
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress on the synthesis and oxygen reduction applications of Fe-based single-atom and double-atom catalysts
Yan
Yan†
a,
Haoyan
Cheng†
b,
Zehua
Qu
c,
Rui
Yu
a,
Fan
Liu
a,
Qianwen
Ma
a,
Shuang
Zhao
 a,
Hao
Hu
*b,
Yu
Cheng
e,
Chongyang
Yang
a,
Zaifeng
Li
*d,
Xu
Wang
a,
Shaoyu
Hao
a,
Yuyu
Chen
a and
Mingkai
Liu
*ad
a,
Hao
Hu
*b,
Yu
Cheng
e,
Chongyang
Yang
a,
Zaifeng
Li
*d,
Xu
Wang
a,
Shaoyu
Hao
a,
Yuyu
Chen
a and
Mingkai
Liu
*ad
aSchool of Chemistry & Materials Science, Jiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, Jiangsu Normal University, Xuzhou 221116, P. R. China. E-mail: liumingkai@jsnu.edu.cn
bCollaborative Innovation Center of Nonferrous Metals, School of Materials Science and Engineering, Henan University of Science and Technology, Luoyang, 471023, China. E-mail: huhao@haust.edu.cn
cState Key Laboratory of Molecular Engineering of Polymers, Department of Macromolecular Science, Fudan University, Shanghai 200433, China
dState Key Laboratory Base of Eco-chemical Engineering, Qingdao University of Science and Technology, No. 53 Zhengzhou Rd, Qingdao, Shandong, China 266042. E-mail: lizfengphd@126.com
eHubei Key Laboratory of Polymer Materials, Ministry-of-Education Key Laboratory for the Green Preparation and Application of Functional Materials, School of Materials Science & Engineering, Hubei University, Wuhan 430062, China
First published on 6th July 2021
Abstract
Single-atom and double-atom catalysts have emerged as a new Frontier in many fields due to their high atom-utilization efficiency, excellent catalytic properties and good durability. In this decade, Fe-based atomic catalysts have exhibited great advantages toward the oxygen reduction reaction (ORR) owing to the high utilization efficiency of their single/dual atomic sites, well-defined active sites and high selectivity toward the four-electron (4e−) pathway. To explore their catalytic behavior, many efforts have been made to synthesize Fe-based atomic catalysts. In addition, the high performance of Fe-based single/dual atoms toward ORR is mainly attributed to the critical coordination condition and the corresponding electronic structures. Currently, it is a great challenge to achieve high activity and good stability with a high content of metal atoms, while maintaining their atomic dispersion. In this review, we begin with a systematic review about the influence of the coordination environment of Fe-based single/dual-atom catalysts on their electrocatalytic properties, followed by an overview of some new strategies to improve their coordination environment. Then, we present a comprehensive summary on the advantages of their unique structures, catalytic activity for the ORR and further applications. Finally, the remaining key challenges and future opportunities in this emerging field are concluded to provide more useful clues for the development of advanced catalysts.
 Yan Yan | Yan Yan is an associate professor in the School of Chemistry and Materials Science at Jiangsu Normal University. She earned her BS degree in Materials Chemistry at Henan University, and later received her PhD in Inorganic Chemistry from Sun Yat-Sen University in 2014. She studied at the Georgia Institute of Technology as a visiting scholar from 2019–2020. Her research interests focus on nanocarbon materials, advanced energy storage/conversion, lithium–sulfur batteries, and catalysis. |
 Hao Hu | Hao Hu is an associate professor in the School of Materials Science and Engineering and director of the Nonferrous Metal and New Energy Materials Lab at Henan University of Science and Technology. He received his BS and PhD at Central China Normal University in 2012 and 2019, respectively. He joined Prof. Meilin Liu's group as a joint PhD student at the Georgia Institute of Technology from November 2016 to November 2018. His research interests mainly focus on efficient energy storage and conversion, including metal-ion batteries, interface electrochemistry, and catalysis. |
 Zaifeng Li | Prof. Zaifeng Li acquired his PhD in the college of chemical engineering from the China University of Petroleum (Beijing) in 1998. Between 1998–2001, he worked at Qingdao Institute of Chemical Engineering, and in 2001–2002 he spent his post-doctoral career at the National Research Council Canada (NRC) in Ottawa. In 2002, he came back to China and has since been working at Qingdao University of Science and Technology. He served as the director of the State Key Laboratory Base of Eco-chemical Engineering and now he is the director of science and technology at Qingdao University of Science and Technology. |
 Mingkai Liu | Mingkai Liu is currently an Associate Professor in the School of Chemistry and Materials Science at Jiangsu Normal University. He received his PhD Degree from Fudan University in 2014. He worked as a Postdoctoral Fellow at the Georgia Institute of Technology from 2018 to 2020. His research focuses on the rational design and synthesis of nanomaterials for applications in energy storage and catalytic processes. |
1. Introduction
Advanced catalysts for electrochemical reactions are critical in the development of renewable energy.1 Many typical chemical reactions such as the oxygen reduction reaction (ORR), electrochemical reduction of CO2 (CO2RR), hydrogen evolution reaction (HER), oxygen evolution reaction (OER), and nitrogen reduction reaction (NRR) involve critical chemical transformation processes, which need catalysts to overcome the reaction energy barrier or effectively enhance the reaction rate.2–5 The activity, selectivity and stability of catalysts play a pivotal role in determining the quality of the product during the processes of reactant adsorption, intermediate conversion and product desorption.1,6–10 However, the high cost and low utilization efficiency of bulk catalysts remain obstacles for their broad commercial application. Substantially, for the catalytic process, the reaction generally happens on the surface or interface of the catalyst, accompanied by the adsorption of reactants, their subsequent conversion into products, and then desorption from the active sites.11 For bulk catalysts, only a small fraction of the surface atoms are involved in the catalytic reactions. Thus, an effective strategy to increase the utilization efficiency of catalysts is to increase the proportion of atoms exposed on the surface by reducing their particle size.12 Catalysts based on ultra-small particles or well-controlled nanostructures exhibited more efficient utilization and enhanced reactivity because of their relatively high surface-to-volume ratio.13 When the size of a catalyst is reduced to the atomic level, its interaction with the reactants is significantly different from that of nanocrystals or nanoclusters, which may originate from its special electronic structure, geometry, as well as its charge transfer with the substrate.14,15 These characteristics of atomically dispersed catalysts make them equipped with high unsaturated coordination and activity.Single-atom catalysts (SACs), possessing isolated active sites, tailored electronic structure, special quantum size effects, coordination environments, and strong metal-substrate interactions, have attracted widespread attention in heterogeneous catalysis due to their 100% atomic utilization efficiency and excellent catalytic performance.16–30 The unique active centers in SACs have been proven to accelerate the thermal catalytic reaction, organic catalytic reaction, ORR, CO2RR, NRR, and OER.31–41 Among the SACs, nonprecious metal catalysts with the advantages of natural abundance and low cost have recently emerged as a new Frontier in the field of catalysis.42,43 For example, a series of SACs with nitrogen-coordinated transition metal sites, such as M–N–C (M = Fe, Co, Ni, etc.), has been widely studied, and their ORR activity follows the order of Fe > Co > Ni.4,44 The special electronic structure of Fe-based SACs can be easily modified using metallic or inorganic coordination structures to improve the adsorption and/or desorption of adsorbates on the catalyst surface, leading to comparably high activity, desirable stability and anti-poisoning effect.45 In addition, Fe-based SACs show good catalytic performance in both acidic and alkaline electrolytes, making them promising catalysts for the ORR in fuel cells.46
Meanwhile, the environment is under severe threat due to the overuse of fossil fuels, resulting in climate change and environmental disruption. The dramatic increase in the carbon dioxide (CO2) concentration in the atmosphere compels us to develop sustainable energy technologies. Proton exchange membrane fuel cells (PEMFCs) and metal–air batteries are considered promising systems for supplying high energy densities and sustainable clean energy with low or zero pollution emissions.47–52 However, both PEMFCs and metal–air batteries suffer from the kinetically sluggish ORR at the oxygen/air cathode, which is the main reaction in the electrochemical conversion process.53–58 Thus, currently, to boost the ORR kinetics for practical applications, low-cost and highly active electrocatalysts are much more desirable than in the last few decades. Besides the expensive platinum (Pt), Fe-based SACs have been widely investigated and reported to possess comparable electrochemical activities toward the ORR, while maintaining their characteristic of ultralow cost. Well-constructed substrates can help develop Fe-based SACs with high activity, while deep investigation of the coordination environment around the metal single atoms can contribute to establishing Fe SACs with a high metal content and good life span.
Among the reported strategies for the construction of Fe-based SACs, Fe–Nx coordinated species in the carbon framework of Fe–N–C catalysts are considered as the most important active centers, as evidenced by structural characterizations and electrochemical results.59,60 Carbon nanomaterials, with good conductivity and excellent porous structures,61–70 are considered desirable loading substrates for guest molecules. Density functional theory (DFT) calculation indicated that the edge-hosted Fe–N4 sites can tailor the bonding configuration of nitrogen (N) and decrease the overall reaction barriers.71 To achieve the optimized binding energy for catalytic activity and improve the active site density of Fe in Fe-based SACs, the design and synthesis of carbon-based Fe–N–C nanomaterials by tuning the coordination number of N atoms has received great attention. Also, introducing heteroatoms (P, S, B and F, etc.) or defects has been confirmed to be a beneficial method to regulate the electronic structure of the center metal atom and control the electronic configuration of the substrate.72–84 These heteroatoms doping strategies can be further used to modify the coordination environment of the Fe sites.
To date, besides introducing heteroatoms for the construction of Fe–N–C SACs, another effective method for improving the catalytic activity of Fe-based catalysts is to exploit a second metal component as dual-atomic catalysts (DACs) to boost their synergistic effect. According to the d-band electronic model, the molecular interaction between adsorbates and the metal surface has a deep relationship with the d-band center shift. The electronic structures of Fe–N–C SACs can be more readily adjusted by introducing heteronuclear metal atoms with different d-band configurations such as Ni, Co, and Zn.85–87 This electronic modification can significantly facilitate the adsorption and/or desorption of adsorbates on the catalyst surface. Therefore, the selective combination of single-atom Fe and other metal sites endow SACs with excellent bi-functional property and synergistic effect. In addition, synergistic metal active sites such as Fe–Ni and Fe–Co are beneficial for complicated electrocatalytic reactions, which include multiple reaction steps and reaction intermediates.88,89 Moreover, the introduction of a second metal further makes the catalysts more adaptable to harsh reaction conditions.
Considering the ever-increasing number of atomic catalysts based on Fe for electrocatalysis, it is highly desirable to provide timely updates on Fe-based SACs and DACs with emphasis on the strategies to optimize their coordination environment and further improve their catalytic activity, which is important for researchers to get a systematic understanding of Fe-based atomic catalysts. In this review, we present the recent progress on the Fe-based atomic catalysts and summarize different synthetic strategies to tune the electronic and geometrical structure of their active centers (Scheme 1), which may promote the development of Fe-based atomic electrocatalysts. Firstly, the coordination of nitrogen atoms with Fe is briefly discussed, followed by the methods for their construction and ORR catalytic performance. Then, other heteroatoms including B, P, S and F that are employed to modify the coordination environment of the loaded Fe atoms are also briefly introduced. The guest metal atoms that are utilized to regulate the coordination environment of Fe atoms are also summarized and discussed. Finally, with an emphasis on practical applications throughout this review, we provide insight into the present challenges and the future opportunities of Fe-based SACs/DACs, which may shed light on the development of more effective and environmentally friendly electrocatalysts for fuel cells and metal–air batteries.
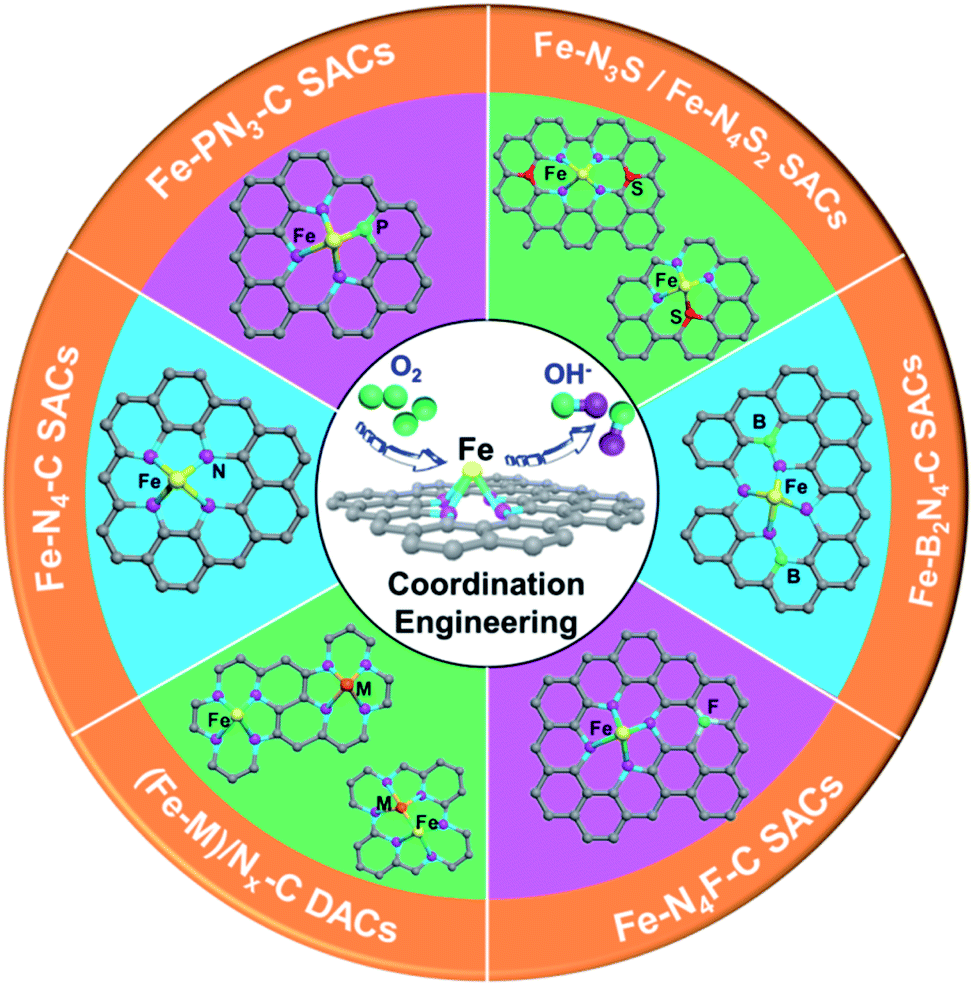 | ||
| Scheme 1 Strategies for regulating the coordination environment of Fe-based SACs/DACs. | ||
2. Fe-based SACs/DACs regulated by coordination engineering
2.1 Fe-based SACs coordinated as Fe–N4
Among the non-Pt group metal catalysts, the metal nitrogen-coordinated carbon nanomaterials, especially Fe (or Co)–N–C, have been confirmed to have good ORR catalytic activity. These active cathode catalysts were first discovered in 1964,90 and following investigations and theoretical calculation have demonstrated that the Fe–Nx species was the primary active sites toward oxygen reduction. Subsequently, many studies showed that Fe–N–C catalysts possess effective ORR activity, which is comparable or even superior to that of commercial Pt/C due to their high activity, resulting from the anchored Fe active sites and their 4e− selectivity.90–95 Besides catalytic activity toward the ORR, Fe–N–C catalysts also show remarkable catalytic activity in other fields, such as the OER,96 HER,97 CO2RR,98,99 N2RR,100 and degradation of organic contaminants.101 By using carbon nanomaterials with high conductivity, specific surface area and abundant defect sites as the loading substrate, Fe–N–C catalysts have made substantial progress in electrocatalysis. Usually, Fe–N–C catalysts can be synthesized via heat treatment of a mixture of iron salt, nitrogen source (aromatic or aliphatic ligand or other nitrogen-rich molecule) and carbon precursor. Heteroatom doping of N between the Fe atoms and the carbon matrix can effectively improve the stability of the anchored Fe atoms and also increase the number of introduced Fe active sites.102 In this section, we mainly focus on the coordination of Fe atoms with pure N elements.The structure of the FeN4 complex is commonly studied in Fe–N–C catalysts. The central Fe atom is coordinated with four N atoms to form an active FeN4 site. These complexes not only can carry out fast redox reactions toward the ORR, but also exhibit excellent electrocatalytic activity in the OER, HER, CO2RR and other reactions.101,103,104 Zhou reported an atomic Fe and N co-doped ordered mesoporous carbon nanosphere (meso-Fe−N–C), which showed excellent ORR activity (Fig. 1a and b).90 Meso-Fe–N–C possesses a hierarchical structure with a high surface area of 494.7 m2 g−1 and high mass loading of Fe atoms (2.9 wt%) and abundant N sites (4.4 wt%) (Fig. 1c and d). With these beneficial structural characteristics, meso-Fe–N–C exhibited excellent activity and durability, outperforming the state-of-the-art Pt/C electrocatalysts toward the ORR (Fig. 1e and f). An efficient three-dimensional (3D) N-doped ordered mesoporous carbon (N-OMC) supported Fe–C–N catalyst (Fe–N–C/NOMC) showing outstanding ORR performance was reported by Han et al. Fe–N–C/N-OMC with Fe–N–C sites was embedded in a three-dimensional (3D) N-doped ordered mesoporous carbon framework. The obtained Fe–N–C/N-OMC showed a high half-wave potential, kinetic current density, turnover frequency, mass activity towards the ORR in alkaline electrolyte, and also exhibited comparable ORR activity to commercial Pt/C in acidic electrolyte, which further achieved a high open-circuit voltage, high power density and remarkable durability in the assembled zinc-air battery (Fig. 1g and h). Experiments and theoretical calculations suggest that the ultra-high ORR activity originated from the boosted intrinsic activity of the FeN4 sites by the graphitic N dopants, high density of accessible active sites generated by the high Fe and N loadings and ordered mesoporous carbon structure, together with facilitated mass and electron transport in its 3D interconnected pores. For PEMFC, the cathode catalyst needs to work for a long period in an acidic environment, and thus it is necessary to develop ORR catalysts with good acid resistance.105 Zhang and co-authors reported the synthesis of a high-performance atomic Fe catalyst derived from chemically Fe-doped zeolitic imidazolate frameworks (ZIFs) by directly bonding Fe ions to imidazolate ligands within 3D frameworks. The new atomic Fe catalyst achieved acceptable ORR activity in challenging acidic medium (0.5 M H2SO4), showing a half-wave potential of 0.85 V (vs. reversible hydrogen electrode, RHE), only leaving a 30 mV gap with Pt/C (60 μgPt/cm2). Enhanced stability was attained with the same catalyst with the loss of only 20 mV after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 potential cycles (0.6 to 1.0 V) in O2 saturated acid solution (Fig. 1i and j).106 The high-performance atomic Fe PGM-free catalyst has great potential to replace Pt in future PEMFCs.
