
Radical addition reaction between chromenols and toluene derivatives initiated by Brønsted acid catalyst under light irradiation†
Jun
Kikuchi
 ,
Shota
Kodama
and
Masahiro
Terada
*
,
Shota
Kodama
and
Masahiro
Terada
*
Department of Chemistry, Graduate School of Science, Tohoku University, Aoba-ku, Sendai 980-8578, Japan. E-mail: mterada@tohoku.ac.jp; Fax: +81-22-795-6602; Tel: +81-22-795-6602
First published on 19th May 2021
Abstract
A Brønsted acid-catalyzed benzylic C(sp3)–H functionalization of toluene and its derivatives was accomplished through the photo-excitation of benzopyrylium cation intermediates. Light irradiation promoted the reaction of chromenols with toluene and its derivatives in the presence of a Brønsted acid catalyst, affording the corresponding benzylated chromene derivatives having tetrasubstituted carbon centers. Experimental and theoretical studies elucidated that the reaction proceeds through a radical addition pathway in which the benzopyrylium cation intermediate acts as both an electrophile and a photo-redox catalyst. The use of a chiral phosphoric acid catalyst enables the enantioselective radical addition reaction through a chiral anion-controlled asymmetric induction via an ion-pairing interaction to provide an addition product in an enantioenriched form.
Introduction
Brønsted acid catalysis is one of the fundamental and versatile catalytic processes in organic chemistry. In addition to traditional textbook transformations, such as the hydrolysis and formation of esters and acetals, Brønsted acid catalysts have been widely utilized for carbon–carbon bond forming reactions.1 Over the past few decades, Brønsted acid catalysis has drawn much attention as one of the metal-free organocatalytic processes.2 In particular, the development of enantioselective catalysis using a chiral Brønsted acid as a representative organocatalyst has evolved into an active research field.3,4 Transformations under Brønsted acid catalysis are initiated by (i) protonation of a substrate to generate a cationic species, followed by (ii) bond formation with a nucleophile, and (iii) deprotonation of the formed species by the conjugate base of the Brønsted acid to afford a product along with the regeneration of the Brønsted acid catalyst. A wide range of transformations have been accomplished through this fundamental catalytic process. However, applicable functional substrates are still limited because Brønsted acid catalysts are effective primarily for polarized functional substrates, for example representative carbon nucleophiles, electronically enriched (hetero)aromatics, and olefins having an oxygen or a nitrogen substituent. In fact, the direct functionalization of nonpolarized chemical bonds, such as the benzylic C(sp3)–H bonds of toluene derivatives, has received sparse attention in Brønsted acid catalysis5 despite the fact that the direct conversion of rich carbon resources enables the derivatization of value-added carbon frameworks without pre-functionalization. Therefore, if such a transformation could be achieved in Brønsted acid catalysis, it would become valuable not only with regard to expanding the scope of transformations, but also from the viewpoint of atom-economical and environmentally benign synthesis.Photo-excited compounds exhibit redox properties, thereby enabling reactions that are difficult to achieve in the ground state. For example, a photo-excited cationic species would function as a strong oxidant because of its intrinsic high electron affinity. Hence, photochemical radical transformations of cationic species have been elaborated through single-electron transfer (SET) from donor molecules such as toluene derivatives to the photo-excited cationic species.6–8 In the reported reaction systems, preformed cationic substrates or cationic intermediates generated using an excess of strong acids were employed in most cases. Meanwhile, the catalytic variant is strictly limited to the photo-excitation of iminium intermediates generated from α,β-unsaturated aldehydes and amine catalysts.7 The scope of cationic species generated under the catalytic conditions is still narrow. In fact, radical reactions of photo-excited cationic intermediates as strong oxidants using a Brønsted acid catalyst have never been reported despite the fact that cationic species are readily available through the protonation of substrates, which is one of the representative approaches for the generation of these species. In this context, we envisioned the development of a radical transformation of nonpolarized C(sp3)–H bonds through the generation of photo-excited cationic intermediates using a Brønsted acid catalyst (Fig. 1). The intended transformation is initiated as follows: (i) protonation of a substrate using a Brønsted acid catalyst generates a cationic intermediate that can function as a strong oxidant upon photo-excitation; (ii) SET from the donor molecule to photo-excited cationic intermediate I forms radical cation II; and (iii) deprotonation by the conjugate base of the Brønsted acid proceeds smoothly to generate nucleophilic radical III along with the regeneration of the Brønsted acid catalyst because the acidity of the adjacent C(sp3)–H bond of radical cation II is dramatically enhanced compared with that of the parent donor molecule. Subsequently, (iv) generated radical III undergoes further transformation based on its nucleophilic nature.9
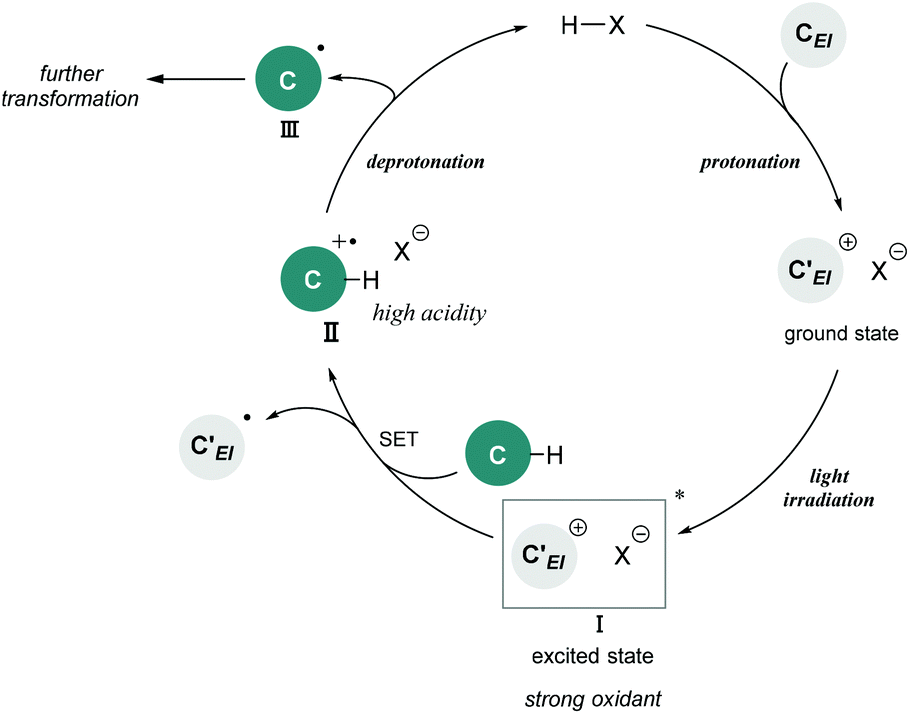 | ||
| Fig. 1 Brønsted acid-catalyzed radical transformation utilizing photo-excitation of the cationic intermediate as a strong oxidant. | ||
As shown in Fig. 1, the combination of fundamental Brønsted acid catalytic processes, namely, protonation and deprotonation, and light irradiation would allow the transformation of nonpolarized C(sp3)–H bonds in a catalytic fashion. Herein, we report a benzylic C(sp3)–H functionalization of toluene and its derivatives 2 as donor molecules utilizing benzopyrylium cation intermediates, which are generated by the dehydration of chromenols 1 using Brønsted acid catalysts as strong oxidants under light irradiation (Scheme 1). The reaction provided the corresponding benzylated chromene derivatives 3 and 3′ having tetrasubstituted carbon centers that are generally difficult to construct. Experimental and theoretical studies elucidated that the reaction proceeds not through a radical coupling pathway but through a radical addition pathway in which the nucleophilic benzylic radical attacks the benzopyrylium cation intermediate. Furthermore, the use of a chiral Brønsted acid catalyst enabled the enantioselective radical addition reaction to proceed, thereby providing chromene derivatives in an enantioenriched form.
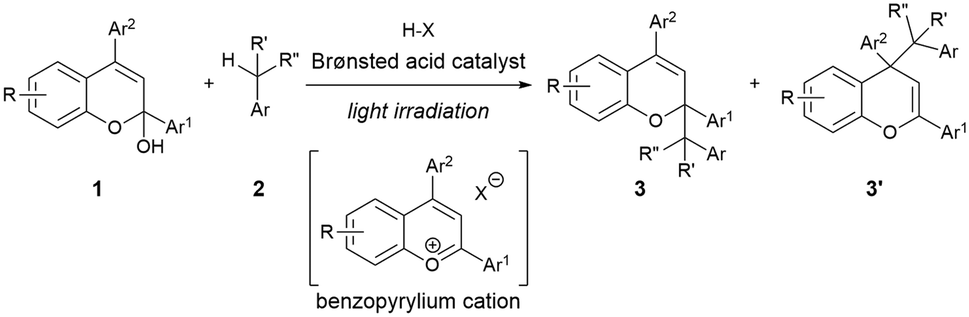 | ||
| Scheme 1 Brønsted acid-catalyzed radical reaction between chromenols and toluene derivatives. | ||
Results and discussion
Optimization of reaction conditions
As an initial attempt, the reaction of 1a with toluene 2a, which serves as both the solvent and substrate, was performed under LED irradiation (λmax = 405 nm) in the presence of a series of Brønsted acid catalysts at 0 °C (Table 1, entries 1–4). The reaction using diphenyl phosphate afforded a small amount of the desired benzyl adducts as a mixture of 3aa and 3′aa (entry 1). The use of AcOH and BzOH did not provide the products probably due to the low acidity of the catalysts (entries 2 and 3). The reaction using TFA resulted in an improvement of yield (entry 4).|
|
|||||
|---|---|---|---|---|---|
| Entry | Acid | Solvent | Time (h) | Yieldb (%) | 3aa/3′aa |
| a Unless otherwise noted, all reactions were carried out using 0.1 mmol of 1a and 0.01 mmol of catalyst (10 mol%) in the indicated solvent. b NMR yield. Isolated yield is shown in parentheses. c 3.0 mmol of 2a was used. d Reaction was carried out in air. e 0.1 mmol TEMPO was used. f No light irradiation. | |||||
| 1 | (PhO)2PO2H | 2a (0.025 M) | 8 | 11 | 75/25 |
| 2 | AcOH | 2a (0.025 M) | 8 | <1 | — |
| 3 | BzOH | 2a (0.025 M) | 8 | <1 | — |
| 4 | TFA | 2a (0.025 M) | 2 | 47 | 83/17 |
| 5 | TfOH | 2a (0.025 M) | 4 | 14 | 84/16 |
| 6c | TFA | CH2Cl2 (0.1 M) | 2 | 84 (81) | 95/5 |
| 7c,d | TFA | CH2Cl2 (0.1 M) | 3 | 20 | 93/7 |
| 8c,e | TFA | CH2Cl2 (0.1 M) | 2 | <1 | — |
| 9c,f | TFA | CH2Cl2 (0.1 M) | 3 | 0 | — |
| 10c | — | CH2Cl2 (0.1 M) | 13 | 0 | — |
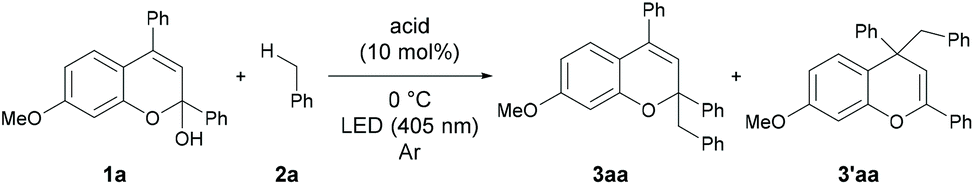
In contrast, a strong Brønsted acid, TfOH, afforded the products with a decrease in yield (entry 5). This result suggests that the basicity of the conjugate base of the Brønsted acid is important presumably because of the efficient deprotonation from the benzyl radical cation. Consequently, TFA was identified as the optimal catalyst. Gratifyingly, when the reaction was performed with 30 equivalents of toluene in CH2Cl2 as the solvent, the desired product was obtained in 81% isolated yield with high regioselectivity (3aa/3′aa = 95/5) (entry 6).10 The inhibition of the reactivity was observed under an aerobic atmosphere or in the presence of TEMPO (entries 7 and 8). No product formation was detected in the absence of a Brønsted acid catalyst (entry 9) or light irradiation (entry 10).11 The results indicate that the generation of photo-excited benzopyrylium cation intermediates is essential to promote the present reaction.
Scope of substrates
With the optimized reaction conditions in hand, the substrate scope of the present reaction was demonstrated using a series of chromenols (Table 2). The reaction of chromenols having a substituent at the para- or meta-position of Ar1 (1b–1e) afforded 3ba–3ea and 3′ba–3′ea in moderate yields with high regioselectivities. The introduction of a methyl substituent at the ortho-position of Ar1 did not retard the reaction at all and high yield and regioselectivity were observed. Further investigation of the substrate scope with respect to chromenols having a series of aryl substituents Ar2 was carried out. Chromenols having the 4-methylphenyl group 1g and the 4-chlorophenyl group 1h at the 4-position underwent the reaction efficiently, affording products in good yields with high regioselectivities. The reaction of chromenol having the 4-methoxyphenyl group 1i did not provide the desired products under the optimized reaction conditions. In contrast, the use of an LED with λmax = 448 nm instead of an LED with λmax = 405 nm resulted in the formation of products with high regioselectivities, albeit in low yields. This is probably because the introduction of the methoxy group induces a redshift in the absorption spectrum of the generated benzopyrylium cation intermediate, resulting in low excitation efficiency at 405 nm.12,13 Chromenols having meta- or ortho-substituted aryl moieties, 1j–1l, also underwent the reaction to afford products with high regioselectivities. The scope with respect to chromenols having a series of substituents R on the chromenol framework was further investigated. As shown in the bottom of Table 2, the regioselectivity of products was markedly dependent on the electronic nature of the R substituent. The use of 1m having a methoxy group at the 6-position resulted in the formation of products 3ma and 3′ma in moderate yield with moderate regioselectivity. In contrast, the reaction of 1n having a methoxy group at the 5-position did not afford any desired product. Chromenol having a methyl group at the 7-position (1o) underwent the reaction and provided products in moderate yield with fairly good regioselectivity. The introduction of the fluorine atom at the 7-position retarded the reaction with moderate regioselectivity.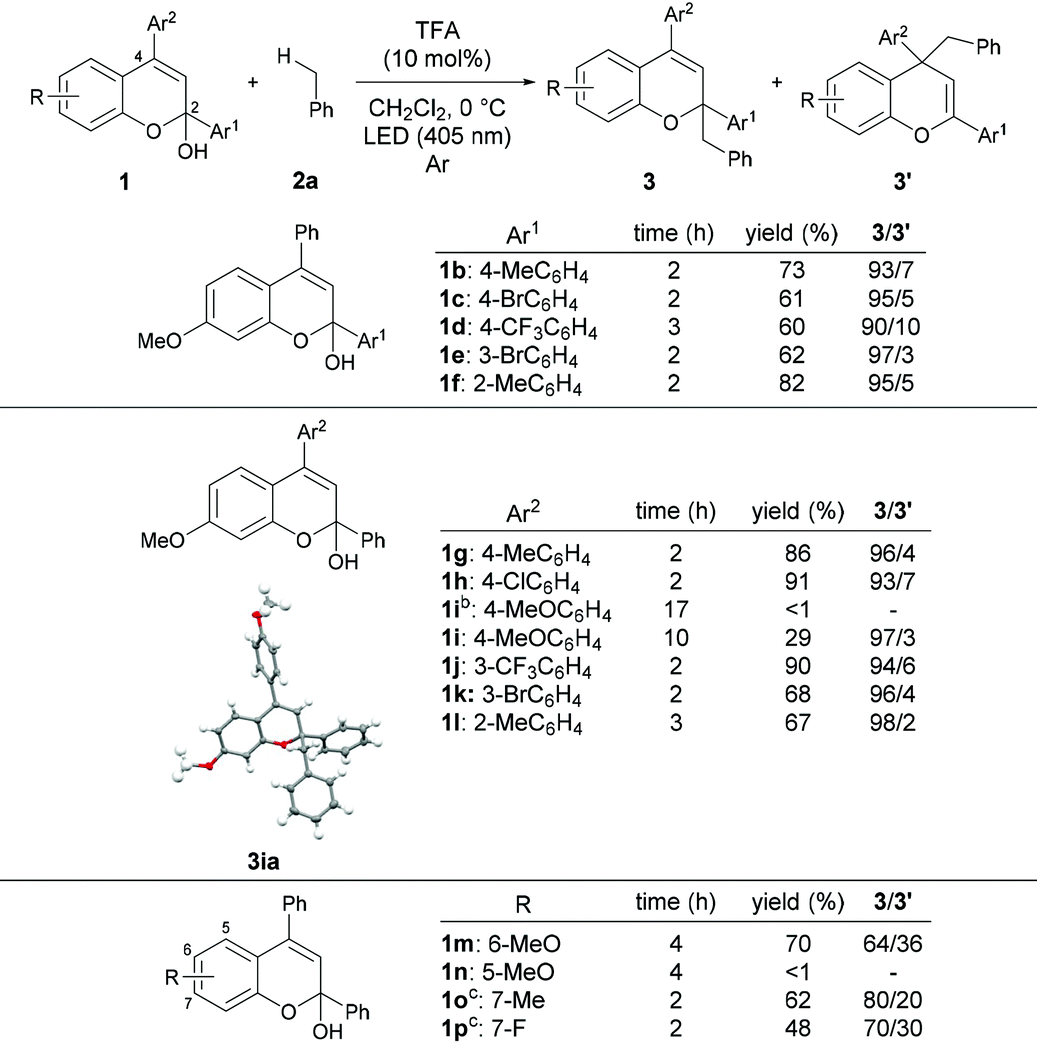
We next investigated the scope of toluene derivatives (Scheme 2). The use of a series of xylenes (2b–2d) and mesitylene (2e) afforded the corresponding products in good yields with high regioselectivities. Ethylbenzene (2f) as a secondary toluene derivative reacted smoothly to afford the product in high yield with good regioselectivity, albeit with low diastereoselectivity. In the reaction of cumene (2g), the product having vicinal tetrasubstituted carbon centres was formed in moderate yield with high regioselectivity using 20 mol% of TFA. The reaction of 4-isopropyl toluene (2h), which has two reaction sites, namely a primary carbon on the methyl group and a tertiary carbon on the isopropyl group, proceeded only on the methyl group, even though the tertiary carbon radical is more stable than the primary one. This result indicates that the deprotonation of the corresponding radical cation was influenced by steric effects. Electron-withdrawing halogenated toluene derivatives (2i and 2j) were also applicable in this reaction and the desired products were obtained. However, the reaction of the toluene derivative 2k having a trifluoromethyl group as a strong electron-withdrawing group did not afford the desired product.
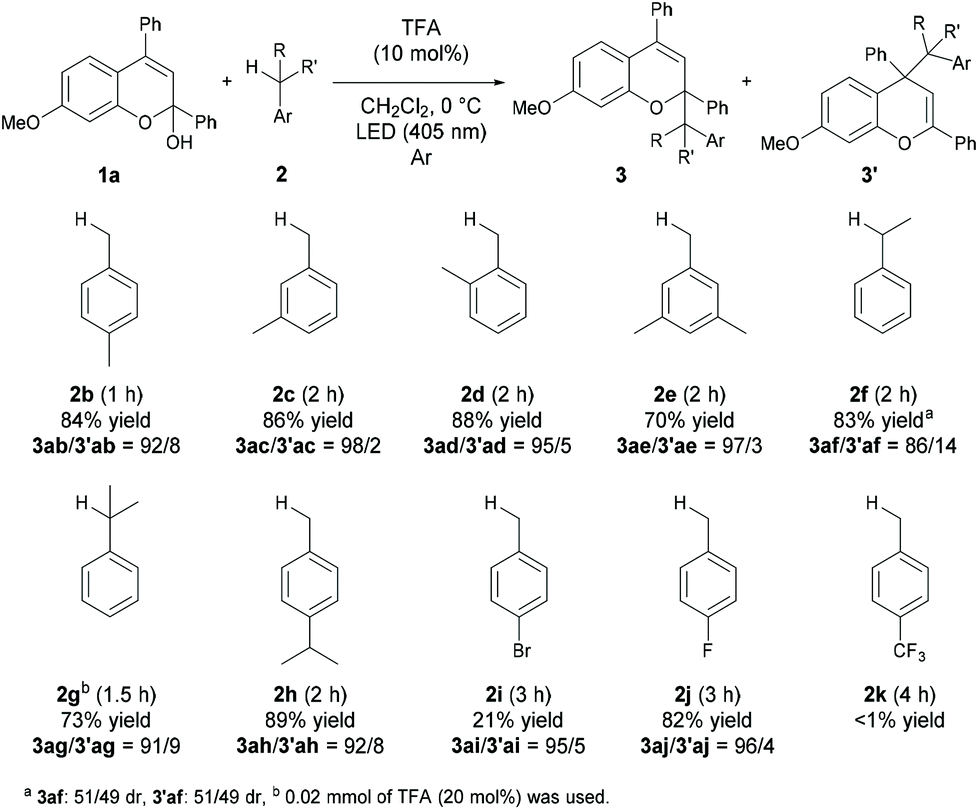 | ||
| Scheme 2 Scope of toluene derivatives. | ||
Mechanistic studies
The mechanistic elucidation of the present reaction was our next focus. At the outset of mechanistic studies, we conducted Stern–Volmer luminescence quenching experiments with benzopyrylium cation salt 1′a+BF4− to find out that the photo-excited benzopyrylium cation could be quenched with toluene (Eox = +2.26 V vs. SCE).14,15 As a result, the emission intensity of benzopyrylium cation salt 1′a+BF4− decreased as the amount of toluene in the CH2Cl2 solution increased (Fig. 2). This observation indicated that SET from toluene to the photo-excited benzopyrylium cation occurs, generating benzopyrylium-derived radical A and benzyl radical cation B having the highly acidic benzylic C(sp3)–H bond (pKa of B was estimated to be −13 in CH3CN),9a which can be deprotonated by the conjugate base of the Brønsted acid catalyst to generate the benzyl radical.16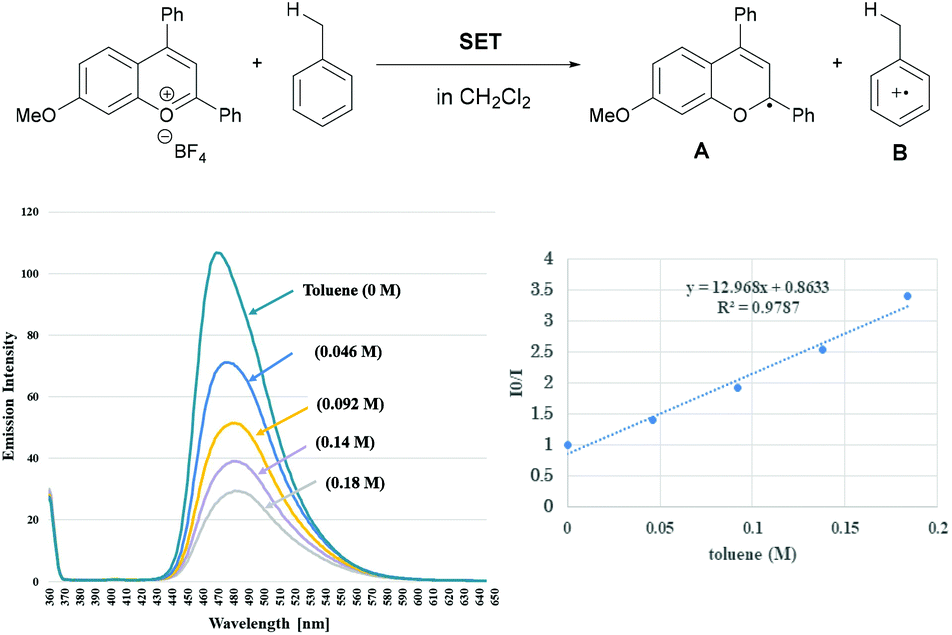 | ||
| Fig. 2 Stern–Volmer luminescence quenching experiments. | ||
We next conducted DFT calculations to determine the actual reaction pathway in the carbon–carbon bond forming step. All calculations were performed with the Gaussian 16 package (Revision B.01).17,18 Geometries were optimized and characterized using frequency calculations at the UM06-2X/6-311+G(d,p) level. Gibbs free energies in the solution phase were calculated using single-point energy calculations at the same level according to the solvation model based on the solute electron density (SMD) (dichloromethane: ε = 8.93) of the optimized structures. First, the energy profile for the radical coupling reaction of radical A generated by a single electron reduction of the benzopyrylium cation with the benzyl radical C was identified (Fig. 3a). The energy barrier of TS-1 (ΔG≠ = + 36.9 kcal mol−1) is too high for the reaction to proceed under the present reaction conditions. This result suggests that the radical coupling reaction is an unfavorable pathway. We then explored another reaction pathway. As shown in Fig. 3b, the energy profile for the radical addition reaction of benzyl radical C with benzopyrylium cation intermediate D was identified. As a result, the radical addition pathway exhibits a smaller energy barrier of TS-2 (ΔG≠ = + 12.6 kcal mol−1) than that of TS-1 in the radical coupling pathway.19 This radical addition reaction of benzyl radical C with benzopyrylium cation intermediate D through TS-2 generates benzylated radical cation E. SET from radical A to radical cation E gives product 3aa along with the generation of benzopyrylium cation D. This process is exergonic by 34.6 kcal mol−1 and therefore thermodynamically favorable (Fig. 3c).
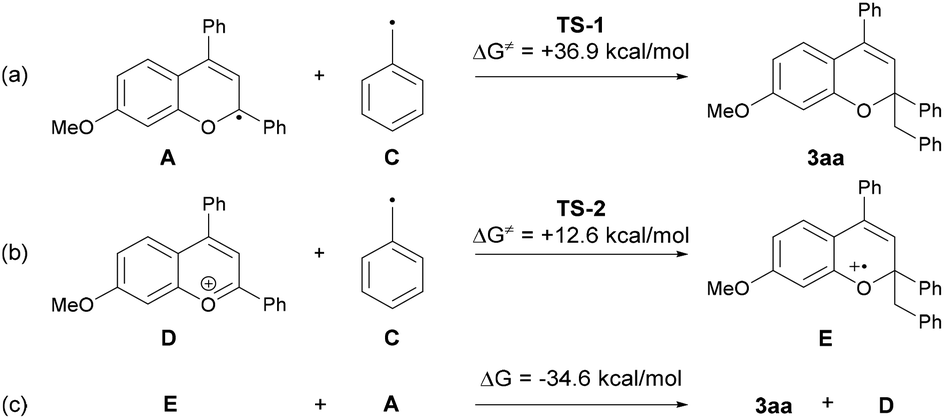 | ||
| Fig. 3 Calculated energy barriers. | ||
From these experimental and theoretical studies, we propose a mechanism for the present radical addition reaction (Fig. 4). First, the Brønsted acid catalyst protonates the hydroxy group of chromenol 1 to generate benzopyrylium cation intermediate D, which can reach an excited state under light irradiation. Photo-excited benzopyrylium cation intermediate D* oxidizes toluene to form benzyl radical cation B and radical A. Radical cation B is deprotonated by the conjugate base of the Brønsted acid to form benzyl radical C while regenerating the Brønsted acid catalyst. The radical addition of benzyl radical C to another benzopyrylium cation intermediate D generates radical cation E. SET from radical A to radical cation E forms the corresponding product 3 along with the regeneration of benzopyrylium cation D. Thus, this reaction mechanism indicates that the present reaction proceeds in a dual catalytic system, namely, Brønsted acid catalysis (protonation and deprotonation) and photo-redox catalysis of the benzopyrylium cation intermediate.20,21
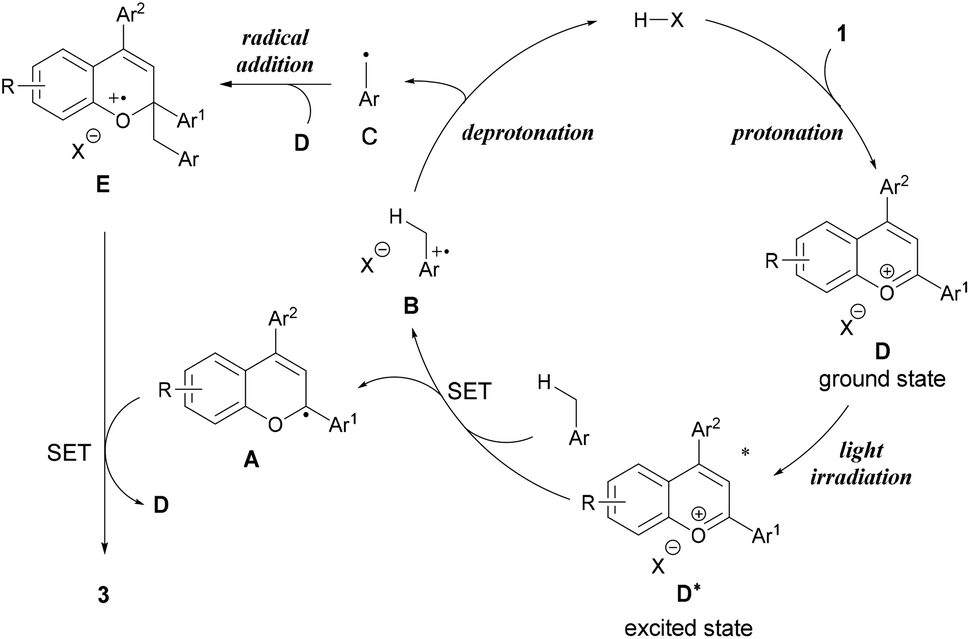 | ||
| Fig. 4 Plausible reaction mechanism. | ||
Enantioselective reaction with a chiral phosphoric acid catalyst
From the present reaction mechanism, we expected that the radical addition reaction would proceed in an enantioselective manner if the benzyl radical attacks the benzopyrylium cation intermediate in the chiral environment created by the conjugate base of the chiral Brønsted acid catalyst.22–24 Therefore, we finally investigated an enantioselective radical addition reaction using a chiral Brønsted acid catalyst. The reaction of 1m with toluene 2a, which functions as both the solvent and substrate, using chiral phosphoric acid (S)-4 at 0 °C afforded a mixture of addition products 3ma and 3′ma with moderate regioselectivity (Table 3, entry 1).25 As expected, the desired products were formed in an enantioselective manner. Gratifyingly, when the reaction temperature was decreased to −75 °C, the addition product 3ma was obtained with an increase in enantioselectivity to 60% ee, albeit in moderate yield (entry 2). Although the stereochemical outcome is still unsatisfactory, this is the first example of the enantioselective radical addition reaction through a chiral anion-controlled asymmetric induction via an ion-pairing interaction. In the present system, the cationic intermediate does not have any H–X hydrogen-bonding interaction site (X = NR, O, etc.) and therefore, the present achievement deserves high marks.26|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Conditions | Time (h) | Yield (%) | 3ma/3′ma | ee (%) | |
| 3ma | 3′ma | |||||
| a Unless otherwise noted, all reactions were carried out using 0.1 mmol of 1m, 4 mL of 2 (0.05 M), and 0.01 mmol (S)-4 (10 mol%). Isolated yield. The ee value was determined by chiral stationary phase HPLC analysis. | ||||||
| 1 | 0 °C, 1 h | 1 | 72 | 67/33 | 31 | 15 |
| 2 | −75 °C, 12 h | 12 | 35 | 74/26 | 60 | 26 |
Conclusions
In conclusion, we have developed a radical transformation between chromenols and toluene derivatives using a Brønsted acid catalyst under light irradiation. The reaction that was initiated through the photo-excitation of the benzopyrylium cation intermediate converted the benzylic C(sp3)–H bonds of toluene and its derivatives to afford benzylated chromene derivatives having tetrasubstituted carbon centers in good yields with high regioselectivities in most cases. Experimental and theoretical studies of the present reaction mechanism suggested that the reaction proceeds through a radical addition pathway where the benzopyrylium cation intermediate acts as both an electrophile and a photo-redox catalyst. The investigation of enantioselective reactions using chiral phosphoric acid revealed that the chiral anion-controlled asymmetric induction via an ion-pairing interaction is effective even for the enantiocontrol in the addition of radical species to cationic intermediates. The present catalytic reaction system utilizing the photo-excitation of cationic intermediates enables the expansion of the scope of functional substrates that are generally difficult to transform in Brønsted acid catalysis and is an environmentally benign synthetic method for the metal-free conversion of abundant and inexpensive carbon resources into high value-added compounds. Further studies of the development of other radical addition reactions, especially in an enantioselective manner, utilizing a variety of photo-excited cationic intermediates and Brønsted acid catalysts will be conducted in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Hybrid Catalysis for Enabling Molecular Synthesis on Demand” (JP17H06447) from MEXT, Japan (M. T.), and a Grant-in-Aid for Young Scientists (JP19K15552) from JSPS, Japan (J. K.).Notes and references
- (a) C. H. Cheon and H. Yamamoto, Super Brønsted acid catalysis, Chem. Commun., 2011, 47, 3043–3056 Search PubMed; (b) T. Akiyama and K. Mori, Stronger Brønsted Acids: Recent Progress, Chem. Rev., 2015, 115, 9277–9306 Search PubMed; (c) H. Yamamoto and K. Ishihara, Acid Catalysis in Modern Organic Synthesis, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) P. I. Dalko, Enantioselective Organocatalysis: Reactions and Experimental Procedures, Wiley-VCH, New York, 2007 Search PubMed; (b) B. List, Science of Synthesis: Asymmetric Organocatalysis 1: Lewis Base and Acid Catalysts, Georg Thieme Verlag KG, New York, 2012 Search PubMed; (c) K. Maruoka, Science of Synthesis, Asymmetric Organocatalysis 2: Brønsted Base and Acid Catalysts, and Additional Topics, Georg Thieme Verlag KG, New York, 2012 Search PubMed.
- For selected reviews on chiral Brønsted acid catalysis, see: (a) T. Akiyama, Stronger Brønsted Acids, Chem. Rev., 2007, 107, 5744–5758 Search PubMed; (b) M. Terada, Chiral Phosphoric Acids as Versatile Catalysts for Enantioselective Transformations, Synthesis, 2010, 1929–1982 Search PubMed; (c) D. Kampen, C. M. Reisinger and B. List, Chiral Brønsted acids for asymmetric organocatalysis, Top. Curr. Chem., 2010, 291, 395–456 Search PubMed; (d) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Complete Field Guide to Asymmetric BINOL-Phosphate Derived Brønsted Acid and Metal Catalysis: History and Classification by Mode of Activation; Brønsted Acidity, Hydrogen Bonding, Ion Pairing, and Metal Phosphates, Chem. Rev., 2014, 114, 9047–9153 Search PubMed.
- For seminal studies of chiral phosphoric acid catalysts, see: (a) T. Akiyama, J. Itoh, K. Yokota and K. Fuchibe, Enantioselective Mannich–Type Reaction Catalyzed by a Chiral Brønsted Acid, Angew. Chem. Int. Ed., 2004, 43, 1566–1568 Search PubMed; (b) D. Uraguchi and M. Terada, Chiral Brønsted Acid-Catalyzed Direct Mannich Reactions via Electrophilic Activation, J. Am. Chem. Soc., 2004, 126, 5356–5357 Search PubMed.
- F. Li, D. Tian, Y. Fan, R. Lee, G. Lu, Y. Yin, B. Qiao, X. Zhao, Z. Xiao and Z. Jiang, Chiral acid-catalysed enantioselective C−H functionalization of toluene and its derivatives driven by visible light, Nat. Commun., 2019, 10, 1774 Search PubMed.
- (a) P. S. Mariano, Electron-transfer mechanisms in photochemical transformations of iminium salts, Acc. Chem. Res., 1983, 16, 130–137 Search PubMed; (b) P. S. Mariano, The photochemistry of iminium salts and related heteroaromatic systems, Tetrahedron, 1983, 39, 3845–3879 Search PubMed; (c) R. M. Borg, R. O. Heuckeroth, A. J. Y. Lan, S. L. Quillen and P. S. Mariano, Arene-iminium salt electron-transfer photochemistry. Mechanistically interesting photoaddition processes, J. Am. Chem. Soc., 1987, 109, 2728–2737 Search PubMed.
- (a) M. Silvi, C. Verrier, Y. P. Rey, L. Buzzetti and P. Melchiorre, Visible-light excitation of iminium ions enables the enantioselective catalytic β-alkylation of enals, Nat. Chem., 2017, 9, 868–873 Search PubMed; (b) D. Mazzarella, G. E. M. Crisenza and P. Melchiorre, Asymmetric Photocatalytic C–H Functionalization of Toluene and Derivatives, J. Am. Chem. Soc., 2018, 140, 8439–8443 Search PubMed.
- (a) R. Zhou, H. Liu, H. Tao, X. Yua and J. Wu, Metal-free direct alkylation of unfunctionalized allylic/benzylic sp3 C–H bonds via photoredox induced radical cation deprotonation, Chem. Sci., 2017, 8, 4654–4659 Search PubMed; (b) H. Liu, L. Ma, R. Zhou, X. Chen, W. Fang and J. Wu, One-Pot Photomediated Giese Reaction/Friedel–Crafts Hydroxyalkylation/Oxidative Aromatization To Access Naphthalene Derivatives from Toluenes and Enones, ACS Catal., 2018, 8, 6224–6229 Search PubMed.
- (a) A. M. De Nicholas and D. A. Arnold, Thermochemical parameters for organic radicals and radical ions. Part 1. The estimation of the pKa of radical cations based on thermochemical calculations, Can. J. Chem., 1982, 60, 2165–2179 Search PubMed; (b) F. G. Bordwell and J.-P. Cheng, Radical-cation acidities in solution and in the gas phase, J. Am. Chem. Soc., 1989, 111, 1792–1795 Search PubMed.
- The reactions of preformed benzopyrylium cation salts resulted in the formation of only small amounts of products (see the ESI† for details).
- For the UV-vis absorption spectra of 1a and the mixture of 1a and TFA, see the ESI† for details.
- For the UV-vis absorption spectra of 1i and the mixture of 1i and TFA, see the ESI† for details.
- For the crystallographic data of 3ia, see: J. Kikuchi, CCDC 2064775: CSD Communication, 2021, DOI: DOI:10.5517/ccdc.csd.cc279kns.
- The reduction potential of 1′a+*BF4− (singlet state) was determined to be
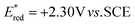 in CH3CN (see the ESI† for details).
in CH3CN (see the ESI† for details). - P. B. Merkel, P. Luo, J. P. Dinnocenzo and S. Farid, Accurate Oxidation Potentials of Benzene and Biphenyl Derivatives via Electron-Transfer Equilibria and Transient Kinetics, J. Org. Chem., 2009, 74, 5163–5173 Search PubMed.
- Because of the high oxidation potential of the TFA anion (F3CCO2Na: Eox = >+2.4 V vs. SCE), a benzyl radical is generated through SET from toluene to the benzopyrylium cation intermediate, followed by deprotonation, rather than through SET from the conjugate base of TFA, followed by hydrogen atom transfer (HAT) from toluene. See: C. Depecker, H. Marzouk, S. Trevin and J. Devynckb, Trifluoromethylation of aromatic compounds via Kolbe electrolysis in pure organic solvent. Study on laboratory and pilot scale, New J. Chem., 1999, 23, 739–742 Search PubMed.
- M. J. Frisch, et al., Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed, see the ESI† for the full citation.
- For Gaussian basis sets, see: (a) M. J. Frisch, J. A. Pople and J. S. Binkley, Self–consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets, J. Chem. Phys., 1984, 80, 3265–3269 Search PubMed; (b) W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory; John Wiley, New York, USA, 1986, and references cited therein Search PubMed.
- The radical coupling pathway was proposed in previously reported catalytic radical reactions utilizing the photo-exited cationic intermediates as strong oxidants (see ref. 7).
- T. McCallum, S. P. Pitre, M. Morin, J. C. Scaiano and L. Barriault, The photochemical alkylation and reduction of heteroarenes, Chem. Sci., 2017, 8, 7412–7418 Search PubMed.
- M. A. Miranda and H. Garcia, 2,4,6-Triphenylpyrylium Tetrafluoroborate as an Electron-Transfer Photosensitizer, Chem. Rev., 1994, 94, 1063–1089 Search PubMed.
- For selected reviews about chiral anion controlled asymmetric catalysis, see: (a) J. Lacour and D. Moraleda, Chiral anion-mediated asymmetric ion pairing chemistry, Chem. Commun., 2009, 7073–7089 Search PubMed; (b) R. J. Phipps, G. L. Hamilton and F. D. Toste, The progression of chiral anions from concepts to applications in asymmetric catalysis, Nat. Chem., 2012, 4, 603–614 Search PubMed; (c) M. Mahlau and B. List, Asymmetric Counteranion–Directed Catalysis: Concept, Definition, and Applications, Angew. Chem., Int. Ed., 2013, 52, 518–533 Search PubMed; (d) K. Brak and E. N. Jacobsen, Asymmetric Ion–Pairing Catalysis, Angew, Chem., Int. Ed., 2013, 52, 534–561 Search PubMed.
- (a) M. Terada, T. Yamanaka and Y. Toda, Chiral Anion Catalysis in the Enantioselective 1,4−Reduction of the 1−Benzopyrylium Ion as a Reactive Intermediate, Chem. – Eur. J., 2013, 19, 13658–13662 Search PubMed; (b) C.-C. Hsiao, H.-H. Liao, E. Sugiono, I. Atodiresei and M. Rueping, Shedding Light on Organocatalysis—Light–Assisted Asymmetric Ion–Pair Catalysis for the Enantioselective Hydrogenation of Pyrylium Ions, Chem. – Eur. J., 2013, 19, 9775–9779 Search PubMed.
- For the reviews about enantioselective radical reactions in cooperative photoredox and chiral Brønsted acid catalysis, see: (a) S. Li, S.-H. Xiang and B. Tan, Chiral Phosphoric Acid Creates Promising Opportunities for Enantioselective Photoredox Catalysis, Chin. J. Chem., 2020, 38, 213–214 Search PubMed; (b) R. S. J. Proctor, A. C. Colgan and R. J. Phipps, Exploiting attractive non-covalent interactions for the enantioselective catalysis of reactions involving radical intermediates, Nat. Chem., 2020, 12, 990–1004 Search PubMed.
- A series of catalyst screenings are summarized in the ESI (see Table S1).†.
- (a) Z. Yang, H. Li, S. Li, M.-T. Zhang and S. Luo, A chiral ion-pair photoredox organocatalyst: enantioselective anti-Markovnikov hydroetherification of alkenols, Org. Chem. Front., 2017, 4, 1037–1041 Search PubMed; (b) P. D. Morse, T. M. Nguyen, C. L. Cruz and D. A. Nicewicz, Enantioselective counter-anions in photoredox catalysis: The asymmetric cation radical Diels-Alder reaction, Tetrahedron, 2018, 74, 3266–3272 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qo00657f |
| This journal is © the Partner Organisations 2021 |