
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePicomolar FKBP inhibitors enabled by a single water-displacing methyl group in bicyclic [4.3.1] aza-amides†
Jürgen M.
Kolos‡
 ab,
Sebastian
Pomplun‡§
b,
Sascha
Jung
c,
Benedikt
Rie߶
b,
Patrick L.
Purder
a,
Andreas M.
Voll
a,
Stephanie
Merz
a,
Monika
Gnatzy
a,
Thomas M.
Geiger
a,
Ingrid
Quist-Løkken
def,
Jerome
Jatzlau
g,
Petra
Knaus
g,
Toril
Holien
def,
Andreas
Bracher
h,
Christian
Meyners
a,
Paul
Czodrowski
c,
Vera
Krewald
a and
Felix
Hausch
*a
ab,
Sebastian
Pomplun‡§
b,
Sascha
Jung
c,
Benedikt
Rie߶
b,
Patrick L.
Purder
a,
Andreas M.
Voll
a,
Stephanie
Merz
a,
Monika
Gnatzy
a,
Thomas M.
Geiger
a,
Ingrid
Quist-Løkken
def,
Jerome
Jatzlau
g,
Petra
Knaus
g,
Toril
Holien
def,
Andreas
Bracher
h,
Christian
Meyners
a,
Paul
Czodrowski
c,
Vera
Krewald
a and
Felix
Hausch
*a
aDepartment of Chemistry, Technical University of Darmstadt, Alarich-Weiss-Straße 4, 64293 Darmstadt, Germany. E-mail: felix.hausch@tu-darmstadt.de
bMax Planck Institute of Psychiatry, Kraepelinstr. 2-10, 80804 München, Germany
cTechnische Universität Dortmund, Fakultät für Chemie und Chemische Biologie, Otto-Hahn-Straße 6, 44227 Dortmund, Germany
dDepartment of Clinical and Molecular Medicine, Norwegian University of Science and Technology, 7491 Trondheim, Norway
eDepartment of Immunology and Transfusion Medicine, St. Olav's University Hospital, 7030 Trondheim, Norway
fDepartment of Hematology, St. Olav's University Hospital, 7030 Trondheim, Norway
gInstitute for Chemistry and Biochemistry, Freie Universität Berlin, 14195 Berlin, Germany
hResearch Department Cellular Biochemistry, Max Planck Institute of Biochemistry, Am Klopferspitz 18, 82152 Planegg, Germany
First published on 3rd November 2021
Abstract
Methyl groups can have profound effects in drug discovery but the underlying mechanisms are diverse and incompletely understood. Here we report the stereospecific effect of a single, solvent-exposed methyl group in bicyclic [4.3.1] aza-amides, robustly leading to a 2 to 10-fold increase in binding affinity for FK506-binding proteins (FKBPs). This resulted in the most potent and efficient FKBP ligands known to date. By a combination of co-crystal structures, isothermal titration calorimetry (ITC), density-functional theory (DFT), and 3D reference interaction site model (3D-RISM) calculations we elucidated the origin of the observed affinity boost, which was purely entropically driven and relied on the displacement of a water molecule at the protein–ligand–bulk solvent interface. The best compounds potently occupied FKBPs in cells and enhanced bone morphogenic protein (BMP) signaling. Our results show how subtle manipulation of the solvent network can be used to design atom-efficient ligands for difficult, solvent-exposed binding pockets.
Introduction
Replacement of a hydrogen by a methyl group is the smallest chemical modification in drug design. Although minimal in size, this perturbation can have profound effects on drug-like properties.1–4 In favorable cases, the addition of a methyl group can enhance affinity to the target protein ten-fold and in extreme cases – also known as magic methyl effect – by a factor >100.2,3 The possibilities to utilize strategically positioned methyl groups have recently been substantially expanded by the development of sophisticated late-stage methylation reactions.4,5 Mechanistically, the methyl-induced affinity gain can be achieved by filling a complementary small hydrophobic pocket (Fig. 1A) or by stabilizing an otherwise unfavorable active conformation (Fig. 1B). Unfortunately, these options are not always available in drug design projects, especially for shallow, solvent-exposed binding pockets as exemplified for FK506-binding proteins (FKBPs). FKBPs belong to the immunophilin family, possess cis–trans peptidyl-prolyl isomerase (PPIase) activity and are potential drug targets for several human diseases.6,7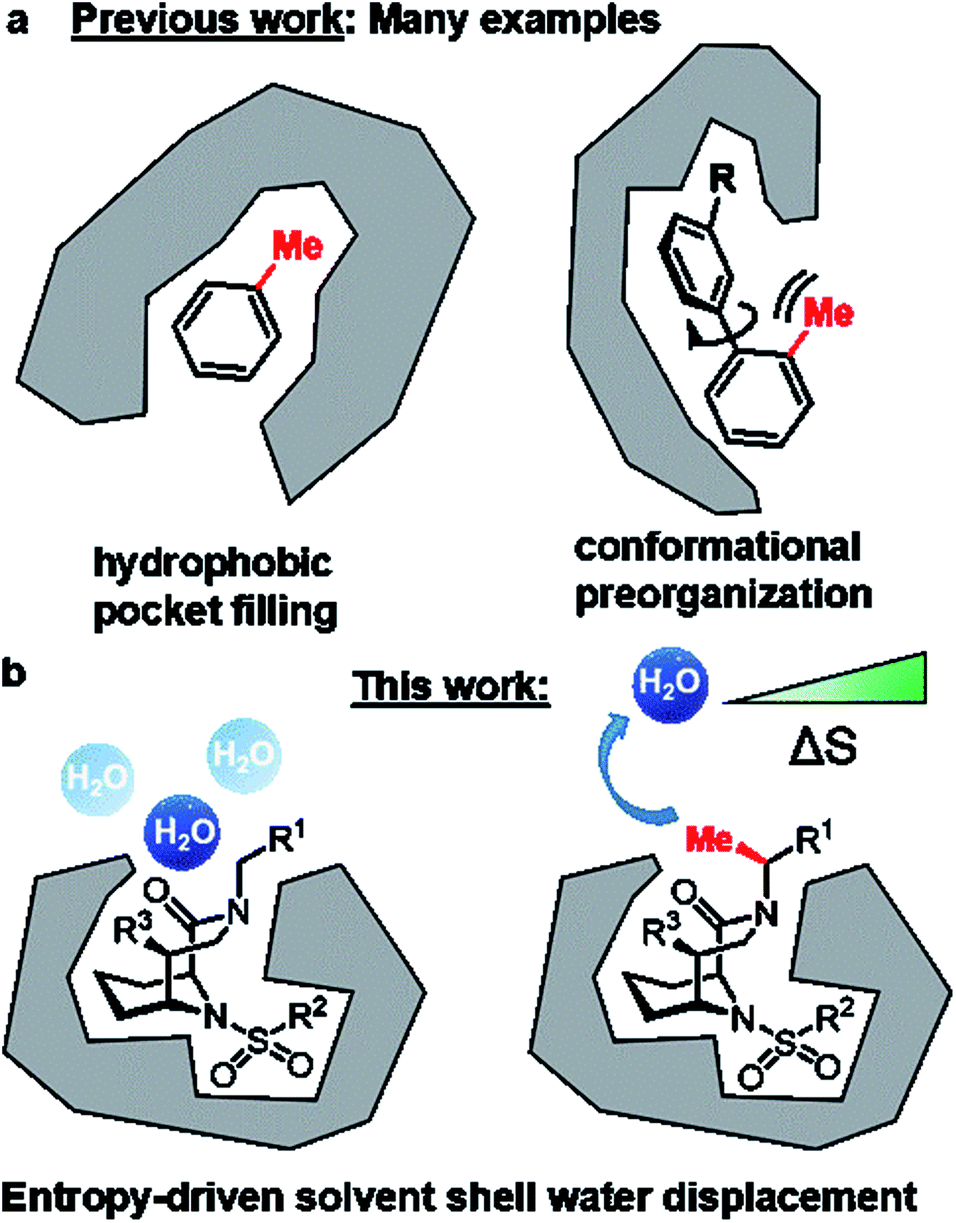 | ||
| Fig. 1 Different roles for methyl groups in ligand–protein interactions and in rational drug design. (A) Established affinity enhancements by hydrogen/methyl replacements. (B) The bicyclic [4.3.1] aza-amide scaffold allows the precise positioning of a methyl group in the binding site of FK506-binding proteins to displace an unfavorable, solvent-exposed water molecule. | ||
FKBP12 represses BMP-signaling8 and drug-like FKBP12 inhibitors are potential treatments for pulmonary arterial hypertension, hereditary hemorrhagic telangiectasia, wound healing,9 and acute kidney injury.10 Moreover, FKBP51 has emerged as a promising target for depression, obesity, and chronic pain.6,11–14 Microbial or parasitic FKBPs (sometimes also called Macrophage infectivity potentiators, Mips) are important for replication of the pathogens and FKBP/Mip inhibitors in turn have anti-infective potential.15 Finally, FKBPs are the key enabling adaptors for the clinically approved immunosuppressants FK506 and rapamycin as well as for several recently identified molecular glues.16–22 Previously, we developed bicyclic [4.3.1] sulfonamides,23–27 which mimic the core of the natural products FK506 and rapamycin, retain the anti-infective23 and BMP-stimulating properties6,10 of these natural products, but lack their immunosuppressive properties. However, the shallow FKBP binding site generally makes the development of ligands with drug-like properties challenging.7,15,28 Here, we exploit the defined rigid architecture of the bicyclic scaffold to strategically install a solvent exposed chiral methyl group that displaces a conserved water molecule, robustly leading to a boost in binding affinity to FK506-binding proteins (FKBPs).
Results
We started our ligand optimization with the [4.3.1] bicyclic scaffold, which efficiently locked the active conformation necessary for binding.23–26 The remaining protein surface as well as appending moieties on the ligands are all highly solvent-exposed, without any promising features such as nearby hydrophobic cavities.27 However, upon analysis of nine high resolution co-crystal structures of FKBP51 with bicyclic ligands we identified several reoccurring water molecules (Fig. S1†), including a water close to the R1 or R2 substituent that was conserved in all or most available co-crystal structures (Fig. 2A and S2†). A computational assessment by 3D-RISM calculations confirmed these water sites (distance to crystallographic water ∼1 Å, Fig. 3B and S4–S7†). A water in proximity of the R2 substituent was found to be energetically favorable (ΔGlocal = −11.79 kJ mol−1, Table S1, Fig. S2 & S7†) and therefore not considered for displacement. In contrast, the water close to the R1 substituent was predicted to be energetically highly unfavorable (local free energy ΔGlocal = +28.18 kJ mol−1 for 1, +26.03 kJ mol−1 for 22). The predicated interaction energy between this water site and the receptor (ΔGinteract = +2.77 kJ mol−1 for 1, +3.22 kJ mol−1 for 22) was considerably low, suggesting a stabilization of the ligand–FKBP complex upon displacement of this water molecule. The strongly preserved occupancy of an energetically unfavorable water site, which is highly solvent-exposed and has only minimal contact with the protein, is highly unusual.29,30 We thus set out to explore the role of this water molecule in more detail.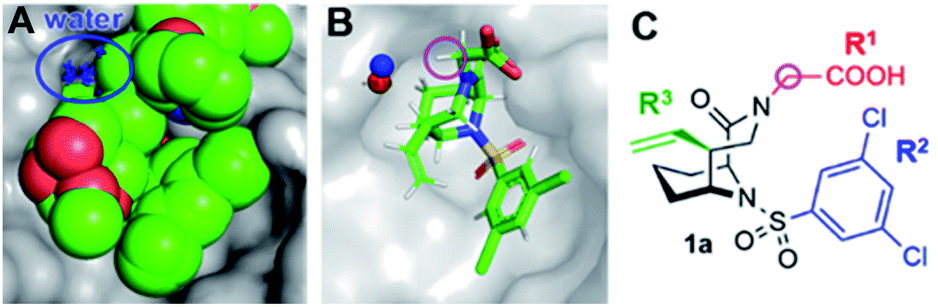 | ||
| Fig. 2 Overview of bicycles in the binding pocket of FKBP51. (A) Superposition of nine cocrystal structures of bicyclic [4.3.1] aza-amide ligands (green spheres) in complex with FKBP51 (gray surface from PDB-ID 5OBK). The position of the conserved water molecule is highlighted in blue. (B) Cocrystal structures of 1 (PDB-ID: 7APT) in complex with FKBP51, with the conserved water molecule highlighted as blue spheres. The water site predicted via 3D-RISM calculations is superimposed as red sphere. (C) Chemical structure of the representative bicyclic [4.3.1] aza-amide 1. The C-α-position is highlighted by a pink circle. | ||
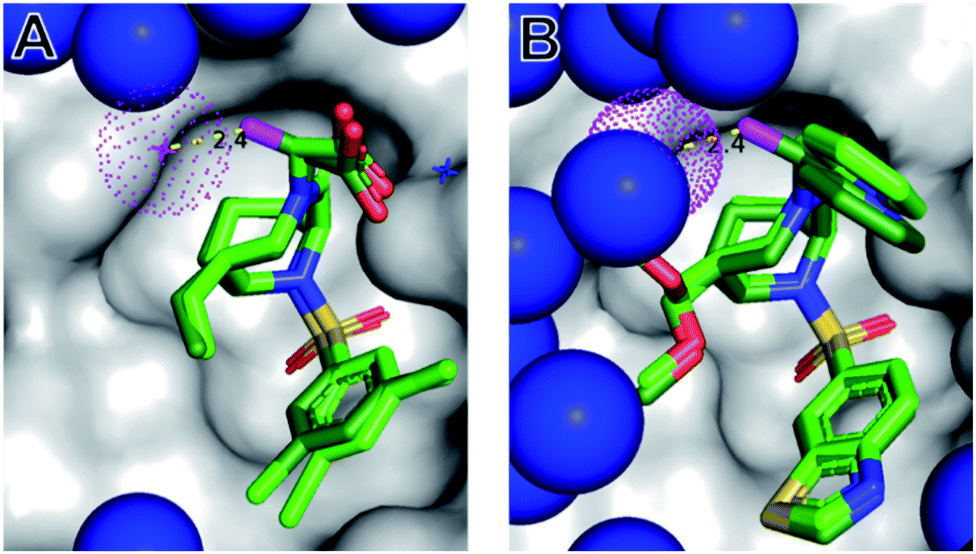 | ||
| Fig. 3 The (S)-αMe displaces a conserved water molecule from the protein surface. Cocrystal structures of 1(S)-Me (A, PDB-ID: 7APS) and 22(S)-Me (B, 7APW), each shown as dark green sticks, in complex with FKBP51 (grey surface). The water molecules are indicated as blue spheres, with the conserved water close to the α-position highlighted in red. The binding modes of the corresponding parent compounds 1 (from 7APT) and 22 (from 7APQ) derived from the superimposed structures are shown as light green sticks. Nitrogens, oxygens, sulfur, and chlorines are depicted in blue, red, yellow and green, respectively. The key methyl in the α-position of 1(S)-Me and 22(S)-Me is highlighted in magenta. | ||
To explore possible extension vectors of the bicyclic [4.3.1] aza-amide motif, we focused on the C-α position of the R1 substituent (highlighted in pink in Fig. 2B & C), which is in proximity to the conserved unfavorable water site. We decided to start with an additional methyl as the smallest perturbation. Based on our structural analysis, an (S)-methyl group would most likely displace the water molecule of interest. The synthesis of R- and S-α-methyl analogs of the known compounds 1 (R1 = CH2COOH, Fig. 2) and 2 (R1 = C2H4OH, Fig. S11†)27 commenced with the readily available amino acid derivatives (R)- or (S)-2-amino-1-propanol 3, which were sequentially benzyl- and nosyl-protected (Scheme 1). Allylation followed by metathesis with Grubbs 1st generation catalyst gave (R)-5 and (S)-5, which were nosyl-deprotected, coupled with (S)-6-oxo-2-piperidinecarboxylic acid and Boc-protected to furnish amides (R)-6/(S)-6. Reduction with DIBAL-H and treatment with HF-pyridine gave the bicyclic [4.3.1] aza-amide intermediates 7(R)-Me/7(S)-Me. Reaction with the respective sulfonyl chlorides gave sulfonamides 8(R)-Me/8(S)-Me and 10(R)-Me/10(S)-Me, which after treatment with boron trichloride yielded alcohols 9(R)-Me/9(S)-Me and 11(R)-Me/11(S)-Me, respectively. The latter two were further oxidized by Jones oxidation to furnish carboxylic acids 1(R)-Me/1(S)-Me. When testing the binding affinity of the resulting bicycles to FKBP12, 12.6, 51 and 52 via a competitive FP-assay (Table S3†), we were delighted to see that the introduction of a methyl group in S-configuration (1(S)-Me) enhanced affinity for FKBP12 and especially for FKBP51 compared to 1 (Table 1). A similar trend was also observed for the alcohols 2 and 11(S)-Me. Here, the R-configured methyl group in 11(R)-Me dramatically compromised affinity, similar to the alcohols 9(R)-Me/9(S)-Me.
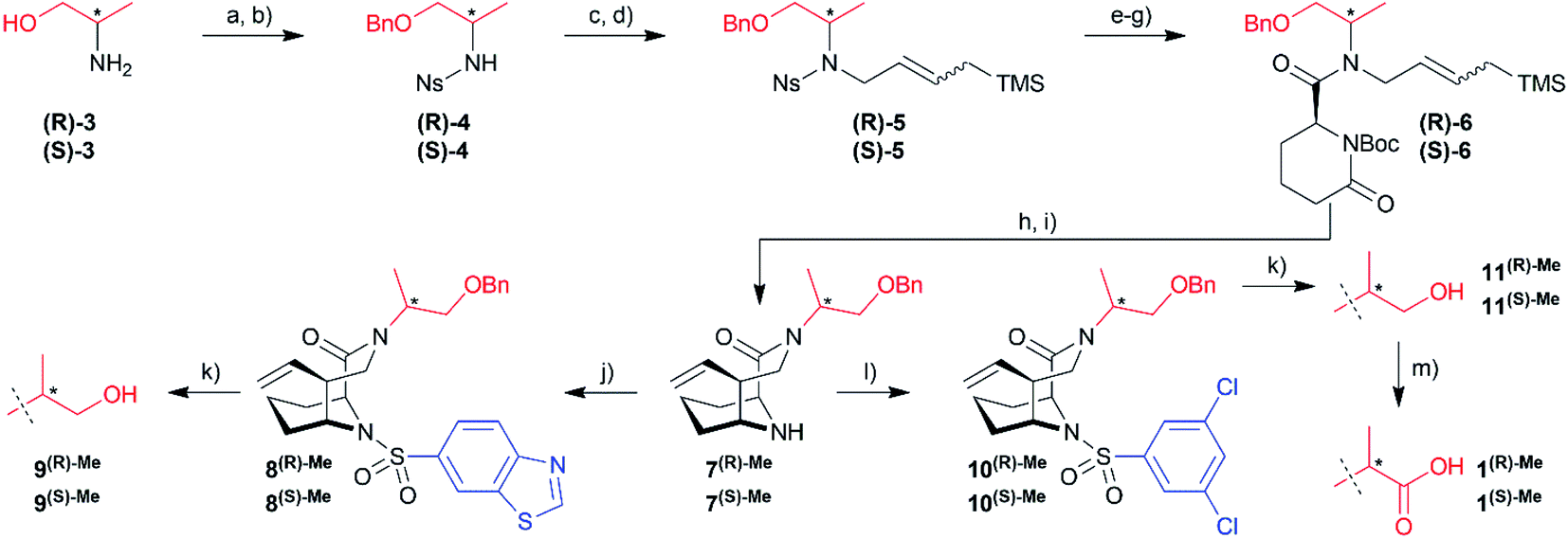 | ||
| Scheme 1 Reagent and conditions: (a) NaH, Bn-Cl, THF, reflux, 3 h; (b) Ns-Cl, DIPEA, DCM, 1 h, (R)-4: 62%, (S)-4: 66% (over 2 steps); (c) allyl bromide, K2CO3, DMF, 2 h; (d) allyltrimethylsilane, Grubbs II, p-benzoquinone, DCM, reflux, 6 h, (R)-5: 53%, (S)-5: 32% (over 2 steps); (e) thiophenol, K2CO3, DMF, rt, 16 h; (f) (S)-6-oxopiperidine-2-carboxylic acid, HATU, rt, DMF, 2 h; (g) Boc2O, DIPEA, DMAP, DCM, 48 h, (R)-6: 61%, (S)-6: 48% (over 3 steps); (h) DIBAL, THF, −78 °C, 15 min; (i) HF-pyridine, DCM, −78 °C, 1 h, 7(R)-Me: 69%, 7(S)-Me: 63% (over 2 steps); (j) benzo[d]thiazole-6-sulfonyl chloride, DIPEA, DMAP, DCM, rt, 16 h, 8(R)-Me: 42%, 8(S)-Me: 33%; (k) BCl3*SMe2, DCM, rt, 16 h, 9(R)-Me: 70%, 9(S)-Me: 58%, 11(R)-Me: 76%, 11(S)-Me: 47%; (l) 3,5-dichlorobenzene-1-sulfonyl chloride, DIPEA, DMAP, DCM, rt, 16 h, 10(R)-Me: 46%, 10(S)-Me: 47%; (m) Jones-reagent, acetone, rt, 5 h, 1(R)-Me: 35%, 1(S)-Me: 56%. | ||
| No. | FKBP 12 [nM] | FKBP 51 [nM] | R1 | R2 | R3 |
|---|---|---|---|---|---|
| a DCB = 3,5-dichlorobenzene. b BTZ = 6-benzothiazole. Values for FKBP52 are similiar to FKBP51 and shown in Table S3. 22(R)-Me and 22(S)-Me were synthesized via a different route (Scheme S1). | |||||
| FK506 | 0.55 ± 0.08 | 405 ± 81 | |||
| 1(R)-Me | 48 | 123 | CH((R)-CH3)COOH | DCBa | Vinyl |
| 1 | 33 | 172 | CH2COOH | ||
| 1(S)-Me | 13 | 22 | CH((S)-CH3)COOH | ||
| 9(R)-Me | >5000 | >5000 | CH((R)-CH3)CH2OH | BTZb | Vinyl |
| 9(S)-Me | 21 | 110 | CH((S)-CH3)CH2OH | ||
| 11(R)-Me | >5000 | >5000 | CH((R)-CH3)CH2OH | DCBa | Vinyl |
| 11(S)-Me | 3.5 | 107 | CH((S)-CH3)CH2OH | ||
| 17(R)-Me | 240 | 4546 | CH((R)-CH3)Py | BTZb | Vinyl |
| 17 | 7.5 | 294 | CH2Py | ||
| 17(S)-Me | 2.2 | 33 | CH((S)-CH3)Py | ||
| 18 | 0.52 | 33 | CH2Py | DCBa | CH2–OH |
| 18(S)-Me | 0.29 | 2.6 | CH((S)-CH3)Py | ||
| 19 | 0.65 | 12 | CH2Py | DCBa | CH2–OMe |
| 19(S)-Me | 0.33 | 2.9 | CH((S)-CH3)Py | ||
| 20 | 6.5 | 410 | CH2Py | BTZb | Ethyl |
| 20(S)-Me | 1.9 | 27 | CH((S)-CH3)Py | ||
| 21 | 5.5 | 283 | CH2Py | BTZb | CH2–OH |
| 21(S)-Me | 1.5 | 27.2 | CH((S)-CH3)Py | ||
| 22(R)-Me | 52 | 696 | CH((R)-CH3)Py | BTZb | CH2–OMe |
| 22 | 2.5 | 104 | CH2Py | ||
| 22(S)-Me | 2.2 | 32 | CH((S)-CH3)Py | ||
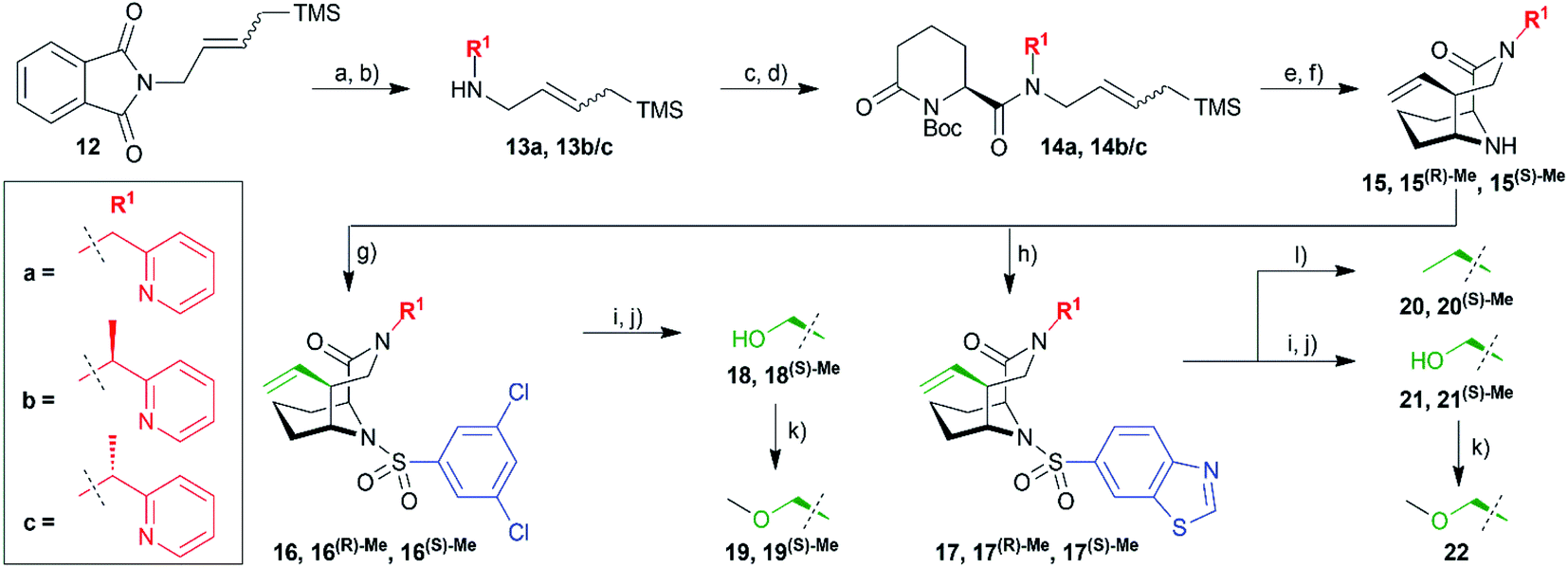 | ||
| Scheme 2 Reagent and conditions: (a) hydrazine, MeOH, 70 °C, 24 h; (b) 13a: pyridine-2-carbaldehyde, NaBH4, EtOH, rt, 4 h, 76% (over 2 steps), 13b/c: 1 (pyridin-2-yl)ethan-1-one, TTIP, NaBH4, EtOH, rt, 4 h, 62% (over 2 steps), (c) (S)-6-oxopiperidine-2-carboxylic acid, HATU, rt, DMF, 2 h; (d) Boc2O, DIPEA, DMAP, DCM, 48 h, 14a: 60%, 14b/c: 50%, (all yields over 2 steps); (e) DIBAL, THF, −78 °C, 15 min; (f) HF-pyridine, DCM, −78 °C, 1 h, 15: 39%, separation of diastereomers: 15(R)-Me: 39%, 15(S)-Me: 34% (all yields over 2 steps); (g) 3,5-dichlorobenzene-1-sulfonyl chloride, DIPEA, MeCN, rt, 24 h, 16: 66%, 16(R)-Me: 42%, 16(S)-Me: 42%; (h) benzo[d]thiazole-6-sulfonyl chloride, DIPEA, MeCN, rt, 24 h, 17: 29%, 17(R)-Me: 43%, 17(S)-Me: 43%; (i) OsO4, NaIO4, 2,6-lutidine, dioxane/H2O, rt, 20 h; (j) NaBH4, EtOH, 1 h, rt, 18: 47%, 18(S)-Me: 58%, 21: 27%, 21(S)-Me: 70% (all yields over 2 steps); (k) MeI, NaH, rt, 1 h, 19: 89%, 19(S)-Me: 92%, 22: 89%; (l) H2, Pd/C, 1 h, 20: quant., 20(S)-Me: quant. | ||
Hydrazinolysis of 12 (ref. 27) and reductive amination afforded the secondary amines 13a and a racemic mixture of 13b/c, which was coupled to commercially available (S)-6-oxo-2-piperidinecarboxylic acid followed by Boc-protection (Scheme 2). Reduction with DIBAL-H followed by HF-mediated N-acyliminium cyclization yielded the bicyclic [4.3.1] aza-amide building blocks 15 in good overall yields of 10–19% in 8 steps. At this stage, it was possible to separate the two diastereomers 15(R)-Me and 15(S)-Me by column chromatography. Reaction with the respective sulfonyl chlorides gave the sulfonamides 16/16(R)-Me/16(S)-Me and 17/17(R)-Me/17(S)-Me ready for testing. Lemieux–Johnson oxidation followed by reduction with sodium borohydride furnished alcohols 18/18(S)-Me and 21/21(S)-Me. The former three were methylated with methyl iodide yielding methyl ethers 19/19(S)-Me and 22. Reduction of the vinyl group with palladium-catalyzed hydrogenation gave compounds 20/20(S)-Me.
The obtained α-methylated sulfonamides as well as the respective parent analogs were tested for binding to three human FKBPs (FKBP12, FKBP51, FKBP52) by a fluorescence polarization assay (Tables 1 and S3†). We found that binding affinities were consistently stronger for the (S)-Me diastereomers compared to the parent analogs (R1 = CH2R). Conversely, the (R)-Me diastereomers displayed reduced affinities in most cases, compared to the (S)-Me and to the unsubstituted parent compounds. This trend was consistent for all tested FKBPs. Compounds 18(S)-Me and 19(S)-Me are picomolar FKBP12 ligands and low nanomolar ligands for FKBP51 and FKBP52 (up to 50-fold better than the prototypical ligand FK506), representing the most potent and ligand-efficient FKBP ligands known to date. They will be provided to the scientific community as donated chemical probes via the Structural Genomics Consortium at the Goethe University Frankfurt (SGC Frankfurt) to enable researchers to pharmacologically probe the role of FKBPs in well-defined manner (https://www.sgc-ffm.uni-frankfurt.de/).
In order to explore the structural basis for the enhanced binding affinity induced by the (S)-Me group, we co-crystallized the (S)-diastereomers 1(S)-Me and 22(S)-Me as well as the respective parent compounds 1 and 22 with the FK1 domain of FKBP51 (Fig. 3). All crystal structures had atomic resolution (1: 1.09 Å, 22(S)-Me: 0.89 Å) allowing a detailed analysis of the binding mode and solvation shell. A superposition revealed only minimal differences between the (S)-diastereomers and the parent compounds, with a slight rearrangement of the R1 group as the only apparent difference. Both conformations enable a hydrogen bond between the pyridine nitrogen or the carboxy group to Tyr113, which is 0.1 Å shorter for the high-affinity (S)-Me analogs compared to the non-α-Me derivatives. As expected, the key additional methyl group is largely solvent-exposed and makes only a single van-der-Waals interaction (3.2 Å) with the backbone carbonyl of Gln85. Most importantly, the S-methyl indeed displaces the conserved water molecule observed before. This was confirmed by 3D-RISM calculations, which also indicated that no new unfavorable water site appeared (Fig. S2 & S5†). Since methyl groups can profoundly affect binding energies by conformational effects, we investigated the influence of the α-methyl group on intrinsic conformational preferences. The rotational barriers for the NCCN and CNNC bonds were calculated by DFT, using compounds 22, 22(S)-Me and 22(R)-Me as a model system (Fig. 4).
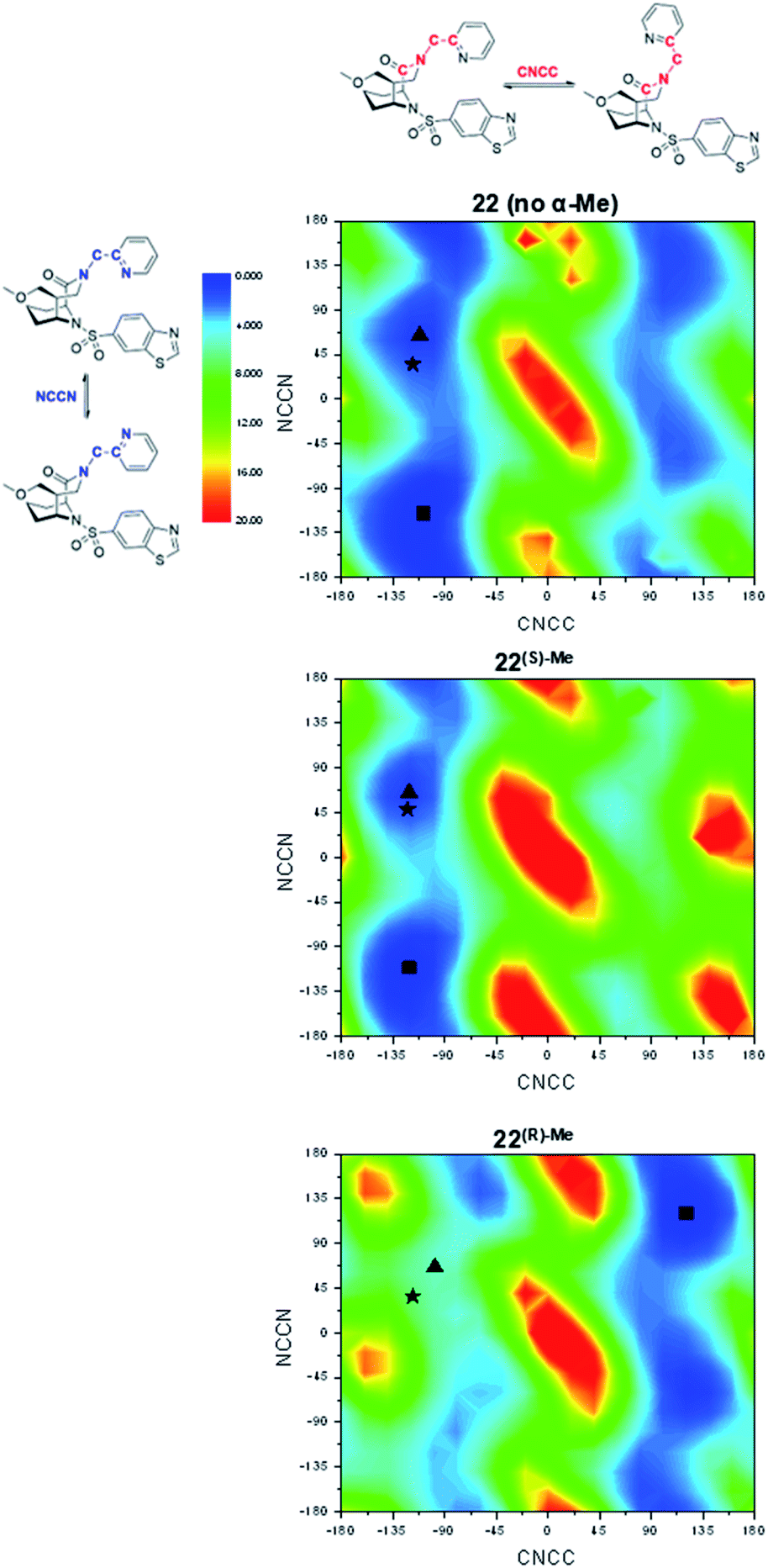 | ||
| Fig. 4 Conformational preorganization explains the affinity loss for the (R)-α-Me analogs. DFT evaluation of conformations of the CNCC and NCCN dihedral angles next to the α-Me carbon for compounds 22, 22(S)-Me and 22(R)-Me [grid of 18 × 18]. Energy maps are colored according to energy (blue: low energy; red: high energy; energies in kcal mol−1). The black asterisk indicates the conformation found in the respective crystal structure (for 22(R)-Me: adopted from 22), a black triangle indicates the nearest local minimum, a black square indicates the global minimum. | ||
Two areas of low energy were identified for the CNCC dihedral angle (approx. −100° and 100°, corresponding to the pyridyl group pointing towards or away from Tyr113 when bound to FKBPs). These two orientations are strongly preferred by the S-α-Me and R-α-Me group, respectively. The pyridyl ring itself can rotate more freely, with slight preferences for the NCCN dihedral angle at ≈70° and −112°, corresponding to the pyridyl nitrogen point to and away from the Tyr113-ε-OH. In the cocrystal structures, 22 and 22(S)-Me adopt conformations close to the local minima around CNCC ≈100° and NCCN ≈70° respectively.
To refine the energy landscape, thermodynamic corrections were computed for selected minima and for the conformations observed in the crystal structure (Table S6†). For 22 and 22(S)-Me, the crystal structure conformations were found to be isoenergetic to their respective local minimum (ΔΔG = 0.2 kcal mol−1) and slightly disfavored compared to the global minimum (ΔΔG = 2.8–2.9 kcal mol−1). While the energetic penalties between the global minima and the active conformations are similar for 22 and 22(S)-Me, the energy landscape of 22 indicates a higher degree of conformational freedom with two approximately equally accessible valleys at CNCC dihedral angles around −110° and +90° approximately equally accessible. This effect may account for a difference in binding affinity of at most a factor of two. Therefore, conformational preorganization cannot fully explain the enhanced affinity of (S)-Me-compounds over their respective non-methylated analogs. For 22(R)-Me, however, the predicted key hydrogen bond-enabling conformation is ΔΔG = 5.7 kcal mol−1 less favorable compared to the global minimum, explaining the reduced affinities of the R-isomers.
To explore the thermodynamic signature for the enhanced affinity of α-methyl-containing bicyclic [4.3.1] aza amides we used isothermal titration calorimetry (ITC)13,31 (Table S4†). The affinities measured with ITC were fully consistent with the FP-assay data (Fig. S13†). Overall, the bicyclic [4.3.1] aza-amide FKBP ligands were highly enthalpy-driven, as observed before.12,23 Strikingly, the comparison of the (S)-α-Me derivatives with their non-methylated analogs revealed that the affinity gain imparted by the additional methyl group was based exclusively on a gain in entropy (Fig. 5 & S14†), without any entropy-enthalpy compensation. This is consistent with enhanced degrees of freedom for the solvent shell of the (S)-α-Me derivatives-FKBP51 complexes compared to their non-methylated counterparts.
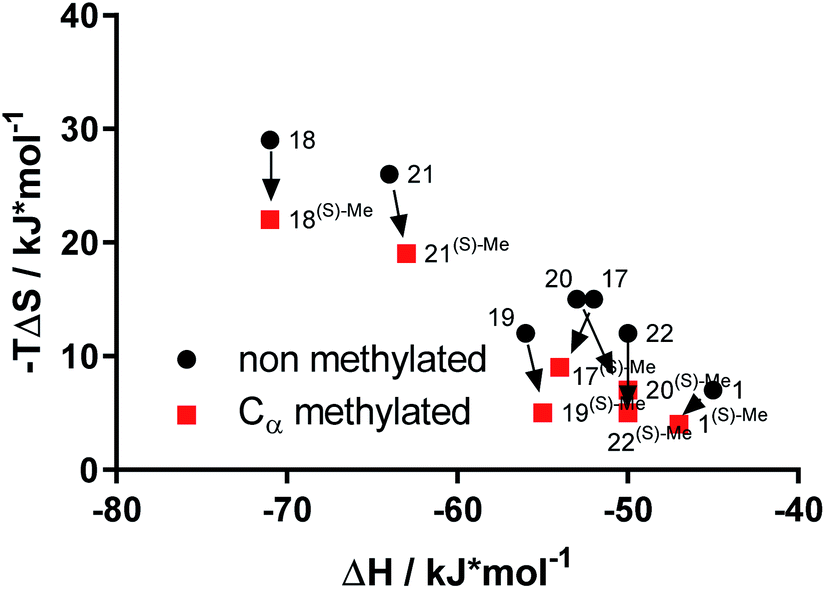 | ||
| Fig. 5 The affinity boost of the (S)-Me is purely driven by entropy. Thermodynamic signature of bicyclic [4.3.1] aza-amides with and without α-methyl group for binding to FKBP51, as determined by ITC. | ||
Finally, we tested if the high ligand efficiency of the α-Me derivatives translated into cellular potencies. We therefore performed a NanoBRET assay32 for the best compound 19(S)-Me, which competitively bound to FKBP12 in HEK cells with an apparent Kappi of 2.9 nM (Fig. 6A). A competitive inhibition was likewise observed for compounds 18, 18(S)-Me, 19, and 19(S)-Me for FKBP51 (Fig. S18†), which also reflected the substantially enhanced potency of the α-methyl derivatives compared to their corresponding non-methylated analogs. For FKBP12, all tested compounds bound substantially better to intracellular FBKP12 compared to the prototypic FKBP ligand FK506 (Fig. 6B).
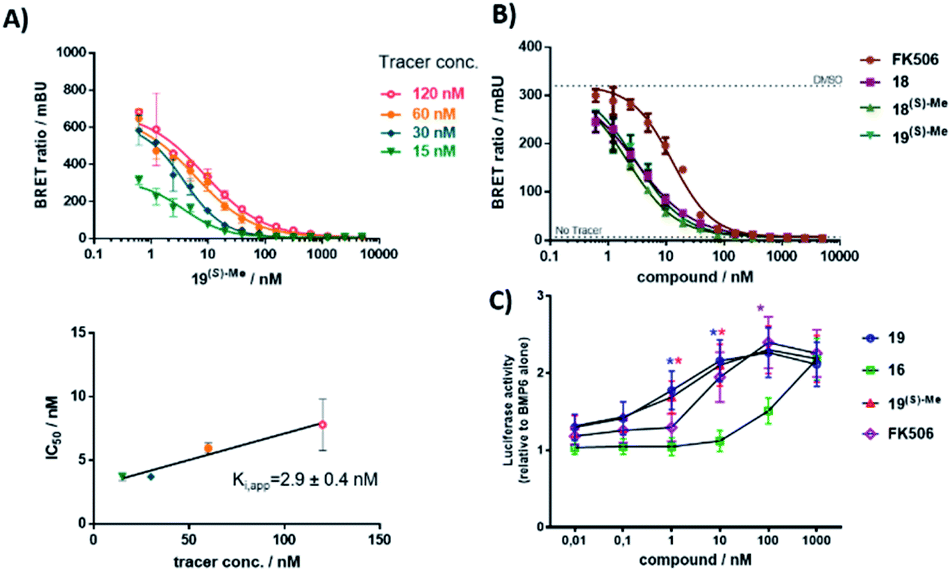 | ||
| Fig. 6 (S)-Me-substituted [4.3.1] bicycles potently block FKBPs in cells. (A) Intracellular FKBP12-NLuc engagement of compound 19(S)-Me determined by competitive NanoBRET assay. HEK293T cells stably expressing FKBP12-NLuc constructs were treated with increasing concentrations of 19(S)-Me in the presence of varying concentrations of NanoBRET tracer as shown in the upper panel. Each curve was fitted to determine an IC50, which was plotted in the lower panel in dependence of the tracer concentration to determine the Kappi by Cheng–Prusoff analysis. (B) Competitive NanoBRET assay for FK506 and three bicyclic compounds. NanoBRET experiments were performed in three independent cellular assays for all samples. (C) Dose responses for FK506 and three bicyclic compounds determined in an INA-6 BRE-luc reporter assay. All cells were treated with BMP-6 (7.5 ng mL−1) and indicated compounds for 18 hours. The measured luciferase activities were normalized to BMP-6 alone. Shown are the averages and standard error of the mean (SEM) of n = 4 biological replicates. A two-way ANOVA with Tukey's multiple comparison test and Geisser–Greenhouse correction was performed. The asterisks indicate statistical significance (*p < 0.05) for compound 16vs.19 (blue asterisks) or 19(S)-Me (red asterisks) at 1 nM and 10 nM, and compound 16vs. FK506 (pink asterisk) at 100 nM. The other comparisons were not significant (p > 0.05). | ||
FKBP12 has been shown to repress BMP-signaling by binding to receptors of the ALK family and inhibition of FKBPs has been suggested as a potential treatment option for ALK-associated diseases.8–10,33 To accesses the potential of the α-Me [4.3.1] bicycles for these indications, we used a reporter assay based on the INA-6 myeloma cell line, INA-6 BRE-luc, that responds well to treatment with bone morphogenetic proteins (BMPs).34 The BRE-luc construct employed has a BMP-responsive element derived from a mouse Id1 promoter, which is stimulated by BMP-activated SMADs.35 All compounds dose-dependently potentiated BMP-6-induced SMAD signaling (Fig. 6C), with 19, 19(S-Me) and FK506 being significantly more potent than compound 16, consistent with the reduced biochemical affinity of the latter (Table S3†).
Conclusion
Taken together, we have identified a solvent-exposed methyl group that robustly enhances affinity by a purely entropic mechanism. Methyl groups have been repeatedly observed to profoundly boost affinity of protein ligands, e.g., by filling buried hydrophobic cavities or by conformational preorganization. Neither of these mechanisms seem to be the major driver of affinity enhancement in our case. Instead, we identified displacement of a conserved, energetically unfavorable and unusually solvent-exposed water molecule as the major origin for the enhanced affinity. Water displacement or replacement strategies have been used for ligand optimization before36–40 and are especially effective for buried water molecules. However, the role of exposed solvent shell water – although clearly important for binding – is still poorly understood,29,30,41 and the manipulation of water molecules that predominantly face the solvent have been rarely used for ligand optimization.40 We propose that the observed water position (although energetically unfavorable) is still strongly populated because other water arrangements are even worse. It is exactly this situation that makes the displacement of this water very favorable.Our series of C-α-Me substituted [4.3.1]-bicycles represents one of the most sophisticated model systems known to date, where affinity gain can be traced back to a purely entropically driven solvent shell reorganization with minimal other confounding factors such as conformational changes or additional ligand–protein interactions. Our findings provide a framework how solvent-exposed ligand moieties in shallow, notoriously difficult protein cavities can be used to gain binding affinity in a ligand-efficient manner. In our specific case, the introduction of a strategically positioned single methyl group improved binding affinities for all tested FKBPs up to 10-fold, yielding the most potent FKBP ligands known to date, which potently enhanced BMP-signaling in a relevant cell line.
Data availability
All associated experimental and computational details are provided in the ESI.† Crystallographic data for compounds number 1, 1S-Me, 22, and 22S-Me have been deposited at the PBD under accession numbers 7APT (for 1), 7APS (for 1S-Me), 7APQ (for 22), and 7APW (for 22S-Me). Cartesian Coordinates of protein pockets and ligands and associated data obtained from the DFT calculations are available at TU repository (https://tudatalib.ulb.tu-darmstadt.de/handle/tudatalib/2998).Author contributions
Jürgen M. Kolos: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing; Sebastian Pomplun: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – review & editing; Sascha Jung: data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review & editing; Benedikt Rieß: data curation, investigation; Patrick L. Purder: data curation, investigation; Andreas M. Voll: data curation, investigation; Stephanie Merz: data curation, investigation; Monika Gnatzy: data curation, investigation; Thomas M. Geiger: data curation, investigation; Ingrid Quist-Løkken: data curation, investigation; Jerome Jatzlau: data curation, investigation; Petra Knaus: data curation, investigation; Toril Holien: funding acquisition, resources, validation; Andreas Bracher: resources, validation; Christian Meyners: formal analysis, investigation, methodology, validation, visualization; aul Czodrowski: funding acquisition, resources, validation; Vera Krewald: data curation, investigation, funding acquisition, resources, software, supervision, validation, writing – original draft, writing – review & editing; Felix Hausch: conceptualization, data curation, formal analysis, funding acquisition, project administration, resources, software, supervision, validation, visualization, writing – original draft, writing – review & editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Jerome Basquin, MPI of Biochemistry, Martinsried, Germany, for collecting diffraction data at SLS beamline X10SA, and the staff of ESRF and of EMBL-Grenoble for assistance and support in using beamlines ID29 and ID30B. The Lichtenberg HPC of TU Darmstadt, the Center for Scientific Computing Frankfurt and the Goethe-HLR HPC are gratefully acknowledged. This work was supported by the BMBF grants 51TaValP (16GW0290K) and iMIP (16GW0211K), the DFG grant (HA-5655-5/1) and the LOEWE cluster TRABITA.References
- E. J. Barreiro, A. E. Kümmerle and C. A. M. Fraga, Chem. Rev., 2011, 111, 5215–5246 CrossRef CAS PubMed.
- C. S. Leung, S. S. Leung, J. Tirado-Rives and W. L. Jorgensen, J. Med. Chem., 2012, 55, 4489–4500 CrossRef CAS PubMed.
- H. Schönherr and T. Cernak, Angew. Chem., Int. Ed., 2013, 52, 12256–12267 CrossRef PubMed.
- K. Feng, R. E. Quevedo, J. T. Kohrt, M. S. Oderinde, U. Reilly and M. C. White, Nature, 2020, 580, 621–627 CrossRef CAS PubMed.
- A. Vasilopoulos, S. W. Krska and S. S. Stahl, Science, 2021, 372, 398 CrossRef CAS PubMed.
- M. V. Schmidt, M. Paez-Pereda, F. Holsboer and F. Hausch, ChemMedChem, 2012, 7, 1351–1359 CrossRef CAS PubMed.
- J. M. Kolos, A. M. Voll, M. Bauder and F. Hausch, Front. Pharmacol., 2018, 9, 1425 CrossRef CAS PubMed.
- A. Chaikuad, I. Alfano, G. Kerr, C. E. Sanvitale, J. H. Boergermann, J. T. Triffitt, F. von Delft, S. Knapp, P. Knaus and A. N. Bullock, J. Biol. Chem., 2012, 287, 36990–36998 CrossRef CAS PubMed.
- B. J. Peiffer, L. Qi, A. R. Ahmadi, Y. Wang, Z. Guo, H. Peng, Z. Sun and J. O. Liu, Cell Chem. Biol., 2019, 26, 652–661 CrossRef CAS PubMed.
- M.-H. Larraufie, X. Gao, X. Xia, P. J. Devine, J. Kallen, D. Liu, G. Michaud, A. Harsch, N. Savage, J. Ding, K. Tan, M. Mihalic, S. Roggo, S. M. Canham, S. M. Bushell, P. Krastel, J. Gao, A. Izaac, E. Altinoglu, P. Lustenberger, M. Salcius, F. Harbinski, E. T. Williams, L. Zeng, J. Loureiro, F. Cong, C. J. Fryer, L. Klickstein, J. A. Tallarico, R. K. Jain, D. M. Rothman and S. Wang, Cell Chem. Biol., 2021, 28, 1271–1282 CrossRef CAS PubMed.
- S. Gaali, A. Kirschner, S. Cuboni, J. Hartmann, C. Kozany, G. Balsevich, C. Namendorf, P. Fernandez-Vizarra, C. Sippel, A. S. Zannas, R. Draenert, E. B. Binder, O. F. Almeida, G. Ruhter, M. Uhr, M. V. Schmidt, C. Touma, A. Bracher and F. Hausch, Nat. Chem. Biol., 2015, 11, 33–37 CrossRef CAS PubMed.
- P. K. A. Jagtap, S. Asami, C. Sippel, V. R. I. Kaila, F. Hausch and M. Sattler, Angew. Chem., Int. Ed., 2019, 58, 9429–9433 CrossRef CAS PubMed.
- A. M. Voll, C. Meyners, M. C. Taubert, T. Bajaj, T. Heymann, S. Merz, A. Charalampidou, J. Kolos, P. L. Purder, T. M. Geiger, P. Wessig, N. C. Gassen, A. Bracher and F. Hausch, Angew. Chem., Int. Ed., 2021, 60, 13257–13263 CrossRef CAS PubMed.
- S. Martinelli, E. A. Anderzhanova, T. Bajaj, S. Wiechmann, F. Dethloff, K. Weckmann, D. E. Heinz, T. Ebert, J. Hartmann, T. M. Geiger, M. Döng, K. Hafner, M. L. Pöhlmann, L. Jollans, A. Philipsen, S. V. Schmidt, U. Schmidt, G. Maccarrone, V. Stein, F. Hausch, C. W. Turck, M. V. Schmidt, A. Gellner, B. Kuster and N. C. Gassen, Nat. Commun., 2021, 12, 4643 CrossRef CAS PubMed.
- N. J. Scheuplein, N. M. Bzdyl, E. A. Kibble, T. Lohr, U. Holzgrabe and M. Sarkar-Tyson, J. Med. Chem., 2020, 63, 13355–13388 CrossRef CAS PubMed.
- R. J. Deshaies, Nature, 2020, 580, 329–338 CrossRef CAS PubMed.
- Z. Guo, S. Y. Hong, J. Wang, S. Rehan, W. Liu, H. Peng, M. Das, W. Li, S. Bhat, B. Peiffer, B. R. Ullman, C.-M. Tse, Z. Tarmakova, C. Schiene-Fischer, G. Fischer, I. Coe, V. O. Paavilainen, Z. Sun and J. O. Liu, Nat. Chem., 2019, 11, 254–263 CrossRef CAS PubMed.
- U. K. Shigdel, S.-J. Lee, M. E. Sowa, B. R. Bowman, K. Robison, M. Zhou, K. H. Pua, D. T. Stiles, J. A. V. Blodgett, D. W. Udwary, A. T. Rajczewski, A. S. Mann, S. Mostafavi, T. Hardy, S. Arya, Z. Weng, M. Stewart, K. Kenyon, J. P. Morgenstern, E. Pan, D. C. Gray, R. M. Pollock, A. M. Fry, R. D. Klausner, S. A. Townson and G. L. Verdine, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 17195 CrossRef CAS PubMed.
- H. A. Flaxman, C.-F. Chang, H.-Y. Wu, C. H. Nakamoto and C. M. Woo, J. Am. Chem. Soc., 2019, 141, 11759–11764 CrossRef CAS PubMed.
- Z. Zhang and K. M. Shokat, Angew. Chem., Int. Ed., 2019, 58, 16314–16319 CrossRef CAS PubMed.
- Z. Guo, Z. Cheng, J. Wang, W. Liu, H. Peng, Y. Wang, A. V. S. Rao, R. J. Li, X. Ying, P. Korangath, M. V. Liberti, Y. Li, Y. Xie, S. Y. Hong, C. Schiene-Fischer, G. Fischer, J. W. Locasale, S. Sukumar, H. Zhu and J. O. Liu, Angew. Chem., Int. Ed., 2019, 58, 17158–17162 CrossRef CAS PubMed.
- X. Zhang, L. M. Luukkonen, C. L. Eissler, V. M. Crowley, Y. Yamashita, M. A. Schafroth, S. Kikuchi, D. S. Weinstein, K. T. Symons, B. E. Nordin, J. L. Rodriguez, T. G. Wucherpfennig, L. G. Bauer, M. M. Dix, D. Stamos, T. M. Kinsella, G. M. Simon, K. A. Baltgalvis and B. F. Cravatt, J. Am. Chem. Soc., 2021, 143, 5141–5149 CrossRef CAS PubMed.
- Y. S. Wang, A. Kirschner, A. K. Fabian, R. Gopalakrishnan, C. Kress, B. Hoogeland, U. Koch, C. Kozany, A. Bracher and F. Hausch, J. Med. Chem., 2013, 56, 3922–3935 CrossRef CAS PubMed.
- M. Bischoff, C. Sippel, A. Bracher and F. Hausch, Org. Lett., 2014, 16, 5254–5257 CrossRef CAS PubMed.
- M. Bischoff, P. Mayer, C. Meyners and F. Hausch, Chemistry, 2020, 26, 4677–4681 CrossRef CAS PubMed.
- S. Pomplun, Y. S. Wang, A. Kirschner, C. Kozany, A. Bracher and F. Hausch, Angew. Chem., Int. Ed., 2015, 54, 345–348 CrossRef CAS PubMed.
- S. Pomplun, C. Sippel, A. Hahle, D. Tay, K. Shima, A. Klages, C. M. Unal, B. Riess, H. T. Toh, G. Hansen, H. S. Yoon, A. Bracher, P. Preiser, J. Rupp, M. Steinert and F. Hausch, J. Med. Chem., 2018, 61, 3660–3673 CrossRef CAS PubMed.
- B. M. Dunyak and J. E. Gestwicki, J. Med. Chem., 2016, 59, 9622–9644 CrossRef CAS PubMed.
- F. Spyrakis, M. H. Ahmed, A. S. Bayden, P. Cozzini, A. Mozzarelli and G. E. Kellogg, J. Med. Chem., 2017, 60, 6781–6827 CrossRef CAS PubMed.
- S. Geschwindner and J. Ulander, Expert Opin. Drug Discovery, 2019, 14, 1221–1225 CrossRef PubMed.
- M. Bauder, C. Meyners, P. L. Purder, S. Merz, W. O. Sugiarto, A. M. Voll, T. Heymann and F. Hausch, J. Med. Chem., 2021, 64, 3320–3349 CrossRef CAS PubMed.
- M. T. Gnatzy, T. M. Geiger, A. Kuehn, N. Gutfreund, M. Walz, J. M. Kolos and F. Hausch, ChemBioChem, 2021, 22, 2257–2261 CrossRef CAS PubMed.
- M. C. Taubert and F. Hausch, Cell Chem. Biol., 2021, 28, 1253–1255 CrossRef CAS PubMed.
- O. E. Olsen, M. Sankar, S. Elsaadi, H. Hella, G. Buene, S. R. Darvekar, K. Misund, T. Katagiri, P. Knaus and T. Holien, J. Cell Sci., 2018, 131(11), jcs220731 CrossRef.
- O. Korchynskyi and P. ten Dijke, J. Biol. Chem., 2002, 277, 4883–4891 CrossRef CAS PubMed.
- P. Matricon, R. R. Suresh, Z.-G. Gao, N. Panel, K. A. Jacobson and J. Carlsson, Chem. Sci., 2021, 12, 960–968 RSC.
- J. Schiebel, R. Gaspari, T. Wulsdorf, K. Ngo, C. Sohn, T. E. Schrader, A. Cavalli, A. Ostermann, A. Heine and G. Klebe, Nat. Commun., 2018, 9, 3559 CrossRef PubMed.
- S. G. Krimmer, J. Cramer, M. Betz, V. Fridh, R. Karlsson, A. Heine and G. Klebe, J. Med. Chem., 2016, 59, 10530–10548 CrossRef CAS PubMed.
- E. L. Ratkova, M. Dawidowski, V. Napolitano, G. Dubin, R. Fino, M. S. Ostertag, M. Sattler, G. Popowicz and I. V. Tetko, Chem. Commun., 2020, 56, 4360–4363 RSC.
- M. S. Bodnarchuk, Drug Discovery Today, 2016, 21, 1139–1146 CrossRef CAS PubMed.
- M. Betz, T. Wulsdorf, S. G. Krimmer and G. Klebe, J. Chem. Inf. Model., 2016, 56, 223–233 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc04638a |
| ‡ These authors contributed equally. |
| § Sebastian Pomplun: Leiden Academic Centre for Drug Research (LACDR), Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. |
| ¶ Benedikt Rieß: Department of Chemistry, Technical University of Munich, Lichtenbergstraße 4,85748 Garching, Germany |
| This journal is © The Royal Society of Chemistry 2021 |