
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereodivergent synthesis via iridium-catalyzed asymmetric double allylic alkylation of cyanoacetate†
Chong
Shen
a,
Xiang
Cheng
a,
Liang
Wei
 a,
Ruo-Qing
Wang
a and
Chun-Jiang
Wang
*ab
a,
Ruo-Qing
Wang
a and
Chun-Jiang
Wang
*ab
aEngineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, China. E-mail: cjwang@whu.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Shanghai, 230021, China
First published on 20th November 2021
Abstract
Methods that enable the rapid construction of multiple C–C bonds using a single catalyst with high diastereo- and enantio-control are particularly valuable in organic synthesis. Here, we report an Ir-catalyzed double allylic alkylation reaction in which bisnucleophilic cyanoacetate reacted successionally with electrophilic π-allyl-Ir species, producing various pseudo-C2-symmetrical cyanoacetate derivatives in high yield with excellent stereocontrol. More challenging sequential allylic alkylation/allylic alkylation with two distinct allylic carbonates that can deliver the corresponding products bearing three contiguous tertiary–quaternary–tertiary stereocenters was also developed by using a modified catalytic system, which is revealed to be associated with the quasi-dynamic kinetic resolution of the initially formed diastereomeric monoallylation intermediates. Notably, stereodivergence for this sequential process depending on a single iridium catalyst was successfully realized, and up to six stereoisomers could be predictably prepared by combining the appropriate enantiomer of the chiral ligand for the iridium catalyst and adjusting the adding sequence of two distinct allylic precursors.
Introduction
The physiological or pharmacological properties of chiral compounds mainly depend on the interactions between such molecules and chiral environments created by enzymes/receptors in biological systems.1 Hence, for biologically important compounds bearing multiple stereogenic centers, developing selective access to different enantio- and diastereomers to evaluate the corresponding biological activity is of great practical importance for pharmaceutical applications.2 Although the efficacy of asymmetric catalysis has been thoroughly demonstrated for the construction of a tremendous amount of chiral skeletons, stereodivergent synthesis of as many as possible stereoisomers from the same starting material remains extremely challenging.3 For a given asymmetric transformation, while enantiodivergence could be realized through employing an enantiomeric pair of the chiral catalyst, controlling the relative configuration of the product to produce individual diastereomers is much more difficult. Specifically, one typical diastereomer is usually inherently preferred and the complementary diastereomer(s) cannot be effectively constructed via the same strategy.4 To overcome the diastereoselective bias in the chiral induction process, diverse methodologies, such as the modulation of the structure of the catalyst and substrate, changing the reaction conditions (solvent, temperature, pressure, and additives) or using an entirely different catalytic system, have been developed in the past twenty years.5,6 Despite many important advances, serendipity is the most important factor in these findings and such strategies cannot be readily extended to the development of other stereodivergent transformations.In comparison, the recently developed stereodivergent synthesis that can predictably induce stereodivergence has demonstrated to be a superior strategic alternative to traditional methods (Scheme 1) since the groundbreaking research work of Carreira in 2013.7,8 Two conceptually distinct methodologies have been disclosed for stereodivergent synthesis: dual catalysis involves the simultaneous activation of prochiral nucleophiles and allylic carbonates/alkynes with a synergistic catalytic system, exerting full control over the absolute/relative configuration in the coupling step (Scheme 1a);7,9 and single catalysis allows the use of a single catalyst to introduce multiple stereogenic centers sequentially in a multistep transformation (Scheme 1b).10,11 Numerous elegant studies have been reported in the field of stereodivergent synthesis via dual catalysis for the construction of biologically important chiral molecules.7–9 In sharp contrast, several daunting challenges such as the need for a multitasking catalyst and the potential matched/mismatched effects between the substrate and catalyst resulted in the applications of single catalysis in stereodivergent synthesis being largely unexplored. In 2016, Buchwald et al. developed the first example of a sequential reduction/hydroamination of enals/enones catalyzed by a chiral copper hydride catalyst for the stereodivergent synthesis of 1,3-aminoalcohols.10 Recently, Trost et al. developed a bimetallic Zn–ProPhenol-catalyzed enantioselective Mannich/aldol-type reaction of branched aldehydes, allowing the preparation of four stereoisomers of the corresponding 1,3-aminoalcohols.11 Nonetheless, stereodivergent synthesis of highly functionalized and diversified chiral scaffolds with single catalysis is still in its infancy and remains significantly challenging.
 | ||
| Scheme 1 Two conceptually distinct stereodivergent syntheses with dual catalysis (a) and single catalysis (b). | ||
Iridium-catalyzed enantioselective allylic alkylation is a powerful methodology for carbon–carbon and carbon–heteroatom bond formation and constitutes great contributions in the field of stereodivergent synthesis via dual catalysis (Scheme 2a).12 To date, most of the investigations focus on the incorporation of one allyl moiety into diverse nucleophiles with high branched selectivity. We anticipated that an Ir-catalyzed sequential allylic alkylation/allylic alkylation of one-site C-based bisnucleophilic reagents would provide a rapid access to complex structures with multiple stereogenic centers and functional groups that can be easily manipulated in downstream transformations. In addition, the use of an enantiomeric pair of the chiral iridium catalyst for each allylation step might permit the stereodivergent construction of the target products. However, such a process has only been sporadically realized by using bisnucleophiles with minimum steric hindrance such as Na2S and NH3,13 and the double allylation of prochiral bisnucleophiles remains undeveloped due to the inherent steric repulsion in the formation of an acyclic quaternary stereogenic center.14 Furthermore, it is unpredictable how the matched/mismatched effects of the chiral intermediate formed in the first allylation step with a chiral π-allyl-Ir complex facilitate/deteriorate the catalytic efficiency and the stereoselective control in the second step. Herein, we successfully alleviate these problems by presenting a single iridium-catalyzed stereodivergent synthesis of highly functionalized cyanoacetate bearing two tertiary and one all-carbon quaternary stereogenic centers, and the quasi-dynamic kinetic resolution15 of the monoallylated intermediate made the designed sequential allylic alkylation/allylic alkylation process highly diastereoselective. More importantly, up to six stereoisomers of the corresponding products could be efficiently obtained in high regio-, diastereo- and enantio-selectivity (Scheme 2b).