000 potential cycles (0.6 to 1.0 V) in O2 saturated acid solution (Fig. 1i and j).106 The high-performance atomic Fe PGM-free catalyst has great potential to replace Pt in future PEMFCs.
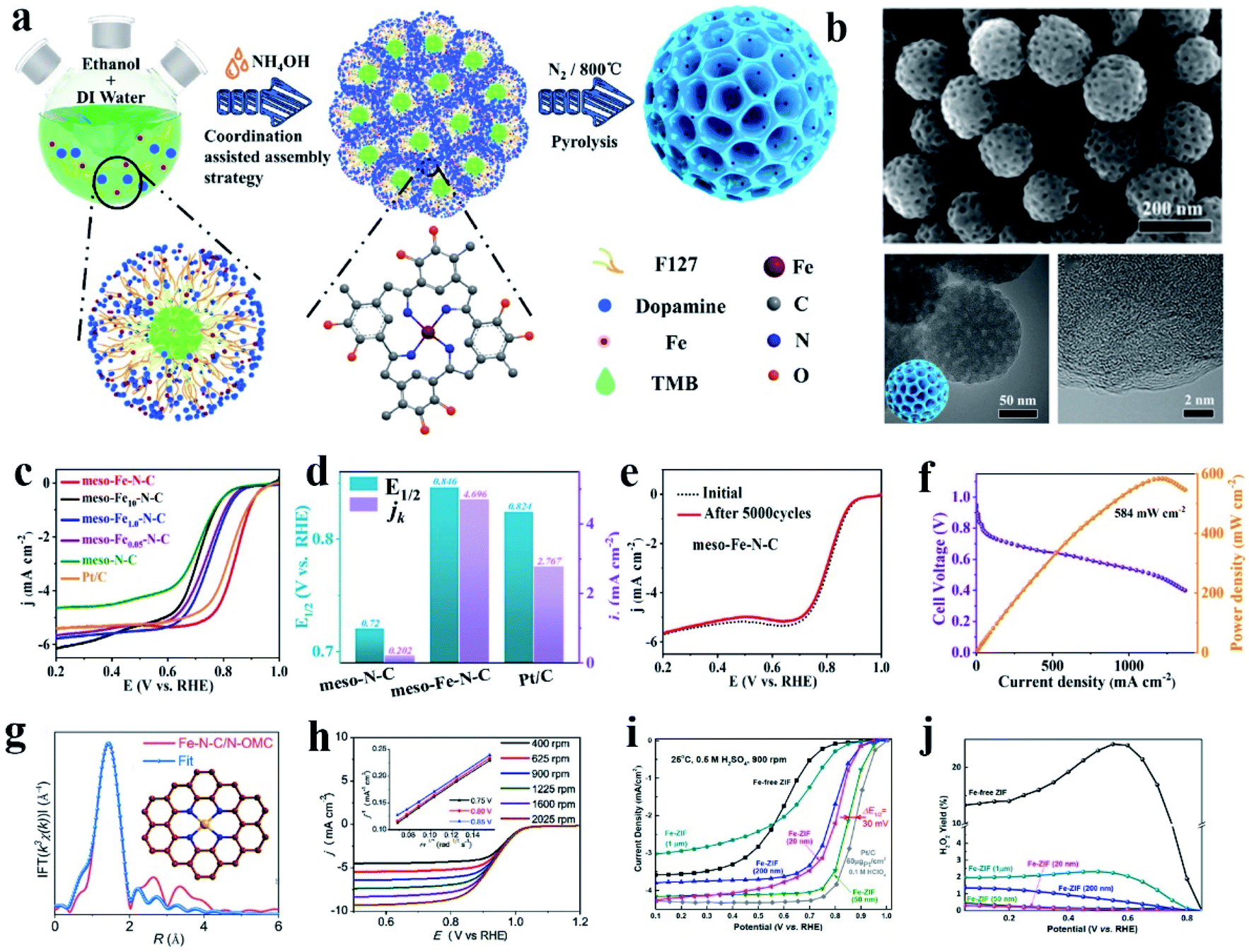 | ||
| Fig. 1 (a) Procedure for the synthesis of an atomic Fe-decorated N-doped mesoporous carbon sphere (meso-Fe–N–C). (b) SEM and TEM images of meso-Fe–N–C materials. (c) ORR curves of different materials, with meso-Fe–N–C loading of 0.4 mg cm−2. (d) Half-wave potential, E1/2, and kinetic current density, jk, for meso-N–C, meso-Fe–N–C, and Pt/C. (e) ORR curves of meso-Fe–N–C before and after 5000 cycles, loading: 0.2 mg cm−2. (f) H2–O2 fuel cell polarization curves and power density plots of MEA using meso-Fe–N–C (loading of 4.0 mg cm−2) as cathode catalysts. Membrane: Alkymer W-25, temperature: 80 °C, electrode area: 1 cm2. Reproduced with permission.90 Copyright 2021, the American Chemical Society. (g) Corresponding FT-EXAFS fitting curves of Fe–N–C/N-OMC with the inset showing the schematic model of the FeN4 center: Fe (yellow), N (blue) and C (brown). (h) Polarization curves of Fe–N–C/N-OMC catalyst at different rotating speeds in the range of 400 to 2025 rpm with the inset showing the K–L plots at different potentials. Reproduced with permission.105 Copyright 2020, Elsevier. (i) ORR polarization plots for Fe-ZIF-derived catalysts in 0.5 M H2SO4 and Pt/C catalysts (60 gPt/cm2) in 0.1 M HClO4 at 25 °C and 900 rpm. (j) Calculated H2O2 yield for Fe-ZIF catalysts as a function of particle size. Reproduced with permission.106 Copyright 2017, the American Chemical Society. | ||
2.2 Fe-based SACs coordinated by phosphorus (P) heteroatom
In recent decades, many strategies have been developed to tune the surrounding environment of atomic metal centers. A suitable coordination structure can help optimize the ORR performance of SACs. Besides N atoms, several other heteroatoms including P, S, and B can be employed as coordination atoms for improving the catalytic activity and selectivity of SACs/DACs by tuning their electronic structures.107,108 Recent research has found that the ORR capability of single metallic sites can be greatly enhanced by introducing P or S coordination atoms, which has been attributed to the discrepancy in electron spin density and electronegativity between carbon and P/S and the charge polarization.109–111 The electron cloud distribution induced by the incorporation of heteroatoms and metal can result in much improved oxygen adsorption, and subsequent O–O double bond cleavage.112 It has been confirmed that the introduction of dual dopants tends to improve the ORR catalytic activity substantially compared to mono-doping.P, as a dopant, has attracted significant attention because it belongs to the same group as nitrogen in the periodic table, and thus has the same number of valence electrons and similar chemical properties as N.107 Compared with N, P has lower electronegativity and a larger covalent radius. P doping can change the structure and activity of carbon more effectively, and thus P-doped carbon has attracted great interest.113–115 Theoretical calculations show that P-doped carbon materials have higher electron–donor properties, higher adsorption capacity for O2 and higher oxidation potential than N atoms, indicating that P-doped carbon materials are promising ORR catalysts.116,117 In recent years, theoretical and experimental studies on phosphorus coordination active sites have been carried out, including P coordination metal (M–Px)112,118,119 and N, P double-coordination metal (M–NxPy).120,121 However, the activity of Fe–P–C catalysts is still insufficient due to the poor control of the dispersion and distribution of Fe and P sources, and the local pore structure. Therefore, Li et al. prepared ordered mesoporous N-free electrocatalysts with Fe–Px active sites via a wet impregnation nanocasting method using (Ph3P)2Fe(CO)3 containing Fe–P coordination centers as precursors.122 The results showed that the catalytic performance of the prepared Fe–P–C for the ORR was superior to that of other Fe–P–C catalysts in alkaline and acidic media. Deconvolution of the P 2p signal gave the binding energy peak centered at 132.5 eV, which can be assigned to P–C, indicating that the P atom was successfully bonded to carbon. Furthermore, the pore size of the mesoporous Fe–P–C increased with an increase in the treatment temperature. The FT-IR spectrum of Fe–P–C-900 showed absorption bands at 581 cm−1 and 632 cm−1, corresponding to the stretching vibration of the Fe–P and Fe–C species, respectively, which further confirmed the formation of Fe–P–C active sites in the catalyst. In addition, a series of ORR catalysts composed of metal and P-doped carbon was synthesized via an in situ polymerization process coupled with carbonization. Relevant research showed that the pyrolysis temperature has an important impact on the microstructure of Fe–P–C and the doping content of P and Fe in carbon, and further affects the catalytic activity of Fe–P–C in the ORR.123 For example, using resorcinol and formaldehyde as carbon sources, tetraphenylphosphine bromide as a phosphorus source, and iron nitrate as an iron source, P, Fe co-doped carbon was prepared via in situ polymerization.119 Using Raman spectroscopy, it was found that an increase in ID/IG indicates the an increase in the amount of defects in carbon, which is the result of the incorporation of heteroatoms into the carbon aerogels. Due to the high doping and large specific surface area of P and Fe, the prepared Fe–P–C-900 electrocatalyst showed good catalytic activity towards the ORR, and its long-term stability was better than that of commercial Pt/C (20 wt%).
It has been reported that P and N co-doped carbon display enhanced acidic ORR activity in comparison to N-doped carbon and B, N-co-doped carbon124 because the P-induced asymmetric charge density can increase the charge delocalization of the nearby carbon atoms, resulting in the 4e− pathway required for the ORR. Afterwards, researchers further developed a series of SACs supported by P–N-doped carbon (M–N/P–C) for catalyzing the ORR process.125–127
For the catalyst with the M–NxPy configuration, the P atoms can connect with the M–Nx moieties in the carbon structure to form a P–M–Nx bond. Replacing N with P will significantly change the electrocatalytic properties of the central M atom because P disrupts the original N–M coordination bond length and electropositivity of the M atom.128,129 At present, the investigation on Fe coupled with N, P-doped carbon-based catalysts (Fe–N, P–C) has become a research hotspot. Firstly, viewed from the intrinsic structure of the heteroatoms, P has a larger radius and lower electronegativity than N and C (N (3.04) > C (2.55) > P (2.19)). Also, P-doped Fe SACs show superior catalytic activity in comparison with N-coordinated samples.130 The configuration of the active centers of N/P dual-coordination Fe are favorable for the adsorption/desorption of oxygen intermediates, which can accelerate the reaction kinetics and improve the catalytic ORR performance. Simultaneously, the optimized Fe–N/P–C catalyst shows good ORR catalytic activity due to its abundant active sites, large specific surface area and hierarchical porous structure.131 For example, Yuan et al. synthesized carbon nanosheets embedded with N and P dual-coordinated SACs via high-temperature pyrolysis treatment, denoted as Fe–N/P–C (FeN3P) (Fig. 2a and b).132 Extended XAFS (EXAFS) data indicates that the Fe atom in Fe–N/P–C-700 was coordinated with three N atoms and one P atom, resulting in the formation of a square-planar configuration (as shown in Fig. 2c). It has been found that when O2 is adsorbed on the Fe–N3P site, one oxygen atom is bound to the Fe atom and the other oxygen atom is bound to the P atom. This structure will promote the cleavage of the O–O bond. The adsorption energy of O2 on the Fe–N3P active site is −1.01 eV, which is lower than that on Fe–N4. The unique structure of FeN3P resulted in an excellent ORR performance in 0.1 M KOH with the most positive onset potential (Eonset = 0.941 V) and half-wave potential (E1/2 = 0.867 V vs. reversible hydrogen electrode) (Fig. 2d and e). Compared with Fe–N4 (endothermic by 1.02 eV), Fe–N3P (endothermic by 0.85 eV) is more thermodynamically conducive to the reduction of *OH to OH−. In addition, Fe–N/P–C-700 also showed good catalytic activity for oxygen evolution in alkaline solution. Fe–N/P–C-700 showed a small potential of 1.66 V at 10 mA cm−2 (Ej = 10) (Fig. 2f), which is comparable to that of RuO2 (1.61 V). Similarly, ZIF-8 was treated with triphenylphosphine and ferrous sulfate to form a new C–P–Fe–Nx–P–C system as an ORR catalyst (Fig. 2g). Different from Fe–N–C, the CPFeNPC curve moved to a larger R space, which was ascribed to the longer bond length of Fe–P than that of Fe–N. This result confirms the effect of the incorporation of P on the Fe binding environment. Research results demonstrated that regardless of alkaline or acidic conditions, CPFeNPC possesses greatly enhanced ORR activity, and its activity is higher than that of Fe–N–C and comparable to that of commercial Pt/C (Fig. 2h).133
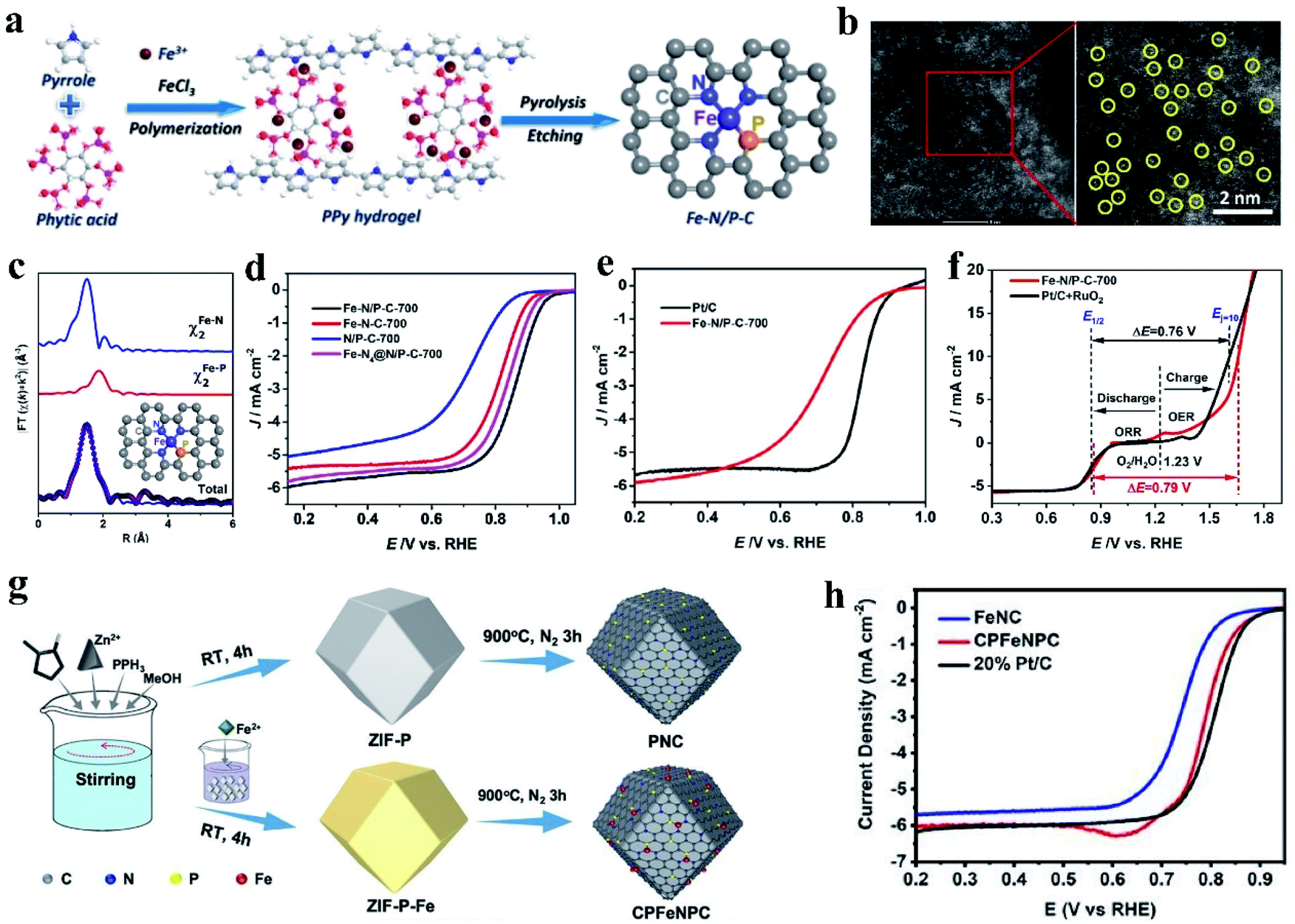 | ||
| Fig. 2 (a) Schematic of the synthesis of the Fe–N/P–C catalyst. (b) Aberration-corrected HAADF-STEM image and enlarged image of Fe–N/P–C-700, partial single Fe atoms are circled in yellow. (c) EXAFS analysis of Fe–N/P–C-700 at R space. (d) ORR polarization curves of Fe–N/P–C-700, Fe–N–C-700, N/P–C-700, and Fe–N4@N/P–C-700 catalysts at a rotating speed of 1600 rpm in 0.1 M KOH solution. (e) LSV curves of Fe–N/P–C-700 and Pt/C. (f) Overall polarization plots (1600 rpm) in the ORR and OER potential window for Fe–N/P–C-700 and commercial Pt/C + RuO2 catalysts, and the inset depicts the overall electrocatalytic oxygen performance (ΔE = Ej = 10 − E1/2). Reproduced with permission.131 Copyright 2020, the American Chemical Society. (g) Preparation of PNC and CPFeNPC. (h) ORR activities of the PNC and CPFeNPC catalysts in 0.1 M KOH electrolyte. Reproduced with permission.133 Copyright 2020, The Royal Society of Chemistry. | ||
For catalysts with the M–NxPy configuration, P may also dope in the carbon matrix by replacing an arbitrary carbon. P-Doped single-atom Fe–N–C catalysts were synthesized by employing a self-assembly strategy using phytic acid-modified polypyrrole nanowire. DFT calculation showed that P doping can adjust the electronic structure around the FeN4 group, thus reducing the energy barrier in the rate-determining step and improving the ORR activity. The ORR processes on the FeN4CP1 and FeN4CP3 models have a lower theoretical overpotential and thermodynamic overpotential than that of the FeN4C model, which indicates that P-doped Fe–N–C catalysts are more conducive to the ORR process in terms of energy.134
2.3 Fe-based SACs coordinated by sulfur (S) heteroatom
In addition to introducing P into M–N catalysts, S doping can also effectively change the electron distribution of doped carbon materials, which will contribute to their catalytic property in the reaction process.135,136 The relatively large atomic radius of S made it possible to induce defects on the carbon support and the lower electronegativity of S (N (3.04) > S (2.58) ≈ C (2.55)) was expected to modify the electronic structures of the Fe–N active center.137,138 S is considered an excellent dopant to provide positive effects on catalytic activity for the ORR and OER.110 In the presence of S heteroatoms, coordination bonding with M (Ni, Co, Cu)–N–C catalysts occurs with M–S bonds, resulting in NiN3S, CoN3S, and CuN3S atomic sites.139,140 For example, Yang et al. synthesized Ni–N3S1 by pyrolyzing a mixture of Ni salt and small organic molecules as the N and S sources. Compared with NiN4, NiN3S1 substantiated the positive effect of S coordination for improving the electrochemical catalytic activity for the CO2RR.141 In the case of Fe–N–C, the formed coordination bond between the introduced S and the single metal is different. Zhang and co-workers synthesized FeN4S2 (Fe–SAs/NSC), CoN3S1 (Co–SAs/NSC) and NiN3S1 (Ni–SAs/NSC) by mixing metal ions (Fe3+, Co2+ and Ni2+) with a single carbon precursor of 1-allyl-2thiourea, which contains N and S, followed by pyrolysis at 900 °C under an inset atmosphere for 3 h.136 The XANES spectra indicated that no Fe–Fe bonds, Co–Co bonds, and Ni–Ni bonds existed in Fe SAC, Co SAC, and Ni SAC, respectively. In addition, Co–S bonds and Ni–S bonds were observed in the Fourier transformed (FT) k2-weighted χ(k) function of the EXAFS spectra. However, no Fe–S bond was observed. The above-mentioned results showed that Fe SAC remained as FeN4S2, while Co SAC and Ni SAC remained as CoN3S1 and NiN3S1, respectively.142 The different affinities of Fe and Co/Ni ions towards S-containing ligands result in different organic complex intermediates, thus generating distinct final structures and products.In the case of Fe-NSC SACs, one of the effective coordination strategies is to coordinate each Fe atom with four N atoms, and the S atoms are not directly bonded to the Fe atom (Fig. 3a). For instance, Jia et al. synthesized an S-doped Fe-NC SAC via solvothermal treatment and annealing, which was denoted as Fe-NSC (Fig. 3b and c).143 The electrochemical test results showed that the ORR performance of Fe-NSC was significantly improved compared with that of the Fe–N–C catalyst without S. In 0.1 M KOH, the initial potential was 1.09 V and the half-wave potential was 0.92 V (Fig. 3d). Simultaneously, the acid ORR performance of Fe-NSC was equivalent to that of Pt/C, which is 0.78 V E1/2. DFT simulation showed that the significant increase in ORR activity can be attributed to the charge enrichment induced by S doping with S substituting an N atom, which can achieve the best O2 binding and rapid electron transfer. Similarly, Li et al. reported a novel pyrrole–thiophene copolymer pyrolysis strategy to synthesize Fe-isolated single atoms on S and N co-doped carbon (Fe-ISA/SNC).37 The Fe atom is coordinated by four N atoms and partial S coordination with N in Fe-ISA/SNC. The catalytic efficiency of Fe-ISA/SNC increased with S doping. The half-wave potential of Fe-ISA/SNC is 0.896 V (vs. RHE), which is higher than that of single Fe atom on nitrogen-codoped carbon (Fe-ISA/NC, 0.839 V), commercial Pt/C (0.841 V) and most reported non-noble metal catalysts. Moreover, Fe-ISA/SNC exhibited outstanding methanol tolerance, and its activity decay during 15000 voltage cycles under alkaline conditions was negligible. The doped S changes the charge on the N atoms around the Fe reaction center. The enriched charge promotes the rate-limiting reduction release of OH*, showing a high ORR catalytic performance.
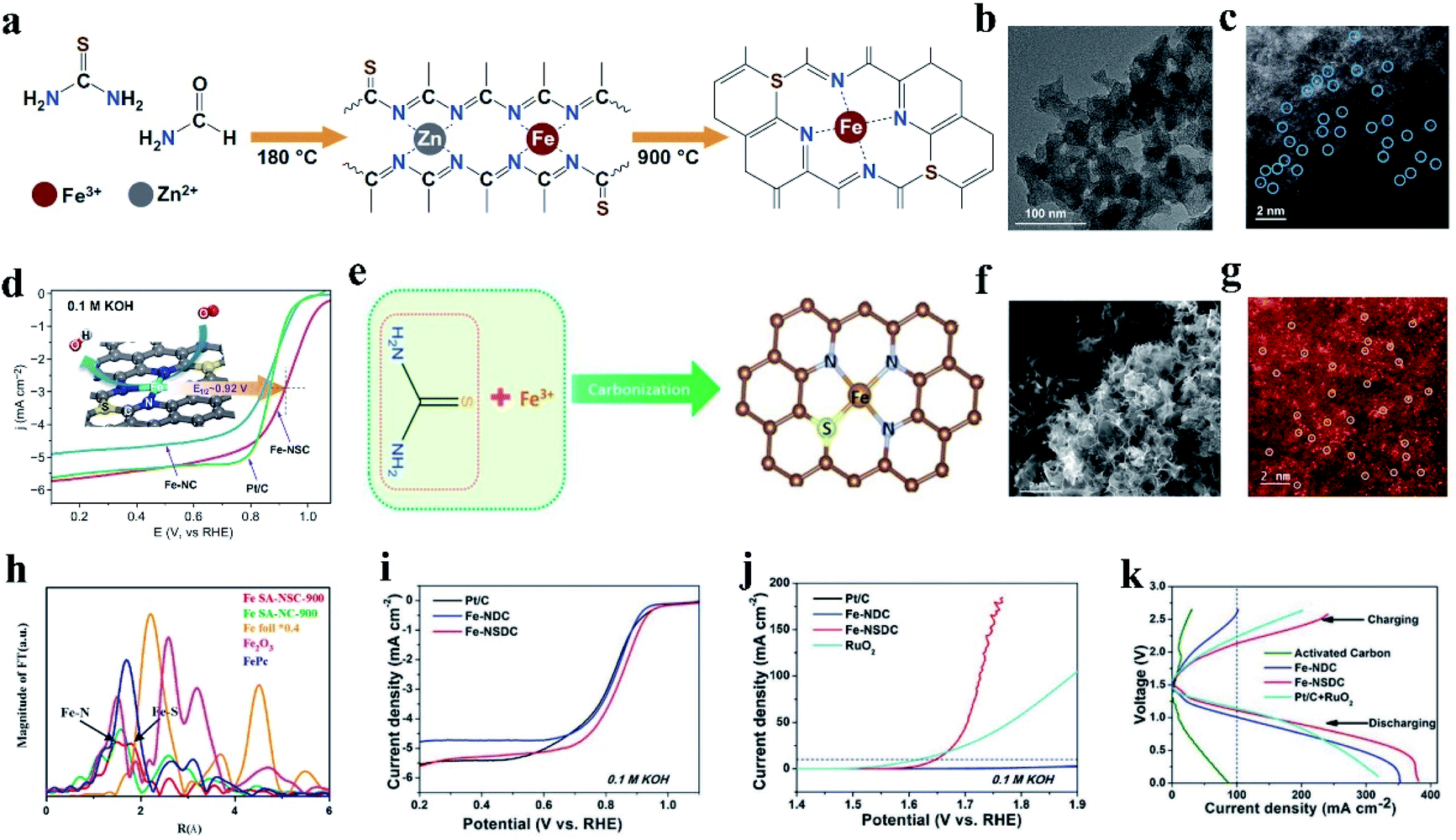 | ||
| Fig. 3 (a) Schematic illustration of the synthesis of Fe-NSC. (b) HRTEM and (c) element mapping images of Fe-NSC. (c) HAADF-STEM image of Fe-NSC, where the bright dots correspond to the atomically dispersed Fe atoms. (d) ORR performance in 0.1 M KOH at a sweep rate of 5.0 mV s−1 and 1600 rpm. Reproduced with permission.143 Copyright 2020, Springer. (e) Schematic illustration of the formation of Fe SA-NSC-900 structure. (f) SEM image of Fe SA-NSC-900. (g) HAADF-STEM images of Fe SA-NSC-900 showing the single Fe atoms (white circles) embedded in a carbon matrix. (h) Corresponding Fourier transform of EXAFS curves of Fe SA-NSC-900. Reproduced with permission.144 Copyright 2021, the American Chemical Society. (i) ORR polarization LSV curves of Fe-NSDC. (j) OER polarization LSV curves of Fe-NSDC. (k) Charge and discharge polarization curves of rechargeable Zn–air batteries using different catalysts as air electrode. Reproduced with permission.110 Copyright 2019, Wiley-VCH. | ||
Another effective way to coordinate S with Fe–N–C SACs is to bond S directly with the anchored Fe active atoms. As shown in (Fig. 3e), Wang et al. reported a NaCl-template pyrolysis method to fabricate single Fe atom catalysts with atomically dispersed Fe-heteroatom (N, S) bridge sites anchored on carbon nanosheets (Fig. 3f). The HAADF-STEM image exhibited atomically dispersed Fe atoms, which can be clearly identified as bright spots highlighted by white circles (Fig. 3g). The two different types of Fe-heteroatom bridge sites of Fe SA-NSC-900 can be observed from the two main peaks in the EXAFS spectrum (Fig. 3h). One peak is centered at ∼1.57 Å, which can be assigned to the Fe–N bond, similar to the main peak in FePc. The other peak located at ∼1.8 Å can be attributed to the Fe–S coordination structure. The structural characterization and experimental results confirmed that the N and S-coordinated Fe atomic sites (FeN3S) induced charge redistribution, lowering the binding strength of the oxygenated reaction intermediates and leading to fast reaction kinetics and good ORR activity. Moreover, Fe–SAs/NSC exhibited better stability and higher tolerance towards methanol compared with Pt/C. Both the electrochemical results and theoretical calculations indicate the positive role of introducing S in the single Fe atom microenvironment, which effectively regulates the local charge density distribution of the single Fe atomic sites, and thus promotes their interactions with the oxygenated reactive intermediates in the ORR.144
Similarly, Zhang et al. developed a porphyra-derived S-doped Fe–N–C effective bifunctional electrocatalyst, denoted as Fe-NSDC. An increase in the S content in Fe-NSDC triggered the most advanced ORR performance, impressive OER performance, and lower ΔE (ΔE = 0.8 V), which was due to the large number of monoatomic dispersed active sites and enhanced mass/electron adsorption transport performance (Fig. 3i and j). Simultaneously, an Fe-NSDC-driven Zn–air battery showed an ultrahigh open circuit potential of 1.53 V and outstanding discharge behaviors with a high peak power density of 225.1 mW cm−2, implying its enormous feasibility to act as an efficient air cathode catalyst (Fig. 3k).110 In conclusion, S-doping can optimize the charge and spin distribution in Fe–N–C materials, resulting in superior ORR activities.
It is also a feasible way to boost the catalytic performance of Fe–N–C materials by doping S and P simultaneously. Li and co-authors synthesized hollow carbon polyhedron Fe–N–C materials co-doped with N, P and S from a metal–organic framework@polymer composite, denoted as Fe–SAs/NPS-HC, which exhibited excellent ORR properties in both acid and alkaline media. In alkaline media, a positive half-wave potential (E1/2) of 0.912 V (vs. RHE) was obtained. Also, an E1/2 of 0.791 V can be achieved in acid media. The DFT calculations show that the excellent kinetic behavior of Fe–SAs/NPS-HC resulted from the atomic dispersion of N-coordinated Fe and the electronic effect of the surrounding S and P atoms. This can provide electrons to the single atom Fe center, which reduces the positive charge of Fe (Feδ+) and weakens the binding force of the adsorbed OH species. Meanwhile, Fe–SAs/NPS-HC exhibited a superior performance to Pt/C in the Zn–air battery and H2–air fuel cell test.145
2.4 Fe-based SACs coordinated by boron (B) coordination
The high overpotential of Fe SACs during the ORR process can be linked to the difficulty in adsorbing oxygen molecules, or due to the fact that the reaction intermediates can be strongly bonded at the reversible potential.110,146,147 It has been reported that the adsorption behavior of the active Fe sites can be regulated by changing the bond length of Fe SACs, the oxidation states of Fe and the chemical property of the nearest heterogeneous doped atoms.148 Boron (B) heteroatom doping is an effective way to realize the electron modulation of active sites.149–152 The B heteroatom holds great potential to redistribute the inhomogeneous spin and charge densities induced by the coordination of metal and N atoms, making the metal center more favorable for intermediate adsorption, thus resulting in enhanced catalytic activity.156Yuan and co-workers developed a general high-temperature pyrolysis approach using polypyrrole (PPy) hydrogel as the C/N source and boric acid/FeCl3 as the B and Fe sources to generate porous carbons decorated with B centers and atomically dispersed Fe–Nx species, denoted as FeBNC (Fig. 4a). The Fe K-edge XANES spectra (Fig. 4b) and Fourier transforms of the Fe K-edge EXAFS (Fig. 4c) demonstrate the presence of atomically dispersed Fe–Nx sites. This FeBNC catalyst exhibited excellent electrocatalytic properties towards the ORR with an ultra-low overpotential as low as 0.851 V (vs. RHE), which is comparable to that of the state-of-the-art porous carbon catalysts and the benchmark system Pt/C. Rotating ring-disk electrode (RRDE) measurements confirmed that the FeBNC-800 catalyst exhibited a low ring current density of approximately 0.008 mA cm−2 for peroxide oxidation and a much higher disk current density of approximately 5.5 mA cm−2 for the ORR. Theoretical calculation revealed that the incorporation of B dopant into traditional Fe–Nx species-enriched porous carbons significantly lowers the energy barrier for the ORR, and therefore boosts the overall performance.153 Saputro et al. studied the dissociative ORR mechanism on the neighboring active sites of a B-doped pyrolyzed Fe–N–C catalyst using a combination of DFT calculations and microkinetics simulations (Fig. 4d). The structure of the neighboring FeN4 and B-doped active sites facilitates the side-on adsorption of O2 for a facile dissociation process. The dissociative ORR mechanism analyzed based on the Gibbs free energy diagram, which was constructed by compiling the lowest activation energy for each reaction step (Fig. 4e), confirmed that the presence of neighboring FeN4 and Bsubs sites in the FeN4G-BN system greatly facilitates the O2 dissociation process through the formation of an O2 side-on configuration.154 Moreover, Yan group designed a B dopant for a single-atom Fe–N–C system to modulate the Fe d-band center for favored adsorption kinetics, therefore boosting the ORR process (Fig. 4f and g). The Fe K-edge XANES spectra indicated that the single Fe atoms in FeSA/B,N-CNT were much more positively charged than B-doped catalysts (Fig. 4h). The calculation based on first-principles theory indicated significant redistribution occurred because of B doping, which can be demonstrated by the isosurface plots of charge density variation with a smaller electron cloud shift away from N to B, leading to the modulation of the d-band center of Fe. The optimized FeSA/B,N-CNT catalyst outperformed pure Fe–N–C and commercial Pt/C in 0.1 M KOH medium, showing a half-wave potential of 0.933 V versus the reversible hydrogen electrode (vs. RHE) (Fig. 4i). The increased valence electrons and decreased magnetic moment of the single-atom site after B-doping can also be obtained from theoretical calculation. The modulated d-band center provides the system with favorable adsorption energy for oxygen and a much lower overpotential, thus greatly boosting the ORR performance.155
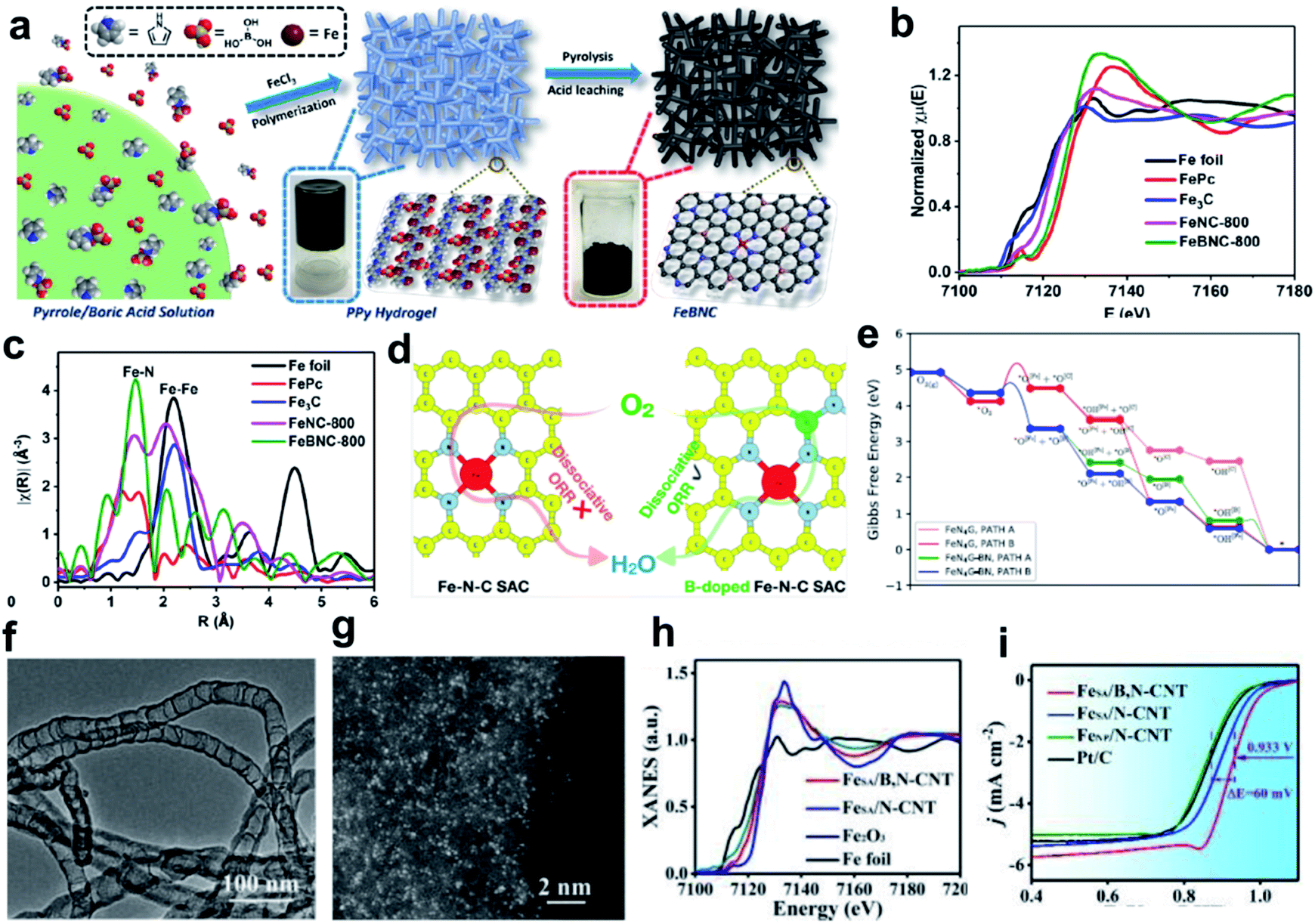 | ||
| Fig. 4 (a) Schematic of the overall procedure for the synthesis of Fe-, N-, and B-doped FeBNC catalysts. (b) Fe K-edge XANES spectra and (c) Fourier transforms of the Fe K-edge EXAFS oscillations of the FeBNC-800 and FeNC-800 samples, with FePc, Fe3C and iron foil as references. Reproduced with permission.153 Copyright 2017, the American Chemical Society. (d) DFT calculation and microkinetic simulation of the ORR mechanism on the neighboring active sites of B-doped pyrolyzed Fe–N–C catalyst. (e) ORR energy profiles for the dissociative mechanism on the neighboring FeN4 and B subs-sites of the FeN4G-BN system compared to that of the undoped FeN4G system at U = 0 V. Reproduced with permission.154 Copyright 2020, the American Chemical Society. (f) TEM image of FeSA/B,N-CNT. Scale bar, 100 nm. (g) AC HAADF-STEM image of FeSA/B,N-CNT. Scale bar, 2 nm. (h) XANES spectra of FeSA/N-CNT, FeSA/B,N-CNT, Fe2O3, and Fe foil. (i) ORR polarization curves of FeSA/B,N-CNT and other samples. Reproduced with permission.155 Copyright 2019, The Royal Society of Chemistry. | ||
2.5 Fe SACs coordinated by fluorine (F) coordination
In addition, fluorine (F) can also be used to tune the electron structure of guest atoms because the incorporation of non-metal atoms (N: 3.04; P: 2.19; S: 2.58; F: 3.98; and B: 2.04) with greatly different electronegativity into a carbon matrix can effectively tailor the distribution of the electronic and charge density of carbon materials, indicating that the electronegativity of the nonmetal atoms induces a positive charge in the surrounding carbon atoms, and further promotes the ORR performance.157–159 Due to the highest electronegativity and the suitable atomic radius of F, when the F− anion is incorporated into metal oxides or phosphates, it can effectively increase the polarity of their chemical bonds, thus greatly affecting the adsorption and dissociation of intermediate molecules.160–163 Meanwhile, its strong electron-absorbing ability increases the metal valence state of the host catalyst, thus enhancing the intrinsic activity of the active sites. Therefore, designing and developing F− anion-doped metal compound catalysts may be a promising method to promote the catalytic efficiencies of catalysts.164Zhou and coworkers created a multilayer stabilization strategy for the construction of M-SACs in N-, S- and F-co-doped graphitized carbons (M = Fe, Co, Ru, Ir and Pt), in which metal precursors are embedded in perfluorotetradecanoic acid multilayers, and are further coated with polypyrrole prior to pyrolysis, thus efficiently inhibiting the aggregation of the metal atoms to achieve M-SACs with a high metal of loading up to ∼16%. For the ORR, Fe-SA-NSFC exhibited the highest kinetic current density (Jk) of 61.5 mA cm−2 at 0.85 V compared to that for the Fe-SA-NC (5.3 mA cm−2), Fe-SA-NSC (13.6 mA cm−2), and Pt/C catalysts (5.2 mA cm−2). The Tafel slope of Fe-SA-NSFC was measured to be 53 mV dec−1, which was lower than that of 70 mV dec−1 for Fe-SANC, 69 mV dec−1 for Fe-SA-NSC, and 72 mV dec−1 for Pt/C. Consequently, Fe-SA-NSFC served as an efficient oxygen reduction catalyst with half-wave potentials of 0.91 and 0.82 V (vs. RHE) in alkaline and acid solutions, respectively. Moreover, as an air electrode in zinc-air batteries, Fe-SAC demonstrated a large peak power density of 247.7 mW cm−2 and superior long-term stability.165
2.6 Fe-based DACs
Besides SACs, polynary metals consisting of two types of metals, while maintaining their atomic dispersion also emerged as promising alternative catalysts to the commercial Pt/C toward the ORR. To accurately modulate each metal catalytic site, the polynary doping strategy has been proposed, which was inspired from the synergistic function of alloyed metal catalysts. In this section, we particularly focus on Fe-based dual metal SACs with one component of Fe and the other metal atoms as active centers, which may have the advantage of the potential synergistic interactions of the two types of active centers and inherit the merits of high exposure and high atom utilization. The interaction between the two anchored metal atoms can alter the availability of active sites on the surface or interface, modulate the binding energy to achieve a suitable metal state between the reactants and catalytic sites, as well as the intermediates, and further affect the final catalytic performance. Optimizing the active sites of DACs by modulating their electronic structures via the introduction of metal coordination is beneficial for enhancing the activity and selectivity of the products. Taking Zn and Co DACs as an example, Zn, Co polynary metal SACs supported on N-doped carbon (ZnCo-NC SACs) exhibited outstanding ORR performance in both alkaline and acid conditions with a half-wave potential of 0.861 and 0.796 V, respectively. Undoubtedly, the ZnCo-NC SACs exhibited superior catalytic activity for the ORR with a 4e− pathway compared to their monometallic counterparts (Zn-NC and Co-NC SACs).166The formation of M1–M2 dual sites not only favors the cost/time-efficient assembly of integrated energy utilization devices, but also significantly boosts the electrocatalytic performance by virtue of creating a more efficient catalytic route compared to SACs.85 According to the bonding condition between the anchored metal atoms and the coordination heteroatoms, DACs can be further classified into two subcategories, Fe-based DACs coordinated by heteroatoms together or separately.
In the first category, Fe and other metal atoms are bonded together, forming an Fe–M active site, which is further coordinated by N atoms, as shown in Fig. 5a. DACs with this geometric configuration can effectively enhance the mass loading of SACs, which was firstly proposed to resolve the problem of a catalyst loading of less than 1.5%. Simultaneously, these DACs have been demonstrated to be more active than single-center sites, and the synergistic effect between M1 and M2 in DACs can significantly improve the catalytic performance.167,168 In the second category, two metal atoms are coordinated by N atoms separately without any binding connection between M1 and M2, as illustrated in Fig. 5b. In this geometric configuration, the Fe atom and the introduced guest metal atoms are coordinated by four N atoms in the case of Fe–N4/M–N4. This DAC can effectively functionalize the catalysts, such as atomically dispersed Fe and Ni co-anchored to a microsized trimodal porous-structured nitrogen-doped graphitic carbon support with highly ordered macropores (denoted as Fe/Ni–Nx/OC). The extended X-ray absorption fine structure spectra confirmed that Fe- and Ni–SAs are affixed to the carbon support via Fe–N4 and Ni–N4 coordination bonds, respectively. The electrochemical results demonstrated that the outstanding ORR activity of Fe/Ni–Nx/OC results from the synergistic enhancement induced by the coexisting Fe–N4 and Ni–N4 sites.169
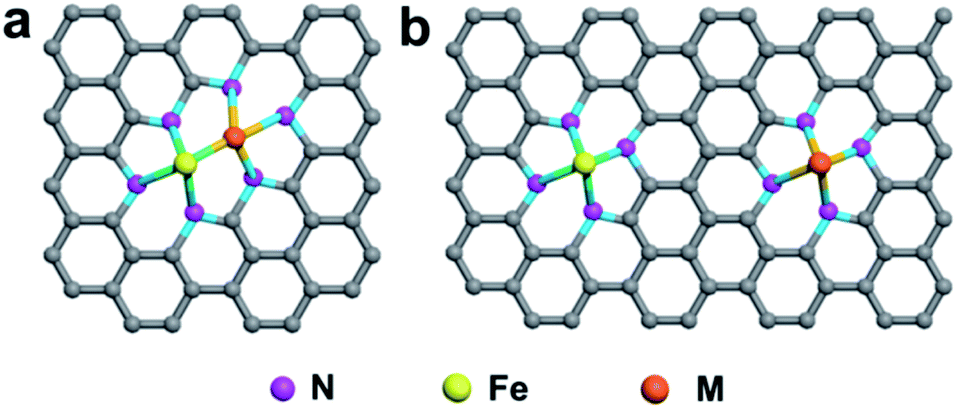 | ||
| Fig. 5 Schematic illustration of DACs with different coordination environments. (a) Fe and M are bonded and further coordinated by N atoms. (b) Fe and M are coordinated by N atoms separately. | ||
(i) Fe–CoNx DACs. Multiple attempts to create Fe–Co dual atomic catalysts resulted in their successful synthesis, with methods including high-temperature pyrolysis, host–guest strategy, in situ electrochemical method, and vesicle strategies.170–172 For example, Bai et al. fabricated Fe–Co DACs in iron-containing alkaline electrolyte through an in situ electrochemical method. The electrochemical activation of the Co species whose atoms are dispersed on nitrogen-doped carbon (Co–N–C) resulted in the accidental combination of Co–Fe and formation of a Co–Fe double-atomic catalyst (Co–Fe–N–C). The OER performance was due to the significant synergistic effect of Fe and Co, together with a decrease in the overpotential from 443 to 309 mV at a current density of 10 mA cm2.173
Wang et al. constructed Fe–Co dual sites embedded in N-doped carbon nanotubes ((Fe,Co)/CNT) by precisely controlling the bonding between Fe3+ precursors and Zn/Co bimetal organic frameworks (BMOFs) in pristine bimetallic MOFs (Fig. 6a and b).174 In their synthetic process, a bimetallic (Zn/Co) MOF was used as the host material, and FeCl3 was encapsulated in the cavity of the MOF, and further used as a guest metal source to produce Fel sites using a double-solvent method. Next, the organic ligands in the BMOFs were annealed at 900 °C under an N2 atmosphere to convert the initial rhombohedral dodecahedrons into n-doped carbon nanotubes. The Fe–Co dual sites on the surface of BMOFs catalyzed the growth of the carbon nanotubes. Finally, the Zn species could be removed by volatilization, and (Fe, Co)/N–C DACs were formed, and the corresponding morphology is shown in Fig. 6c and d. This unique structure facilitated the rapid transport of ORR-associated ions and showed a state-of-the-art ORR performance with an admirable onset potential (Eonset, 1.15 V vs. 1.05 V) and halfwave potential (E1/2, 0.954 V vs. 0.842 V), outperforming the commercial Pt/C.174 Using a model of N-doped graphene-supported dual Co–Fe sites, DFT calculation found that the dissociation energy barriers of the steps of O2 and OOH into O and OH were 0.25 eV and 0.02 eV, respectively, which are much lower than that of the respective single-atom Co and Fe catalysts, manifesting that the dual-atom Fe–Co sites can significantly lower the cleavage barrier of the O–O bond for enhanced ORR activity.175 Moreover, Yang et al. proposed isolated diatomic Fe–Co anchored on N-doped porous carbon as an efficient Fenton-like catalyst through a cage-encapsulated-precursor method. Zeolitic imidazolate frameworks (ZIF-8), possessing abundant pores and cavity, were used as cages to separate and encapsulate single Fe(acac)3 and Co(acac)2 molecules via the micropore confinement strategy. The constructed Fe(acac)3/Co(acac)2-1@ZIF-8 precursor was pyrolyzed, which resulted in the formation of isolated diatomic Fe–Co encapsulated by N-doped carbon. Further, the experimental and DFT calculation results both demonstrated that the isolated diatomic metal-nitrogen sites (FeCoN6) with a moderate adsorption energy greatly facilitate electron transport for the activation of peroxymonosulfate.176 Similarly, with Co(NO3)2·6H2O and Zn(NO3)2·6H2O as the metal precursor, Wang and co-workers developed a host–guest strategy to construct an electrocatalyst of porphyrin-like Fe–Co dual sites ((Fe, Co)/N–C) with Fe–Co dual sites embedded on N-doped porous carbon. They found that several connected transformations occurred during the pyrolysis, and the Fe3+ moieties were gradually reduced by the as-generated carbon and bonded with the neighboring Co atoms. Mössbauer spectroscopic analysis, in which a minor amount of iron species was found, effectively demonstrated the presence of a Fe–Co bond. The synthesized (Fe, Co)/N–C exhibited the maximum power density values of ∼0.85 and 0.98 W cm−2 at back pressures of 0.1 and 0.2 MPa with O2 as the oxidant, respectively. The DFT calculation results manifested that cleavage of the O–O bond on the Fe–Co dual site was extremely easy to occur. Also, the dissociation barrier of O2 and OOH into O and OH was calculated to be as low as 0.25 eV and 0.02 eV, respectively, much lower than that on the single Fe SAs/N–C and Co SAs/N–C sites.177
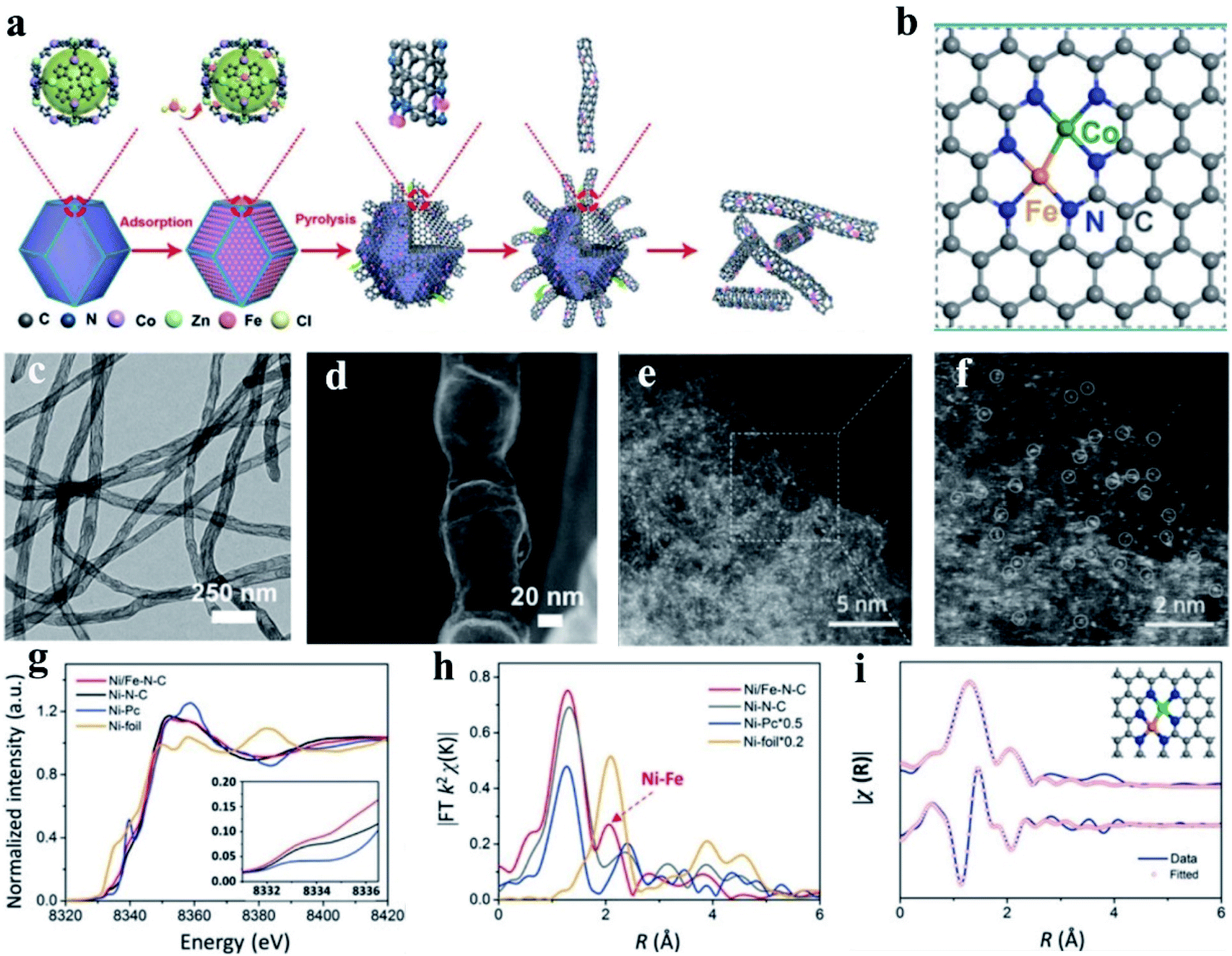 | ||
| Fig. 6 (a) Schematic illustration of the preparation of (Fe, Co)/CNT. (b) Proposed architecture of Fe–Co dual sites. (c) TEM image and (d) HAADF-STEM image of (Fe, Co)/CNT. Reproduced with permission.174 Copyright 2018, The Royal Society of Chemistry. (e and f) Magnified HAADF-STEM images of Ni/Fe–N–C. (g) Ni K-edge XANES spectra of Ni/Fe–N–C, Ni–N–C, Fe–N–C, and Ni-foil. (h) Fourier transformation of the EXAFS spectra in the R space. (i) First two-shell (Ni–N, Ni–Fe) fitting of the Fourier transformations of the EXAFS spectra for Ni/Fe–N–C. Reproduced with permission.178 Copyright 2019, Wiley-VCH. | ||
(ii) Fe–NiNx-DACs. Some catalytic processes such as the CO2RR always involve complex reaction processes. Previous research evidenced that Ni–N sites exhibit high current density for CO production, but suffer from sluggish kinetics of the first proton-coupled electron transfer.141 The Fe–N site shows a low onset potential for the CO2RR, whereas the desorption of *CO into the gas phase lowers its reactivity due to the strong binding of CO to the single Fe atom site.179 Thus Fe–Ni-based DACs have been confirmed to be promising electrocatalysts combined with the advantages of homogeneous and heterogeneous catalysts, providing synergistic functions based on different molecules and their interfaces. Some Fe–Ni bimetallic N-doped carbon frameworks achieved excellent electrocatalytic performances and outstanding durability in acidic (259 mV, 10 mA cm−2) and alkaline (219 mV, 10 mA cm−2) solutions towards the HER.180
There are many ways to form Fe–Ni dual sites, such as solvothermal and one-pot methods. Ren et al. reported the synthesis of a diatomic metal–nitrogen site (Ni/Fe–N–C) through an ionic exchange strategy based on the pyrolysis of the Zn/Ni/Fe zeolitic imidazolate framework for efficient CO2RR (Fig. 6e and f). The inset in Fig. 6g highlights the pre-edge features at approximately 8334 eV, which correspond to the 3d and 4p orbital hybridization of the Ni central atoms. Fig. 6h displays the Fourier transform (FT) k2-weighted χ(k) function of the EXAFS spectra. The calculated Ni–Fe path in Fig. 6i fits well with the experimental spectra at 2.06 Å. The Ni–Fe dual center reduced the energy barrier for the desorption of the COOH* and CO intermediates in comparison to the simple Ni or Fe center, thereby significantly enhancing the CO2RR activity.178 In another study, N-doped carbon hollow spheres with atomic Fe–Ni dual-metal pairs (Fe–NiNC) were fabricated via a dual solvent route. Benefiting from the porous substrate and electrochemical interactions between the Fe–Ni dual-metal pairs, the resultant catalyst showed exceptional bifunctional catalytic performances towards both the OER and ORR in rechargeable Zn–air batteries. Theoretical and experimental results revealed that the Fe and Ni atoms act as effective active sites for the ORR and OER, respectively.181
(iii) Fe–RhNx-DACs. The dual single-atom Fe–Rh catalyst exhibits an excellent electrocatalytic performance for the HER in acidic electrolyte. Wang et al. obtained a dual single-atom Rh–Fe catalyst using a top-down process, which starts from iron nanoparticles, using dual-metal interbonds (Rh–Fe bonding) as a chemical facilitator to spontaneously convert the Fe nanoparticles to single atoms at low temperatures. The presence of Fe–Rh bonding between adjacent Fe and Rh single atoms contributes to the thermodynamic stability, which facilitates the stripping of a single Fe atom from the Fe nanoparticles, leading to a stabilized single atom. This discovery of dual-metal interbonding as a chemical facilitator paves a novel way for the atomic dispersion of chemical metals and the design of efficient catalysts at the atomic scale.182
(i) FeN–Ni–Nx DACs. Hetero-SACs consisting of Fe and Ni sites separately co-anchored to carbon substrates present desirable activity toward the ORR by taking the advantage of two different activity sites through long-range coupling to alter their coordination environments and electronic structures. Zhu et al. prepared a hetero-single-atom ORR electrocatalyst with Fe and Ni co-anchored to a microsized nitrogen-doped graphitic carbon support through a template method combining wet chemical metal ion impregnation and pyrolysis (denoted as Fe/Ni–Nx/OC) (Fig. 7a). The obtained materials presented a cuboctahedron-shaped structure with 3D-ordered micropores (≈180 nm) interconnected by 3-D ordered mesopores channels (Fig. 7b). Based on the N2 adsorption–desorption isotherms (Fig. 7c) and EXAFS result (Fig. 7d and e, respectively), they concluded that Fe- and Ni–SAs are affixed to the carbon support via Fe–N4 and Ni–N4 coordination bonds (Fig. 7f). The resultant Fe/Ni–Nx/OC electrocatalyst exhibited outstanding ORR activity, outperforming SAC electrocatalysts with only Fe–N4 or Ni–N4 sites, which can be derived from the synergistic enhancement induced by the coexisting F–N4 and Ni–N4 sites and the superior mass-transfer capability promoted by the trimodal porous-structured carbon support.169 In another work, a binuclear FeNi–N6 site embedded in porous nitrogen-doped carbon was fabricated, as shown in Fig. 7g and h. The active sites and catalytic mechanism in the N-coordinated Fe, Ni dual-doped carbon toward the ORR were revealed with the assistance of XANES fitting and DFT calculations.183 This report exposed the fact that FeNi–N6 with a type I (each metal atom coordinated with four nitrogen atoms) instead of type II configuration (each metal atom coordinated with three nitrogen atoms) dominates the catalytic activity of the noble metal-free catalyst, as shown in Fig. 7i and j. This study is an important step in the development of bimetallic materials by developing dual-metal doping for the ORR in terms of revealing new structural configurations and their correlation with catalytic activity.183
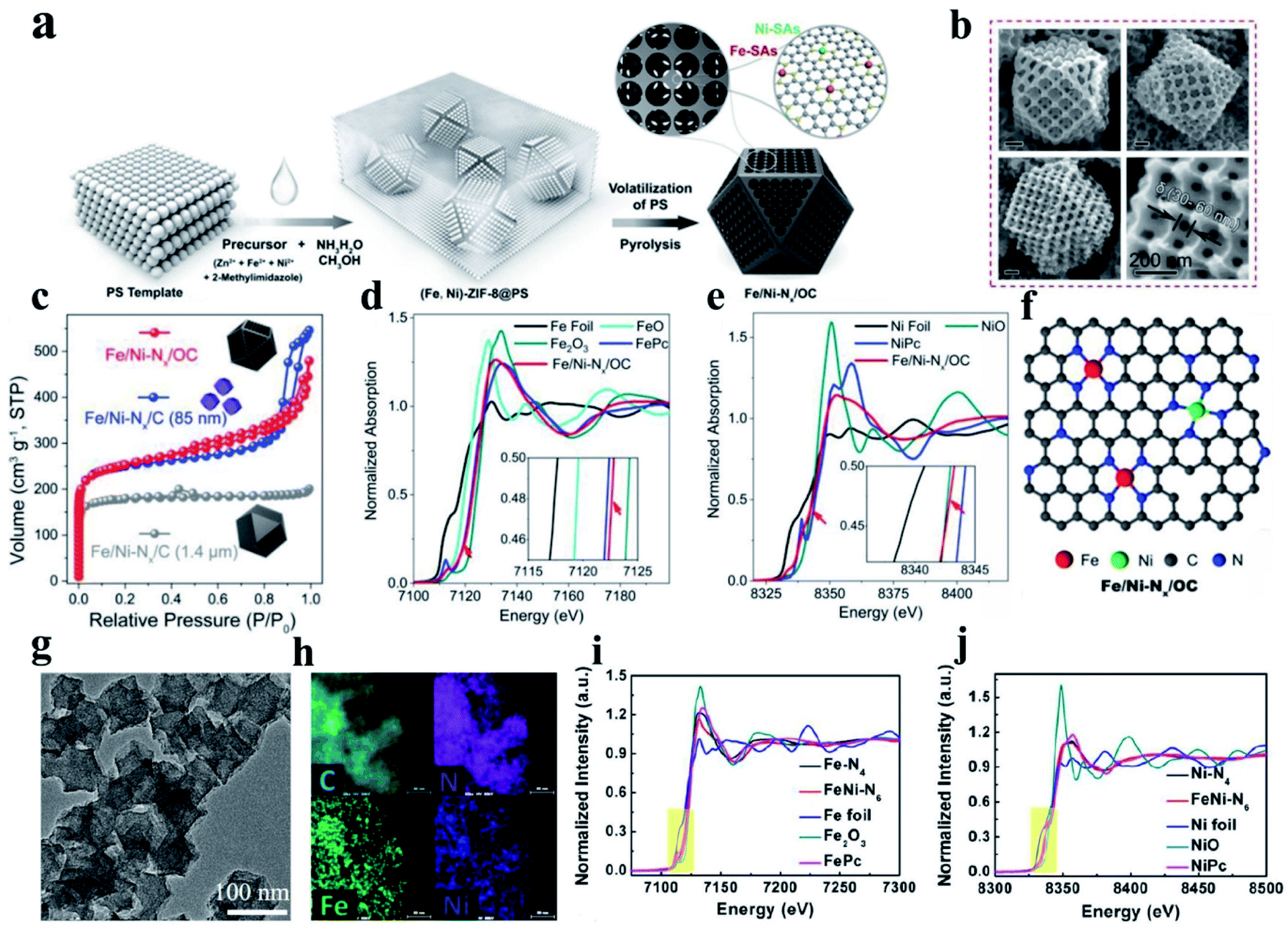 | ||
| Fig. 7 (a) Schematic diagram illustrating the procedure for the synthesis of Fe/Ni–Nx/OC. (b) SEM images of Fe/Ni–Nx/OC. (c) N2 adsorption–desorption isotherms. (d) Fe K-edge XANES spectra. (e) Ni K-edge XANES spectra with a magnified view in the inset. (f) Proposed structural model for Fe/Ni–Nx/OC. Reproduced with permission.169 Copyright 2020, Wiley-VCH. (g) TEM and (h) corresponding element mapping patterns of FeNi–N6. (i and j) Fe k-edge fittings curves of FeNi–N6 and the structural model (insets) of FeNi–N6 (type I) and FeNi–N6 (type II). Reproduced with permission.183 Copyright 2020, the American Chemical Society. | ||
(ii) FeN–Co–Nx DACs. Another metal, Co, is widely investigated as an active SAC for electrochemical reactions. Recently, it has been reported that the corrosion of the highly active FeNx species towards the ORR can be effectively prevented by the neighboring Co species,184,185 which provides a good future for Fe–Co DACs with promising electrochemical activities. For example, metalloporphyrinic MOFs, PCN-224-FeCox, with an adjustable molar ratio of FeII/CoII alternatively residing inside the porphyrin center, were employed by Fang et al. as precursors to prepare FeCo-N-doped porous carbon (denoted as FeCo-NPC) after pyrolysis. Upon pyrolysis and acid etching, PCN-224-FeCox was converted to N-doped porous nanotubes with a hollow structure, consisting of highly dispersed FeNx and CoNx species. For oxygen reduction, the electron transfer number based on this Fe–Co–N-900 catalyst was calculated to be 3.9 via the Koutecky–Levich equation, indicating the ideal 4 e− ORR process. Also, the Tafel slope was calculated to be −78 mV dec−1, which is much lower than that of −86 mV dec−1 for commercial Pt/C, indicating favorable kinetic behavior.186 Meanwhile, Deng and co-workers developed a bimetal nitride of (Co, Fe)3N_R as an electrocatalyst for the ORR and further as an electrode for Zn–air batteries. A bimetal-layered double hydroxide was firstly prepared via a cation-carving strategy with FeII as an etching agent to attack the Co-based nanocuboid precursor, which resulted in a series of ligand release and re-coordination to obtain a secondary hollow structure with a tunable Co/Fe ratio controlled by the stoichiometric factor of the FeII source. XANES was used to reveal the electronic structure of (Co, Fe)3N_R, which presented a slightly weakened pre-edge hump as the nitride feature and an enlarged white-line crest, suggesting higher electron occupancies at the Co 3d orbits and lower electron allocation at the 4p orbits. This observation was ascribed to the weaker 4p–3d hybridization at the Co sites and the altered net charge distribution. (Co, Fe)3N_R was utilized in a Zn–air battery for the ORR/OER, and the assembled Zn–air battery delivered a comparable performance to precious benchmarks in its initial stage, and also demonstrated a maturation process, which led to a maximum power density of 234 mW cm−2 and a discharge–charge voltage gap of 0.85 V at 30 mA cm−2.187
(iii) FeN–Pt–Nx DACs. The high activity of bimetallic composite single-atom electrocatalysts, which is enhanced by the synergistic effect, is desirable for the ORR and zinc-air batteries. Cao et al. initially fabricated Fe/Pt composite single-atom bifunctional electrocatalysts (Fe1Pt1/NC) via the pyrolysis of porphyra and adsorbed urea, where a unique electronic structure and coordination environment were created in the presence of Pt–N4 and Fe–N4 structures, leading to a favorable kinetic current density nearly five times that of Pt/C.188 Meanwhile, Zhong et al. proposed a new method of embedding Pt species in an Fe-doped imidazole zeolite framework to increase the density of active metal atoms and increase the activity through simple pyrolysis, where both isolated Pt and Fe single atoms were anchored on the nitrogen-doped carbon. The synergist effect of the Pt–N4 and Fe–N4 structure is facile to break the O2 molecule and form OOH*, resulting in high ORR activity and excellent performance in zinc–air batteries.189
(iv) FeN–Pd–C DACs. Pd is highly active towards many catalytic reactions, including C–C coupling, selective hydrogenation, and some oxidation reactions.2 In a recent study, Zhao et al. reported a biomimetic Pd1@Fe1 yolk–shell featuring two compatible single-atom systems with atomically dispersed Fe1 sites in the N-doped carbon shell and Pd1 sites in the yolk derived from a metal–organic framework. By coating PdCl2/UiO-66-NH2 with stable PdCl2/NH2 coordination configuration (denoted as PdCl2/UiO-66-NH2), the surface of PdCl2/UiO-66-NH2 was coated with an SiO2 layer, resulting in the formation of core–shell PdCl2/UiO-66-NH2@SiO2. Fe-Tipp (5,10,15,20-tetra(4-(imidazole-1-yl)phenyl)porphyria) was subsequently polymerized on the surface of PdCl2/Na to generate PdCl2/SiO2@Fe-Tipp via a quaternary mechanism. The resulting PdCl2/UiO-66-NH2@SiO2@Fe-Tipp was pyrolyzed at 700 °C directly (N2, 3 h) to reduce the entry of Pd and Fe cations in the Pd1 and Fe1 sites, respectively. Finally, the SiO2 template was removed with sodium hydroxide to obtain the yolk shell Pd1@Fe1. The synthesized biomimetic composite featuring two compatible single-atom systems with Fe1 sites in an N-doped carbon shell and Pd1 sites in an MOF-derived yolk could simultaneously catalyze the hydrogenation of nitroaromatics and alkene epoxidation reactions, leading to the cascade synthesis of amino alcohols.190
3. Summary and perspective
Recent years have witnessed great progress in exploiting SACs and DACs with regard to their synthesis, characterization, electronic structures and exceptional performances. This review article outlined the research progress on Fe-based SACs and DACs with different coordination environments. On one hand, we reviewed the influence of different heteroatoms (N, B, P, and S) in substrate on the intrinsic activity of the Fe active sites, as well as the catalytic performance of Fe-based SACs. Due to their different electronegativity and atomic radii, heteroatoms can affect the charge density distribution around the carbon atom and change the adsorption state of oxygen molecules. The coordination between Fe atoms and heteroatoms leads to a large number of new active sites and improved catalytic activity. However, a single atomic site for one reaction results in its low selectivity for other reactions. For some complex electrocatalytic reactions, they not only require a multi-step electrocatalytic process, but also the intermediate products are complex, and the simple single atomic site cannot maintain high efficiency and selectivity for each reaction step. Therefore, based on the synergistic effect between different single metal atoms, it is very important to develop new diverse SACs to achieve high selectivity for complex reactions. On the other hand, based the aspects of material synthesis and property enhancement, synergistic Fe-metal dual active sites such as Fe–Ni, Fe–Co and Fe–Rh for the ORR were also discussed in this review. The formation of Fe–metal dual sites not only enhances the loading capacity of the active materials on the carbon substrate,174,191,192 but also significantly boosts their electrocatalytic performance towards energy utilization devices by virtue of inventing more efficient catalytic routes compared to single-site Fe–N–C.193Although tremendous achievements have been made in the development of Fe-based SACs and DACs towards the ORR in recent years, there are still several issues that should be carefully considered for the future design of more effective Fe-based electrocatalysts. For example, how to improve the loading capacity without losing the characteristic molecular structure of SACs and DACs and high activity of catalysts, and how to scale up their synthesis also needs to be considered. Having a deep understanding of these characteristics also plays an important role in the rational design of Fe-based catalysts for commercial application. In this section, the associated challenges are summarized and perspectives regarding the future development of Fe-based SACs and DACs are also proposed.
(i) The unfortunate shortcoming of Fe-based SACs and DACs is their chemical instability. The carbon substrate always undergoes detrimental corrosion and shape changes, leading to a decrease in metal active sites and reduced charge and mass transfer rates, which will drastically change their overall performance.194 Amorphous carbon is more susceptible to corrosion; however, carbon with increased long-range order may result in fewer defects and decreased capability to anchor metals and/or heteroatoms (such as S, P, and B atoms), thus reducing the catalytic activity. Therefore, it is necessary to balance the catalytic activity and stability of catalysts. Besides, the morphology, structure and atomic coordination state of Fe-based SAC/DAC catalysts should be accurately regulated to improve their stability.
(ii) Although the structure and intrinsic atomic and electronic structures of Fe-based SACs/DACs have been successfully characterized using modern characterization methods such as HAADF-STEM and SANES together with theoretical modeling, a quantitative understanding of the evolution of the catalyst structure and electrode material is still lacking for the operation process in the cell device. Deep understanding of the microstructure evolution of the catalyst performance and the reaction mechanisms at the atomic level with the assistance of in situ technology will be beneficial for theoretical guidance for the rational design and application of fuel cell catalysts in the future.
(iii) Most of the current materials show excellent catalytic activity on the laboratory scale. However, as reported by some theoretical calculation results, the dissociation energy barrier value of the O–O bond on the surface of carbon is usually high, which is not conducive to the 4e− dissociation pathway.195 Therefore, for almost all carbon materials, the electron transfer number measured by experiment much less than four, indicating the generation of H2O2 in the ORR reaction.196 For Fe-based SACs/DACs, Fe2+ resulting from the dissolution of Fe in the electrolyte can react with H2O2 through Fenton reactions and generate free radicals, which will attack the PEMFC membrane, resulting in a serious degradation in battery performance. In addition, monatomic catalysts are prone to aggregate during the reaction process due to their high free energy and complex working environment.23 Thus, it is necessary to design and evaluate their catalytic activities considering their commercialization in energy applications in the future.
(iv) Essentially, most SACs/DACs are prepared using complex methods involving the deposition of a salt precursor on a support, followed by pyrolysis, and then reduction at an elevated temperature under a suitable gaseous environment. The synthesis of SACs/DACs with a generally low loading capacity on the support using this method is complex, which makes it difficult to realize their large-scale commercial application. Thus, to successfully use Fe-based SACs/DACs in future renewable energy systems, it is necessary to develop techno-economic and convenient technology for preparing catalysts, while increasing their loading capability.
Author contributions
Yan Yan, Hao Hu, Zaifeng Li and Mingkai Liu created the idea and discussed the summary fields. Zaifeng, Haoyan Cheng, Zehua Qu, Rui Yu, Fan Liu and Qianwen Ma wrote the manuscript. Shuang Zhao, Yu Cheng, Chongyang Yang, Xu Wang, Yuyu Chen and Shaoyu Hao drew the pictures. Mingakai Liu, Zaifeng Li and Hao Hu revised the manuscript. All the authors contributed to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51908256, 52002119), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (19KJA460004), the Priority Academic Program Development of Jiangsu Higher Education Institutions, and the State Key Laboratory Base of Eco-chemical Engineering (STHG1902).Notes and references
- Y. Ying, X. Luo, J. Qiao and H. Huang, Adv. Funct. Mater., 2021, 31, 2007423 CrossRef CAS.
- Y. Shi, Z. Lyu, M. Zhao, R. Chen, Q. N. Nguyen and Y. Xia, Chem. Rev., 2021, 121, 649–735 CrossRef CAS PubMed.
- X. Zhu, C. Hu, R. Amal, L. Dai and X. Lu, Energy Environ. Sci., 2020, 13, 4536–4563 RSC.
- X. Luo, X. Wei, H. Wang, Y. Wu, W. Gu and C. Zhu, ACS Sustainable Chem. Eng., 2020, 8, 9721–9730 CrossRef CAS.
- K. Yuan, C. Lu, S. Sfaelou, X. Liao, X. Zhuang, Y. Chen, U. Scherf and X. Feng, Nano Energy, 2019, 59, 207–215 CrossRef CAS.
- J. Hao, W. Zhan, L. Sun, G. Zhuang, X. Wang and X. Han, Inorg. Chem., 2020, 59, 937–942 CrossRef CAS PubMed.
- L. Sun, X. He, S. Zeng, Y. Yuan, R. Li, W. Zhan, J. Chen, X. Wang and X. Han, Inorg. Chem., 2021, 60, 1160–1171 CrossRef CAS PubMed.
- L. Sun, R. Li, W. Zhan, Y. Yuan, X. Wang, X. Han and Y. Zhao, Nat. Commun., 2019, 10, 2270 CrossRef PubMed.
- R. Li, L. Sun, W. Zhan, Y.-A. Li, X. Wang and X. Han, J. Mater. Chem. A, 2018, 6, 15747–15754 RSC.
- Y. Yuan, K. Sheng, S. Zeng, X. Han, L. Sun, I. Lončarić, W. Zhan and D. Sun, Inorg. Chem., 2020, 59, 5456–5462 CrossRef CAS PubMed.
- P. Ball, Natl. Sci. Rev., 2018, 5, 690–693 CrossRef CAS.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S.-I. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412 CrossRef CAS PubMed.
- H. Cheng, Z. Cao, Z. Chen, M. Zhao, M. Xie, Z. Lyu, Z. Zhu, M. Chi and Y. Xia, Nano Lett., 2019, 19, 4997–5002 CrossRef CAS PubMed.
- X. Wei, S. Song, N. Wu, X. Luo, L. Zheng, L. Jiao, H. Wang, Q. Fang, L. Hu, W. Gu, W. Song and C. Zhu, Nano Energy, 2021, 84, 105840 CrossRef CAS.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- T. Sun, L. Xu, D. Wang and Y. Li, Nano Res., 2019, 12, 2067–2080 CrossRef CAS.
- Z. Zhang, C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si, S. Zhou and J. Zeng, Nat. Commun., 2020, 11, 1215 CrossRef CAS PubMed.
- L. Wang, W. Zhang, S. Wang, Z. Gao, Z. Luo, X. Wang, R. Zeng, A. Li, H. Li, M. Wang, X. Zheng, J. Zhu, W. Zhang, C. Ma, R. Si and J. Zeng, Nat. Commun., 2016, 7, 14036 CrossRef CAS PubMed.
- H. Li, M. Wang, L. Luo and J. Zeng, Adv. Sci., 2019, 6, 1801471 CrossRef PubMed.
- X. Luo, X. Wei, H. Wang, W. Gu, T. Kaneko, Y. Yoshida, X. Zhao and C. Zhu, Nano-Micro Lett., 2020, 12, 163 CrossRef CAS PubMed.
- X. Luo, X. Wei, H. Zhong, H. Wang, Y. Wu, Q. Wang, W. Gu, M. Gu, S. P. Beckman and C. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 3539–3546 CrossRef CAS PubMed.
- X. Zhao, P. Gao, Y. Yan, X. Li, Y. Xing, H. Li, Z. Peng, J. Yang and J. Zeng, J. Mater. Chem. A, 2017, 5, 20202–20207 RSC.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, 1803498 CrossRef PubMed.
- Y. Peng, Z. Geng, S. Zhao, L. Wang, H. Li, X. Wang, X. Zheng, J. Zhu, Z. Li, R. Si and J. Zeng, Nano Lett., 2018, 18, 3785–3791 CrossRef CAS PubMed.
- Y. Pan, Y. Chen, K. Wu, Z. Chen, S. Liu, X. Cao, W.-C. Cheong, T. Meng, J. Luo, L. Zheng, C. Liu, D. Wang, Q. Peng, J. Li and C. Chen, Nat. Commun., 2019, 10, 4290 CrossRef PubMed.
- Y. Han, Y. Wang, R. Xu, W. Chen, L. Zheng, A. Han, Y. Zhu, J. Zhang, H. Zhang, J. Luo, C. Chen, Q. Peng, D. Wang and Y. Li, Energy Environ. Sci., 2018, 11, 2348–2352 RSC.
- L. Luo, J. Luo, H. Li, F. Ren, Y. Zhang, A. Liu, W.-X. Li and J. Zeng, Nat. Commun., 2021, 12, 1218 CrossRef CAS PubMed.
- F. Wu, C. Pan, C.-T. He, Y. Han, W. Ma, H. Wei, W. Ji, W. Chen, J. Mao, P. Yu, D. Wang, L. Mao and Y. Li, J. Am. Chem. Soc., 2020, 142, 16861–16867 CrossRef CAS PubMed.
- L. Li, Y. Li, Y. Xiao, R. Zeng, X. Tang, W. Yang, J. Huang, K. Yuan and Y. Chen, Chem. Commun., 2019, 55, 7538–7541 RSC.
- L. Wang, H. Li, W. Zhang, X. Zhao, J. Qiu, A. Li, X. Zheng, Z. Hu, R. Si and J. Zeng, Angew. Chem., Int. Ed., 2017, 56, 4712–4718 CrossRef CAS PubMed.
- Y. Zhong, X. Kong, Z. Geng, J. Zeng, X. Luo and L. Zhang, ChemPhysChem, 2020, 21, 2051–2055 CrossRef CAS PubMed.
- H. Chen, X. Guo, X. Kong, Y. Xing, Y. Liu, B. Yu, Q.-X. Li, Z. Geng, R. Si and J. Zeng, Green Chem., 2020, 22, 7529–7536 RSC.
- X. Yang, Y. Chen, L. Qin, X. Wu, Y. Wu, T. Yan, Z. Geng and J. Zeng, ChemSusChem, 2020, 13, 6307–6311 CAS.
- Z. Zhang, C. Liu, C. Feng, P. Gao, Y. Liu, F. Ren, Y. Zhu, C. Cao, W. Yan, R. Si, S. Zhou and J. Zeng, Nano Lett., 2019, 19, 8774–8779 CrossRef CAS PubMed.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- Q. Li, W. Chen, H. Xiao, Y. Gong, Z. Li, L. Zheng, X. Zheng, W. Yan, W.-C. Cheong, R. Shen, N. Fu, L. Gu, Z. Zhuang, C. Chen, D. Wang, Q. Peng, J. Li and Y. Li, Adv. Mater., 2018, 30, 1800588 CrossRef PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- S. Tian, M. Hu, Q. Xu, W. Gong, W. Chen, J. Yang, Y. Zhu, C. Chen, J. He, Q. Liu, H. Zhao, D. Wang and Y. Li, Sci. China Mater., 2021, 64, 642–650 CrossRef CAS.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji, X. Zheng, Y. Wang, L. Zheng, C. Chen, D. Wang, J. Zhang and Y. Li, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS PubMed.
- D. Zhao, Z. Chen, W. Yang, S. Liu, X. Zhang, Y. Yu, W.-C. Cheong, L. Zheng, F. Ren, G. Ying, X. Cao, D. Wang, Q. Peng, G. Wang and C. Chen, J. Am. Chem. Soc., 2019, 141, 4086–4093 CrossRef CAS PubMed.
- P.-H. Li, M. Yang, Y.-X. Li, Z.-Y. Song, J.-H. Liu, C.-H. Lin, J. Zeng and X.-J. Huang, Anal. Chem., 2020, 92, 6128–6135 CrossRef CAS PubMed.
- D. W. Su, J. Ran, Z. W. Zhuang, C. Chen, S. Z. Qiao, Y. D. Li and G. X. Wang, Sci. Adv., 2020, 6, eaaz8447 CrossRef CAS PubMed.
- X. Wei, X. Luo, H. Wang, W. Gu, W. Cai, Y. Lin and C. Zhu, Appl. Catal., B, 2020, 263, 118347 CrossRef CAS.
- X. X. Wang, M. T. Swihart and G. Wu, Nat. Catal., 2019, 2, 578–589 CrossRef CAS.
- T. Sharifi, E. Gracia-Espino, A. Chen, G. Hu and T. Wågberg, Adv. Energy Mater., 2020, 10, 1902084 CrossRef CAS.
- Y. Li, R. Cao, L. Li, X. Tang, T. Chu, B. Huang, K. Yuan and Y. Chen, Small, 2020, 16, 1906735 CrossRef CAS PubMed.
- X. Shen, S. Dai, Y. Pan, L. Yao, J. Yang, X. Pan, J. Zeng and Z. Peng, ACS Catal., 2019, 9, 11431–11437 CrossRef CAS.
- S. Zhao, L. Ban, J. Zhang, W. Yi, W. Sun and Z. Zhu, Chem. Eng. J., 2021, 409, 128171 CrossRef CAS.
- M. Liu, Z. Lyu, Y. Zhang, R. Chen, M. Xie and Y. Xia, Nano Lett., 2021, 21, 2248–2254 CrossRef CAS PubMed.
- L. Zhang, S. Chen, Y. Dai, Z. Shen, M. Wei, R. Huang, H. Li, T. Zheng, Y. Zhang, S. Zhou and J. Zeng, ChemCatChem, 2018, 10, 925–930 CrossRef CAS.
- X. Tang, R. Cao, L. Li, B. Huang, W. Zhai, K. Yuan and Y. Chen, J. Mater. Chem. A, 2020, 8, 25919–25930 RSC.
- J. Zhang, M. Zhang, L. Qiu, Y. Zeng, J. Chen, C. Zhu, Y. Yu and Z. Zhu, J. Mater. Chem. A, 2019, 7, 19045–19059 RSC.
- X. Zhao, S. Chen, Z. Fang, J. Ding, W. Sang, Y. Wang, J. Zhao, Z. Peng and J. Zeng, J. Am. Chem. Soc., 2015, 137, 2804–2807 CrossRef CAS PubMed.
- H. Huang, K. Li, Z. Chen, L. Luo, Y. Gu, D. Zhang, C. Ma, R. Si, J. Yang, Z. Peng and J. Zeng, J. Am. Chem. Soc., 2017, 139, 8152–8159 CrossRef CAS PubMed.
- S. Chen, J. Zhao, H. Su, H. Li, H. Wang, Z. Hu, J. Bao and J. Zeng, J. Am. Chem. Soc., 2021, 143, 496–503 CrossRef CAS PubMed.
- X. Li, X. Li, C. Liu, H. Huang, P. Gao, F. Ahmad, L. Luo, Y. Ye, Z. Geng, G. Wang, R. Si, C. Ma, J. Yang and J. Zeng, Nano Lett., 2020, 20, 1403–1409 CrossRef CAS PubMed.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D.-j. Liu, L. Zhuang, C. Ma, J. Zeng, B. Zhang, D. Su, P. Song, W. Xing, W. Xu, Y. Wang, Z. Jiang and G. Sun, Nat. Commun., 2017, 8, 15938 CrossRef CAS PubMed.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- J. L. Kneebone, S. L. Daifuku, J. A. Kehl, G. Wu, H. T. Chung, M. Y. Hu, E. E. Alp, K. L. More, P. Zelenay, E. F. Holby and M. L. Neidig, J. Phys. Chem. C, 2017, 121, 16283–16290 CrossRef CAS.
- M. K. Liu, Q. H. Meng, Z. Yang, X. S. Zhao and T. X. Liu, Chem. Commun., 2018, 54, 5090–5093 RSC.
- Y. Yuan, L. Sun, Y. Li, W. Zhan, X. Wang and X. Han, Inorg. Chem., 2020, 59, 4080–4089 CrossRef CAS PubMed.
- M. K. Liu, Y. Q. Liu, Y. Yan, F. S. Wang, J. H. Liu and T. X. Liu, Chem. Commun., 2017, 53, 9097–9100 RSC.
- M. K. Liu, Z. B. Yang, H. Sun, C. Lai, X. S. Zhao, H. S. Peng and T. X. Liu, Nano Res., 2016, 9, 3735–3746 CrossRef CAS.
- Z. Y. Yang, P. Zhang, J. Wang, Y. Yan, Y. Yu, Q. H. Wang and M. K. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 37434–37444 CrossRef CAS PubMed.
- P. Zhang, Y. Q. Liu, Y. Yan, Y. Yu, Q. H. Wang and M. K. Liu, ACS Appl. Energy Mater., 2018, 1, 4814–4823 CrossRef CAS.
- M. K. Liu, B. M. Li, H. Zhou, C. Chen, Y. Q. Liu and T. X. Liu, Chem. Commun., 2017, 53, 2810–2813 RSC.
- B. M. Li, Y. Yan, C. T. Shen, Y. Yu, Q. H. Wang and M. K. Liu, Nanoscale, 2018, 10, 16217–16230 RSC.
- M. Liu, P. Zhang, Z. Qu, Y. Yan, C. Lai, T. Liu and S. Zhang, Nat. Commun., 2019, 10, 3917 CrossRef PubMed.
- Y. Yan, P. Zhang, Z. Qu, M. Tong, S. Zhao, Z. Li, M. Liu and Z. Lin, Nano Lett., 2020, 20, 7662–7669 CrossRef CAS PubMed.
- S. Chen, M. Cui, Z. Yin, J. Xiong, L. Mi and Y. Li, ChemSusChem, 2021, 14, 73–93 CrossRef CAS PubMed.
- B. Dou, J. Yan, Q. Chen, X. Han, Q. Feng, X. Miao and P. Wang, Sens. Actuators, B, 2021, 328, 129082 CrossRef CAS.
- Y. Zhuang, L. Sun, S. Zeng, W. Zhan, X.-J. Wang, Y. Zhao and X. Han, Chem.–Eur. J., 2019, 25, 14133–14139 CrossRef CAS PubMed.
- Y. Yuan, L. Sun, G. Wu, Y. Yuan, W. Zhan, X. Wang and X. Han, Inorg. Chem., 2020, 59, 2104–2110 CrossRef CAS PubMed.
- W. Zhan, Y. Yuan, L. Sun, Y. Yuan, X. Han and Y. Zhao, Small, 2019, 15, 1901024 CrossRef PubMed.
- Q. Shen, L. Sun, Y. Zhuang, W. Zhan, X. Wang and X. Han, Inorg. Chem., 2020, 59, 17650–17658 CrossRef CAS PubMed.
- Y. Yuan, L. Sun, S. Zeng, W. Zhan, X. Wang and X. Han, Chem.–Eur. J., 2020, 26, 921–926 CrossRef CAS PubMed.
- Y. Yang, G. Zhuang, L. Sun, X. Zhang, X. Yan, W. Zhan, X. Wang and X. Han, J. Mater. Chem. A, 2020, 8, 17449–17453 RSC.
- L. Sun, R. Li, W. Zhan, F. Wang, Y. Zhuang, X. Wang and X. Han, Chem.–Eur. J., 2019, 25, 3053–3060 CrossRef CAS PubMed.
- W. Zhan, L. Sun and X. Han, Nano-Micro Lett., 2019, 11, 1 CrossRef CAS PubMed.
- J. Tan, D. Li, Y. Liu, P. Zhang, Z. Qu, Y. Yan, H. Hu, H. Cheng, J. Zhang, M. Dong, C. Wang, J. Fan, Z. Li, Z. Guo and M. Liu, J. Mater. Chem. A, 2020, 8, 7980–7990 RSC.
- Y. Yang, L. Sun, W. Zhan, X. Wang and X. Han, J. Mater. Chem. A, 2021, 9, 4310–4316 RSC.
- L. Sun, Y. Yuan, R. Li, W. Zhan, X.-J. Wang, Y. Zhao and X. Han, J. Mater. Chem. A, 2019, 7, 25423–25432 RSC.
- L. Wang, W. Zhang, X. Zheng, Y. Chen, W. Wu, J. Qiu, X. Zhao, X. Zhao, Y. Dai and J. Zeng, Nat. Energy, 2017, 2, 869–876 CrossRef CAS.
- M. Ma, A. Kumar, D. Wang, Y. Wang, Y. Jia, Y. Zhang, G. Zhang, Z. Yan and X. Sun, Appl. Catal., B, 2020, 274, 119091 CrossRef CAS.
- H.-X. Zhong, J. Wang, Q. Zhang, F. Meng, D. Bao, T. Liu, X.-Y. Yang, Z.-W. Chang, J.-M. Yan and X.-B. Zhang, Adv. Sustainable Syst., 2017, 1, 1700020 CrossRef.
- Y. Li, R. Hu, Z. Chen, X. Wan, J.-X. Shang, F.-H. Wang and J. Shui, Nano Res., 2021, 14, 611–619 CrossRef CAS.
- C. Zhu, H. Li, S. Fu, D. Du and Y. Lin, Chem. Soc. Rev., 2016, 45, 517–531 RSC.
- W. He, Y. Wang, C. Jiang and L. Lu, Chem. Soc. Rev., 2016, 45, 2396–2409 RSC.
- Y. Zhou, Y. Yu, D. Ma, A. C. Foucher, L. Xiong, J. Zhang, E. A. Stach, Q. Yue and Y. Kang, ACS Catal., 2021, 11, 74–81 CrossRef CAS.
- T. Chen, J. Wu, C. Zhu, Z. Liu, W. Zhou, C. Zhu, C. Guan and G. Fang, Chem. Eng. J., 2021, 405, 125956 CrossRef CAS.
- X. Xu, X. Zhang, Z. Xia, R. Sun, H. Li, J. Wang, S. Yu, S. Wang and G. Sun, J. Energy Chem., 2021, 54, 579–586 CrossRef.
- J. Huo, L. Lu, Z. Shen, Y. Liu, J. Guo, Q. Liu, Y. Wang, H. Liu, M. Wu and G. Wang, J. Mater. Chem. A, 2020, 8, 16271–16282 RSC.
- R. Ding, Y. Liu, Z. Rui, J. Li, J. Liu and Z. Zou, Nano Res., 2020, 13, 1519–1526 CrossRef CAS.
- G. Zhu, Y. Qi, F. Liu, S. Ma, G. Xiang, F. Jin, Z. Liu and W. Wang, ChemSusChem, 2021, 14, 866–875 CrossRef CAS PubMed.
- C. Lei, H. Chen, J. Cao, J. Yang, M. Qiu, Y. Xia, C. Yuan, B. Yang, Z. Li, X. Zhang, L. Lei, J. Abbott, Y. Zhong, X. Xia, G. Wu, Q. He and Y. Hou, Adv. Energy Mater., 2018, 8, 1801912 CrossRef.
- L. Wang, X. Liu, L. Cao, W. Zhang, T. Chen, Y. Lin, H. Wang, Y. Wang and T. Yao, J. Phys. Chem. Lett., 2020, 11, 6691–6696 CrossRef CAS PubMed.
- J. Tuo, Y. Zhu, H. Jiang, J. Shen and C. Li, ChemElectroChem, 2020, 7, 4767–4772 CrossRef CAS.
- S. Wu, X. Lv, D. Ping, G. Zhang, S. Wang, H. Wang, X. Yang, D. Guo and S. Fang, Electrochim. Acta, 2020, 340, 135930 CrossRef CAS.
- C. He, Z.-Y. Wu, L. Zhao, M. Ming, Y. Zhang, Y. Yi and J.-S. Hu, ACS Catal., 2019, 9, 7311–7317 CrossRef CAS.
- N. Du, Y. Liu, Q. Li, W. Miao, D. Wang and S. Mao, Chem. Eng. J., 2021, 413, 127545 CrossRef CAS.
- Y. Xu, L. Zhu, X. Cui, M. Zhao, Y. Li, L. Chen, W. Jiang, T. Jiang, S. Yang and Y. Wang, Nano Res., 2020, 13, 752–758 CrossRef CAS.
- Q. Ding, Y. Yu, F. Huang, L. Zhang, J.-G. Zheng, M. Xu, J. B. Baell and H. Huang, Chem.–Eur. J., 2020, 26, 4592–4598 CrossRef CAS PubMed.
- J. Lei, H. Liu, D. Yin, L. Zhou, J.-A. Liu, Q. Chen, X. Cui, R. He, T. Duan and W. Zhu, Small, 2020, 16, 1905920 CrossRef CAS PubMed.
- J. Han, H. Bao, J.-Q. Wang, L. Zheng, S. Sun, Z. L. Wang and C. Sun, Appl. Catal., B, 2021, 280, 119411 CrossRef CAS.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- H. Peng, F. Liu, X. Liu, S. Liao, C. You, X. Tian, H. Nan, F. Luo, H. Song, Z. Fu and P. Huang, ACS Catal., 2014, 4, 3797–3805 CrossRef CAS.
- J. Masa, A. Zhao, W. Xia, M. Muhler and W. Schuhmann, Electrochim. Acta, 2014, 128, 271–278 CrossRef CAS.
- W. Fan, Z. Li, C. You, X. Zong, X. Tian, S. Miao, T. Shu, C. Li and S. Liao, Nano Energy, 2017, 37, 187–194 CrossRef CAS.
- J. Zhang, M. Zhang, Y. Zeng, J. Chen, L. Qiu, H. Zhou, C. Sun, Y. Yu, C. Zhu and Z. Zhu, Small, 2019, 15, 1900307 CrossRef PubMed.
- F. Razmjooei, K. P. Singh, D.-S. Yang, W. Cui, Y. H. Jang and J.-S. Yu, ACS Catal., 2017, 7, 2381–2391 CrossRef CAS.
- F. Razmjooei, K. P. Singh, E. J. Bae and J.-S. Yu, J. Mater. Chem. A, 2015, 3, 11031–11039 RSC.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- J. Xu and L. Guan, RSC Adv., 2013, 3, 5577–5582 RSC.
- D. Yu, Y. Xue and L. Dai, J. Phys. Chem. Lett., 2012, 3, 2863–2870 CrossRef CAS PubMed.
- V. V. Strelko, V. S. Kuts and P. A. Thrower, Carbon, 2000, 38, 1499–1503 CrossRef CAS.
- E. Cruz-Silva, F. Lopez-Urias, E. Munoz-Sandoval, B. G. Sumpter, H. Terrones, J.-C. Charlier, V. Meunier and M. Terrones, Nanoscale, 2011, 3, 1008–1013 RSC.
- F. He, K. Li, G. Xie, Y. Wang, M. Jiao, H. Tang and Z. Wu, Phys. Chem. Chem. Phys., 2016, 18, 12675–12681 RSC.
- Z. Yang, J. Wu, X. Zheng, Z. Wang and R. Yang, J. Power Sources, 2015, 277, 161–168 CrossRef CAS.
- Y. Li, B. Chen, X. Duan, S. Chen, D. Liu, K. Zang, R. Si, F. Lou, X. Wang, M. Rønning, L. Song, J. Luo and D. Chen, Appl. Catal., B, 2019, 249, 306–315 CrossRef CAS.
- X. Zhu, X. Tan, K.-H. Wu, C.-L. Chiang, Y.-C. Lin, Y.-G. Lin, D.-W. Wang, S. Smith, X. Lu and R. Amal, J. Mater. Chem. A, 2019, 7, 14732–14742 RSC.
- C. Li, Z. Chen, Y. Ni, F. Kong, A. Kong and Y. Shan, J. Mater. Chem. A, 2016, 4, 14291–14297 RSC.
- N. Wang, Y. Li, Z. Guo, H. Li, S. Hayase and T. Ma, J. Electron. Mater., 2018, 165, G3080–G3086 CAS.
- H.-H. Wang, L.-B. Lv, S.-N. Zhang, H. Su, G.-Y. Zhai, W.-W. Lei, X.-H. Li and J.-S. Chen, Nano Res., 2020, 13, 2079–2084 CrossRef CAS.
- D. Liu, L. Dai, X. Lin, J.-F. Chen, J. Zhang, X. Feng, K. Müllen, X. Zhu and S. Dai, Adv. Mater., 2019, 31, 1804863 CrossRef PubMed.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- X. Zhu, R. Amal and X. Lu, Small, 2019, 15, 1804524 CrossRef PubMed.
- S. Guo, P. Yuan, J. Zhang, P. Jin, H. Sun, K. Lei, X. Pang, Q. Xu and F. Cheng, Chem. Commun., 2017, 53, 9862–9865 RSC.
- F. Ning, X. Wan, X. Liu, R. Yu and J. Shui, ChemNanoMat, 2020, 6, 1601–1610 CrossRef CAS.
- K. Yuan, D. Lützenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479 CrossRef CAS PubMed.
- H. Jin, Z. Kou, W. Cai, H. Zhou, P. Ji, B. Liu, A. Radwan, D. He and S. Mu, J. Mater. Chem. A, 2020, 8, 9121–9127 RSC.
- J.-C. Li, H. Zhong, M. Xu, T. Li, L. Wang, Q. Shi, S. Feng, Z. Lyu, D. Liu, D. Du, S. P. Beckman, X. Pan, Y. Lin and M. Shao, Sci. China Mater., 2020, 63, 965–971 CrossRef CAS.
- K. Hu, L. Tao, D. Liu, J. Huo and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 19379–19385 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, C. Chen, Y.-C. Huang, C.-L. Dong, C.-J. Chen, R.-S. Liu, C. Wang, K. Yan, Y. Li and G. Wang, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed.
- K. Qu, Y. Zheng, S. Dai and S. Z. Qiao, Nano Energy, 2016, 19, 373–381 CrossRef CAS.
- R. Zhang, Y. Fang, T. Chen, F. Qu, Z. Liu, G. Du, A. M. Asiri, T. Gao and X. Sun, ACS Sustainable Chem. Eng., 2017, 5, 7502–7506 CrossRef CAS.
- Y. Hou, M. Qiu, M. G. Kim, P. Liu, G. Nam, T. Zhang, X. Zhuang, B. Yang, J. Cho, M. Chen, C. Yuan, L. Lei and X. Feng, Nat. Commun., 2019, 10, 1392 CrossRef PubMed.
- H. Shang, X. Zhou, J. Dong, A. Li, X. Zhao, Q. Liu, Y. Lin, J. Pei, Z. Li, Z. Jiang, D. Zhou, L. Zheng, Y. Wang, J. Zhou, Z. Yang, R. Cao, R. Sarangi, T. Sun, X. Yang, X. Zheng, W. Yan, Z. Zhuang, J. Li, W. Chen, D. Wang, J. Zhang and Y. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- J. Miao, F.-X. Xiao, H. B. Yang, S. Y. Khoo, J. Chen, Z. Fan, Y.-Y. Hsu, H. M. Chen, H. Zhang and B. Liu, Sci. Adv., 2015, 1, e1500259 CrossRef PubMed.
- Y. Jia, X. Xiong, D. Wang, X. Duan, K. Sun, Y. Li, L. Zheng, W. Lin, M. Dong, G. Zhang, W. Liu and X. Sun, Nano-Micro Lett., 2020, 12, 116 CrossRef CAS PubMed.
- M. Wang, W. Yang, X. Li, Y. Xu, L. Zheng, C. Su and B. Liu, ACS Energy Lett., 2021, 6, 379–386 CrossRef CAS.
- Y. Chen, S. Ji, S. Zhao, W. Chen, J. Dong, W.-C. Cheong, R. Shen, X. Wen, L. Zheng, A. I. Rykov, S. Cai, H. Tang, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 5422 CrossRef CAS PubMed.
- S. Chen, N. Zhang, C. W. Narváez Villarrubia, X. Huang, L. Xie, X. Wang, X. Kong, H. Xu, G. Wu, J. Zeng and H.-L. Wang, Nano Energy, 2019, 66, 104164 CrossRef CAS.
- M. Zhang, J. Zhang, S. Ran, L. Qiu, W. Sun, Y. Yu, J. Chen and Z. Zhu, Nano Res., 2021, 14, 1175–1186 CrossRef CAS.
- H. Su, L. Chen, Y. Chen, R. Si, Y. Wu, X. Wu, Z. Geng, W. Zhang and J. Zeng, Angew. Chem., Int. Ed., 2020, 59, 20411–20416 CrossRef CAS PubMed.
- X. Lv, W. Wei, F. Li, B. Huang and Y. Dai, Nano Lett., 2019, 19, 6391–6399 CrossRef CAS PubMed.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132–7135 CrossRef CAS PubMed.
- L. Jiao, W. Xu, Y. Zhang, Y. Wu, W. Gu, X. Ge, B. Chen, C. Zhu and S. Guo, Nano Today, 2020, 35, 100971 CrossRef CAS.
- Z. Yao, M. Hu, Z. Iqbal and X. Wang, ACS Catal., 2020, 10, 160–167 CrossRef CAS.
- K. Yuan, S. Sfaelou, M. Qiu, D. Lützenkirchen-Hecht, X. Zhuang, Y. Chen, C. Yuan, X. Feng and U. Scherf, ACS Energy Lett., 2018, 3, 252–260 CrossRef CAS.
- A. G. Saputro, A. K. Fajrial, A. L. Maulana, F. Fathurrahman, M. K. Agusta, F. T. Akbar and H. K. Dipojono, J. Phys. Chem. C, 2020, 124, 11383–11391 CrossRef CAS.
- H. Sun, M. Wang, X. Du, Y. Jiao, S. Liu, T. Qian, Y. Yan, C. Liu, M. Liao, Q. Zhang, L. Meng, L. Gu, J. Xiong and C. Yan, J. Mater. Chem. A, 2019, 7, 20952–20957 RSC.
- B. He, J. Shen, D. Ma, Z. Lu and Z. Yang, J. Phys. Chem. C, 2018, 122, 20312–20322 CrossRef CAS.
- L. Yang, X. Zeng, W. Wang and D. Cao, Adv. Funct. Mater., 2018, 28, 1704537 CrossRef.
- X. Sun, Y. Zhang, P. Song, J. Pan, L. Zhuang, W. Xu and W. Xing, ACS Catal., 2013, 3, 1726–1729 CrossRef CAS.
- S. G. Peera, A. Arunchander and A. K. Sahu, Nanoscale, 2016, 8, 14650–14664 RSC.
- L. Chen, J. Chang, Y. Zhang, Z. Gao, D. Wu, F. Xu, Y. Guo and K. Jiang, Chem. Commun., 2019, 55, 3406–3409 RSC.
- Y. Xue, Y. Wang, H. Liu, X. Yu, H. Xue and L. Feng, Chem. Commun., 2018, 54, 6204–6207 RSC.
- M. Karuppannan, J. E. Park, H. E. Bae, Y.-H. Cho and O. J. Kwon, Nanoscale, 2020, 12, 2542–2554 RSC.
- S.-G. Han, D.-D. Ma, S.-H. Zhou, K. Zhang, W.-B. Wei, Y. Du, X.-T. Wu, Q. Xu, R. Zou and Q.-L. Zhu, Appl. Catal., B, 2021, 283, 119591 CrossRef CAS.
- X.-Y. Zhang, Y.-R. Zhu, Y. Chen, S.-Y. Dou, X.-Y. Chen, B. Dong, B.-Y. Guo, D.-P. Liu, C.-G. Liu and Y.-M. Chai, Chem. Eng. J., 2020, 399, 125831 CrossRef CAS.
- Y. Zhou, X. Tao, G. Chen, R. Lu, D. Wang, M.-X. Chen, E. Jin, J. Yang, H.-W. Liang, Y. Zhao, X. Feng, A. Narita and K. Müllen, Nat. Commun., 2020, 11, 5892 CrossRef CAS PubMed.
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi, Z. Jiang, M. Li, S. Mu, S. Liao, J. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 2622–2626 CrossRef CAS PubMed.
- J. Zhang, Q.-a. Huang, J. Wang, J. Wang, J. Zhang and Y. Zhao, Chinese J. Catal., 2020, 41, 783–798 CrossRef CAS.
- N. R. Sahraie, U. I. Kramm, J. Steinberg, Y. Zhang, A. Thomas, T. Reier, J.-P. Paraknowitsch and P. Strasser, Nat. Commun., 2015, 6, 8618 CrossRef CAS PubMed.
- Z. Zhu, H. Yin, Y. Wang, C.-H. Chuang, L. Xing, M. Dong, Y.-R. Lu, G. Casillas-Garcia, Y. Zheng, S. Chen, Y. Dou, P. Liu, Q. Cheng and H. Zhao, Adv. Mater., 2020, 32, 2004670 CrossRef CAS PubMed.
- L. Cao, Y. Shao, H. Pan and Z. Lu, J. Phys. Chem. C, 2020, 124, 11301–11307 CrossRef CAS.
- K. Chen, S. Kim, M. Je, H. Choi, Z. Shi, N. Vladimir, K. H. Kim and O. L. Li, Nano-Micro Lett., 2021, 13, 60 CrossRef PubMed.
- J. Guo, J. Huo, Y. Liu, W. Wu, Y. Wang, M. Wu, H. Liu and G. Wang, Small Methods, 2019, 3, 1900159 CrossRef.
- L. Bai, C.-S. Hsu, D. T. L. Alexander, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2019, 141, 14190–14199 CrossRef CAS PubMed.
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang, C. Zhao, Y. Qu, Z. Yang, T. Yao, Y. Li, Y. Lin, Y. Wu and Y. Li, Energy Environ. Sci., 2018, 11, 3375–3379 RSC.
- J. Wang, Z. Li, Y. Wu and Y. Li, Adv. Mater., 2018, 30, 1801649 CrossRef PubMed.
- J. Yang, D. Zeng, J. Li, L. Dong, W.-J. Ong and Y. He, Chem. Eng. J., 2021, 404, 126376 CrossRef CAS.
- J. Wang, Z. Huang, W. Liu, C. Chang, H. Tang, Z. Li, W. Chen, C. Jia, T. Yao, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 17281–17284 CrossRef CAS PubMed.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- Z. Zhang, L. Cong, Z. Yu, L. Qu and W. Huang, Mater. Today Energy, 2020, 16, 100387 CrossRef.
- X. Zhu, D. Zhang, C.-J. Chen, Q. Zhang, R.-S. Liu, Z. Xia, L. Dai, R. Amal and X. Lu, Nano Energy, 2020, 71, 104597 CrossRef CAS.
- Y. Zhou, E. Song, W. Chen, C. U. Segre, J. Zhou, Y.-C. Lin, C. Zhu, R. Ma, P. Liu, S. Chu, T. Thomas, M. Yang, Q. Liu, K. Suenaga, Z. Liu, J. Liu and J. Wang, Adv. Mater., 2020, 32, 2003484 CrossRef CAS PubMed.
- Y. Zhou, W. Yang, W. Utetiwabo, Y.-m. Lian, X. Yin, L. Zhou, P. Yu, R. Chen and S. Sun, J. Phys. Chem. Lett., 2020, 11, 1404–1410 CrossRef CAS PubMed.
- Q. Lin, X. Bu, A. Kong, C. Mao, F. Bu and P. Feng, Adv. Mater., 2015, 27, 3431–3436 CrossRef CAS PubMed.
- T. Palaniselvam, V. Kashyap, S. N. Bhange, J.-B. Baek and S. Kurungot, Adv. Funct. Mater., 2016, 26, 2150–2162 CrossRef CAS.
- X. Fang, L. Jiao, S.-H. Yu and H.-L. Jiang, ChemSusChem, 2017, 10, 3019–3024 CrossRef CAS PubMed.
- Y.-P. Deng, Y. Jiang, R. Liang, S.-J. Zhang, D. Luo, Y. Hu, X. Wang, J.-T. Li, A. Yu and Z. Chen, Nat. Commun., 2020, 11, 1952 CrossRef CAS PubMed.
- L. Cao, X. Wang, C. Yang, J. Lu, X. Shi, H. Zhu and H.-P. Liang, ACS Sustainable Chem. Eng., 2021, 9, 189–196 CrossRef CAS.
- X. Zhong, S. Ye, J. Tang, Y. Zhu, D. Wu, M. Gu, H. Pan and B. Xu, Appl. Catal., B, 2021, 286, 119891 CrossRef CAS.
- Y. Zhao, H. Zhou, X. Zhu, Y. Qu, C. Xiong, Z. Xue, Q. Zhang, X. Liu, F. Zhou, X. Mou, W. Wang, M. Chen, Y. Xiong, X. Lin, Y. Lin, W. Chen, H.-J. Wang, Z. Jiang, L. Zheng, T. Yao, J. Dong, S. Wei, W. Huang, L. Gu, J. Luo, Y. Li and Y. Wu, Nat. Catal., 2021, 4, 134–143 CrossRef CAS.
- S. Li, C. Cheng, X. Zhao, J. Schmidt and A. Thomas, Angew. Chem., Int. Ed., 2018, 57, 1856–1862 CrossRef CAS PubMed.
- Y. Pan, S. Liu, K. Sun, X. Chen, B. Wang, K. Wu, X. Cao, W.-C. Cheong, R. Shen, A. Han, Z. Chen, L. Zheng, J. Luo, Y. Lin, Y. Liu, D. Wang, Q. Peng, Q. Zhang, C. Chen and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 8614–8618 CrossRef CAS PubMed.
- F. Yang, J. Ye, Q. Yuan, X. Yang, Z. Xie, F. Zhao, Z. Zhou, L. Gu and X. Wang, Adv. Funct. Mater., 2020, 30, 1908235 CrossRef CAS.
- C. H. Choi, C. Baldizzone, J.-P. Grote, A. K. Schuppert, F. Jaouen and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2015, 54, 12753–12757 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- E. Luo, H. Zhang, X. Wang, L. Gao, L. Gong, T. Zhao, Z. Jin, J. Ge, Z. Jiang, C. Liu and W. Xing, Angew. Chem., Int. Ed., 2019, 58, 12469–12475 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |