
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning light-driven rotary molecular motors
Daisy R. S.
Pooler
 ,
Anouk S.
Lubbe
,
Stefano
Crespi
and
Ben L.
Feringa
*
,
Anouk S.
Lubbe
,
Stefano
Crespi
and
Ben L.
Feringa
*
Stratingh Institute for Chemistry, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
First published on 20th October 2021
Abstract
The ability to induce and amplify motion at the molecular scale has seen tremendous progress ranging from simple molecular rotors to responsive materials. In the two decades since the discovery of light-driven rotary molecular motors, the development of these molecules has been extensive; moving from the realm of molecular chemistry to integration into dynamic molecular systems. They have been identified as actuators holding great potential to precisely control the dynamics of nanoscale devices, but integrating molecular motors effectively into evermore complex artificial molecular machinery is not trivial. Maximising efficiency without compromising function requires conscious and judicious selection of the structures used. In this perspective, we focus on the key aspects of motor design and discuss how to manipulate these properties without impeding motor integrity. Herein, we describe these principles in the context of molecular rotary motors featuring a central double bond axle and emphasise the strengths and weaknesses of each design, providing a comprehensive evaluation of all artificial light-driven rotary motor scaffolds currently present in the literature. Based on this discussion, we will explore the trajectory of research into the field of molecular motors in the coming years, including challenges to be addressed, potential applications, and future prospects.
 Anouk S. Lubbe, Daisy R. S. Pooler, Stefano Crespi and Ben L. Feringa (from left to right). | Anouk S. Lubbe obtained her PhD in 2017 from the University of Groningen under the supervision of Prof. Ben Feringa and Prof. Wiktor Szymanski. After travelling, she returned to the Feringa group in 2019 as a research manager. Daisy R. S. Pooler received her MChem degree from the University of Manchester in 2018, where she worked under the supervision of Prof. David Leigh. She is currently pursuing her PhD in the Feringa group in the field of molecular motors. Stefano Crespi received his PhD in 2017 from the University of Pavia and won a two-year postdoc fellowship there. He then joined the group of Prof. Burkhard König at the University of Regensburg. In 2019, he joined the Feringa group as a Marie Skłodowska-Curie fellow. His research interests include the combination of reaction design in organic (photo)chemistry with computational models. Ben L. Feringa obtained his PhD in 1978 at the University of Groningen under Prof. Hans Wynberg, where he was appointed full professor in 1988, after working as a research scientist for Shell. He was named the Jacobus H. van't Hoff Professor of Molecular Sciences in 2004. His research interests include organic synthesis, asymmetric catalysis, nanotechnology, and molecular switches and motors. |
Introduction
Motion is omnipresent at the macroscopic, microscopic and nanoscopic levels. From planes and trains in the macroscopic world to ATP synthase in our bodies and flagellar motors in the bacterial cell wall, both natural and artificial mechanical systems utilise motion to carry out essential tasks.1–5 As technology progresses, the requirement for engineering on the molecular scale to create interactive and stimuli-responsive smart materials becomes apparent.6 Molecular engineers will require a vast diversity of actuators to generate an array of functional and responsive systems to control motion at the nanoscale, hence there is a necessity for molecular machines which exhibit a variety of properties to be developed and added to the molecular engineer's toolbox.The development of artificial molecular machines and switches brings chemists further towards this goal.1–3,7–12 To this end, the field has exploded in recent years, including the development of molecular switches,13–17 shuttles,2,18 pumps,19–22 transporters,23,24 muscles,25,26 walkers,27–29 and, most relevant to this perspective, motors.1,12,30–38 Driving molecular machines and switches photochemically is inherently advantageous due to light being a bountiful energy source able to impart high spatiotemporal control, while leaving no waste products behind.39 Since the first developed light-driven rotary molecular motors based on overcrowded alkenes,1,36 extensive fundamental work has been carried out on these scaffolds in the two decennia following this breakthrough. By designing new families of rotary motors, there are now highly accurate and tuneable tools suited to a myriad of applications involving the control and exploitation of nanoscale dynamics.40–42
As the field has progressed, the fundamental principles behind the effective design of molecular motors have been better understood. However, the development of molecular motors still faces several challenges, particularly when rotary motors are embedded into more complex multifunctional systems,43,44 which opens up ample opportunity for novel motor designs to take centre stage.
Overcrowded alkene-based molecular motors
 | ||
| Fig. 1 Key examples of the various families of artificial light-driven molecular motors discussed in this perspective. (A) Overcrowded alkene-based motors; (B) hemithioindigo-based motors; and (C) imine motors. | ||
Hemithioindigo (HTI)-based molecular motors
Imine-based molecular motors
Other functional groups in addition to carbon–carbon double bonds have been exploited in photoswitchable molecules13–16 and unidirectional rotary motors. In 2006, Lehn envisioned a design based on imines, in which the motors employ two sets of sequential photochemical E–Z isomerisations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond followed by a thermal nitrogen inversion.74 It was postulated that in chiral imines bearing the stereogenic centre adjacent to the CN bond, the photochemical E–Z isomerisation would occur preferentially in one direction over the other. Combined with the thermal inversion of the nitrogen atom through its lone pair of electrons, this motif would offer two-stroke unidirectional rotation about the central double bond axle, representing the simplest example of a small-molecule rotary motor to date. Later in 2014, these hypotheses were realised with the development of a series of imine-based motors (8, Fig. 1C).35 These structures could act as either two- or four-stroke motors, depending on the flexibility of the lower half and the thermal nitrogen inversion barrier of the upper half.
N bond followed by a thermal nitrogen inversion.74 It was postulated that in chiral imines bearing the stereogenic centre adjacent to the CN bond, the photochemical E–Z isomerisation would occur preferentially in one direction over the other. Combined with the thermal inversion of the nitrogen atom through its lone pair of electrons, this motif would offer two-stroke unidirectional rotation about the central double bond axle, representing the simplest example of a small-molecule rotary motor to date. Later in 2014, these hypotheses were realised with the development of a series of imine-based motors (8, Fig. 1C).35 These structures could act as either two- or four-stroke motors, depending on the flexibility of the lower half and the thermal nitrogen inversion barrier of the upper half.
Scope
In this perspective, we will discuss the different molecular motor designs, touch upon their properties and compare these with the overcrowded alkene-based molecular motors, which have been a source of inspiration for the development of the alternative designs. By looking at the corpus of literature present on these systems, we can gain significant insight into which features are necessary to build genuine rotary molecular motors, and how tweaking these key parameters can change the intrinsic properties of molecular motors, making them evermore adaptable for application in nanotechnology.Rotation cycle
The rotation cycle of any molecular motor describes the sequential isomerisations of the molecule that enforce continuous unidirectional motion, as opposed to randomised Brownian motion.7 The idea is that the motor has one unit that revolves with respect to the rest of the molecule over multiple steps, resulting overall in a progressive winding motion.† For this to be possible, the motor must undergo sequential steps that are biased to proceed in one direction. Here, it is important to distinguish between the compounding motion of molecular motors as opposed to the stochastic motion observed in binary photoswitches. For continuous work to be carried out by a molecular motor, the rotational cycle must progress back to its initial state through a closed path composed of an oriented sequence of unique steps, therefore establishing unidirectionality, regardless of the number of steps involved in the process. In other words, the motion must continuously progress forwards, rather than allowing movement back and forth via the same pathway. The constituent steps in the rotation cycle can be fully unidirectional – as in the entirety of the previous state progresses to the following state in the rotation cycle – or it can be that a majority of the molecules progress in a forwards motion, known as preferred directionality. Either way, the result is net unidirectional rotation.The aforementioned families of light-driven molecular motors (Fig. 1) undergo diverse rotational mechanisms within their cycles, which will be outlined in this section. The constituent transformations of these cycles are either photochemical or thermal reactions, and it is important to distinguish that the photochemical steps are not constrained by microscopic reversibility. As a consequence of this, photochemical irradiation allows the population of high energy states that are otherwise thermally inaccessible or energetically disfavoured. Therefore, light is the energy used to fuel motor rotation.75 There can be two limiting cases for light-driven rotary molecular motors: (i) molecules operating under the limit of thermal steps and (ii) molecules operating under the limit of photon supply. For the former, the processes comprising the rotation cycle can be quantitatively described by a photoequilibrium between a stable and a metastable state – which is characterised by a photostationary distribution of these isomers (PSD) at the corresponding photostationary states (PSSs) – and the thermal ratcheting steps from the metastable state to the following stable state – which is associated with a thermal Gibbs free energy of activation (ΔG‡). For motors running under the limit of photon supply, the presence of consecutive photoreactions leads to the overall unidirectionality of the motion, without the need for ratcheting thermal steps in the ground state. During the transformation between two consecutive species, the back-reaction of the product formed is negligible and therefore the light stimulus ensures progression of the rotation cycle.71
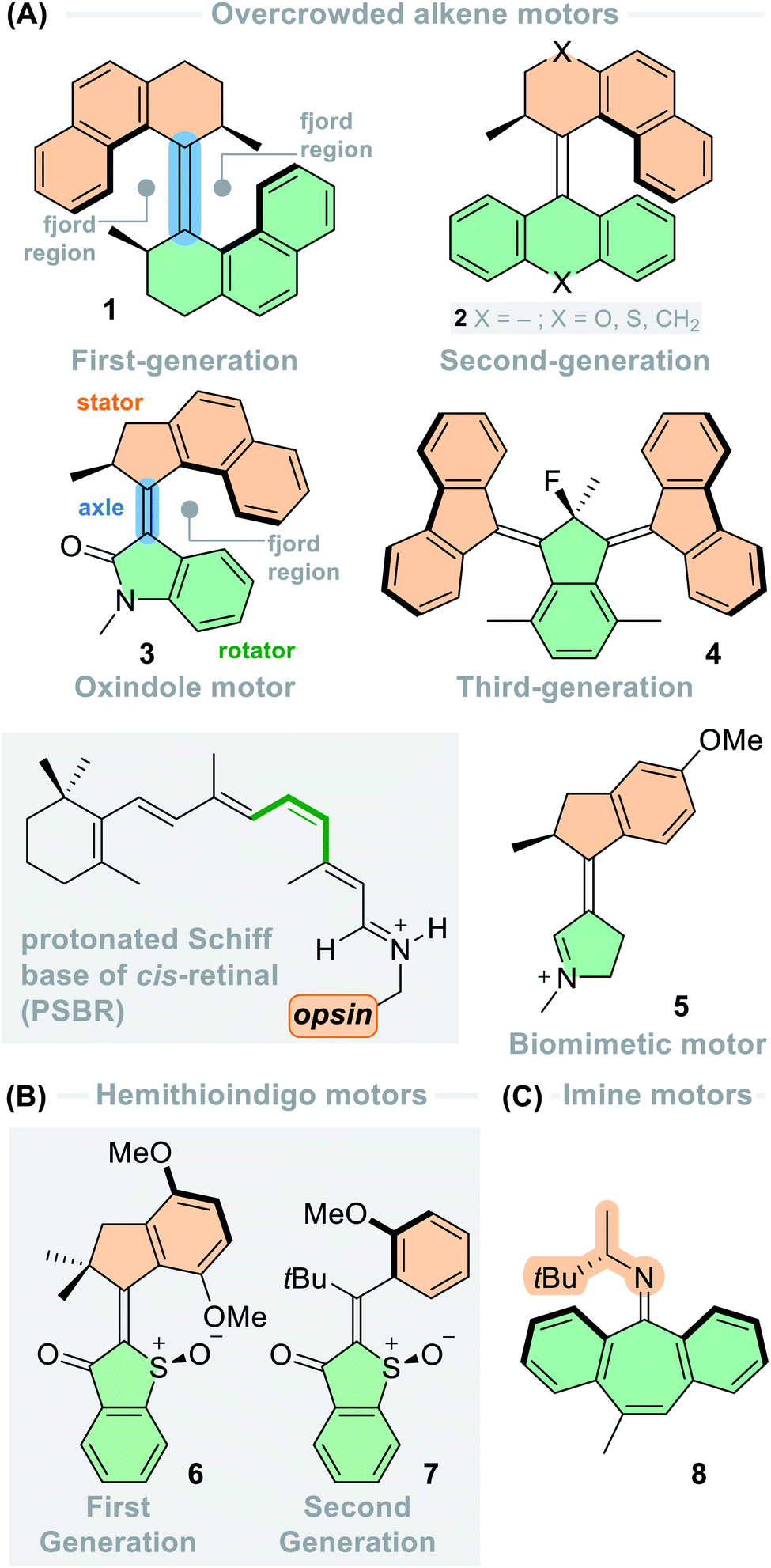
At the core of the overcrowded alkene-based molecular motors are the photochemical E–Z (PEZ) isomerisation and thermal helix inversion (THI) steps. The first-generation molecular motor presented by our group is based on an overcrowded alkene, which is forced out of plane into a helical shape due to significant steric strain between the two identical halves of the molecule.36 Each half bears a stereogenic methyl group in the allylic position next to the central double bond axle, which governs the direction of the rotation. The motor can adopt one of two helicities, M or P, in both stereoisomers of the motor, E and Z, resulting in four distinct isomeric forms. The rotation cycle of the (R,R) enantiomer of first-generation motor 1 is shown in Fig. 2A. Starting from the thermally stable ES form, (P,P)-E-1, possessing pseudo-axial methyl substituents. The molecule is irradiated with UV light which promotes it into the excited state; here the rotator can move due to the reduced bond order of the central alkene axle following the excited state potential energy surface gradient which imparts directionality to the motion. In the excited state, the helix inversion is hindered. Once the molecule relaxes to the ground state, the metastable ZM isomer, (M,M)-Z-1, is populated by the molecule. During the photoisomerisation both halves of the motor invert their helicity forcing the stereogenic methyl groups into relatively strained pseudo-equatorial positions, hence the term “metastable”. From the metastable state, the barrier for the THI is much lower than that of backwards E–Z isomerisation (Fig. 2C). This energy discrepancy offers a favoured, alternative relaxation pathway of helix inversion, as opposed to relaxation via rotation of the alkene bond. Hence, the thermal relaxation from the metastable state to the corresponding stable state has a distinctly different pathway from that of the photochemical reaction, thus establishing unidirectionality.75 During the THI, the methyl groups can adopt their sterically favoured pseudo-axial positions which affords the stable ZS form, (P,P)-Z-1. The photoisomerisation is repeated once more to yield the metastable EM state, (M,M)-E-1, which then relaxes by THI to regenerate the ES isomer, (P,P)-E-1, completing the 360° rotation cycle. The photochemical E–Z isomerisation is the fundamental motion that fuels the rotation in a power stroke process, and the THI biases the unidirectionality of the motor in a subsequent ratcheting step.75
 | ||
| Fig. 2 (A) Rotation cycle of overcrowded alkene-based first-generation motor 1; (B) rotation cycle of overcrowded alkene-based second-generation motor 9; (C) potential energy surface of the rotation cycle of overcrowded alkene-based motors, with photochemical E–Z (PEZ) and thermal helix inversion (THI) steps highlighted. | ||
One defining feature of the first-generation motors is the difference in the energy barriers of the two THI steps, due to the different structures of the E and Z isomers. The thermal energy required to overcome the THI barrier for the EM isomer (25.6 kcal mol−1) is higher than for the ZM isomer (21.7 kcal mol−1) due to the increased steric strain induced by the methyl groups clashing with the naphthyl moieties on both sides. In the case of the THI for ZM, the steric interaction between the two methyl groups is minimal and the two naphthyl moieties can simply slide past each other, resulting in a lower free energy barrier.76 Consequently, both 180° cranks of first-generation overcrowded alkene-based motors have different rotation speeds, introducing inherent complexity into the rotation cycle.
Both photoisomerisations are not completely selective due to overlap of absorption bands of the stable and metastable forms. Therefore, after cycling through a single rotation there will always be a mixture of isomers present, so the PSD at the PSS (the ratio between stable![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :metastable states) will never be 0:100. Nevertheless, this discrepancy does not affect the overall unidirectionality of the motors (i.e. net unidirectional rotation) because the THI step will convert all of the molecules populating a metastable state to the following stable state (e.g. ZM → ZS). Any unreacted stable state can then be excited during the next photoisomerisation. Thermal E–Z isomerisation (TEZI) backwards reactions are possible at elevated temperatures, however, this reaction only becomes a competing process when the barriers for the THI are sufficiently high (>26 kcal mol−1).77
:metastable states) will never be 0:100. Nevertheless, this discrepancy does not affect the overall unidirectionality of the motors (i.e. net unidirectional rotation) because the THI step will convert all of the molecules populating a metastable state to the following stable state (e.g. ZM → ZS). Any unreacted stable state can then be excited during the next photoisomerisation. Thermal E–Z isomerisation (TEZI) backwards reactions are possible at elevated temperatures, however, this reaction only becomes a competing process when the barriers for the THI are sufficiently high (>26 kcal mol−1).77
A structurally related first-generation molecular motor with iso-propyl groups at the stereogenic centres was found to undergo THI from the metastable (M,M)-E to the stable (P,P)-E state so slowly that it revealed a two-step mechanism and a fifth state with mixed helicity, (P,M)-E.76 This finding suggests that the THI could also be a stepwise rather than a concerted process, a hypothesis supported by DFT calculations for certain first- and second-generation motors.41,78
The rotation cycles of second-generation motors follow an identical pathway, with the distinction that the E and Z isomers are interchangeable provided a symmetrical lower half (9, Fig. 2B, R = H) is present, so that there is only one stable and one metastable isomer. There are some practical advantages to this design, such as simpler characterisation and THI rates that are identical for each half of the cycle.79 However, since any PEZ/THI cycle will recover the stable isomer quantitatively, unidirectionality can only be proven when the lower half is desymmetrised. The introduction of E and Z configurations reveals four isomeric states, which can be identified as the two stable and two metastable states (9, Fig. 2B, R = OMe).37
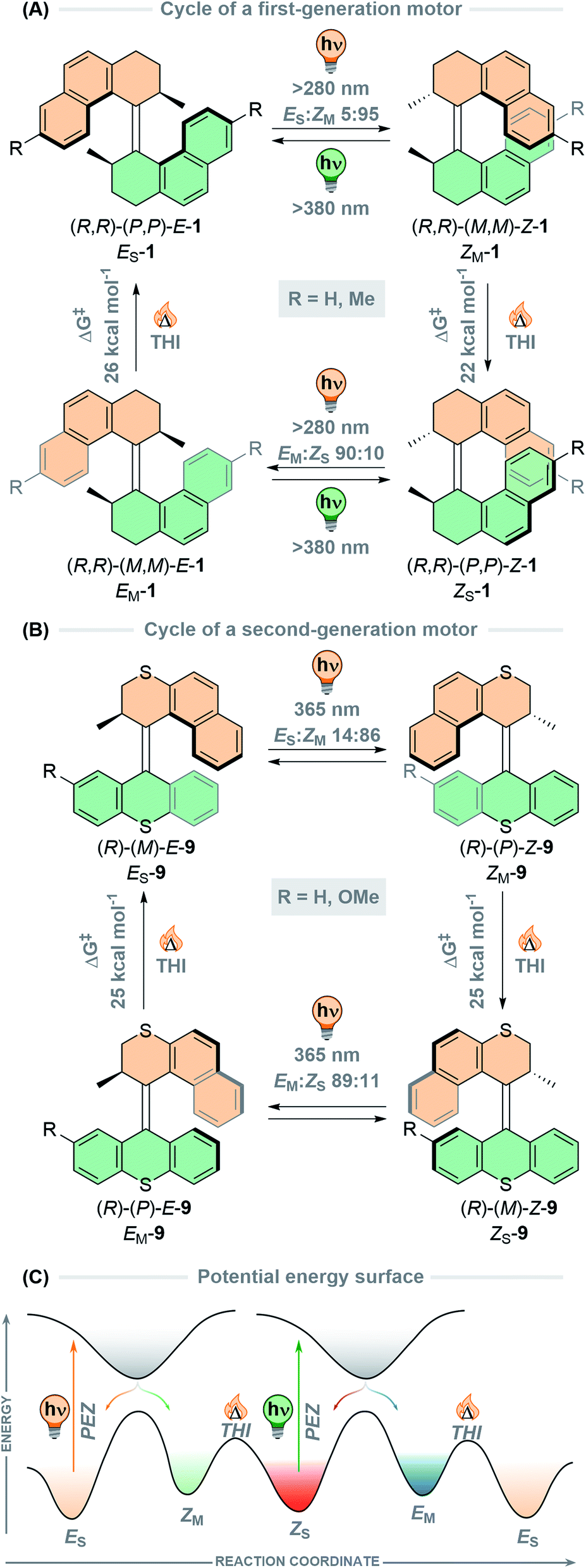
Recently, our group has developed a new class of second-generation molecular motors with a phosphorus stereoelement in the lower half of the molecule (Fig. 3).80 The lone pair on the phosphine stereocentre allows binding to AuCl forming a gold(I) phosphine complex, or it can be oxidised to form a phosphine oxide. It was found that these motors undergo a full 360° rotation through the four states using photonic energy alone, contrasting to other motors which require both light and heat input. When irradiating a pure sample of either stable isomer (a or c) of motor 11 or 12 at reduced temperatures, a PSS with four components was obtained. Irradiation of motor 12 with 365 nm light at −50 °C generates a PSD of 13:75:5:7 (12a:12b:12c:12d), but at longer wavelengths the PSS favours almost exclusively the red-shifted metastable states, b and d. Interestingly, the most noteworthy stereoelement implemented in this new family of motors is the free phosphine of motor 10, which can undergo epimerisation at elevated temperatures allowing a shortcut during rotation, transitioning through three isomeric states rather than the typical four-step cycle (Fig. 3). Epimerisation has been a practical handle used to alter the rotation cycles of molecular motors, with base-catalysed epimerisation of an amide-based stereocentre of a second-generation motor being utilised to precisely switch between clockwise and anticlockwise unidirectional rotation via chemical stimuli.81
 | ||
| Fig. 3 All-photochemical rotation cycle of phosphine-based motors 10–12, including thermal phosphorus inversion “shortcut” cycle of motor 10. | ||
Third-generation overcrowded alkene-based motors also follow a similar rotation pathway, with the distinction being that the stereoelement is a pseudo-asymmetric centre; this is formed by the fusion of two chiral second-generation motors at the same point, rendering the molecule itself a chiral (4, Fig. 1A). Here, the fluorene moieties act as two ‘wheels’ both rotating in the same direction. Absorption of a UV photon can lead to two processes of equal probability: either a 180° clockwise rotation of the upper rotor or a 180° clockwise rotation of the lower rotor. To date, this mesomeric centre, in conjunction with the folded axial chirality of the system, is the lowest chiral requirement necessary to direct the unidirectional rotation of a molecular motor.38 The motors can readily be functionalised with groups of different sizes at the pseudo-asymmetric centre and substitution patterns on the lower half.82,83 So far, the only other example of a molecular motor lacking point chirality is theoretical motor 19 (Fig. 6, vide infra) from Durbeej and co-workers. Instead, this motor has induced asymmetry by the puckering of a cyclohexenylidene upper half. The introduction of bulky bromine substituents on the upper half helps to increase the barrier for ring-flipping, a crucial point necessary to maintain the directionality of motor 19.64
The first of many HTI-based molecular motors was presented in 2015, featuring an intrinsically red-shifted backbone and elevated rotation speeds (6, Fig. 1).34 These overcrowded alkene-based motors consist of a stilbene upper half and a hemithioindigo (HTI) lower half, equipped with a sulfoxide stereogenic centre. Like the overcrowded alkene-based motors, the rotation cycle consists of two uphill photochemical E–Z isomerisations to generate the metastable isomers, and two downhill thermal helix inversion steps which regenerate their stable counterparts, resulting in unidirectional 360° rotation. Although the first-generation HTI-based motors are quite different in structure to that of a second-generation overcrowded alkene-based motor, there is only one stereocentre adjacent to the central alkene axle, so the function is essentially the same.
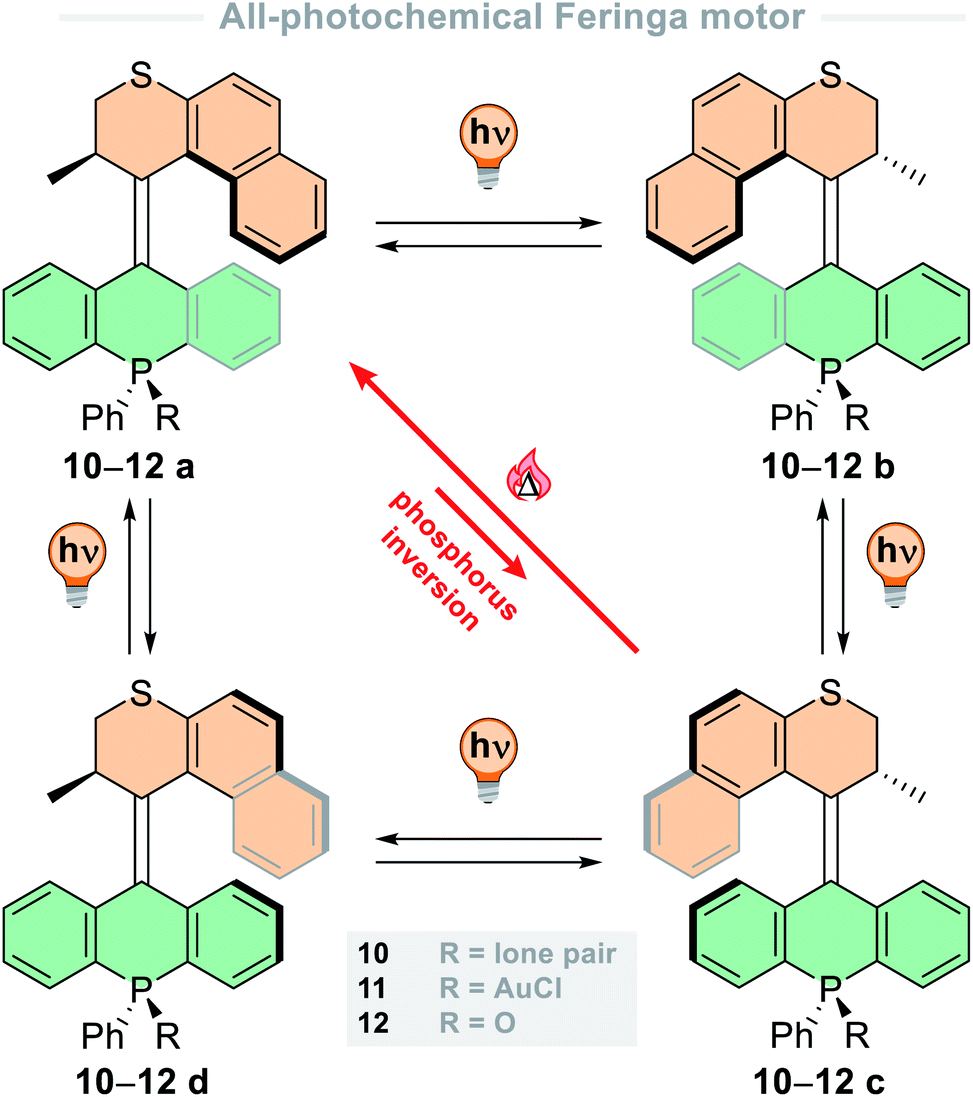
Since then, these motors have been used in switchable catalysis,84 functionalised for attachment to metal surfaces,85 and the rotation of the motor has been used to bias and actively accelerate the rotation of a remotely attached biaryl axis.86,87 In addition to their general design (vide supra), the Dube group has developed a few HTI-based motors that have an entirely different mode of rotation; these structures operate through the light-driven rotation of a single-bonded group about a central alkene bond.71–73 The key structural difference between these new second-generation HTI motors (7, R = H, Fig. 4A) and first-generation HTI motors is the absence of confinement of the central alkene bond by two neighbouring cyclic moieties. Due to the nature of alkene bonds with neighbouring single carbon–carbon bonds, different isomerisation reactions can arise: single bond rotation (SBR), photochemical E–Z isomerization (PEZ), and the hula twist (HT) motion, whose outcome is the formal combination of the two transformations mentioned thereof.88 The mechanism of sequential photoreactions is notoriously challenging to elucidate, due to the high instability of the primary photoproducts generated, which rapidly react thermally to produce more stable intermediates. To tackle this challenge, the Dube group developed a molecular motor in which its four diastereomers were isolable and did not interconvert at room temperature (ΔG‡ ≤ 30 kcal mol−1). This effectively decouples thermal motion from the system so solely photochemical changes were observed, representing the first example of a photon-only motor described in the literature.72 The four states are geometrically distinct and could be analysed by X-ray crystallography, providing an explicit assignment of the primary photoproducts for each interconversion. From these findings, unequivocal evidence for the occurrence of SBR and HT photoreactions was unearthed. Furthermore, the influence of temperature and solvent medium were probed to bias the different photoreactions, so from this work, the photochemical control of complex motion at the nanoscale can be precisely employed.
 | ||
| Fig. 4 (A) Rotation cycle of a second-generation hemithioindigo-based motor. The single bond rotation (SBR), double bond isomerization (DBI) and hula twist motion (HT) of 7 are highlighted; (B) second-generation hemithioindigo motor 13 with a figure-of-eight rotation cycle. | ||
This study led to further development of the HTI-based molecular motors,71 including a design that undergoes a peculiar figure-of-eight motion with a single crossing point (13, Fig. 4B).73 This design marked a revolutionary discovery in the field of molecular machines, since before this work only circular and linear locomotions had been realised.9,12 The motor consists of an HTI lower half, with a tBu group and julolidine unit attached to the other end of the alkene. The julolidine unit possesses a methyl group on one side, making it unsymmetrical, and is axially chiral due to the steric demand of the tBu group which hinders atropisomerisation at ambient temperatures (ΔG‡ > 30 kcal mol−1). The rotation cycle progresses through four isomeric states, which are interchanged through alternating photochemical E–Z steps and thermal HT steps. The E–Z photoisomerisations are activated by 520 nm light and are highly selective in apolar solvents, with quantum yields indicating all other photoreactions to be at least one order of magnitude less efficient. The thermal HT was assessed in acetonitrile/water mixtures and the process was >95% efficient, but in polar solvents the photoreactions were strongly hindered. For optimal efficiency and high directional preference (82% over one cycle), the photoisomerisation steps are carried out in cyclohexane, and the thermal steps in acetonitrile/water mixtures. The directionality is ratcheted by the thermal HT steps which are almost quantitative processes under these conditions. However, the motor can be continuously powered in 1,2-dichlorobenzene at 130 °C under irradiation of green light, at the cost of diminished directional preference (47% over one cycle). The use of a relatively apolar solvent is necessary for the photochemical DBI steps to occur, but it also significantly increases the barriers of the thermal HT steps by ∼6 kcal mol−1, which hinders their progressivity.
Carbon–nitrogen (CN) double bonds possess the same photochemical characteristics as alkene bonds, but with added stereochemical mutability at the nitrogen atom.74 The sequential photoactivated and thermoactivated E–Z isomerisation steps that CN bonds can undergo (both of which follow distinct pathways) represent imines and their related compounds as the simplest unidirectional light-driven molecular motors to date.
Following these design principles, Lehn and co-workers developed the first imine-based molecular motors in 2014, bearing a stereogenic centre adjacent to the nitrogen atom of the central imine axle.35 The motor exists as four stereoisomers due to the chirality of the imine and the folded nature of the lower half (Fig. 5A). Irradiation of the stable (P)-Z form of motor 8 promotes the molecule into the excited state, where it undergoes an out-of-plane rotation about the CN axis via a perpendicular diradical structure (PSROT), and consequently relaxes back to the ground state to form the first metastable state, (M)-E-7. The thermal reactions that can occur in these compounds are two-fold: nitrogen inversion (NI) via a planar transition state (TSNI), or ring inversion (RI) in which the folded configuration of the lower half inverts through a flipping motion of the aryl rings on either side of the cycloheptatriene moiety, due to its inherent flexibility. In the case of four-step motor 8, the thermal step occurs via RI, due to the relatively low thermal barrier (20 kcal mol−1) in comparison to nitrogen inversion (>23 kcal mol−1). Subsequent ring inversion of (M)-E-8 inverts the axial chirality of the CN axis, ratcheting the motor forwards to yield (P)-E-8. This state then undergoes a second PEZ step via the PSROT excited state structure to give the metastable state (M)-Z-8, followed by a second RI step to reform stable (P)-Z-8 and completing the 360° rotation about the central CN axis.
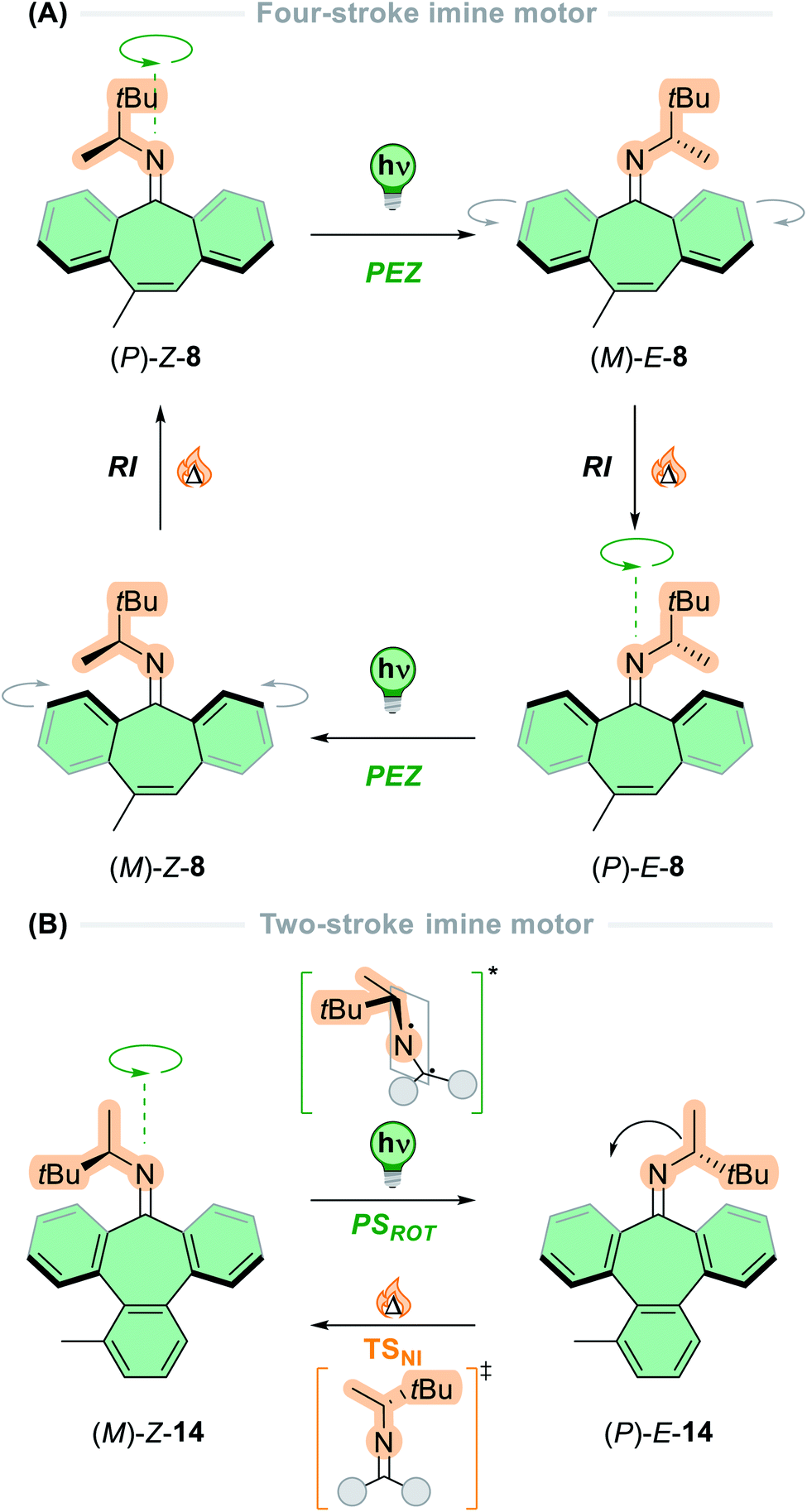 | ||
| Fig. 5 Schematic representation of the rotation cycle for (A) four-stroke imine-based motor 8 and (B) two-stroke imine-based motor 14. | ||
Due to the discrepancy between the energy barriers for the NI and RI thermal pathways, the question of what would happen if the NI inversion pathway was favoured over the RI pathway was raised.35 To bias the NI pathway, an aryl ring was fused to the cycloheptatriene moiety to yield motor 14 in which the flexibility of the lower half is compromised significantly, so the RI barrier increases to 38 kcal mol−1. This motor undergoes a two-step rotation cycle: first by irradiating the stable (M)-Z-14 isomer via the same PSROT excited state structure to form the metastable (P)-E-14 state, then subsequent heating to 60 °C reforms the stable (M)-Z-14 state via TSNI through nitrogen inversion (Fig. 5B). Here, the directionality of the rotation is governed by the photochemical step, contrasting to other light-driven molecular motors which are ratcheted by the thermal isomerisation step. A follow-up paper described entrapment of the photochemically generated species from both diastereomers of newly synthesised imine motors based on camphorquinone.89 This result experimentally demonstrates the directional preference of the excited state pathways, as presented in the original conjecture.74
Motors based on biologically relevant chromophores have been developed by the group of Olivucci using the biomimetic approach – this involves integrating the π-skeleton of the desired photoefficient chromophore into small molecular photoswitches, and then introducing point chirality adjacent to the double bond axle of these switches to bias unidirectional rotation.57,59 These motors are powered solely by photonic energy, due to their lack of steric bulk eliminating thermal steps in the ground state. Initial non-adiabatic molecular dynamics (NAMD) simulations on a very minimal N-alkyl indanylidene-pyrrolinium (NAIP) structure showed that a small 2° angle of helicity of the double bond induced by the stereogenic centre is enough to promote a degree of directionality of ∼70%.57 Later, biomimetic NAIP-based motor 5 (Fig. 1) was prepared and was found to have a similar degree of directionality, 76%;59 it seems that the methyl stereocentre alone does not impose enough steric demand on the system to fully dictate directional motion. Similar results were observed for a structurally related p-HDBI motor.60 These studies provide a basis for the growth of a novel generation of photochemically-driven rotary motors, fortified with a biomimetic framework.
In this section, we have explored the modus operandi of several families of artificial light-driven molecular motors.37,79 Their ability to move continuously in one direction following a unique trajectory (that does not intersect with the corresponding backwards pathway) drives compounding, progressive motion, which can allow the system to carry out work on a secondary coupled process.90 Although almost all motors rotate in a circular motion – the exception being Dube's revolutionary figure-of-eight motor 1373 – there are distinct differences in their rotation cycles that make them unique from each other, and therefore suited to different potential applications. For example in the phosphine-based second-generation molecular motors, the introduction of a three-step rotation cycle by heating the system offers a simple way to bypass one of the four states.80 For the imine motors, the opportunity to tune whether the motor operates via a two- or four-step rotation cycle is a unique aspect.35 When comparing first- and second-generation overcrowded alkene-based motors, the energy difference in the thermal barriers for the first-generation motors brings inherent complexity into the system,36,76 whereas this characteristic is not present in second-generation motors. One can imagine different areas where these features may find application – triggering, multistate switching, adaptation, sensing, information processing, feedback loops.6 The exploration of novel rotational mechanisms, particularly in cases where the nature, speed or efficiency of the motion can be easily tuned and/or is responsive to external stimuli, provides more options to help move molecular motors towards integration into functional, responsive, and interactive molecular systems.
Absorption wavelength
Visible light comprises almost half of the solar spectrum that reaches the Earth's surface and benefits from a large window of low energy wavelengths.13 Typically, light-driven molecular motors are small molecules which necessitate the use of high energy photons i.e. ultraviolet (UV) light, to drive their photochemical isomerisations. This has a number of drawbacks: (i) UV light is non-selectively absorbed by most chromophores which is undesirable for constructing modular photoresponsive systems, (ii) due to its high energy it is invasive to materials where molecular motors can be installed – for instance, some polymeric materials are prone to degradation under UV light91 – and (iii) the absorption wavelength must be within the so-called “phototherapeutic window” (650–850 nm) in biomedical applications, for optimal penetration at the correct tissue depth and to avoid necrosis.92 It has therefore been identified as a key challenge in the field of light-responsive systems to shift the absorption band of chromophores towards the visible and near-infrared (NIR) areas of the electromagnetic spectrum.There are two theoretically feasible ways to populate the excited state of photochromic molecules: through direct or indirect photoexcitation. Direct photoexcitation is the process of the photoswitchable molecule being promoted to the excited state through absorption of photons by the molecule itself, whereas indirect photoexcitation involves the use of sensitisers to absorb low energy photons which can then be used to power photoisomerisation of the molecule via energy transfer, for example. In this section of the review, recent advances in both of these techniques will be outlined in the context of light-driven rotary molecular motors. It is important to note that both of these principles have been extensively studied with various photoswitches,13,93 and the motor community can further build upon this research and apply it to unidirectional rotary systems. The absorption wavelength can be quantified in two ways: (i) the absorption band (i.e. the range of wavelengths at which the compound absorbs) or (ii) the maximum absorption wavelength, λmax (i.e. the wavelength at which absorption by the molecule is highest). When discussing the absorption band, the wavelength quoted will be the longest wavelength at which the molecule absorbs i.e. the end of the absorption band. When red-shift is discussed, the value quoted will be the difference between the λmax of the two molecules being compared. The irradiation wavelength, λirr, is an experimental choice – by selecting a long wavelength towards the end of the absorption band of the motor (where the absorbance is relatively low) may come at the cost of increased irradiation time to reach the PSS and the ratio of its related PSD.
Direct photoexcitation has been studied extensively, but the design principles employed to use it are not always straightforward in practice. To red-shift the absorption wavelength of the compound, the light absorbed must be of a lower energy. This can be done by tuning the HOMO–LUMO band gap intrinsic to the molecule itself,79,94,95 or by two-photon absorption (2PA).96 2PA involves the simultaneous absorption of two photons of shorter-wavelength which add up to a single longer-wavelength excitation, more than or equal to the energy of the HOMO–LUMO gap. 2PA is a non-linear optical process with absorption cross-sections typically being a few orders of magnitude smaller than that of single-photon absorptions, and therefore only high intensity, confocal light can be used to trigger 2PA processes. Direct photoexcitation of structurally unmodified Feringa-type second-generation molecular motor 15 has been achieved through 2PA (Fig. 6).97 NIR (λ = 710 nm) light was absorbed by the motor to power its rotation, although the high intensity light (∼TW cm−2) necessary to access the small 2PA cross-section of the motor is disruptive to biological media.98
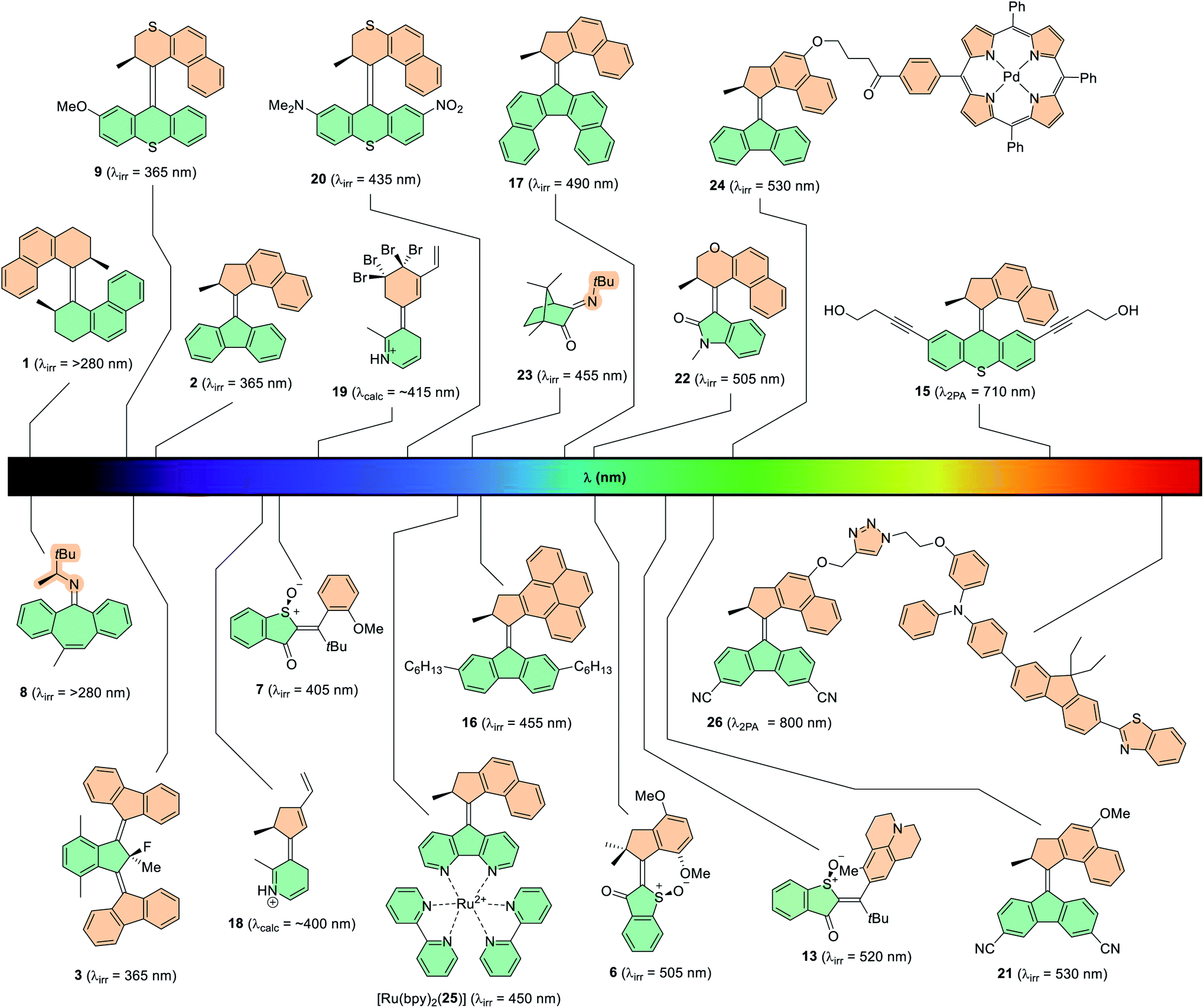 | ||
| Fig. 6 Various light-driven molecular motors with their irradiation wavelength quoted in brackets. | ||
Tuning the HOMO–LUMO gap of a given molecule involves the modification of molecular design, for example by extending the π-system to increase conjugation of the chromophore. This additional π-conjugation corresponds to an increase in energy of the HOMO, and a decrease in energy of the LUMO. This technique has been employed in various photoswitches,13 and also for motors through structural modification of parent second generation motor 2 (Fig. 6).99 By building for instance a pyrene moiety into the upper half (motor 16)100 or by annealing two aryl rings to the lower half to create a dibenzofluorene moiety (motor 17),94 the increase in π-conjugation led to a bathochromic shift in the λmax values from 385 nm of parent motor 2, to 414 nm and 419 nm respectively for motors 16 and 17. In the case of motor 17 a broad absorption band (≤550 nm) is observed in the UV-vis absorption spectrum, and hence it could be operated with wavelengths of up to 490 nm. The pyrene-based molecular motor 16 has a narrower absorption band, but functioned under irradiation with 455 nm light. This shows promising results with bathochromic shifts of up to 35 nm for this technique, however it was found that these motors suffer from depleted quantum yields and PSD ratios. Another variation on motor 2 was the development of a system functionalised with a diarylethene (DAE) photoswitch which could be operated at 455 nm light, but only when the DAE was in its open form. This boasted bathochromically shifted absorption bands due to the extended conjugation of the DAE photoswitch, and marked the first instance of light-gated rotation in a unidirectional rotary motor.101 Calculations also support this theory,64,102 with Durbeej and co-workers investigating theoretical N-protonated IP-based motors 18 and 19 (Fig. 6) which possess conjugated vinyl groups on the upper halves. The increased π-conjugation within the scaffolds shifted their calculated absorption wavelengths from the near-UV region into the visible region of the spectrum.
An approach that has shown greater success involves installing electron-withdrawing and electron-donating groups conjugated to each other through the π-system of the molecule, contracting the HOMO–LUMO gap via a push–pull mechanism. This technique was first exploited by molecular motors in our group in 2003, by installing a donor–acceptor system across the lower half of second generation molecular motor 20 using dimethylamino and nitro groups, which could be irradiated with wavelengths of up to 435 nm,95 giving a red-shift of 70 nm compared to parent motor 9 (Fig. 6).37 More recently, push–pull systems have been implemented to resonate through the central alkene axle of second generation molecular motors, using methoxy and cyano groups.79 These motors, including 21, present absorption bands stretching into the green section of the electromagnetic spectrum and can be operated with up to 530 nm light, in addition to having good isomer distributions at the PSS and quantum yields of up to 12%. The push–pull method proves to be advantageous because photochemical efficiency is uncompromised. In addition, the additional moieties are relatively small so they can be installed with only a few additional synthetic steps, also providing opportunities for further functionalisation.103
Over the past five years, a new field has emerged which involves the exploration of new motor scaffolds based on heterocyclic visible-light dyes. Here, the heterocyclic backbone is intrinsically red shifted in comparison to purely hydrocarbon-based molecular motors (e.g.2, Fig. 6). The main candidates explored for this purpose are the indigoid dyes, which consist of two indole-based fragments connected by a central alkene bond.68,104,105 They exist as a resonance hybrid between many canonical forms, which are relatively unstable due to the charge separation on the electron-withdrawing carbonyl group and the electron-donating heterocyclic nitrogen atom. This inherent push–pull character causes a partial loss of aromaticity, resulting in a more conjugated scaffold and a substantial decrease of the HOMO–LUMO energy gap, owing to their absorption bands extending upwards of 600 nm.106 Of the indigo derivatives, the archetypal chromophore that has been investigated in the field of artificial molecular motors is hemithioindigo (HTI).71–73 First-generation HTI-based motor 6 (Fig. 6) absorbs light of wavelengths up to 505 nm, and perhaps more interestingly, it can be powered simply by irradiation under sunlight.34 The thioindoxyl lower half acts as an electron acceptor and the stilbene fragment as the electron donor, whose donating ability is enhanced by two additional electron-donating methoxy groups on the moiety. This new system paved the way for more elaborate HTI-based motors to be developed, including that of the aforementioned figure-of-eight motor, 13 (Fig. 6). Due to the increased electron-donating power of the julolidine moiety and its position para- to the central alkene bond, this motor is powered at 520 nm and absorbs light extending towards the orange section of the visible light spectrum (≤600 nm).73 Isoindigo is an indigoid dye which consists of two oxindole subunits connected by a central carbon–carbon double bond axle. Oxindole has been employed in a molecular motor framework recently introduced by our group, as the resulting photoactuators feature intrinsically red-shifted absorption maxima (λmax ≈ 400 nm) and can function at wavelengths up to 505 nm, in the case of motor 22 (Fig. 6).46
Photon-only two-stroke imine motors based on camphorquinone (23, Fig. 6) have been shown to function under irradiation of visible 455 nm light for the Z → E photochemical isomerisation, but the E → Z isomerisation requires higher energy light of 365 nm.89 The 90 nm difference between the two isomerisations may originate from the increased steric strain of the Z isomer giving rise to a twisted CN bond and subsequently destabilising the HOMO, a phenomenon which has been noted for structurally related photoswitches.17 The camphorquinone-based imine motors have red-shifted absorption bands in comparison to the previously synthesised imine motors (8 and 14, Fig. 5), which could only be operated with UV light up to 280 nm.35 The red-shift may be due to the extra degree of π-conjugation derived from the neighbouring carbonyl moiety.
The second method to power photoswitchable molecules with light of longer wavelengths is through indirect photoexcitation, which is typically achieved through use of triplet sensitisers or upconverting sensitisers which make use of two-photon absorption processes. These sensitisers absorb light in the visible/NIR region of the electromagnetic spectrum and can transfer the harvested light energy to light-driven molecular motors, either inter- or intramolecularly, powering their isomerisation. The low-energy light harvested by the sensitiser can be passed on to the motor by energy transfer, or simply by stimulated emission of UV photons which are then reuptaken by the motor. Triplet energy states are usually inadequately populated due to the limited efficiency of intersystem crossing (ISC) processes, necessitating the use of triplet sensitisers. If the energy of triplet states can be harnessed, they can unlock the use of longer wavelength light because triplet states are lower in energy than their singlet state counterparts. The triplet state of the sensitiser (donor) must be higher in energy than the triplet state of the molecular motor (acceptor) to allow energy transfer to take place.13,107
Transition metal complexes can act as photosensitisers, and this has successfully been employed as a tool to red-shift the absorption wavelength of overcrowded alkene-based molecular motors. This was achieved through covalent attachment of a Pd(II) porphyrin triplet sensitiser moiety (Fig. 6, motor 24), which absorbs 530 nm light and relays the photonic energy to the motor, populating its low-lying triplet energy state.108 The photoisomerisation proceeded with comparable quantum yield and PSD values to direct UV photoexcitation, proving that rotation via a triplet state mechanism does not diminish motor rotation. Additionally, the motor could be operated by intermolecular energy transfer, that is without the porphyrin moiety being covalently tethered to it. A related approach was used to bathochromically shift the absorption wavelength through ligation of a motor fitted with a 4,5-diazafluorenyl lower half to a Ru(II) complex, ([Ru(bpy)2(25)], Fig. 6).109 The motor could be operated through irradiation of the metal-to-ligand charge-transfer (MCLT) dπ → π* bands of the [Ru(bpy)2(25)] complex, with absorption maxima centred at 425 and 450 nm, the latter of which extends upwards of 500 nm. The motor was operated through irradiation of 450 nm light, again without compromising the efficiency of motor rotation in comparison to the decomplexated parent motor 25.
More recently, Feringa-type second generation molecular motor 26 with prominent push–pull character was powered by NIR (λ = 800 nm) light through use of a tethered 2PA sensitiser.103 The methoxy group of parent motor 279 was a good handle to attach the 2PA sensitiser to the upper half whilst offering a nine-bond degree of spatial separation between the 2PA and the motor. The sensitiser chosen has a large 2PA cross-section, as well as a good match between its fluorescence spectra and the excited state absorption spectra of parent motor 21. In comparison to unsubstituted motor 15 which can be powered by direct 2PA,97 motor 26 can be powered with light of a much lower intensity (0.15 W cm−2), which is several orders of magnitude lower than the threshold at which damage to biological media has been observed.98
In this section, various efforts into red-shifting the absorption wavelength of molecular motors have been explored. Structural modifications of the motor core, such as extension of π-conjugation94,100,101 and the integration of push–pull substituents,79,95,110 have been proven to induce bathochromic shifting effectively, yet to a limited degree. Another valid method is to use inherently red-shifted motor cores, such as those based on indigoid dyes, like thio- or iso-indigo.34,46,73 Evidently, heterocyclic scaffolds hold more promise than adaptation of current hydrocarbon-based overcrowded alkene motor designs. Indirect excitation of motors using various photosensitisers has proven to be a powerful tool,108,109 particularly the use of 2PA sensitisers, which can shift the absorption wavelength into the near-IR region of the electromagnetic spectrum.103 Thus far only extremely high intensity light has been used to power unsensitised molecular motors with 2PA,97 therefore further research into increasing the 2PA cross-sections of molecular motors should be executed. Implementing push–pull systems and extending π-conjugation can contribute towards this goal,96 however the correlations between molecular structure and increased 2PA cross-section are not immediately obvious. Clearly, red-shifting the absorption wavelength is a significant venture as the use of relatively high energy UV light is a bottleneck for implementation of motors into sensitive media, such as biological systems and various smart materials. Nevertheless, it is important to recognise that red-shifting the absorption wavelength often comes at the cost of photochemical efficiency i.e. low quantum yields (vide infra) and low photostationary distributions at the PSS.46,94,100 For many applications, simpler, highly efficient UV light-driven systems will fulfil the role perfectly well.
Quantum efficiency
In contrast to the ground state thermal steps of the rotation cycle, studying the excited state dynamics of the elusive photochemical steps is considerably more difficult. Excited state processes occur on the order of femto- to picoseconds and therefore ultrafast transient spectroscopies are required to investigate them experimentally, often supplemented by quantum chemical calculations. Although progress in this field has been rapid, the photochemical behaviour of light-driven rotary molecular motors is not yet fully understood. Exploration of the challenges still present in this field, such as (i) the relationship between molecular structure and excited state dynamics, (ii) the geometrical changes occurring during the photorearrangement, and (iii) control of the photochemical quantum yield, will further advance the design of molecular motors.To illustrate the various processes occurring during the photochemical excitation of a typical second-generation overcrowded alkene-based molecular motor, we will use symmetric second-generation motor 2 (Fig. 7).111,112 When the stable isomer absorbs a photon of an appropriate wavelength a vertical transition from the S0 ground state to the S1 Franck–Condon (FC) state occurs, which usually corresponds to a π → π* transition. In this process, the central olefinic bond elongates and decreases in bond order, allowing free rotation about the axle as the molecule traverses the S1 potential energy surface (PES). From the optically bright FC state, the motor undergoes an ultrafast barrierless (or almost barrierless) relaxation on the order of 100 fs towards the region of the global minimum of the excited state, signified by a quenching of fluorescence and/or stimulated emission.112,113 Due to this peculiarity, the region near the excited state minimum is usually referred as “dark”, a feature shared with stilbenes and stiff-stilbenes.111,114–116 This state has a relatively long excited state lifetime, and relaxes back to the ground state in the order of 1 ps.112 However, the exact origin and structure of this dark state remains unclear, especially in the presence of substituents with different electronic features installed on the motor, but it is proposed to possess a perpendicular arrangement of upper half and lower half.111,112,117–120
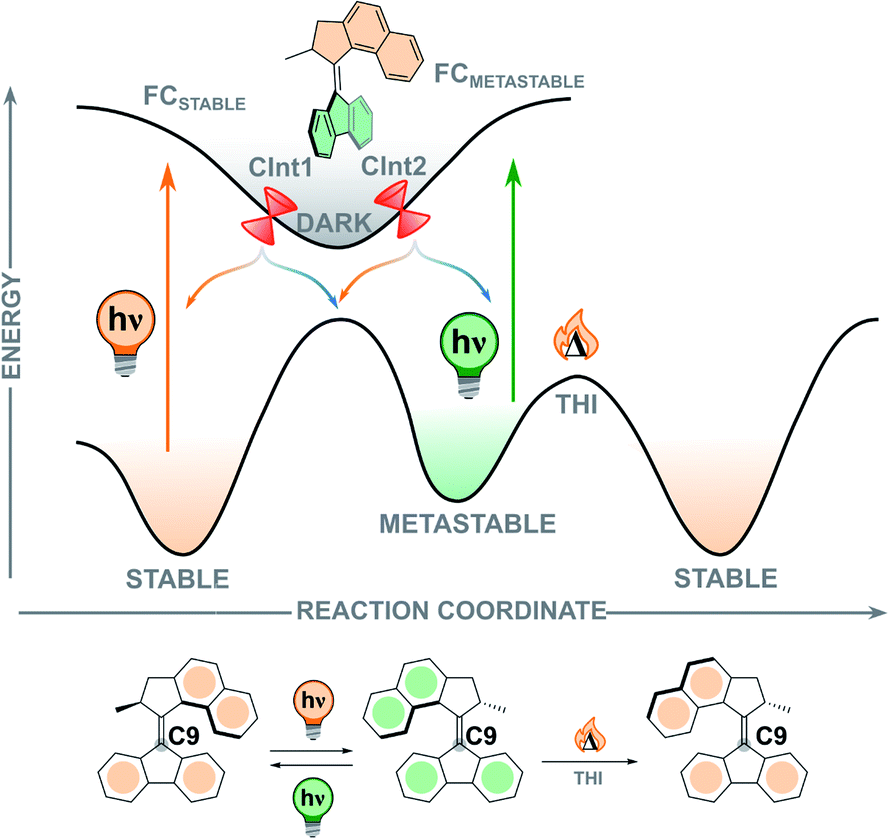 | ||
| Fig. 7 Simplified potential energy surface (PES) along the isomerisation coordinate, showing the excited state processes occurring during the photochemical E–Z isomerisation.117 The region of the perpendicular minimum is marked as “dark”. | ||
In second-generation motors, the excited state minimum is computed to be an avoided crossing.113 The transition to the ground state is supposed to occur via sloped conical intersections (CInts) which are energetically uphill compared to the global S1 minimum. At the CInt(s), the carbon atom on the lower half of the molecule (C9, Fig. 7) undergoes significant pyramidalisation paired with twisting of the central double bond axis, a phenomenon which is also observed in the photorearrangements of related ethylene and stilbene compounds.121,122 This twist-pyramidalisation distortion (Fig. 8A) corresponds to an inherently asymmetric PES in which rotation occurs during the excited state relaxation of the motor. A nonadiabatic hopping event (S1 → S0) occurs where the motor relaxes back down to form the vibrationally hot metastable isomer in the ground state.
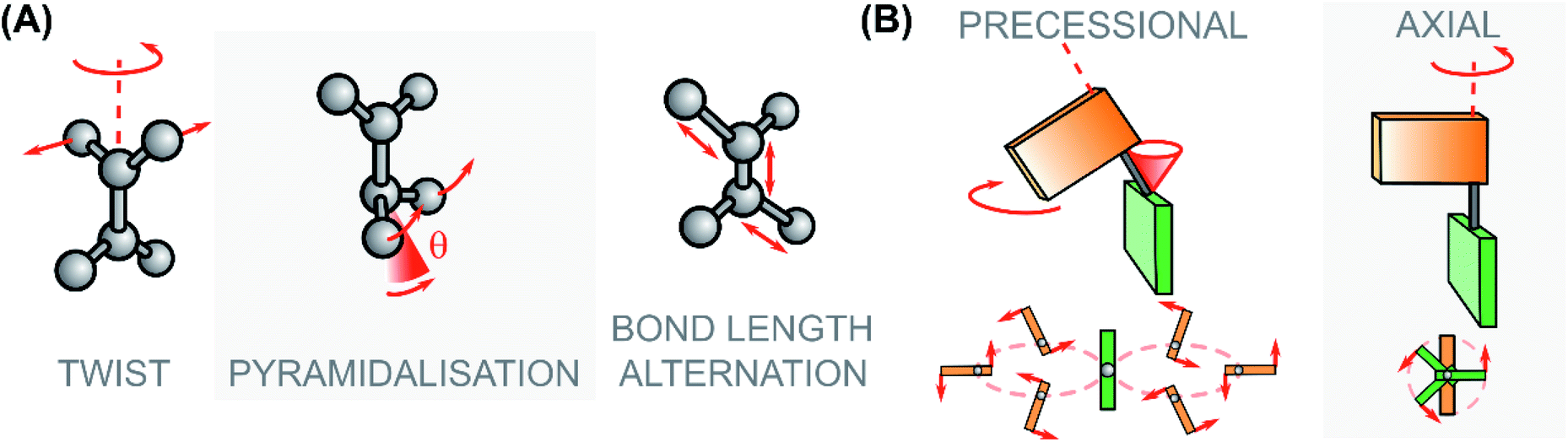 | ||
| Fig. 8 (A) Modes associated to the excited state rotational movement of overcrowded alkenes and Rho-like compounds: the pyramidalization angle θ is highlighted. (B) Paradigmatic rotational motions: on the left the precessional (or hippopede-like) type, typical of switches and motors with zwitterionic character around the S1 global minimum/S1 → S0 CInt; on the right the axial type, typical of photoactuators with diradical character at the S1 global minimum/S1 → S0 CInt.58,113 | ||
The excited state processes occurring in structurally related first-generation overcrowded alkene-based motors have been studied by ultrafast spectroscopies, and show slightly different dynamics to second-generation motors.120,123 Starting from the stable Z form, ZS, excitation promotes the molecule to a bright FC state, which undergoes ultrafast relaxation (ca. 100 fs) to a second red-shifted emissive state, which has a lifetime of 5–10 ps, significantly longer than was observed for second-generation motors (1–2 ps).40,112 This increase in excited state lifetime was ascribed to the presence of a barrier along the reaction co-ordinate on the S1 PES, a feature which is absent in second-generation motors. This barrier effectively traps the excited state population in a strongly radiative part of the PES, and when the barrier is finally overcome, the population is led to a region of the S1 PES where the CInt funnels the population directly back to the S0 PES to form the metastable ZM state.120 An alternative pathway relaxation featuring a dark state in first-generation motors for both E → Z and Z → E transitions has been reported, from which decays to the corresponding ZM and EM metastable states were 13 and 71 ps, respectively.123
As demonstrated by non-adiabatic molecular dynamics (NAMD) simulations41,121 and ultrafast fluorescence spectroscopy,112 the S1/S0 CInts play a dominant role in the photorearrangement of second-generation overcrowded alkene molecular motors. To this end, engineering of the energetic and geometric parameters at the S1/S0 CInts is necessary to fully harness the efficiency of the photochemical isomerisation step. In overcrowded alkenes, the π-bond breaking during excitation occurs in a homolytic mechanism which forms a diradical FC state. As the molecule traverses the S1 PES the electron pair of the π-bond axle is shifted towards one side of the bond, which polarises it towards a zwitterionic, charge-transfer conformation. The electron pair of the carbanion then deforms by pyramidalisation to stabilise the lone-pair as the axle rotates via the twist-pyramidalisation plane (Fig. 8A), resulting in precessional motion about the C2 axis.58,112,121,124 In the case of motors bearing heteroatoms or substituents which are strongly electron-withdrawing, heterolytic breaking of the π-bond upon excitation becomes favourable as the carbanionic lone pair is now stabilised. Consequently, the pyramidalisation distortion is no longer required and the molecule relaxes through bond length alternation (BLA) instead, via the twist-BLA plane (Fig. 8A). Thus, molecular geometry at the CInt is markedly more planar, and the rotation of the motor occurs in an axial (rather than a precessional) fashion (Fig. 8B).58
The structural deformations occurring at the S1/S0 CInts – particularly the pyramidalisation angle – may have some influence on the quantum yield (QY) of photoisomerisation, although the precise relationship between these two factors has not yet been established.58 Recently in our group, an oxindole-based molecular motor with push–pull character was developed. The pyramidalisation distortion at the S1/S0 CInt was significantly reduced in comparison to typical fluorene-based Feringa motors, due to the push–pull system offering stabilisation for the zwitterionic excited state conformation. This revealed rotational properties that are intermediate between precessional and axial motion, as well as a four-fold increase in QY compared to other reported oxindole motors.113 Experimentally determined QYs of second-generation overcrowded alkene-based molecular motors range from 0.55–20%,40,41,46,79 and the theoretical maximum QY is 20–30%.124 Typically, higher values of QYs are observed in motors with electron-withdrawing substituents conjugated to the axle,40,79 although this is not a fixed rule.46 First-generation overcrowded alkene-based motors benefit from much higher quantum yields, of up to 85%. In addition to this, the quantum yields of the back reactions are much lower (ca. 10%) meaning that the motor is effectively “primed” for efficient forwards rotation.123
Along the lines of these design principles, the development of biomimetic photoswitches54,56,125 and motors57–59 based on indanylidene pyrrolinium (IP) has been carried out by Olivucci and co-workers. These scaffolds contain N-protonated57 or N-alkylated59 moieties in the lower half, benefitting the motors with intrinsic electron-withdrawing properties. This takes inspiration from the protonated Schiff base of 11-cis-retinal (rPSB, Fig. 1A), which boasts the highest QY of E–Z photoisomerisation engineered by Nature (67%).48 The main structural distinction between IP-based photoswitches and motors is that the latter bear stereogenic centres at the allylic position, which offer a small degree of axial pre-twist, governing the directionality.57 The cationic N atom in the lower half biases the evolution of the charge-transfer FC state and subsequent relaxation to S0via the twist-BLA plane, which diminishes the requirement for pyramidalisation at the S1/S0 CInt.58 The photoisomerisation QY of the NAIP-based molecular motor (5, Fig. 1) is 20% for the E → Z and 24% for the Z → E directions,59 providing some evidence for the tentative link between decline of the pyramidalisation parameter at the S1/S0 CInt and an enhanced photoisomerisation QY. Biomimetic motors based on p-HDBI have also been prepared, with similar values for the photoisomerisation QY (E → Z = 23%).60
The photochemical processes of first-generation HTI-based motor 6 (Fig. 1B) has been examined by excited state calculations and transient absorption spectroscopy.69 Excitation of the (S)-(P)-E isomer leads to population of the S1 FC state, where it consequently relaxes to the first S1 minimum. Following this, the molecule overcomes a barrier and afterwards it can go to the S2/S1 CInt directly, or via a second S1 minimum. At the S2/S1 CInt, the molecule is funnelled back to the ground state forming the metastable Z-(S)-(M) isomer (QY = 7%). In contrast to second-generation motors, a second pathway is possible where the molecule moves from the S1 FC state to reach the T2 (n–π*) state via intersystem crossing (ISC). From here, the molecule moves towards T2/T1 CInt and the T1 (π–π*) state, and subsequently relaxes back to reform the stable (S)-(P)-E isomer via ISC, resulting in a unproductive isomerisation (QY = 8%). In comparison, the Z → E photoisomerisation is barrierless in the S1 state, and the triplet pathway is unfavoured due to the absence of S1 → T2 ISC (however, S1 → T1 ISC is possible, but unlikely). The relatively high QY for the Z → E photoisomerisation (24%) and no obvious formation of a triplet state suggests that the photoisomerisation QY is somewhat diminished by the unproductive triplet state pathway. When studying structurally related first-generation HTI motors, it was found that the Z → E photoisomerisation QYs were significantly decreased (by ca. 10%) when the methoxy groups on the upper half were exchanged for methyl or iso-propyl groups, however only small differences in the QYs for the E → Z and triplet state pathways were observed.126
Extensive computational research into the quantum chemical processes of light-driven rotary molecular motors in recent years has proven to be a powerful tool in studying the elusive photoisomerisation step.49,65,66,69,124 These calculations have led to the development of several theoretical motor scaffolds (Fig. 9), in which a major topic of interest is engineering the photoisomerisation steps to be fast and efficient, with high QYs. In 2016, Filatov and co-workers unveiled N-alkylated indanylidene benzopyrrole (NAIBP) motor 27 (Fig. 9).66 It was reported that this motor favoured heterolytic π-bond breaking (leading to the species with charge-transfer character) over homolytic π-bond breaking by 22 kcal mol−1, inferring that axial (rather than precessional) rotation would occur.58 Indeed, the NAMD simulations confirm this hypothesis, and reveal QYs of between 57–67%,124 over three times the value of hydrocarbon-based second-generation motor 2.40
 | ||
| Fig. 9 Novel motor scaffolds investigated via computational methods. | ||
In 2017, Durbeej and co-workers reported bicyclic alkene-based molecular motor 28, fitted with an electron-donating chiral N-methylpyrrolidine lower half (Fig. 9).65 In the ground state, the cyclopentadiene upper half is not aromatic, however when excited to the S2 state it exhibits cyclic electron delocalisation and becomes aromatic. Through NAMD simulations, they found that the motor possesses high calculated QYs of between 75–77%, albeit with a relatively low net directionality of 58%. The strategy of excited state aromaticity relies on the theory that favouring heterolytic, charge-transfer cleavage of the olefinic axle may be a viable approach to increase the photoisomerisation QY.58 Here, heterolytic cleavage is favoured, because during the formation of the excited state one half of the motor (here, the cyclopentadienyl upper half) receives an electron from the other half of the motor to become negatively charged and aromatic.127
In 2019, Filatov and co-workers reported motor 29 with a chiral upper half based on cyclopentathiophene, bearing two methyl groups (Fig. 9). The first is at the stereogenic centre adjacent to the central alkene bond, and the second is on the thiophene ring pointing into the fjord region of the motor, introducing some steric bulk into the system.62 This motor operates in two photochemical isomerisations and possesses unprecedented calculated QY values upwards of 90%. Perhaps more interestingly, it was also reported that the S1/S0 CInts of this motor have a peaked topography.
In other words, the S1 PES has a substantial gradient moving from the FC state towards the CInt, so the movement in this direction is strongly favoured. At the CInt, the molecule undergoes a non-adiabatic S1 → S0 transition, funnelling the molecule towards a successful photoisomerisation i.e. moving from Z → E or E → Z, rather than back towards its original ground state configuration. In addition to this, the S0 PES is inclined in the direction of increasing dihedral angle (which defines the twist of the central alkene axle) near the CInts, which also biases the motor to continue rotating unidirectionally. This combination of sloped ground and excited state PESs gives the motor a degree of directionality of 94%. Motor 30 with fulgide-based lower half was also postulated (Fig. 9), demonstrating similar excited state lifetimes and sloped S1/S0 CInt topographies, although with reduced (but still high!) QYs of 55–68%.61
Dube and co-workers have reported a second-generation HTI-based molecular motor that runs entirely on photonic energy.71 The motor exists as four isomeric states, yet the favoured rotation cycle includes just three of these isomers. Thermal interconversion between these isomers is highly disfavoured, with their thermal half-lives being at least 0.7 years at 27 °C. The motor rotation operates through sequential PEZ, SBR and HT photoreactions (7, R = OMe, Fig. 4A). Each isomer of the motor can undergo each type of photoreaction, resulting in conversion to each of the other three isomers – however, not all of these photoreactions are equally likely; the QYs of the photoreactions range from <0.1–17%. The degree of directionality can be verified by calculating the probability of each photochemical process occurring (against all other possible processes) in the desired direction from the QYs of the constituent steps in the rotation cycle. From this calculation, it was found that the most probable reaction cycle was ACB via7a → 7c SBR (QY = 10.0%), 7c → 7b HT (QY = 2.3%), and 7b → 7a PEZ (QY = 0.27%); at 20 °C the relative probability of this reaction cycle compared to all others was 81%. However, a second competing rotation cycle occurs at this temperature at 16% probability – ACD. This is due to the relatively small difference between the QYs of the transformations between 7c → 7d (PEZ, QY = 1.3%) and 7d → 7a (HT, QY = 0.2%), in comparison to the QYs of the transformations from 7c → 7b and 7b → 7a at this temperature. This competing cycle clearly affects the “monodirectionality” of the motor, which is defined as “a preference for one cycle against all others”.71 When cooled to −50 °C, it was found that the QY of the 7c → 7b HT transition was almost doubled to 3.6%, whereas the QY of the unproductive 7c → 7d transition was halved to 0.54%. As a result, the monodirectionality of the cycle ACB is boosted to 99% at reduced temperatures, demonstrating increased speed and quantum efficiency. This finding contrasts with typical overcrowded alkene-based scaffolds for which efficiency completely declines at sufficiently low temperatures, because the THI steps used to ensure the unidirectionality are frozen out.
In this section, we have explored the excited state processes and QY values of various families of light-driven molecular motors. The QY of any given molecular motor is a vital characteristic, as it quantifies the efficiency of the photochemical step – higher QY values equate to more efficient motors. First-generation overcrowded alkene-based motors have the highest QY values overall, up to 85%.119,123 Theoretical simulations may further decipher the excited state dynamics of first-generation motors and shed light on the underlying reasons for these high quantum yields; up to now, no such simulations have been reported. For second-generation overcrowded alkene-based motors, theoretical scaffolds boast the highest QY values, but so far none of these values have been experimentally verified.61–63,65,66 However, studying the excited state processes of theoretical motors has revealed multiple factors at play which could lead to improved QY values, such as peaked vs. sloped topography of the S1/S0 CInts on the excited state PES.61,62,66 The highest experimentally determined QY values for second-generation motors reported so far are roughly 20%.40,59,60 A solid method towards improving QY values is the biomimetic approach, where molecular motor scaffolds are based on photoactive molecules designed by Nature. These natural compounds already have high QY values for E–Z isomerisation which can be engineered into the π-backbones of the molecular motors, providing high experimental QYs.59,60 Quantum chemical studies of these compounds and related theoretical scaffolds has revealed that there may be a link between high QY values and a preference for axial rotation during the photoisomerisation,58–60,65,66,113 which is defined by the geometrical changes occurring on the excited state PES, and in particular by the molecular structure at the S1/S0 CInts.
Rotation speed
The rotation speed of any given light-driven molecular motor is influenced by all of the individual steps comprising its rotation cycle, and the rate-limiting step determines the overall rotation speed. Clearly this varies vastly between different motor scaffolds, due to their distinct structures. Motors with thermal ratcheting steps in the ground state are limited by the half-lives of these steps, which can range from years to nanoseconds.46,128,129 For comparison, only the half-lives at room temperature (293.15 K) and the Gibbs free energy barriers (ΔG‡) will be discussed. Photonically fuelled motors are limited by the much faster photoexcitation steps, which operate on the order of femto- to picoseconds.112In our group, considerable research effort has been dedicated to establishing the relationship between molecular structure and the rate of the THI for overcrowded alkene-based motors. Before we continue, it is important to distinguish between first- and second-generation motors. In the case of first-generation motors, the structures of the metastable E and Z isomers are intrinsically asymmetric, giving rise to two individual E and Z THI rates (Fig. 2A). Due to this asymmetry, even subtle structural modification can have a starkly different effect on both of these rates e.g. one barrier may increase drastically while the other remains unchanged, or even decreases. For second-generation motors with symmetric lower halves there is only one THI step to consider, and for those with asymmetric lower halves the barriers for the two THI steps are essentially equal. This makes the trend between structural modification in second-generation motors simpler to interpret, so this is what we will discuss in this section. Reviews discussing the rate acceleration of first-generation motors can be found elsewhere.129,130
There are two ways in which the thermal barrier can be decreased (and therefore the rotation speed increased): (i) reducing steric hindrance in the fjord region or (ii) increasing the energy of the metastable state relative to the transition state (Fig. 10). Decreasing the degree of steric hindrance in the fjord region can be achieved by reducing the size of the rings directly attached to the double bond axle; this has been achieved by simply swapping the 6-membered ring for a 5-membered one (compare 31 with 2, and 39 with 40),46,99,131 through introduction of different bridging heteroatoms in the rings (compare 32 with 22),46,132 or by substituting the naphthalene moieties on the upper half for less bulky xylene ones (compare 2 with 34, Fig. 10).133,134 On the other hand, introducing steric hindrance in the fjord region can be used as a tool to increase the THI barriers.135 For the first-generation HTI motor 6 the highest thermal barrier (for the EM → ES conversion) was 13.1 kcal mol−1, equating to a half-life of 662 μs. The thermal barrier for the ZM → ZS was calculated to be 5.5 kcal mol−1 (t1/2 = 1.5 ns), and for this reason the ZM isomer could not be observed empirically.34 To this end, the more sterically hindered analogue 36 was synthesised with an iso-propyl group pointing into the fjord region, which increased the ZM → ZS barrier to 11.3 kcal mol−1 (t1/2 = 30 μs) allowing unequivocal observation of the elusive fourth state, operating with an overall speed of 34 ms (EM → ES barrier = 15.4 kcal mol−1).70 Similar observations were reported in a later study when comparing motors 35 and 37 with ethyl and methyl groups on the aromatic ring of the upper half, giving thermal half-lives of 36 ms and 19 ms, respectively.135
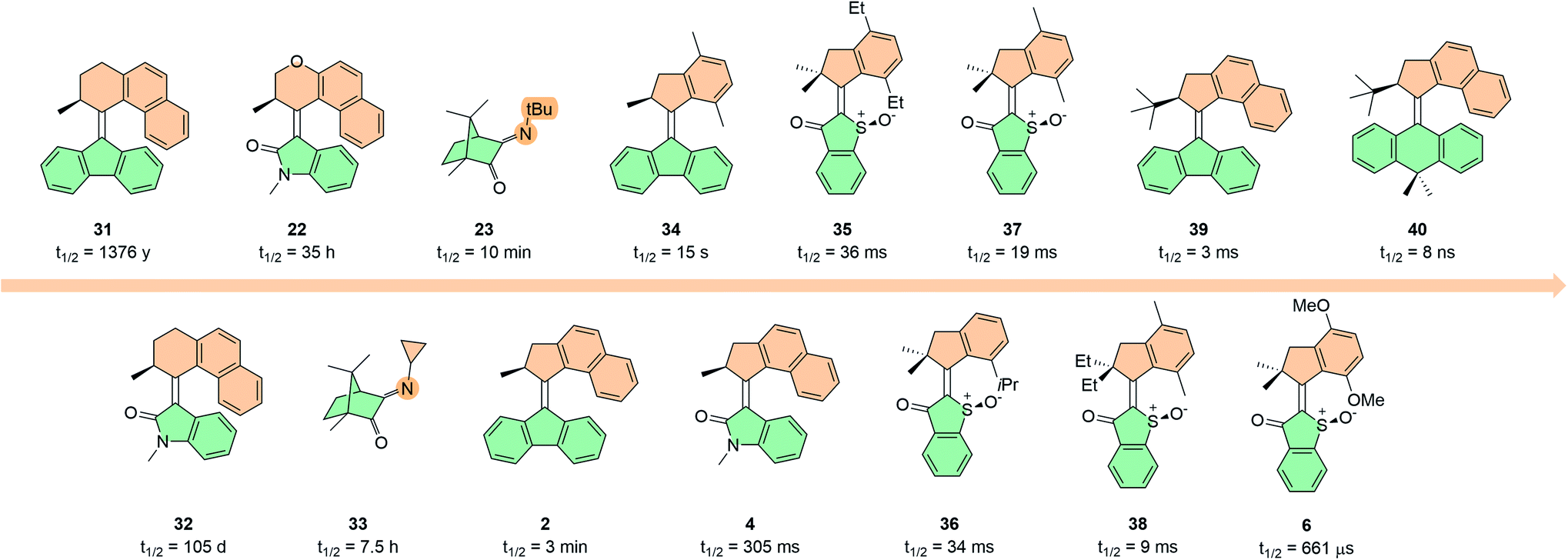 | ||
| Fig. 10 Half-lives of various molecular motors with thermal steps in the ground state at 293.15 K. | ||
Thermal barriers can also be decreased by increasing the steric demand of the stereogenic centre, so that the metastable state of the second-generation overcrowded alkene-based motors is destabilised with respect to the transition state. This is because the unfavourable pseudo-equatorial orientation at the stereogenic centre generates substantial steric strain with the lower half in the metastable state, which is relieved in the THI. For this reason, an increase in rotation speed was observed with increasing substituent size.136 Replacing the stereogenic methyl groups in the allylic position of first-generation HTI motor 37 with more sterically demanding ethyl groups in motor 38 was coupled with an acceleration of 10 ms.135 A similar trend was observed in imine motors based on camphorquinone. It was found that the half-lives for the NI step decreased from 7.5 h to 10 min when moving from substitution of a cyclopropyl group in the motor to a larger tBu group on the N atom (compare 23 and 33) indicating a ground state destabilisation of the metastable Z state with respect to the transition state.89
Multiple systems have been developed in which the rotation speed of light-driven molecular motors can be modulated through allosteric binding. With 4,5-diazafluorenyl functionalised motor 25 (Fig. 11A), the rotation speed could be modulated through chelation of various transition metal complexes.137 The half-life of naked parent motor 25 is 1.4 min, which upon complexation speeds up to 30 times faster in the case of the [Pt(Cl)2(25)] complex (t1/2 = 2.7 s). This phenomenon was also observed in [Ru(bpy)2(25)] which has a rotation speed that is 50 times faster than parent motor 25 (t1/2 = 1.7 s).109 The acceleration is due to contraction of the lower half of the motor complex, which reduces the degree of steric crowding in the fjord region. Allosteric binding was used to modulate the speed of biphenol motor 41 (Fig. 11B), but in this case the speed decreased upon binding. Through supramolecular hydrogen bonding to a cyclic diamine the half-life of motor 41 increased from 0.3 s to 30 s, and upon covalent bonding to a methylene tether the half-life further increased to 1.4 h – effectively acting as a molecular brake. In addition to structural modifications having an impact on the thermal barriers, the environment can also have an effect. A comprehensive study of a second-generation molecular motor substituted with n-butyl chains carried out in over 50 solvents and solvent mixtures showed weak correlation between rotation speed and most solvent effects, suggesting that the influence of solvent effects on the THI is complex and multi-faceted.42
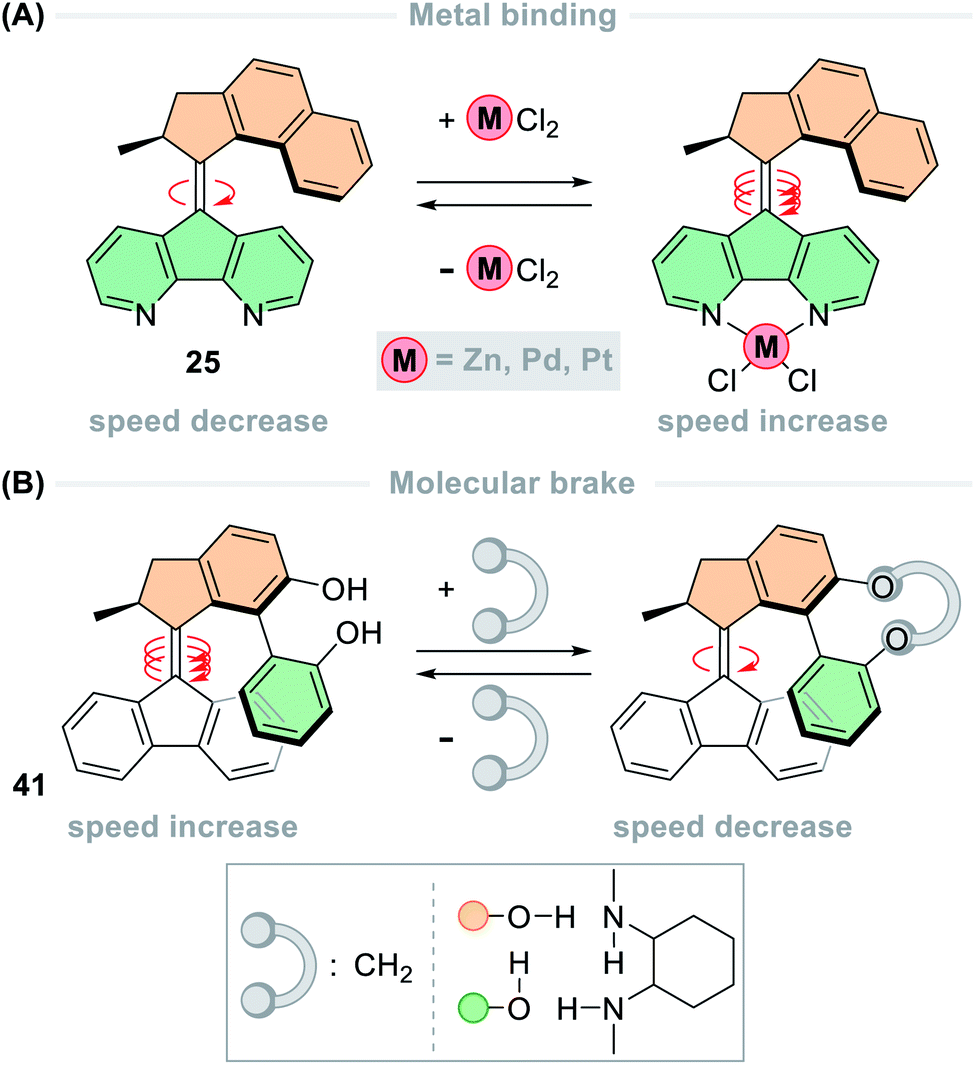 | ||
| Fig. 11 Modulation of rotation speed of second-generation overcrowded alkene-based motors by allosteric binding. (A) Binding of transition metal ions to the lower half increases the rotation speed; (B) non-covalent binding of molecular tethers to the upper half decreases the rotation speed. | ||
There was no significant correlation found between the effect of solvent polarity on the THI, however this may be partially due to the intrinsic apolarity of the motor studied. However, it was shown that the rate of the THI step has a large dependence on solvent viscosity,42,138 a phenomenon that was also observed in first-generation motors fitted with rod-like aromatic arms up to 32 Å in length – so-called molecular stirrers.139 In this study, it was shown that longer and more rigid stirrer arm substituents increase the entropic barrier for rotation, leading to more pronounced viscosity dependency. Trends between other solvent effects showed only weak correlation. In the case of the figure-of-eight HTI motor 13, the rate-limiting step is the thermal HT step, and depending on the solvent mixtures used to operate the motor, the rotational speed also changes. The half-life of the rate-limiting thermal HT step (13c → 13b, Fig. 4B) in polar MeCN:D2O is 11 days, compared to 1094 years in relatively apolar 1,2-dichlorobenzene.73 This factor of 3.6 × 104 difference clearly shows that solvent polarity can have a large influence on thermal barriers.
It is important to mention that for motors with low THI barriers, factors such as the light intensity, quantum yields and absorptivity of the molecule can influence the rate limiting step for the overall rotation cycle.69,126,140 Indeed, there is an optimal temperature range at a given light intensity at a certain wavelength for optimal rotation speed; increasing the temperature over this range reduces the motor performance.140
Molecular motors that are powered entirely by photonic energy are free from the restraints of the thermal steps in the ground state, and therefore can have much faster rotation speeds. With this blueprint in mind, fully photochemically powered motors could harness faster, temperature-independent rotation. As described in the previous section of this review, the photoisomerisations of overcrowded alkene-based molecular motors occur on the scale of picoseconds,40,112,113,117 which 103 times faster than even the fastest THI steps.133 First-generation HTI-based motor 6 similarly experiences photoisomerisations on the scale of picoseconds, but interestingly when substituting the methoxy groups on the aromatic ring of the upper half for more sterically demanding iso-propyl groups, these photoisomerisations can accelerate up to 270 fs (30 times faster).126 But of course here the speed is limited by the thermal steps in the ground state (t1/2 = 34 ms).70,135
In a similar way that removal of steric bulk in the fjord region can reduce the barriers of the thermal steps in the ground state, if enough bulk is removed, thermal barriers can be eliminated completely. This is the approach employed in many theoretical motor scaffolds, which focus on increasing quantum efficiency, as described in the previous section. NAMD simulations reveal fast photoisomerisation processes at work on the scale of femtoseconds, up to 103 times faster than is typical for second-generation motors.112 Ultrafast photoisomerisations have been reported for theoretical motors 27–30 (Fig. 9) and motors 18 and 19 (Fig. 6) of between 200–300 fs.61,62,64–66,102 For enamine-based motor 28, the excited state aromaticity proved to be a key design principle for this. It enabled ultrafast photoisomerisation (∼200 fs) by initial excitation to the S2 state, followed by barrierless relaxation along the twist-BLA plane towards an S2/S1 CInt which funnels almost immediately to the S0 state through an S1/S0 CInt, with very few additional geometric distortions required.65 In a follow-up paper, it was found that an isotopically chiral variant of this motor – fitted with a deuterated stereocentre rather than a methylated one – undergoes faster photoisomerisations (∼150 fs) than its chemically chiral counterpart (28, Fig. 9). This permitted the motor to have the fastest photoisomerisation lifetime reported for any molecular motor to date.141 Although not confirmed experimentally yet, this finding may prove to be an important tool in reducing the excited state lifetimes of molecular motors from the order of picoseconds to femtoseconds, leading to faster motors.
In the case of the experimentally derived photon-only N-alkylated IP-based motor 5 (Fig. 1), the excited state lifetimes were probed by transient absorption spectroscopy in addition to NAMD simulations, and it was found that complete decay of this motor from S1 occurred within 500 fs.59 Other theoretical motors based on N-protonated or N-alkylated IP scaffolds have been found to have excited state lifetimes of between 175–300 fs.57,64,66,102 From these examples, a link between faster photoisomerisation steps and a preference for axial rotation can be implied.58 Presumably, the acceleration is due to the relatively small geometrical changes necessary to traverse the S1 PES along the twist-BLA plane to the S1/S0 CInt in comparison to the twist-pyramidalisation plane.
The rotation speed of molecular motors differs drastically,129 from thousands of years128 to hundreds of femtoseconds.141 For first- and second-generation overcrowded alkene-based molecular motors, the links between molecular structure and the size of the thermal barriers are well described128–130 – namely reducing steric hindrance in the fjord region and destabilising the metastable state in relation to the transition state. These rules translate well to other motors with thermal steps, such as first-generation HTI motors126 and imine motors.35 It is possible for the molecular engineer to fine-tune the rotation speed of a given motor through tweaking its molecular architecture, as the field has now gained enough insight to predict the delicate balance between the ground state and transition state energies of the rate-determining thermal step. This prospect offers plenty of customisability within these photoactuators, which can suit them towards a multitude of different purposes. Modulation of rotation speed through orthogonal (photo)chemical triggers is a powerful tool to control molecular motion at the nanoscale, particularly in the development of multi-responsive molecular systems.6 Of course, the most effective way to accelerate the rotation of light-driven molecular motors is by eliminating thermal steps in the ground state, as these are several orders of magnitude slower than photochemical isomerisations.112 The topologies of the excited state PESs can be better understood with further studies combining ultrafast spectroscopy and predictions with excited state quantum chemical methods, which will pave the way towards faster and more photoefficient molecular motors.
Synthetic accessibility
Typically, light-driven rotary molecular motors are sterically crowded structures with highly strained central alkene bonds, meaning that their synthesis can be tricky and low yielding. Overcrowded alkene-based molecular motors are generally synthesised through either a McMurry reaction for first-generation motors (Fig. 12A), or through a Barton–Kellogg olefination for second-generation motors (Fig. 12B). Both McMurry and Barton–Kellogg couplings have widely variable yields, but they are generally scalable and the ketone-based starting materials are easy to synthesise, depending on the substituent.83 Both McMurry and Barton–Kellogg coupling procedures involve the generation of a cyclic intermediate (a 1,2-diol with low-valent titanium for the McMurry,142 or a sulfaphosphatane for the Barton–Kellogg143) which subsequently undergoes elimination to yield the overcrowded alkene. In the case of heterocouplings, McMurry reactions yield a statistical mixture of isomers.144 Hence, the McMurry reaction is predominantly useful for the synthesis of symmetric first-generation motors, yielding from between <1% to >90%.76,145 There is correlation between more sterically demanding groups at the stereocentre of first-generation motors and lower yielding McMurry reactions.76 Like the McMurry reaction, Barton–Kellogg couplings also have widely varying yields (from <20% to >75%).‡,79,146,147 Recently, a comprehensive computational investigation has revealed a reliable method for estimating the feasibility of Barton–Kellogg reactions – a tool which may prove useful for the synthesis of second-generation overcrowded alkene-based molecular motors.143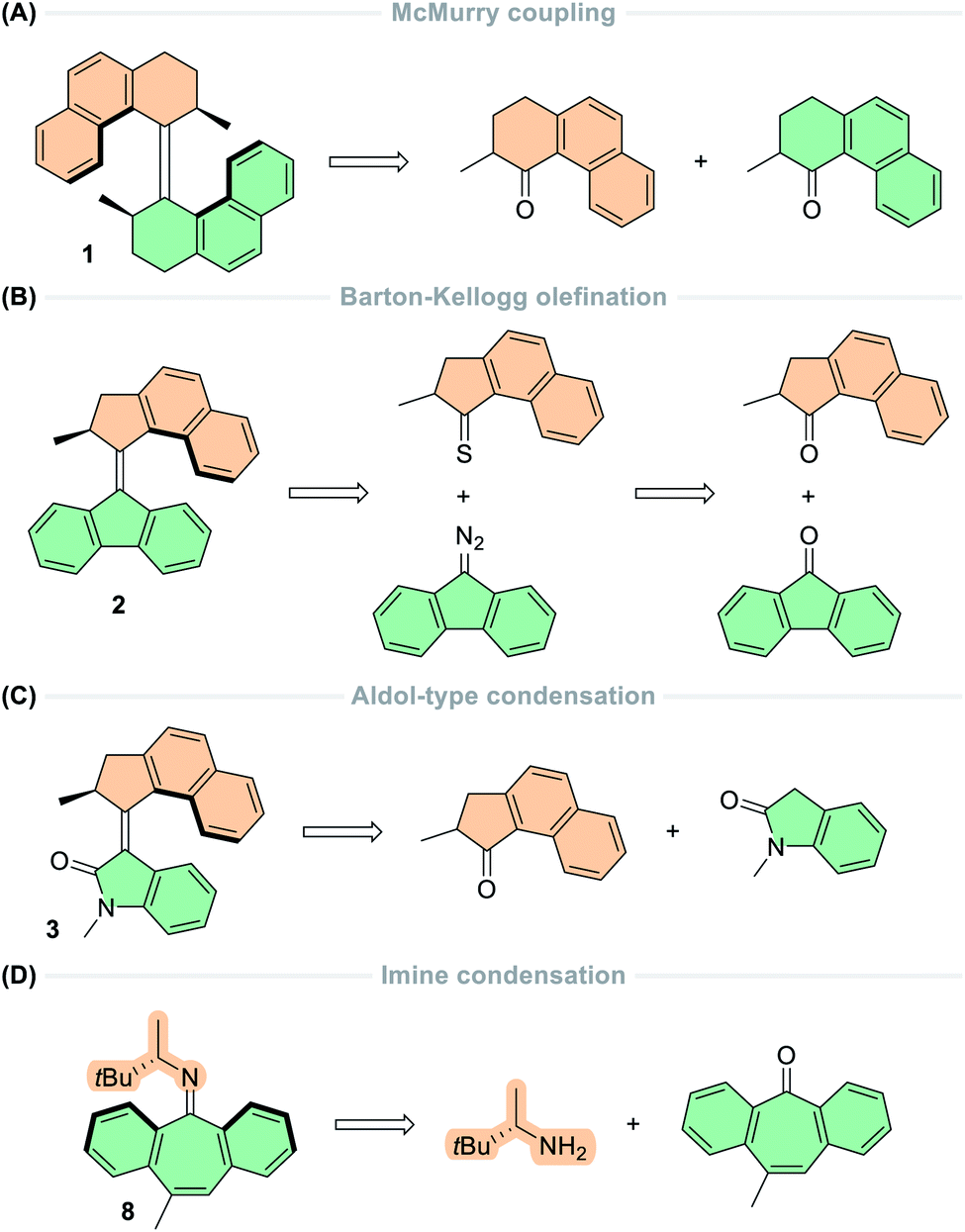 | ||
| Fig. 12 General synthetic methods for synthesising artificial light-driven molecular motors. (A) McMurry coupling for first-generation overcrowded alkene-based motors; (B) Barton–Kellogg coupling for second- and third-generation overcrowded alkene-based motors; (C) aldol-type condensation chemistry for oxindole-based motors, biomimetic motors, first-generation HTI-based motors; (D) imine condensation for imine-based motors. | ||
The development of alternative motor scaffolds has offered new, facile synthetic methods of molecular motors, often from commercially available starting materials. For many of these more recent motors, these synthetic methods utilise condensation chemistry (Fig. 12C).34,46,70,71,73,85 In our group, oxindole motors are prepared by a Knoevenagel-type condensation using titanium tetrachloride as a Lewis acid which activates the respective top-half ketone to the attack from a relatively weak nucleophile, the oxindole enolate. This synthetic method is a one-pot, two-step reaction which yields a single stereoisomer of the given motor in as little as two hours, in yields ranging from 22–68%, depending on the rigidity of the ketone used.46,113
The first-generation HTI motors are synthesised in an aldol condensation reaction mediated by boron trifluoride etherate, in 12% overall yield.34 This result was later improved to 85% when using boron trichloride as the Lewis acid and carrying out the reaction at 0 °C rather than at room temperature, and this method also decreased the reaction time from two days to 50 min.85
First-generation HTI motors have also been synthesised in 67% yield over a five-step method, involving the addition of a lithiated acetylide species to the top-half indanone followed by a Meyer–Schuster rearrangement and oxidation of the sulfur atom, then subsequent intramolecular cyclisation forming the HTI chromophore.70
Second-generation HTI motors can be synthesised in five steps from commercially available thiosalicylic acid derivatives and α-bromoketones, a method that is high yielding, modular, and with a broad substrate scope.72,148 The key step is the formation of the HTI chromophore, which is achieved through a one-pot intramolecular cyclisation mediated by sodium acetate and thionyl chloride. This step also establishes a chloride substituent as the fourth substituent on the double bond, which can be readily outfitted with a large range of various substituents via nucleophilic substitutions or cross-coupling chemistry.148 The final step involves oxidation of the sulfur atom on the HTI chromophore to a chiral sulfoxide group, converting the compound from a binary switch to a unidirectional motor.71–73
Imine-based motors can be synthesised by a one-step imine formation from the corresponding chiral primary amine and ketone fragments mediated by titanium tetrachloride, with quantitative yields on a gram scale from commercially available, inexpensive starting materials (Fig. 12D).35,89
A key aspect of unidirectional motor rotation is the absolute configuration at the stereogenic centre, this controls the direction of the rotation. A racemic mixture of enantiomers at the stereogenic centre will result in no net directionality of the motor rotation – as 50% will be rotating clockwise, and the other 50% will be rotating anti-clockwise. Therefore, access to enantiomerically pure motors is vital to produce bulk rotation of molecular motors in one direction. All of the synthetic methods described in this section up to now produce racemic mixtures of molecular motors, from which enantiomerically pure material can be obtained by preparative chiral chromatography, such as HPLC or SFC (Fig. 13A).34,113,131 However, this is hardly feasible for generating optically pure motors on a multi-gram scale, so alternative methods have been developed.
 | ||
| Fig. 13 Various methods used to access enantiomerically pure molecular motors. (A) Preparative chiral chromatography; (B) chiral resolution processes; (C) asymmetric catalysis; (D) use of a chiral auxiliary. | ||
Both first- and second-generation overcrowded alkene-based molecular motors have been obtained via asymmetric synthesis in various ways.147,149–152 In the case of first-generation motors, the chiral Evans auxiliary was used to facilitate enantioselective methylation of a carboxylic acid derivative, which could later be cyclised to form enantiopure indanone in seven steps overall. The following McMurry coupling generated 30% of enantiomerically pure material.149 A switchable chiral resolution process using supramolecular complexation of a first-generation motor equipped with hydroxyl moieties to a chiral ammonium derivative (N-benzylcinchodinium chloride) yields enantiopure motor in up to 81% yield and >99% ee (Fig. 13B).152 Optically pure first-generation motors have also been synthesised through an enantioselective protonation of the silyl enol ethers of indanones by a Au(I)BINAP complex, then using lithium aluminium hydride to increase diastereoselectivity. This method yielded up to 78% over three steps and up to >98% enantiomeric excess (Fig. 13C).150
For second-generation motors, it is key to note that conversion of the precursor ketone into the thioketone or diazo compounds necessary for the Barton–Kellogg olefination, or the Barton–Kellogg coupling itself, leads to racemisation of the stereogenic centre at the α-position when starting from enantiopure ketones. Therefore, epimerisation at this position must be circumvented to generate optically pure motors. This result was achieved by the introduction of an additional group on the indanone at the β-position, which disfavours epimerisation of the α-stereogenic centre, while not compromising the unidirectional rotation of the motor.151 Alternatively, diastereoselective synthesis developed by Giuseppone and co-workers using lactic ethyl esters as chiral auxiliaries on the upper half, provides a facile method to chiral ketones which could be converted to the corresponding thioketone to be used in the Barton–Kellogg coupling with a thioxanthone-based lower half (Fig. 13D). The chiral auxiliaries on the upper half facilitate separation of the diastereomers simply through column chromatography, producing optically pure motor on a gram scale.147
In this section, we have reviewed various methods used to synthesise light-driven molecular motors. The sterically hindered nature of molecular motors makes them intrinsically difficult to synthesise, yet it is this exact structure that generates the helical chirality necessary for their operation. Typical methods of synthesising molecular motors e.g. the McMurry coupling,76,145 the Barton–Kellogg olefination79,146 and various aldol-type reactions34,46,85 result in widely varying yields, and are seemingly highly dependent on the molecular structure of the two motor halves coming together. Nevertheless, these methods are reliable for forming sterically crowded alkene and imine bonds for a range of motors with different structures and functionalities. Use of orthogonal anchoring groups is particularly useful as it allows different parts of the motor to be independently connected to any other components, which is clearly desirable for the integration of motors into various functional systems.146,147 Intramolecular cyclisation to form the HTI chromophore is an elegant and high yielding method of making both first- and second-generation HTI motors.70,148 Imine-based molecular motors can be synthesised on a gram scale from inexpensive starting materials in one step.35 Considerable progress has been made towards the asymmetric synthesis of overcrowded alkene-based motors,147,150–152 but current options are still limited. So far there have been no methods reported for synthesising optically pure material for other motor scaffolds, which limits their versatility in applications requiring unidirectional bulk rotation.
Conclusions and outlook
The emergence of artificial molecular motors and machines has signified a huge step towards the development of responsive and dynamic molecular systems, the so-called “synthesis of function”.1 The field is moving swiftly from a proof of concept to application of molecular machines in responsive and adaptive systems – the “control and use of function”. Since the revolutionary discovery of the first light-driven unidirectional rotary molecular motor in 1999,36 great progress has been made in controlling the rotation about double bonds. The development of new and improved motor scaffolds has thrived in the past decades, proving molecular motors to be a highly diverse, tunable and robust tool for the molecular engineer's toolbox. Light-driven rotary molecular motors have been used in many fields already, ranging from smart materials and surface anchoring, to catalysis and biomolecular technology.43,84,85,129,145,153–155 For effective integration into more complex, functional molecular systems, it is important to have an idea of which motor scaffold would be best tailored for each potential purpose.In this perspective, we have discussed the recent advances in the development of light-driven molecular motors, highlighting the main design principles underpinning motor function by reviewing several examples from current literature. We have explored in detail the differences in rotational mechanism, as well as how to effectively tune various motor properties, such as rotational speed, quantum efficiency and absorption wavelength. By discussing the key aspects of general motor design, we offer some insights into the various families of light-driven molecular motors, in addition to aiming to provide guidelines on selecting and applying molecular motors for future explorations.
Application of molecular motors in chemical biology and materials science is hindered by the necessity to use UV light, which can be harmful to the media associated with these applications, such as polymers or cells. Motors with intrinsically red-shifted absorption maxima such as oxindole-based and HTI-based motors may find use in these applications,34,46 and recent advances with 2PA sensitisers has extended this window to the near-IR region.103 Further exploration into multi-photon absorption should be carried out, as this offers the largest red-shift observed in the absorption wavelength of molecular motors to date. Additional methods that may offer large bathochromic shifts are photon upconversion and triplet–triplet annihilation processes, which so far have not been applied to molecular motors.
Significant research effort has been invested into deciphering the link between quantum efficiency and the geometrical changes occurring during the photoisomerisations of molecular motors, and this continues to remain a challenge today. Although more efficient motors are being developed at a steady pace, the specific details behind the increased quantum efficiency remain difficult to diagnose. Recently, a first-generation overcrowded alkene-based motor scaffold was reported to have quantum yields of 85%, a result which up to now is unprecedented.123 Further research into the excited state processes of first-generation motors, particularly through computational simulations, is required to further establish the links between high quantum yields and motor structure. The influence of structural changes on the rotational speed of molecular motors involving rate-determining thermal steps is established,129,130 however increasing the speed of the photochemical steps is less well understood. The prospect of photon-only powered molecular motors is a very promising one,71,80 however the unidirectionality of such systems is not so simple to control without thermal ratcheting steps in the ground state. Further development of molecular motors powered solely by photonic energy, particularly with orthogonal switching wavelengths, may be particularly interesting for multi-motor systems and information processing.6
Amplification of motion from the nanoscale to the macroscale requires the transition from disordered motion to ordered motion to be navigated. For this, molecular motors must be precisely assembled along length scales in materials, and within these systems, the motors must work together in unison (i.e. cooperatively) to create bulk rotation. The operation of molecular motors in solution is well understood, but the integration of motors into larger molecular systems is not trivial.43,44 Numerous motorised architectures can be envisioned: liquid crystals, MOFs/COFs, surfaces, gels, polymers, membranes and interfaces, for example. Integration of molecular motors into several of these smart materials has already been realised, and has been used to generate macroscopic movement – such as the contraction of a motor doped gel,90 and the movement of a glass rod by a liquid crystal film doped with molecular motors.156 In both of these cases, enantiopure molecular motor was integrated into these materials which enforces bulk rotation in one direction,147 however this is not entirely necessary to make useful, smart materials. For instance, a hydrogel containing only 5 wt% of a racemic mixture of molecular motors was used as a molecular “muscle” to even pick up a small piece of paper,26 and 3D organised molecular motor-based MOFs have been developed.146 Nevertheless, the field is limited by the fact that synthesising large amounts of enantiomerically pure motor is difficult; more research effort should be invested into methodologies for simple, gram-scale synthesis of enantiopure motors. This exploration will make integrating molecular motors as the core dynamic unit into (supra)molecular frameworks (e.g. polymers or porous materials) more practical, which can then perform as functional actuators in responsive materials.
One can speculate over what the “perfect motor scaffold” might look like – it would be easy to (stereoselectively) synthesise, intrinsically red-shifted, have high QYs and quantitative PSD ratios, in addition to being easily functionalised with various anchoring groups for integration into materials. Of course there is no such thing as one perfect motor – but if there is one lesson to be learnt from this perspective, it is that there are many excellent options. Building on twenty years of experience, the ideal wavelength, speed, functionalisation and rotation cycle can be combined into a different preferred design for each new application. The fundamental challenges outlined in this perspective must be considered for the future prospect of molecular machines: moving towards responsive, interactive molecular systems by means of intentional molecular engineering. Use of molecular motors in such systems will require amplification of their motion across length scales, development of emergent and adaptive properties, coupled motion, cooperativity and synchronisation, molecular sensing and information processing, amongst other things. Addressing these challenges head on will allow ample opportunities to manufacture sensitive, cooperative, and precisely organised motion from the nanoscale to the macroscale – a thoroughly exciting prospect for future molecular engineers.
Author contributions
All authors contributed to the bibliographic research, discussion, writing, editing, and revision of the article.Conflicts of interest
The authors declare there to be no conflicts of interest.Acknowledgements
D. R. S. P. gratefully acknowledges Dr Romain Costil, Dr Michael Kathan, Brian Corbet and Ainoa Guinart for fruitful discussions. Financial support from the Horizon 2020 Framework Programme (ERC Advanced Investigator Grant No. 694345 to B. L. F.), the Netherlands Ministry of Education, Culture and Science (Gravitation Programme 024.001.035 to B. L. F.) and the Marie Skłodowska-Curie Action (Individual Fellowship No. 838280 to S. C.) is gratefully acknowledged.Notes and references
- B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11060–11078 CrossRef CAS PubMed.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- J. P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed.
- W. R. Browne and B. L. Feringa, Molecular Switches, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- D. S. Goodsell, The Machinery of Life, Springer, New York, 2nd edn, 2009 Search PubMed.
- B. L. Feringa, Adv. Mater., 2020, 32, 1–3 CrossRef PubMed.
- R. D. Astumian, Phys. Chem. Chem. Phys., 2007, 9, 5067–5083 RSC.
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS PubMed.
- V. Balzani, A. Credi and M. Venturi, Chem. Soc. Rev., 2009, 38, 1542–1550 RSC.
- F. Lancia, A. Ryabchun and N. Katsonis, Nat. Rev. Chem., 2019, 3, 536–551 CrossRef CAS.
- A. W. Heard and S. M. Goldup, ACS Cent. Sci., 2020, 6, 117–128 CrossRef CAS PubMed.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- D. Bléger and S. Hecht, Angew. Chem., Int. Ed., 2015, 54, 11338–11349 CrossRef PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- J. D. Harris, M. J. Moran and I. Aprahamian, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9414–9422 CrossRef CAS PubMed.
- S. Crespi, N. A. Simeth and B. König, Nat. Rev. Chem., 2019, 3, 133–146 CrossRef CAS.
- M. W. H. Hoorens, M. Medved’, A. D. Laurent, M. Di Donato, S. Fanetti, L. Slappendel, M. Hilbers, B. L. Feringa, W. J. Buma and W. Szymanski, Nat. Commun., 2019, 10, 2390 CrossRef PubMed.
- S. Silvi, M. Venturi and A. Credi, J. Mater. Chem., 2009, 19, 2279–2294 RSC.
- C. Cheng, P. R. McGonigal, S. T. Schneebeli, H. Li, N. A. Vermeulen, C. Ke and J. F. Stoddart, Nat. Nanotechnol., 2015, 10, 547–553 CrossRef CAS PubMed.
- Y. Qiu, L. Zhang, C. Pezzato, Y. Feng, W. Li, M. T. Nguyen, C. Cheng, D. Shen, Q. H. Guo, Y. Shi, K. Cai, F. M. Alsubaie, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2019, 141, 17472–17476 CrossRef CAS PubMed.
- G. Ragazzon, M. Baroncini, S. Silvi, M. Venturi and A. Credi, Beilstein J. Nanotechnol., 2015, 6, 2096–2104 CrossRef CAS PubMed.
- Y. Feng, M. Ovalle, J. S. W. Seale, C. K. Lee, D. J. Kim, R. D. Astumian and J. F. Stoddart, J. Am. Chem. Soc., 2021, 143, 5569–5591 CrossRef CAS PubMed.
- S. Kassem, A. T. L. Lee, D. A. Leigh, A. Markevicius and J. Solà, Nat. Chem., 2016, 8, 138–143 CrossRef CAS PubMed.
- J. Chen, S. J. Wezenberg and B. L. Feringa, Chem. Commun., 2016, 52, 6765–6768 RSC.
- C. J. Bruns and J. F. Stoddart, Acc. Chem. Res., 2014, 47, 2186–2199 CrossRef CAS PubMed.
- J. Chen, F. K.-C. Leung, M. C. A. Stuart, T. Kajitani, T. Fukushima, E. van der Giessen and B. L. Feringa, Nat. Chem., 2018, 10, 132–138 CrossRef CAS PubMed.
- M. von Delius and D. A. Leigh, Chem. Soc. Rev., 2011, 40, 3656–3676 RSC.
- M. von Delius, E. M. Geertsema and D. A. Leigh, Nat. Chem., 2010, 2, 96–101 CrossRef CAS PubMed.
- C. J. Martin, A. T. L. Lee, R. W. Adams and D. A. Leigh, J. Am. Chem. Soc., 2017, 139, 11998–112002 CrossRef CAS PubMed.
- M. Baroncini, S. Silvi and A. Credi, Chem. Rev., 2020, 120, 200–268 CrossRef CAS PubMed.
- V. García-López, D. Liu and J. M. Tour, Chem. Rev., 2020, 120, 79–124 CrossRef PubMed.
- T. R. Kelly, H. De Silva and R. A. Silva, Nature, 1999, 401, 150–152 CrossRef CAS PubMed.
- D. Dattler, G. Fuks, J. Heiser, E. Moulin, A. Perrot, X. Yao and N. Giuseppone, Chem. Rev., 2020, 120, 310–433 CrossRef CAS PubMed.
- M. Guentner, M. Schildhauer, S. Thumser, P. Mayer, D. Stephenson, P. J. Mayer and H. Dube, Nat. Commun., 2015, 6, 8406 CrossRef CAS PubMed.
- L. Greb and J. M. Lehn, J. Am. Chem. Soc., 2014, 136, 13114–13117 CrossRef CAS PubMed.
- N. Koumura, R. W. J. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- N. Koumura, E. M. Geertsema, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2000, 122, 12005–12006 CrossRef CAS.
- J. C. M. Kistemaker, P. Štacko, J. Visser and B. L. Feringa, Nat. Chem., 2015, 7, 890–896 CrossRef CAS PubMed.
- A. Albini and M. Fagnoni, ChemSusChem, 2008, 1, 63–66 CrossRef CAS PubMed.
- J. Conyard, A. Cnossen, W. R. Browne, B. L. Feringa and S. R. Meech, J. Am. Chem. Soc., 2014, 136, 9692–9700 CrossRef CAS PubMed.
- A. Cnossen, J. C. M. Kistemaker, T. Kojima and B. L. Feringa, J. Org. Chem., 2014, 79, 927–935 CrossRef CAS PubMed.
- A. S. Lubbe, J. C. M. Kistemaker, E. J. Smits and B. L. Feringa, Phys. Chem. Chem. Phys., 2016, 18, 26725–26735 RSC.
- Q. Zhang, D. H. Qu, H. Tian and B. L. Feringa, Matter, 2020, 3, 355–370 CrossRef.
- S. Krause and B. L. Feringa, Nat. Rev. Chem., 2020, 4, 550–562 CrossRef CAS.
- G. S. Kottas, L. I. Clarke, D. Horinek and J. Michl, Chem. Rev., 2005, 105, 1281–1376 CrossRef CAS PubMed.
- D. Roke, M. Sen, W. Danowski, S. J. Wezenberg and B. L. Feringa, J. Am. Chem. Soc., 2019, 141, 7622–7627 CrossRef CAS PubMed.
- G. Wald, Nature, 1968, 219, 800–807 CrossRef CAS PubMed.
- H. J. A. Dartnall, Vision Research, 1968, 8, 339–358 CrossRef CAS PubMed.
- A. Strambi, B. Durbeej, N. Ferré and M. Olivucci, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21322–21326 CrossRef CAS PubMed.
- R. S. H. Liu and G. S. Hammond, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11153–11158 CrossRef CAS PubMed.
- J. Léonard, I. Schapiro, J. Briand, S. Fusi, R. R. Paccani, M. Olivucci and S. Haacke, Chem.–Eur. J., 2012, 18, 15296–15304 CrossRef PubMed.
- M. Gueye, M. Manathunga, D. Agathangelou, Y. Orozco, M. Paolino, S. Fusi, S. Haacke, M. Olivucci and J. Leónard, Nat. Commun., 2018, 9, 1–8 CrossRef CAS PubMed.
- M. Gueye, M. Paolino, E. Gindensperger, S. Haacke, M. Olivucci and J. Léonard, Faraday Discuss., 2020, 221, 299–321 RSC.
- J. Briand, O. Bräm, J. Réhault, J. Léonard, A. Cannizzo, M. Chergui, V. Zanirato, M. Olivucci, J. Helbing and S. Haacke, Phys. Chem. Chem. Phys., 2010, 12, 3178–3187 RSC.
- A. Sinicropi, E. Martin, M. Ryazantsev, J. Helbing, J. Briand, D. Sharma, J. Léonard, S. Haacke, A. Cannizzo, M. Chergui, V. Zanirato, S. Fusi, F. Santoro, R. Basosi, N. Ferré and M. Olivucci, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17642–17647 CrossRef CAS PubMed.
- A. D. Dunkelberger, R. D. Kieda, J. Y. Shin, R. R. Paccani, S. Fusi, M. Olivucci and F. F. Crim, J. Phys. Chem. A, 2012, 116, 3527–3533 CrossRef CAS PubMed.
- G. Marchand, J. Eng, I. Schapiro, A. Valentini, L. M. Frutos, E. Pieri, M. Olivucci, J. Léonard and E. Gindensperger, J. Phys. Chem. Lett., 2015, 6, 599–604 CrossRef CAS PubMed.
- M. Filatov and M. Olivucci, J. Org. Chem., 2014, 79, 3587–3600 CrossRef CAS PubMed.
- I. Schapiro, M. Gueye, M. Paolino, S. Fusi, G. Marchand, S. Haacke, M. E. Martin, M. Huntress, V. P. Vysotskiy, V. Veryazov, J. Léonard and M. Olivucci, Photochem. Photobiol. Sci., 2019, 18, 2259–2269 CrossRef CAS PubMed.
- M. Paolino, T. Giovannini, M. Manathunga, L. Latterini, G. Zampini, R. Pierron, J. Léonard, S. Fusi, G. Giorgi, G. Giuliani, A. Cappelli, C. Cappelli and M. Olivucci, J. Phys. Chem. Lett., 2021, 12, 3875–3884 CrossRef CAS PubMed.
- M. Filatov, M. Paolino, S. K. Min and K. S. Kim, J. Phys. Chem. Lett., 2018, 9, 4995–5001 CrossRef CAS PubMed.
- M. Filatov, M. Paolino, S. K. Min and C. H. Choi, Chem. Commun., 2019, 55, 5247–5250 RSC.
- J. Wang, B. Oruganti and B. Durbeej, Phys. Chem. Chem. Phys., 2017, 19, 6952–6956 RSC.
- J. Wang and B. Durbeej, ChemistryOpen, 2018, 7, 583–589 CrossRef CAS PubMed.
- B. Oruganti, J. Wang and B. Durbeej, Org. Lett., 2017, 19, 4818–4821 CrossRef CAS PubMed.
- A. Nikiforov, J. A. Gamez, W. Thiel and M. Filatov, J. Phys. Chem. Lett., 2016, 7, 105–110 CrossRef CAS PubMed.
- W. Steinle and K. Ruck-Braun, Org. Lett., 2003, 5, 141–144 CrossRef CAS PubMed.
- S. Wiedbrauk and H. Dube, Tetrahedron Lett., 2015, 56, 4266–4274 CrossRef CAS.
- R. Wilcken, M. Schildhauer, F. Rott, L. A. Huber, M. Guentner, S. Thumser, K. Hoffmann, S. Oesterling, R. De Vivie-Riedle, E. Riedle and H. Dube, J. Am. Chem. Soc., 2018, 140, 5311–5318 CrossRef CAS PubMed.
- L. A. Huber, K. Hoffmann, S. Thumser, N. Böcher, P. Mayer and H. Dube, Angew. Chem., Int. Ed., 2017, 56, 14536–14539 CrossRef CAS PubMed.
- A. Gerwien, P. Mayer and H. Dube, J. Am. Chem. Soc., 2018, 140, 16442–16445 CrossRef CAS PubMed.
- A. Gerwien, M. Schildhauer, S. Thumser, P. Mayer and H. Dube, Nat. Commun., 2018, 9, 2510 CrossRef PubMed.
- A. Gerwien, P. Mayer and H. Dube, Nat. Commun., 2019, 10, 4449 CrossRef PubMed.
- J. M. Lehn, Chem.–Eur. J., 2006, 12, 5910–5915 CrossRef CAS.
- R. D. Astumian, Chem. Sci., 2017, 8, 840–845 RSC.
- M. K. J. ter Wiel, R. A. van Delden, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2005, 127, 14208–14222 CrossRef CAS PubMed.
- J. C. M. Kistemaker, S. F. Pizzolato, T. van Leeuwen, T. C. Pijper and B. L. Feringa, Chem.–Eur. J., 2016, 22, 13478–13487 CrossRef CAS PubMed.
- C. Fang, B. Oruganti and B. Durbeej, RSC Adv., 2014, 4, 10240–10251 RSC.
- L. Pfeifer, M. Scherübl, M. Fellert, W. Danowski, J. Cheng, J. Pol and B. L. Feringa, Chem. Sci., 2019, 10, 8768–8773 RSC.
- G. B. Boursalian, E. R. Nijboer, R. Dorel, L. Pfeifer, O. Markovitch, A. Blokhuis and B. L. Feringa, J. Am. Chem. Soc., 2020, 142, 16868–16876 CrossRef CAS PubMed.
- N. Ruangsupapichat, M. M. Pollard, S. R. Harutyunyan and B. L. Feringa, Nat. Chem., 2011, 3, 53–60 CrossRef CAS PubMed.
- J. A. Berrocal, L. Pfeifer, D. Heijnen and B. L. Feringa, J. Org. Chem., 2020, 85, 10670–10680 CrossRef CAS PubMed.
- J. C. M. Kistemaker, P. Štacko, D. Roke, A. T. Wolters, G. H. Heideman, M. C. Chang, P. van der Meulen, J. Visser, E. Otten and B. L. Feringa, J. Am. Chem. Soc., 2017, 139, 9650–9661 CrossRef CAS PubMed.
- K. Grill and H. Dube, J. Am. Chem. Soc., 2020, 142, 19300–19307 CrossRef CAS PubMed.
- K. Hoffmann, P. Mayer and H. Dube, Org. Biomol. Chem., 2019, 17, 1979–1983 RSC.
- E. Uhl, S. Thumser, P. Mayer and H. Dube, Angew. Chem., Int. Ed., 2018, 57, 11064–11068 CrossRef CAS PubMed.
- E. Uhl, P. Mayer and H. Dube, Angew. Chem., Int. Ed., 2020, 59, 5730–5737 CrossRef CAS PubMed.
- R. S. H. Liu and G. S. Hammond, Photochem. Photobiol. Sci., 2003, 2, 835–844 CrossRef CAS PubMed.
- L. Greb, A. Eichhöfer and J. M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 14345–14348 CrossRef CAS PubMed.
- Q. Li, G. Fuks, E. Moulin, M. Maaloum, M. Rawiso, I. Kulic, J. T. Foy and N. Giuseppone, Nat. Nanotechnol., 2015, 10, 161–165 CrossRef CAS PubMed.
- E. Yousif and R. Haddad, SpringerPlus, 2013, 2, 398 CrossRef PubMed.
- I. Yoon, J. Z. Li and Y. K. Shim, Clin. Endosc., 2013, 46, 7–23 CrossRef PubMed.
- I. M. Welleman, M. W. H. Hoorens, B. L. Feringa, H. H. Boersma and W. Szymanski, Chem. Sci., 2020, 11, 11672–11691 RSC.
- T. van Leeuwen, J. Pol, D. Roke, S. J. Wezenberg and B. L. Feringa, Org. Lett., 2017, 19, 1402–1405 CrossRef CAS PubMed.
- R. A. van Delden, N. Koumura, A. Schoevaars, A. Meetsma and B. L. Feringa, Org. Biomol. Chem., 2003, 1, 33–35 RSC.
- M. Albota, D. Beljonne, J. L. Brédas, J. E. Ehrlich, J. Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Röckel, M. Rumi, G. Subramaniam, W. W. Webb, X. L. Wu and C. Xu, Science, 1998, 281, 1653–1656 CrossRef CAS PubMed.
- D. Liu, V. Garcia-Lopez, R. S. Gunasekera, L. Greer Nilewski, L. B. Alemany, A. Aliyan, T. Jin, G. Wang, J. M. Tour and R. Pal, ACS Nano, 2019, 13, 6813–6823 CrossRef CAS PubMed.
- A. Vogel, J. Noack, G. Hüttman and G. Paltauf, Appl. Phys. B, 2005, 81, 1015–1047 CrossRef CAS.
- J. Vicario, A. Meetsma and B. L. Feringa, Chem. Commun., 2005, 5910–5912 RSC.
- D. Roke, B. L. Feringa and S. J. Wezenberg, Helv. Chim. Acta, 2019, 102, e1800221 CrossRef.
- D. Roke, C. Stuckhardt, W. Danowski, S. J. Wezenberg and B. L. Feringa, Angew. Chem., Int. Ed., 2018, 57, 10515–10519 CrossRef CAS PubMed.
- J. Wang and B. Durbeej, Comput. Theor. Chem., 2019, 1148, 27–32 CrossRef CAS.
- L. Pfeifer, N. V. Hoang, M. Scherübl, M. S. Pshenichnikov and B. L. Feringa, Sci. Adv., 2020, 6, eabb6165 CrossRef CAS PubMed.
- C. Y. Huang, A. Bonasera, L. Hristov, Y. Garmshausen, B. M. Schmidt, D. Jacquemin and S. Hecht, J. Am. Chem. Soc., 2017, 139, 15205–15211 CrossRef CAS PubMed.
- C. Petermayer and H. Dube, Acc. Chem. Res., 2018, 51, 1153–1163 CrossRef CAS PubMed.
- P. W. Sadler, J. Org. Chem., 1956, 21, 316–318 CrossRef CAS.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, 1st edn, 2010 Search PubMed.
- A. Cnossen, L. Hou, M. M. Pollard, P. V. Wesenhagen, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2012, 134, 17613–17619 CrossRef CAS PubMed.
- S. J. Wezenberg, K. Y. Chen and B. L. Feringa, Angew. Chem., Int. Ed., 2015, 54, 11457–11461 CrossRef CAS PubMed.
- D. Pijper, R. A. van Delden, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2005, 127, 17612–17613 CrossRef CAS PubMed.
- C. R. Hall, W. R. Browne, B. L. Feringa and S. R. Meech, Angew. Chem., Int. Ed., 2018, 57, 6203–6207 CrossRef CAS PubMed.
- J. Conyard, K. Addison, I. A. Heisler, A. Cnossen, W. R. Browne, B. L. Feringa and S. R. Meech, Nat. Chem., 2012, 4, 547–551 CrossRef CAS PubMed.
- D. R. S. Pooler, R. Pierron, S. Crespi, R. Costil, L. Pfeifer, J. Léonard, M. Olivucci and B. L. Feringa, Chem. Sci., 2021, 12, 7486–7497 RSC.
- N. Minezawa and M. S. Gordon, J. Phys. Chem. A, 2011, 115, 7901–7911 CrossRef CAS PubMed.
- J. Satiel, J. Am. Chem. Soc., 1967, 89, 1036–1037 CrossRef.
- M. Quick, A. L. Dobryakov, I. N. Ioffe, A. A. Granovsky, S. A. Kovalenko and N. P. Ernsting, J. Phys. Chem. Lett., 2016, 7, 4047–4052 CrossRef CAS PubMed.
- C. R. Hall, J. Conyard, I. A. Heisler, G. Jones, J. Frost, W. R. Browne, B. L. Feringa and S. R. Meech, J. Am. Chem. Soc., 2017, 139, 7408–7414 CrossRef CAS PubMed.
- S. Amirjalayer, A. Cnossen, W. R. Browne, B. L. Feringa, W. J. Buma and S. Woutersen, J. Phys. Chem. A, 2016, 120, 8606–8612 CrossRef CAS PubMed.
- P. Roy, A. S. Sardjan, W. Danowski, W. R. Browne, B. L. Feringa and S. R. Meech, J. Phys. Chem. A, 2021, 125, 1711–1719 CrossRef CAS PubMed.
- A. S. Sardjan, P. Roy, W. Danowski, G. Bressan, L. Nunes dos Santos Comprido, W. R. Browne, B. L. Feringa and S. R. Meech, ChemPhysChem, 2020, 21, 594–599 CrossRef CAS PubMed.
- A. Kazaryan, J. C. M. Kistemaker, L. V. Schäfer, W. R. Browne, B. L. Feringa and M. Filatov, J. Phys. Chem. A, 2010, 114, 5058–5067 CrossRef CAS PubMed.
- R. W. J. Zijlstra, P. T. van Duijnen, B. L. Feringa, T. Steffen, K. Duppen and D. A. Wiersma, J. Phys. Chem. A, 1997, 101, 9828–9836 CrossRef CAS.
- T. E. Wiley, A. Konar, N. A. Miller, K. G. Spears and R. J. Sension, J. Phys. Chem. A, 2018, 122, 7548–7558 CrossRef CAS PubMed.
- A. Kazaryan, Z. Lan, L. V. Schäfer, W. Thiel and M. Filatov, J. Chem. Theory Comput., 2011, 7, 2189–2199 CrossRef CAS PubMed.
- J. Léonard, J. Briand, S. Fusi, V. Zanirato, M. Olivucci and S. Haacke, New J. Phys., 2013, 15, 105022 CrossRef.
- R. Wilcken, L. Huber, K. Grill, M. Guentner, M. Schildhauer, S. Thumser, E. Riedle and H. Dube, Chem.–Eur. J., 2020, 26, 13507–13512 CrossRef CAS PubMed.
- B. Oruganti, J. Wang and B. Durbeej, ChemPhysChem, 2016, 17, 3399–3408 CrossRef CAS PubMed.
- N. Giuseppone and A. Walther, Out-of-Equilibrium (Supra)molecular Systems and Materials, Wiley, Hoboken, NJ, 1st edn, 2021 Search PubMed.
- D. Roke, S. J. Wezenberg and B. L. Feringa, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9423–9431 CrossRef CAS PubMed.
- M. M. Pollard, M. Klok, D. Pijper and B. L. Feringa, Adv. Funct. Mater., 2007, 17, 718–729 CrossRef CAS.
- M. K. J. ter Wiel, R. A. van Delden, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2003, 125, 15076–15086 CrossRef CAS PubMed.
- N. Koumura, E. M. Geertsema, M. B. van Gelder, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2002, 124, 5037–5051 CrossRef CAS PubMed.
- J. Bauer, L. Hou, J. C. M. Kistemaker and B. L. Feringa, J. Org. Chem., 2014, 79, 4446–4455 CrossRef CAS PubMed.
- M. M. Pollard, A. Meetsma and B. L. Feringa, Org. Biomol. Chem., 2008, 6, 507–512 RSC.
- L. A. Huber, S. Thumser, K. Grill, D. Voßiek, N. N. Bach, P. Mayer and H. Dube, Chem.–Eur. J., 2021, 27, 10758–10765 CrossRef CAS PubMed.
- J. Vicario, M. Walko, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 5127–5135 CrossRef CAS PubMed.
- A. Faulkner, T. van Leeuwen, B. L. Feringa and S. J. Wezenberg, J. Am. Chem. Soc., 2016, 138, 13597–13603 CrossRef CAS PubMed.
- J. C. M. Kistemaker, A. S. Lubbe, E. A. Bloemsma and B. L. Feringa, ChemPhysChem, 2016, 17, 1819–1822 CrossRef CAS PubMed.
- J. Chen, J. C. M. Kistemaker, J. Robertus and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 14924–14932 CrossRef CAS PubMed.
- E. M. Geertsema, S. J. van der Molen, M. Martens and B. L. Feringa, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16919–16924 CrossRef CAS PubMed.
- J. Wang, B. Oruganti and B. Durbeej, J. Org. Chem., 2021, 86, 5552–5559 CrossRef CAS PubMed.
- C. Villiers and M. Ephritikhine, Angew. Chem., Int. Ed., 1997, 36, 2380–2382 CrossRef CAS.
- J. M. Burns, T. Clark and C. M. Williams, J. Org. Chem., 2021, 86, 7515–7528 CrossRef CAS PubMed.
- J. E. McMurry and R. G. Dushin, J. Am. Chem. Soc., 1989, 111, 8928–8929 CrossRef CAS.
- R. Dorel and B. L. Feringa, Angew. Chem., Int. Ed., 2020, 59, 785–789 CrossRef CAS PubMed.
- W. Danowski, T. van Leeuwen, S. Abdolahzadeh, D. Roke, W. R. Browne, S. J. Wezenberg and B. L. Feringa, Nat. Nanotechnol., 2019, 14, 488–494 CrossRef CAS PubMed.
- Q. Li, J. T. Foy, J. R. Colard-Itté, A. Goujon, D. Dattler, G. Fuks, E. Moulin and N. Giuseppone, Tetrahedron, 2017, 73, 4874–4882 CrossRef CAS.
- A. Gerwien, T. Reinhardt, P. Mayer and H. Dube, Org. Lett., 2018, 20, 232–235 CrossRef CAS PubMed.
- M. K. J. ter Wiel, N. Koumura, R. A. van Delden, A. Meetsma, N. Harada and B. L. Feringa, Chirality, 2000, 12, 734–741 CrossRef CAS PubMed.
- T. M. Neubauer, T. van Leeuwen, D. Zhao, A. S. Lubbe, J. C. M. Kistemaker and B. L. Feringa, Org. Lett., 2014, 16, 4220–4223 CrossRef CAS PubMed.
- T. van Leeuwen, W. Danowski, E. Otten, S. J. Wezenberg and B. L. Feringa, J. Org. Chem., 2017, 82, 5027–5033 CrossRef CAS PubMed.
- T. van Leeuwen, J. Gan, J. C. M. Kistemaker, S. F. Pizzolato, M.-C. Chang and B. L. Feringa, Chem.–Eur. J., 2016, 22, 7054–7058 CrossRef CAS PubMed.
- T. van Leeuwen, A. S. Lubbe, P. Štacko, S. J. Wezenberg and B. L. Feringa, Nat. Rev. Chem., 2017, 1, 0096 CrossRef CAS.
- V. García-López, F. Chen, L. G. Nilewski, G. Duret, A. Aliyan, A. B. Kolomeisky, J. T. Robinson, G. Wang, R. Pal and J. M. Tour, Nature, 2017, 548, 567–572 CrossRef PubMed.
- R. A. van Delden, M. K. J. ter Wiel, M. M. Pollard, J. Vicario, N. Koumura and B. L. Feringa, Nature, 2005, 437, 1337–1340 CrossRef CAS PubMed.
- R. Eelkema, M. M. Pollard, J. Vicario, N. Katsonis, B. S. Ramon, C. W. M. Bastiaansen, D. J. Broer and B. L. Feringa, Nature, 2006, 440, 163 CrossRef CAS PubMed.
- P. Štacko, J. C. M. Kistemaker and B. L. Feringa, Chem.–Eur. J., 2017, 23, 6643–6653 CrossRef PubMed.
Footnotes |
| † The word “winding” was used intentionally here to illustrate the progressive and continuous nature of the rotation, likening it to a macroscopic object such as a winch on the reel of a fishing rod. |
| ‡ Barton–Kellogg couplings involve the synthesis of relatively unstable diazo and thioketone reagents, which can be air, light and moisture sensitive.157 |
| This journal is © The Royal Society of Chemistry 2021 |