 | ||
| Scheme 2 Ir-catalyzed double-asymmetric allylic allylation ((a) previous work) and stereodivergent doble allylaltion ((b) this work). | ||
Results and discussion
In initial attempts to implement the proposed allylic alkylation/allylic alkylation sequence, we first evaluated the reaction of glycine-derived imine ester162 with 2.2 equivalents of methyl cinnamyl carbonate 1a. A synergistic Cu/Ir system which has proved to be efficient for stereodivergent allylation in our previous work9i was utilized; however, none of the desired bisallylation product 6 could be detected. Instead, the reaction finished exclusively in the first allylation step, providing the corresponding 4 in 85% yield with poor diastereoselectivity (Table 1, entry 1). The variation of reaction parameters including increasing the reaction temperature or using stronger bases was unable to promote the generation of 6. We speculated that the unfavorable steric congestion completely suppressed the second allylation step and a suitable bisnucleophile with sufficient nucleophilicity and less steric hindrance would be the linchpin for the design process. To verify this hypothesis, various potential bisnucleophiles were subjected to reaction with 1a. Delightfully, when cyanoacetate 3 was selected as the reactant, the reaction proceeded smoothly using a single Ir(I)/(R,R,R)-L2 catalyst along with triethylamine as the base, affording the bisallylation product 7a in good yield with a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of di/mono, 10:1 dr and 98% ee (entry 2). Switching the base to Cs2CO3 further advanced the catalytic efficiency, leading to full conversion of starting materials to desired 7a quantitatively as an exclusive diastereomer (>20:1 dr) with maintained enantioselectivity (entry 3).
:1 ratio of di/mono, 10:1 dr and 98% ee (entry 2). Switching the base to Cs2CO3 further advanced the catalytic efficiency, leading to full conversion of starting materials to desired 7a quantitatively as an exclusive diastereomer (>20:1 dr) with maintained enantioselectivity (entry 3).
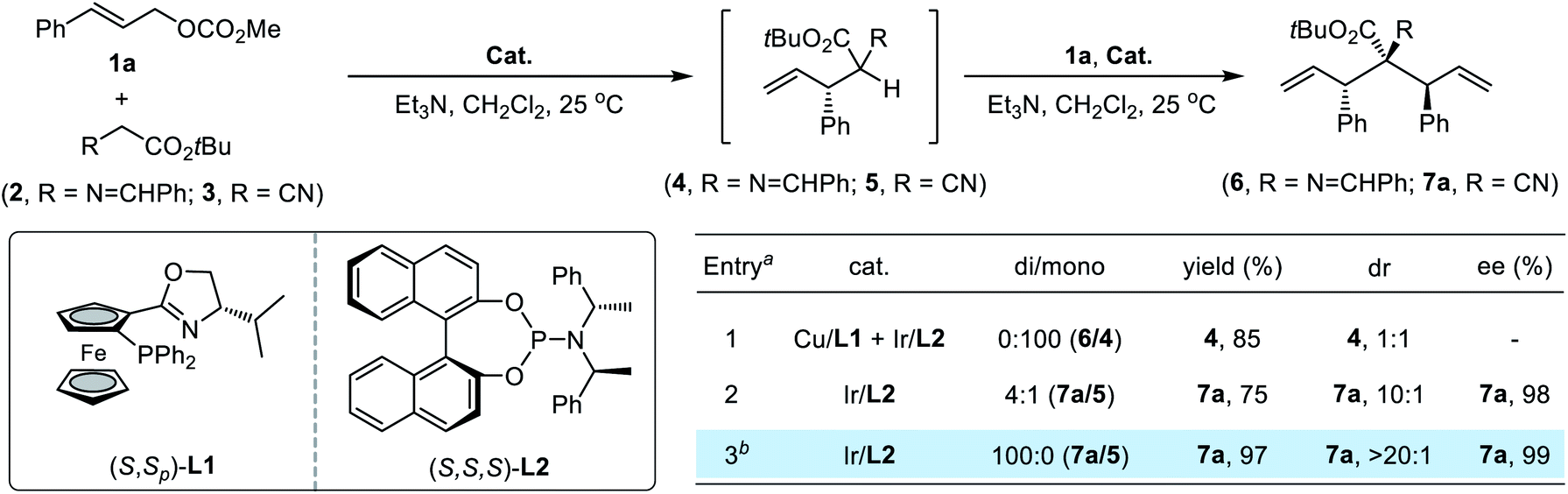
With reaction conditions for double allylic alkylation of cyanoacetate established, we first examined the substrate scope of this transformation (Table 2). A range of para- and meta-substituted cinnamyl carbonates bearing diverse functional groups were efficient Ir-π-allyl precursors, providing the desired bisallylated cyanoacetate derivatives 7a–n in high yield with excellent stereoselectivity (entries 1–14). Ortho-substituted cinnamyl carbonate was not a viable substrate, which is a known issue associated with Feringa-type ligands in Ir-catalyzed allylic alkylation reactions.12 2-Naphthyl allylic carbonate 1o also reacted successfully with 3 to produce 7o in 97% yield with >20:1 dr and 99% ee. A moderate yield and diastereoselectivity were observed for bulky 1-naphthyl substituted 1p under the standard conditions due to the disfavored steric congestion. Using You's (S,S)-Me-THQphos17 as the chiral ligand instead of (R,R,R)-L2 effectively improved the catalytic efficacy, giving 7p in 81% yield with exclusive diastereoselectivity (>20:1 dr) and excellent enantioselectivity (>99% ee). In addition, the iridium catalysis showed a comparable level of stereoselectivity towards allylic carbonates bearing heteroaromatic groups, and the corresponding products 7q and 7r were achieved in >20:1 dr and 99% ee albeit with a moderate yield. Furthermore, the current system could be further extended to less reactive crotyl carbonate 1s, affording 7s in 72% yield with 7:1 dr and 95% ee. Switching the iridium precursor to [Ir(DBCOT)Cl]2 significantly enhanced the reactivity and diastereoselectivity. The absolute configuration of ent-7j using Ir(I)/(S,S,S)-L2 as the catalyst was unambiguously determined to be (S,S) by X-ray crystallographic analysis (CCDC 2096475†).

Having developed an efficient method to produce cyanoacetate derivatives bearing two identical allyl moieties, we attempted to further investigate the more challenging double allylic alkylation using different allylic carbonates for each step (Scheme 3). 4-Bromo-substituted cinnamyl carbonate 1d and 2-naphthyl allyl carbonate 1o were selected as the representative allyl precursors to react sequentially with cyanoacetate 3. We anticipated that the selective interruption of the reaction process to avoid the above-mentioned gem-allylation is key to realizing the designed process. Based on this surmise, we first tested the reaction of 3 with 1 equivalent of 1d under the standard conditions except for changing the base to weaker basic Et3N. Indeed, the mono-allylation product could be isolated in 47% yield. The same catalytic system further initiated the following allylic alkylation with 1 equivalent of 1o and thus converted the isolated mono-allylation intermediate into the desired bisallylation cyanoacetate (R,R,R)-8A bearing three contiguous tertiary–quaternary–tertiary stereogenic centers in 92% yield. However, a mixture of inseparable diastereomers with a 1:0.25:0.1:0.08 diastereomeric ratio was detected through NMR analysis. We reasoned the unsatisfactory overall yield and diastereoselectivity to be as a result of (1) incomplete suppression of double allylation in the first step; (2) insufficient stereocontrol over the allyl stereogenic center by the Ir(I)/(R,R,R)-L2 complex under slightly modified reaction conditions; (3) the mutual effects of substrate- and catalyst-controlled chiral induction on the quaternary stereogenic center in the second allylation step.
 | ||
| Scheme 3 Initial test for sequential allylic alkylation with two different allyl carbonates. | ||
To validate our conjecture, the stereocontrol efficacy of iridium catalysis for the first allylation step was then systematically investigated (Table 3). We first isolated the mixture of diastereomeric monoallylation intermediates 5d in 47% yield with an unsatisfactory diastereoselectivity (1.1:1 dr), which was caused by the easy epimerization of the tertiary stereogenic center at the α-position of compound 5d under basic conditions. Furthermore, the enantioselectivity of the allyl stereogenic center was determined to be 90% ee by transforming 5d into nitrile 9 through a LiCl-promoted Krapcho reaction (entry 1).18 An imperfect chiral induction by the Ir(I)/(R,R,R)-L2 catalyst in the first allylation step would dramatically deteriorate the diastereoselectivity in the sequential process, and thus we set out to re-optimize the reaction conditions for this monoallylation reaction aiming to improve the chemical yield and enantioselectivity (see the ESI† for details). After screening various solvents and chiral phosphoramidite ligands, a moderate yield with a slightly higher enantioselectivity (93% ee for 9) was obtained using Ir(I)/(R,R,R)-L3 as the catalyst in THF at room temperature (entry 2). Replacing the base with DABCO and using two equivalents of 3 slightly enhanced the reactivity, giving compound 5d in 56% yield (entry 3). Further variation of the reaction temperature indicated that a lower temperature was beneficial to the current transformation, leading to a significant improvement in terms of both yield and enantioselectivity (entry 4). To conclude, the [Ir(cod)Cl]2/(R,R,R)-L3/DABCO/THF system was chosen for the first allylic alkylation step. Additionally, the subsequent optimization of the second step identified the [Ir(cod)Cl]2/(R,R,R)-L3/Cs2CO3/DCM system to be optimal (see the ESI† for details).

With the modified reaction conditions for the sequential double allylic alkylation in hand, we were particularly interested in advancing this method to stereodivergently assemble the stereoisomers of the cyanoacetate derivatives (Scheme 4). As described above, we have prepared (R,R,R)-8A by using Ir(I)/(R,R,R)-L3 as the chiral catalyst for both steps (Scheme 4h). The enantiomeric (S,S,S)-8A could be easily obtained via the use of the (S,S,S)-L3 ligand for the iridium catalyst (Scheme 4e). To prepare (R,S,S)-8A, we developed a modified protocol involving one chiral ligand switch in the sequential double allylation, that is, cyanoacetate 3 was first monoallylated with para-bromo cinnamyl carbonate 1d using (S,S,S)-L3 as the ligand, and the resultant α-monoallylation intermediate was isolated and then subjected to the second allylation conditions with 2-naphthyl allylic carbonate 1o catalyzed by Ir(I)/(R,R,R)-L3 (Scheme 4f). (S,R,R)-8A was then constructed efficiently by employing the same ligand-switch strategy except for using (R,R,R)-L3 and (S,S,S)-L3 as the chiral ligand for each step, respectively (Scheme 4g). Moreover, reversing the adding sequence of 1d/1o along with the utilization of the same chiral ligand (S,S,S)-L3 or (R,R,R)-L3 for two sequential allylations could afford enantiomeric (S,R,S)-8A (X-ray, CCDC 2096476†) or (R,S,R)-8A, respectively, which are the opposite enantiomers of each other (Scheme 4a and d). Interestingly, combining the reversed adding sequence with the ligand-switch strategy as shown in Scheme 4b and c led to the generation of (S,R,R)-8A and (R,S,S)-8A, respectively, which could also be obtained with the protocol shown in Scheme 4g and f. Ultimately, by pairwise selection of the enantiomeric ligand and adding sequence of two distinct allylic carbonates, six of all eight stereoisomers of bisallylated cyanoacetate 8A were predictably constructed in good overall yields (around 60%) with excellent stereoselectivities. The generality of the current stereodivergent synthesis via the single catalysis strategy was further extended to the preparation of six stereoisomers of 8B (from 3, 1d and 1n) and 8C (from 3, 1n and 1o) in synthetically useful yield with comparable levels of stereoselectivities.
 | ||
| Scheme 4 Stereodivergent synthesis of six accessible stereoisomers using an appropriate enantiomer of the chiral ligand for the iridium catalyst and adding sequence of two different allylic carbonates. | ||
Based on the experimental results, a plausible reaction pathway and transition states were proposed to rationalize the origin of the stereochemical outcome. As depicted in Scheme 5, Ir(I)/(S,S,S)-L3-catalyzed monoallylation with 1o would produce a mixture of diastereomeric syn-(2R,3R)-5o (in which the bulky substituents, Ar and CO2tBu groups, reside in a syn-fashion) and anti-(2S,3R)-5o (in which the bulky substituents, Ar and CO2tBu groups, reside in an anti-fashion), both of which could be readily interconverted under basic conditions through the enol form (S)-10 due to the strong α-H acidity of 5o. When using the Ir(I)/(S,S,S)-L3 complex as the chiral catalyst in the second step, both syn-(2R,3R)-5o and anti-(2S,3R)-5o were diastereo-convergently converted into the corresponding syn-anti-(S,R,S)-8A in a quasi-dynamic kinetic resolution (quasi-DKR) manner via the attack of the Si-face of the nucleophilic (S)-10 to the Re-face of the electrophilic (R)-(S,S,S)-π-allyl-Ir species.19a,b Consequently, in the second carbon–carbon bond-forming step, the newly generated allylic stereogenic center had (S)-configuration (catalyst-control), and the original labile tertiary stereocenter in 5o was transformed into a stable non-epimerized quaternary stereogenic center as (R)-configuration (substrate-control) (Scheme 5a). We reasoned that the energy-favored transition state TS-1 dominated the generation of the relative syn-configuration between the first allylic tertiary stereocenter and the quaternary stereocenter. When using the Ir(I)/(R,R,R)-L3 complex as the chiral catalyst in the second step, the corresponding diastereomer syn–syn-(S,R,R)-8A was obtained in a similar quasi-DKR manner via the attack of the Si-face of (S)-10 to the Si-face of (S)-(R,R,R)-π-allyl-Ir species,19c,d in which the relative configuration between the first allylic tertiary stereocenter and the quaternary stereocenter also maintained as the observed syn-configuration (Scheme 5b). Similarly, the stereochemical outcome of syn–syn-(R,S,S)-8A and syn–anti-(R,S,R)-8A generated from the manipulation procedures shown in Scheme 5c and d, respectively, could also be rationalized as a result of quasi-DKR of (2R,3S)-5 and (2S,3S)-5via the transition state TS-2. In addition, the stereochemical outcomes of syn–anti-(S,S,S)-8A, syn–syn-(R,S,S)-8A, syn–syn-(S,R,R)-8A, and syn–anti-(R,R,R)-8A, which were obtained as shown in Scheme 4e–h using a reversed adding sequence of the two allylic carbonates, can be explained similarly (not shown in Scheme 5). Notably, the protocols in Scheme 4b/g and 4c/f provided the same stereoisomers syn–syn-(S,R,R)-8A and syn–syn-(R,S,S)-8A, respectively. The reaction model and the related plausible transition states shown in Scheme 5 also revealed that two additional isomers anti–anti-(S,S,R)-8A and anti–anti-(R,R,S)-8A cannot be generated via the current methodology probably due to the kinetically disfavored anti–anti-configuration, which is consistent with the experimental results.
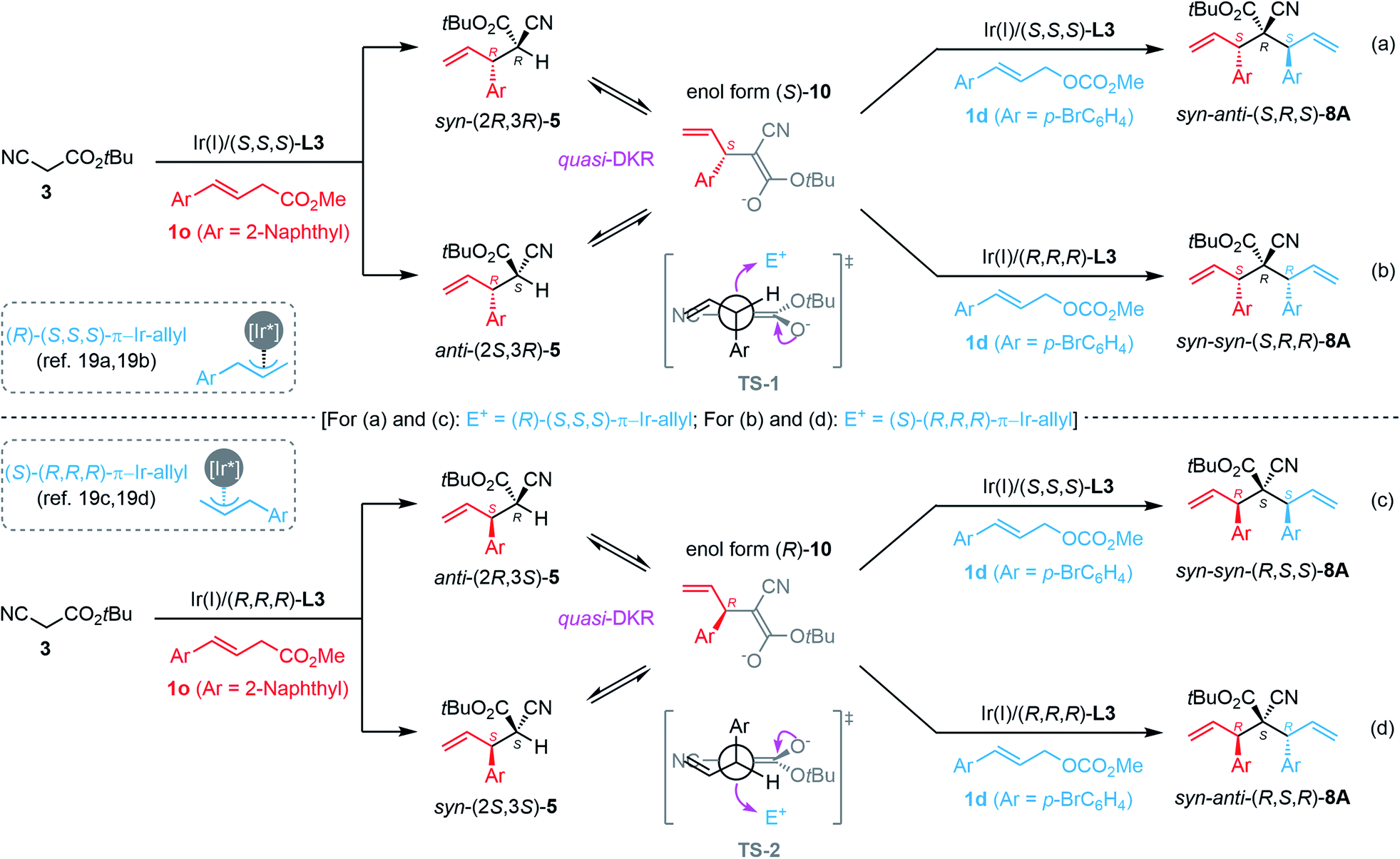 | ||
| Scheme 5 The rationale of the stereochemical outcome of stereodivergent synthesis. | ||
According to the experimental results and rationale shown in Scheme 5b and c, we envisioned that a meso-bisallylation compound could be prepared through a sequential double allylation process in which two enantiomeric chiral ligands were respectively used in the first and the second allylation reaction with the same allylic carbonate as the π-allyl-Ir precursor. As expected, starting from cyanoacetate 3, the relay Ir(I)/(S,S,S)-L3 and Ir(I)/(R,R,R)-L3 catalyzed double allylic alkylation with allylic carbonate 1d produced syn–syn-selective meso-(S,s,R)-7d (X-ray, CCDC 2096474†) in 86% yield with exclusive diastereoselectivity (>20:1 dr), and no anti–anti-selective meso-isomer (S,r,R)-7d was observed (Scheme 6), which is fully consistent with the above analysis (Scheme 5b and c).
 | ||
| Scheme 6 Preparation of meso-(S,S,R)-7d using the sequential double allylation protocol. | ||
Next, gram-scale double allylic alkylation of 3 (3.0 mmol, 0.42 g) with 1n (6.0 mmol, 1.57 g) catalyzed by the 5 mol% of Ir(I)/(S,S,S)-L2 complex was conducted to show the practicability of the current method, and the desired product (S,S)-7n could be isolated in high yield with excellent yield and stereoselectivity (1.38 g, 90% yield, >20:1 dr and 99% ee) (Scheme 7a). The presence of multiple synthetically useful functional groups in bisallylated cyanoacetate enabled rapid access to a variety of highly functionalized complex molecules. As summarized in Scheme 7b, (S,S)-7 could be converted to diverse enantioenriched building blocks 11–14 under standard chemistry. The hydrolysis of (S,S)-7n by TFA provided α-cyano carboxylic acid 11 in 81% yield, which could then be stereospecifically transformed into γ-butyrolactone derivative 12 in 57% yield with >20:1 dr and 99% ee through I2-promoted lactonization. The absolute configuration of 12 was determined by X-ray analysis (CCDC 2096478†). Nitrile 13 was efficiently generated from 7nvia a Krapcho reaction in good yield with excellent stereoselectivity. In addition, the [Ir(COD)Cl]2/DPPM catalyzed double hydroboration of (S,S)-7o successfully delivered the desired boranate 14.
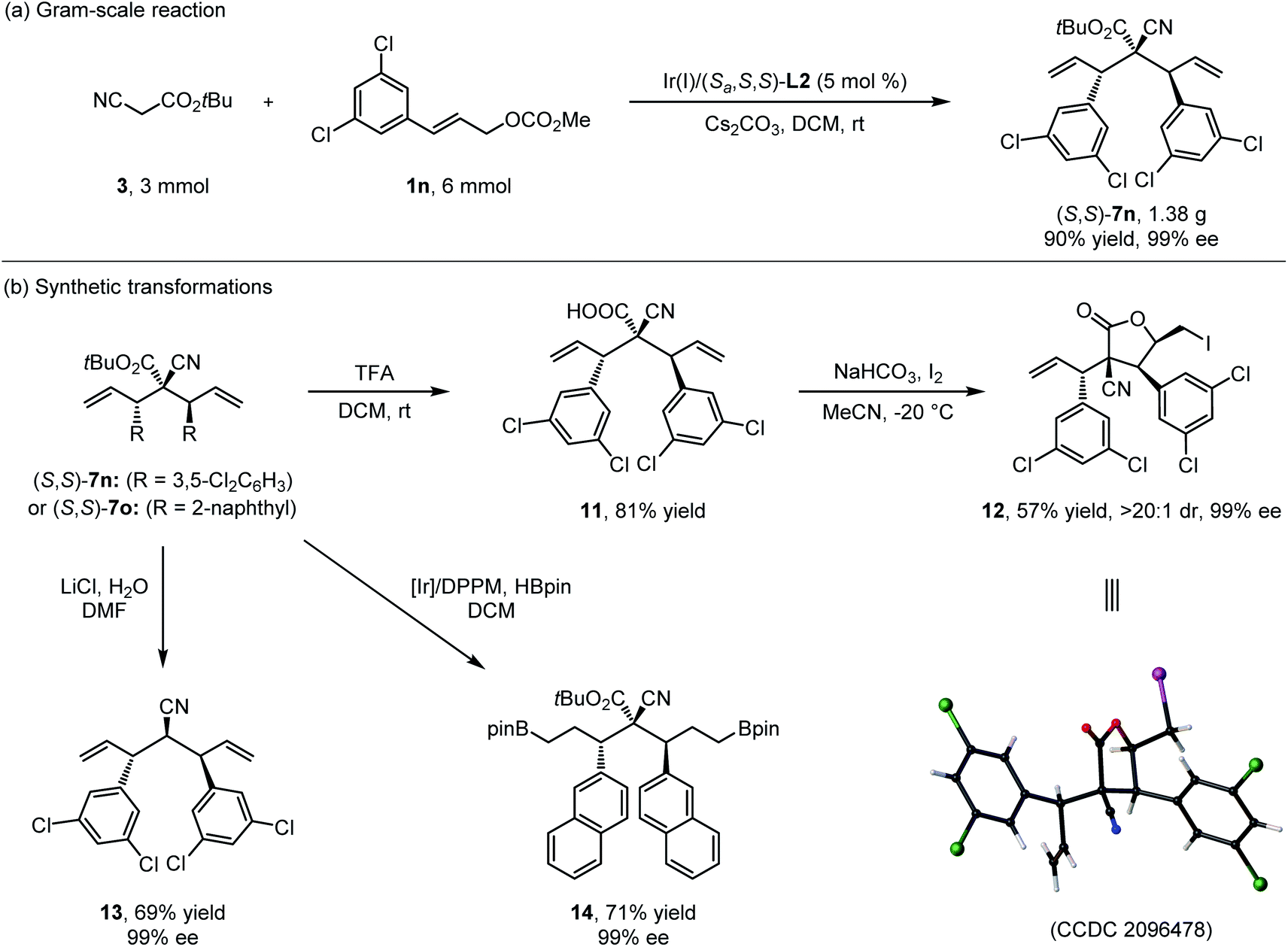 | ||
| Scheme 7 Gram scale preparation of (S,S)-7n (a) and synthetic transformations (b). | ||
Conclusions
In conclusion, we have presented a highly efficient sequential allylic alkylation/allylic alkylation of cyanoacetate empowered by single iridium catalysis. Using an identical allylic carbonate as the electrophile in succession, a range of pseudo-C2-symmetrical bisallylated cyanoacetates were constructed in high yield with excellent diastereo- and enantio-selectivity. The careful investigation and analysis of double allylic alkylation using two different allyl carbonates further demonstrated the single iridium catalysis in stereodivergent synthesis. By combining the appropriate enantiomer of the chiral ligand for the iridium catalyst with adjusting the adding sequence of two distinct allylic precursors, we were able to predictably prepare six of all eight stereoisomers of the corresponding products containing three contiguous tertiary–quaternary–tertiary stereocenters in good yield with excellent stereoselective control. In addition, a quasi-dynamic kinetic resolution process of the initially formed diastereomeric monoallylation intermediates was also clearly revealed in the ensuing allylic alkylation step. Further investigations on the stereodivergent synthesis via single iridium catalysis, as well as the synthetic applications of this method to high-value molecules, are ongoing in our laboratory.Data availability
All experimental and characterization data in this manuscript are available in the ESI.† Crystallographic data for compound (S,S)-7j, (S,R,S)-8A, meso-(S,s,R)-7d, (3S,4S,5R)-12 have been deposited at the CCDC 2096474-2096476 and 2096478, respectively.Author contributions
C.-J. W. conceived and designed the research. C. S., X. C., and R.-Q. W. performed the research. C.-J. W., L. W., and C. S. co-wrote the paper. All authors analysed the data, discussed the results, and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21772147, 220711186) and Hubei Province NSF (2020CFA036). The Program of Introducing Talents of Discipline to Universities of China (111 Project) is also appreciated.Notes and references
- B. Waldeck, Chirality, 1993, 5, 350 CrossRef CAS PubMed.
- (a) K. Jozwiak, W. J. Lough, I. W. Wainer, Drug Stereochemistry: Analytical Methods and Pharmacology, Informa, 3rd edn, 2012 CrossRef; (b) C. G. Wermuth, The Practice of Medicinal Chemistry, Elsevier, 3rd edition, 2008 Search PubMed; (c) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752 CrossRef CAS PubMed; (d) K. M. Rentsch, J. Biochem. Biophys. Methods, 2002, 54, 1 CrossRef CAS PubMed.
- E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, 1999, vol. I–III, Suppl. I–II Search PubMed.
- E. M. Carreira, L. Kvaerno, Classics in Stereoselective Synthesis.Wiley, Weinheim, 2009 Search PubMed.
- For reviews on stereodivergent synthesis, see: (a) M. Bihani and J. C.-G. Zhao, Adv. Synth. Catal., 2017, 359, 534 CrossRef CAS; (b) L. Lin and X. Feng, Chem. -Eur. J., 2017, 23, 6464 CrossRef CAS PubMed; (c) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Rev., 2018, 118, 5080 CrossRef CAS PubMed; (d) Y. Wu, X. Huo and W. Zhang, Chem. -Eur. J., 2020, 26, 4895 CrossRef CAS PubMed; (e) U. B. Kim, D. J. Jung, H. J. Jeon, K. Rathwell and S.-G. Lee, Chem. Rev., 2020, 120, 13382 CrossRef CAS PubMed; (f) L. Wei and C.-J. Wang, Chin. J. Chem., 2021, 39, 15 CrossRef CAS.
- For representative examples on stereodivergent synthesis, see: (a) Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051 CrossRef CAS PubMed; (b) B. Wang, F. Wu, Y. Wang, X. Liu and L. Deng, J. Am. Chem. Soc., 2007, 129, 768 CrossRef CAS PubMed; (c) X.-X. Yan, Q. Peng, Q. Li, K. Zhang, J. Yao, X.-L. Hou and Y.-D. Wu, J. Am. Chem. Soc., 2008, 130, 14362 CrossRef CAS PubMed; (d) A. Nojiri, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 3779 CrossRef CAS PubMed; (e) M. Luparia, M. T. Oliveira, D. Audisio, F. Frébault, R. Goddard and N. Maulide, Angew. Chem., Int. Ed., 2011, 50, 12631 CrossRef CAS PubMed; (f) X. Tian, C. Cassani, Y. Liu, A. Moran, A. Urakawa, P. Galzerano, E. Arceo and P. Melchiorre, J. Am. Chem. Soc., 2011, 133, 17934 CrossRef CAS PubMed; (g) D. Uraguchi, K. Yoshioka and T. Ooi, Nat. Commun., 2017, 8, 14793 CrossRef PubMed; (h) D. Kaldre, I. Klose and N. Maulide, Science, 2018, 361, 664 CrossRef CAS PubMed; (i) H. Zheng, Y. Wang, C. Xu, X. Xu, L. Lin, X. Liu and X. Feng, Nat. Commun., 2018, 9, 1968 CrossRef PubMed; (j) T.-T. Gao, W.-W. Zhang, X. Sun, H.-X. Lu and B.-J. Li, J. Am. Chem. Soc., 2019, 141, 4670 CrossRef CAS PubMed; (k) X.-J. Liu, S. Jin, W.-Y. Zhang, Q.-Q. Liu, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2020, 59, 2039 CrossRef CAS PubMed; (l) D.-X. Zhu, J.-G. Liu and M.-H. Xu, J. Am. Chem. Soc., 2021, 143, 8583 CrossRef CAS PubMed.
- S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 CrossRef CAS PubMed.
- S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627 CrossRef CAS PubMed.
- For selected representative stereodivergent synthesis using dual metal catalysts, metal/organocatalysts or dual organocatalysts, see: (a) S. Krautwald, M. A. Schafroth, D. Sarlah and E. M. Carreira, J. Am. Chem. Soc., 2014, 136, 3020 CrossRef CAS PubMed; (b) T. Sandmeier, S. Krautwald, H. F. Zipfel and E. M. Carreira, Angew. Chem., Int. Ed., 2015, 54, 14363 CrossRef CAS PubMed; (c) F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1029 CrossRef CAS PubMed; (d) X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87 CrossRef CAS PubMed; (e) X. Huo, R. He, X. Zhang and W. Zhang, J. Am. Chem. Soc., 2016, 138, 11093 CrossRef CAS PubMed; (f) X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87 CrossRef CAS PubMed; (g) X. Huo, R. He, J. Fu, J. Zhang, G. Yang and W. Zhang, J. Am. Chem. Soc., 2017, 139, 9819 CrossRef CAS PubMed; (h) L. Wei, S.-M. Xu, Q. Zhu, C. Che and C.-J. Wang, Angew. Chem., Int. Ed., 2017, 56, 12312 CrossRef CAS PubMed; (i) L. Wei, Q. Zhu, S.-M. Xu, X. Chang and C.-J. Wang, J. Am. Chem. Soc., 2018, 140, 1508 CrossRef CAS PubMed; (j) X. Huo, J. Zhang, J. Fu, R. He and W. Zhang, J. Am. Chem. Soc., 2018, 140, 2080 CrossRef CAS PubMed; (k) X. Jiang, P. Boehm and J. H. Hartwig, J. Am. Chem. Soc., 2018, 140, 1239 CrossRef CAS PubMed; (l) Z.-T. He, X. Jiang and J. H. Hartwig, J. Am. Chem. Soc., 2019, 141, 13066 CrossRef CAS PubMed; (m) L. Wei, Q. Zhu, L. Xiao, H.-Y. Tao and C.-J. Wang, Nat. Commun., 2019, 10, 1594 CrossRef PubMed; (n) Q. Zhang, H. Yu, L. Shen, T. Tang, D. Dong, W. Chai and W. Zi, J. Am. Chem. Soc., 2019, 141, 14554 CrossRef CAS PubMed; (o) R. He, X. Huo, L. Zhao, F. Wang, L. Jiang, J. Liao and W. Zhang, J. Am. Chem. Soc., 2020, 142, 8097 CrossRef CAS PubMed; (p) L. Peng, Z. He, X. Xu and C. Guo, Angew. Chem., Int. Ed., 2020, 59, 14270 CrossRef CAS PubMed; (q) S. Singha, E. Serrano, S. Mondal, C. G. Daniliuc and F. Glorius, Nat. Catal., 2020, 3, 48 CrossRef CAS; (r) M. Zhu, Q. Zhang and W. Zi, Angew. Chem., Int. Ed., 2021, 60, 6545 CrossRef CAS PubMed; (s) S.-Q. Yang, Y.-F. Wang, W.-C. Zhao, G.-Q. Lin and Z.-T. He, J. Am. Chem. Soc., 2021, 143, 7285 CrossRef CAS PubMed; (t) B. Kim, Y. Kim and S. Y. Lee, J. Am. Chem. Soc., 2021, 143, 73 CrossRef CAS PubMed; (u) J. Zhang, X. Huo, J. Xiao, L. Zhao, S. Ma and W. Zhang, J. Am. Chem. Soc., 2021, 143, 12622 CrossRef CAS PubMed; (v) Y. Peng, X. Hong, Y. Luo, L. Wu and W. Zhang, Angew. Chem., Int. Ed., 2021, 60, 24941 CrossRef CAS PubMed; (w) L. Xiao, L. Wei and C.-J. Wang, Angew. Chem., Int. Ed., 2021, 60, 24930 CrossRef CAS PubMed; (x) M. Ke, Z. Liu, K. Zhang, S. Zuo and F. Chen, Green Synth. Catal., 2021, 2, 228–232 CrossRef; (y) Z. Sun, T. Zha and Z. Shao, Green Synth. Catal., 2021, 2, 241–245 CrossRef.
- S.-L. Shi, Z. L. Wong and S. L. Buchwald, Nature, 2016, 532, 353 CrossRef CAS PubMed.
- B. M. Trost, C.-I. Hung, T. Saget and E. Gnanamani, Nat. Catal., 2018, 1, 523 CrossRef CAS.
- (a) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461 CrossRef CAS PubMed; (b) J. Qu and G. Helmchen, Acc. Chem. Res., 2017, 50, 2539 CrossRef CAS PubMed; (c) Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855 CrossRef CAS PubMed.
- (a) M. J. Pouy, A. Leitner, D. J. Weix, S. Ueno and J. F. Hartwig, Org. Lett., 2007, 9, 3949 CrossRef CAS PubMed; (b) S. Zheng, W. Huang, N. Gao, R. Cui, M. Zhang and X. Zhao, Chem. Commun., 2011, 47, 6969 RSC.
- (a) J. P. Das and I. Marek, Chem. Commun., 2011, 47, 4593 RSC; (b) I. Marek, Y. Minko, M. Pasco, T. Mejuch, N. Gilboa, H. Chechik and J. P. Das, J. Am. Chem. Soc., 2014, 136, 2682 CrossRef CAS PubMed; (c) J. Christoffers and A. Baro, Quaternary Stereocenters: Challenges and Solution for Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2005 CrossRef.
- P. J. Walsh and M. C. Kozlowski, Fundamentals of Asymmetric Catalysis; University Science Books, USA, 2009 Search PubMed.
- L. Wei, X. Chang and C.-J. Wang, Acc. Chem. Res., 2020, 53, 1084 CrossRef CAS PubMed.
- W.-B. Liu, C. Zheng, C.-X. Zhuo, L.-X. Dai and S.-L. You, J. Am. Chem. Soc., 2012, 134, 4812 CrossRef CAS PubMed.
- A. Matsunami, K. Takizawa, S. Sugano, Y. Yano, H. Sato and R. Takeuchi, J. Org. Chem., 2018, 83, 12239 CrossRef CAS PubMed.
- (a) S. Spiess, J. A. Raskatov, C. Gnamm, K. Brödner and G. Helmchen, Chem. -Eur. J., 2009, 15, 11087 CrossRef CAS PubMed; (b) J. A. Raskatov, S. Spiess, C. Gnamm, K. Brödner, F. Rominger and G. Helmchen, Chem. -Eur. J., 2010, 16, 6601 CrossRef CAS PubMed; (c) S. T. Madrahimov, D. Markovic and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 7228 CrossRef CAS PubMed; (d) S. Rieckhoff, J. Meisner, J. Kästner, W. Frey and R. Peters, Angew. Chem., Int. Ed., 2018, 57, 1404 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2096474–2096476 and 2096478. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06115a |
| This journal is © The Royal Society of Chemistry 2021 |