651 CrossRef CAS.
- A. Venkatesh, M. P. Hanrahan and A. J. Rossini, Solid State Nucl. Magn. Reson., 2017, 84, 171–181 CrossRef CAS.
- H. Nagashima, J. Trébosc, Y. Kon, K. Sato, O. Lafon and J.-P. Amoureux, J. Am. Chem. Soc., 2020, 142, 10659–10672 CrossRef CAS PubMed.
- C. E. Avalos, B. J. Walder, J. Viger-Gravel, A. Magrez and L. Emsley, Phys. Chem. Chem. Phys., 2019, 21, 1100–1109 RSC.
- K. Takeda, Solid State Nucl. Magn. Reson., 2012, 47–48, 1–9 CrossRef CAS PubMed.
- H. Al-Johani, E. Abou-Hamad, A. Jedidi, C. M. Widdifield, J. Viger-Gravel, S. S. Sangaru, D. Gajan, D. H. Anjum, S. Ould-Chikh, M. N. Hedhili, A. Gurinov, M. J. Kelly, M. El Eter, L. Cavallo, L. Emsley and J.-M. Basset, Nat. Chem., 2017, 9, 890–895 CrossRef CAS.
- A. R. Oganov, C. J. Pickard, Q. Zhu and R. J. Needs, Nat. Rev. Mater., 2019, 4, 331–348 CrossRef.
- P. Hodgkinson, Prog. Nucl. Magn. Reson. Spectrosc., 2020, 118–119, 10–53 CrossRef CAS PubMed.
- D. L. Bryce, IUCrJ, 2017, 4, 350–359 CrossRef CAS.
- The Cambridge Structural Database (CSD), https://www.ccdc.cam.ac.uk/solutions/csd-core/components/csd/ Search PubMed.
- Inorganic Crystal Structure Database, https://icsd.products.fiz-karlsruhe.de/ Search PubMed.
- The Materials Project, https://materialsproject.org/ Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |