
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progresses in the synthesis of MnO2 nanowire and its application in environmental catalysis
Huikang Song†
a,
Leilei Xu† *a,
Mindong Chen*a,
Yan Cuia,
Cai-e Wub,
Jian Qiuc,
Liang Xuc,
Ge Chenga and
Xun Hud
*a,
Mindong Chen*a,
Yan Cuia,
Cai-e Wub,
Jian Qiuc,
Liang Xuc,
Ge Chenga and
Xun Hud
aCollaborative Innovation Centre of the Atmospheric Environment and Equipment Technology, School of Environmental Science and Engineering, Nanjing University of Information Science & Technology, Jiangsu Key Laboratory of Atmospheric Environment Monitoring and Pollution Control, Nanjing, 210044, P. R. China. E-mail: leileixu88@gmail.com; chenmdnuist@163.com
bCollege of Light Industry and Food Engineering, Nanjing Forestry University, Nanjing 210037, P. R. China
cJiangsu ShuangLiang Environmental Technology Co., Ltd, Jiangyin, 214400, P. R. China
dSchool of Material Science and Engineering, University of Jinan, Jinan, 250022, P. R. China
First published on 3rd November 2021
Abstract
Nanostructured MnO2 with various morphologies exhibits excellent performance in environmental catalysis owing to its large specific surface area, low density, and adjustable chemical properties. The one-dimensional MnO2 nanowire has been proved to be the dominant morphology among various nanostructures, such as nanorods, nanofibers, nanoflowers, etc. The syntheses and applications of MnO2-based nanowires also have become a research hotspot in environmental catalytic materials over the last two decades. With the continuous deepening of the research, the control of morphology and crystal facet exposure in the synthesis of MnO2 nanowire materials have gradually matured, and the catalytic performance also has been greatly improved. Differences in the crystalline phase structure, preferably exposed crystal facets, and even the length of the MnO2 nanowires will evidently affect the final catalytic performances. Besides, the modifications by doping or loading will also significantly affect their catalytic performances. This review carefully summarizes the synthesis strategies of MnO2 nanowires developed in recent years as well as the influences of the phase structure, crystal facet, morphology, dopant, and loading amount on the catalytic performance. Besides, the cutting-edge applications of MnO2 nanowires in the field of environmental catalysis, such as CO oxidation, the removal of VOCs, denitrification, etc., have been also summarized. The application of MnO2 nanowire in environmental catalysis is still in the early exploratory stage. The gigantic gap between theoretical investigation and industrial application is still a great challenge. Compared with noble metal based traditional environmental catalytic materials, the lower cost of MnO2 has injected new momentum and promising potential into this research field.
1. Introduction
The worldwide emissions of atmospheric pollutants, such as CO and volatile organic compounds (VOCs), have greatly increased year by year with the rapid development of the global economy.1,2 For example, CO is generally regarded as a flammable, explosive, and toxic air pollutant, which is extremely harmful to human health and the living environment.3–6 Besides, it is also a weak direct greenhouse gas and has an important indirect effect on global warming.7–10 Generally, CO is mainly derived from the incomplete combustion of fuels.11,12 As for the VOCs, they commonly include formaldehyde, benzene, toluene, xylene, etc.2 They can enter into the human body through breathing and skin permeation, causing discomfort to the body and even inducing cancer. Besides, VOCs are also considered as important precursors of high concentrations of PM2.5 and O3 in the atmosphere. Therefore, it is greatly necessary to develop efficient technologies to control the emissions of CO, VOCs, and other atmospheric pollutants. The current technologies mainly include adsorption, separation, biodegradation, catalytic combustion, plasma coordinated catalysis, photocatalysis, etc. Among these strategies, the catalytic oxidation or combustion has attracted increasing attention due to the advantages of high purification efficiency, low reaction temperature, and low cost. Therefore, designing and developing the catalysts with advanced performances are the key solutions to the catalytic oxidation of these air pollutants.Noble metals (Au,13,14 Pt,15–17 Pd,18 and Rh19) supported on particular metal oxides (CeO2, CexZr1−xO2 and Fe2O3) with excellent oxygen storage capacities usually perform high catalytic activity toward CO oxidation. However, they are extremely restricted in wide and large-scale application due to the high price and scarcity. Thus, it is very important to design and explore the low-cost catalysts with identical catalytic performance to the noble metals. It was reported that various transition metal oxides (Co3O4,20,21 CuO,22 MnO2,23 and CeO2 (ref. 24)) performed excellent catalytic activity for CO oxidation in the past decades. Among them, MnO2 has been extensively investigated due to various advantages, such as low price, environmental friendliness, and relatively high activity.25,26 With the continuous studies on MnO2 based materials, it has been found that MnO2 based catalysts usually perform good oxidation activities for the VOCs, such as formaldehyde, toluene, benzene, dimethyl ether, etc. Besides, the catalytic oxidation ability of MnO2 can be greatly improved through crystal facet exposure engineering and regulating the loading methods.
Compared with the traditional bulk MnO2, the nanostructured MnO2 materials usually perform better physiochemical characteristics, such as the higher specific area, lower density, and more adjustable chemical nature. Furthermore, various studies have shown that MnO2 with nanowire morphology usually perform high surface areas and strong metal–support interactions.27–31 Therefore, the nanowire is the dominant morphology among the MnO2-based nanostructured materials. Compared with MnO2 nanorods, MnO2 nanowires commonly possesses much more adsorbed surface oxygen, greater reducibility, higher specific area, and lower Mn–O bond strength, accounting for the better catalytic performances in the catalytic combustion of dimethyl ether27 and plasma catalytic oxidation of toluene.28 Saputra et al.29 found that MnO2 nanowire displayed much higher activity than the MnO2 nanorod and nanofiber counterparts in activating oxygen ketones to degrade phenol. In addition to this, compared with rod-shaped and tubular MnO2, the MnO2 nanowire as the support behaved stronger interaction with Ag and displayed the much higher reactivity of toluene oxidation.30 The α-MnO2 nanowire also displayed the best catalytic activity toward gaseous benzene oxidation among the MnO2-based nanomaterials with different morphologies (e.g., wire and rod-like α-MnO2, β-MnO2 microrods) even after decorating Co3O4 nanoparticles.31
In recent years, more and more advanced and important materials have been developed for the applications of MnO2-based nanomaterials in the removal of environmental pollutants,32,33 biosensing,34 theranostic,35 especially in the field of electrochemistry.36–38 For example, Li et al.37,38 synthesized α-MnO2 nanowires with outstanding circulation stability, demonstrated rate performance, and high specific capacitance for supercapacitor electrode. Therefore, the previous reviews of MnO2-based nanomaterials were mainly focused on their applications in electrochemistry.39–43 However, there were few reviews paid close attention to their environmental applications. Yang et al.44 systematically summarized the environmental applications of MnO2-based materials. But they did not specially highlight the unique advantages of MnO2-based nanowire materials, which have been considered as the basic assembly unit of two-dimensional or three-dimensional structure complex/heterostructure materials of MnO2 (ref. 45) and normally have higher catalytic activity compared with MnO2-based nanomaterials in other morphologies.27–30 Meanwhile, the previous discussions about MnO2-based nanowire materials were very scattered and their applications were mainly focused on the topic in the field of the electrochemistry. There has been few comprehensive reviews targeting the applications of MnO2-based nanowire materials in the field of environmental catalysis. A systematic overview of frontier scientific research on the modulation of MnO2-based nanowire materials for environmental catalysis application is urgent for the rational design and fabrication of high-efficiency MnO2-based nanowire materials. Therefore, this review comprehensively summarizes the synthesis methods of MnO2-based nanowires, such as hydrothermal, reflux, sol–gel, template method, etc., and analyses the effects of various influencing factors, such as the crystalline phase structure, crystal facet, morphology, doping, and loading, on the performances of catalytic oxidation of CO, VOCs, and other environmental pollutants. In addition, the latest research progresses of MnO2-based nanowire materials in the field of environmental catalysis are also covered in this review. With the in-depth research on MnO2-based nanowire materials, their environmental applications have been greatly improved. However, there is still big gap between the theoretical study and practical application. Therefore, it is of great necessity to summarize the recent progresses for better understanding the real surface reaction mechanism, which will promise the future environmental application in the fields of catalytic oxidation, combustion, purification, and other related research fields.
2. Synthesis of MnO2-based nanowires
There are various preparation methods of MnO2 nanomaterials, such as the liquid phase precipitation method,46,47 reflux method,48 electrodeposition method,49 hydrothermal method,50–53 sol–gel method,54,55 template method,56 thermal decomposition method,57 and so on. As a result, the phase structures and chemical characteristics of MnO2 nanomaterials fabricated by different methods will be completely different. Similarly, the synthesis of MnO2-based nanowires also mainly employs the hydrothermal method, reflux method, sol–gel, and template method. Herein, the synthesis method, reaction condition, morphology parameters, and structural properties of MnO2-based nanowires have been summarized in Table 1.| MnO2 nanowire | Preparation method | Raw materials | Synthesis temperature (°C) | Synthesis time (h) | Length | Diameter (nm) | BET (m2 g−1) | Reference |
|---|---|---|---|---|---|---|---|---|
| α-MnO2 | Hydrothermal method | MnSO4·H2O, KMnO4 | 150 | 12 | 2.5 μm | 20–40 | 61.0 | 82 |
| Hydrothermal method | KMnO4, NH4Cl | 200 | 48 | Several μm | 100–200 | 53.1 | 61 | |
| Hydrothermal method | MnSO4·H2O, (NH4)2S2O8 | 140 | 12 | 0.4–5 μm | 5–20 | 148.0 | 29 | |
| Hydrothermal method | δ-MnO2, H2SO4 | 100 | 24 | 250–500 nm | 30–60 | 93.1 | 27 | |
| Hydrothermal method | KMnO4, CH3COOH | 140 | 12 | 6–10 μm | 30–60 | 39.9 | 83 | |
| Reflux method | δ-MnO2 nanosheets | 80–90 | 90–120 | >10 μm | 20–30 | 53.2 | 58 | |
| Sol–gel template method | AAO template, Mn (CH3COO)2, citric acid | 400 | 12 | 500–700 nm | 70 | — | 54 | |
| β-MnO2 | Hydrothermal method | MnSO4·H2O, (NH4)2S2O8 | 140 | 12 | 1.5–2.5 μm | 60–100 | 55.7 | 84 |
| Hydrothermal method | Mn (NO3)2, O3 | 200 | 8 | 2–5 μm | 6–12 | 73.5 | 58 | |
| γ-MnO2 | Hydrothermal method | MnSO4·H2O, (NH4)2S2O8 | 90 | 24 | — | — | — | 85 |
| δ-MnO2 | Hydrothermal method | MnSO4·H2O, KMnO4 | 240 | 24 | — | — | — | |
| α-MnO2-100 | Hydrothermal method | MnSO4·H2O, (NH4)2S2O8, (NH4)2SO4, KNO3 | 120 | 20 | Several μm | 15–20 | 87.9 | 86 |
| α-MnO2-110 | Hydrothermal method | KMnO4, (NH4)2SO4 | 180 | 24 | — | 45–50 | 119.5 | |
| Hydrothermal method | KMnO4, (NH4)2C2O4·H2O | 180 | 24 | 100–500 nm | 10–50 | — | 87 | |
| α-MnO2-210 | Hydrothermal method | MnCO3, HNO3, (NH4)2S2O8, H2SO4 | 140 | 2 | 100–500 nm | 10–50 | — | |
| α-MnO2-310 | Hydrothermal method | KMnO4, (NH4)2SO4 | 180 | 24 | 100–500 nm | 10–50 | — | |
| Hydrothermal method | KMnO4, (NH4)2C2O4 | 180 | 24 | Several μm | 15–20 | 135.3 | 86 |
2.1 Hydrothermal synthesis method
The hydrothermal synthesis of MnO2-based nanowires mainly involves oxidation–reduction reaction of MnO4− and/or Mn2+ or the phase transition of MnO2 precursor. The hydrothermal method usually involves several key parameters, including the reagent amount, reaction time, hydrothermal temperature, pH, and so on. Therefore, the MnO2-based nanowires with different phase structures can be synthesized in a precisely controllable way by tuning these key parameters in the hydrothermal process.DeGuzman et al.50 pointed out that pH and temperature could affect the final phase structure of MnO2 in the synthesis of fibrous α-MnO2 nanomaterials and α-MnO2 would transit into β-MnO2 when the temperature was kept above 120 °C. However, Wang et al.51 found that the effects of pH and temperature were not significant. In their study, 1D MnO2 nanomaterials with different phase structures could be obtained in a wide temperature range (100–180 °C) just by varying the molar ratio of raw reactants (KMnO4 and MnSO4) without adjusting the pH (∼2.5) of the system. Besides, the type of the raw reactants greatly determines the hydrothermal reaction temperature of preparing the α-MnO2 nanowire. For example, the optimal hydrothermal temperature was located between 120 °C and 160 °C when using KMnO4 and MnSO4·H2O as raw materials;58–60 whereas, the optimal hydrothermal temperature was 200 °C when using KMnO4 and NH4Cl as the precursors.61–63 The effect of pH on the crystalline phase structure of the final product was also largely derived from the ion concentration as the tunnels of different MnO2 crystal forms have different sizes and need different amounts of cations to stabilize.64 Zhang et al.53 showed that the phase structure of MnO2 and the aspect ratio of the final product were greatly depended on the concentration of ions in the reaction system, especially K+. Because the high concentration of K+ could be used as the effective template, which was more conducive to the synthesis of α-MnO2 nanowire.53 Wang et al.51 believed that the layer structure of δ-MnO2 tended to curl under the high temperature and pressure, which was considered as the determinant step for the formation of 1D nanostructures of MnO2. Zhang et al.53 also indicated that prolongating hydrothermal time could drive the self-assembly of α-MnO2 into bundles and then transform into β-MnO2. Therefore, the possible synthesis route of MnO2-based nanostructures was described in Fig. 1. First, the MnOx units appear in the solution and the δ-MnO2 with the layer structure is then formed through a series of condensation reactions. δ-MnO2 tends to curl under high temperature and high pressure. When K+ concentration is high, α-MnO2 can be formed; otherwise, β-MnO2 can be formed. With the prolonging of hydrothermal time, the [2 × 2] tunnel structure of α-MnO2 directly collapses into the [1 × 1] tunnel structure of β-MnO2.53 Wei et al.52 studied the hydrothermal synthesis of MnO2-based nanowires through the phase transition of granular MnO2 precursor. They synthesized α-MnO2 and β-MnO2 single crystal nanowires by the hydrothermal treatment of γ-MnO2 at different temperatures. This method not employing catalysts or templates could be easily reproduced. Therefore, the hydrothermal synthesis of different crystal types of MnO2 nanowires with potential application prospects in large-scale could be realized.
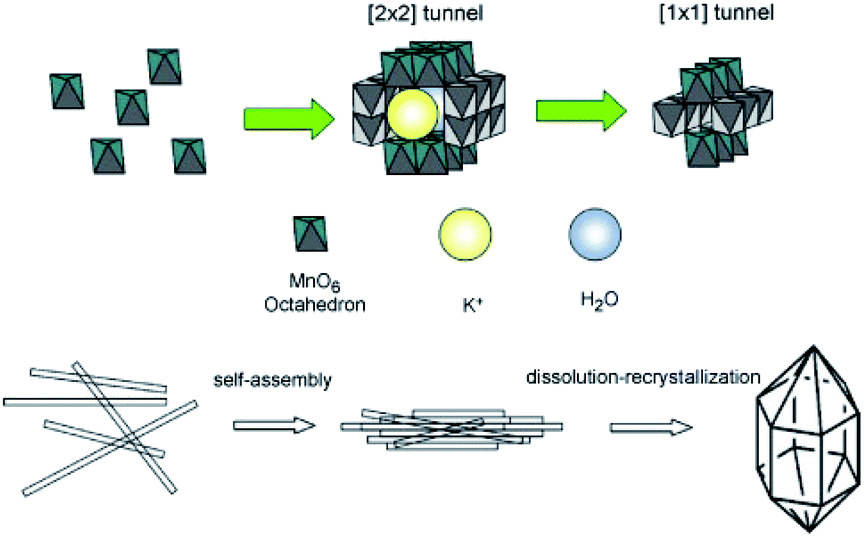 | ||
| Fig. 1 Schematic illustration of the self-assembly of α-MnO2 nanowires into bundles and their subsequent phase transformation to β-MnO2 microcrystals under prolonged hydrothermal conditions. Reproduced by Zhang et al.53 with permission from Elsevier. | ||
Generally, the hydrothermal method of preparing MnO2-based nanowire has various advantages, such as high product purity, excellent dispersion, good crystallinity, uniform morphology, etc. Besides, the preparation conditions are simple and the fabrication time is short. Therefore, the hydrothermal method has been widely used for the synthesis of MnO2-based nanowires. However, the hydrothermal method is usually limited by the capacity of the reaction system, so it is not suitable for the large-scale industrial production. Furthermore, the tightness of the reaction system will bring safety hazards.65
2.2 Reflux method
The reflux is a kind of wet chemical synthesis method that does not require high-temperature calcination, and directly synthesizes nanomaterials with uniform particle size and high activity. The reflux method is suitable for the large-scale synthesis of MnO2 nanowires owing to various advantages, such as simple operation and mild reaction conditions. Kijima et al.66 synthesized MnO2 with three different phase structures (α-, β-, and γ-) by the acid digestion of Mn2O3 under reflux conditions. The type of polymorph of MnO2 products was strongly dependent on the reaction temperature, type of acid used, and the concentration. When H2SO4 was used as the acid, the α-MnO2 was inclined to form at higher H2SO4 concentrations and lower reaction temperatures. In contrast, the β-MnO2 was obtained at lower H2SO4 concentrations and higher reaction temperatures. The γ-MnO2 were formed at intermediate conditions. When HNO3 was used as the acid, only the β-MnO2 and γ-MnO2 could be formed. The α-MnO2 could not be obtained in the HNO3 solution. Compared with γ-MnO2, the β-MnO2 formation usually demanded higher reaction temperatures (>80 °C) and higher concentrations of HNO3 (>10 mol L−1).66 Chen et al.67 synthesized transition metal doped α-MnO2 nanofibers by the reflux method. Compared to the pristine MnO2, metal cation doped MnO2 materials had slightly higher surface areas and pore volumes, especially when the dopant content was relatively high. Jin et al.68 synthesized γ-MnO2 homogeneous nanofiber with the length of ∼1 μm and the diameter of 50 nm by refluxing and used it as the catalyst for catalysing the atmospheric oxidation of toluene with molecular oxygen. Sinha et al.65 prepared δ-MnO2 nanosheets by the reflux method and further deepened the morphological evolution and shape transformation into ultra-long α-MnO2 nanowires with extremely high purity by reflux. The length of the nanowire was observed to exceed 10 μm. The detailed preparation process and SEM results are shown in the Fig. 2. The reflux method also has the advantage of low chemical cost and avoids using autoclaves under harsh hydrothermal reaction conditions. As a result, the high-quality α-MnO2 nanowires could be generated without using catalysts and stencils. The reflux method provides a promising preparation method of α-MnO2 nanowires in large scale.65 May et al.69 synthesized α-MnO2-based nanowires and further studied the effect of doping methods on low-temperature CO oxidation of CuO/α-MnO2 catalysts based on the nitric acid reflux method of Uematsu et al.70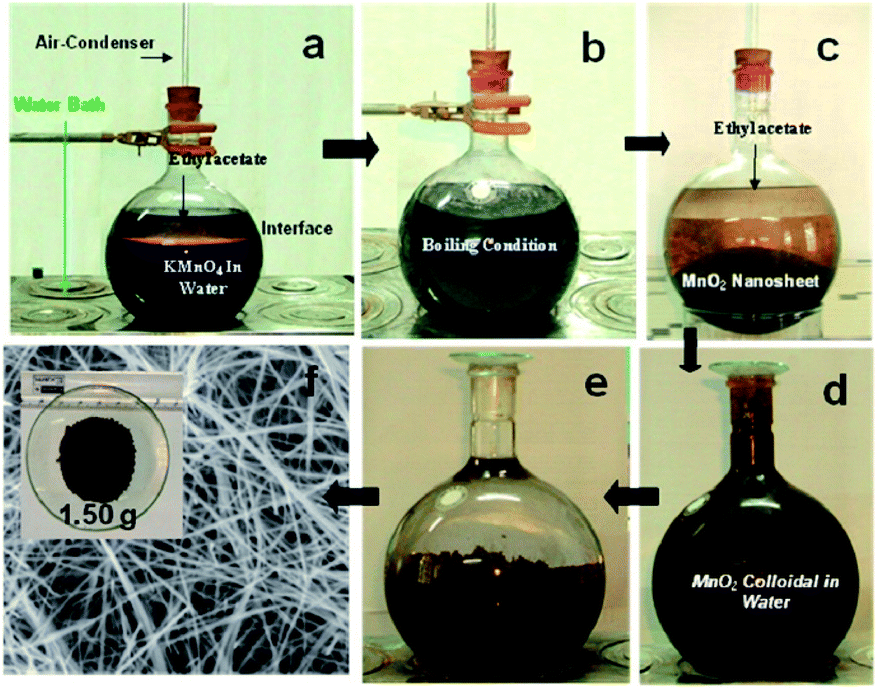 | ||
| Fig. 2 Preparation of gram level α-MnO2 nanowires from aqueous KMnO4/ethyl acetate biphasic system and their different stages (a−f) in digital/FESEM images. Reproduced by Sinha et al.65 with permission from ACS. | ||
2.3 Sol–gel method
As for the sol–gel method, it employs the metal alkoxides or inorganic salts as precursors to achieve gelation through continuous hydrolysis and polycondensation processes. Then, the gel experiences the heat, and other subsequent treatments to obtain the final target product. The sol–gel synthesis of nanostructured MnO2 commonly includes various combinations of hydrated metal cations, such as manganese acetate (MnAc2) and citric acid,71 MnAc2 and n-propyl alcohol,54 tetrabutylammonium permanganate and methanol,72 and KMnO4 and fumaric acid (C4H4O4).73,74 The structures of the obtained MnO2 are strongly depended on the molar ratio between the KMnO4 and the organic reducing agents.75 For example, the 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reactant ratio of KMnO4 to glucose favoured the cryptomelane (2 × 2 tunnel) synthesis, while the 5:1 ratio generated the layered birnessite-type material.72 Hastuti et al.74 synthesized irregular spherical α-MnO2 particles by the sol–gel method and discovered that transition metal (Fe3+, Cu2+) doping did not affect the phase structure but only changed the lattice parameters and morphology of the final product. Hashem et al.76 prepared the α-MnO2 materials by redox reaction of KMnO4 and ascorbic acid using the sol–gel method. The morphology of the final sol–gel samples were long needles with uniform diameter of ∼15 nm and average length of ∼0.8 μm, and further aggregated to urchin-like balls. Hashemzadeh et al.71 prepared γ-MnO2 polymorph using the manganese acetate and citric acid as the raw reagents in the sol–gel way. The obtained γ-MnO2 polymorph was composed of particles with surface covered with needle-like nanorods. The researches have demonstrated that the sol–gel method could prepare 1D nanostructure MnO2 nanowire with different morphologies. Ghorbani et al.77 successfully synthesized MnO2 nanowires with 45 nm in diameter using simple precursors, such as KMnO4 and NaNO3, by sol–gel method. They found that the optimal annealing temperature for achieving crystalline structure and nanowire morphology was 600 °C. Chin et al.78 successfully prepared MnO2 nanowires via the self-assembly of MnO2 nanoparticles using the sol–gel method with KMnO4 and Mn(ClO4)2 as the precursors. The microstructure and morphology of sol–gel derived MnO2 nanowire were affected by the synthesis conditions and post synthesis heat treatment. Acidic pH value (pH 5.7) and calcination temperature at 300 °C were effective on achieving nanowire morphology and high surface area. Liu et al.79 synthesized wire-like ion sieve MnO2·0.5H2O with about 50–200 nm in diameter and 0.5–2 μm in length using the sol–gel method. The ion sieve MnO2·0.5H2O nanowire had excellent adsorption performance for lithium extraction in seawater system and had a good application prospect. Furthermore, the combination of sol–gel and template has been considered as a better way to synthesize highly-ordered MnO2-based nanowires than the sol–gel itself.54,55 As shown in Fig. 3, Tang et al.55 used CTAB as the surfactant to obtain highly dispersed ultrafine MnO2 nanowires with the diameter of only 7 nm by the template directed sol–gel template method.
:1 reactant ratio of KMnO4 to glucose favoured the cryptomelane (2 × 2 tunnel) synthesis, while the 5:1 ratio generated the layered birnessite-type material.72 Hastuti et al.74 synthesized irregular spherical α-MnO2 particles by the sol–gel method and discovered that transition metal (Fe3+, Cu2+) doping did not affect the phase structure but only changed the lattice parameters and morphology of the final product. Hashem et al.76 prepared the α-MnO2 materials by redox reaction of KMnO4 and ascorbic acid using the sol–gel method. The morphology of the final sol–gel samples were long needles with uniform diameter of ∼15 nm and average length of ∼0.8 μm, and further aggregated to urchin-like balls. Hashemzadeh et al.71 prepared γ-MnO2 polymorph using the manganese acetate and citric acid as the raw reagents in the sol–gel way. The obtained γ-MnO2 polymorph was composed of particles with surface covered with needle-like nanorods. The researches have demonstrated that the sol–gel method could prepare 1D nanostructure MnO2 nanowire with different morphologies. Ghorbani et al.77 successfully synthesized MnO2 nanowires with 45 nm in diameter using simple precursors, such as KMnO4 and NaNO3, by sol–gel method. They found that the optimal annealing temperature for achieving crystalline structure and nanowire morphology was 600 °C. Chin et al.78 successfully prepared MnO2 nanowires via the self-assembly of MnO2 nanoparticles using the sol–gel method with KMnO4 and Mn(ClO4)2 as the precursors. The microstructure and morphology of sol–gel derived MnO2 nanowire were affected by the synthesis conditions and post synthesis heat treatment. Acidic pH value (pH 5.7) and calcination temperature at 300 °C were effective on achieving nanowire morphology and high surface area. Liu et al.79 synthesized wire-like ion sieve MnO2·0.5H2O with about 50–200 nm in diameter and 0.5–2 μm in length using the sol–gel method. The ion sieve MnO2·0.5H2O nanowire had excellent adsorption performance for lithium extraction in seawater system and had a good application prospect. Furthermore, the combination of sol–gel and template has been considered as a better way to synthesize highly-ordered MnO2-based nanowires than the sol–gel itself.54,55 As shown in Fig. 3, Tang et al.55 used CTAB as the surfactant to obtain highly dispersed ultrafine MnO2 nanowires with the diameter of only 7 nm by the template directed sol–gel template method.
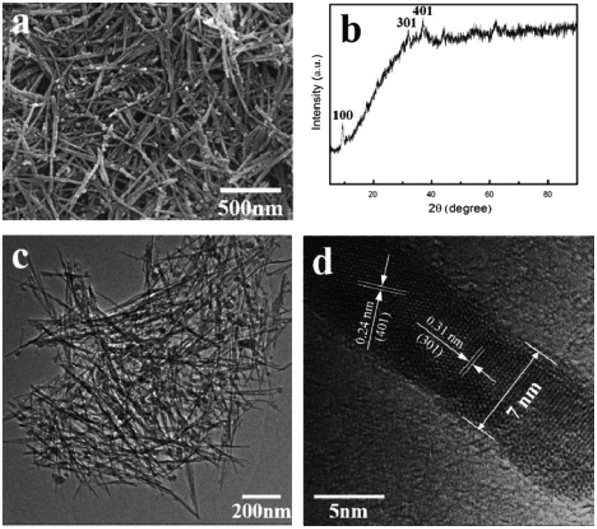 | ||
| Fig. 3 (a) SEM image, (b) XRD pattern, (c) and (d) TEM images of MnO2 nanowire prepared by sol–gel approach. Reproduced by Tang et al.55 with permission from Elsevier. | ||
2.4 Template method
The template method has been considered as a common method for preparing nanomaterials by employing various structure directing agents or templates in recent years. Nanomaterials with ordered structures are generated through the guidance of the template in the form of self-assembly by using the organic molecules as template agents. For example, Wang et al.54 successfully prepared α-MnO2 nanowire with the diameter of about 70 nm and the length between 500 nm and 700 nm using the sol–gel template method. The length and diameter of the nanowires greatly depended on the pore size and thickness of the employed AAO template. Gu et al.56 used mesoporous silica SBA-15 as the hard template to prepare highly ordered array of mesoporous β-MnO2 nanowires by the nano-casting method. Fig. 4 shows the schematic representation of the preparation of MnO2 by using the SBA-15 as the template.80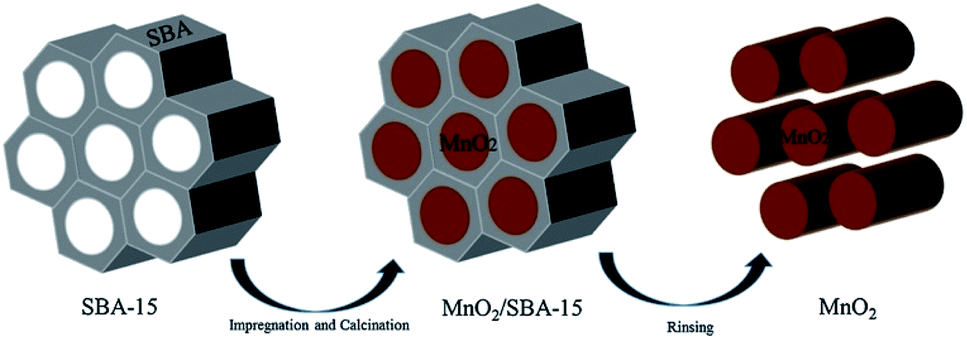 | ||
| Fig. 4 Schematic representation of the preparation of MnO2 by SBA-15 template. Reproduced by Zhang et al.80 with permission from Elsevier. | ||
The morphology of materials synthesized by the template method can be tunable by the choice of the template. However, the use of templates obviously increases the production cost. Therefore, reducing the costs of the templates is still an important concerned issue. Wang et al.81 fabricated hierarchical MnO2 microfibers by using cotton as the template and KMnO4 as the precursor. In comparison with other templates, biological materials are “green” and easy to obtain, and the morphology of cotton fiber is more uniform than other kinds of plant fibers. This green, sustainable, and low-cost biotemplating method can be extended to the synthesis of MnO2-based nanowires and other metal oxide materials in the view of economical concern.
3. The factors affecting the catalytic performances of MnO2 nanowires
3.1 Phase structure
The [MnO6] octahedral unit is the basic building block for crystalline-phase MnO2 materials. As shown in Fig. 5, when [MnO6] octahedral units are linked in different ways, the MnO2 can form a variety of polymorphs, such as α-, β-, γ- and δ-MnO2.88–90 Studies have found that the phase structure can significantly affect the catalytic activity of MnO2, and both α-MnO2 and δ-MnO2 are the dominant phase structures of MnO2-based catalytic nanomaterials.91–94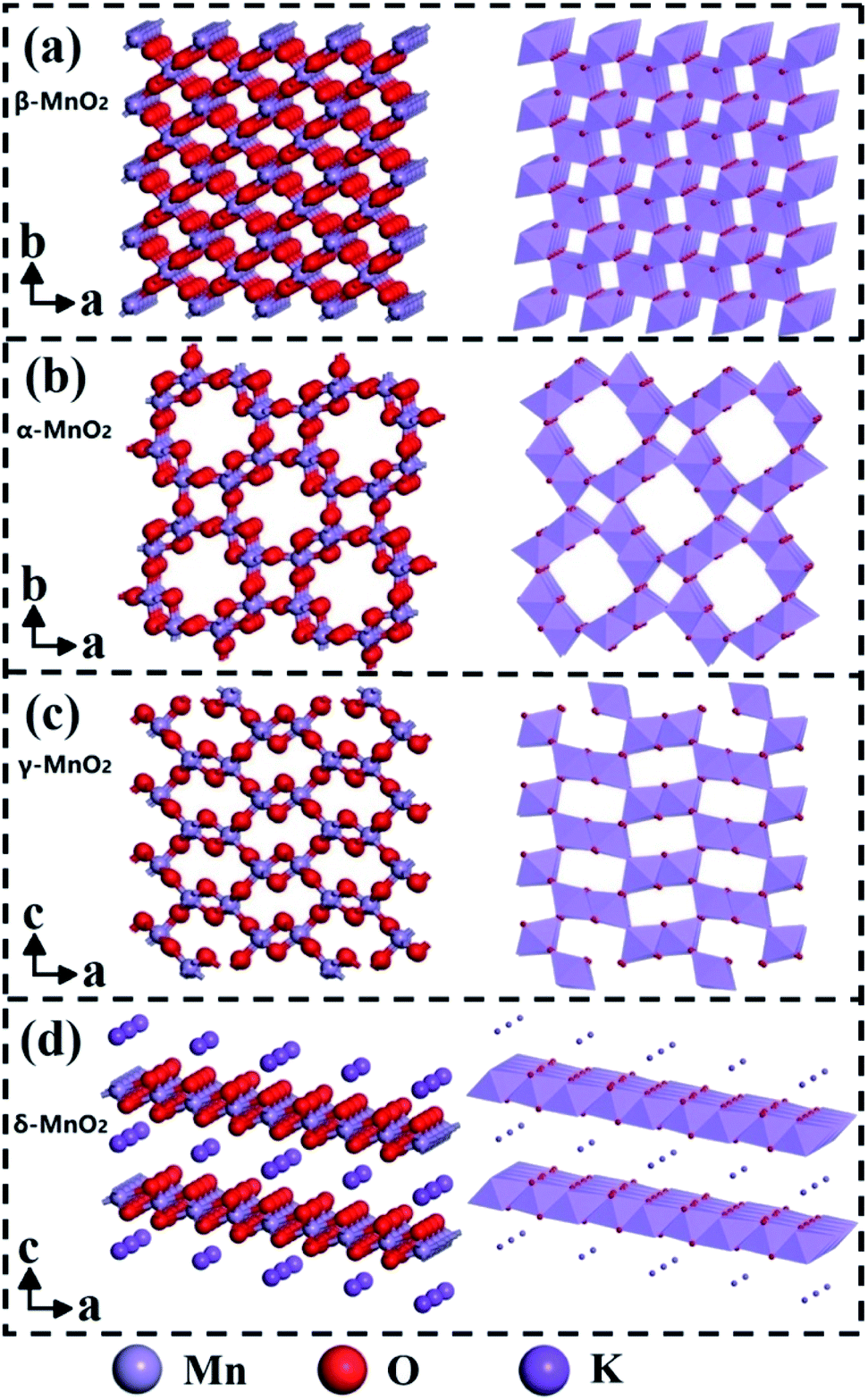 | ||
| Fig. 5 Ball-stick and polyhedron representations of (a) β-, (b) α-, (c) γ- and (d) δ-MnO2. Reproduced by Li et al.91 with permission from Elsevier. | ||
Specifically, α-MnO2 occupies more space than β-, γ-, and δ-MnO2 due to the double tunnel structure of [2 × 2] and [1 × 1]. As a result, it obtains more adsorption sites for the reactants (such as CO and VOCs) than other phases. At the same time, the existence of surface open pore structure can also increase the surface area of α-MnO2 nanowires, thereby providing abundant oxygen adsorption sites for the reactants.92 In addition, α-MnO2 also has the weakest Mn–O bond strength among these crystalline phase structures, which could provide the lattice and absorbed oxygen during the oxidation reaction. Therefore, α-MnO2 performs much better activity than other phase structures in catalytic reactions, such as CO oxidation,26 O3 catalytic degradation,93 plasma oxidation of toluene,28 and degradation of phenol by activating oxyketone.29
δ-MnO2 is composed of [MnO6] octahedrons shared by the double layer edges. The special layer structure makes it easy for toluene molecule to absorb on the surface of δ-MnO2 and thereby behaves excellent activity in the catalytic oxidation of toluene.91 As the comparison, the α-MnO2 performs weaker toluene oxidation activity due to the lower content of Mn4+ and lattice oxygen.91 Besides, δ-MnO2 has also been proved to be the best crystalline phase structure for low-temperature catalytic oxidation of formaldehyde.94
β-MnO2 with [1 × 1] tunnels have the most stable redox ability among these four phase structures. Besides, the surface area of β-MnO2 nanowire materials is usually the smallest among the four phase structures, which will greatly hinder the adsorption and activation of the reactants and oxygen. As a result, it usually performs the lowest activity in the catalytic oxidation of CO and VOCs. The [2 × 1] and [1 × 1] tunnel structures of γ-MnO2 have relatively mediocre catalytic activity in the oxidation of CO and VOCs.
3.2 Crystal facet
Studies have shown that the catalytic activities toward specific substances can be significantly improved by synthesizing nanomaterials with a high proportion of exposure specific crystal facets.95–97 Recently, more and more studies have paid attention to the influence of specific crystal facets on their catalytic effects, such as TiO2,95 Fe2O3,96 CeO2 (ref. 97) and Co3O4,20 because they are closely related with the final catalytic performances. Meanwhile, the great progresses have been made in the application of specific crystal facet of MnO2-based nanowires to the catalytic oxidation of VOCs, such as formaldehyde,86 methylmercury93 and toluene,87 etc.Theoretically, the reactivity and activity of crystal faces are proportional to their surface energy. Therefore, the crystal facets with higher surface energies are usually more reactive in heterogeneous reactions.98 Many studies have been carried out on the α-MnO2 nanowires because α-MnO2 has highest activity among the four phase structures of MnO2-based nanowires. The characteristic crystal facets of α-MnO2 generally include (310), (100), (110), and (210). Among them, (310) crystal facet possesses the highest surface energy, which is beneficial to the formation of oxygen vacancy. Thereby, the adsorption and activation of O2, H2O (ref. 86) and O3,93 etc., could be promoted. As a result, (310) crystal facet performs obviously better activities on the oxidation of formaldehyde and the ozonation of methylmercury than (100) and (110) counterparts (see Fig. 6). Rong et al.86 reported that the 310-α-MnO2 could completely convert 100 ppm formaldehyde to CO2 at 60 °C, which was even better than some silver-based catalysts.99,100 As shown in Fig. 7, the (310) facets with high surface energy could not only facilitate adsorption/activation of O2 and H2O but also be beneficial to the generation of oxygen vacancies, which resulted in significant enhancement of HCHO oxidation activity. He et al.93 reported that the 310-α-MnO2 could completely remove 70 ppm of methyl mercaptan within 20 minutes at room temperature. However, crystal planes with higher surface energy usually disappear in most crystals with the fast growth and the thermodynamically stable crystal planes would preferentially dominate the surface to minimize the total surface energy of the crystal.86 Meanwhile, Huang et al.87 found that (210) crystal plane with the lower surface activation energy (24.75 kJ mol−1) showed better catalysis activity and stability for toluene than (110) and (310) crystal plane. The lower surface activation energy accelerated the activation and oxidation of toluene. The alternating arrangement of Mn–O atoms in the electronic structure also speeds up the oxidation and desorption of intermediate products, thus exhibiting excellent activity.
 | ||
| Fig. 6 TEM and HRTEM images of (a–c) α-MnO2-310, (e–g) α-MnO2-110, and (i–k) α-MnO2-100. Atomic structure of the (d) {310}, (h) {110}, and (l) {100} surface of α-MnO2. Small (red) spheres are oxygen and large (purple) are manganese. Reproduced by Rong et al.86 with permission from ACS. | ||
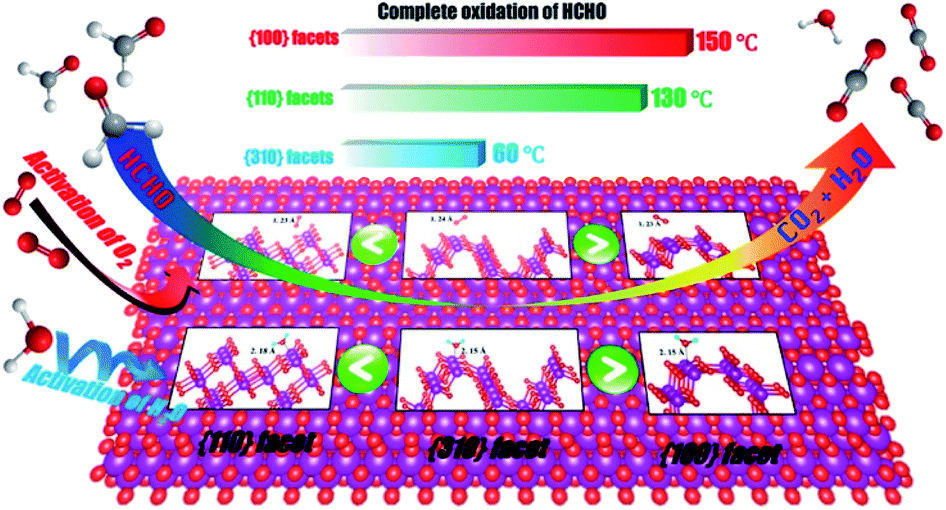 | ||
| Fig. 7 α-MnO2 nanowires with (100), (110) and (310) facets for HCHO oxidation. Reproduced by Rong et al.86 with permission from ACS. | ||
All in all, the specific VOCs oxidation reaction mechanism on the MnO2-based nanowires is the key to improving the oxidation ability of MnO2-based nanowire catalyst to VOCs through crystal facet engineering. It is also greatly necessary to strengthen the research on the mechanism of catalytic oxidation of VOCs because of the specific advantages of each crystal facet of MnO2-based nanowires.
3.3 Morphology
As well known, the physiochemical properties of MnO2 with different morphologies are often different.101,102 The common one-dimensional structure of MnO2 includes nanorods, nanotubes and nanowires. Among these morphologies, the MnO2 nanowires occupies an important place. Because one-dimensional nanowires can be used as the basic assembly unit of two-dimensional or three-dimensional structure materials.45 Furthermore, the composite materials with novel structures and properties can be facilely prepared on the basis of them.One of the basic parameters greatly affecting the performance of nanowires is the length or aspect ratio. For example, the photocatalytic performance of W18O49 nanowires is greatly affected by its aspect ratio;103 the dielectric properties of BaTiO3 nanowires also have close relationship with its length.104 Therefore, the performance of the nanowire can be tuned by controlling the length or aspect ratio. Liao et al.105 investigated the influence of the length of α-MnO2 nanowires on the catalytic combustion performance of dimethyl ether. The results revealed that the length of the nanowires would affect the redox property of α-MnO2 and the final catalytic combustion performance of dimethyl ether. Among them, α-MnO2 nanowires with medium length (4–8 μm) possessed the best catalytic performance with ignition temperature at 167 °C and the complete combustion temperature at 240 °C. The 20 h life test demonstrated that the sample behaved good stability without deactivation.105 Li et al.91 prepared three kinds of δ-MnO2 with different crystallinities and morphologies. The complete toluene conversion was achieved at 250–275 °C for δ-MnO2 and the toluene conversion efficiency of δ-MnO2 would be promoted with the decrease of crystallinity degree and particle size. Although the effect of the crystallinity and morphology were not as significant as the phase structure, they indeed greatly affected the activity of MnO2 in the catalytic oxidation of toluene.
3.4 Metal loading and doping
The metal loading and doping are two common used modification methods when preparing catalyst. Both of them could significantly improve the catalytic activity of the MnO2-based nanowire. The loading method could highly and uniformly disperse metal or metal oxide on the surface of catalytic support and the catalysts with higher activities could be achieved due to the formed strong metal-support interactions.106 The doping method is to replace a small portion of the second metal cations in the host metal oxide with different cations can greatly change the chemical bonding on the surface of the host metal oxide, thereby improving its catalytic performance.107 The active site in such catalytic system could be either the oxygen atoms near the dopant or the dopant itself. As a result, the activity of MnO2-based nanowire can be further improved by dispersing more active ingredients on its surface.The studies related to the metal modification of MnO2 usually starts with the loading of noble metals, such as Au and Ag. For example, Gu et al.56 loaded Au nanoparticles on α-, β-MnO2 nanowires and developed Au/MnO2 catalyst with extremely high activity and stability for CO oxidation at low temperature. The Au/α-MnO2 nanowire catalyst showed the CO conversion of around 37% at −90 °C. As the temperature increased, the CO conversion increased rapidly to 90% below −60 °C, and slowly reached 100% at around −34 °C. The low-temperature (−85 °C) CO oxidation rate over this catalyst (2.8 molCO h−1 gAu−1) was almost 30 times higher than that of the Au/Mn2O3.108 Qin et al.109 investigated the toluene oxidation activity over the α-MnO2 nanowires supported catalysts with different Ag loading amounts, which greatly affected the toluene oxidation performance. As could be seen in Fig. 8, 4% Ag/MnO2 catalyst performed the highest activity of the toluene oxidation and the lowest apparent activation energy (45.5 KJ mol−1) among Ag/MnO2 catalysts. They found that 4% loading of Ag could greatly facilitate the adsorption of toluene molecules and optimally increase the activation capacity of MnO2 lattice oxygen between the interface of Ag and MnO2, thereby increasing the reaction rate and promoting deep oxidation between toluene and lattice oxygen.109 Apart from the loading of noble metals, Sadeghinia et al.84 reported that the catalytic activity of CO oxidation could be enhanced by loading the α-MnO2 nanowires with different contents of CuO. The 30% CuO/α-MnO2 catalyst could achieve the complete CO conversion at about 100 °C, which was nearly 100 °C lower than pristine α-MnO2 nanowire. May et al.69 further confirmed that the loading of CuO by the deposition precipitation (DP) method would make the one-dimensional nanostructure of MnO2 with more excellent CO catalytic oxidation activity than the incipient wetness impregnation. Specifically, the most active 5CuO/MnO2 could achieve 80% CO conversion rate at 30 °C.
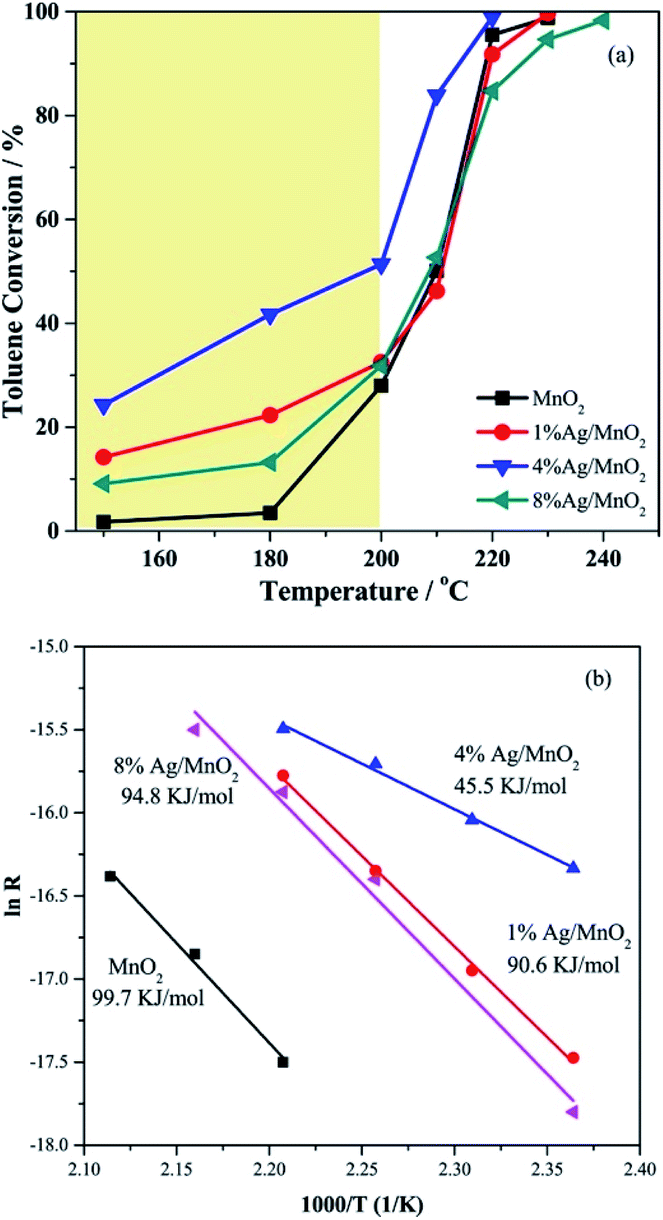 | ||
| Fig. 8 (a) Catalytic performance of the catalysts for toluene oxidation; (b) Arrhenius plot of the reaction rate of the catalysts. Reproduced by Qin et al.109 with permission from Elsevier. | ||
In view of the excellent performance of CuO loaded MnO2-based nanowire catalysts,84 the researches gradually focused on the transition metals doped catalysts because of the low cost and abundant availability. Lambert et al.110 reported that the catalytic activities of α-MnO2 nanowires doped with Cu and Ni were significantly better than that of the pristine α-MnO2 nanowires. Gao et al.61 investigated the effects of doping Fe, Co, Ni, and Cu transition metals on the catalytic performance CO oxidation of α-MnO2 nanowires. As shown in Fig. 9, the appropriate amount of transition metal doping would not apparently change the morphology, phase structure, and specific surface area of MnO2 nanowires. As could be observed in Fig. 10, the CO oxidation activities of the transition metal doped α-MnO2 nanowires were also significantly increased. Among them, Cu doped α-MnO2 nanowires showed the best CO oxidation activity. The density functional theory calculations further revealed that doping Cu could more easily form oxygen vacancies in MnO2 than doping Fe, Co, and Ni, which was the determinant of the CO oxidation reaction rate. Therefore, the Cu-doped nanowires performed the higher CO oxidation catalytic activity than those for noble-metal-free catalysts, such as Cu-substituted Co3O4,111 Cu-doped CeO2,112,113 CuO/Al2O3.114 In addition, Jampaiah et al.62 also studied the MnO2 nanowires doped with two transition metals. Their results showed that the Cu and Co doped MnO2 nanowires could greatly improve the soot oxidation activity and its T50 of soot conversion was as low as 279 °C. The advantage of two transition metals doped MnO2 nanowires was that it enhanced the strong synergistic interaction between the transition metals. However, this study had shown that there was an upper limit for the amount of surface metal doping of MnO2 nanowires. Because the original nanowire morphology would be damaged when the surface metal doping amount reached 20%.62
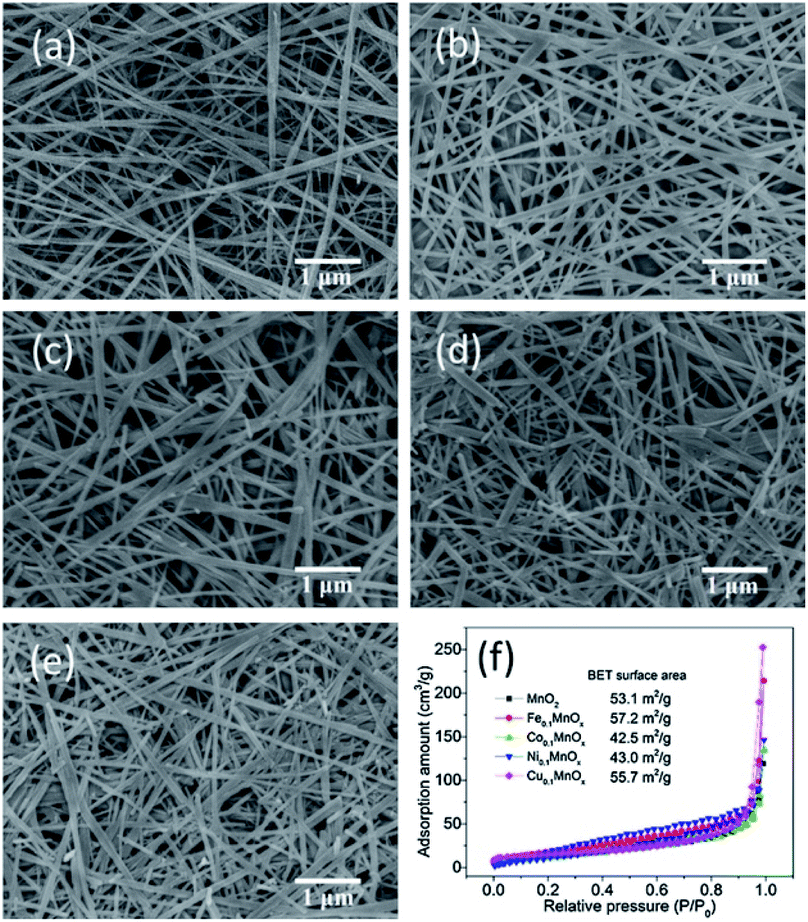 | ||
| Fig. 9 SEM images of (a) MnO2, (b) Fe0.1MnOx, (c) Co0.1MnOx, (d) Ni0.1MnOx, and (e) Cu0.1MnOx nanowires and (f) N2 adsorption–desorption isotherms. Reproduced by Gao et al.61 with permission from Elsevier. | ||
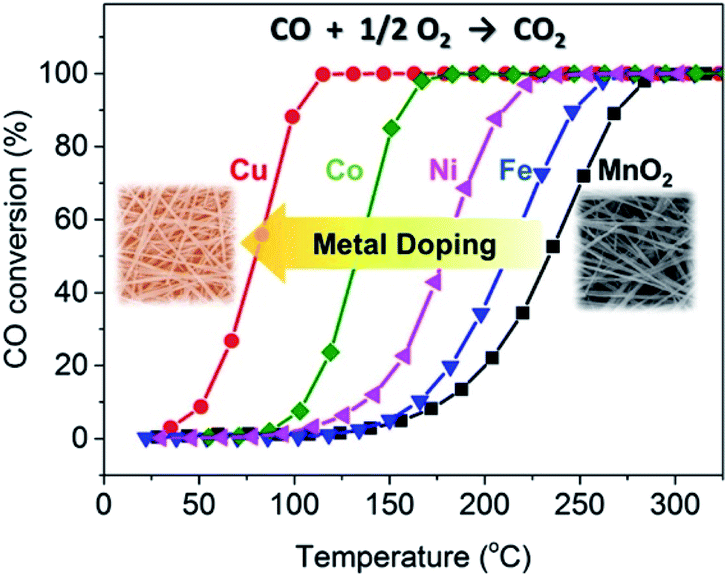 | ||
| Fig. 10 CO oxidation activity tests of the MnO2 and Fe-, Co-, Ni-, and Cu-doped MnO2 catalysts. Reproduced by Gao et al.61 with permission from Elsevier. | ||
3.5 Interface effect
The interface effect derived from nanostructures plays an important role in enhancing the catalytic activity. It is widely believed that multi-component metal oxides with coupling interfaces often exhibit better catalytic activity than single-component metal oxides.115–119 For example, as regards the α-MnO2@Co3O4 catalyst, the heterogeneous interface constructed by MnO2 nanowires and Co3O4 nanoparticles could not only enhance the redox pair of Mn4+/Mn3+ and Co2+/Co3+ but also strengthen the adsorption of oxygen on the surface and accelerate the fluidity of oxygen. As a result, the catalytic activities of α-MnO2@Co3O4 toward the oxidation of benzene31 and toluene120 were much better than that of pristine MnO2 nanowires. The α-MnO2 nanowire@Co3O4 could reduce the T90 of benzene oxidation to 247 °C (see Fig. 11) and the T90 of toluene conversion to 229 °C, which were 100 °C and 47 °C lower than that of the pure α-MnO2 nanowire, respectively.31,120 Deng et al.121 successfully prepared the MnO2 nanowire@NiO nanosheet core–shell hybrid nanostructure by uniformly decorating NiO nanosheets on α-MnO2 nanowires. The obtained catalyst could achieve the complete oxidation of benzene at 320 °C, which was 60 °C lower than the pure MnO2 nanowire. Ma et al.122 prepared MnO2@TiO2 by immobilizing crystalline anatase TiO2 on the surface of α-MnO2 nanowire. The as-prepared MnO2@TiO2 exhibited much higher maximum adsorption capacity (105.3 mg g−1) of the radioactive element U(VI) than the pure MnO2 (13.3 mg g−1), illustrating that the performance of the surface-modified MnO2 was greatly improved.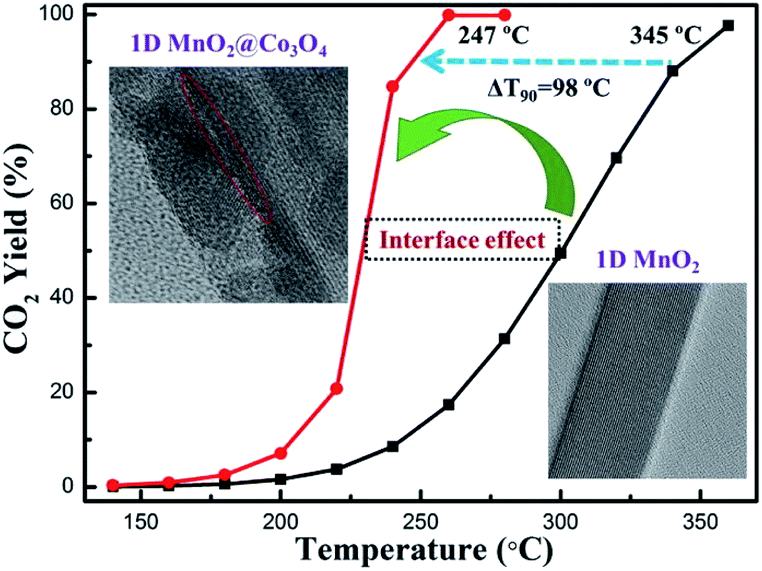 | ||
| Fig. 11 Benzene oxidation activity tests of the 1D MnO2@Co3O4 and 1D MnO2 catalysts. Reproduced by Tang et al.31 with permission from Elsevier. | ||
Homojunctions are junctions layer made of the same semiconductor materials.44 It has been revealed that the construction of homojunctions could enhance the adsorbing and catalytic performance of the MnO2 nanomaterials.123,124 Zhou et al.123 constructed α-MnO2@δ-MnO2 homojunction by loading layered δ-MnO2 nanoflakes on the framework of α-MnO2 nanotubes. The α-MnO2@δ-MnO2 homojunction integrated the exposed surfaces of the α-MnO2 (100) surface and δ-MnO2 (001) surface at the same time and displayed outstanding catalytic performance toward the oxidation of formaldehyde. The α-MnO2 (100) surface was conducive to the adsorption and activation of oxygen molecules and the δ-MnO2 (001) surface was favourable for the desorption of H2O. The synergistic effect of the two surfaces significantly enhanced the catalytic activity for formaldehyde oxidation at room temperature. Huang et al.124 synthesized the special two-phase structure MnO2 catalyst simultaneously with α-MnO2 and β-MnO2 (α-MnO2: β-MnO2 = 1:1). As a result, toluene could be completely oxidized over the α@β-MnO2 two-phase catalyst at about 205 °C, which was lower than the pure α-MnO2 (220 °C) and β-MnO2 (260 °C). Compared with the single-phase MnO2, the α@β-MnO2 with two-phase structure generated more abundant active interface and oxygen vacancy, thus promising enhanced catalytic performances.
4. Application in catalytic oxidation
4.1 CO oxidation
CO is one of the most common atmospheric pollutants harmful to the health of human beings. Its capacity of binding to hemoglobin is about 210 times higher than O2.125 Therefore, the combination of CO and hemoglobin severely hinders the delivery of blood oxygen, which will cause the supply shortage of blood oxygen and further threaten the life safety. Given these drawbacks, the low-temperature catalytic oxidation of CO has been considered as one of the most important environmental catalysis processes. As shown in Fig. 12, the proposed reaction mechanism of CO oxidation on manganese oxide catalyst commonly abides by Langmuir–Hinshelwood reaction mechanism (L–H mechanism) at low temperature (<90 °C) and Mars-van Krevelen reaction mechanism (MvK mechanism) at high temperatures (T > 190 °C).126 When the temperature is low (50–90 °C), gaseous O2 are captured by oxygen vacancies and the active oxygen species (O2− or O22−) could be formed. Subsequently, the adsorbed CO (CO–Mn3+) tent reacts with active oxygen species to generate bidentate carbonate species, and then decompose into CO2. Finally, the dissociated O species could be restrained by oxygen vacancies and react with the adsorbed CO to form CO2 which follows the L–H mechanism. When the temperature is high (T > 190 °C), the surface lattice oxygen species can directly react with adsorbed CO to generate CO2 and oxygen vacancies due to the activation of surface lattice oxygen species. Subsequently, the oxygen vacancy was refilled by gaseous O2 and the redox cycle is completed by following the MvK mechanism. The T10, T50, T90, T100 and BET of MnO2-based nanowire materials on the catalytic oxidation of CO and some VOCs have been summarized in Table 2.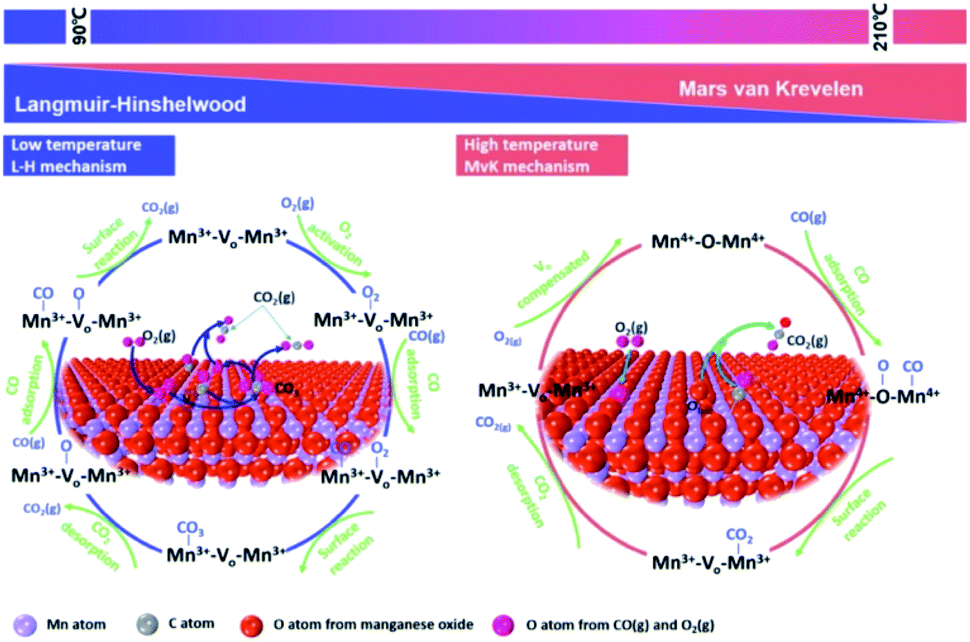 | ||
| Fig. 12 Proposed reaction mechanism of CO oxidation on metal oxide. Reproduced by Tian et al.126 with permission from Elsevier. | ||
| MnO2 based nanowire material | BET (m2 g−1) | Catalytic substance | T10 (°C) | T50 (°C) | T90 (°C) | T100 (°C) | Reference |
|---|---|---|---|---|---|---|---|
| α-MnO2 | 31.3 | CO | — | ∼180 | ∼210 | 260 | 84 |
| 20% CuO/α-MnO2 | 22.0 | — | ∼75 | ∼100 | 150 | ||
| Au/α-MnO2 | 40.0 | — | −83 | −60 | −34 | 56 | |
| α-MnO2 | 53.1 | ∼180 | 234 | ∼275 | ∼300 | 61 | |
| Fe0.1MnOx | 57.2 | ∼170 | 212 | ∼260 | ∼275 | ||
| Co0.1MnOx | 42.5 | ∼110 | 133 | ∼160 | ∼180 | ||
| Ni0.1MnOx | 43.0 | ∼140 | 178 | ∼220 | ∼260 | ||
| Cu0.1MnOx | 55.7 | 50 | 80 | — | 100 | ||
| α-MnO2 | 80.6 | ∼80 | ∼110 | 126 | ∼155 | 127 | |
| β-MnO2 | 55.7 | ∼95 | ∼150 | 169 | ∼185 | ||
| Ag/α-MnO2 | 64.2 | ∼45 | ∼75 | 90 | ∼120 | ||
| Ag/β-MnO2 | 40.0 | ∼35 | ∼65 | 80 | ∼115 | ||
| α-MnO2 | 51.1 | Soot | 492 | 547 | 602 | — | 62 |
| 5Co–MnO2 | 50.2 | 436 | 513 | 567 | — | ||
| 10Co–MnO2 | 51.7 | 420 | 475 | 546 | — | ||
| 20Co–MnO2 | 50.7 | 479 | 528 | 586 | — | ||
| 5Ni5Co–MnO2 | 58.3 | 379 | 445 | 513 | — | ||
| 5Cu5Co–MnO2 | 59.4 | 368 | 431 | 485 | — | ||
| α-MnO2 | 52.7 | Benzene | 243 | 301 | 345 | ∼375 | 31 |
| α-MnO2@Co3O4 | 58.5 | 205 | 227 | 247 | ∼265 | ||
| α-MnO2 | 52.7 | 243 | 301 | ∼350 | 380 | 121 | |
| α-MnO2@NiO | 88.9 | 233 | 273 | ∼300 | 320 | ||
| α-MnO2 | — | Toluene | ∼186 | ∼210 | ∼220 | ∼230 | 109 |
| 4% Ag/α-MnO2 | — | — | ∼195 | ∼215 | ∼220 | ||
| α-MnO2 | 39.9 | 214 | ∼230 | 237 | ∼245 | 83 | |
| α-MnO2-110 | — | — | 142 | 170 | 200 | 87 | |
| α-MnO2-210 | — | — | 88 | 109 | 140 | ||
| α-MnO2-310 | — | — | 130 | 170 | 200 | ||
| α-MnO2 | 60.6 | ∼240 | 264 | 276 | ∼285 | 120 | |
| α-MnO2@Co3O4 | 54.9 | ∼205 | 220 | 229 | ∼235 | ||
| α-MnO2 | 93.1 | Dimethyl ether | 200 | ∼230 | 274 | ∼290 | 27 |
| α-MnO2(1 ∼ 3 μm) | — | 176 | — | — | 250 | 105 | |
| α-MnO2(4 ∼ 8 μm) | — | 167 | — | — | 240 | ||
| α-MnO2(6–10 μm) | — | 170 | — | — | 250 | ||
| α-MnO2-100 | 87.9 | Formaldehyde | ∼80 | 125 | ∼145 | 150 | 86 |
| α-MnO2-110 | 119.5 | ∼50 | 100 | ∼125 | 130 | ||
| α-MnO2-310 | 135.3 | — | 35 | ∼55 | 65 |
Previous studies found that the crystalline phase structure could significantly affect the CO catalytic activity of MnO2.26,92,127 Liang et al.26 synthesized four different crystalline MnO2 nano-oxides. They found that the order of activity for catalytic oxidation of CO was α ≈ δ > γ > β-MnO2. Because the tunnel structure of [2 × 2] and [1 × 1] of α-MnO2 could occupy more space to obtain more CO adsorption sites; whereas, other crystalline MnO2 only involved β-MnO2 with [1 × 1] tunnel, γ-MnO2 with [2 × 1] and [1 × 1] tunnels, and δ-MnO2 composed of [MnO6] octahedrons shared by the double-layer edges. Another reason accounting for the enhanced catalytic CO oxidation performance of α-MnO2 was that the elongated Mn–O bond length stemming from the distorted [MnO6] units could benefit the cracking of the Mn–O bond, promoting CO oxidation.26,127 As a result, α-MnO2 could easily react with CO and thus further improved its catalytic performance.
In addition, various studies have been conducted to improve the catalytic activity of MnO2-based nanowires for CO oxidation. In order to achieve this goal, effective strategies including element doping, surface engineering and combination with other active substances have been extensively conducted. Gao et al.61 found that the hydrothermal doping of Cu would significantly increase CO oxidation activity (TOF = 9.1 × 10−3 s−1 at 70 °C) and water resistance of α-MnO2 nanowires. As for γ-MnO2, Zn-doped MnO2 could achieve the 90% CO conversion at 160 °C and perform the best CO oxidation activity among Cu, Zn, Mg, Co, Ni, Ca, Al, Fe, and La-doped γ-MnO2.128 Besides, the surface engineering is considered as another effective method to improve the catalytic activity of MnO2. Li et al.92 synthesized α-MnO2 with a large number of surface open cells through acid treatment. The open cells were rich in adsorption sites for O2, thus enhancing the catalytic activity of MnO2 for CO oxidation. Moreover, combining Au,56 Ag,127 CuO,69,84 etc. with MnO2 nanomaterials can effectively improve CO oxidation efficiency. Xu et al.127 investigated the CO oxidation activity over the α-MnO2 nanowires supported Ag based catalysts and the T90 of Ag/α-MnO2 below 100 °C could be achieved. Although many studies have been carried out to enhance the catalytic activity of MnO2 for CO oxidation and the complete CO conversion could be achieved at −34 °C,56 the current research hotspot is still focused on the loading or doping noble/transition metals. The further explorations should be concentrated on the surface engineering or the rare earth metals modification.
4.2 Catalytic removal of VOCs
The volatile organic compounds (VOCs) typically refer to organic compounds with boiling points between 50 °C and 260 °C at atmospheric pressure (101.325 kPa) according to the World Health Organization.129 Besides, most VOCs emitted into the atmosphere can cause the formation of secondary pollutants, such as tropospheric ozone, peroxyacetyl nitrate, and secondary organic aerosols.130–132 Various means have been used to remove the VOCs, such as the physical adsorption,133 catalytic oxidation,134,135 photocatalytic degradation,136,137 plasma catalytic oxidation,138 and biological decomposition.139 Among these strategies, the catalytic oxidation has been considered as one of the most effective ways owing to various advantages, such as high efficiency, less secondary pollutants, and low reaction temperature. As for the catalysts of VOCs oxidation, compared with precious metal catalysts, the transition metal oxides catalysts are considered to be more promising candidates owing to their low costs and excellent catalytic performances.140 MnO2 based nanomaterials are widely investigated as the effective catalysts and/or supports in the fields of pollutant removal, energy storage, and water treatment due to the excellent oxygen storage/release capacities, adsorption performances, and redox performances.141Hu et al.146 found that the order of catalytic activities on benzene combustion over MnO2 with different phase structures was γ-MnO2 > β-MnO2 > α-MnO2 > δ-MnO2. The surface adsorbed oxygen species concentration and low temperature O2 desorption of these manganese oxides were in good agreement with the sequence of their catalytic performance on benzene combustion. Besides, the strategies such as metal doping and loading have been investigated to improve the catalytic activities on benzene combustion over MnO2. Liu et al.147 found that the Ce3+ and Cu2+ doped of MnO2 nanosheet could significantly improve the activity of MnO2 for benzene decomposition. For the pristine MnO2, benzene was hardly decomposed below 200 °C and only ∼40% conversion was achieved at 400 °C. Whereas, as for the Ce–MnO2 and Cu–MnO2 catalysts, the benzene started to decompose around 100 °C and nearly all benzene could be removed around 250 °C. Moreover, the Cu–MnO2 catalyst also exhibited high resistance to high-temperature deactivation and water resistance during continuous long-term testing. Recently, Ma et al.148 synthesized the tungsten-doped MnO2 (W–MnO2) with excellent performance of the photo-assisted thermal catalytic oxidation/removal of benzene. They found that doping W could not only generate abundant oxygen vacancies in MnO2, but also produce strong covalent interactions with adjacent O atoms through W–O bridge bonds, thereby promoting O2 polarization and electron transference.
As for the metal loading, Ye et al.149 prepared nanosized α-MnO2-supported silver catalysts for benzene oxidation. They found that the 5Ag/MnO2 catalyst performed the best activity of the benzene oxidation. Tang et al.31 modified one-dimensional MnO2-based catalysts (wire-like, rod-like α-MnO2 and β-MnO2 microrods) by the Co3O4 nanoparticles (see Fig. 13) and investigated them as the catalysts for benzene oxidation. They found that the modified MnO2@Co3O4 composite exhibited better catalytic performance than the pristine MnO2. The specific conversion efficiencies of the modified material to benzene abided by the order of α-MnO2 nanowires > α-MnO2 nanorods > β-MnO2 nanorods. The reason for this could be attributed to the enhanced low-temperature reducibility and abundant active surface adsorption of oxygen derived from the heterogeneous hybrid interface.
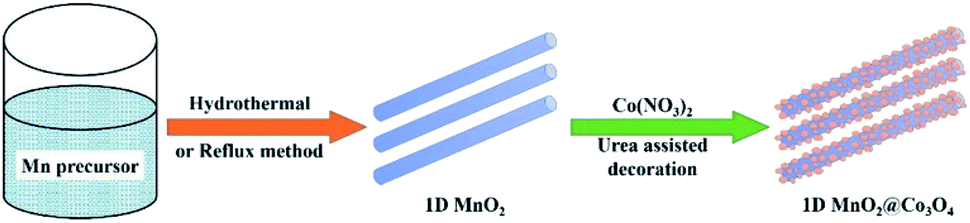 | ||
| Fig. 13 Schematic process for the preparation of 1D MnO2@Co3O4 hetero-nanostructures. Reproduced by Tang et al.31 with permission from Elsevier. | ||
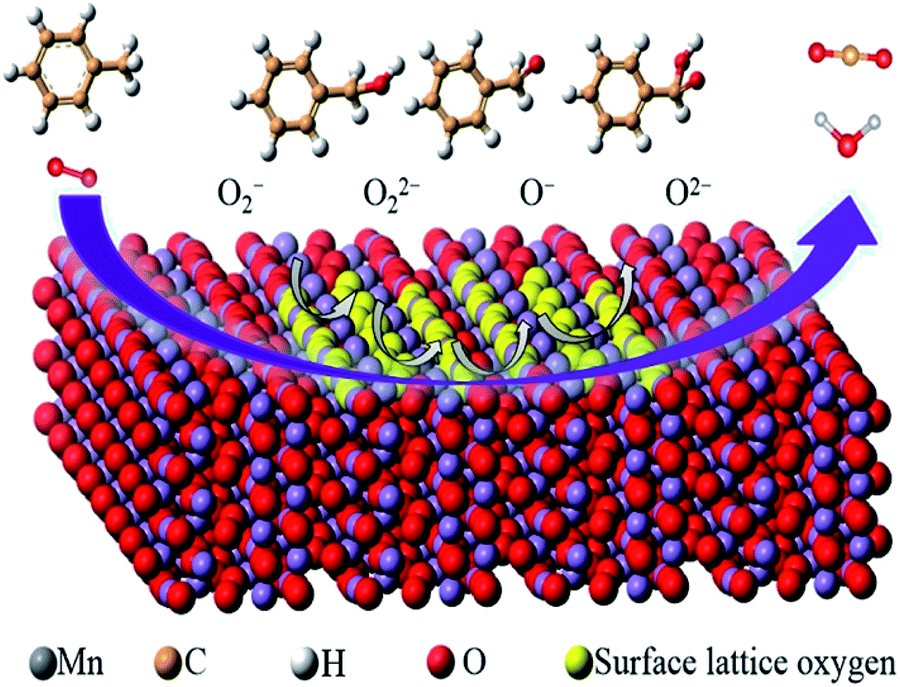 | ||
| Fig. 14 Proposed mechanism of toluene degradation pathway on α-MnO2. Reproduced by Huang et al.87 with permission from Elsevier. | ||
It has been reported that the catalytic performances of the toluene oxidation over the MnO2 nanomaterials were strongly related to the crystalline phase structures.91,124,152 Li et al.91 prepared MnO2 with different crystalline phases (β-, α-, γ-, and δ-). The results showed that catalytic toluene oxidation activities followed the sequence of δ > γ > α > β-MnO2. The morphology of the MnO2 nanostructure also greatly influences the catalytic performance of the toluene oxidation. The previous study showed that the MnO2 nanowire showed the most excellent toluene combustion catalytic activity (T100 = 220 °C) after loading silver among the MnO2-based one-dimensional nanostructures (nanowire, nanorod, and nanotube).30
Besides, the metal loading and doping can also greatly enhance the catalytic performance in toluene oxidation removal.153,154 Dong et al.153 found that the introducing Na+ and Ca2+ into MnO2 promised the higher catalytic activity for toluene oxidation than pristine MnO2. Specifically, the Ca–MnO2 catalyst exhibited the best catalytic activity (T90 = 215 °C) and possessed good water resistance. The outstanding catalytic performance of Ca–MnO2 was closely related to the abundant acidic sites, high oxygen vacancy concentration, and the weak CO2 adsorption capacity. Min et al.154 found that transition metal (Ni and Co) doped α-MnO2 and β-MnO2 exhibited better catalytic performances in toluene oxidation removal owing to their lower crystallinities, more defects, larger specific surface areas, more oxygen vacancies, and better low-temperature redox ability than the pristine α-MnO2 and β-MnO2.
The catalytic performance of the toluene oxidation was also influenced by the interface between different metal oxides. Ren et al.120 fabricated ZIF-derived Co3O4 grown in situ on 1D α-MnO2 materials (as displayed in Fig. 15) and used it as the catalyst for toluene oxidation. They found that the α-MnO2@Co3O4 could reduce the T90 of toluene conversion to 229 °C, which was 47 °C and 28 °C lower than those of the pure α-MnO2 nanowire and Co3O4-b, respectively. The main reason for the enhanced catalytic activity could be attributed to the synergistic effect produced by the coupling interface constructed between α-MnO2 and Co3O4. The specific reaction route of toluene oxidation on α-MnO2@Co3O4 catalyst could be illustrated as follows: toluene → benzoate species → alkanes containing oxygen functional group → CO2 and H2O. In addition, the α-MnO2@Co3O4 catalyst also exhibited excellent stability and water resistance during the toluene oxidation.120 Zhang et al.155 found that the fluorine (F)-doped titanium dioxide (TiO2) coupled with a small number of α-MnO2 nanowires could effectively improve the photocatalytic degradation effect of toluene. Specifically, the toluene degradation efficiency before coupling was about 90%, and the toluene degradation efficiency after coupling was increased to 96%.
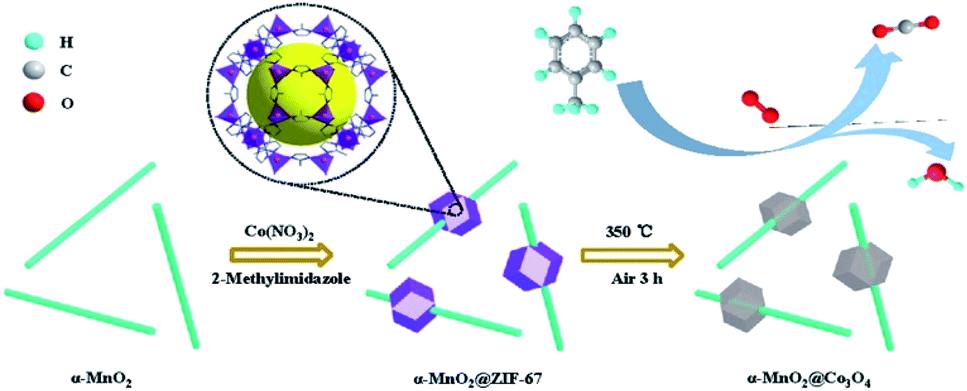 | ||
| Fig. 15 Illustration of the fabrication of α-MnO2@Co3O4 samples. Reproduced by Ren et al.120 with permission from Elsevier. | ||
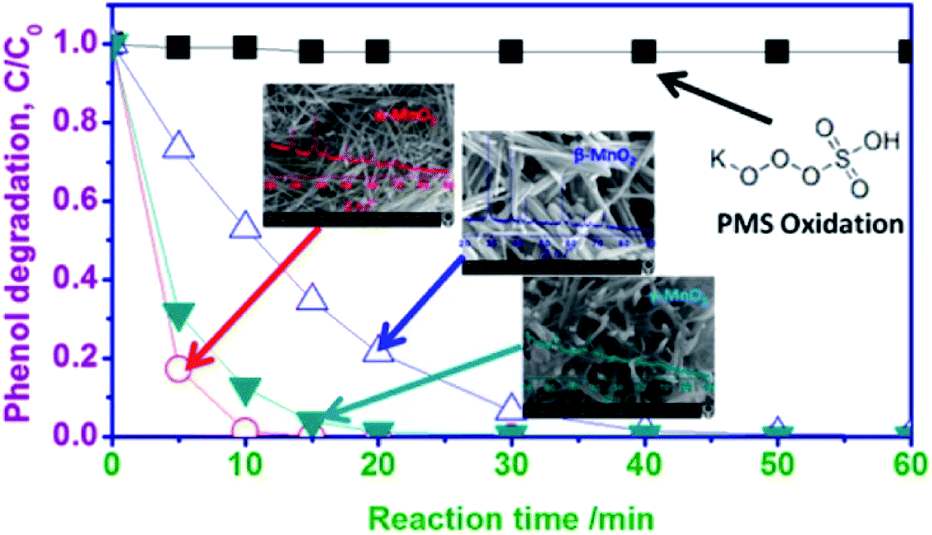 | ||
| Fig. 16 Phenol degradation of MnO2-based nanomaterials with different phase structures. Reproduced by Saputra et al.29 with permission from ACS. | ||
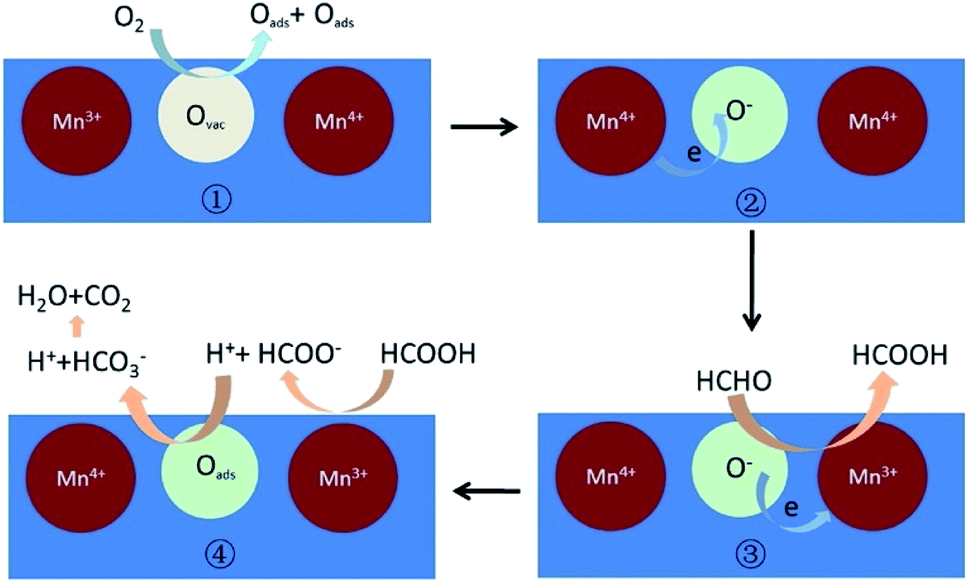 | ||
| Fig. 17 The possible mechanism of catalytic oxidation of HCHO to CO2 on partially crystallized mesoporous MnOx under dry condition. Reproduced by Guan et al.173 with permission from Elsevier. | ||
Li et al.174 reported that the sphere-like mesoporous MnO2 exhibited good catalytic activity and reusability for the formaldehyde gas oxidation and the formaldehyde purification efficiency could reach 99% after reacting at room temperature for 2 hours. Zhang et al.94 studied the catalytic activity of formaldehyde oxidation over the MnO2 nanomaterials with different phase structures (α-, β-, γ- and δ-). It was found that the δ-MnO2 catalyst performed the best activity among these catalysts and could achieve the complete conversion of the HCHO at 80 °C, which was much lower than the α, β, and γ-MnO2 at 125 °C, 200 °C, 150 °C, respectively. Besides, previous researches revealed that the Au-doped175 and Ag-loading176 α-MnO2 nanorods exhibited superior catalytic performances of HCHO oxidation. The 100% conversion of HCHO could be achieved at 75 °C and 80 °C over the Au-doped and Ag-loading α-MnO2 nanorods, respectively. Rong et al.86 prepared (310) facet exposed α-MnO2 nanowire catalyst through crystal facet engineering with superior HCHO oxidation activity. It could completely convert HCHO into CO2 at 60 °C and achieve 86% conversion rate of 0.53 mg m−3 HCHO at room temperature. He et al.177 enhanced the catalytic oxidation of HCHO at ambient temperature over the α-MnO2 with four well-defined morphologies (nanowire, nanotube, nanorod, and nanoflower) with the help of the mechanical vibration. Among them, α-MnO2 nanowires exhibited the best catalytic activity with 100% conversion of 125 ppm HCHO at ambient temperature owing to the high oxygen vacancy concentration.
The oxygen vacancies in MnO2 are usually considered as the active sites for O3 adsorption and catalysing ozonation.188 Fig. 18 shows the adsorption of CH3SH over the MnO2 catalyst and the mechanism of catalytic ozone.93 O3 can be captured and decomposed by the structural defects on the MnO2 surface, then transformed into activate oxygen species (O2−/O22−), and quickly evolved into ˙O2−/˙OH/1O2 through a chain cycle reaction, which can directly participate in CH3SH and adsorbed CH3S− oxidation. It is desirable to obtain highly active crystal facets with abundant oxygen vacancies through surface engineering to improve the catalytic activity. He et al.93 prepared α-MnO2 nanowire with different crystal facets exposed and successfully employed them to catalyse the ozonation of CH3SH. The synthesized 310-MnO2 catalyst performed higher activity than the 110-MnO2 and 100-MnO2. In addition, previous researches found that loading the metal active sites on MnO2 nanomaterials could also enhance the ozonation catalysis of CH3SH.187,189 Xia et al.187 prepared Ag deposited three-dimensional MnO2 porous hollow microspheres (Ag/MnO2 PHMSs). The optimal 0.3% Ag/MnO2 PHMSs could completely degrade 70 ppm CH3SH within 600 seconds, displaying much higher efficiency than that of the pristine MnO2 PHMSs support (79%). Additionally, the catalyst retained long-term stability and could be facilely regenerated to its initial activity with ethanol and HCl. Yang et al.189 synthesized urchin-like oxygen vacancy-rich (VO) hollow-structured MnO2 with the loading of CuO nanoparticles on the surface (CuO/VO-MnO2). The well-dispersed CuO on the surface of urchin-like hollow MnO2 enhanced the chemisorption of CH3SH, thus promoting the efficient decomposition of CH3SH on the CuO/VO-MnO2. Among the as-prepared CuO/VO-MnO2 catalyst, the 5CuO/VO-MnO2 realized the optimum CH3SH removal efficiency (98.5%) within 1200 s reaction under 25 °C, which was obviously higher than that of individual MnO2 (70.7%) and CuO (57.0%).
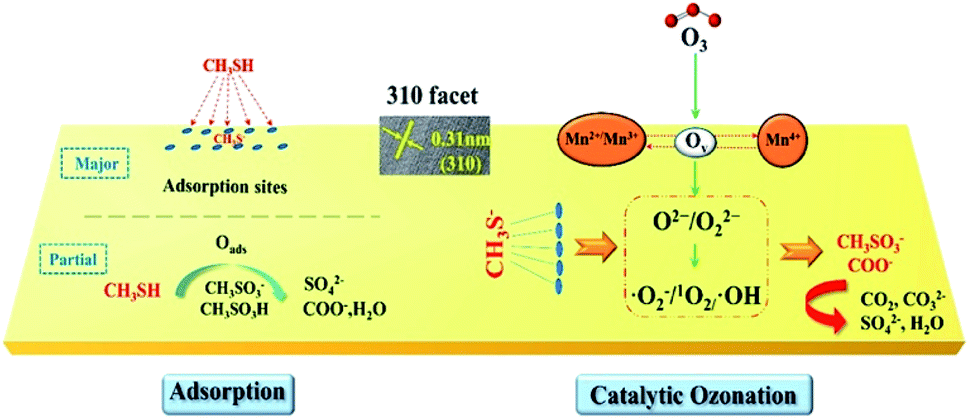 | ||
| Fig. 18 Mechanism of adsorption and catalytic ozonation toward CH3SH by 310-α-MnO2 catalysts. Reproduced by He et al.93 with permission from ACS. | ||
However, there are few researches about the wire-like MnO2 for the ozonation of CH3SH except for the study of He.93 In view of the strong interaction between wire-like MnO2 and noble/transition metals, more attentions will be attributed to the metal loading/doping of MnO2 nanowire for the ozonation of CH3SH in the future research.
4.3 Ozone
Ozone (O3) is a light blue gas with special odor and strong oxidizing ability. O3 usually can accomplish the complete oxidation of VOCs at room temperature. Therefore, the ozone is widely used as the oxidant in environmental treatment. However, when the amount of the pollutants, such as VOCs, are very large, the balance of ozone in the atmosphere will be broken, leading to the ozone pollution. Besides, the exposure to low-level ozone usually can cause serious effects on the human body's heart, lungs, respiratory tract, and cardiovascular system.190–193 The catalysts used for the degradation of ozone can be divided into noble metals,194,195 transition metal oxides196–201 and noble transition compounds.202 In the view of the cost and large-scale application, the studies on the catalytic decomposition of ozone have been mainly focused on transition metal oxides, especially MnO2.α-MnO2 is a newly emerging material for catalytic decomposition of ozone. Jia et al.141 investigated the ozone removal activity over the MnO2 with different phase structures (α, β, and γ). They found that α-MnO2 performed the highest ozone removal efficiency. Zhu et al.203 developed a hierarchical structure consisted of graphene encapsulating α-MnO2 nanofiber to address the challenges of the low stability and water inactivation during ozone decomposition. The optimized catalyst exhibited high ozone conversion efficiency of 80% and excellent stability over 100 h under a relative humidity (RH) of 20%. Even though the RH was as high as 50%, the ozone conversion also could reach 70%, well beyond the performance of α-MnO2 nanofiber. Zhang et al.155 found that introducing α-MnO2 nanowires into the fluorine (F)-doped titanium dioxide (TiO2) could not only improve the photocatalytic degradation of toluene but also remove almost the total O3 in the system. As the comparison, only 15.8% of O3 could be removed by the pristine F–TiO2 catalyst without mixing with α-MnO2. This indicated that α-MnO2 nanowire was an excellent catalyst for O3 decomposition. Previous study showed that the oxygen vacancies on MnO2 nanomaterials were active sites for ozone adsorption and further decomposition,199 which largely determined the catalytic degradation activity of MnO2. Zhu et al.82 developed the α-MnO2 nanowire with high K+ content in tunnel structure through the simple KOH solution post-treatment. As shown in Fig. 19, the substantial increase in K+ content reduced the formation energy of oxygen vacancies, which was further conducive to the formation of oxygen vacancies in the crystal lattice. As a result, the ozone removal performance was improved by doping K+. However, peroxide species (O22−) could accumulate on the surface of the catalyst and further evolve into relatively stable oxygen, resulting in the decrease in the ozone removal rate. Therefore, the accumulation of intermediate oxygen was a key factor limiting the high efficiency and sustainability of ozone decomposition. Liu et al.204 prepared cerium-modified todorokite-type manganese dioxides (Ce–MnO2). The as-obtained catalyst showed greatly improved stability, which could keep ∼98% of removal efficiency for ∼115 ppm of O3 within 5 h under a very high space velocity of 1200 L g−1 h−1 at room temperature. Besides, the Ce–MnO2 catalyst also exhibited highly stable O3 removal efficiency at temperature as low as 0 °C.
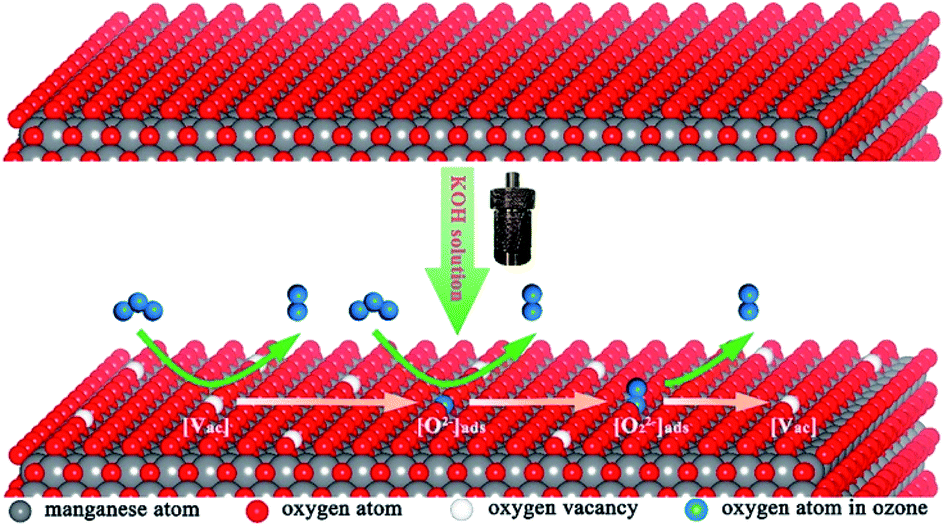 | ||
| Fig. 19 Schematic of ozone catalytic decomposition on the K-rich MnO2. Reproduced by Zhu et al.82 with permission from ACS. | ||
5. Summary and prospect
MnO2-based nanowires are becoming one of the most popular and promising catalysts in environmental applications due to their high catalytic performance, low cost and environmental friendliness. This review systematically summarizes the preparation methods of MnO2-based nanowires, and the effects of phase structure, crystal facet, and morphology on their environmentally catalytic performance. In addition, this review also summarizes their applications in environmental pollution control, such as CO oxidation, removal of VOCs, and the decomposition of O3.(1) Hydrothermal is a flexible and diverse preparation method, which can facilely control the phase structure, morphology, and exposed crystal facets of the product by changing hydrothermal reaction conditions and has been widely used in laboratory research of MnO2 nanowires. As for the reflux method, it has various advantages (e.g. simple operation procedure, mild reaction condition, and low cost) and is suitable for the large-scale industrial synthesis. However, it is difficult for the sol–gel method alone to obtain wire-like MnO2-based nanomaterials. Therefore, it is often used in combination with template method to prepare highly ordered MnO2 nanowires. However, the employment of the templates will increase the cost of the preparation.
(2) Crystal facet engineering is the research hotspot in the field of MnO2 nanowires. Compared with the influences of phase structure and morphology, it has more significant impact on the catalytic activity of MnO2 nanowires in environmental catalysis.
(3) Doping and/or loading the metals or their oxides can effectively improve the chemical bond strength, phase structure, and electronic structure of the MnO2 nanowires. As a result, the catalytic performance will be improved by increasing the fluidity of oxygen.
Up to now, lots of MnO2-based nanowire material modulation strategies have been developed and their performances in the fields of environmental catalysis have also been promoted. However, the environmental applications of MnO2-based nanowire materials are still at the early stage with various challenges and have a long way to go before the practical applications, specifically in the following aspects.
(1) The complex crystal structures and the morphology diversity of MnO2 result in challenging obstacles in obtaining pure phase structures and uniform morphologies of MnO2 nanowires. Therefore, it is urgent to develop stable and efficient strategies to fabricate MnO2 nanowires in single-crystalline solids with uniform morphology. Considering the remarkable improvement of the catalytic oxidation ability of MnO2 nanowire by the crystal facet engineering, novel synthesis strategies for MnO2 nanowire exposed a high proportion of specific crystal facet should be investigated in depth.
(2) The crystal facet engineering of MnO2 nanowire is mostly focused on the catalysis of VOCs. It can be predicted that it will also have great potential application in catalytic CO oxidation and ozone degradation. However, there are few studies in these areas. Different crystal facets have different catalytic oxidation performances toward specific VOCs. Therefore, it is of great urgency and necessity to systematically investigate the special functions of each crystal facet of MnO2, and finally verify the types of VOCs matched.
(3) Previous studies mainly focused on the noble metals (Au and Ag) and transition metals (Fe, Co, Ni, and Cu) loading or doping. However, few studies have focused on the modification of MnO2 nanowire by loading or doping rare earth metals (La, Ce, Pr, and Sm). In view of the improvement of the O3 removal efficiency of MnO2 at low temperature by the cerium doping, the effect of catalytic degradation of other air pollutants by the rare earth metals modification should be explored in the future.
(4) Although MnO2-based nanowire materials as the supports or the efficient catalysts are gradually emerging in the field of environmental catalysis, yet there is still giant gap between the laboratory results and actually industrial requirement in the aspects of the catalytic performance, long-term stability, and water resistance of removing CO and VOCs at relatively low temperature compared with traditional noble metal catalysts.
Author contributions
LX: conceived ideas, made comments, edited; YX: investigation, writing – original draft; GC, CW, NW: assisted YX in literature research and summary; LX, MC, XH: investigation, supervision, funding acquisition. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely acknowledge the financial support from National Natural Science Foundation of China (Grant No. 21503113, 21871144, 21577065, 21976094), the National Key Research and Development Project (Grant No. 2018YFC0213802), Environmental Protection Projects of Jiangsu Province (2017022), a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX20_0309), and the Natural Science Foundation of Jiangsu Province (BK20190786). This study was also supported by Startup Foundation of Nanjing University of Information Science and Technology.References
- S. Dey and G. C. Dhal, Mater. Sci. Energy Technol., 2019, 2, 607–623 Search PubMed.
- M. S. Kamal, S. A. Razzak and M. M. Hossain, Atmos. Environ., 2016, 140, 117–134 CrossRef CAS.
- Q. Liu, C. X. Liu, X. L. Nie, L. Bai and S. H. Wen, Mater. Lett., 2012, 72, 101–103 CrossRef CAS.
- K. J. Cole, A. F. Carley, M. J. Crudace, M. Clarke, S. H. Taylor and G. J. Hutchings, Catal. Lett., 2010, 138, 143–147 CrossRef CAS.
- M. T. Salam, J. Millstein, Y. F. Li, F. W. Lurmann, H. G. Margolis and F. D. Gilliland, Environ. Health Perspect., 2005, 113, 1638–1644 CrossRef PubMed.
- M. Wilhelm and B. Ritz, Environ. Health Perspect., 2005, 113, 1212–1221 CrossRef CAS PubMed.
- K. Guo, W. W. Kong and Z. M. Yang, Plant, Cell Environ., 2009, 32, 1033–1045 CrossRef CAS PubMed.
- K. Guo, K. Xia and Z. M. Yang, J. Exp. Bot., 2008, 59, 3443–3452 CrossRef CAS PubMed.
- Z. Hu, Y. Utiskul, J. G. Quintiere and A. Trouve, Proc. Combust. Inst., 2007, 31, 2537–2545 CrossRef.
- P. Fagone, K. Mangano, S. Mammana, E. Cavalli, R. Di Marco, M. L. Barcellona, L. Salvatorelli, G. Magro and F. Nicoletti, Clin. Immunol., 2015, 157, 198–204 CrossRef CAS PubMed.
- R. N. Colvile, E. J. Hutchinson, J. S. Mindell and R. F. Warren, Atmos. Environ., 2001, 35, 1537–1565 CrossRef CAS.
- Y. C. Yao, J. H. Tsai, H. F. Ye and H. L. Chiang, J. Air Waste Manage. Assoc., 2009, 59, 1339–1346 CrossRef CAS PubMed.
- M. M. Schubert, S. Hackenberg, A. C. van Veen, M. Muhler, V. Plzak and R. J. Behm, J. Catal., 2001, 197, 113–122 CrossRef CAS.
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709–2724 CrossRef CAS PubMed.
- B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- G. X. Chen, Y. Zhao, G. Fu, P. N. Duchesne, L. Gu, Y. P. Zheng, X. F. Weng, M. S. Chen, P. Zhang, C. W. Pao, J. F. Lee and N. F. Zheng, Science, 2014, 344, 495–499 CrossRef CAS PubMed.
- K. Liu, A. Q. Wang and T. Zhang, ACS Catal., 2012, 2, 1165–1178 CrossRef CAS.
- L. Y. Zhang, H. Y. Liu, X. Huang, X. P. Sun, Z. Jiang, R. Schlogl and D. S. Su, Angew. Chem., Int. Ed., 2015, 54, 15823–15826 CrossRef CAS PubMed.
- T. Bunluesin, H. Cordatos and R. J. Gorte, J. Catal., 1995, 157, 222–226 CrossRef CAS.
- X. W. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. J. Shen, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- D. Gu, C. J. Jia, C. Weidenthaler, H. J. Bongard, B. Spliethoff, W. Schmidt and F. Schuth, J. Am. Chem. Soc., 2015, 137, 11407–11418 CrossRef CAS PubMed.
- M. Y. Guo, F. Z. Liu, J. Tsui, A. A. Voskanyan, A. M. C. Ng, A. B. Djurisic, W. K. Chan, K. Y. Chan, C. Z. Liao, K. M. Shih and C. Surya, J. Mater. Chem. A, 2015, 3, 3627–3632 RSC.
- S. Royer and D. Duprez, Chemcatchem, 2011, 3, 24–65 CrossRef CAS.
- Y. Ren, Z. Ma, L. P. Qian, S. Dai, H. Y. He and P. G. Bruce, Catal. Lett., 2009, 131, 146–154 CrossRef CAS.
- K. Ramesh, L. W. Chen, F. X. Chen, Y. Liu, Z. Wang and Y. F. Han, Catal. Today, 2008, 131, 477–482 CrossRef CAS.
- S. H. Liang, F. T. G. Bulgan, R. L. Zong and Y. F. Zhu, J. Phys. Chem. C, 2008, 112, 5307–5315 CrossRef CAS.
- B. B. He, G. Cheng, S. F. Zhao, X. H. Zeng, Y. F. Li, R. N. Yang, M. Sun and L. Yu, J. Solid State Chem., 2019, 269, 305–311 CrossRef CAS.
- T. Wang, S. Chen, H. G. Wang, Z. Liu and Z. B. Wu, Chin. J. Catal., 2017, 38, 793–804 CrossRef CAS.
- E. Saputra, S. Muhammad, H. Q. Sun, H. M. Ang, M. O. Tade and S. B. Wang, Environ. Sci. Technol., 2013, 47, 5882–5887 CrossRef CAS PubMed.
- J. Li, Z. Qu, Y. Qin and H. Wang, Appl. Surf. Sci., 2016, 385, 234–240 CrossRef CAS.
- W. X. Tang, M. S. Yao, Y. Z. Deng, X. F. Li, N. Han, X. F. Wu and Y. F. Chen, Chem. Eng. J., 2016, 306, 709–718 CrossRef CAS.
- S. Zhu, S. H. Ho, C. Jin, X. Duan and S. Wang, Environ. Sci.: Nano, 2020, 7, 368–396 RSC.
- L. Miao, J. L. Wang and P. Y. Zhang, Appl. Surf. Sci., 2019, 466, 441–453 CrossRef CAS.
- J. Chen, H. M. Meng, Y. Tian, R. Yang, D. Du, Z. H. Li, L. B. Qu and Y. H. Lin, Nanoscale Horiz., 2019, 4, 321–338 RSC.
- B. B. Ding, P. Zheng, P. A. Ma and J. Lin, Adv. Mater., 2020, 32, 1905823 CrossRef CAS PubMed.
- W. Li, K. Xu, L. An, F. Jiang, X. Zhou, J. Yang, Z. Chen, R. Zou and J. Hu, J. Mater. Chem. A, 2014, 2, 1443–1447 RSC.
- W. Li, Q. Liu, Y. Sun, J. Sun, R. Zou, G. Li, X. Hu, G. Song, G. Ma, J. Yang, Z. Chen and J. Hu, J. Mater. Chem., 2012, 22, 14864–14867 RSC.
- J. Li, W. Li, L. Huang, C. Ma and T. Ji, J. Nanoelectron. Optoelectron., 2021, 16, 149–156 CrossRef.
- R. A. Davoglio, G. Cabello, J. F. Marco and S. R. Biaggio, Electrochim. Acta, 2018, 261, 428–435 CrossRef CAS.
- J. G. Wang, F. Y. Kang and B. Q. Wei, Prog. Mater. Sci., 2015, 74, 51–124 CrossRef CAS.
- W. Guo, C. Yu, S. F. Li, Z. Wang, J. H. Yu, H. W. Huang and J. S. Qiu, Nano Energy, 2019, 57, 459–472 CrossRef CAS.
- X. Guo, S. Yang, D. Wang, A. Chen, Y. Wang, P. Li, G. Liang and C. Zhi, Curr. Opin. Electrochem., 2021, 30, 100769 CrossRef CAS.
- A. K. Worku, D. W. Ayele, N. G. Habtu, M. A. Teshager and Z. G. Workineh, Materials Today Sustainability, 2021, 13, 100072 CrossRef.
- R. J. Yang, Y. Y. Fan, R. Q. Ye, Y. X. Tang, X. H. Cao, Z. Y. Yin and Z. Y. Zeng, Adv. Mater., 2021, 33, 2004862 CrossRef CAS PubMed.
- Y. Long, J. F. Hui, P. P. Wang, S. Hu, B. Xu, G. L. Xiang, J. Zhuang, X. Q. Lv and X. Wang, Chem. Commun., 2012, 48, 5925–5927 RSC.
- D. Portehault, S. Cassaignon, E. Baudrin and J. P. Jolivet, Chem. Mater., 2008, 20, 6140–6147 CrossRef CAS.
- R. R. Jiang, T. Huang, J. L. Liu, J. H. Zhuang and A. S. Yu, Electrochim. Acta, 2009, 54, 3047–3052 CrossRef CAS.
- J. C. Villegas, L. J. Garces, S. Gomez, J. P. Durand and S. L. Suib, Chem. Mater., 2005, 17, 1910–1918 CrossRef CAS.
- M. S. Wu, J. T. Lee, Y. Y. Wang and C. C. Wan, J. Phys. Chem. B, 2004, 108, 16331–16333 CrossRef CAS.
- R. N. DeGuzman, Y. F. Shen, E. J. Neth, S. L. Suib, C. L. O'Young, S. Levine and J. M. Newsam, Chem. Mater., 1994, 6, 815–821 CrossRef CAS.
- X. Wang and Y. D. Li, Chem. –Eur. J., 2003, 9, 300–306 CrossRef CAS PubMed.
- M. Wei, Y. Konishi, H. Zhou, H. Sugihara and H. Arakawa, Nanotechnology, 2005, 16, 245–249 CrossRef CAS PubMed.
- X. Zhang, W. Yang, J. Yang and D. G. Evans, J. Cryst. Growth, 2008, 310, 716–722 CrossRef CAS.
- X. Y. Wang, X. Y. Wang, W. G. Huang, P. J. Sebastian and S. Gamboa, J. Power Sources, 2005, 140, 211–215 CrossRef CAS.
- W. X. Tang, X. Shan, S. D. Li, H. D. Liu, X. F. Wu and Y. F. Chen, Mater. Lett., 2014, 132, 317–321 CrossRef CAS.
- D. Gu, J. C. Tseng, C. Weidenthaler, H. J. Bongard, B. Spliethoff, W. Schmidt, F. Soulimani, B. M. Weckhuysen and F. Schuth, J. Am. Chem. Soc., 2016, 138, 9572–9580 CrossRef CAS PubMed.
- D. Zitoun, N. Pinna, N. Frolet and C. Belin, J. Am. Chem. Soc., 2005, 127, 15034–15035 CrossRef CAS PubMed.
- X. H. Su, L. Yu, G. Cheng, H. H. Zhang, M. Sun and X. F. Zhang, Appl. Energy, 2015, 153, 94–100 CrossRef CAS.
- D. J. Davis, T. N. Lambert, J. A. Vigil, M. A. Rodriguez, M. T. Brumbach, E. N. Coker and S. J. Limmer, J. Phys. Chem. C, 2014, 118, 17342–17350 CrossRef CAS.
- T. N. Lambert, J. A. Vigil, S. E. White, C. J. Delker, D. J. Davis, M. Kelly, M. T. Brumbach, M. A. Rodriguez and B. S. Swartzentruber, J. Phys. Chem. C, 2017, 121, 2789–2797 CrossRef CAS.
- J. J. Gao, C. M. Jia, L. P. Zhang, H. M. Wang, Y. H. Yang, S. F. Hung, Y. Y. Hsu and B. Liu, J. Catal., 2016, 341, 82–90 CrossRef CAS.
- D. Jampaiah, V. K. Velisoju, P. Venkataswamy, V. E. Coyle, A. Nafady, B. M. Reddy and S. K. Bhargava, ACS Appl. Mater. Interfaces, 2017, 9, 32652–32666 CrossRef CAS PubMed.
- W. Wang, Y. Shi, Y. Su, Y. Wang and H. Sun, Research and Application of Materials Science, 2019, 1, 18–23 CrossRef.
- Y. F. Shen, S. L. Suib and C. L. O'Young, J. Am. Chem. Soc., 1994, 116, 11020–11029 CrossRef CAS.
- A. K. Sinha, M. Pradhan and T. Pal, J. Phys. Chem. C, 2013, 117, 23976–23986 CrossRef CAS.
- N. Kijima, Y. Sakata, Y. Takahashi, J. Akimoto, T. Kumagai, K. Igarashi and T. Shimizu, Solid State Ionics, 2009, 180, 616–620 CrossRef CAS.
- X. Chen, Y. F. Shen, S. L. Suib and C. L. O'Young, Chem. Mater., 2002, 14, 940–948 CrossRef CAS.
- L. Jin, C. H. Chen, V. M. B. Crisostomo, L. P. Xu, Y. C. Son and S. L. Suib, Appl. Catal., A, 2009, 355, 169–175 CrossRef CAS.
- Y. A. May, S. Wei, W. Z. Yu, W. W. Wang and C. J. Jia, Langmuir, 2020, 36, 11196–11206 CrossRef CAS PubMed.
- T. Uematsu, Y. Miyamoto, Y. Ogasawara, K. Suzuki, K. Yamaguchi and N. Mizuno, Catal. Sci. Technol., 2016, 6, 222–233 RSC.
- F. Hashemzadeh, M. M. K. Motlagh and A. Maghsoudipour, J. Sol-Gel Sci. Technol., 2009, 51, 169–174 CrossRef CAS.
- S. Ching, E. J. Welch, S. M. Hughes, A. B. F. Bahadoor and S. L. Suib, Chem. Mater., 2002, 14, 1292–1299 CrossRef CAS.
- S. Ching, D. J. Petrovay, M. L. Jorgensen and S. L. Suib, Inorg. Chem., 1997, 36, 883–890 CrossRef CAS.
- E. Hastuti, W. R. Agustin and I. Yuliana, IOP Conf. Ser., Earth Environ. Sci., 2020, 456, 012017 CrossRef.
- S. Ching, J. L. Roark, N. Duan and S. L. Suib, Chem. Mater., 1997, 9, 750–754 CrossRef CAS.
- A. M. Hashem, A. E. Abdel-Ghany, R. El-Tawil, A. Bhaskar, B. Hunzinger, H. Ehrenberg, A. Mauger and C. M. Julien, Ionics, 2016, 22, 2263–2271 CrossRef CAS.
- M. H. Ghorbani and A. M. Davarpanah, Mod. Phys. Lett. B, 2017, 31, 1750006 CrossRef CAS.
- S. F. Chin, S. C. Pang and M. A. Anderson, Mater. Lett., 2010, 64, 2670–2672 CrossRef CAS.
- L. F. Liu, H. W. Zhang, Y. S. Zhang, D. M. Cao and X. H. Zhao, Colloids Surf., A, 2015, 468, 280–284 CrossRef CAS.
- J. L. Zhang, T. Zhuang, S. J. Liu, G. C. Zhang and K. L. Huo, J. Environ. Chem. Eng., 2020, 8, 103967 CrossRef CAS.
- H. Q. Wang, M. B. Zheng, J. H. Chen, G. B. Ji and J. M. Cao, J. Nanotechnol., 2010, 2010, 479172 Search PubMed.
- G. X. Zhu, J. G. Zhu, W. L. Li, W. Q. Yao, R. L. Zong, Y. F. Zhu and Q. F. Zhang, Environ. Sci. Technol., 2018, 52, 8684–8692 CrossRef CAS PubMed.
- G. Cheng, L. Yu, B. Lan, M. Sun, T. Lin, Z. W. Fu, X. H. Su, M. Q. Qiu, C. H. Guo and B. Xu, Mater. Res. Bull., 2016, 75, 17–24 CrossRef CAS.
- M. Sadeghinia, M. Rezaei and E. Amini, Korean J. Chem. Eng., 2013, 30, 2012–2016 CrossRef CAS.
- Z. Wu, J. Wu, X. Sun, B. Liu and J. Shen, Sci. Sin.: Chim., 2017, 47, 1226–1232 CrossRef.
- S. P. Rong, P. Y. Zhang, F. Liu and Y. J. Yang, ACS Catal., 2018, 8, 3435–3446 CrossRef CAS.
- J. Huang, R. M. Fang, Y. J. Sun, J. Y. Li and F. Dong, Chemosphere, 2021, 263, 128103 CrossRef CAS PubMed.
- X. Wang and Y. D. Li, J. Am. Chem. Soc., 2002, 124, 2880–2881 CrossRef CAS PubMed.
- V. Subramanian, H. W. Zhu, R. Vajtai, P. M. Ajayan and B. Q. Wei, J. Phys. Chem. B, 2005, 109, 20207–20214 CrossRef CAS PubMed.
- D. S. Zheng, S. X. Sun, W. L. Fan, H. Y. Yu, C. H. Fan, G. X. Cao, Z. L. Yin and X. Y. Song, J. Phys. Chem. B, 2005, 109, 16439–16443 CrossRef CAS PubMed.
- K. Li, C. Chen, H. B. Zhang, X. J. Hu, T. H. Sun and J. P. Jia, Appl. Surf. Sci., 2019, 496, 143662 CrossRef CAS.
- K. Z. Li, J. J. Chen, V. Peng, W. C. Lin, T. Yan and J. H. Li, J. Mater. Chem. A, 2017, 5, 20911–20921 RSC.
- C. He, Y. C. Wang, Z. Y. Li, Y. J. Huang, Y. H. Liao, D. H. Xia and S. C. Lee, Environ. Sci. Technol., 2020, 54, 12771–12783 CrossRef CAS PubMed.
- J. H. Zhang, Y. B. Li, L. Wang, C. B. Zhang and H. He, Catal. Sci. Technol., 2015, 5, 2305–2313 RSC.
- X. G. Han, Q. Kuang, M. S. Jin, Z. X. Xie and L. S. Zheng, J. Am. Chem. Soc., 2009, 131, 3152–3153 CrossRef CAS PubMed.
- J. S. Chen, T. Zhu, X. H. Yang, H. G. Yang and X. W. Lou, J. Am. Chem. Soc., 2010, 132, 13162–13164 CrossRef CAS PubMed.
- T. Kropp, J. Paier and J. Sauer, J. Phys. Chem. C, 2017, 121, 21571–21578 CrossRef CAS.
- G. Liu, J. C. Yu, G. Q. Lu and H. M. Cheng, Chem. Commun., 2011, 47, 12889 Search PubMed.
- B. Y. Bai and J. H. Li, ACS Catal., 2014, 4, 2753–2762 CrossRef CAS.
- X. F. Tang, J. L. Chen, Y. G. Li, Y. Li, Y. D. Xu and W. J. Shen, Chem. Eng. J., 2006, 118, 119–125 CrossRef CAS.
- F. Wang, H. X. Dai, J. G. Deng, G. M. Bai, K. M. Ji and Y. X. Liu, Environ. Sci. Technol., 2012, 46, 4034–4041 CrossRef CAS PubMed.
- F. J. Shi, F. Wang, H. X. Dai, J. X. Dai, J. G. Deng, Y. X. Liu, G. M. Bai, K. M. Ji and C. T. Au, Appl. Catal., A, 2012, 433, 206–213 CrossRef.
- J. C. Liu, O. Margeat, W. Dachraoui, X. J. Liu, M. Fahlman and J. Ackermann, Adv. Funct. Mater., 2014, 24, 6029–6037 CrossRef CAS.
- H. X. Tang, Z. Zhou and H. A. Sodano, ACS Appl. Mater. Interfaces, 2014, 6, 5450–5455 CrossRef CAS PubMed.
- X. H. Liao, Y. Jiang, G. K. Jian, G. Cheng and M. Sun, J. Guangdong Univ. Technol., 2018, 35, 75–79 Search PubMed.
- M. Wojciechowska, A. Malczewska, B. Czajka, M. Zieliński and J. Goslar, Appl. Catal., A, 2002, 237, 63–70 CrossRef CAS.
- E. W. McFarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS PubMed.
- L. C. Wang, X. S. Huang, Q. Liu, Y. M. Liu and J. H. Zhuang, J. Catal., 2008, 259, 66–74 CrossRef CAS.
- Y. Qin, Y. Wang, J. M. Li and Z. P. Qu, Surf. Interfaces, 2020, 21, 100657 CrossRef CAS.
- T. N. Lambert, D. J. Davis, W. Lu, S. J. Limmer, P. G. Kotula, A. Thuli, M. Hungate, G. D. Ruan, Z. Jin and J. M. Tour, Chem. Commun., 2012, 48, 7931–7933 RSC.
- M. J. Zhou, L. L. Cai, M. Bajdich, M. Garcia-Melchor, H. Li, J. J. He, J. Wilcox, W. D. Wu, A. Vojvodic and X. L. Zheng, ACS Catal., 2015, 5, 4485–4491 CrossRef CAS.
- J. S. Elias, M. Risch, L. Giordano, A. N. Mansour and Y. Shao-Horn, J. Am. Chem. Soc., 2014, 136, 17193–17200 CrossRef CAS PubMed.
- W. Liu and M. Flytzani-Stephanopoulos, Chem. Eng. J. Biochem. Eng. J., 1996, 64, 283–294 CrossRef CAS.
- Z. Boukha, J. L. Ayastuy, A. Iglesias-Gonzalez, B. Pereda-Ayo, M. A. Gutierrez-Ortiz and J. R. Gonzalez-Velasco, Appl. Catal., B, 2014, 160, 629–640 CrossRef.
- H. L. Jiang and Q. Xu, J. Mater. Chem., 2011, 21, 13705–13725 RSC.
- M. Han, S. L. Liu, L. Y. Zhang, C. Zhang, W. W. Tu, Z. H. Dai and J. C. Bao, ACS Appl. Mater. Interfaces, 2012, 4, 6654–6660 CrossRef CAS PubMed.
- Z. C. Wu, Y. J. Xue, H. Wang, Y. Q. Wu and H. Yu, RSC Adv., 2014, 4, 59009–59016 RSC.
- S. S. Li, J. N. Zheng, A. J. Wang, F. L. Tao, J. J. Feng, J. R. Chen and H. Y. Yu, J. Power Sources, 2014, 272, 1078–1085 CrossRef CAS.
- X. W. Teng, W. Q. Han, Q. Wang, L. Li, A. I. Frenkel and J. C. Yang, J. Phys. Chem. C, 2008, 112, 14696–14701 CrossRef CAS.
- Q. M. Ren, S. P. Mo, J. Fan, Z. T. Feng, M. Y. Zhang, P. R. Chen, J. J. Gao, M. L. Fu, L. M. Chen, J. L. Wu and D. Q. Ye, Chin. J. Catal., 2020, 41, 1873–1883 CrossRef CAS.
- Y. Z. Deng, W. X. Tang, W. H. Li and Y. F. Chen, Catal. Today, 2018, 308, 58–63 CrossRef CAS.
- J. P. Ma, Q. Y. Zhao, D. L. Wei, H. Q. Liu, X. K. Wang, Z. Chen and J. J. Wang, Inorg. Chem. Front., 2019, 6, 1011–1021 RSC.
- J. Zhou, L. F. Qin, W. Xiao, C. Zeng, N. Li, T. Lv and H. Zhu, Appl. Catal., B, 2017, 207, 233–243 CrossRef CAS.
- N. Huang, Z. P. Qu, C. Dong, Y. Qin and X. X. Duan, Appl. Catal., A, 2018, 560, 195–205 CrossRef CAS.
- A. Katsnelson, C&EN Global Enterprise, 2019, 97, 18–21 Search PubMed.
- F. X. Tian, M. Zhu, X. Liu, W. Tu and Y. F. Han, J. Catal., 2021, 401, 115–128 CrossRef CAS.
- R. Xu, X. Wang, D. S. Wang, K. B. Zhou and Y. D. Li, J. Catal., 2006, 237, 426–430 CrossRef CAS.
- P. P. Wang, J. H. Duan, J. Wang, F. M. Mei and P. Liu, Chin. J. Catal., 2020, 41, 1298–1310 CrossRef CAS.
- WHO, Iarc Monographs on the Evaluation of Carcinogenic Risks to Humans, Household Use of Solid Fuels and High-Temperature Frying, 2010, vol. 95, pp. 9–38 Search PubMed.
- Y. Dumanoglu, M. Kara, H. Altiok, M. Odabasi, T. Elbir and A. Bayram, Atmos. Environ., 2014, 98, 168–178 CrossRef CAS.
- H. Guo, S. C. Lee, P. K. K. Louie and K. F. Ho, Chemosphere, 2004, 57, 1363–1372 CrossRef CAS PubMed.
- A. Srivastava, A. E. Joseph, S. Patil, A. More, R. C. Dixit and M. Prakash, Atmos. Environ., 2005, 39, 59–71 CrossRef CAS.
- Y. L. Wang, Z. H. Li, C. Tang, H. X. Ren, Q. Zhang, M. Xue, J. Xiong, D. B. Wang, Q. Yu, Z. Y. He, F. Wei and J. K. Jiang, Environ. Sci.: Nano, 2019, 6, 3113–3122 RSC.
- S. M. Saqer, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2011, 103, 275–286 CrossRef CAS.
- Z. Sihaib, F. Puleo, J. M. Garcia-Vargas, L. Retailleau, C. Descorme, L. F. Liotta, J. L. Valverde, S. Gil and A. Giroir-Fendler, Appl. Catal., B, 2017, 209, 689–700 CrossRef CAS.
- Y. X. Zeng, Y. J. Zhan, R. J. Xie, K. Hu, J. P. Cao, D. X. Lei, B. Y. Liu, M. He and H. B. Huang, Chemosphere, 2020, 244, 125567 CrossRef CAS PubMed.
- S. Weon, F. He and W. Choi, Environ. Sci.: Nano, 2019, 6, 3185–3214 RSC.
- Y. X. Ruan, H. Guo, J. Li, Z. Y. Liu, N. Jiang and Y. Wu, Chemosphere, 2020, 258, 127334 CrossRef CAS PubMed.
- S. Malakar, P. D. Saha, D. Baskaran and R. Rajamanickam, Environ. Technol. Innovation, 2017, 8, 441–461 CrossRef.
- S. C. Kim and W. G. Shim, Appl. Catal., B, 2010, 98, 180–185 CrossRef CAS.
- J. B. Jia, P. Y. Zhang and L. Chen, Appl. Catal., B, 2016, 189, 210–218 CrossRef CAS.
- H. S. Kim, T. W. Kim, H. L. Koh, S. H. Lee and B. R. Min, Appl. Catal., A, 2005, 280, 125–131 CrossRef CAS.
- S. F. Zuo, Y. J. Du, F. J. Liu, D. Han and C. Z. Qi, Appl. Catal., A, 2013, 451, 65–70 CrossRef CAS.
- X. Jiang, J. F. Hua, H. Deng and Z. B. Wu, J. Mol. Catal. A: Chem., 2014, 383, 188–193 CrossRef.
- H. H. Kim, S. M. Oh, A. Ogata and S. Futamura, Catal. Lett., 2004, 96, 189–194 CrossRef CAS.
- Z. Hu, R. L. Mi, X. Yong, S. Liu, D. Li, Y. Li and T. Zhang, Chemistryselect, 2019, 4, 473–480 CrossRef CAS.
- Y. Liu, W. J. Zong, H. Zhou, D. S. Wang, R. R. Cao, J. J. Zhan, L. F. Liu and B. W. L. Jang, Catal. Sci. Technol., 2018, 8, 5344–5358 RSC.
- J. Ma, J. Wang and Y. Dang, Chem. Eng. J., 2020, 388, 124387 CrossRef CAS.
- Q. Ye, J. S. Zhao, F. F. Huo, J. Wang, S. Y. Cheng, T. F. Kang and H. X. Dai, Catal. Today, 2011, 175, 603–609 CrossRef CAS.
- Z. T. Feng, Q. M. Ren, R. S. Peng, S. P. Mo, M. Y. Zhang, M. L. Fu, L. M. Chen and D. Q. Ye, Catal. Today, 2019, 332, 177–182 CrossRef CAS.
- S. Ren, W. J. Liang, Q. L. Li and Y. X. Zhu, Chemosphere, 2020, 251, 126382 CrossRef CAS.
- W. H. Yang, Z. A. Su, Z. H. Xu, W. N. Yang, Y. Peng and J. H. Li, Appl. Catal., B, 2020, 260, 118150 CrossRef CAS.
- C. Dong, H. Wang, Y. W. Ren and Z. P. Qu, J. Environ. Sci., 2021, 104, 102–112 CrossRef PubMed.
- X. Min, M. M. Guo, L. Z. Liu, L. Li, J. N. Gu, J. X. Liang, C. Chen, K. Li, J. P. Jia and T. H. Sun, J. Hazard. Mater., 2021, 406, 124743 CrossRef CAS PubMed.
- Y. Zhang, M. Wu, Y. Wang, H. K. Yu and D. Leung, Appl. Catal., B, 2021, 280, 119388 CrossRef CAS.
- P. K. Arora and H. Bae, J. Environ. Sci., 2014, 13, 31 Search PubMed.
- A. Kumar, S. Kumar and S. Kumar, Biochem. Eng. J., 2005, 22, 151–159 CrossRef CAS.
- Y. M. Dong, H. X. Yang, K. He, S. Q. Song and A. M. Zhang, Appl. Catal., B, 2009, 85, 155–161 CrossRef CAS.
- E. Saputra, S. Muhammad, H. Q. Sun, A. Patel, P. Shukla, Z. H. Zhu and S. B. Wang, Catal. Commun., 2012, 26, 144–148 CrossRef CAS.
- W. C. Peng, S. B. Wang and X. Y. Li, Sep. Purif. Technol., 2016, 163, 15–22 CrossRef CAS.
- H. W. Liang, H. Q. Sun, A. Patel, P. Shukla, Z. H. Zhu and S. B. Wang, Appl. Catal., B, 2012, 127, 330–335 CrossRef CAS.
- R. Maddalena, M. Russell, D. P. Sullivan and M. G. Apte, Environ. Sci. Technol., 2009, 43, 5626–5632 CrossRef CAS PubMed.
- J. J. Pei and J. S. S. Zhang, HVACR Res., 2011, 17, 476–503 CAS.
- J. H. Yang, G. S. Yang and Z. Z. Ye, Appl. Mech. Mater., 2014, 668, 294–297 Search PubMed.
- C. B. Zhang, H. He and K. Tanaka, Appl. Catal., B, 2006, 65, 37–43 CrossRef CAS.
- S. S. Kim, K. H. Park and S. C. Hong, Appl. Catal., A, 2011, 398, 96–103 CrossRef CAS.
- J. X. Peng and S. D. Wang, Appl. Catal., B, 2007, 73, 282–291 CrossRef CAS.
- Y. Sekine, Atmos. Environ., 2002, 36, 5543–5547 CrossRef CAS.
- M. Wang, L. X. Zhang, W. M. Huang, T. P. Xiu, C. G. Zhuang and J. L. Shi, Chem. Eng. J., 2017, 320, 667–676 CrossRef CAS.
- P. Liu, H. P. He, G. L. Wei, X. L. Liang, F. H. Qi, F. D. Tan, W. Tan, J. X. Zhu and R. L. Zhu, Appl. Catal., B, 2016, 182, 476–484 CrossRef CAS.
- J. Wang, B. Z. Yeung, M. J. Cui, C. J. Peer, Z. Lu, W. D. Figg, M. G. Wientjes, S. Woo and J. L. S. Au, J. Controlled Release, 2017, 268, 147–158 CrossRef CAS PubMed.
- J. L. Wang, R. Yunus, J. G. Li, P. L. Li, P. Y. Zhang and J. Kim, Appl. Surf. Sci., 2015, 357, 787–794 CrossRef CAS.
- S. N. Guan, W. Z. Li, J. R. Ma, Y. Y. Lei, Y. S. Zhu, Q. F. Huang and X. M. Dou, J. Ind. Eng. Chem., 2018, 66, 126–140 CrossRef CAS.
- G. Li, X. Ding, Y. Shen and X. Jing, Knitting Industries, 2017, 7, 39–42 Search PubMed.
- J. Chen, D. X. Yan, Z. Xu, X. Chen, W. J. Xu, H. P. Jia and J. Chen, Environ. Sci. Technol., 2018, 52, 4728–4737 CrossRef CAS PubMed.
- F. L. Huang, X. Wang, Q. Y. Zhu, K. L. Li, X. F. Zhou, S. H. Lu, Z. Fan, L. L. He, Y. X. Liu and F. J. Pang, Catal. Surv. Asia, 2019, 23, 33–40 CrossRef CAS.
- T. H. He, D. D. Shao, X. S. Zeng and S. P. Rong, Chemosphere, 2020, 261, 127778 CrossRef CAS.
- J. Huang, S. Zhong, Y. Dai, C. C. Liu and H. Zhang, Environ. Sci. Technol., 2018, 52, 11309–11318 CrossRef CAS.
- J. C. Lu, H. S. Hao, L. M. Zhang, Z. Z. Xu, L. P. Zhong, Y. T. Zhao, D. D. He, J. P. Liu, D. K. Chen, H. P. Pu, S. F. He and Y. M. Luo, Appl. Catal., B, 2018, 237, 185–197 CrossRef CAS.
- J. R. Kastner, K. C. Das, Q. Buquoi and N. D. Melear, Environ. Sci. Technol., 2003, 37, 2568–2574 CrossRef CAS PubMed.
- J. L. Yang, Q. Zhang, F. Zhang, D. H. Xia, H. D. Liu, S. H. Tian, L. P. Sun, D. Shu, C. He and S. Runa, J. Hazard. Mater., 2018, 358, 136–144 CrossRef CAS PubMed.
- S. H. Xie, Y. X. Liu, J. G. Deng, X. T. Zhao, J. Yang, K. F. Zhang, Z. Han, H. Arandiyan and H. X. Dai, Appl. Catal., B, 2017, 206, 221–232 CrossRef CAS.
- H. B. Huang, Y. Xu, Q. Y. Feng and D. Y. C. Leung, Catal. Sci. Technol., 2015, 5, 2649–2669 RSC.
- Y. T. Zhao, D. K. Chen, J. P. Liu, D. D. He, X. H. Cao, C. Y. Han, J. C. Lu and Y. M. Luo, Chem. Eng. J., 2020, 389, 124384 CrossRef CAS.
- D. D. He, G. P. Wan, H. S. Hao, D. K. Chen, J. C. Lu, L. Zhang, F. Liu, L. P. Zhong, S. F. He and Y. M. Luo, Chem. Eng. J., 2016, 289, 161–169 CrossRef CAS.
- T. Nothe, H. Fahlenkamp and C. von Sonntag, Environ. Sci. Technol., 2009, 43, 5990–5995 CrossRef CAS.
- D. H. Xia, W. J. Xu, Y. C. Wang, J. L. Yang, Y. J. Huang, L. L. Hu, C. He, D. Shu, D. Y. C. Leung and Z. H. Pang, Environ. Sci. Technol., 2018, 52, 13399–13409 CrossRef CAS.
- W. Li and G. V. Gibbs, J. Am. Chem. Soc., 1998, 120, 9041–9046 CrossRef CAS.
- J. L. Yang, Y. J. Huang, Y. W. Chen, D. H. Xia, C. Y. Mou, L. L. Hu, J. W. Zeng, C. He, P. K. Wong and H. Y. Zhu, Nano Today, 2020, 35, 100944 CrossRef CAS.
- H. Y. Li, S. W. Wu, L. Pan, J. H. Xu, J. Shan, X. Yang, W. Dong, F. R. Deng, Y. H. Chen, M. Shima and X. B. Guo, Environ. Pollut., 2018, 232, 358–366 CrossRef CAS PubMed.
- S. Magzamen, B. F. Moore, M. G. Yost, R. A. Fenske and C. J. Karr, J. Occup. Environ. Med., 2017, 59, 624–630 CrossRef CAS PubMed.
- J. D. Berman, N. Fann, J. W. Hollingsworth, K. E. Pinkerton, W. N. Rom, A. M. Szema, P. N. Breysse, R. H. White and F. C. Curriero, Environ. Health Perspect., 2012, 120, 1404–1410 CrossRef CAS PubMed.
- B. Hoffmann, H. Luttmann-Gibson, A. Cohen, A. Zanobetti, C. de Souza, C. Foley, H. H. Suh, B. A. Coull, J. Schwartz, M. Mittleman, P. Stone, E. Horton and D. R. Gold, Environ. Health Perspect., 2012, 120, 241–246 CrossRef CAS PubMed.
- Z. P. Hao, D. Y. Cheng, Y. Guo and Y. H. Liang, Appl. Catal., B, 2001, 33, 217–222 CrossRef CAS.
- P. Nikolov, K. Genov, P. Konova, K. Milenova, T. Batakliev, V. Georgiev, N. Kumar, D. K. Sarker, D. Pishev and S. Rakovsky, J. Hazard. Mater., 2010, 184, 16–19 CrossRef CAS PubMed.
- T. Gopi, G. Swetha, S. C. Shekar, C. Ramakrishna, B. Saini, R. Krishna and P. V. L. Rao, Catal. Commun., 2017, 92, 51–55 CrossRef CAS.
- S. Y. Gong, J. Y. Chen, X. F. Wu, N. Han and Y. F. Chen, Catal. Commun., 2018, 106, 25–29 CrossRef CAS.
- J. Z. Ma, C. X. Wang and H. He, Appl. Catal., B, 2017, 201, 503–510 CrossRef CAS.
- G. X. Zhu, J. G. Zhu, W. J. Jiang, Z. J. Zhang, J. Wang, Y. F. Zhu and Q. F. Zhang, Appl. Catal., B, 2017, 209, 729–737 CrossRef CAS.
- W. X. Tang, H. D. Liu, X. F. Wu and Y. F. Chen, Ozone: Sci. Eng., 2014, 36, 502–512 CrossRef CAS.
- D. Mehandjiev, A. Naydenov and G. Ivanov, Appl. Catal., A, 2001, 206, 13–18 CrossRef CAS.
- H. Fan, C. F. Zhao, Z. S. Ma and Y. K. Yang, Environ. Pollut., 2020, 267, 115164 CrossRef CAS PubMed.
- G. X. Zhu, W. Zhu, Y. Lou, J. Ma, W. Q. Yao, R. L. Zong and Y. F. Zhu, Nat. Commun., 2021, 12, 4152 CrossRef CAS PubMed.
- Y. Liu and P. Y. Zhang, J. Phys. Chem. C, 2017, 121, 23488–23497 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |