
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdapting decarbonylation chemistry for the development of prodrugs capable of in vivo delivery of carbon monoxide utilizing sweeteners as carrier molecules†
Ladie Kimberly
De La Cruz‡
a,
Xiaoxiao
Yang‡
a,
Anna
Menshikh
b,
Maya
Brewer
b,
Wen
Lu
a,
Minjia
Wang
c,
Siming
Wang
a,
Xingyue
Ji
a,
Alyssa
Cachuela
a,
Haichun
Yang
d,
David
Gallo
e,
Chalet
Tan
c,
Leo
Otterbein
e,
Mark
de Caestecker
b and
Binghe
Wang
 *a
*a
aDepartment of Chemistry, Georgia State University, Atlanta, GA 30303, USA. E-mail: wang@gsu.edu
bDepartment of Medicine, Division of Nephrology, Vanderbilt University Medical Center, Nashville, TN 37232, USA
cDepartment of Pharmaceutics and Drug Delivery, University of Mississippi, MS 38677, USA
dDepartment of Pathology, Microbiology, and Immunology, Vanderbilt University Medical Center, Nashville, TN 37232, USA
eDepartment of Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA 02115, USA
First published on 1st July 2021
Abstract
Carbon monoxide as an endogenous signaling molecule exhibits pharmacological efficacy in various animal models of organ injury. To address the difficulty in using CO gas as a therapeutic agent for widespread applications, we are interested in developing CO prodrugs through bioreversible caging of CO in an organic compound. Specifically, we have explored the decarboxylation–decarbonylation chemistry of 1,2-dicarbonyl compounds. Examination and optimization of factors favorable for maximal CO release under physiological conditions led to organic CO prodrugs using non-calorific sweeteners as leaving groups attached to the 1,2-dicarbonyl core. Attaching a leaving group with appropriate properties promotes the desired hydrolysis–decarboxylation–decarbonylation sequence of reactions that leads to CO generation. One such CO prodrug was selected to recapitulate the anti-inflammatory effects of CO against LPS-induced TNF-α production in cell culture studies. Oral administration in mice elevated COHb levels to the safe and efficacious levels established in various preclinical and clinical studies. Furthermore, its pharmacological efficacy was demonstrated in mouse models of acute kidney injury. These studies demonstrate the potential of these prodrugs with benign carriers as orally active CO-based therapeutics. This represents the very first example of orally active organic CO prodrugs with a benign carrier that is an FDA-approved sweetener with demonstrated safety profiles in vivo.
Introduction
With the firm establishment of carbon monoxide (CO) as an endogenous signaling molecule, there has been an increasing level of interest in developing CO-based therapeutics by taking advantage of its demonstrated cyto- and organ-protective effects.1–3 Along this line, the organ protective effects of CO have been demonstrated in injury models of the kidneys,4,5 lungs,6 the gastrointestinal tract,7,8 and the liver,9 among others. Because of the gaseous nature of CO, the difficulty in delivering a gas as a therapeutic to patients, and the potential risks it poses to healthcare workers as well as patients in terms of accidental poisoning, there have been intense efforts in developing alternative delivery forms.1,10,11 Earlier efforts focused on metal-based CO releasing molecules (CORMs),1 photo-sensitive-organic CO donors,11–13 CO in solution,14,15 and CO immobilized on modified hemoglobin.16 There are several reviews discussing in detail the various existing delivery forms of CO.1,10,11 We are interested in developing organic CO prodrugs as donors for developing CO-based therapeutics as “CO in a pill” for eventual clinical applications.17 Along this line, we18 and Larsen's lab19 developed cheletropic extrusion-based CO prodrugs with tunable release rates, triggered release, and the ability to target the mitochondria.20Fig. 1 shows some representative CO donors. In terms of drug-like features, each class has its own features that need improvement including metal reactivity for some metal-based CO-RMs (Fig. 1A),21,22 high hydrophobicity for some of the organic prodrugs due to the presence of multiple aryl groups (Fig. 1B),18 and the known and unknown toxicity of transition metals23,24 and the organic “carrier” portions of the organic CO prodrugs.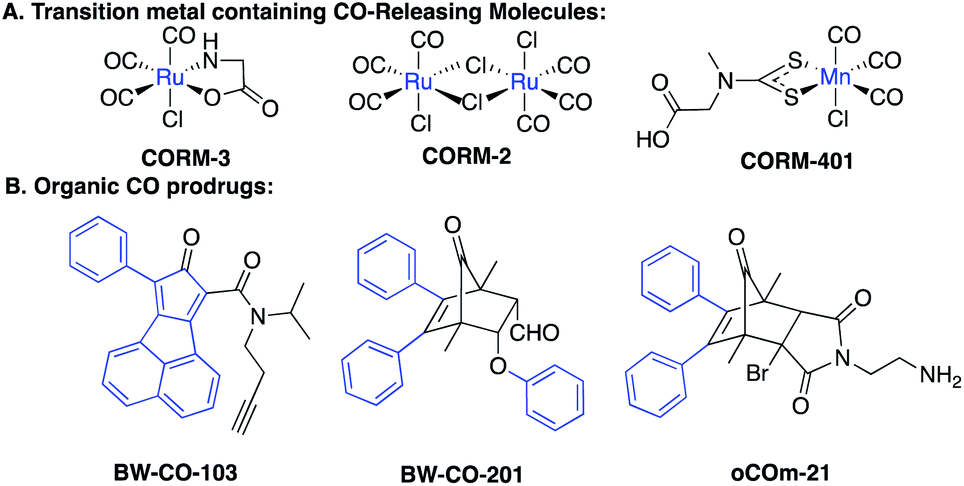 | ||
| Fig. 1 Examples of CO donors that are activated by physiological stimuli – (A) ligand exchange from endogenous nucleophiles, and (B) exposure to an aqueous environment and/or at pH 7.4. | ||
In search of CO prodrugs with improved pharmaceutical properties, we are especially interested in those that use a “carrier” moiety with known safety profiles. Along this line, we look to known decarbonylation chemistry for our design. Several types of decarbonylation chemistry are known in synthetic chemistry in organic solvents, including decarbonylation of formic acid and its derivatives, α-hydroxyacid, 5-(hydroxymethyl)furfural25,26 and other aldehydes and carbonyl-containing compounds.27–33 However, most of these reactions happen under non-physiological conditions or require harsh acids, heating, or metal catalysis for CO release. In most cases, especially those that require metal catalysis, application under near physiological conditions is hard to achieve.
The hydrolysis, decarboxylation, decarbonylation reaction sequence characteristic of 1,2-dicarbonyl compounds with appropriate leaving groups, presented an opportunity to install benign carriers as leaving groups. As a literature precedent, oxalyl chloride, one of the simplest 1,2-dicarbonyl compounds in the form of oxalates, is known to decompose in water to produce CO, HCl, and CO2 through first hydrolysis of one acyl chloride, then decarboxylation, and finally decarbonylation to release CO (Fig. 2A).34 Because of the reliability of this chemistry, oxalyl chloride has been used as a convenient CO gas donor in carbonylation reactions.35,36 However, its use for biological and therapeutic applications presents significant challenges because of the release of HCl, and its corrosive and acutely toxic nature.37 With CO fixed in the 1,2-dicarbonyl moieties, our goal was to design a new class of CO donors involving benign carriers (Fig. 2B).
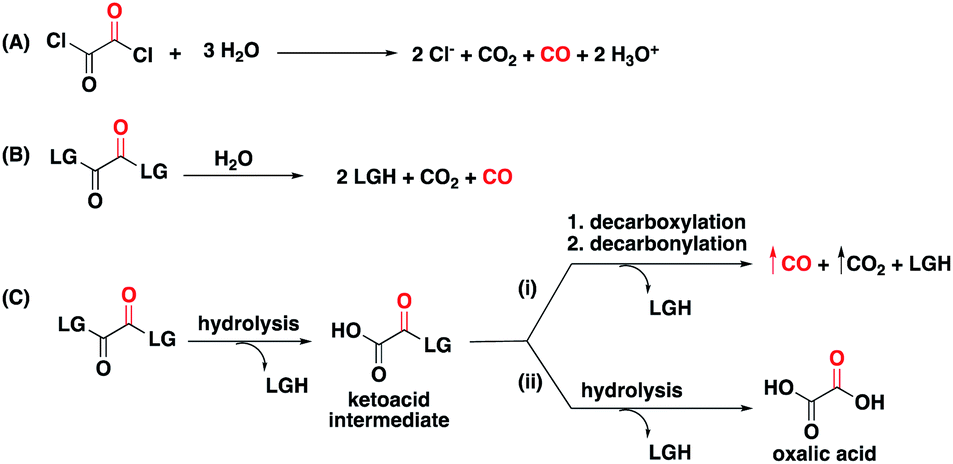 | ||
| Fig. 2 (A) Decomposition of oxalyl chloride to produce CO. (B) Design of CO donors based on the hydrolysis, decarboxylation, decarbonylation chemistry of oxalyl derivatives. (C) Competing paths by which 1,2-dicarbonyl CO prodrugs can breakdown in aqueous solution – (i) a CO-generating pathway; (ii) non-CO producing pathway that generates oxalic acid as the product. | ||
Results and discussion
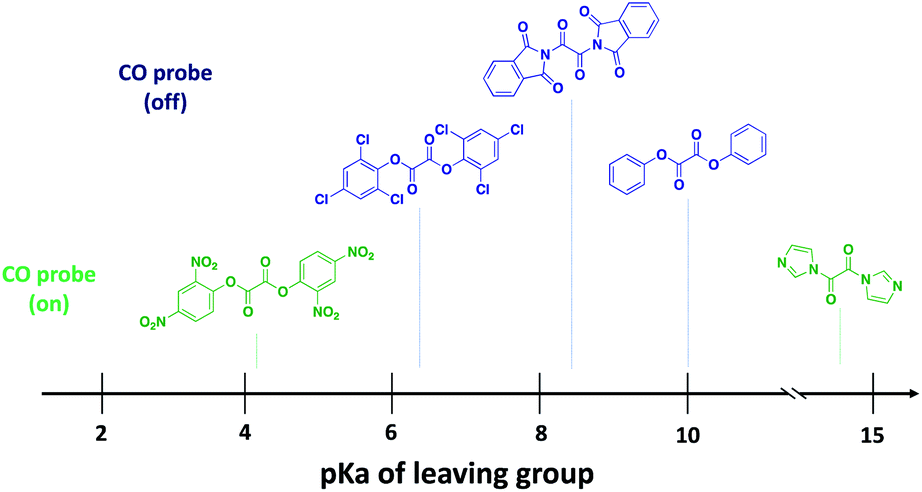
Because of enhanced electrophilicity arising from vicinal keto groups, 1,2-dicarbonyl compounds with appropriate leaving groups are susceptible to hydrolysis, expelling one leaving group and generating a ketoacid intermediate (Fig. 2C). CO is generated once this ketoacid intermediate undergoes a decarboxylation–decarbonylation sequence of reactions. For oxalyl chloride, this path predominates and thus forms CO, CO2, and HCl as end-products when placed in water. However, when the chloride leaving group is replaced by other leaving groups, another decomposition path may compete with the CO generating path.38,39 The ketoacid intermediate can undergo hydrolysis to expel the second leaving group and form oxalic acid without CO generation. Therefore, the identity of the leaving group must be carefully selected to tune the reaction in favor of the CO generating path.For understanding the basic chemistry and parameters that control the partitioning between the two pathways, chloride was replaced by various leaving groups having different pKa values to survey the effect of pKa on CO release profiles (Fig. 3 and Table S1†). In doing so, we utilized a selective fluorescent CO probe for monitoring CO production in situ. Specifically, COP-1 (ref. 40) is a widely tested fluorescent CO probe based on the Pd-mediated carbonylation reaction. We selected a number of leaving groups with different pKa values including phenol, 2,4,6-trichlorophenol, 2,4-dinitrophenol, imidazole, and an imide compound, phthalimide.
 | ||
| Fig. 3 Examination of the effect of pKa of the leaving group on the CO releasing capacity of 1,2-dicarbonyl compounds using a CO probe, COP-1. | ||
A 6-fold increase in fluorescence turn-on intensity was observed when the leaving group was 2,4-dinitrophenol. In general, compounds with leaving groups having pKa greater than 4 were incapable of CO release, except for a marginal 0.1-fold increase with imidazole. According to previous reports, hydrolysis of symmetrical oxalates occurs in two steps with the first step being at least two orders of magnitude faster than the second hydrolysis.38 In certain cases and as is shown in the later section, the ketoacid with a good leaving group is stable enough to be isolated. The nature of the attached leaving group to the keto acid influences the decomposition pathway it goes through (Fig. 2C). Leaving groups with pKa less than 4 favor the CO-releasing pathway. Based on these results, additional leaving groups with pKa less than four were considered. Two “carrier” molecules seemed especially interesting to us for CO delivery under physiological conditions: saccharin and acesulfame. Both are FDA-approved, non-calorific sweeteners, having pKa values that fall within the range of 1.6 and 3.0, respectively (Fig. 4A). The oxalyl derivative with saccharin as a leaving group was previously reported as a coupling reagent for the synthesis of carboxylic acid derivatives.41 In this report it was noted that CO and CO2 are released but under non-physiological conditions.
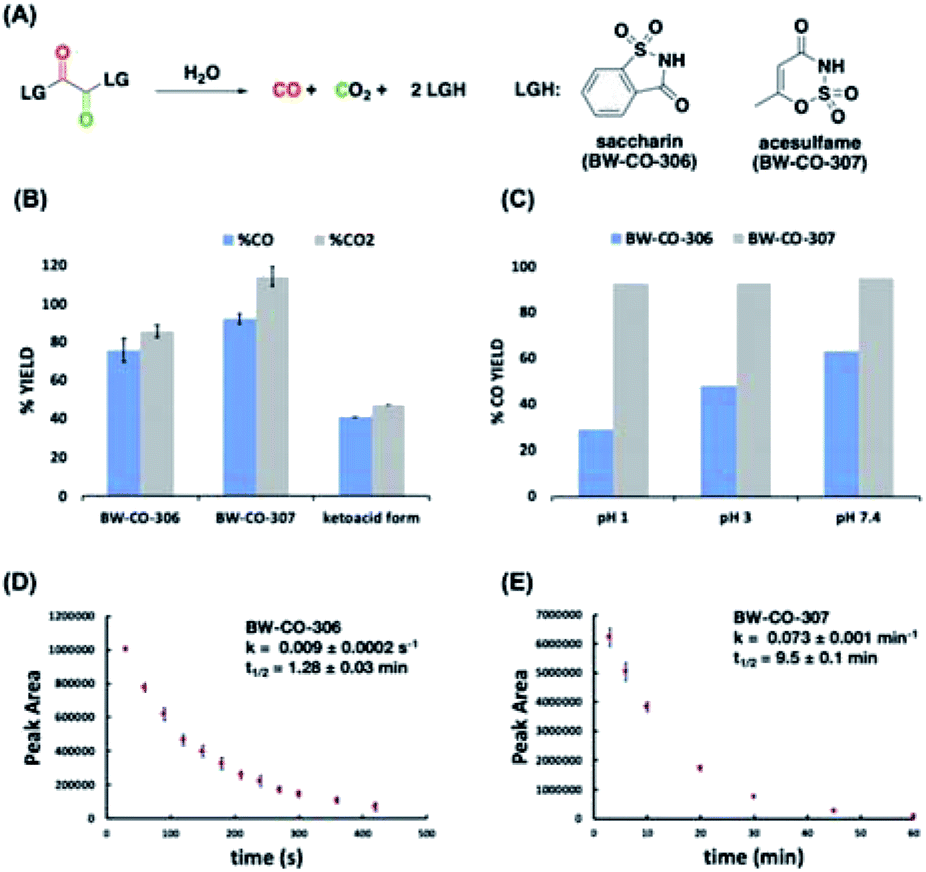 | ||
Fig. 4 (A) Structures of CO prodrugs. (B) GC-TCD analysis of CO and CO2 yields of CO prodrugs in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ACN:H2O at 37 °C, 1 h incubation time. (C) Effect of pH on CO and CO2 yields in 4:1 ACN:buffer (pH 1 – glycine/NaCl/HCl, pH 3 – citric acid/NaOH/HCl, pH 7.4 – phosphate buffer) at 37 °C, 1 h incubation time. (D) and (E) Decomposition profile of CO prodrugs in 60% PBS analyzed by RP-HPLC. :1 ACN:H2O at 37 °C, 1 h incubation time. (C) Effect of pH on CO and CO2 yields in 4:1 ACN:buffer (pH 1 – glycine/NaCl/HCl, pH 3 – citric acid/NaOH/HCl, pH 7.4 – phosphate buffer) at 37 °C, 1 h incubation time. (D) and (E) Decomposition profile of CO prodrugs in 60% PBS analyzed by RP-HPLC. | ||
Therefore, CO prodrugs BW-CO-306 and BW-CO-307 were made in one step from the reaction between commercially available sweeteners saccharin and acesulfame, respectively, with oxalyl chloride in the presence of triethylamine (Scheme S1†). Qualitative determinations using a household CO detector, COP-1 probe, and CO-myoglobin assay indicated CO production with BW-CO-306 (Fig. S1–S3†). The CO release yields were then quantified using an Agilent 7820A GC with a thermal conductivity detector (TCD). CO prodrugs BW-CO-306 and BW-CO-307 released 76% ± 6 and 92% ± 3 CO, respectively when dissolved in 4:1 ACN:H2O and incubated at 37 °C for 1 h (Fig. 4B). The ketoacid form of BW-CO-306 was also synthesized and isolated as a white solid (Scheme S2†). The lower CO yield of only around 40% (Fig. 4B) indicates that installation of two leaving groups with pKa less than 4 into the 1,2-dicarbonyl core favors the CO generating reaction pathway. An LC-MS method was used to analyze the proportion of prodrug BW-CO-306 that is converted to oxalic acid through the non-CO generating pathway. Oxalic acid (47 μM) was produced from 200 μM of prodrug BW-CO-306. Therefore, CO production is estimated to be about 75%, which agrees well with the results generated from the GC-TCD experiments.
Since CO release from the prodrug involves hydrolysis, low pH might enhance the hydrolysis rates and therefore decrease CO yield. This is indeed what was observed with prodrug BW-CO-306 wherein CO yield decreased from 60% to 30% with decreasing pH (Fig. 4C). In contrast, the CO release yield for prodrug BW-CO-307 is pH-independent with CO yields constant at around 90% across a broad pH range. Furthermore, RP-HPLC studies revealed that prodrugs BW-CO-306 and BW-CO-307 are stable in the solid state for at least 24 days exposed to ambient light and temperature. These prodrugs are also stable in solutions of acetonitrile stored at room temperature for at least seven days. Kinetic experiments using HPLC revealed that the half-lives of compound BW-CO-306 and BW-CO-307 in 60% phosphate buffered saline are 1.28 ± 0.03 min and 9.5 ± 0.14 min, respectively (Fig. 4D and E). HPLC and NMR analyses both provide strong evidence that products after CO release are mainly the carriers saccharin and acesulfame (Fig. S8–S11†).
To demonstrate that these compounds are compatible CO donors for biological applications and that these donors can recapitulate CO-associated anti-inflammation activity, an ELISA assay for the pro-inflammatory cytokine TNF-α was conducted. Because the half-life of BW-CO-306 is very short, the produced CO may escape readily in a cell culture experimental setting. Thus, we repeatedly exposed the cells to the prodrug five times. BW-CO-307, with a slightly longer half-life, was added only once. Dose-dependent inhibition of LPS-induced secretion of TNF-α was observed for both prodrugs (Fig. 5A and B). For in vivo applications, the utility of orally administered BW-CO-306 was demonstrated in a pharmacokinetic study wherein COHb levels in the blood were monitored by a CO-oximeter. Elevation of COHb was observed up to around 4–5.5% and remained above the baseline for about an hour (Fig. 5C). In comparison, this COHb elevation is within the limits established by human clinical trials to be safe42,43 and comparable to the efficacy levels established in preclinical animal studies.44,45 Consistent with the in vitro kinetic properties of BW-CO-306 and BW-CO-103 (another CO prodrug which has been shown to be efficacious in various animal models such as colitis),46 with half-lives of 1.3 min and 1.2 h, respectively, BW-CO-103 gave a more sustained elevation of COHb17 with a tmax of 60 min while BW-CO-306 gave a burst of CO resulting in a spike of COHb elevation with a tmax of 10 min that only lasted for around 30 min (Fig. 5D). The dose–response relationship and the increase of COHb above the blank vehicle control after oral administration of BW-CO-306 indicated a facile CO release in vivo and demonstrated the feasibility of systemic CO delivery.
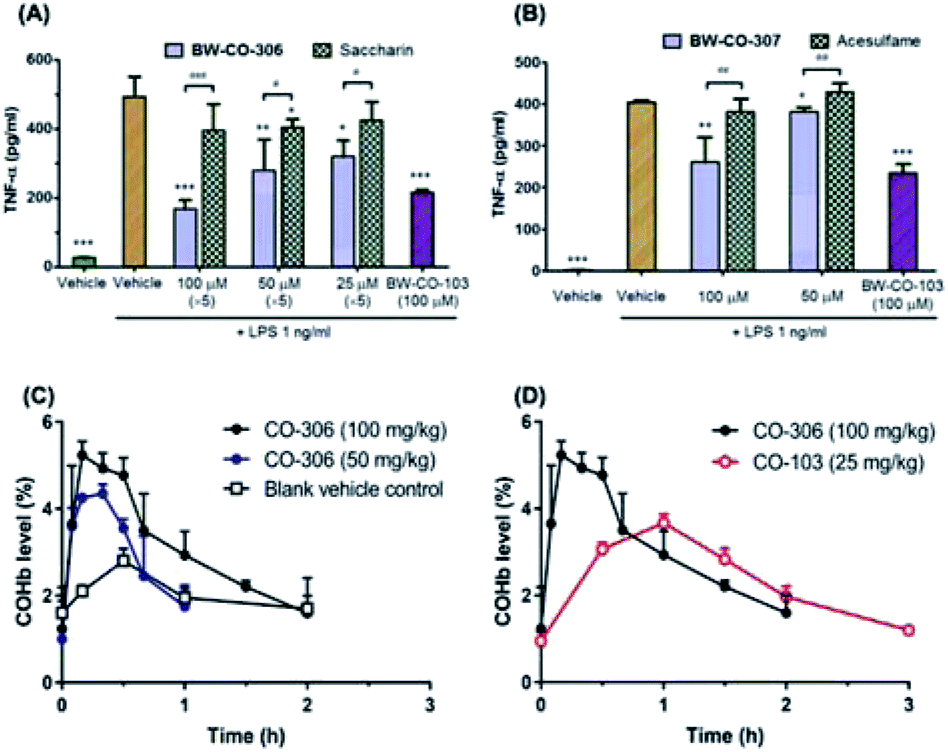 | ||
| Fig. 5 (A) and (B) The anti-inflammatory effects of BW-CO-306 and BW-CO-307 in RAW 264.7 cells. Mean of each concentration vs. LPS 1 μg mL−1: *p < 0.05, **p < 0.01 and ***p < 0.001; mean of each concentration vs. the corresponding saccharin only group: #p < 0.05 and ###p < 0.001 (C) COHb elevation of BW-CO-306 in mice (D) COHb level elevation of BW-CO-306 compared to another CO prodrug, BW-CO-103. | ||
The first in vivo validation of heme oxygenase (HO-1, the enzyme responsible for endogenous CO production through heme catabolism) induced protection in response to tissue injury was conducted using a rhabdomyolysis induced acute kidney injury (AKI) model in rats.47 The model recapitulates the mechanisms of heme-induced kidney injury that occurs in patients with extensive muscle damage, and intravascular hemolysis associated with sickle cell disease, falciparum malaria, cardiopulmonary bypass surgery and severe sepsis.47–50 This pivotal paper demonstrated that the kidneys employ HO-1 coupled with ferritin synthesis (to sequester free iron) to launch a rapid, protective response against acute kidney injury (AKI).51 Since that time, a number of pharmacological and genetic loss and gain of function studies have shown that HO-1 is protective in a number of different models of AKI, including sepsis,52 cisplatin,53–56 and ischemia reperfusion induced (IRI)-AKI.57–59 These data indicate that induction of HO-1 may be a viable therapeutic strategy against AKI.60 However, heme catabolism also produces free iron which if not sequestered by ferritin can also exacerbate AKI.60 A different pharmacological strategy is to employ CO, one of the end-products of heme catabolism as the therapeutic agent. For this reason, we were interested in testing our CO prodrug in this rhabdomyolysis model of kidney injury.
BW-CO-306 was prepared as an oral formulation in activated charcoal and 50 mg kg−1 was administered to groups of 13 week male BALB-c mice injected with 5.4 mL kg−1 of 50% glycerol to the muscle of hindlegs. We utilized BW-CP-306 (i.e. saccharin), the “carrier” portion of BW-CO-306 after CO release, as an inactive control. Intramuscular injection of glycerol-induced AKI was associated with a rapid increase in blood urea nitrogen (BUN) levels 24 hours after injection while CO treatment using BW-CO-306 resulted in significant lowering of BUN levels (Fig. 6A). BW-CO-306 also increased survival after glycerol-induced rhabdomyolysis from 3/7 mice (42%) in BW-CP-306 treated mice to 6/8 (75%) (Fig. 6B), although these changes were not statistically significant (log-rank test, p = 0.3). Seventy-two hours after glycerol injection, histological studies revealed a marked reduction in features of acute tubular injury including flattened, dedifferentiated tubular epithelial cells, and tubular casts (Fig. 6C and S14†) in mice treated with BW-CO-306. In support of the reduced histological evidence of renal injury, both QRT-PCR (Fig. 6D) for kidney injury molecule 1 (Kim1) mRNA, a marker of renal tubular injury, and immunofluorescence staining (Fig. 6E) for Kim1 in the outer stripe of the outer medulla (OSOM) indicated reduced expression in comparison to BW-CP-306 treated mice. As expected, Kim1 expression61 is restricted to LTL binding proximal tubular epithelial cells in the cortex and OSOM after rhabdomyolysis-induced AKI (Fig. 6F). Furthermore, in a kidney ischemia reperfusion injury mice model, a similar protection was observed from the p.o. administration of BW-CO-306 (50 mg kg−1) as indicated by the significant lowering of creatinine levels (Fig. S15†). These findings indicate that in lieu of direct HO-1 induction strategies, oral administration of a CO prodrug protects from AKI and may represent a more straightforward strategy of harnessing HO-1 cytoprotective effects in kidneys for different types of AKI.
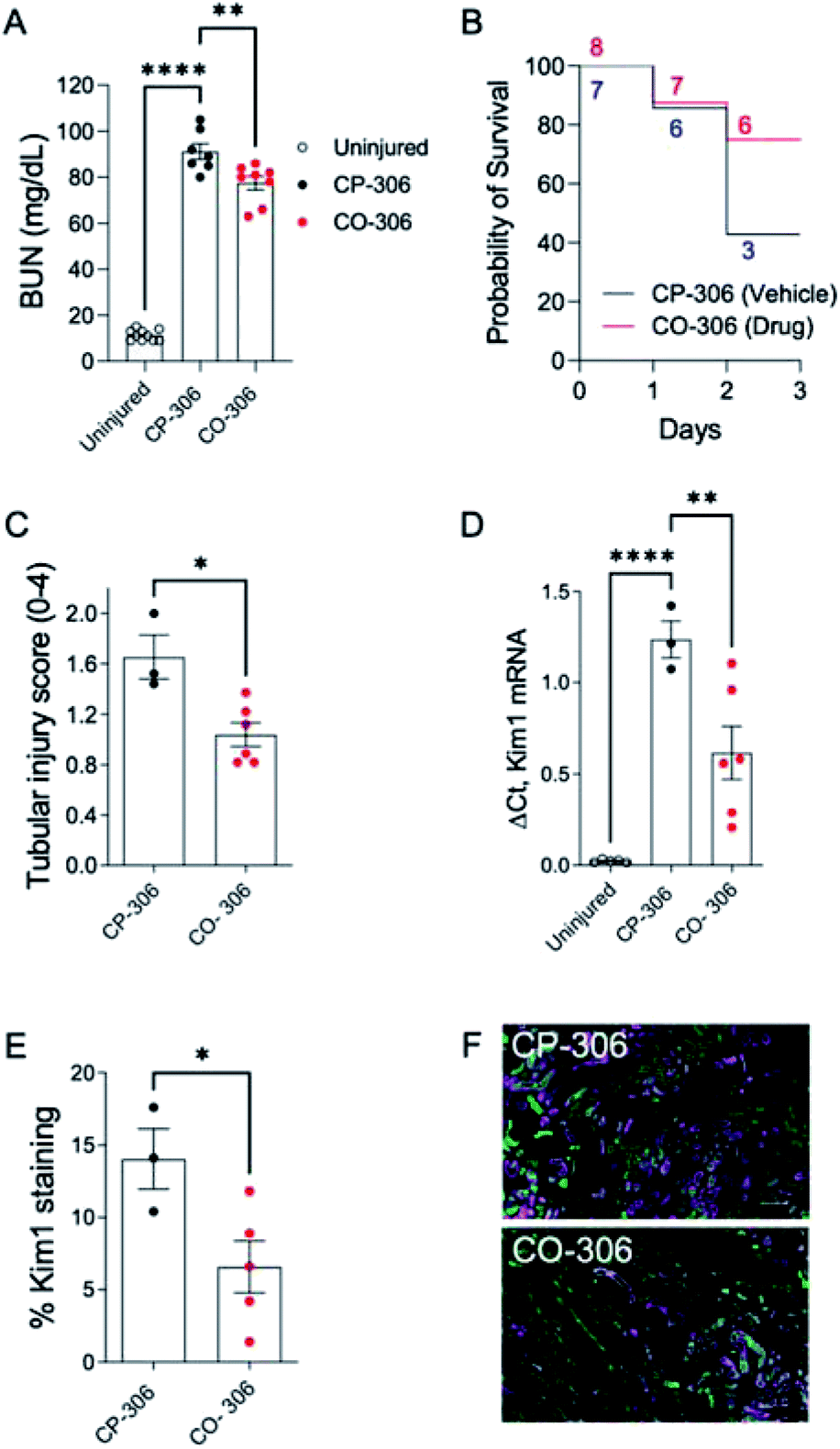 | ||
| Fig. 6 BW-CO-306 protects against rhabdomyolysis-induced acute kidney injury (AKI). Male BALB/c mice were treated with 50 mg kg−1BW-CO-306 (or release product saccharin) by oral gavage starting 24 hours before and then daily after intramuscular injection of 5.4 mL kg−1 50% glycerol in water to induce rhabdomyolysis, as described.62 Blood was collected for BUN measurements 24 hours after glycerol injection, and tissues were collected after euthanasia at day 3. (A) Blood urea nitrogen (BUN) 24 hours after glycerol injection; (B) survival curves with numbers of mice at each time point indicated; (C) semiquantitative analysis of renal tubular injury (tubular injury score 0–4). Representative images of PAS-stained sections shown in Fig. S14;† (D) QRT-PCR for Kim1 mRNA in the kidneys. (E) and (F) Quantification of the % surface area immunofluorescence staining for Kim1 with representative images of Kim1 (magenta), and LTL (green), staining in the OSOM of CP-306 and CO-306 treated mouse kidneys 3 days after rhabdomyolysis-induced AKI, as indicated. Scale bars, 100 μM. Mouse numbers as indicated, with individual data points, mean ± SEM indicated. One-way ANOVA was used to determine statistical significance of differences between multiple groups, and a post-hoc Dunnett's test used to compare pairwise significance, as shown (A) and (C). A two-sided unpaired T-test was used to compare two groups (C). *p < 0.05; **p < 0.01; ****p < 0.0001. | ||
Conclusions
In conclusion, we have designed a CO prodrug with benign, FDA-approved carrier molecules. This CO prodrug strategy relies on the adaptation of decarbonylation chemistry used in organic solvents for application under physiological conditions through a hydrolysis, decarboxylation, decarbonylation reaction sequence to release CO. In vitro anti-inflammation assays and in vivo pharmacokinetics and acute kidney injury mouse model (rhabdomyolysis-induced and renal IRI) studies validate the use of this prodrug system to deliver CO for therapeutic applications.Data availability
Supporting data for this article have been uploaded as part of the ESI material.Author contributions
Conceptualization – LKDLC, XY, XJ, CT, LO, MDC, BW; investigation and methodology – LKDLC, XY, AM, MB, WL, MW, SW, AC, HY, DG, LO, CT, MDC, BW; funding acquisition – CT, LO, MDC, BW; supervision – CT, LO, MDC, BW; writing, original draft – LKDLC, BW; writing, reviewing & editing – LKDLC, XY, AM, MW, CT, LO, MDC, BW.Conflicts of interest
There are no conflicts to declare.Acknowledgements
All animal procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal procedure protocols were approved by the respective Institutional Animal Care and Use Committees at the performing institutions (University of Mississippi, Vanderbilt University, and BIDMC/Harvard Medical School). The authors are grateful for financial support from the National Institutes of Health (R01DK119202, R01DK112688), the Georgia Research Alliance, Eminent Scholar fund, and internal resources at Georgia State University.References
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728 CrossRef CAS PubMed.
- L. Wu and R. Wang, Pharmacol. Rev., 2005, 57, 585 CrossRef CAS PubMed.
- X. Yang, W. Lu, C. P. Hopper, B. Ke and B. Wang, Acta Pharm. Sin. B, 2021, 11, 1434 CrossRef PubMed.
- E. Csongradi, L. A. Juncos, H. A. Drummond, T. Vera and D. E. Stec, Curr. Pharm. Biotechnol., 2012, 13, 819 CAS.
- X. Yang, M. de Caestecker, L. E. Otterbein and B. Wang, Med. Res. Rev., 2020, 40, 1147 CrossRef CAS PubMed.
- S. W. Ryter and A. M. Choi, Curr. Opin. Pharmacol., 2006, 6, 257 CrossRef CAS PubMed.
- T. Takagi, K. Uchiyama and Y. Naito, Digestion, 2015, 91, 13 CrossRef CAS PubMed.
- D. Bakalarz, M. Surmiak, X. Yang, D. Wójcik, E. Korbut, Z. Śliwowski, G. Ginter, G. Buszewicz, T. Brzozowski, J. Cieszkowski, U. Głowacka, K. Magierowska, Z. Pan, B. Wang and M. Magierowski, Acta Pharm. Sin. B, 2021, 11, 456 CrossRef CAS PubMed.
- B. Florian and T. Tung Yu, Curr. Pharm. Biotechnol., 2012, 13, 803 Search PubMed.
- X. X. Yang, B. W. Ke, W. Lu and B. H. Wang, Chin. J. Nat. Med., 2020, 18, 284 Search PubMed.
- L. S. Lazarus, A. D. Benninghoff and L. M. Berreau, Acc. Chem. Res., 2020, 53, 2273 CrossRef CAS PubMed.
- P. Peng, C. Wang, Z. Shi, V. K. Johns, L. Ma, J. Oyer, A. Copik, R. Igarashi and Y. Liao, Org. Biomol. Chem., 2013, 11, 6671 RSC.
- L. Štacková, M. Russo, L. Muchová, V. Orel, L. Vítek, P. Štacko and P. Klán, Chemistry, 2020, 26, 13184 CrossRef PubMed.
- T. Takagi, Y. Naito, K. Uchiyama, K. Mizuhima, T. Suzuki, R. Horie, I. Hirata, H. Tsuboi and T. Yoshikawa, Free Radical Res., 2016, 50, 1098 CrossRef CAS PubMed.
- J. D. Belcher, E. Gomperts, J. Nguyen, C. Chen, F. Abdulla, Z. M. Kiser, D. Gallo, H. Levy, L. E. Otterbein and G. M. Vercellotti, PLoS One, 2018, 13, e0205194 CrossRef PubMed.
- W. H. Nugent, R. F. Cestero, K. Ward, R. Jubin, A. Abuchowski and B. K. Song, Shock, 2019, 52, 108 CrossRef CAS PubMed.
- M. Wang, X. Yang, Z. Pan, Y. Wang, L. K. De La Cruz, B. Wang and C. Tan, J. Controlled Release, 2020, 327, 174 CrossRef CAS PubMed.
- X. Ji and B. Wang, Acc. Chem. Res., 2018, 51, 1377 CrossRef CAS PubMed.
- J. T. B. Kueh, N. J. Stanley, R. J. Hewitt, L. M. Woods, L. Larsen, J. C. Harrison, D. Rennison, M. A. Brimble, I. A. Sammut and D. S. Larsen, Chem. Sci., 2017, 8, 5454 RSC.
- Y. Zheng, X. Ji, B. Yu, K. Ji, D. Gallo, E. Csizmadia, M. Zhu, M. R. Choudhury, L. K. C. De La Cruz, V. Chittavong, Z. Pan, Z. Yuan, L. E. Otterbein and B. Wang, Nat. Chem., 2018, 10, 787 CrossRef CAS PubMed.
- Z. Yuan, X. Yang, L. K. De La Cruz and B. Wang, Chem. Commun., 2020, 56, 2190 RSC.
- Z. Yuan, X. Yang, Y. Ye, R. Tripathi and B. Wang, Anal. Chem., 2021, 93, 5317 CrossRef CAS PubMed.
- R. E. Yasbin, C. R. Matthews and M. J. Clarke, Chem.–Biol. Interact., 1980, 31, 355 CrossRef CAS PubMed.
- V. Brabec and O. Nováková, Drug Resist. Updates, 2006, 9, 111 CrossRef CAS PubMed.
- F. M. Geilen, T. vom Stein, B. Engendahl, S. Winterle, M. A. Liauw, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed. Engl., 2011, 50, 6831 CrossRef CAS PubMed.
- Y. B. Huang, Z. Yang, M. Y. Chen, J. J. Dai, Q. X. Guo and Y. Fu, ChemSusChem, 2013, 6, 1348 CrossRef CAS PubMed.
- M. Kreis, A. Palmelund, L. Bunch and R. Madsen, Adv. Synth. Catal., 2006, 348, 2148 CrossRef CAS.
- R. N. Monrad and R. Madsen, J. Org. Chem., 2007, 72, 9782 CrossRef PubMed.
- K. Ohno and J. Tsuji, J. Am. Chem. Soc., 1968, 90, 99 CrossRef CAS.
- J. Tsuji and K. Ohno, Tetrahedron Lett., 1965, 6, 3969 CrossRef.
- J. Blum, Tetrahedron Lett., 1966, 15, 1605 CrossRef.
- N. E. Hoffman and T. Puthenpurackal, J. Org. Chem., 1965, 30, 420 CrossRef CAS.
- J. O. Hawthorne and M. H. Wilt, J. Org. Chem., 1960, 25, 2215 CrossRef CAS.
- H. Staudinger, Ber. Dtsch. Chem. Ges., 1908, 41, 3558 CrossRef CAS.
- S. V. F. Hansen and T. Ulven, Org. Lett., 2015, 17, 2832 CrossRef CAS PubMed.
- M. L. N. Rao, V. Venkatesh and P. Dasgupta, Tetrahedron Lett., 2010, 51, 4975 CrossRef CAS.
- Sigma-Aldrich, Oxylyl Chloride: Material Safety Data Sheet, 2014 Search PubMed.
- G. Orosz, Tetrahedron, 1989, 45, 3493 CrossRef CAS.
- G. Orosz and E. Dudar, Anal. Chim. Acta, 1991, 247, 141 CrossRef CAS.
- B. W. Michel, A. R. Lippert and C. J. Chang, J. Am. Chem. Soc., 2012, 134, 15668 CrossRef CAS PubMed.
- T. Kitagawa, H. Kuroda, K. Iida, M. Ito and M. Nakamura, Chem. Pharm. Bull., 1989, 37, 3225 CrossRef CAS.
- L. E. Fredenburgh, M. A. Perrella, D. Barragan-Bradford, D. R. Hess, E. Peters, K. E. Welty-Wolf, B. D. Kraft, R. S. Harris, R. Maurer, K. Nakahira, C. Oromendia, J. D. Davies, A. Higuera, K. T. Schiffer, J. A. Englert, P. B. Dieffenbach, D. A. Berlin, S. Lagambina, M. Bouthot, A. I. Sullivan, P. F. Nuccio, M. T. Kone, M. J. Malik, M. A. P. Porras, E. Finkelsztein, T. Winkler, S. Hurwitz, C. N. Serhan, C. A. Piantadosi, R. M. Baron, B. T. Thompson and A. M. K. Choi, JCI Insight, 2018, 3, 23 CrossRef PubMed.
- I. O. Rosas, H. J. Goldberg, H. R. Collard, S. El-Chemaly, K. Flaherty, G. M. Hunninghake, J. A. Lasky, D. J. Lederer, R. Machado, F. J. Martinez, R. Maurer, D. Teller, I. Noth, E. Peters, G. Raghu, J. G. N. Garcia and A. M. K. Choi, Chest, 2018, 153, 94 CrossRef PubMed.
- L. E. Fredenburgh, B. D. Kraft, D. R. Hess, R. S. Harris, M. A. Wolf, H. B. Suliman, V. L. Roggli, J. D. Davies, T. Winkler, A. Stenzler, R. M. Baron, B. T. Thompson, A. M. Choi, K. E. Welty-Wolf and C. A. Piantadosi, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2015, 309, L834 CrossRef CAS.
- E. Bathoorn, D.-J. Slebos, D. S. Postma, G. H. Koeter, A. J. M. van Oosterhout, M. van der Toorn, H. M. Boezen and H. A. M. Kerstjens, Eur. Respir. J., 2007, 30, 1131 CrossRef CAS.
- X. Ji, C. Zhou, K. Ji, R. E. Aghoghovbia, Z. Pan, V. Chittavong, B. Ke and B. Wang, Angew. Chem., Int. Ed. Engl., 2016, 55, 15846 CrossRef CAS.
- K. A. Nath, Kidney Int., 2006, 70, 432 CrossRef CAS PubMed.
- V. E. Kerchberger and L. B. Ware, Semin. Nephrol., 2020, 40, 148 CrossRef CAS PubMed.
- J. B. O'Neal, A. D. Shaw and F. T. t. Billings, Crit. Care, 2016, 20, 187 CrossRef PubMed.
- X. Bosch, E. Poch and J. M. Grau, N. Engl. J. Med., 2009, 361, 62 CrossRef CAS PubMed.
- K. A. Nath, G. Balla, G. M. Vercellotti, J. Balla, H. S. Jacob, M. D. Levitt and M. E. Rosenberg, J. Clin. Invest., 1992, 90, 267 CrossRef CAS PubMed.
- K. Kang, C. Nan, D. Fei, X. Meng, W. Liu, W. Zhang, L. Jiang, M. Zhao, S. Pan and M. Zhao, Shock, 2013, 40, 136 CrossRef CAS PubMed.
- S. Bolisetty, A. M. Traylor, J. Kim, R. Joseph, K. Ricart, A. Landar and A. Agarwal, J. Am. Soc. Nephrol., 2010, 21, 1702 CrossRef CAS PubMed.
- S. Bolisetty, A. Traylor, R. Joseph, A. Zarjou and A. Agarwal, Am. J. Physiol.: Renal Physiol., 2016, 310, F385 CrossRef CAS PubMed.
- J. Kim, A. Zarjou, A. M. Traylor, S. Bolisetty, E. A. Jaimes, T. D. Hull, J. F. George, F. M. Mikhail and A. Agarwal, Kidney Int., 2012, 82, 278 CrossRef CAS PubMed.
- A. Agarwal, J. Balla, J. Alam, A. J. Croatt and K. A. Nath, Kidney Int., 1995, 48, 1298 CrossRef CAS.
- T. D. Blydt-Hansen, M. Katori, C. Lassman, B. Ke, A. J. Coito, S. Iyer, R. Buelow, R. Ettenger, R. W. Busuttil and J. W. Kupiec-Weglinski, J. Am. Soc. Nephrol., 2003, 14, 745 CrossRef CAS PubMed.
- S. T. Pittock, S. M. Norby, J. P. Grande, A. J. Croatt, G. D. Bren, A. D. Badley, N. M. Caplice, M. D. Griffin and K. A. Nath, Kidney Int., 2005, 68, 611 CrossRef CAS PubMed.
- T. D. Hull, A. I. Kamal, R. Boddu, S. Bolisetty, L. Guo, C. C. Tisher, S. Rangarajan, B. Chen, L. M. Curtis, J. F. George and A. Agarwal, J. Am. Soc. Nephrol., 2015, 26, 2139 CrossRef CAS PubMed.
- D. E. Leaf, S. C. Body, J. D. Muehlschlegel, G. M. McMahon, P. Lichtner, C. D. Collard, S. K. Shernan, A. A. Fox and S. S. Waikar, J. Am. Soc. Nephrol., 2016, 27, 3291 CrossRef CAS PubMed.
- T. Ichimura, J. V. Bonventre, V. Bailly, H. Wei, C. A. Hession, R. L. Cate and M. Sanicola, J. Biol. Chem., 1998, 273, 4135 CrossRef CAS PubMed.
- A. Menshikh, L. Scarfe, R. Delgado, C. Finney, Y. Zhu, H. Yang and M. P. de Caestecker, Am. J. Physiol.: Renal Physiol., 2019, 317, F1383 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02711e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |