
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New chemistry for enhanced carbon capture: beyond ammonium carbamates
Alexander C.
Forse
*a and
Phillip J.
Milner
 *b
*b
aDepartment of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK. E-mail: acf50@cam.ac.uk
bDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA. E-mail: pjm347@cornell.edu
First published on 7th December 2020
Abstract
Carbon capture and sequestration is necessary to tackle one of the biggest problems facing society: global climate change resulting from anthropogenic carbon dioxide (CO2) emissions. Despite this pressing need, we still rely on century-old technology—aqueous amine scrubbers—to selectively remove CO2 from emission streams. Amine scrubbers are effective due to their exquisite chemoselectivity towards CO2 to form ammonium carbamates and (bi)carbonates, but suffer from several unavoidable limitations. In this perspective, we highlight the need for CO2 capture via new chemistry that goes beyond the traditional formation of ammonium carbamates. In particular, we demonstrate how ionic liquid and metal–organic framework sorbents can give rise to capture products that are not favourable for aqueous amines, including carbamic acids, carbamate–carbamic acid adducts, metal bicarbonates, alkyl carbonates, and carbonic acids. These new CO2 binding modes may offer advantages including higher sorption capacities and lower regeneration energies, though additional research is needed to fully explore their utility for practical applications. Overall, we outline the unique challenges and opportunities involved in engineering new CO2 capture chemistry into next-generation technologies.
Introduction
Rising atmospheric levels of carbon dioxide (CO2) are the major contributor to global climate change, with annual emissions approaching 40 billion tonnes.1 Nearly two-thirds of anthropogenic CO2 emissions result from the combustion of fossil fuels, including coal and natural gas, for the global production of electricity.1 In addition, CO2 emissions are an inevitable by-product of other industrial processes, including the production of cement, steel, and natural gas.1 As a result, new technologies are needed to mitigate emissions from these industrial point sources during the gradual transition to cleaner fuels and building materials. One such proposed technology is carbon capture and sequestration or utilization, in which CO2 is selectively removed from low-concentration emission streams (4–15% CO2) prior to its permanent storage underground or conversion into more valuable products.2Building upon technology developed in the 1930s to purify crude natural gas, many have shown that aqueous amine scrubbers are currently the most technology-ready sorbents for CO2 capture from flue emissions on large scale (Fig. 1a).3 Aqueous amine scrubbers are effective because amines react selectively with CO2 to produce carbamic acid intermediates, which rapidly react with a second equivalent of amine to produce ammonium carbamates; under aqueous conditions, ammonium carbamates and carbamic acids can further react with water to produce ammonium (bi)carbonates.4 The captured CO2 is then desorbed using heat and/or vacuum (temperature and/or vacuum swing), thereby regenerating free amines. Over the last ninety years, there has been significant optimisation of the amine structure to maximize working capacities (i.e. the usable amount of CO2 captured in an actual process) while minimising regeneration energies (i.e. the total energy input needed to heat the material and desorb CO2).5 However, aqueous amine scrubbers are still faced with several challenges, including: (1) low capacities (<3 mol CO2 per kg solution or <15 wt%) due to dilution of the corrosive amines with water;6 (2) poor oxidative stability of amines towards O2; and (3) degradation in the presence of contaminants such as SO2, which reacts with amines similarly to CO2.7 In addition, one aspect of aqueous amine scrubbers has remained largely constant: the products of their reaction with CO2.8 This restriction generally leads to high regeneration energies (≥2.4 MJ kg−1 CO2) and CO2 desorption temperatures (>100 °C), greatly increasing the cost of carbon capture from flue emissions.9–11
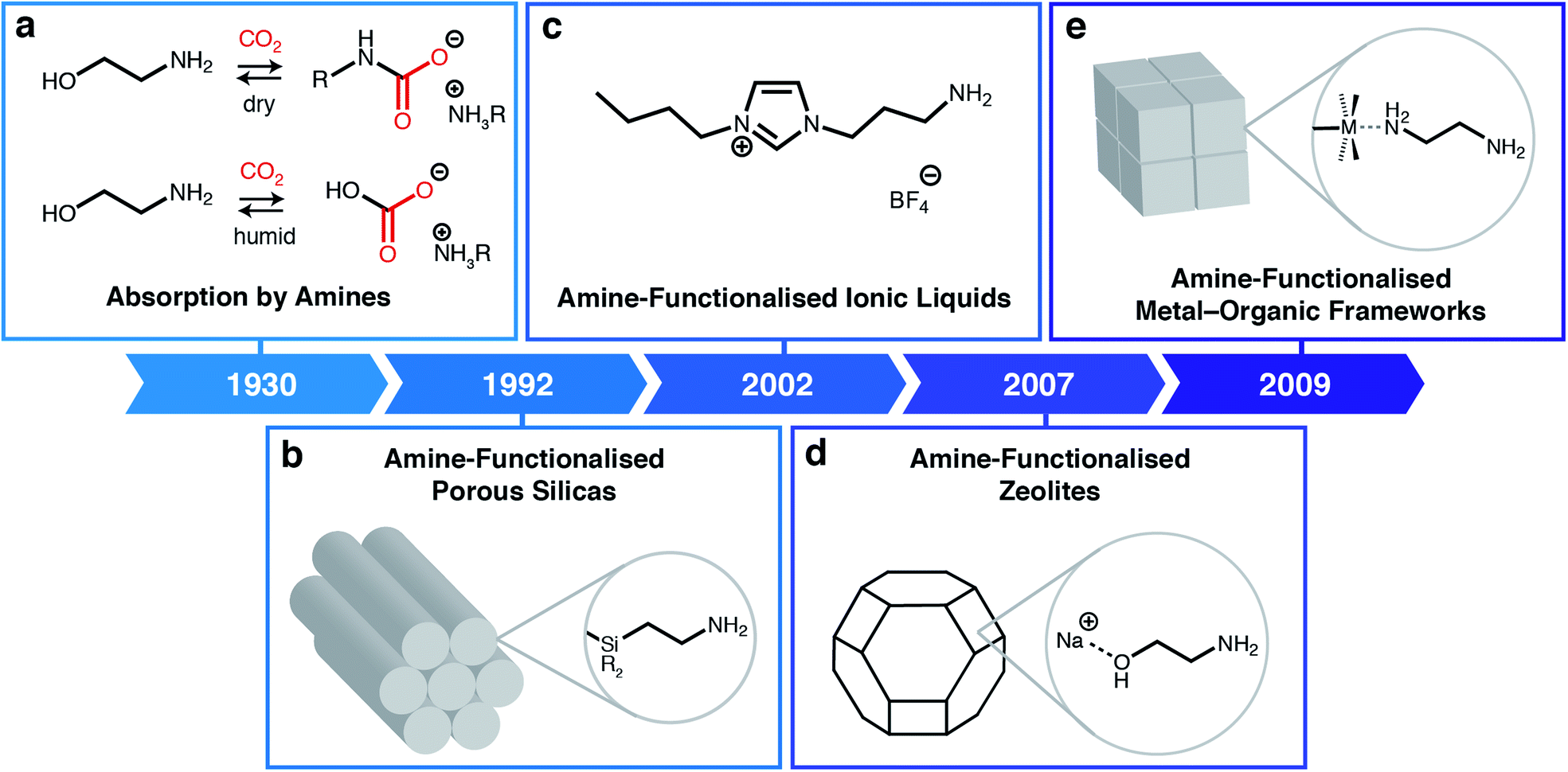 | ||
| Fig. 1 Classes of amine-based materials for CO2 capture. (a) Traditional amine chemistry for CO2 absorption via the formation of ammonium carbamates under dry conditions and ammonium (bi)carbonates under humid conditions.3 (b) Amine-functionalised porous silicas, where amines are either impregnated into or covalently attached to silica supports.12 (c) Amine-functionalised ionic liquids.13 (d) Post-synthetically amine-functionalised zeolites.14 (e) Post-synthetically amine-functionalised metal–organic frameworks.15 | ||
Amine-based materials for CO2 capture
One promising avenue to overcome the challenges associated with CO2 capture by aqueous amine scrubbers is to employ other types of sorbents, such as porous solids or ionic liquids (ILs). Porous materials such as silicas, carbons, zeolites, metal–organic frameworks (MOFs), porous organic polymers (POPs), and covalent-organic frameworks (COFs), have the potential advantages of higher thermal stabilities and lower heat capacities compared to aqueous amine scrubbers.16–21 Likewise, ILs are low-melting ionic salts that offer advantages over aqueous amines including non-volatility (preventing release into the atmosphere) and structural tunability. Although hydrophobic porous solids such as silicon-rich zeolites and carbons are capable of scrubbing CO2 from high-concentration streams (e.g. crude biogas),22 many of these materials cannot remove CO2 from humid low-concentration streams such as flue gas emissions.23 This limitation arises because CO2 and water directly compete for the same physisorption sites in these sorbents. An additional general challenge for porous solid adsorbents that remains to be addressed is their poor thermal conductivity, which complicates adsorbent heating and cooling during adsorption/desorption cycling.A powerful approach to overcome the poor selectivities of typical sorbents towards CO2 under humid conditions is to leverage the favourable reactivity of aqueous amine scrubbers in the form of amine-functionalised sorbents (Fig. 1).24,25 Beginning with the first report of amine-functionalised silicas in 1992 (Fig. 1b),12,24 a range of amine-functionalized solid adsorbents, including zeolites (Fig. 1d),14 MOFs (Fig. 1e),15,19 and carbons26 have been prepared. Researchers have demonstrated that amine-functionalised porous solids possess the high CO2 selectivities native to aqueous amines while generally evidencing improved thermal and chemical stabilities. For example, confining amines within a porous support largely eliminates oxidation pathways that are catalyzed by leached metal ions from the absorption columns.7,27 Similarly, ionic liquids (ILs) can also be functionalised with amine groups to achieve high CO2 capacities and selectivities without the need for dilution with water (Fig. 1c).28,29 Numerous in situ spectroscopic studies using solution- and solid-state nuclear magnetic resonance (SSNMR) and infrared (IR) spectroscopy combined with theoretical calculations suggest that in most cases amine-functionalised materials produce similar sorption products as aqueous amine scrubbers, namely, ammonium carbamates under dry conditions13,30,31 and, as confirmed recently, ammonium bicarbonates under humid conditions.32 As such, the majority of these materials still require high temperatures (>120 °C) to fully desorb CO2, resulting in high regeneration penalties.33 In addition, amine-functionalised silicas suffer from oxidative degradation by distinct bimolecular pathways,34 as well as the irreversible formation of ureas under dry conditions.35 Overcoming these fundamental limitations is critical to enabling the widespread adoption of carbon capture technologies.
New CO2 chemisorption pathways in solution and the solid state
An underexplored approach to overcome the fundamental limitations of amine-based materials is not to focus on the development of new materials, but on new chemisorptive pathways for selective carbon dioxide capture. For example, the formation of carbamic acids by CO2 capture at amine sites is potentially desirable because it involves reaction with CO2 at only a single amine site, increasing the CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :amine sorption ratio to 1:1.36 Indeed, unlocking 1:1 reaction stoichiometries in general should produce higher gravimetric and volumetric sorption capacities by enabling a higher density of reactive sites within a given volume. Additionally, mechanisms beyond ammonium carbamate formation have been shown to lead to lower CO2 desorption temperatures in some cases (see below). Importantly, each combination of CO2 partial pressure (P) and temperature (T) for a given separation (e.g. 400 ppm, 25 °C for capture directly from the atmosphere) leads to an ideal differential free energy of sorption (−ΔG) for that separation (e.g. −19 kJ mol−1 for direct air capture), which is critical to maximising sorption capacities while minimising regeneration energies.41 New chemisorption pathways should enable more dramatic tuning of the differential enthalpies (−ΔH) and entropies (−ΔS) of sorption to achieve these optimal values. Last, moving away from amines entirely could lead to adsorbents with improved oxidative stabilities, a recurring challenge associated with amine-based materials, although more work is required to characterize the oxidative stability of promising sorbents.42 Here, we highlight examples of new CO2 adsorption pathways beyond ammonium carbamates that may ultimately lead to enhanced CO2 capture.
:amine sorption ratio to 1:1.36 Indeed, unlocking 1:1 reaction stoichiometries in general should produce higher gravimetric and volumetric sorption capacities by enabling a higher density of reactive sites within a given volume. Additionally, mechanisms beyond ammonium carbamate formation have been shown to lead to lower CO2 desorption temperatures in some cases (see below). Importantly, each combination of CO2 partial pressure (P) and temperature (T) for a given separation (e.g. 400 ppm, 25 °C for capture directly from the atmosphere) leads to an ideal differential free energy of sorption (−ΔG) for that separation (e.g. −19 kJ mol−1 for direct air capture), which is critical to maximising sorption capacities while minimising regeneration energies.41 New chemisorption pathways should enable more dramatic tuning of the differential enthalpies (−ΔH) and entropies (−ΔS) of sorption to achieve these optimal values. Last, moving away from amines entirely could lead to adsorbents with improved oxidative stabilities, a recurring challenge associated with amine-based materials, although more work is required to characterize the oxidative stability of promising sorbents.42 Here, we highlight examples of new CO2 adsorption pathways beyond ammonium carbamates that may ultimately lead to enhanced CO2 capture.
The unique, highly-charged environment within ILs makes them an ideal setting to unlock new CO2 reactivity. For example, although carbamic acids are normally disfavoured outside of polar aprotic solvents (e.g. dimethyl sulphoxide),36,43 Schneider, Brennecke, and coworkers found that installing amines onto the anions of amino acid-derived ILs favours CO2 capture via the formation of carbamic acids stabilized by hydrogen-bonding (Fig. 2a).37 This change in mechanism doubled the molar absorption capacity of these ILs compared to those bearing amine-functionalized cations, which operate by the traditional ammonium carbamate mechanism.13 In addition, the strong binding of CO2 within a proline-derived IL (−ΔHabs = 80 kJ mol−1) led to nearly complete saturation at low pressures of CO2 (<0.1 bar at 25 °C). Therefore, this switch in chemisorption products demonstrates that the local environment of an amine is a crucial design element for controlling its reactivity towards CO2.44
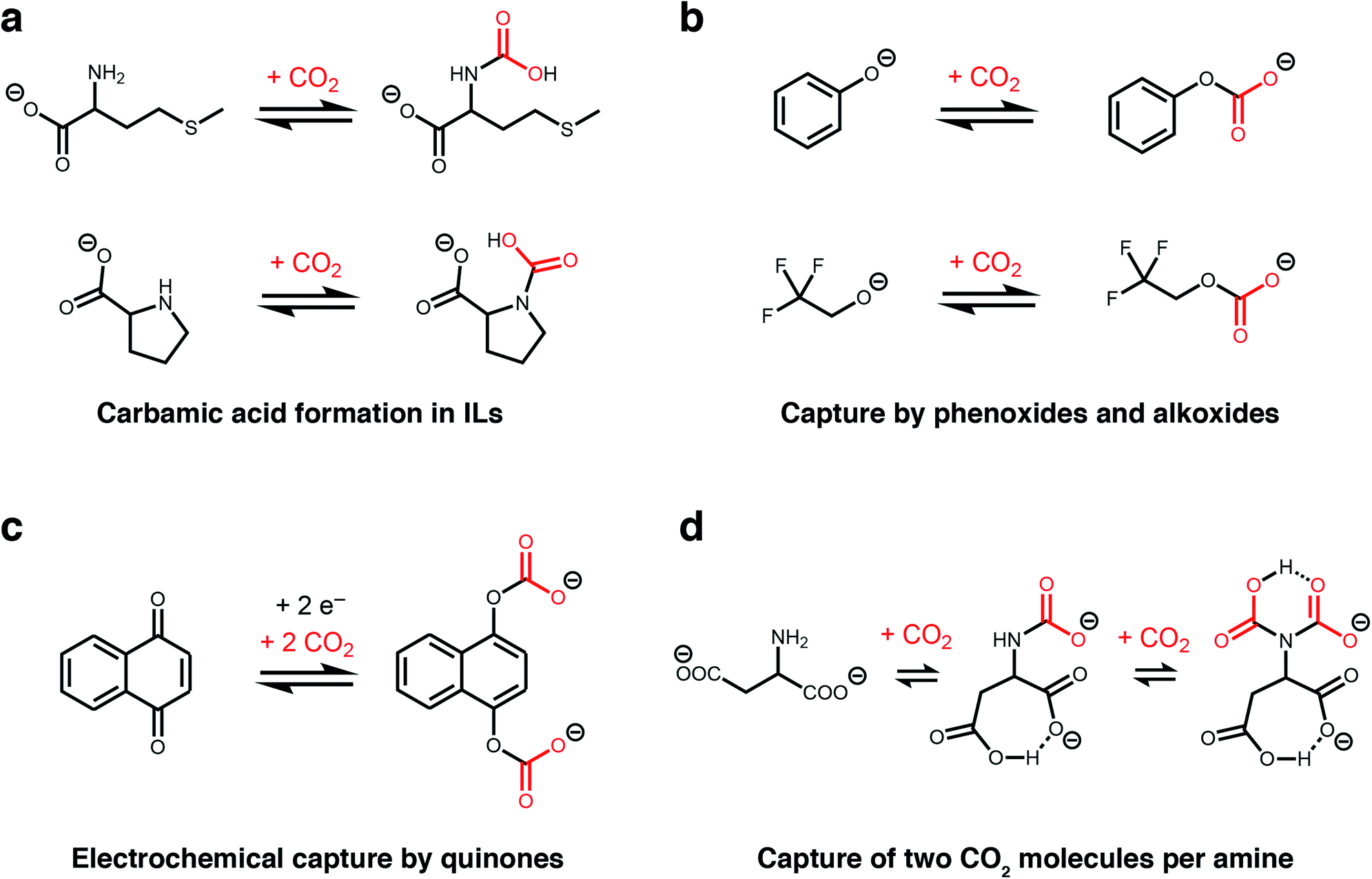 | ||
| Fig. 2 New CO2 absorption mechanisms in solution. (a) Proposed formation of carbamic acid in ILs with amine-functionalised anions.37 (b) Proposed absorption mechanism by phenoxide and alkoxide ILs.38 (c) Proposed mechanism for electrochemical CO2 capture by 1,4-naphthoquinone.39 (d) Proposed absorption mechanism for an IL with an aspartate dianion.40 In all cases, the corresponding cations are omitted for clarity. | ||
Following these initial studies, even more unconventional CO2 absorption pathways began to emerge in ILs. Building upon previous reports,45 Li, Dai, and coworkers demonstrated that amines can be completely bypassed by capturing CO2 in ILs bearing alkoxide or phenoxide anions and organic superbase-derived cations, which reversibly capture CO2via alkylcarbonate formation (Fig. 2b).38 Similar to carbamic acids (Fig. 2a), this chemistry gives rise to a 1:1 reaction stoichiometry and thus higher gravimetric capacities (up to 20 wt%) compared to traditional IL sorbents (<10 wt%). Importantly, alkoxide-based ILs also possess low viscosities and rapid absorption kinetics (saturation in less than 5 minutes at room temperature), overcoming common challenges that plague traditional amine-functionalized ILs.38 Subsequently, Kim and coworkers demonstrated that similar reactivity at oxygen could be achieved in water-lean alcoholamines bearing sterically-hindered amines and that the resulting ammonium alkylcarbonates desorb CO2 more readily than ammonium carbamates.50
Another route to generate oxyanion nucleophiles for rapid CO2 capture via carbonate formation is by the electrochemical reduction of quinones, as demonstrated by Hatton and others (Fig. 2c).39 Promising results with electrochemically-reduced quinones have been observed in the presence of water and oxygen, although some loss in capacity was observed due to re-oxidation of the nucleophile by oxygen.51 This electrochemical approach has subsequently been expanded to other nucleophiles, such as reduced sulphides, suggesting it may be a general strategy to expand the scope of nucleophiles for CO2 capture.52 An advantage of this approach is that electrochemical regeneration of the quinone (electrochemical swing adsorption) leads to energy savings over traditional temperature or pressure swing processes.
Recent work has revealed that CO2 capacities approaching a remarkable 2:1 reaction stoichiometry can be accessed in ILs, representing a four-fold increase compared to the traditional ammonium carbamate mechanism (Fig. 2d).40 Specifically, Wang and coworkers found that an ionic liquid with an aspartate dianion was able to reversibly bind 1.96 mol CO2 per mol IL at 30 °C and 1 atmosphere CO2, which was hypothesised to occur via two subsequent reactions at a single amine site to form both a carbamate (calculated ΔEabs = −69 kJ mol−1) and a carbamic acid (calculated ΔEabs = −54 kJ mol−1).40 This proposed absorption pathway was supported by 13C NMR measurements as well as density functional theory (DFT) calculations, with the latter ruling out reaction of CO2 at the carboxylate groups as proposed for related ILs.53 A similar 2:1 absorption mode was also evidenced in an earlier organic chemistry study. The observation of a triplet in solution 15N NMR studies of selected primary amines in the presence of 13CO2 and a base confirmed the reaction of 2 CO2 molecules with a single amine group at −30 °C.53 The high capacity offered by this absorption mode makes it a very attractive target for CO2 capture applications.
Although ILs and water-lean solvents represent a unique platform for the discovery of new CO2 capture products, they are not without their own challenges. For example, the absorption capacities of most ILs are relatively low (<20 wt%) compared to amine-functionalized solids.13,28 In addition, the viscosities of ionic liquids are relatively high and tend to increase upon CO2 adsorption (in some cases up to 200-fold), which represents a significant process challenge.28,54 While molecular engineering allows access to CO2-loaded ILs with viscosities as low as 650 mPa s,55 these values are still significantly higher than CO2-loaded 30% aqueous monoethanolamine solution (4 mPa s).56 Last, the CO2/N2 absorption selectivities, kinetics, desorption conditions, and long-term cycling stabilities of ILs remain poorly characterized in many cases. Addressing these challenges is critical to advancing the commercial viability of IL-based sorbents.
An emerging alternative approach is to engineer new CO2 capture mechanisms within the controlled pore environments of crystalline porous materials, such as MOFs. The arrangement of functional groups in an ordered fashion within the pores of MOFs presents a potential opportunity for unlocking new CO2 capture chemistry.
One of the earliest demonstrations of CO2 chemisorption in MOF adsorbents involved CD-MOFs (Fig. 3a; CD = γ-cyclodextrin).46,57 These MOFs demonstrate strong adsorption of CO2 at low partial pressures (<2 mbar), leading to excellent CO2/CH4 selectivity (estimated to be >3000) in this regime.46 Using SSNMR measurements, the authors proposed the formation of carbonic acids or alkylcarbonates; however, the exact chemisorption pathway in this material remains unclear. Nonetheless, the strong bonding of CO2 in CD-MOF-2 (>1 mmol CO2 per g MOF adsorbed at 10 mbar and 30 °C) makes this a promising potential material for flue gas capture applications. Analysis of the thermodynamics of CO2 chemisorption in this material by calorimetry revealed a moderate enthalpy of adsorption at intermediate loadings (−ΔHads = 65 kJ mol−1), enabling easier desorption of CO2 from the strong-binding sites compared to amines.58,59 However, the poor water stability of these MOFs necessitates the translation of this chemisorption mechanism to more stable materials for practical applications.46
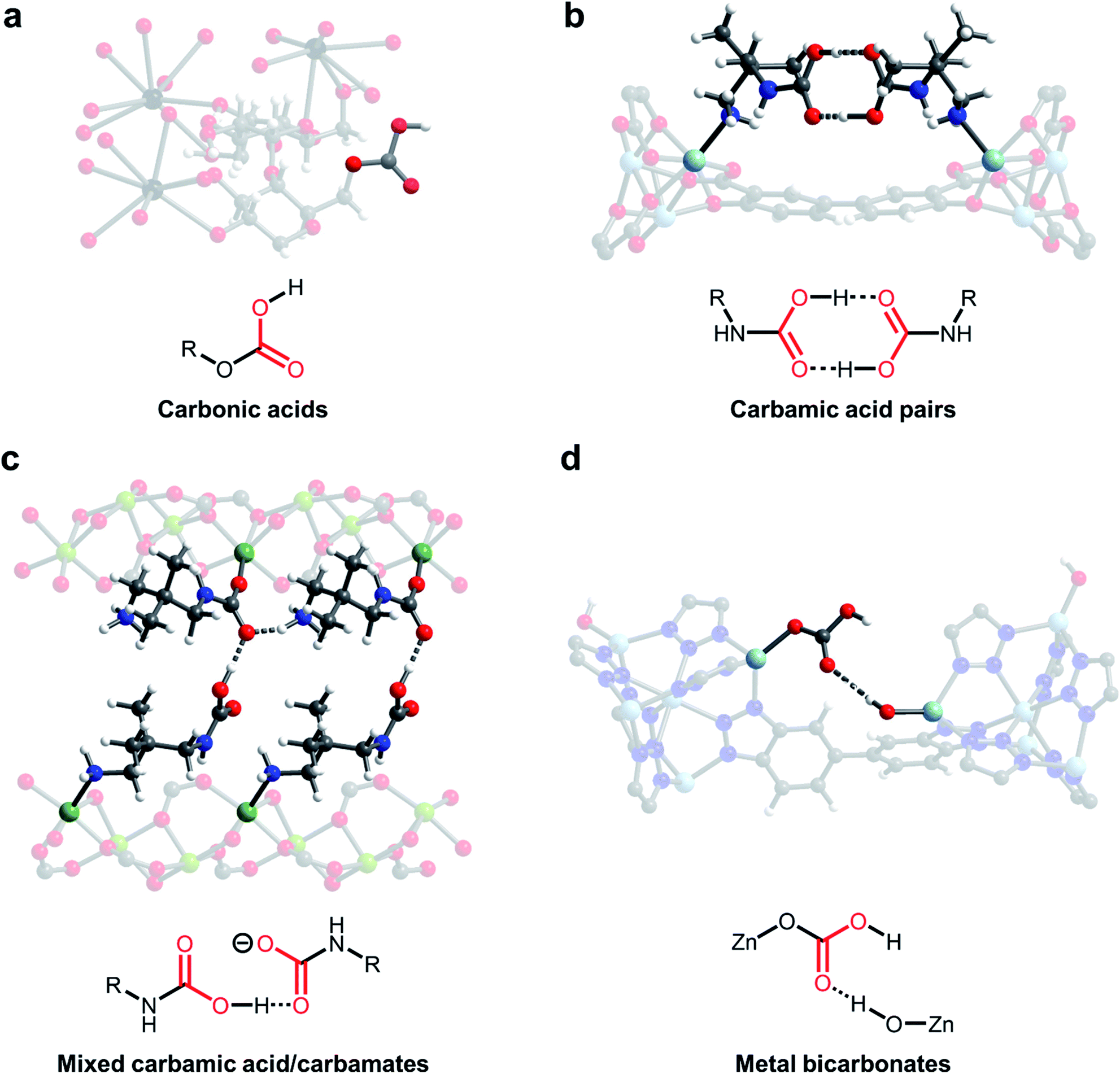 | ||
| Fig. 3 New CO2 adsorption mechanisms unlocked in MOFs. (a) Proposed formation of carbonic acids in CD-MOF-2 (CD = cyclodextrin).46 (b) Crystallographically confirmed formation of carbamic acid pairs in dmpn–Zn2(dobpdc) (dmpn = 2,2-dimethyl-1,3-diaminopropane; dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate).47 (c) Proposed formation of mixed carbamic acids and ammonium carbamates in dmpn–Mg2(dobpdc).48 (d) Proposed formation of metal bicarbonates in Zn(ZnOH)4(bibta)3 (bibta2− = 5,5′-bibenzotriazolate).49 Gray, white, red, black, dark blue, sky blue, and green spheres correspond to carbon, hydrogen, oxygen, rubidium, nitrogen, zinc, and magnesium, respectively. | ||
Carbamic acids have long been invoked as intermediates and products upon CO2 capture in amine scrubbers,4 amine-functionalized silicas30,60 and amine-functionalized MOFs,61,62 as suggested by NMR and IR spectroscopies. For example, Ho and coworkers found that hydrazine-functionalized variants of the MOF Mg2(dobdc) (dobdc4− = 2,5-dioxido-1,4-benzenedicarboxylate) exhibit incredibly strong and selective binding of CO2 (3.89 mmol g−1 at 25 °C and 0.4 mbar of CO2), which they ascribe to highly favourable carbamic acid formation (−ΔHads = 90 kJ mol−1) within the framework pores.62 However, until recently there remained little crystallographic evidence for this elusive adsorption product in the solid state. Long and coworkers identified variants of the MOF M2(dobpdc) (dobpdc4− = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate) functionalised with the diamine 2,2-dimethyl-1,3-diaminopropane (dmpn) as promising adsorbents for post-combustion CO2 capture owing to their exceptional hydrothermal and oxidative stability (Fig. 3b and c).47,48 Exposure of single crystals of dmpn–Zn2(dobpdc) to 1 bar of CO2 induced the formation of carbamic acid pairs bridging two adjacent amine sites in the framework, as confirmed by SCXRD and SSNMR (Fig. 3b).47,48 In this structure, the normally disfavoured formation of carbamic acids is facilitated by well-defined hydrogen-bonding interactions, corroborated by the presence of strong 1H(COOH)⋯13C correlations in 2-dimensional SSNMR experiments. Notably, carbamic acid pairs were actually predicted computationally in related frameworks before they were observed experimentally.63
Building upon this work, the same group demonstrated that dmpn–Mg2(dobpdc) chemisorbs CO2 by another distinct pathway: the formation of both ammonium carbamates and carbamic acids (Fig. 3c).48 In-depth DFT calculations and 2-dimensional SSNMR experiments support the formation of ammonium carbamate chains that interact with carbamic acids via hydrogen-bonding in this material. The advantage of this mechanism lies in its high enthalpy of adsorption (ΔHads = −74 kJ mol−1) coupled with a large entropic penalty (−ΔSads = 204 J mol−1 K−1), which reduces the temperature required to desorb CO2 in a temperature-swing adsorption process to <100 °C, potentially enabling adsorbent regeneration with low-grade steam.47 These thermodynamic parameters enable adsorbent regeneration with an estimated energy of 2.5 MJ kg−1 CO2, comparable to the best-in-class aqueous amine scrubbers such as Mitsubishi KS-1 (2.4 MJ kg−1 CO2).10,64 Therefore, this finding highlights the potential to overcome thermodynamic trade-offs of carbon capture processes by tuning the adsorption pathway. In addition, this adsorption mode leads to faster adsorption kinetics than ammonium carbamate formation in related materials and a high non-competitive CO2/N2 selectivity (880) under the conditions relevant for CO2 capture from coal flue emissions (150 mbar CO2, 750 mbar N2, 40 °C).65
A further promising avenue to unlock new selective CO2 capture reactivities in porous materials is to look to nature for inspiration. For example, carbonic anhydrase enzymes are responsible for the transport of CO2 in the human body. Many members of this family operate by the reversible reaction of a zinc-hydroxide species (Zn–OH) with CO2 to form a zinc-bound bicarbonate species (Zn–OCO2H).66 In an early study, Zhang and coworkers demonstrated that high-valent monodentate metal hydroxides in the water-stable MOFs MnIIMnIII(OH)Cl2(bbta) and [CoIICoIII(OH)Cl2(bbta)] (bbta2− = dihydrobenzo[1,2-d:4,5-d′]bis([1,2,3]triazolate)) strongly bind CO2 with high CO2/N2 selectivity (>250), even under humid conditions.67 These materials exhibit highly exothermic capture of CO2 at low loadings (−ΔHads > 100 kJ mol−1), necessitating regeneration using flowing N2 at 85 °C (simulating a temperature-vacuum swing process). A regeneration energy of 2.7 MJ kg−1 CO2 was calculated for [CoIICoIII(OH)Cl2(bbta)], which is comparable with best-in-class aqueous amines. Closely mimicking the mechanism of carbonic anhydrase enzymes, Wade and coworkers subsequently found that Zn–OH centers in the air-stable MOF Zn(ZnOH)4(bibta)3 (bibta2− = 5,5′-bibenzotriazolate) strongly bind CO2 to form metal-bound bicarbonates with adsorption capacities of 2.2 mmol g−1 at 27 °C and 0.4 mbar of CO2, suitable for direct air capture (Fig. 3d).49 Interestingly, DFT calculations suggest that these metal-bound bicarbonates hydrogen-bond with adjacent Zn–OH centers, stabilizing the adsorption product (−ΔHads = 71 kJ mol−1) and leading to steep uptake of CO2 at low pressures. Dincă and coworkers demonstrated a similar bioinspired approach to CO2 capture in (Zn5(OH)4(btdd)3) (btdd = bis(1,2,3-triazolo[4,5-b],[4′,5′-i])dibenzo[1,4]dioxin), a hydroxide-substituted variant of the MOF MFU-4l.68 This framework was found to exhibit stronger CO2 binding (−ΔHads = 81 kJ mol−1) compared to Zn(ZnOH)4(bibta)3, albeit with a lower adsorption capacity at low pressures (0.9 mmol g−1 at 25 °C and 23 mbar of CO2). In all of these studies, the formation of metal-bound bicarbonates was validated primarily by in situ IR spectroscopy and DFT calculations. Subsequent work by Wade and coworkers has highlighted the importance of metal identity on CO2 adsorption in M–OH MOFs, unveiling a potential handle for tuning the thermodynamics of chemisorption.69,70 In a similar vein, Wang and Lackner have found that hydroxide-functionalised ion-exchange membranes are promising for energy-efficient moisture swing sorption processes, demonstrating that ammonium cations can be used as an alternative to metal ions to prepare hydroxide-rich materials.71 The capture of CO2 with oxygen-based nucleophiles in both adsorbents (Fig. 3a and d) and solution (Fig. 2b and c) represents a promising solution to overcome the inherent limitations of amine-functionalised materials; however, more work is required to map out the stability of these materials and their performance under realistic conditions.
The vast majority of CO2 capture processes discussed above operate via the addition of nitrogen- or oxygen-based nucleophiles to CO2. Recently, the range of nucleophiles that can reversibly react with CO2 has been expanded to include electrochemically-generated sulphides,52 frustrated Lewis pairs,72 and N-heterocyclic carbenes,73 among others.74 In addition, electrochemistry has emerged as a powerful tool to expand the scope of CO2 capture processes. For example, Hamelers and coworkers have shown that capacitive charging and migration of bicarbonate/carbonate ions through ion exchange membranes can drive a CO2 capture process with a low energy requirement of 40 kJ mol−1 CO2 captured.75 Similarly, Landskron and coworkers have developed a related process in which a supercapacitor device reversibly adsorbs CO2 (<0.1 mmol g−1), although the exact adsorption mechanism remains unclear.76 Finally, electrochemically driven pH swings are also being investigated as a new energy-efficient CO2 capture strategy.77 These recent directions represent an exciting opportunity to unlock new carbon capture chemistry.
Opportunities and challenges for next-generation CO2 capture
The foregoing examples highlight the unique opportunities offered by new CO2 capture pathways. Potential advantages of new sorption modes include lower regeneration energies, higher working capacities, and access to a wider range of sorption enthalpies compared to traditional ammonium carbamate formation. Despite this promise, there remains a great need for additional research to assess the application of novel sorption pathways in industrial processes. Key materials challenges (Fig. 4) that remain critically underappreciated include: (i) rapid sorption kinetics, (ii) large working capacities under realistic mixed-gas conditions, (iii) sufficient material stability to survive long-term exposure to reactive contaminants in target gas streams, such as water, oxygen, sulphur dioxide, and hydrogen sulphide, among others depending on the process,23,24 (iv) adsorbent development and structuring to overcome issues with low thermal conductivities and heat management during highly exothermic sorption processes, and (v) sustainable and scalable materials synthesis. As an example, the hydroxide-based MOF [CoIICoIII(OH)Cl2(bbta)] has a promising working capacity and regeneration energy for a flue gas capture process, but its stability in the presence of oxygen and other contaminants remains unknown.67 Moreover the very large heat of CO2 adsorption at low loadings (−ΔHads > 100 kJ mol−1) suggests that heat management will be an important challenge for this material. Promising strategies to aid heat management include the development of structured hollow fibre adsorbents and the design of carbon-based materials that have inherently larger thermal conductivities.78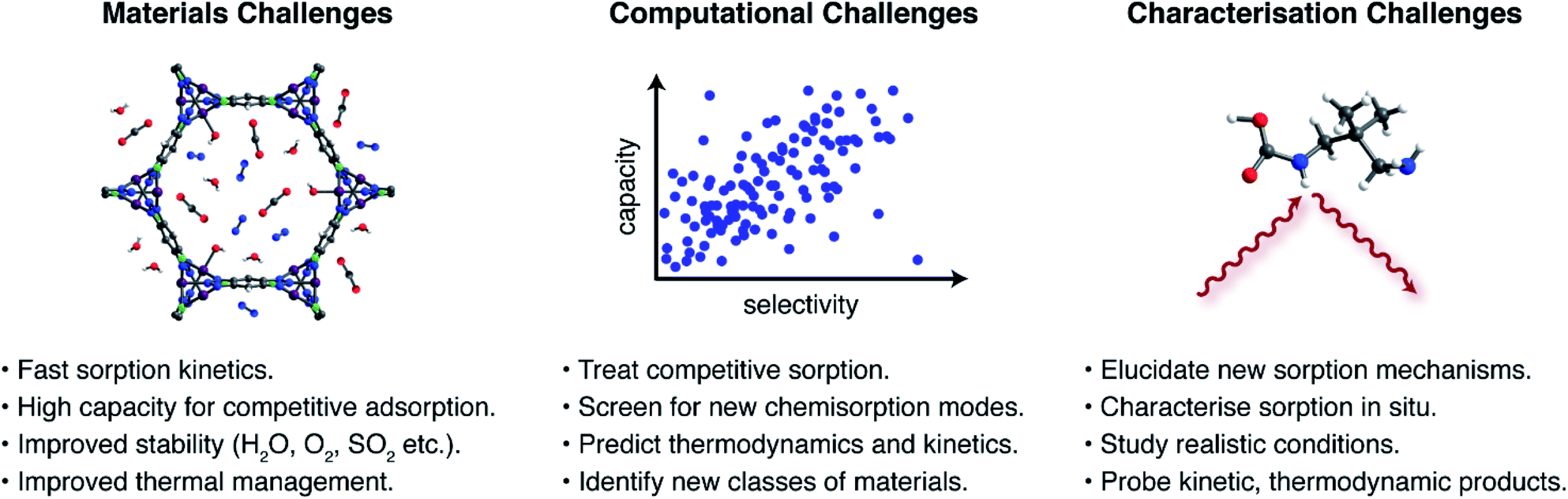 | ||
| Fig. 4 Grand challenges for next-generation sorbents for CO2 capture. | ||
Exploration of entirely new CO2 capture pathways beyond those described in this perspective should also lead to further advances. Recent studies have highlighted the promise of large-scale computational screenings in the search for new CO2 capture materials. For example, Smit and coworkers recently reported the screening of over 300000 theoretical MOFs and identified classes of physisorption sites, termed “adsorbophores”, that endow high CO2 selectivities to frameworks.79 The guided synthesis of optimised materials for operation under humid conditions was achieved by selecting candidates with hydrophobic adsorbophores to maximize the adsorption of CO2 under humid conditions. However, the capacities and CO2/N2 selectivities reported for best-in-class physisorbents are typically lower than those reported for chemisorptive materials. Similar computational screens to predict chemisorption—for example, using a higher level of theory to account for bond-breaking and – forming processes—remain rare but have the potential to be transformative.63,80 Similarly, calculations that can predict chemisorption thermodynamics under realistic mixed gas conditions should lead to promising materials for real-world applications.79 Due to the complex processes inherent to chemisorption, an additional challenge for computational analyses is to predict transition states relevant to sorption kinetics. A promising strategy to address these computational challenges may be to use machine learning to guide the search for new chemisorbent materials.81
In order to elucidate and ultimately build upon new CO2 capture chemistry, advanced characterisation methods are also needed. These methods serve to both validate and discover new chemisorption products when unexpected sorption properties arise. Recent years have seen significant advances in the characterisation of CO2 capture pathways through in situ spectroscopic and X-ray diffraction experiments.30,32,47,48 These experiments must now be adapted to study conditions that more closely mimic envisaged industrial applications and, in particular, must address mixed gas conditions rather than pure CO2.32,48 Furthermore, experiments must not be restricted to studying static/equilibrium conditions and should probe the dynamic conditions associated with practical sorption processes.
Conclusions
New CO2 sorption pathways such as those recently uncovered in appropriately-functionalised ILs and MOFs may offer improved performance for CO2 capture compared to traditional sorbents, including higher capacities and lower regeneration costs. Many of these binding modes do not readily occur in aqueous solution and instead arise from the unique opportunity to precisely install chemical functional groups with a controlled spatial arrangement and carefully tuned local environment. For many of these prospective sorbents, more work is needed to assess their sorption kinetics, selectivities, stabilities, and thermal conductivities. This mechanistically-focused line of sorbent discovery is still in its infancy, and a new generation of computational, analytical, and synthetic chemistry is needed to design transformative materials – and sorption mechanisms – for reducing anthropogenic CO2 emissions.Conflicts of interest
The authors declare the following competing interest: P. J. M. is listed as an inventor on several patents related to the preparation of metal–organic frameworks for CO2 capture.Acknowledgements
A. C. F. thanks the University of Cambridge for financial support of this work. P. J. M. thanks Cornell University for initial support of this work. Additional support for P. J. M. was provided by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences Energy Frontier Research Centers program under Award Number DE-SC0021000. We thank Drs Rebecca Siegelman, Sylvia Britto, and David Halat for helpful feedback.Notes and references
- R. K. Pachauri and L. Mayer, Intergovernmental Panel on Climate Change, Climate change 2014: synthesis report, Intergovernmental Panel on Climate Change, Geneva, Switzerland, 2015 Search PubMed.
- S. Chu, Science, 2009, 325, 1599 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1647–1652 CrossRef.
- J. Wilcox, Carbon Capture, Springer New York, New York, NY, 2012 Search PubMed.
- I. M. Bernhardsen and H. K. Knuutila, Int. J. Greenhouse Gas Control, 2017, 61, 27–48 CrossRef CAS.
- P. Brœder and H. F. Svendsen, Energy Procedia, 2012, 23, 45–54 CrossRef.
- S. B. Fredriksen and K.-J. Jens, Energy Procedia, 2013, 37, 1770–1777 CrossRef CAS.
- M. Caplow, J. Am. Chem. Soc., 1968, 90, 6795–6803 CrossRef CAS.
- A. S. Bhown and B. C. Freeman, Environ. Sci. Technol., 2011, 45, 8624–8632 CrossRef CAS.
- K. Morimura, C. Kuwada, H. Nagai, H. Matsumoto and N. Mikam, Service Technology for Improving Product Reliability, 2011, 48, 7 Search PubMed.
- H.-J. Song, S. Lee, K. Park, J. Lee, D. Chand Spah, J.-W. Park and T. P. Filburn, Ind. Eng. Chem. Res., 2008, 47, 9925–9930 CrossRef CAS.
- T. Tsuda and T. Fujiwara, J. Chem. Soc., Chem. Commun., 1992, 1659 RSC.
- E. D. Bates, R. D. Mayton, I. Ntai and J. H. Davis, J. Am. Chem. Soc., 2002, 124, 926–927 CrossRef CAS.
- P. D. Jadhav, R. V. Chatti, R. B. Biniwale, N. K. Labhsetwar, S. Devotta and S. S. Rayalu, Energy Fuels, 2007, 21, 3555–3559 CrossRef CAS.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8786 CrossRef CAS.
- J. Liu, Y. Wei and Y. Zhao, ACS Sustainable Chem. Eng., 2019, 7, 82–93 CrossRef CAS.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- J. Ozdemir, I. Mosleh, M. Abolhassani, L. F. Greenlee, R. R. Beitle and M. H. Beyzavi, Front. Energy Res., 2019, 7, 77 CrossRef.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- Y. Belmabkhout, V. Guillerm and M. Eddaoudi, Chem. Eng. J., 2016, 296, 386–397 CrossRef CAS.
- S.-Y. Lee and S.-J. Park, J. Ind. Eng. Chem., 2015, 23, 1–11 CrossRef CAS.
- Y. Jiang, J. Ling, P. Xiao, Y. He, Q. Zhao, Z. Chu, Y. Liu, Z. Li and P. A. Webley, Chem. Eng. J., 2018, 334, 2593–2602 CrossRef CAS.
- T. C. Drage, C. E. Snape, L. A. Stevens, J. Wood, J. Wang, A. I. Cooper, R. Dawson, X. Guo, C. Satterley and R. Irons, J. Mater. Chem., 2012, 22, 2815–2823 RSC.
- P. Bollini, S. A. Didas and C. W. Jones, J. Mater. Chem., 2011, 21, 15100–15120 RSC.
- E. E. Ünveren, B. Ö. Monkul, Ş. Sarıoğlan, N. Karademir and E. Alper, Petroleum, 2017, 3, 37–50 CrossRef.
- J. Przepiórski, M. Skrodzewicz and A. W. Morawski, Appl. Surf. Sci., 2004, 225, 235–242 CrossRef.
- K. Min, W. Choi, C. Kim and M. Choi, Nat. Commun., 2018, 9, 726 CrossRef.
- S. Zeng, X. Zhang, L. Bai, X. Zhang, H. Wang, J. Wang, D. Bao, M. Li, X. Liu and S. Zhang, Chem. Rev., 2017, 117, 9625–9673 CrossRef CAS.
- D. J. Heldebrant, P. K. Koech, V.-A. Glezakou, R. Rousseau, D. Malhotra and D. C. Cantu, Chem. Rev., 2017, 117, 9594–9624 CrossRef CAS.
- L. Mafra, T. Čendak, S. Schneider, P. V. Wiper, J. Pires, J. R. B. Gomes and M. L. Pinto, J. Am. Chem. Soc., 2017, 139, 389–408 CrossRef CAS.
- A. Danon, P. C. Stair and E. Weitz, J. Phys. Chem. C, 2011, 115, 11540–11549 CrossRef CAS.
- C.-H. Chen, D. Shimon, J. J. Lee, F. Mentink-Vigier, I. Hung, C. Sievers, C. W. Jones and S. E. Hayes, J. Am. Chem. Soc., 2018, 140, 8648–8651 CrossRef CAS.
- J. W. Dijkstra, S. Walspurger, G. D. Elzinga, J. A. Z. Pieterse, J. Boon and W. G. Haije, Ind. Eng. Chem. Res., 2018, 57, 1245–1261 CrossRef CAS.
- M. Jahandar Lashaki, S. Khiavi and A. Sayari, Chem. Soc. Rev., 2019, 48, 3320–3405 RSC.
- T. C. Drage, A. Arenillas, K. M. Smith and C. E. Snape, Microporous Mesoporous Mater., 2008, 116, 504–512 CrossRef CAS.
- M. Aresta, D. Ballivet-Tkatchenko, D. B. Dell'Amico, D. Boschi, F. Calderazzo, L. Labella, M. C. Bonnet, R. Faure and F. Marchetti, Chem. Commun., 2000, 1099–1100 RSC.
- B. E. Gurkan, J. C. de la Fuente, E. M. Mindrup, L. E. Ficke, B. F. Goodrich, E. A. Price, W. F. Schneider and J. F. Brennecke, J. Am. Chem. Soc., 2010, 132, 2116–2117 CrossRef CAS.
- C. Wang, H. Luo, D. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2010, 49, 5978–5981 CrossRef CAS.
- B. Gurkan, F. Simeon and T. A. Hatton, ACS Sustainable Chem. Eng., 2015, 3, 1394–1405 CrossRef CAS.
- X. Y. Luo, X. Y. Lv, G. L. Shi, Q. Meng, H. R. Li and C. M. Wang, AIChE J., 2019, 65, 230–238 CrossRef CAS.
- K. Huang, Y.-T. Wu and S. Dai, Ind. Eng. Chem. Res., 2015, 54, 10126–10133 CrossRef CAS.
- A. Ahmadalinezhad and A. Sayari, Phys. Chem. Chem. Phys., 2014, 16, 1529–1535 RSC.
- K. Masuda, Y. Ito, M. Horiguchi and H. Fujita, Tetrahedron, 2005, 61, 213–229 CrossRef CAS.
- Q. Yang, Z. Wang, Z. Bao, Z. Zhang, Y. Yang, Q. Ren, H. Xing and S. Dai, ChemSusChem, 2016, 9, 806–812 CrossRef CAS.
- P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102 CrossRef CAS.
- J. J. Gassensmith, H. Furukawa, R. A. Smaldone, R. S. Forgan, Y. Y. Botros, O. M. Yaghi and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 15312–15315 CrossRef CAS.
- P. J. Milner, R. L. Siegelman, A. C. Forse, M. I. Gonzalez, T. Runčevski, J. D. Martell, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2017, 139, 13541–13553 CrossRef CAS.
- A. C. Forse, P. J. Milner, J.-H. Lee, H. N. Redfearn, J. Oktawiec, R. L. Siegelman, J. D. Martell, B. Dinakar, L. B. Porter-Zasada, M. I. Gonzalez, J. B. Neaton, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2018, 140, 18016–18031 CrossRef CAS.
- C. E. Bien, K. K. Chen, S.-C. Chien, B. R. Reiner, L.-C. Lin, C. R. Wade and W. S. W. Ho, J. Am. Chem. Soc., 2018, 140, 12662–12666 CrossRef CAS.
- J. Im, S. Y. Hong, Y. Cheon, J. Lee, J. S. Lee, H. S. Kim, M. Cheong and H. Park, Energy Environ. Sci., 2011, 4, 4284 RSC.
- Y. Liu, H.-Z. Ye, K. M. Diederichsen, T. Van Voorhis and T. A. Hatton, Nat. Commun., 2020, 11, 2278 CrossRef CAS.
- J. H. Rheinhardt, P. Singh, P. Tarakeshwar and D. A. Buttry, ACS Energy Lett., 2017, 2, 454–461 CrossRef CAS.
- F.-F. Chen, K. Huang, Y. Zhou, Z.-Q. Tian, X. Zhu, D.-J. Tao, D. Jiang and S. Dai, Angew. Chem., Int. Ed., 2016, 55, 7166–7170 CrossRef CAS.
- S. Jiang, Y. Hu, Y. Wang and X. Wang, J. Phys. Chem. Ref. Data, 2019, 48, 033101 CrossRef.
- C. Wang, X. Luo, H. Luo, D. Jiang, H. Li and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 4918–4922 CrossRef CAS.
- T. G. Amundsen, L. E. Øi and D. A. Eimer, J. Chem. Eng. Data, 2009, 54, 3096–3100 CrossRef CAS.
- H. A. Patel, T. Islamoglu, Z. Liu, S. K. M. Nalluri, A. Samanta, O. Anamimoghadam, C. D. Malliakas, O. K. Farha and J. F. Stoddart, J. Am. Chem. Soc., 2017, 139, 11020–11023 CrossRef CAS.
- D. Wu, J. J. Gassensmith, D. Gouvêa, S. Ushakov, J. F. Stoddart and A. Navrotsky, J. Am. Chem. Soc., 2013, 135, 6790–6793 CrossRef CAS.
- K. J. Hartlieb, A. W. Peters, T. C. Wang, P. Deria, O. K. Farha, J. T. Hupp and J. F. Stoddart, Chem. Commun., 2017, 53, 7561–7564 RSC.
- M. L. Pinto, L. Mafra, J. M. Guil, J. Pires and J. Rocha, Chem. Mater., 2011, 23, 1387–1395 CrossRef CAS.
- R. W. Flaig, T. M. Osborn Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef CAS.
- P.-Q. Liao, X.-W. Chen, S.-Y. Liu, X.-Y. Li, Y.-T. Xu, M. Tang, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2016, 7, 6528–6533 RSC.
- N. Planas, A. L. Dzubak, R. Poloni, L.-C. Lin, A. McManus, T. M. McDonald, J. B. Neaton, J. R. Long, B. Smit and L. Gagliardi, J. Am. Chem. Soc., 2013, 135, 7402–7405 CrossRef CAS.
- R. L. Siegelman, P. J. Milner, A. C. Forse, J.-H. Lee, K. A. Colwell, J. B. Neaton, J. A. Reimer, S. C. Weston and J. R. Long, J. Am. Chem. Soc., 2019, 141, 13171–13186 CrossRef CAS.
- J. D. Martell, P. J. Milner, R. L. Siegelman and J. R. Long, Chem. Sci., 2020, 11, 6457–6471 RSC.
- S. Lindskog, Pharmacol. Ther., 1997, 74, 1–20 CrossRef CAS.
- P.-Q. Liao, H. Chen, D.-D. Zhou, S.-Y. Liu, C.-T. He, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Energy Environ. Sci., 2015, 8, 1011–1016 RSC.
- A. M. Wright, Z. Wu, G. Zhang, J. L. Mancuso, R. J. Comito, R. W. Day, C. H. Hendon, J. T. Miller and M. Dincă, Chem, 2018, 4, 2894–2901 CAS.
- Z. Cai, C. E. Bien, Q. Liu and C. R. Wade, Chem. Mater., 2020, 32, 4257–4264 CrossRef CAS.
- C. E. Bien, Q. Liu and C. R. Wade, Chem. Mater., 2020, 32, 489–497 CrossRef CAS.
- T. Wang, K. S. Lackner and A. Wright, Environ. Sci. Technol., 2011, 45, 6670–6675 CrossRef CAS.
- C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef.
- H. A. Duong, T. N. Tekavec, A. M. Arif and J. Louie, Chem. Commun., 2004, 112 RSC.
- L. J. Murphy, K. N. Robertson, R. A. Kemp, H. M. Tuononen and J. A. C. Clyburne, Chem. Commun., 2015, 51, 3942–3956 RSC.
- L. Legrand, O. Schaetzle, R. C. F. de Kler and H. V. M. Hamelers, Environ. Sci. Technol., 2018, 52, 9478–9485 CrossRef CAS.
- S. Zhu, J. Li, A. Toth and K. Landskron, ACS Appl. Energy Mater., 2019, 2, 7449–7456 CrossRef CAS.
- M. Rahimi, G. Catalini, S. Hariharan, M. Wang, M. Puccini and T. A. Hatton, Cell Rep. Phys. Sci., 2020, 1, 100033 CrossRef.
- Y. Ma, F. Zhang and R. P. Lively, in Sustainable Nanoscale Engineering, Elsevier, 2020, pp. 33–81 Search PubMed.
- P. G. Boyd, A. Chidambaram, E. García-Díez, C. P. Ireland, T. D. Daff, R. Bounds, A. Gładysiak, P. Schouwink, S. M. Moosavi, M. M. Maroto-Valer, J. A. Reimer, J. A. R. Navarro, T. K. Woo, S. Garcia, K. C. Stylianou and B. Smit, Nature, 2019, 576, 253–256 CrossRef CAS.
- B. Vlaisavljevich, S. O. Odoh, S. K. Schnell, A. L. Dzubak, K. Lee, N. Planas, J. B. Neaton, L. Gagliardi and B. Smit, Chem. Sci., 2015, 6, 5177–5185 RSC.
- Z. Zhang, J. A. Schott, M. Liu, H. Chen, X. Lu, B. G. Sumpter, J. Fu and S. Dai, Angew. Chem., Int. Ed., 2019, 58, 259–263 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |