
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of a photoswitchable DNA glue enabling high regulatory function and supramolecular chirality transfer†
Nadja A.
Simeth
 a,
Shotaro
Kobayashi
b,
Piermichele
Kobauri
a,
Stefano
Crespi
a,
Wiktor
Szymanski
ac,
Kazuhiko
Nakatani
*b,
Chikara
Dohno
*b and
Ben L.
Feringa
*a
a,
Shotaro
Kobayashi
b,
Piermichele
Kobauri
a,
Stefano
Crespi
a,
Wiktor
Szymanski
ac,
Kazuhiko
Nakatani
*b,
Chikara
Dohno
*b and
Ben L.
Feringa
*a
aCentre for Systems Chemistry, Stratingh Institute for Chemistry, Faculty for Science and Engineering, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
bDepartment of Regulatory Bioorganic Chemistry, The Institute of Scientific and Industrial Research, Osaka University, 8-1 Mihogaoka, Ibaraki 567-0047, Japan. E-mail: cdohno@sanken.osaka-u.ac.jp; nakatani@sanken.osaka-u.ac.jp
cDepartment of Radiology, Medical Imaging Center, University of Groningen, University Medical Centre Groningen, Hanzeplein 1, 9713 GZ Groningen, The Netherlands
First published on 27th May 2021
Abstract
Short, complementary DNA single strands with mismatched base pairs cannot undergo spontaneous formation of duplex DNA (dsDNA). Mismatch binding ligands (MBLs) can compensate this effect, inducing the formation of the double helix and thereby acting as a molecular glue. Here, we present the rational design of photoswitchable MBLs that allow for reversible dsDNA assembly by light. Careful choice of the azobenzene core structure results in excellent band separation of the E and Z isomers of the involved chromophores. This effect allows for efficient use of light as an external control element for duplex DNA formation and for an in-depth study of the DNA–ligand interaction by UV-Vis, SPR, and CD spectroscopy, revealing a tight mutual interaction and complementarity between the photoswitchable ligand and the mismatched DNA. We also show that the configuration of the switch reversibly dictates the conformation of the DNA strands, while the dsDNA serves as a chiral clamp and translates its chiral information onto the ligand inducing a preference in helical chirality of the Z isomer of the MBLs.
Introduction
Light is a convenient control element in biology,1–6 supramolecular chemistry,7,8 and material science.9–11 At the molecular level, such non-invasive control is enabled by photoresponsive elements, i.e. optical molecular switches.9,10,12–14 These photoswitches, when integrated into a system of choice, can affect its properties dynamically and reversibly.7,9 A crucial requirement in the photochemical control of a functional entity being part of a biological system or a material is the ability to address the photoactuator selectively in the complex spectral context of the framework that it is supposed to regulate. In other words, the key challenge in designing light-responsive systems is to ensure the separation of the spectral region in which the photoresponsive unit is addressed from those potentially photoactive elements or structures in its surroundings.15–17Molecular photoswitches have emerged as the addressable unit of choice due to the reversible nature of their light-stimulated isomerization.9,13,14 They have been employed to control, for instance, the shape of polymer films,18 surface properties,19 nano architectures,20 probes in chemical biology,21,22 and drug activity.2 DNA binders based on various photoswitches,23,24 such as overcrowded alkenes,25–27 diarylethenes,28,29 spiropyrans,30–32 hemiindigos,33,34 and azobenzenes,35–42 among others, have been investigated. The photoisomerization needs to be highly efficient to dynamically regulate such systems with light.15,16 In biological and biohybrid systems, photons are often filtered by other chromophores, like amino acids, nucleotides, and ligands interfering with the biomolecules (Fig. 1A, top) due to spectral overlap, compromising effective switching.43 The challenge is to spectrally separate the absorption band of the switch from those of all other components of the system (Fig. 1A, bottom). Here, we show how careful design of azobenzene photoswitches enables not only efficient photochemical interconversion, but also light-induced control of supramolecular assemblies. Specifically, light-induced isomerization between the non-binding E and the stabilizing Z isomer allows for reversible DNA ligand binding and duplex formation (Fig. 1B).
 | ||
| Fig. 1 (A) Schematic representation of the different contributions to the UV-Vis spectrum in biological molecules or biohybrid systems with small chromophores as ligands and azobenzene as photoactuator. (Top) Typical scenario where the ππ* and nπ* transitions of both photoisomers, and the spectrum of the ligands are extensively overlapping. (Bottom) Idealized scenario with high band separation of the respective electronic transitions of ligands, and E, Z photoisomers. (B) E–Z photoisomerization of an azobenzene-based mismatch binding ligand (MBL) facilitates reversible DNA hybridization. | ||
Deoxyribonucleic acid (DNA) is a biomacromolecule with a defined supramolecular architecture, determined i.e. by selective hydrogen bond interactions and π stacking.44 Formation of a three-dimensional helical structure is crucial for its biological function, frequently involved in disease-related malfunctions, and represents a key organization element in DNA-based nanomaterials.45–47 Small molecules, decorated with suitable substituents, can interact with DNA,36 and consequently affect its properties, such as sequence-specific hybridization, without introducing chemical modifications onto DNA. For example, mismatch-binding ligands (MBLs) influence DNA hybridization and its secondary structure by enhancing the stability of the duplex DNA.24 Several selective MBLs have been developed that discriminate, for instance, AA48 or GG homo mismatches.49,50 Such MBLs are typically DNA intercalators, which possess hydrogen-bonding groups complementary to the mismatched bases. A molecule with this property is 2-carbonylamino-1,8-naphthyridine (Fig. 2A), a small chromophore that recognizes guanine (G), and its covalent dimer was the first ligand reported for GG mismatch binding.49 A naphthyridine carbamate (NC, Fig. 2A) was found to be a better G recognition unit in terms of binding affinity to a GG homo mismatch and chemical stability of the molecule. Fig. 2B displays a few examples of MBLs based on NC units introduced by Nakatani and coworkers. Specifically, a naphthyridine carbamate dimer (NCD) was the first MBL reported as a molecular glue for DNA.51 A 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of NCD to dsDNA stabilized a GG mismatch by ΔTm (difference in melting temperature of DNA) of more than 30 °C in an appropriate 11-mer duplex DNA.51 Hence, it can function as molecular glue between DNA strands that do not hybridize spontaneously in a certain range of temperatures.51 Subsequently, both TD (a thermally degradable NCD analog) and NCDA3 (a photoswitchable derivative of NCD) were developed (Fig. 2B).52,53 In NCDA3, an azobenzene scaffold was employed as the photochromic core structure.53,54
:1 ratio of NCD to dsDNA stabilized a GG mismatch by ΔTm (difference in melting temperature of DNA) of more than 30 °C in an appropriate 11-mer duplex DNA.51 Hence, it can function as molecular glue between DNA strands that do not hybridize spontaneously in a certain range of temperatures.51 Subsequently, both TD (a thermally degradable NCD analog) and NCDA3 (a photoswitchable derivative of NCD) were developed (Fig. 2B).52,53 In NCDA3, an azobenzene scaffold was employed as the photochromic core structure.53,54
 | ||
| Fig. 2 (A) Naphthyridine carbamate (NC) units selectively bind to GG homo-mismatches. (B) Structures of diverse (activatable) MBLs. (C) Overview of the current work. | ||
Azobenzenes are light-responsive molecules that have a thermodynamically stable E and a photochemically generated, metastable Z isomer. While the E isomer is apolar and planar, the Z isomer has a dipole moment of ca. 3 D and a strongly twisted, helical shape.12,55,56E–Z isomerization can be triggered photochemically in both directions in a reversible manner. Moreover, Z → E interconversion occurs spontaneously in a thermal process and is associated with a defined thermal lifetime τ of the Z form.12,55 Azobenzenes are frequently used as photoswitchable motif to design light-responsive DNA–ligands37–42 or nucleobase surrogates57–61 due to their facile synthesis, tunability, and pronounced geometrical change upon photoisomerization.12 Indeed, in MBLs, the helical conformation of the Z isomer has been shown to have the ideal geometry to bind to DNA mismatches while the E form had lower affinity.24
In NCDA3, the E → Z photoisomerization was inefficient and several minutes of irradiation with UV light were required to induce sufficient isomerization. Moreover, extensive spectral overlap of the various chromophores involved prevented detailed spectroscopic studies and effective isomerization.53,54 These drawbacks impair the use of NCDA3 in rapidly responding DNA-based systems. Development of photoswitchable MBLs with precisely tuned photoisomerization properties would pave the way toward regulation of biological processes by targeting endogenous DNAs and RNAs.62
Here, we focus on tuning the photochemical properties of the azobenzene core structure to adjust them to the requirements of the system (cf. Fig. 1A and B), which enabled fast dynamic control of DNA double helix formation and real-time studies. Careful design of a suitable photoactuator furnished a highly sensitive ligand (Fig. 2C) featuring (i) excellent band separation, (ii) quantitative photoisomerization, (iii) high quantum yield (QY), (iv) fast response, and (v) excellent dsDNA assembly ability. Additionally, we provide in-depth insight into mutual interaction of the photoswitchable MBLs and the dsDNA, based on extensive spectroscopic and computational studies. In particular, we present the discovery of a highly effective photoswitchable DNA glue. This work highlights major steps towards the development of photoresponsive, fully reversible MBLs and important advances towards the rational design of light-responsive systems for future non-invasive control of biological function with potential for nanomaterial assembly.
Results and discussion
Computationally supported molecular design
To analyze the various contributions to the UV-Vis absorption spectrum of our existing photoswitchable MBLs, and to design superb variants, we simulated the spectra of individual components in water using time-dependent density function theory (TD-DFT) at the PBE0/6-311+G(2d,p) level of theory, a method proven to be reliable for azoswitches (with SMD implicit model used for water as a solvent, for details cf. Section 6 in the ESI†).63 The calculated spectra of naphthyridine methylcarbamate and the azobenzene core structure in NCDA3 are depicted in Fig. 3 (black and red solid lines, respectively). The first allowed transition of the naphthyridine methylcarbamate at 294 nm overlaps substantially with the main band of E-CH2NHCH3, namely its ππ* transition, at 355 nm. Due to the spectral overlap, selective irradiation of the ππ* transition of azobenzene might not be possible and photons needed to trigger the E–Z isomerization might be filtered by the NC moiety. With this result in hand, we focused on finding azobenzenes in which the ππ* transitions are more bathochromically shifted and the separation between isomer bands is larger. Anticipating that 4-alkyloxy substituted azobenzenes would absorb at longer wavelengths than alkyl-substituted analogs and show good photostationary distributions (PSDs),64–66 we focused on 4,4′-di-methoxy azobenzene E-OCH3. Using TD-DFT we established that the ππ* transition of the E isomer is bathochromically shifted (λmax = 382 nm, cyan solid line, Fig. 3) and consequently, the overlap with the naphthyridine carbamate absorption band is significantly reduced. We also found that in 4,4′-di-methoxy azobenzene the absorption maximum of the E isomer corresponds to an absorption minimum of the Z form at 388 nm (Fig. 3A). In contrast, the maximum of the E-CH2NHCH3 (λmax = 355 nm) and the minimum of the Z form (λmin = 388 nm) are more than 30 nm apart, leading to a substantial overlap of the spectra. Hence, the bands of both photoisomers in the alkoxy-substituted model are predicted to be better separated than in the alkyl-substituted core of NCDA3, which will facilitate higher PSDs and most likely highly selective switching. | ||
| Fig. 3 (A) Computed UV-Vis spectra of the photoactive core structures involved employing TD-DFT at the PBE0/6-311+G(2d,p) level of theory using the SMD model for water. Vertical lines indicating the position of the ππ* transition bands allow to better appreciate the spectral separation of E-CH3NHCH3 and E-OCH3 with their respective Z isomers. (B) Studied core structures. | ||
Synthesis
The synthesis of 4,4′-bisalkoxy based MBLs started from the azo-coupling between 4-hydroxy aniline (5) and phenol to result in 4,4′-di-hydroxy-azobenzene 6 (Scheme 1).67 Alkylation of 6 with 1,2-dibromoethane and 1,3-dibromopropane, provided 7 and 8, respectively (59% and 56% yield). Treatment with Ns-protected amine 9, under basic conditions, yielded compounds 10 and 11. Subsequent deprotection using thiophenol gave target compounds 2 and 3 in 62% and 79% yield, respectively. MBL 4 was obtained using reductive amination as the key step for the introduction of the NC groups onto the azobenzene core (see Scheme S2, ESI, Sections 1.2 and 7,† together with a full characterization and spectral data of 2–4). | ||
| Scheme 1 Synthesis of target compounds 2 and 3via alkylation as key step. | ||
Photophysical and photochemical studies
We investigated the photophysical and photochemical properties of compounds 2–4 and compared them to the bis-alkyl analogue NCDA3, as well as to the simulated spectra (Fig. 3A). An overview of the characteristics in the presence and absence of DNA is given in Table 1 and representative measurements are displayed in Fig. 4 (see ESI† for the full set of spectra and measurements). The parent alkyl substituted NCDA3 has an absorption maximum at 320 nm in buffer, which lies between the predicted transition maxima of naphthyridine methylcarbamate (294 nm) and E-CH2NHCH3 (355 nm). Due to the overlap with an absorption band of the NC moiety, the exact position of the ππ* transition of the azobenzene moiety in E-NCDA3 could not be assigned (Fig. 4A).53 In contrast, the E isomers of the 4,4′-alkoxy azobenzene derivatives 2–4 exhibit a well-defined ππ* transition that is significantly bathochromically shifted to ca. 365 nm. This shift leads to a clear separation from the absorption band of the NC moiety (λNC, Fig. 4C and ESI, Section 2.2†) and is in agreement with the TD-DFT calculations; the calculations predict the position of the transition maximum ca. 20 nm bathochromically shifted compared to measurements (Fig. 3). Gratifyingly, the addition of dsDNA containing a d(CGG)/d(CGG) mismatch does not significantly influence the positions of the ππ* transition bands of the azobenzene core structures (see Table 1 and ESI, Section 2.2†). However, λNC experiences a slight bathochromic shift of 1–5 nm in compounds 2, 3, and 4, an effect that can be attributed to the intercalation of the corresponding chromophore,68 here the NC.| Compound | λ ππ*(E) (λNC) [nm] | ε (λππ*E) [M−1‧cm−1] | λ nπ*(Z) [nm] | Φ E→Z [%] | τ at 25 °Ch | PSD365nm, [E:Z] |
PSD430nm (PSD460nm) {PSD470nm} [E:Z] |
|
|---|---|---|---|---|---|---|---|---|
| a Aq. tris buffer (50 mM, 150 mM NaCl, pH = 7.5). b Aq. phosphate buffer (0.01 M, 100 mM NaCl, pH = 7.0). c Water. d Absorption maximum consists of contributions from both the azobenzene core and the NC moiety. e At λ = 320 nm. f Not determined. g Aq. phosphate buffer (5 mM, 50 mM NaCl, pH = 7.0). The photostationary state distribution (PSD) differs between. h Assigned by 1H-NMR using a 365 nm lamp or a 430 nm LED, respectively, 1 mM in D2O, see ESI. i Data from ref. 53. j Assigned by peak area integration at the isosbestic point in HPLC analysis after irradiation with a 365 nm lamp, or a 460 nm LED (values in “( )”), or a 470 nm LED (values in “{ }”), respectively, see ESI. | ||||||||
| Without DNA | NCDA3a | 320d | 28850e |
432 | 0.7 | 5.6 | 86:14h, 50:50i |
88:12h |
| 2 | 365 (332) | 12830 |
439 | 42 | 3.2 | 21:79h, 17:83j |
80:20h, (87:13)j |
|
| 3 | 365 (332) | 11900 |
438 | 53 | 7.4 (23)c | 15:85h, 6:94g |
65:35h, (80:20)j |
|
| 4 | 367 (332) | 11540 |
437 | 27 | 18.5 | 12:88h, 3:97j |
49:51h, (71:29)j |
|
| With DNA | NCDA3b | 321d | n.d.f | 432 | 5 | 9.4g | 25:75i |
n.d.h |
| 2 | 365 (334) | n.d.f | 440 | 15 | 12.2 | 32:68j |
79:21j, {91:9}j |
|
| 3 | 365 (337) | n.d.f | 440 | 11 | 15.2 (13)g | 38:62j |
62:38j, {88:12}j |
|
| 4 | 363 (333) | n.d.f | 443 | 14 | 13g | 31:69j |
57:43j, {82:18}j |
|
 | ||
| Fig. 4 (A) The UV-Vis spectrum of compound NCDA3 (25 μM in tris buffer) is shown for comparison at the thermal equilibrium (cyan), the PSS365nm (red), and the PSS420nm (grey). (B) Photoinduced E–Z isomerization of compounds 2–4. (C) UV-Vis spectrum of compound 3 (50 μM in phosphate buffer) at the thermal equilibrium (cyan), the PSS365nm (red), and the PSS445nm (grey). (D) Excerpt of the 1H-NMR spectrum showing the protons of the azo-core moiety of compound 3 at the thermal equilibrium (cyan), the PSS365nm (red), and the PSS430nm (grey). (E) Repeated irradiation of compound 3 with 365 nm and 445 nm light in the presence (red) and absence (cyan) of duplex DNA (5′-d(CTAACGGAATG)-3′/3′-d(GATTGGCTTAC)-5′). | ||
Irradiation of the thermally equilibrated samples with 365 nm light induced photochemical E → Z isomerization in all investigated compounds. The photochemical process is accompanied by characteristic changes in the UV-Vis absorption spectra: upon irradiation, the ππ* transition of the E disappears leaving an absorption minimum and the nπ* band of the Z form around 440 nm increases as predicted by TD-DFT (Fig. 4C and ESI, Section 2.2†). 1H-NMR measurements and the presence of isosbestic points in the UV-Vis spectra indicate a clean two-component isomerization process (cf.Fig. 4D and ESI, Section 2.6†). The QY for this photoreaction is around 0.7% for NCDA3, a quite low value for classical azobenzene scaffolds, which show usually QYs ΦE→Z of around 10–20% in polar protic solvents.55 We hypothesize that, in this case, overlap with the NC unit diminishes the isomerization efficiency of the azobenzene moiety in the MBL, which is supported by the TD-DFT calculations (vide supra). Much to our delight, the spectral separation as well as the isomerization was found to be drastically improved in compounds 2–4 with QYs between 27% and 53%. These high photoisomerization efficiencies allowed for rapid E → Z isomerization in the seconds range at spectroscopic concentrations using a standard UV-LED (cf. ESI, Section 2.3†). The presence of duplex DNA influences the photoisomerization efficiencies (ca. 5% for NCDA3 and 11–15% for 2–4, cf.Table 1) suggesting a tight interaction of the MBL motif with the DNA strands.
The photostationary state distribution (PSD) differs between the various azobenzene-based MBLs. Notably, in the parent NCDA3 only ca. 50% of Z isomer could be observed at the PSS365nm,53 while in compounds 2–4 up to 97% of the metastable Z was detected (HPLC analysis, cf. Table 1 and ESI, Sections 2.6 and 2.7†). The relatively low PSD365nm in NCDA3 can be rationalized by an overlap of the UV-Vis absorption spectra of the E and Z photoisomer according to TD-DFT calculations (vide supra). The alkoxy-substituted derivatives show significantly higher PSD365nm, most likely due to a better separation of the transition bands of E and Z form, as predicted by computational analysis (Fig. 3). Photochemical Z → E isomerization was facilitated by irradiation with blue light (430 nm, 460 nm, or 470 nm depending on the setup used, see ESI, Section 2†) for NCDA3 and compounds 2–4, respectively. In NCDA3, 88% of the E form could be regenerated,53 while for 2–4 between 71 and 87% E isomer was observed (HPLC analysis, cf.Table 1, Fig. 4D, and ESI, Sections 2.6 and 2.7†).
The Z → E isomerization could also be achieved thermally. The thermal lifetimes of all compounds were mostly found to be prolonged in the presence of dsDNA, being an indication that the Z isomer is energetically stabilized in the presence of DNA (cf.Table 1 and ESI, Section 2.5†).
The photochemically induced E–Z isomerization was reversible for all switchable ligands 2–4 and could be repeated over several cycles both in the presence and absence of DNA. We found that the isolated switches suffered fatigue after multiple repetitions. In the presence of DNA, though, photochemical isomerization appeared to be very robust as exemplified for 3 in Fig. 4E (see also ESI, Section 2.4†). This behavior, together with the prolonged thermal stability (increased lifetimes of the Z isomers, vide supra), indicates a tight interaction of the photoswitchable MBLs and the dsDNA, albeit without compromising the photoswitching, which offered intriguing opportunities for a more detailed investigation, as outlined below.
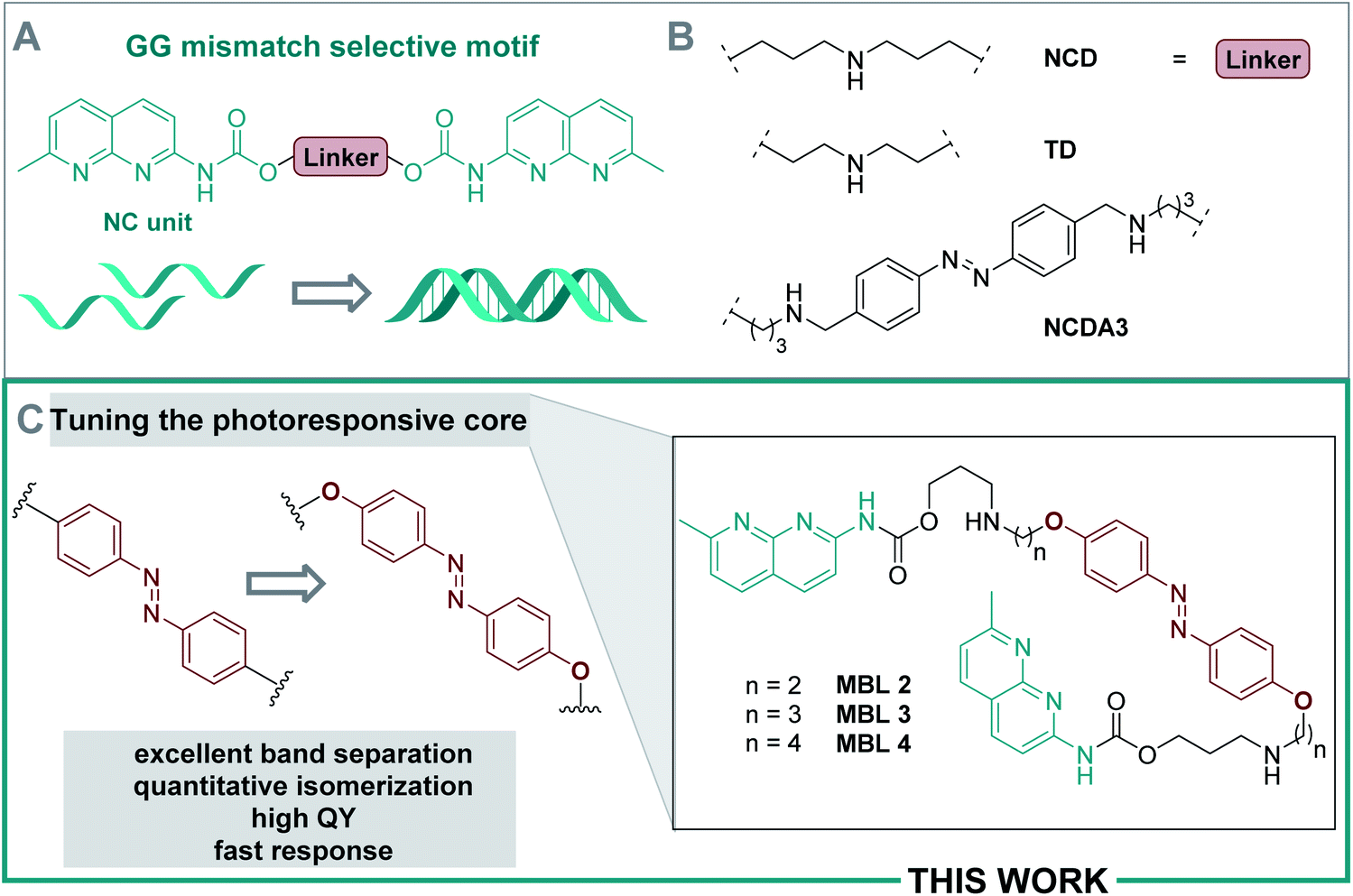
DNA melting experiments
Thermal melting curves were measured for an 11-mer dsDNA (5 μM) containing a d(CGG)/d(CGG) mismatch in the presence of compounds 2, 3, and 4 (20 μM) with and without photoirradiation (365 nm for 20 s). The absorbance of the sample was monitored at 260 nm in the temperature range 2–80 °C with a heating rate of 1 °C min−1. Melting temperatures (Tm, °C) were calculated by a median method except for Tm*, where the differential method was applied. Ligands were preincubated at 60 °C before the thermal melting experiments to reach the thermally equilibrium where the E isomers were exclusive. The presence of the E isomers had neglectable effect on the Tm values under the investigated conditions; the increase in Tms was less than 1 °C compared to the ones in the absence of the ligand (Tm1, Fig. 5A–C, blue filled circles). In contrast, all ligands showed a stabilization effect on the dsDNA after irradiation with 365 nm light indicated by increasing Tm (Tm2, Fig. 5A–C, red filled diamond). The use of compound 3, which contains a C3 linker, resulted in the highest ΔTm value. After photoirradiation, Z-3 stabilized the target DNA duplex by up to ca. 16 °C (from 27.5 to 43.3 °C, Fig. 5A), while the Z isomers of 2 and 4 showed smaller ΔTm values than for 3, raising the temperature by 11 °C (from 27.6 to 38.4 °C, Fig. 5A) and 9 °C (from 27.8 to 37.0 °C, Fig. 5C), respectively. The observations are supported by molecular dynamics (MD) simulations which are in agreement indicating that the DNA double helix in the Z-3-dsDNA complex is consistently the most stable one over 300 ns in temperature-dependent simulations (cf. Section 6.3 in the ESI†). In previous studies NCDA3 was found to exhibit a ΔTm of ca. 15 °C (from 32.7 to 48.0 °C).53 However, E-NCDA3 showed background activity and stabilized the mismatched DNA to some extent (Tm1 = 32.7 °C),53 while E-3 shows hardly any effect on Tm1. This does not only make 3 the MBL providing the largest Tm change before and after irradiation (ΔTm = 16 °C), but also the photoswitch with the clearest difference between the inactive E and the stabilizing Z isomer. The stabilization effect is fully reversible by photoswitching upon irradiation with different wavelengths as expected from Fig. 4E. Melting profiles in the presence of 3 after repeated irradiations with 365 nm and 460 nm light, are shown in Fig. 5D. Tm without irradiation was around 30 °C (cyan half-filled circle), which is slightly higher than that in Fig. 5B because of residual Z isomer existing under ambient light. Irradiation at 365 nm increased Tm (red filled diamond), blue-light irradiation decreased Tm back to 30 °C (blue filled circle), and subsequent irradiation at 365 nm again increased Tm (red half-filled diamond) exhibiting a profile almost identical to the one after the first UV-light irradiation. The reversibility of Tm demonstrates that the light-mediated photoisomerization of MBL 3 (Fig. 5D) can be fully translated to govern the stability of the mismatch containing dsDNA.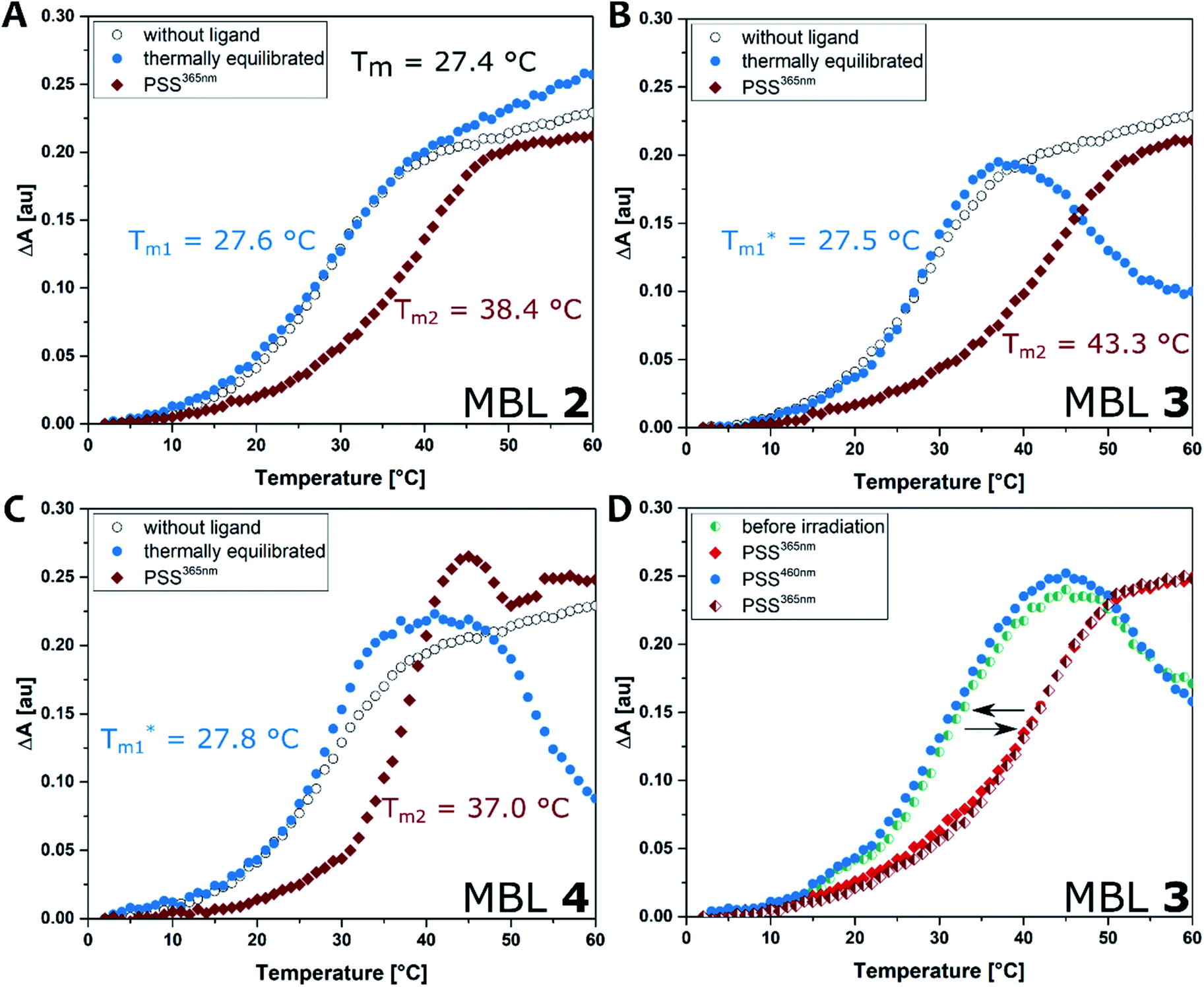 | ||
| Fig. 5 DNA melting experiment: the absorbance of the sample was monitored at 260 nm from 2 to 80 °C with a heating rate of 1 °C min−1. Conditions: dsDNA (5′-d(CTAACGGAATG)-3′/3′-d(GATTGGCTTAC)-5′, 5 μM) in phosphate buffer (10 mM, 100 mM NaCl, pH 7.0), in absence (black open circle) and presence of each ligand (compounds 2–4, 20 μM). (A) Compound 2; (B) 3; (C). 4. Ligands were either pretreated with heat at 60 °C in the dark to reach thermally equilibrated state (blue filled circle) or photo-irradiated at 365 nm for 20 s (red filled diamond). (D) Repeated irradiation of 3 with dsDNA. The sample before irradiation (cyan half-filled circle) was irradiated at 365 nm (red filled diamond), subsequent irradiation at 460 nm (blue filled circle), followed by irradiation at 365 nm (red half-filled diamond). | ||
Surface plasmon resonance
Binding of the ligands to DNA was further evaluated by surface plasmon resonance (SPR). Hairpin forming dsDNA (5′-CTAA CNG AATG TTTT CATT CMG TTAG-3′) containing a d(CNG)/d(CMG) site was immobilized onto a sensor chip surface. The dsDNA containing a GG mismatch (N = M = G), CC mismatch (N = M = C), and GC match (N = C, M = G) were used to compare the binding selectivity. Solutions of each ligand at concentrations ranging from 0.031 to 0.5 μM, with and without irradiation, were injected to the surface. Fig. 6 shows SPR curves for the GG mismatch dsDNA. Increasing concentrations of ligands at the PSS365 (main isomers Z-2–4) showed an increasingly strong response signal (Fig. 6D–F), while ligands at the thermal equilibrium (main isomer E-2–4) exhibited response of much smaller intensities (Fig. 6A–C). It is evident that at similar concentrations, the Z isomer shows drastically enhanced response and as a consequence the SPR curves are convex upward in the association step beginning from 57 to 117 s. This feature suggests the contribution of aggregation/dissociation of ligands and/or conformational changes of the dsDNA–ligand complexes on the surface. Although it is difficult to estimate the binding constants because of the uncommon shape of the curves, ligand Z-3 (Fig. 6E) provided the highest response, which is well-correlated with stabilization effects on dsDNA as observed in Tm (Fig. 5).54 Compounds with linkers that are one methylene unit shorter (2) or longer (4) than in 3 resulted not only in weaker binding affinities but also in smaller changes before and after irradiation. We also compared the binding of the different photoirradiated ligands towards GG and CC homo mismatches and a GC match. The results confirm the marked selectivity towards the GG mismatch, as expected from the recognition moieties in MBLs consisting of dimeric NC (Fig. S25 and S26†).24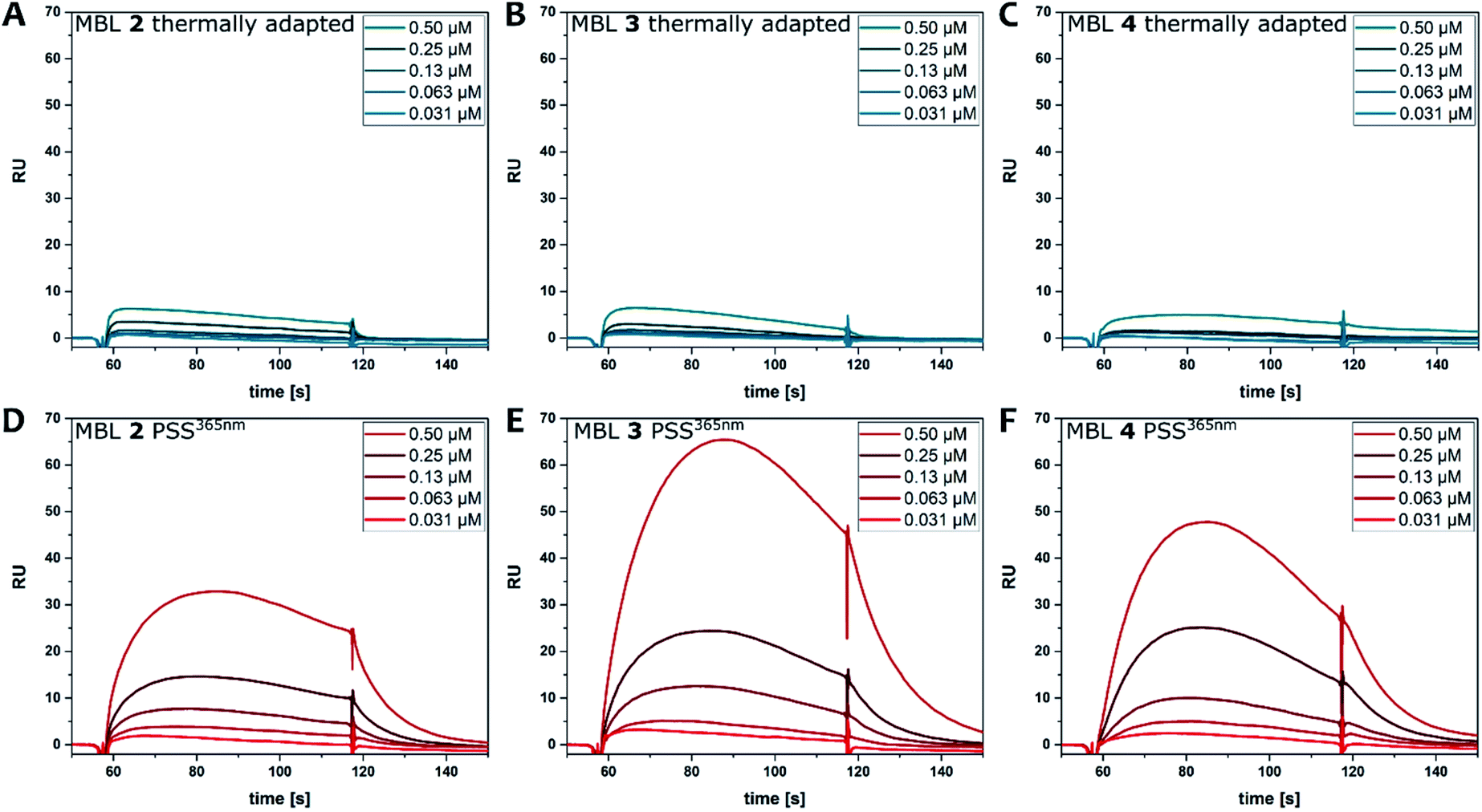 | ||
| Fig. 6 SPR analysis of the binding of ligands 2 (A and D), 3 (B and E), and 4 (C and F) to hairpin dsDNA containing a CGG/CGG site. Ligands at a series of concentrations (0.031, 0.063, 0.13, 0.25, 0.5 μM) were injected during association steps (from 57 to 117 s) followed by dissociation step where running buffer was injected. Either ligands at thermal equilibrium (A–C) or ligands after irradiation at 365 nm (D–F) were injected to surface immobilized with the dsDNA containing a GG mismatch. | ||
CD spectroscopy
CD spectroscopy is frequently used to probe ligand–DNA interactions37,38,40,69–72 and was previously employed to study the affinity of MBLs and their effect on DNA hybridization.48,73,74 Due to the excellent separation of the absorption bands of DNA, NC, and azobenzene our system allows for clear identification of the different transitions contributing to the CD spectrum. The part of the spectrum below 300 nm is mainly attributed to the DNA, while the one above 300 nm can be assigned to the different chromophores in the MBLs. While the compounds in their E form did not show any Cotton effect in the absence of DNA, an increasing CD signal is observed by titrating MBLs to the GG mismatch-containing DNA 11-mer for compounds E-2 and E-4 (Fig. 7A, and ESI, Section 5†). In particular, the positive induced CD (ICD) signal at ca. 340 nm corresponds well with the NC absorption band and the one at 365 nm with the ππ* transition band of the azobenzene core indicating an interaction of the MBL with the DNA (Fig. 7A). In contrast, E-3 hardly showed any ICD (Fig. 7C). While addition of increasing amounts of E-3 and E-4 led to a decrease in intensity of the signal in the DNA region of the CD spectrum (Fig. 7B and S31†), a larger change was found for E-2 with the formation of an additional negative signal at 228 nm (Fig. 7A). We mainly observed this characteristic structural change for Z forms of the MBLs (vide infra). | ||
| Fig. 7 Binding studies using CD spectroscopy by titrating increasing amounts of MBLs 2 (A), and 3 (B) to dsDNA (5′-d(CTAACGGAATG)-3′/3′-d(GATTGGCTTAC)-5′, 5 μM) in phosphate buffer at their thermal equilibrium and of 3 at the PSS365nm (C). (D) CD spectra of compound 3 (10 μM) in the presence of dsDNA (5 μM) in phosphate buffer. Altering irradiation with 365 nm and 470 nm light and reversible structural change in DNA and of the ICD of 3. (E) Excerpt showing the reversible structural change in the DNA-region of the CD spectra. (F) Irradiation of MBL 3 with different colors of light: CD spectrum at the thermal equilibrium (black), the PSS365nm (red), the PSS430nm (grey), and the PSS470nm (cyan) containing varying amounts of Z isomer (a wider spectral range can be found in Fig. S30†). | ||
Irradiation of the samples with 365 nm light induces E → Z isomerization and results in higher Tms (vide supra), greater response in SPR (vide supra), and more pronounced changes in the CD spectrum (exemplarily shown for MBL 3 in Fig. 7C, for further measurements see ESI, Section 5†). The latter shows a significant change of the Cotton effect below 300 nm, which indicates a structural change of the dsDNA and additional contribution originated from NC embedded in the helix.75 As already described for E-2, formation of a new signal at 228 nm is observed for Z-2–4 with Z-3 inducing the most prominent change. In the MBL region, photoisomerization is accompanied by a decrease of the ππ* absorption band of the E isomer in the UV-Vis absorption spectrum (vide supra) and the corresponding signal in the CD spectrum practically disappears. A pronounced negative signal around 440 nm is observed (Fig. 7C and ESI, Section 5†), while the positive signal at ca. 320 nm intensifies. The position of λ = 440 nm the signal is in good accordance with the λmax values of the nπ* transition band of the Z isomer in 4,4′-di-alkoxy azobenzene (cf. TD-DFT spectrum in Fig. 3). Also, in this case, the strongest signals originate from MBL Z-3. Both the structural change in the DNA and the CD signal in the MBL region can be reverted by irradiation with blue light (Fig. 7D and E and ESI, Section 5†). Specifically, the ratio of the two photoisomers can be adjusted using different wavelengths of light and the composition in the dark and at the PSS365nm, PSS430nm, and the PSS470nm directly correlates with the observed CD response and the degree of stabilization of the dsDNA (Fig. 7F, a wider spectral range can be seen in Fig. S30†).
Repeated irradiation with 365 nm and 470 nm light of the same sample showed reversible stabilization and destabilization of the mismatched DNA as exemplarily by MBL 3 shown in Fig. 7D and E. This behavior demonstrates a reversible mutual change in DNA structure and binding of the ligands. Analyzing the titration experiments by plotting the CD signal at 341 nm of the ligand versus the ligand concentration allowed for a qualitative description of the binding affinities of the photochromic MBLs (ESI, Section 5.5†). Using a single set of independent binding sites model and assuming a 2:1 ligand:dsDNA stoichiometry as reported previously,53,54 we found an apparent KD of 0.7 μM for compound 3 as the strongest binder, followed by compound 2 with 15.2 μM and compound 4 with 23.6 μM. This is in agreement with the order observed from DNA melting experiments (vide supra). Also, in SPR compound 3 showed the strongest response while compound 4 and 2 gave weaker signals (vide supra). Though the response of MBL 4 is stronger than the one of 2 in SPR, this could be also due to unspecific binding to DNA and is consequently not necessarily contradictory to the Tm and CD studies. Altogether, compound 3, featuring a C3 linkage, emerged throughout our studies to be both the strongest binder and simultaneously the photoswitch with the largest differences in affinity between the two isomers. A pronounced influence of linker length on the binding to DNA was already observed for photoswitchable MBL in our earlier studies.54 We hypothesize that a combination of factors contribute to the observed behavior, such as relative stabilities of conformations of MBL in both bound and unbound states, and the entropic costs as well as the solvation changes upon binding.
Chirality transfer
Photoswitches such as azobenzenes have been studied previously in combination with DNA, either as nucleobase surrogates23,57–61 or as non-covalent ligands.23,37–40 Commonly, the E isomer showed the stronger interaction with DNA due to its π-stacking ability employing dsDNA38,39,60,61,76 or a G-quadruplex.41,42 In sharp contrast, in our system it is the Z isomer of compound-3 that shows the stronger CD signal (Fig. 7B and C). In the more common designs, E → Z photoisomerization decreased the affinity to DNA by loss of the π–π-interaction and thus allowed for photocontrol of the system's architecture.39,41,61,76 These reports highlight the possibility of using CD spectroscopy both to follow the process, usually showing a decrease of the Cotton effect for the Z isomer38,58,76 to assign the binding mode of both photoisomers, if they are used as ligands, by the pattern of the ICD signal.38,39Furthermore, we and others have reported that the chiral environment provided by DNA can influence the chiroptical properties of organic chromophores77–80 as well as the stereochemical outcome of a photoisomerization reaction as shown, for instance, for DNA-binding diarylethenes.29 The same experiment, however, cannot easily be repeated for azobenzenes, as the E isomer is achiral and the Z(P) and Z(M) enantiomers (interconverting helical conformers, Fig. 8A) undergo fast isomerization (racemization) in solution81i.e. the dynamical helicity of Z-azobenzene as shown in Fig. 8A. As a comparison, the photoinduced closure reaction in diarylethenes forms a new covalent bond and hence, the enantiomers are configurationally stable and can be isolated.29 For this reason, a chirality transfer onto the Z-azobenzene unit has been reported for azobenzenes after covalently attaching a stereogenic element,82,83 often in cyclic systems to induce additional strain,81,84,85 but not for supramolecular systems based on DNA as described here.
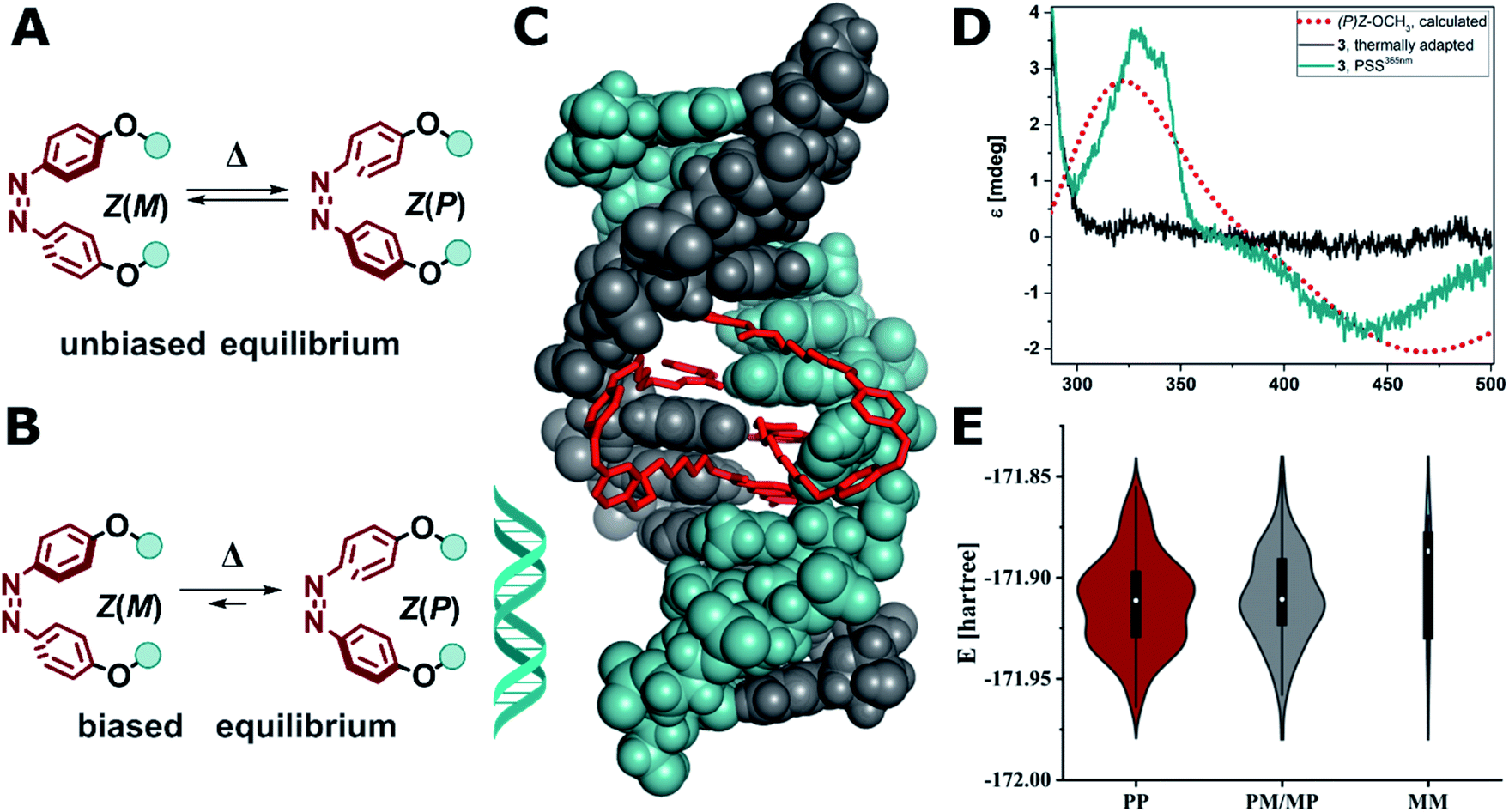 | ||
| Fig. 8 (A) Unbiased equilibrium of interconverting helical conformers. (B) DNA-induced biased equilibrium favoring Z(P) configuration in the alkoxy azobenzene-based MBLs 2–4. (C) Calculated structure of Z-3 in DNA (note two molecules of Z-3 bind to form stable dsDNA). (D) Experimental CD spectrum of compound 3 (10 μM) in the presence dsDNA (5′-d(CTAACGGAATG)-3′/3′-d(GATTGGCTTAC)-5′, 5 μM) in phosphate buffer at the thermal equilibrium (grey solid line) and after E → Z isomerization at the PSS365nm (cyan solid line), which corresponds well with the computed CD spectrum of Z(P)-OCH3 (red dotted line, TD(25 states)-PBE0/6-311+G(2d,p) level of theory using the SMD implicit model for water). (E) Violin plot of the energy and distribution of two molecules of Z-3 in a 2:1 complex to mismatched DNA adopting (PP)Z, (PM/MP)Z, and (MM)Z configuration (for details see ESI, Section 6.3†). | ||
Conversely, in our system the functionalized azobenzenes do not serve as a mere intercalators or groove-binding ligands towards a canonical DNA, but as photocontrollable glue molecule to promote double helix formation in a mismatched dsDNA. Due to the tight binding of the NC moieties, which was previously demonstrated by the necessity to use a thermally degradable variant (TD in Fig. 2B) to make the system reversible,52 our system is closer to a macrocyclic structure, where the dsDNA serves as a chiral clamp, than to a classical DNA–ligand interaction. Hence, we focused on studying the origin of the observed Cotton effect around 440 nm in the Z isomers (Fig. 8D) in more detail aiming to elucidate whether the signal is derived from the excitonic coupling between the chromophores or from DNA imprinting its chiral information in the Z azobenzenes. The transfer of chirality works through energetical discrimination of the enantiomers of the Z-azobenzene inducing a bias in the equilibrium between Z(M)and Z(P) helical configuration (Fig. 8B) and can be approximated by theoretical calculations.81,83,84 Consequently, we performed a thorough computational investigation models of MBLs 2–4 in a 2:1 complex with the DNA 11-mer. The DNA complexes were built by modifying a similar structure that was previously released (PDB ID: 1X26).48 To minimize any bias originated by the selection of the initial structures, we performed a preliminary energy minimization with the OPLS3e force field and subsequently we performed multiple 300 ns MD simulations with the parmbsc1 force field. Randomly sampled structures were finally optimized with Grimme's GFN force field.86Fig. 8C shows as an example the optimized geometry of 3 bound to dsDNA (note that two molecules of 3 are bound to dsDNA). We analyzed the frequency and energy distributions of both Z(P) and Z(M) configurations present in the two MBLs. As two ligands are required to stabilize the mismatched DNA, the two azobenzenes can either both exhibit P (PP), both M (MM), or a mixed configuration (PM or MP). In most cases, the two azobenzenes in the MBLs adopted a PP or a mixed MP/PM helicity in the Z configuration over the MM one (see Fig. 8E and Section 6.3 in the ESI†). These results suggest that the system biases the formation of PP helicity over MM helicity in the two azobenzenes (Fig. 8B). To support the interpretation of a preference in the helicity of the switches, we calculated the CD spectrum of Z-OCH3 in its P form, that matched the Cotton effect observed experimentally (cf.Fig. 8D, red dotted line for the simulation, cyan line for the experiment).
These results indicate that the dsDNA and the MBL show a highly mutual supramolecular interaction: on the one hand, the Z form of the MBL can provide enhanced stabilization of the helical structure of DNA. On the other hand, the DNA is translating its chirality onto the Z isomer of the azoswitch (inducing right-handed (P) helicity in the dynamic helical system). To the best of our knowledge this has only been observed so far in azobenzenes with a covalently attached stereogenic element,9,81–85 and in supramolecular host–guest assemblies based on anion binding stilbenes.87 DNA was up to now only used to template the formation a preferred enantiomer during an electrocyclic reaction in DAE photoswitches,29 but never to induce a biased equilibrium in rapidly interconverting helical conformers such as in Z(P)-3 discussed in this work.
Conclusions
In summary, we used rational design aided by computational analysis to tune the photochemical properties of the azobenzene core structure for the next generation of photoswitchable DNA mismatch binding ligands. TD-DFT calculations suggested employing the 4,4′-di-alkoxy substituted azobenzene core to improve the spectral band separation of the involved chromophores. UV-Vis spectroscopy of the synthesized compounds confirmed the predicted λ-orthogonality between the absorption band of the naphthyridine carbamate moiety and the one corresponding to the ππ* transition of the E isomers in compounds 2–4. Compared to the MBLs based on classical azobenzene, the present ligands exhibit improved photophysical and photochemical properties, such as up to 75 times higher E → Z photoisomerization quantum yields. Moreover, the improved band separation in the UV-Vis absorption spectra between the various chromophores involved resulted in PSDs up to >95% for the new system we developed (in contrast to ca. 50% in NCDAs). These beneficial properties allowed to induce close to quantitative E → Z photoisomerization within a few seconds, a central premise for rapidly responding systems.Detailed studies using various complementary techniques revealed that the Z isomer of the new MBLs stabilized dsDNA more than the E form. Among the tested ligands, compound 3 with a C3 linker showed the best stabilization effect on dsDNA containing a GG mismatch showing a light-induced ΔTm of 16 °C. While in previous studies both photoisomers had affinity to mismatched DNA,53 the E isomer in MBL 3 does hardly show any stabilizing effect on the dsDNA. In stark contrast, the photochemically generated Z form of the same compound rapidly induces DNA duplex formation as seen CD spectroscopy. A strong response in SPR and titration studies revealing an apparent KD of 0.7 μM for Z-3 confirming its high affinity. Both the lack of activity of the thermodynamically stable E form and the high activity of the rapidly light-generated meta-stable Z isomer make MBL 3 and excellent candidate for further studies in real time.
Additionally, binding of the photoswitchable MBLs to dsDNA facilitates a unique supramolecular transfer of chirality onto the helically shaped Z form of the azo-switch as demonstrated by CD spectroscopy supported by an extensive computational analysis. Specifically, the presence of the dsDNA induced a bias in the dynamic equilibrium between Z(M) and Z(P) configuration, favoring an overall P configuration. This shows that photoswitchable MBL and mismatched DNA strands have a distinctive mutual interaction in which the photoswitches serve as DNA glue while the nucleic acid induces a transfer of chirality onto the switch. These findings present an important step towards fully dynamic and external control in reversibly photo-responsive DNA-based architectures, with prospects for adaptive nano systems and genetic regulation, and demonstrate the potential of rational design in the context of photo-responsive systems.
Data availability
The datasets supporting this article have been uploaded as part of the supplementary material.Author contributions
WS, KN, CD, and BLF designed the study. NAS and SK synthesised the compounds investigated. NAS performed UV-Vis and CD spectroscopy, TD-DFT calculations, NMR irradiation experiments. SK and CD performed SPR and DNA melting studies. NAS, SK and CD performed HPLC studies. PK and SC performed structure optimization and MD simulations on the DNA-MBL complex and data analysis. KN, CD and BLF supervised the work. NAS and CD wrote the paper. All authors discussed and commented on the manuscript. NAS, SC, WS, CD, and BLF acquired funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NAS thanks Prof. Dr Wesley R. Browne (University of Groningen) for fruitful discussion and the Humboldt Foundation for a Feodor-Lynen postdoctoral fellowship. PK thanks Dr Sebastian Thallmair (Frankfurt Institute for Advanced Studies, Germany) for his advice regarding the MD simulations. We gratefully acknowledge the generous support from the Horizon 2020 Framework Program (ERC Advanced Investigator grant no. 694345 to BLF), the Marie Skłodowska-Curie Actions (Individual Fellowship 838280 to SC; Co-funding of regional, national and international programmes 713482 to BLF), the Ministry of Education, Culture and Science of the Netherlands (Gravitation Program no. 024.001.035 to BLF), and the Netherlands Organization for Scientific Research (NWO, VIDI grant no. 723.014.001 for WS). This work was partially supported by Grants in Aid for Scientific Research (B) (18H02107) to CD from the Japan Society for the Promotion of Science (JSPS).References
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Reversible photocontrol of biological systems by the incorporation of molecular photoswitches, Chem. Rev., 2013, 113, 6114–6178 Search PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, Photopharmacology: Beyond proof of principle, J. Am. Chem. Soc., 2014, 136, 2178–2191 Search PubMed.
- M. M. Lerch, M. J. Hansen, G. M. van Dam, W. Szymanski and B. L. Feringa, Emerging Targets in Photopharmacology, Angew. Chem., Int. Ed., 2016, 55, 10978–10999 Search PubMed.
- K. Hüll, J. Morstein and D. Trauner, In Vivo Photopharmacology, Chem. Rev., 2018, 118, 10710–10747 Search PubMed.
- L. Albert and O. Vázquez, Photoswitchable peptides for spatiotemporal control of biological functions, Chem. Commun., 2019, 55, 10192–10213 Search PubMed.
- A. A. Beharry and G. A. Woolley, Azobenzene photoswitches for biomolecules, Chem. Soc. Rev., 2011, 40, 4422–4437 Search PubMed.
- M. Kathan and S. Hecht, Photoswitchable molecules as key ingredients to drive systems away from the global thermodynamic minimum, Chem. Soc. Rev., 2017, 46, 5536–5550 Search PubMed.
- J. H. Van Esch, R. Klajn and S. Otto, Chemical systems out of equilibrium, Chem. Soc. Rev., 2017, 46, 5474–5475 Search PubMed.
- Molecular Switches, ed. B. L. Feringa and W. R. Browne, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed.
- A. Goulet-Hanssens, F. Eisenreich and S. Hecht, Enlightening Materials with Photoswitches, Adv. Mater., 2020, 32, 1905966 Search PubMed.
- S. Krause and B. L. Feringa, Towards artificial molecular factories from framework-embedded molecular machines, Nat. Rev. Chem., 2020, 4, 550–562 Search PubMed.
- N. A. Simeth and S. Crespi, Photochemistry, The Royal Society of Chemistry, 2021, vol. 48, pp. 344–375 Search PubMed.
- S. Crespi, N. A. Simeth and B. König, Heteroaryl azo dyes as molecular photoswitches, Nat. Rev. Chem., 2019, 3, 133–146 Search PubMed.
- Z. L. Pianowski, Recent Implementations of Molecular Photoswitches into Smart Materials and Biological Systems, Chem.–Eur. J., 2019, 25, 5128–5144 Search PubMed.
- M. M. Lerch, M. J. Hansen, W. A. Velema, W. Szymanski and B. L. Feringa, Orthogonal photoswitching in a multifunctional molecular system, Nat. Commun., 2016, 7, 12054 Search PubMed.
- D. Wang, F. Schellenberger, J. T. Pham, H. J. Butt and S. Wu, Orthogonal photo-switching of supramolecular patterned surfaces, Chem. Commun., 2018, 54, 3403–3406 Search PubMed.
- M. Škugor, J. Valero, K. Murayama, M. Centola, H. Asanuma and M. Famulok, Orthogonally Photocontrolled Non-Autonomous DNA Walker, Angew. Chem., Int. Ed., 2019, 58, 6948–6951 Search PubMed.
- A. H. Gelebart, D. Jan Mulder, M. Varga, A. Konya, G. Vantomme, E. W. Meijer, R. L. B. Selinger and D. J. Broer, Making waves in a photoactive polymer film, Nature, 2017, 546, 632–636 Search PubMed.
- J. Zhang and H. Tian, Photochromic Materials: Preparation, Properties and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016, pp. 195–242 Search PubMed.
- J. W. Fredy, A. Méndez-Ardoy, S. Kwangmettatam, D. Bochicchio, B. Matt, M. C. A. Stuart, J. Huskens, N. Katsonis, G. M. Pavan and T. Kudernac, Molecular photoswitches mediating the strain-driven disassembly of supramolecular tubules, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11850–11855 Search PubMed.
- C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Light-Controlled Tools, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 Search PubMed.
- N. Yasuike, K. M. Blacklock, H. Lu, A. S. I. Jaikaran, S. McDonald, M. Uppalapati, S. D. Khare and G. A. Woolley, Photoswitchable affinity reagents: Computational design and efficient red-light switching, ChemPhotoChem, 2019, 3, 431–440 Search PubMed.
- A. S. Lubbe, W. Szymanski and B. L. Feringa, Recent developments in reversible photoregulation of oligonucleotide structure and function-1, Chem. Soc. Rev., 2017, 46, 1052–1079 Search PubMed.
- C. Dohno and K. Nakatani, Control of DNA hybridization by photoswitchable molecular glue, Chem. Soc. Rev., 2011, 40, 5718–5729 Search PubMed.
- A. S. Lubbe, Q. Liu, S. J. Smith, J. W. de Vries, J. C. M. Kistemaker, A. H. de Vries, I. Faustino, Z. Meng, W. Szymanski, A. Herrmann and B. L. Feringa, Photoswitching of DNA Hybridization Using a Molecular Motor, J. Am. Chem. Soc., 2018, 140, 5069–5076 Search PubMed.
- M. P. O'Hagan, P. Peñalver, R. S. L. Gibson, J. C. Morales and M. C. Galan, Stiff-Stilbene Ligands Target G-Quadruplex DNA and Exhibit Selective Anticancer and Antiparasitic Activity, Chem.–Eur. J., 2020, 26, 6224–6233 Search PubMed.
- M. P. O'Hagan, S. Haldar, M. Duchi, T. A. A. Oliver, A. J. Mulholland, J. C. Morales and M. C. Galan, A Photoresponsive Stiff-Stilbene Ligand Fuels the Reversible Unfolding of G-Quadruplex DNA, Angew. Chem., Int. Ed., 2019, 58, 4334–4338 Search PubMed.
- M. P. O'Hagan, J. Ramos-Soriano, S. Haldar, S. Sheikh, J. C. Morales, A. J. Mulholland and M. C. Galan, Visible-light photoswitching of ligand binding mode suggests G-quadruplex DNA as a target for photopharmacology, Chem. Commun., 2020, 56, 5186–5189 Search PubMed.
- T. C. S. Pace, V. Müller, S. Li, P. Lincoln and J. Andréasson, Enantioselective Cyclization of Photochromic Dithienylethenes Bound to DNA, Angew. Chem., Int. Ed., 2013, 52, 4393–4396 Search PubMed.
- J. Andersson, S. Li, P. Lincoln and J. Andréasson, Photoswitched DNA-Binding of a Photochromic Spiropyran, J. Am. Chem. Soc., 2008, 130, 11836–11837 Search PubMed.
- C. Brieke and A. Heckel, Spiropyran Photoswitches in the Context of DNA: Synthesis and Photochromic Properties, Chem.–Eur. J., 2013, 19, 15726–15734 Search PubMed.
- D. Avagliano, P. A. Sánchez-Murcia and L. González, Spiropyran Meets Guanine Quadruplexes: Isomerization Mechanism and DNA Binding Modes of Quinolizidine-Substituted Spiropyran Probes, Chem.–Eur. J., 2020, 26, 13039–13045 Search PubMed.
- D. V. Berdnikova, Design, synthesis and investigation of water-soluble hemi-indigo photoswitches for bioapplications, Beilstein J. Org. Chem., 2019, 15, 2822–2829 Search PubMed.
- D. V. Berdnikova, Visible-range hemi-indigo photoswitch: ON–OFF fluorescent binder for HIV-1 RNA, Chem. Commun., 2019, 55, 8402–8405 Search PubMed.
- G. M. Murawska, C. Poloni, N. A. Simeth, W. Szymanski and B. L. Feringa, Comparative Study of Photoswitchable Zinc-Finger Domain and AT-Hook Motif for Light-Controlled Peptide–DNA Binding, Chem.–Eur. J., 2019, 25, 4965–4973 Search PubMed.
- J. Rodriguez, J. Mosquera, S. Learte-Aymamí, M. E. Vázquez and J. L. Mascareñas, Stimuli-Responsive DNA Binding by Synthetic Systems, Acc. Chem. Res., 2020, 53, 2286–2298 Search PubMed.
- P. M. Pithan, C. Kuhlmann, C. Engelhard and H. Ihmels, Synthesis of 5-Alkyl- and 5-Phenylamino-Substituted Azothiazole Dyes with Solvatochromic and DNA-Binding Properties, Chem.–Eur. J., 2019, 25, 16088–16098 Search PubMed.
- B. Heinrich, K. Bouazoune, M. Wojcik, U. Bakowsky and O. Vázquez, Ortho-Fluoroazobenzene derivatives as DNA intercalators for photocontrol of DNA and nucleosome binding by visible light, Org. Biomol. Chem., 2019, 17, 1827–1833 Search PubMed.
- A. Bergen, S. Rudiuk, M. Morel, T. Le Saux, H. Ihmels and D. Baigl, Photodependent Melting of Unmodified DNA Using a Photosensitive Intercalator: A New and Generic Tool for Photoreversible Assembly of DNA Nanostructures at Constant Temperature, Nano Lett., 2016, 16, 773–780 Search PubMed.
- M. Deiana, Z. Pokladek, K. Matczyszyn, P. Mlynarz, M. Buckle and M. Samoc, Effective control of the intrinsic DNA morphology by photosensitive polyamines, J. Mater. Chem. B, 2017, 5, 1028–1038 Search PubMed.
- X. Wang, J. Huang, Y. Zhou, S. Yan, X. Weng, X. Wu, M. Deng and X. Zhou, Conformational Switching of G-Quadruplex DNA by Photoregulation, Angew. Chem., Int. Ed., 2010, 49, 5305–5309 Search PubMed.
- X. Xing, X. Wang, L. Xu, Y. Tai, L. Dai, X. Zheng, W. Mao, X. Xu and X. Zhou, Light-driven conformational regulation of human telomeric G-quadruplex DNA in physiological conditions, Org. Biomol. Chem., 2011, 9, 6639–6645 Search PubMed.
- A. Thomson, An Introduction to Spectroscopy for Biochemists, Biochem. Educ., 1981, 9, 35 Search PubMed.
- B. Alberts, A. Johnson, J. Lewis, D. Morgan, M. Raff, R. Keith and P. Walter, Molecular biology of the cell, Garland Science, New York, 6th edn, 2002 Search PubMed.
- S. C. Raghavan, DNA structure and human diseases, Front. Biosci., 2007, 12, 4402 Search PubMed.
- M. H. M. Schmidt and C. E. Pearson, Disease-associated repeat instability and mismatch repair, DNA Repair, 2016, 38, 117–126 Search PubMed.
- N. C. Seeman, Nanomaterials Based on DNA, Annu. Rev. Biochem., 2010, 79, 65–87 Search PubMed.
- K. Nakatani, S. Hagihara, Y. Goto, A. Kobori, M. Hagihara, G. Hayashi, M. Kyo, M. Nomura, M. Mishima and C. Kojima, Small-Molecule Ligand Induces Nucleotide Flipping in (Cag)n Trinucleotide Repeats, Nat. Chem. Biol., 2005, 1, 39–43 Search PubMed.
- K. Nakatani, S. Sando and I. Saito, Scanning of guanine-guanine mismatches in DNA by synthetic ligands using surface plasmon resonance, Nat. Biotechnol., 2001, 19, 51–55 Search PubMed.
- C. Wang, F. Pu, Y. Lin, J. Ren, C. Dohno, K. Nakatani and X. Qu, Molecular-glue-triggered DNA assembly to form a robust and photoresponsive nano-network, Chem.–Eur. J., 2011, 17, 8189–8194 Search PubMed.
- T. Peng, C. Dohno and K. Nakatani, Mismatch-binding ligands function as a molecular glue for DNA, Angew. Chem., Int. Ed., 2006, 45, 5623–5626 Search PubMed.
- T. Peng, C. Dohno and K. Nakatani, Bidirectional Control of Gold Nanoparticle Assembly by Turning On and Off DNA Hybridization with Thermally Degradable Molecular Glue, ChemBioChem, 2007, 8, 483–485 Search PubMed.
- C. Dohno, S. Uno and K. Nakatani, Photoswitchable Molecular Glue for DNA, J. Am. Chem. Soc., 2007, 129, 11898–11899 Search PubMed.
- C. Dohno, S. Uno, S. Sakai, M. Oku and K. Nakatani, The effect of linker length on binding affinity of a photoswitchable molecular glue for DNA, Bioorg. Med. Chem., 2009, 17, 2536–2543 Search PubMed.
- H. M. D. Bandara and S. C. Burdette, Photoisomerization in different classes of azobenzene, Chem. Soc. Rev., 2012, 41, 1809–1825 Search PubMed.
- O. Weingart, Z. Lan, A. Koslowski and W. Thiel, Chiral Pathways and Periodic Decay in cis -Azobenzene Photodynamics, J. Phys. Chem. Lett., 2011, 2, 1506–1509 Search PubMed.
- T. Takarada, D. Tamaru, X. Liang, H. Asanuma and M. Komiyama, L-Threoninol as a Chiral Linker of Azobenzene for the Effective Photo-regulation of DNA Triplex Formation, Chem. Lett., 2001, 30, 732–733 Search PubMed.
- H. Asanuma, T. Yoshida, T. Ito and M. Komiyama, Photo-responsive oligonucleotides carrying azobenzene at the 2′-position of uridine, Tetrahedron Lett., 1999, 40, 7995–7998 Search PubMed.
- X. Liang, H. Asanuma, H. Kashida, A. Takasu, T. Sakamoto, G. Kawai and M. Komiyama, NMR Study on the Photoresponsive DNA Tethering an Azobenzene. Assignment of the Absolute Configuration of Two Diastereomers and Structure Determination of Their Duplexes in the trans-Form, J. Am. Chem. Soc., 2003, 125, 16408–16415 Search PubMed.
- X. Liang, H. Asanuma and M. Komiyama, Photoregulation of DNA Triplex Formation by Azobenzene, J. Am. Chem. Soc., 2002, 124, 1877–1883 Search PubMed.
- Y. Kamiya and H. Asanuma, Light-Driven DNA Nanomachine with a Photoresponsive Molecular Engine, Acc. Chem. Res., 2014, 47, 1663–1672 Search PubMed.
- C. Dohno and K. Nakatani, Molecular Glue for RNA: Regulating RNA Structure and Function through Synthetic RNA Binding Molecules, ChemBioChem, 2019, 20, 2903–2910 Search PubMed.
- N. A. Simeth, A. Bellisario, S. Crespi, M. Fagnoni and B. König, Substituent Effects on 3-Arylazoindole Photoswitches, J. Org. Chem., 2019, 84, 6565–6575 Search PubMed.
- W. Szymański, B. Wu, C. Poloni, D. B. Janssen and B. L. Feringa, Azobenzene Photoswitches for Staudinger–Bertozzi Ligation, Angew. Chem., Int. Ed., 2013, 52, 2068–2072 Search PubMed.
- W. Szymanski, M. E. Ourailidou, W. A. Velema, F. J. Dekker and B. L. Feringa, Light-Controlled Histone Deacetylase (HDAC) Inhibitors: Towards Photopharmacological Chemotherapy, Chem.–Eur. J., 2015, 21, 16517–16524 Search PubMed.
- M. W. H. Hoorens, H. Fu, R. H. Duurkens, G. Trinco, V. Arkhipova, B. L. Feringa, G. J. Poelarends, D. J. Slotboom and W. Szymanski, Glutamate Transporter Inhibitors with Photo-Controlled Activity, Adv. Ther., 2018, 1, 1800028 Search PubMed.
- O. K. Rasheed, J. Raftery and P. Quayle, A New Benzannulation Reaction of Azoaromatics, Synlett, 2015, 26, 2806–2810 Search PubMed.
- F. Livolant and M. F. Maestre, Circular dichroism microscopy of compact forms of DNA and chromatin in vivo and in vitro: cholesteric liquid-crystalline phases of DNA and single dinoflagellate nuclei, Biochemistry, 1988, 27, 3056–3068 Search PubMed.
- M. Zama and S. Ichimura, Circular dichroism of acridine orange bound to DNA, Biopolymers, 1970, 9, 53–63 Search PubMed.
- D. Fornasiero and T. Kurucsev, Circular dichroism spectra and the interaction between acridine dyes and deoxyribonucleic acid, J. Phys. Chem., 1981, 85, 613–618 Search PubMed.
- B. Nordén and F. Tjerneld, Structure of methylene blue-DNA complexes studied by linear and circular dichroism spectroscopy, Biopolymers, 1982, 21, 1713–1734 Search PubMed.
- J. Rubio-Magnieto, T.-A. Phan, M. Fossépré, V. Matot, J. Knoops, T. Jarrosson, P. Dumy, F. Serein-Spirau, C. Niebel, S. Ulrich and M. Surin, Photomodulation of DNA-Templated Supramolecular Assemblies, Chem.–Eur. J., 2018, 24, 706–714 Search PubMed.
- C. Dohno, T. Yamamoto and K. Nakatani, Photoswitchable unsymmetrical ligand for DNA hetero-mismatches, Eur. J. Org. Chem., 2009, 4051–4058 Search PubMed.
- A. Murata, M. Nakamori and K. Nakatani, Modulating RNA secondary and tertiary structures by mismatch binding ligands, Methods, 2019, 167, 78–91 Search PubMed.
- J. Kypr, I. Kejnovská, K. Bednářová and M. Vorlíčková, Comprehensive Chiroptical Spectroscopy, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2012, vol. 2, pp. 575–586 Search PubMed.
- M. Deiana, Z. Pokladek, J. Olesiak-Banska, P. Młynarz, M. Samoc and K. Matczyszyn, Photochromic switching of the DNA helicity induced by azobenzene derivatives, Sci. Rep., 2016, 6, 28605 Search PubMed.
- D. Wenger, V. L. Malinovskii and R. Häner, Modulation of chiroptical properties by DNA-guided assembly of fluorenes, Chem. Commun., 2011, 47, 3168–3170 Search PubMed.
- A. Mammana, G. T. Carroll, J. Areephong and B. L. Feringa, A Chiroptical Photoswitchable DNA Complex, J. Phys. Chem. B, 2011, 115, 11581–11587 Search PubMed.
- S. Tannir, L. Levintov, M. A. Townley, B. M. Leonard, J. Kubelka, H. Vashisth, K. Varga and M. Balaz, Functional Nanoassemblies with Mirror-Image Chiroptical Properties Templated by a Single Homochiral DNA Strand, Chem. Mater., 2020, 32, 2272–2281 Search PubMed.
- H. Minami, N. Itamoto, W. Watanabe, Z. Li, K. Nakamura and N. Kobayashi, Chiroptical property enhancement of chiral Eu(III) complex upon association with DNA-CTMA, Sci. Rep., 2020, 10, 18917 Search PubMed.
- G. Haberhauer and C. Kallweit, A Bridged Azobenzene Derivative as a Reversible, Light-Induced Chirality Switch, Angew. Chem., Int. Ed., 2010, 49, 2418–2421 Search PubMed.
- R. A. van Delden, T. Mecca, C. Rosini and B. L. Feringa, A Chiroptical Molecular Switch with Distinct Chiral and Photochromic Entities and Its Application in Optical Switching of a Cholesteric Liquid Crystal, Chem.–Eur. J., 2004, 10, 61–70 Search PubMed.
- K. Takaishi, M. Kawamoto, K. Tsubaki, T. Furuyama, A. Muranaka and M. Uchiyama, Helical Chirality of Azobenzenes Induced by an Intramolecular Chiral Axis and Potential as Chiroptical Switches, Chem.–Eur. J., 2011, 17, 1778–1782 Search PubMed.
- C. Lin, S. Maisonneuve, R. Métivier and J. Xie, Photoswitchable Carbohydrate-Based Macrocyclic Azobenzene: Synthesis, Chiroptical Switching, and Multistimuli-Responsive Self-Assembly, Chem.–Eur. J., 2017, 23, 14996–15001 Search PubMed.
- T. Muraoka, K. Kinbara and T. Aida, Mechanical twisting of a guest by a photoresponsive host, Nature, 2006, 440, 512–515 Search PubMed.
- S. Spicher and S. Grimme, Robust Atomistic Modeling of Materials, Organometallic, and Biochemical Systems, Angew. Chem., Int. Ed., 2020, 59, 15665–15673 Search PubMed.
- S. J. Wezenberg and B. L. Feringa, Supramolecularly directed rotary motion in a photoresponsive receptor, Nat. Commun., 2018, 9, 1984 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds, photophysical and chemical studies, CD experiments, Tm and SPR measurements, detailed protocols of the computational calculation. See DOI: 10.1039/d1sc02194j |
| This journal is © The Royal Society of Chemistry 2021 |