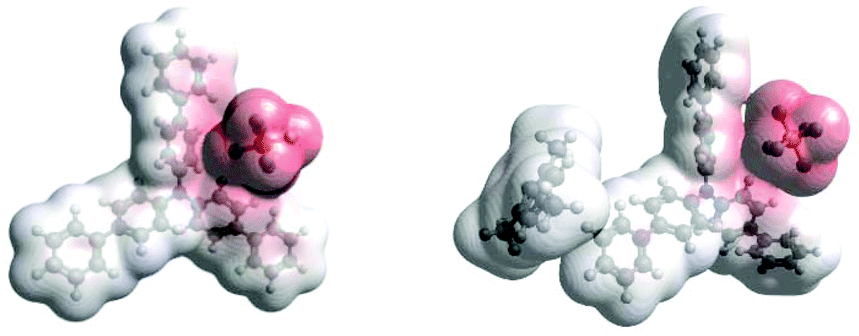DOI:
10.1039/D0QO01609H
(Research Article)
Org. Chem. Front., 2021,
8, 1132-1142
Hole-mediated photoredox catalysis: tris(p-substituted)biarylaminium radical cations as tunable, precomplexing and potent photooxidants†
Received
21st December 2020
, Accepted 17th February 2021
First published on 3rd March 2021
Abstract
As a combination of visible light photoredox catalysis and synthetic organic electrochemistry, electrochemically-mediated photoredox catalysis emerged as a powerful synthetic technique in recent years, overcoming fundamental limitations of electrochemistry and photoredox catalysis in the single electron transfer activation of small organic molecules. Herein we report a tunable class of electroactivated photoredox catalyst, tri(para-substituted)biarylamines, that become superoxidants in their photoexcited states even able to oxidize molecules beyond the solvent window limits of cyclic voltammetry (such as polyfluorobenzene and trifluorotoluene). Furthermore, we demonstrate that precomplexation not only permits the excited state photochemistry of tris(para-substituted)biarylaminium cations to overcome picosecond lifetime, but enables and rationalizes the surprising photochemistry of their higher-order doublet (Dn) excited states, unlocking extremely high oxidative potentials (up to a record-breaking ∼+4.4 V vs. SCE).
Introduction
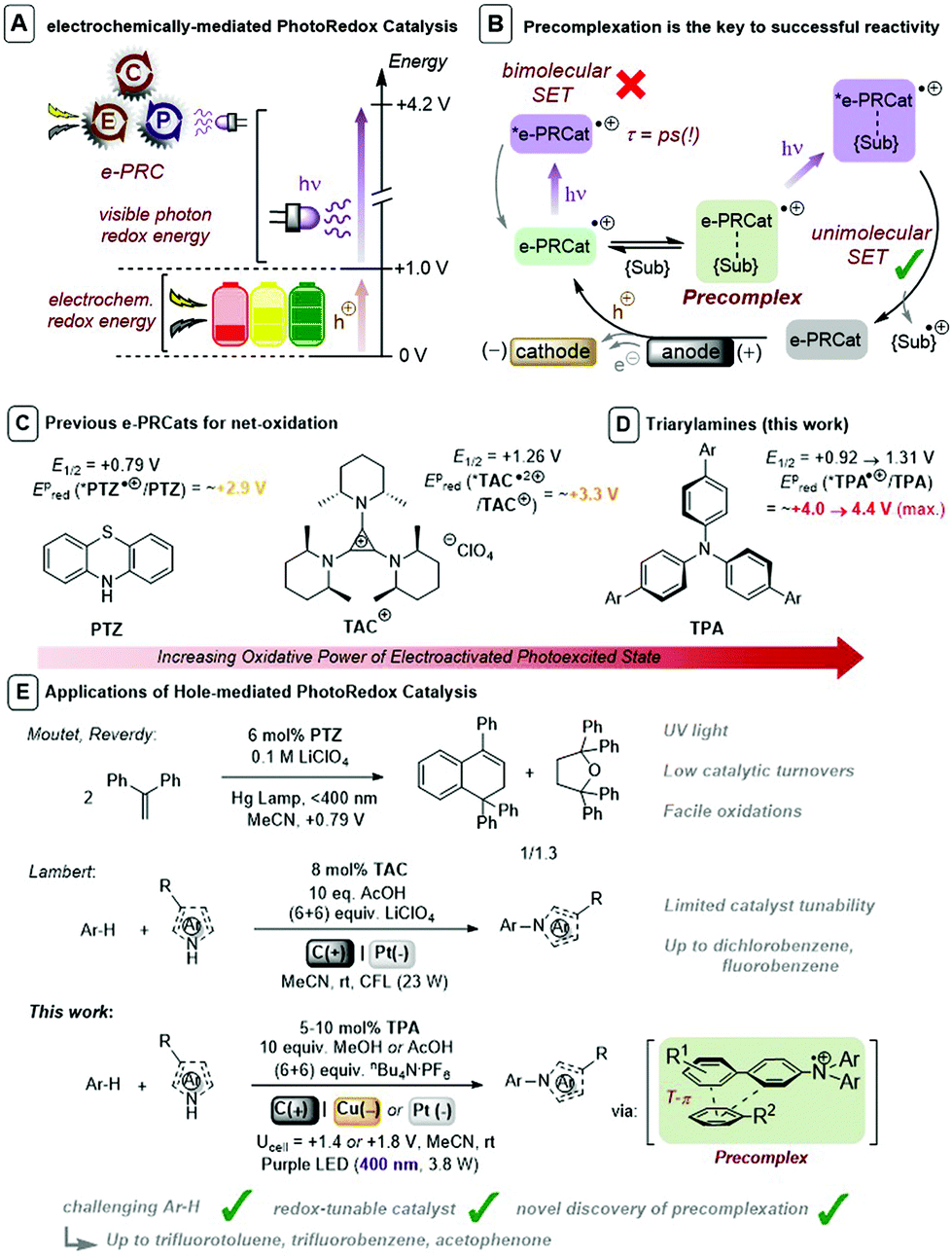
Synthetic Organic Electrochemistry (SOE)1 and visible light PhotoRedox Catalysis (PRC),2 which offer entries to single electron transfer (SET) chemistry and radical intermediates under mild conditions, have risen to the fore of contemporary organic synthesis. A key factor underpinning the success of PRC is the host of available photocatalyst structures with well-characterized photophysical and redox data, allowing chemists to ‘tune’ a given excited state to a desired process. Although PRC exhibits a selectivity benefit in transferring visible light photon energy to a colored transition metal-based or organic dye photocatalysts, its scope of applications are partially redox potential-limited by this photon energy (ca. 1.8–3.1 eV). Multiple-photon-accumulation strategies such as consecutive photoelectron transfer (conPET)3 and triplet–triplet annihilation upconversion (TTA-UC)4 have proven elegant means to achieve powerful SET reductions, but their use in oxidations has eluded chemists. A limiting restriction in conPET is that both ground and radical ion states must be photoactive. Furthermore, net-oxidative/reductive PRC processes employ excess of a sacrificial oxidant/reductant which is necessary for photocatalyst turnover, but which may (or whose by-products may) (i) interfere with downstream chemistry and (ii) require separation.
In comparison to PRC processes, SOE can employ uncapped potentials to chemical redox reactions at the turn of a dial. However, electrode surfaces typically5 cannot discriminate between organic molecules aside from their innate order of thermodynamic redox potentials. Moreover, low electrical conductivity in organic solvents typically require applied potentials to be much higher than the target substrate's redox potential.6 This encourages deleterious redox processes, such as those involving solvent especially if target SET processes lie near the electroactive window (typically ca. +3 to −3 V).6,7 In addition, mechanistic characterization of the heterogeneous electrolysis step (heterogeneous SET at the electrode surface)8 has remained a key challenge in SOE. Screening of electrode materials is inevitable, despite efforts to characterize materials by overpotential, resistivity, surface area, stability and cost.9
As a result of these limitations, synthetic photoelectochemistry is emerging as a state-of-the-art in SET-mediated chemistry.10–12 Different categories for the merger of photo- and electrochemistry have been reported, including interfacial photoelectrochemistry (iPEC) involving photoelectrodes13 and decoupled photoelectrochemistry (dPEC) where photo- and electrochemical steps serve distinct mechanistic roles.14 A third category involves an intimate and synergistic relationship of photo- and electrochemical steps within the same catalytic cycle. Xu,15a,b Lambert,15c Lin15d and Ackermann15e reported elegant examples using electrochemistry to turnover spent photoredox catalysts thus obviating the need for sacrificial oxidants/reductants. This category can also electrogenerate photocatalysts to achieve super-oxidations or super-reductions beyond those accessible via photocatalysis alone.16 A variety of nomenclature has been coined for this sub-category: “electrophotocatalysis”16e “photoelectrocatalysis”16f and “electron-primed photoredox catalysis”.16g We coined nomenclature “electrochemically-mediated PhotoRedox Catalysis (e-PRC)” as a blanket term to cover both net-oxidative/net-reductive variants10 and to avoid any misunderstanding with iPEC (Fig. 1A). In the net-oxidative direction, Moutet and Reverdy achieved the first e-PRC bimolecular cyclization of 1,1-diphenylethylene using phenothiazine (PTZ) as catalyst (Fig. 1C and E), but utilized UV light and only a few catalyst turnovers were reported.16a Recently, Lambert reported an exotic trisaminocyclopropenium cation (TAC) as an e-PRCat for Nicewicz-type oxidative coupling. Chloro-, dichloro- and fluorobenzene, typically beyond reach of photoredox catalysis alone, could be engaged (Fig. 1C and E).16e In context of these seminal reports, a class of e-PRC catalyst that offers tunability, to provide more powerful potentials or new handles for selectivity, is highly attractive.
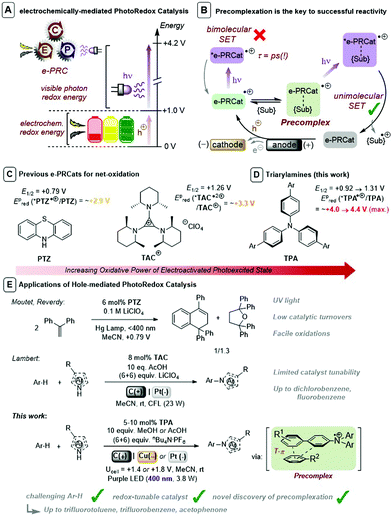 |
| | Fig. 1 (A) Concept of electrochemically-mediated photoredox catalysis. (B) Precomplexation rationalizes successful reactivity of picosecond lifetime photoexcited radical ions. (C) Previous e-PRCats for net oxidation. (D) Triarylamines as e-PRCats. (E) Applications of hole-mediated photoredox catalysis and contextualization of this work. | |
Tri(p-substituted)arylamines, discovered by Walter and co-workers17 are renowned for their photophysical properties as hole-transport materials in OLEDs and in photovoltaics,18 as oxidative mediators in SOE19,20 and as oxidants in radical chain reactions.21 They exhibit fully reversible 1e− oxidations by cyclic voltammetry. Generally colorless in their neutral state, their oxidized tris(para-substituted)aminium radical cation forms are intensely colored. While the commercial tris(4-bromophenyl)aminium radical cation presents issues in synthesis,22 better behaved radical cations derive from tri(para-substituted)biarylamines (TPAs) that are easily be prepared in a single/few steps from Pd-catalyzed cross-coupling reactions of tri-p-bromophenylamine with appropriate partners or after appropriate activation. Barham reported the use of tris(para-substituted)arylaminium radical cations bearing p-Me or p-Ph substituents that exhibited good solution stability and excellent stability as isolated solids, that mediated highly selective N-CH3 oxidative functionalizations.23 Given their favorable properties and facile synthetic accessibility, we foresaw TPAs as a suitable class of customizable e-PRC catalyst that would open new avenues to customize redox and photophysical parameters of e-PRCats. Moreover, the excellent stability of isolated TPA˙+s could be leveraged in photophysical and mechanistic characterization. Herein, we disclose TPAs as powerful and effective e-PRCats (up to Epox (*Ar3N˙+/Ar3N) ∼ +4.3 V vs. SCE) that engage unactivated or electron-poor aromatic systems (Epox = +2.1 V to >3.0 V) in SET oxidation (Fig. 1D and E). Of key importance is the first discovery of precomplexation in the ground state (Fig. 1B), which (i) circumvents ultrashort lifetimes of excited states, (ii) allows engagement of higher order excited states and (iii) enables contra-thermodynamic redox selectivity. Photoexcited triarylaminium radical cations are demonstrated as novel superoxidants in organic synthesis.
Results and discussion
Synthetic results
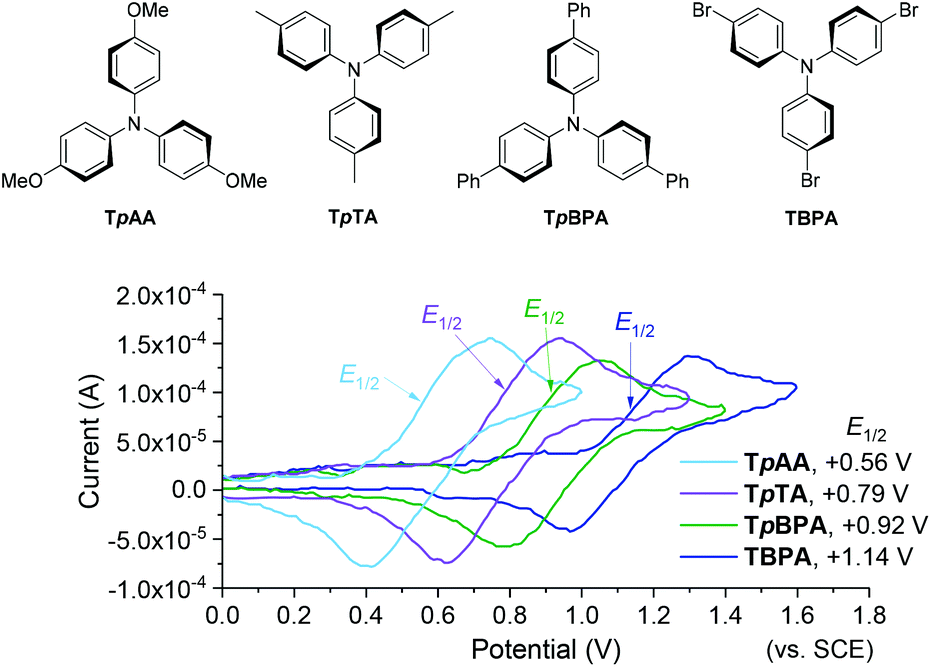
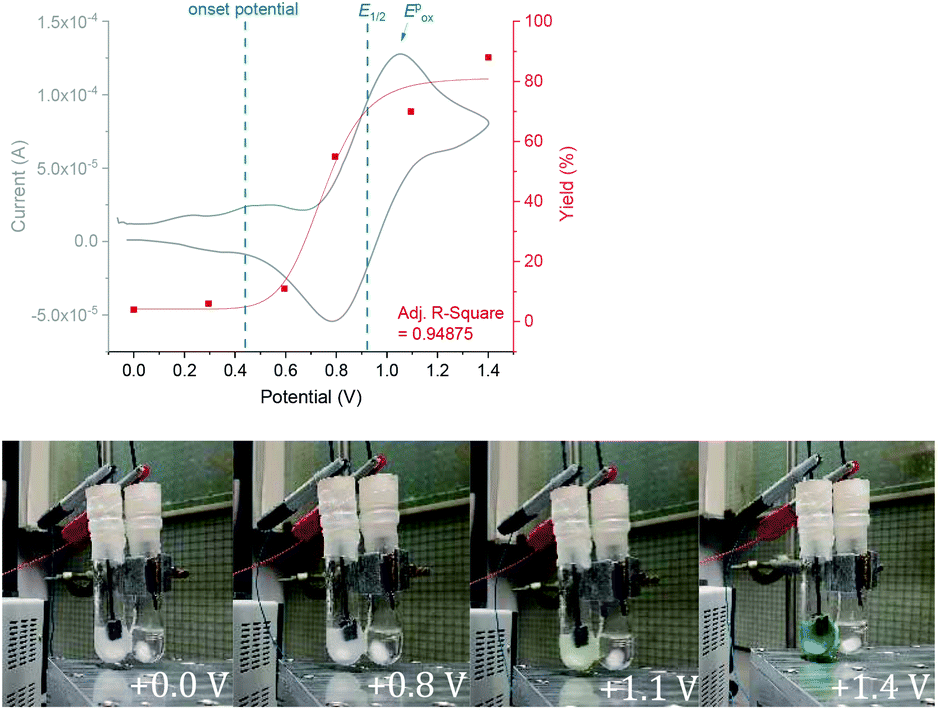
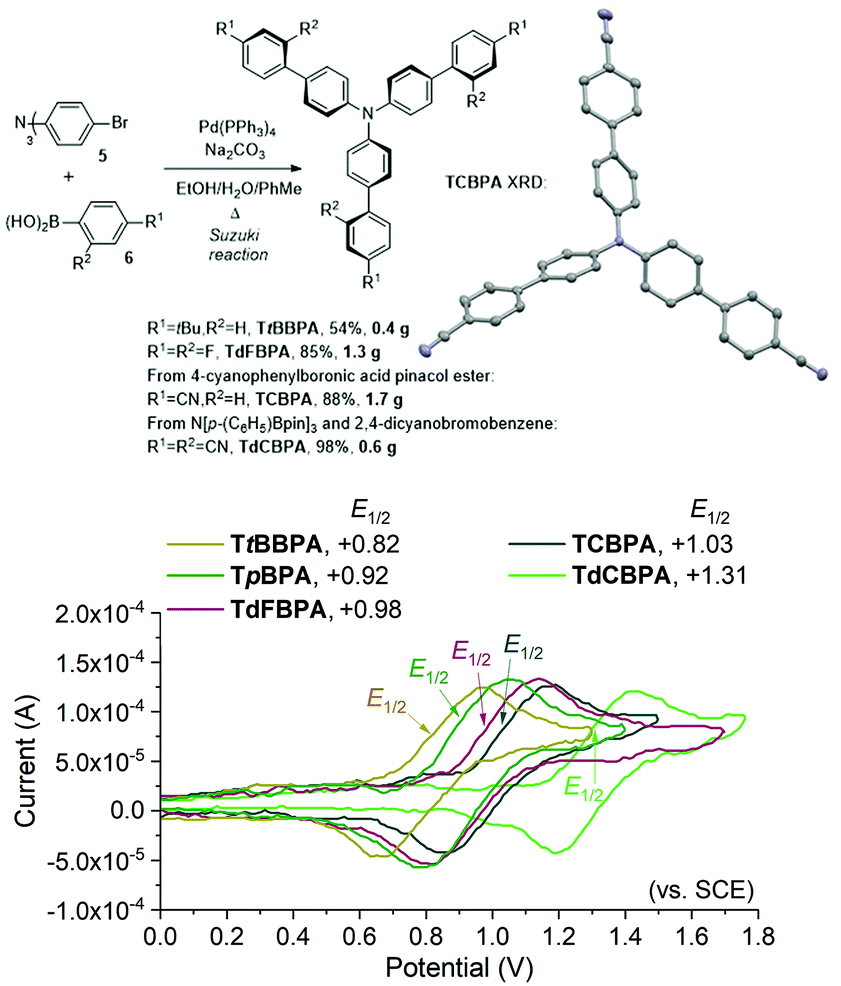
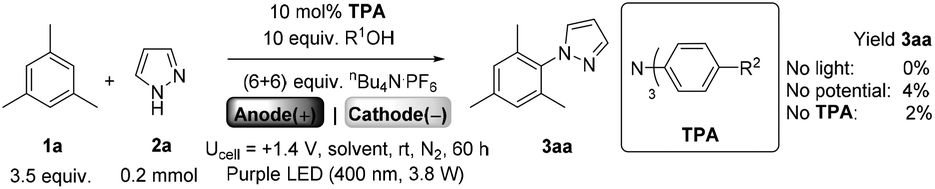
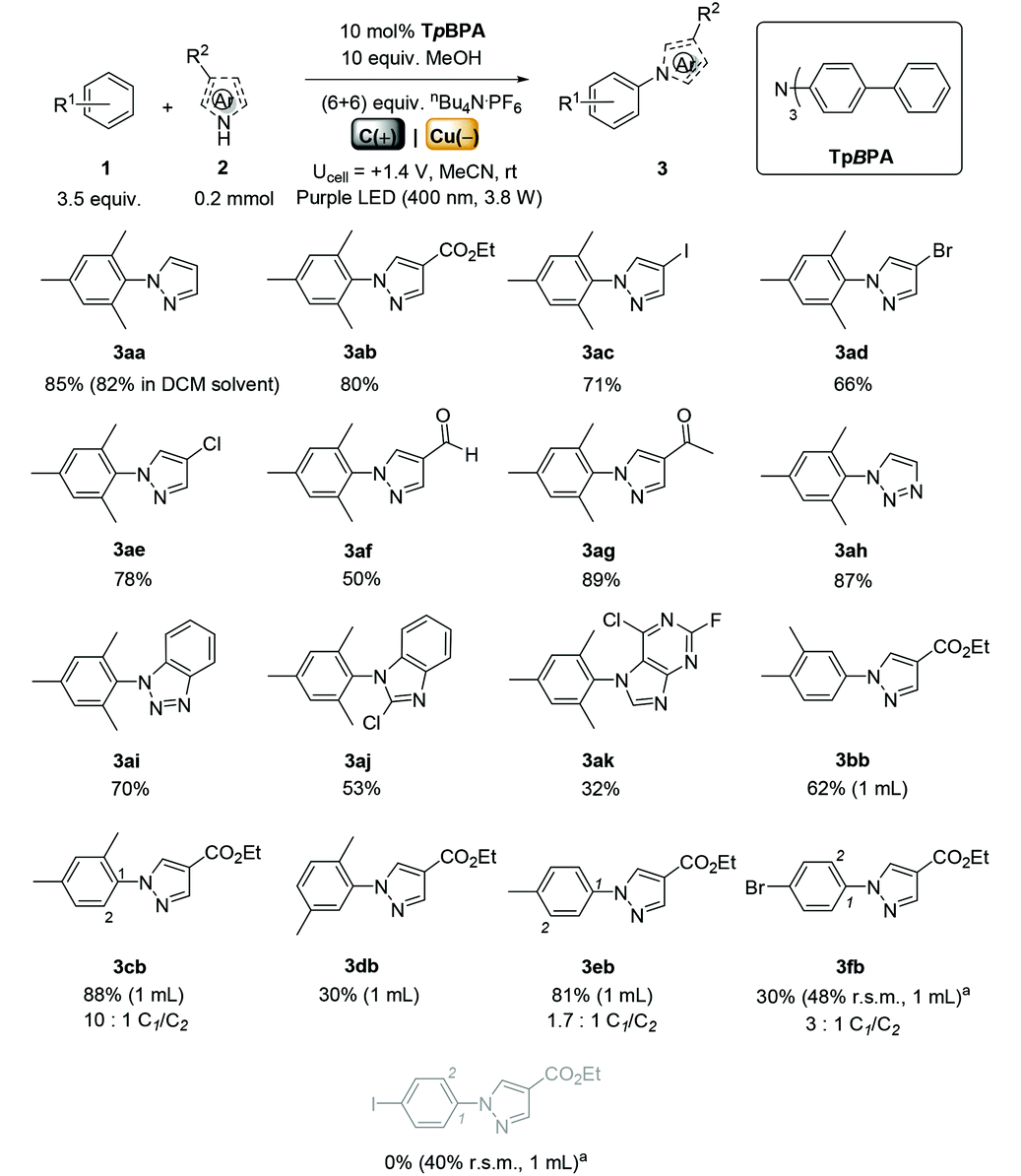
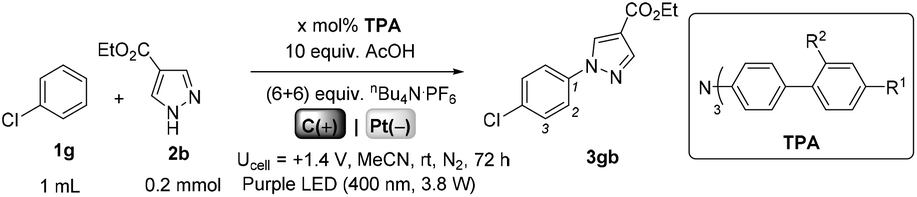
We began by screening different TPA e-PRCats (Generation 1, Fig. 2) with different half-wave oxidation potentials in the Nicewicz model reaction;24 the oxidative C–H amination of mesitylene (Epox = +2.1 V vs. SCE)24 with pyrazole (Table 1). After setting an anodic constant potential of Ucell = +1.4 V (Fig. 3), color developed in the anodic compartments for each TPA e-PRCat, consistent with the expected formation of their TPA˙+s. Gratifyingly, irradiation under the specified conditions afforded C–H activation product 3aa. Control reactions in absence of light, potential or e-PRCat confirmed the operation of e-PRC. Interestingly, tri([1,1′-biphenyl]-4-yl)amine (TpBPA) afforded a notably higher yield of 3aa than the commercial tris(4-bromophenyl)amine (entry 4) despite having an appreciably lower E1/2. After further optimization, using MeOH as proton source, carbon foam as WE and copper as CE gave the best result (entry 8). The yield of 3aa tracked well with increasing applied constant potential (Fig. 3). For full optimization studies, see the ESI.† With optimal conditions in hand, the amination of arenes with a variety of pharmaceutically-relevant N-heterocycles was explored (Table 2). Halide-bearing and carbonyl (aldehyde, ketone and ester)-bearing pyrazoles, triazole, benzotriazole, and a functionalized derivative of benzimidazole afforded generally good to excellent (50–89%) yields of aminated mesitylenes 3aa–3aj. We note that benzimidazole derivatives have not been reported as nucleophiles in previous photoelectrochemical arene amination or conPET photocatalytic methods.13d,16e,25 6-Chloro-2-fluoropurine afforded a modest yield of 3ak (32%). Xylenes and toluene were tolerated to afford 3bb–3fb in moderate to excellent (30–88%) yields. Interestingly, the reactivity trend of xylenes followed the order meta- > ortho- > para-xylene, despite Epox following the opposite trend (see Mechanistic Study).26 Toluene has a higher Epox than xylenes but reacted to give 81% of 3eb.26 Bromobenzene afforded a 30% yield of 3fb with notable r.s.m., while iodobenzene gave no reaction (60% r.s.m.). Benzene and PhCl were unsuccessful, presumably due to their notably higher Epox (only a 10% yield of 3gb was obtained, even with 72 h and an excess of PhCl). CE Substitution of Cu for Pt wire cathode and substitution of MeOH for AcOH increased the yield to 35% (Table 3). We could not improve the yield beyond this threshold. Leveraging the facile customization of TPAs, we synthesized TpBPA derivatives (Fig. 4) with electron-withdrawing groups to bolster their TPA˙+ excited state potentials (entries 1–4). Of these, tris(4′-cyano-[1,1′-biphenyl]-4-yl)amine (TCBPA) increased the yield of 3gb to 46% (Table 3). In contrast to TpBPA, the optimal TCBPA catalyst loading was 5 mol% (entries 4–8), increasing the yield of 3gb to 69% (entry 6). Notably, the reaction was still efficient with only 1.5 mol% of TCBPA (entry 8). Under optimal conditions, reactions of PhCl, benzene and even fluorobenzene were enabled, affording 3gb–3ib in modest to good (30–65%) yields (Table 4). Our conditions tolerated free carboxylic acids (3hm), prone to decarboxylation under typical PRC. Interestingly, under the same applied constant potential Ucell as in Table 2 and in contrast to the use of TpBPA, here toluene underwent benzylic oxidation instead of amination, while a pyrazole-4-carboxaldehyde underwent oxidation to give ultimately 3hm. Bromobenzene gave a lower yield of 3fb than in Table 2. These observations indicate a less oxidizing TPA˙+ excited state (*TpBPA˙+) is beneficial for certain substrates and demonstrates the value of tunability presented by this class of e-PRCats. We further probed the limits of arene SET oxidations with TCBPA˙+, by targeting 1,2-dichlorobenzene and trifluorotoluene, and were encouraged to detect products, albeit in low yields, when using TCBPA (17% of 3jb and 7%, respectively). Yields did not increase with extended time (96 h) or higher potential (Ucell = +1.8 V).
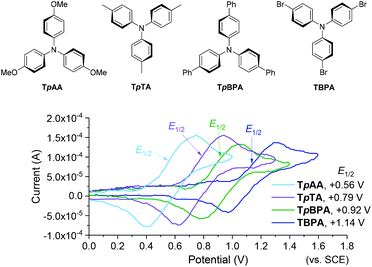 |
| | Fig. 2 First generation TPA e-PRCats (top). Cyclic voltammograms (bottom). | |
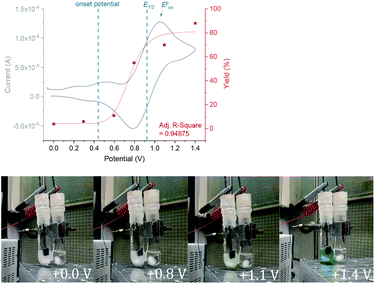 |
| | Fig. 3 Yield as a function of increasing applied Ucell(top). Development of color with increasing applied Ucell(bottom). | |
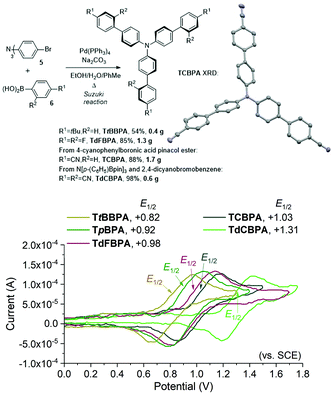 |
| | Fig. 4 Second Generation of TPA e-PRCats, and the XRD structure of TCPBA (top). Thermal ellipsoids are set at the 50% probability level. H atoms omitted for clarity. C atoms (grey), N atoms (blue). Cyclic voltammetry (bottom). | |
Table 1 Optimization of e-PRC C–H heteroamination using first generation TPAs
|
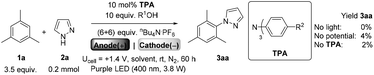
|
| Entry |
R1 |
R2 |
Anode/cathode |
Yield 3aa/r.s.m.a |
Solvent |
| r.s.m., returned starting material. Yields determined by 1H NMR using dibromomethane as an internal standard. 0.35 W 400 nm LED. Average of two replicates. 5 mol% TPA. LiClO4 as electrolyte. |
| 1 |
Ac |
OMe |
Cfoam/Pt |
21/— |
MeCN |
| 2 |
Ac |
Me |
Cfoam/Pt |
23/14 |
MeCN |
| 3 |
Ac |
Br |
Cfoam/Pt |
35/— |
MeCN |
| 4 |
Ac |
Ph |
Cfoam/Pt |
69/15 |
MeCN |
| 5b |
Ac |
Ph |
Cfoam/Pt |
18/8 |
MeCN |
| 6 |
Me |
Ph |
Cfoam/Pt |
72/— |
MeCN |
| 7 |
Me |
Ph |
Cfelt/Pt |
53/— |
MeCN |
| 8 |
Me |
Ph |
Cfoam/Fe |
70/— |
MeCN |
| 9 |
Me |
Ph |
Cfoam/Cu |
88c/— |
MeCN |
| 10 |
Me |
Phd |
Cfoam/Cu |
75/— |
MeCN |
| 11 |
Me |
Ph |
Cfoam/Cu |
60/— |
MeCNe |
| 12 |
Me |
Ph |
Cfoam/Cu |
—/— |
DMF |
| 13 |
Me |
Ph |
Cfoam/Cu |
82/— |
DCM |
Table 2 e-PRC C–H heteroaminations using TpBPA
| Unless otherwise stated, all reactions used 3.5 eq. arene; isolated yields. Yields in parenthesis determined by 1H NMR. Pt CE and AcOH were used. |
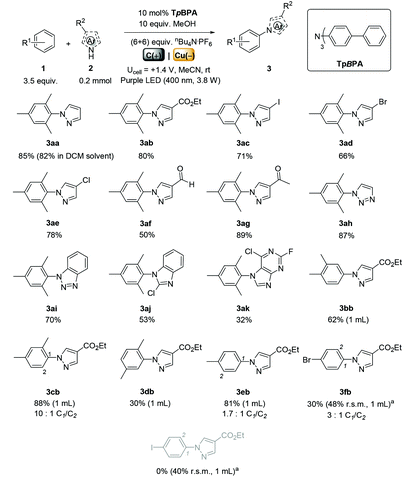
|
Table 3 e-PRC C–H heteroamination optimization with second generation TPA e-PRCats
|
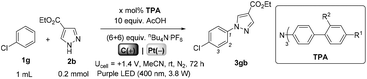
|
| Entry |
R1 |
R2 |
Yield 3gb/r.s.m.a |
Catalyst loading ‘x’ |
| r.s.m., returned starting material. Yields determined by 1H NMR using dibromomethane as an internal standard. Average of two replicates. Reaction conducted with 1g (1 mL), 2b (0.4 mmol), TCPBA (0.006 mmol), AcOH (4 mmol), nBu4N·PF6 ((1.2 + 1.2) mmol), MeCN (3.5 + 3.5 mL). |
| 1 |
H |
H |
35/33 |
10 |
| 2 |
tBu |
H |
38/22 |
10 |
| 3 |
F |
F |
36/18 |
10 |
| 4 |
CN |
H |
46b/25 |
10 |
| 5 |
CN |
H |
40/18 |
20 |
| 6 |
CN |
H |
69/— |
5 |
| 7 |
CN |
H |
52/— |
3 |
| 8c |
CN |
H |
45/25 |
1.5 |
Table 4 e-PRC C–H heteroamination using TCBPA
| N.D. not determined. Unless otherwise stated, reactions used 1 mL arene; isolated yields. Yields in parenthesis determined by 1H NMR. |
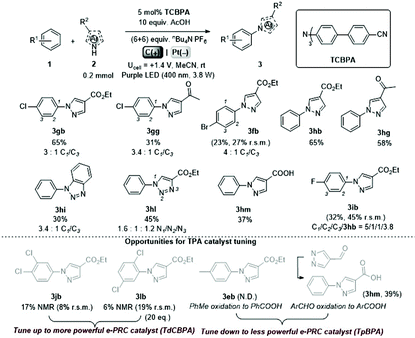
|
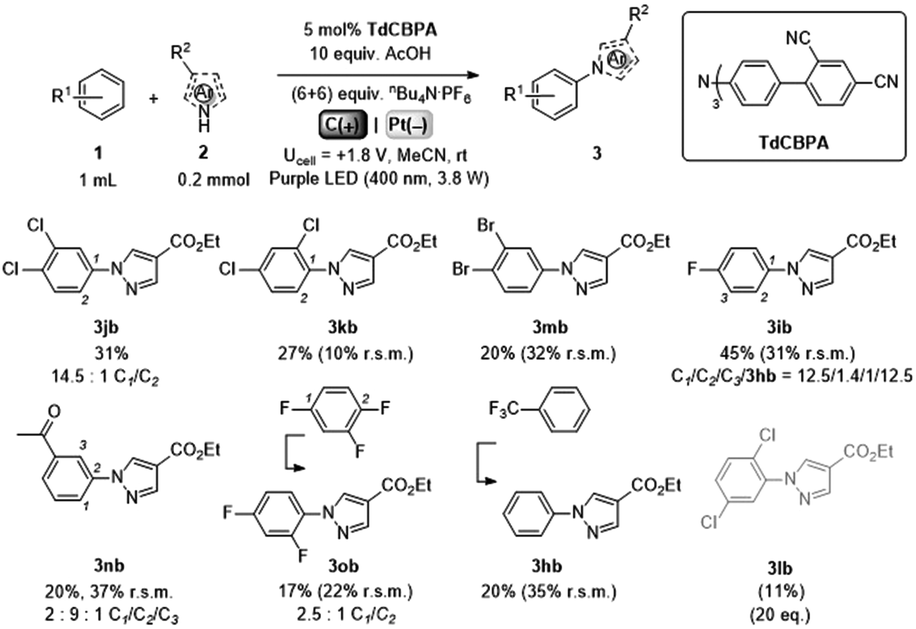
Gratifyingly, further e-PRCat tuning in the form of the even more electron-deficient tris(2′,4′-dicyano-[1,1′-biphenyl]-4-yl)amine (TdCBPA) increased the yield of 3jb to a satisfactory 31% (Table 5). Acetophenone could also be oxidized giving 3nb in 20% yield. Although polyfluorinated arenes gave 3ob in modest yields (∼20%), oxidative SNAr-type activation of such a challenging substrate has not been previously accomplished. In competition with C–H activation, due to the role of F atom as good leaving group in SNAr, net redox-neutral C–F substitution occurred to give 3ob.15c,27 Interestingly, C6H5-bearing compound 3hb was isolated from the reaction of trifluorotoluene, indicating SNAr of a CF3 group. While the role of CF3 groups in promoting SNAr reactions is well-known, no prior examples of a formal C(sp2)–CF3 to C(sp2)–N(Het-Ar) substitution exist. The potentials (Epox) of most arenes in this study have been measured by previous reports15c,16e while those of acetophenone, trifluorotoluene and 1,2,4-trifluorobenzene exceed the solvent windows of MeCN and DMF (>+3.0 V vs. SCE). This reflects the remarkable oxidizing power of TdCBPA˙+'s excited state. In contrast to a previous report16e and in line with reactivity of xylenes herein, product yields increased as a function of the substitution pattern on dichloroarenes (1,4- < 1,3- ≤ 1,2-dichlorobenzene). This was surprising, given the corresponding increasing Epox (1,4- < 1,2- ≤ 1,3-dichlorobenzene).16e
Table 5 e-PRC activation of highly electron-deficient arenes using TdCBPA
| Isolated yields. Yields in parenthesis determined by 1H NMR. |
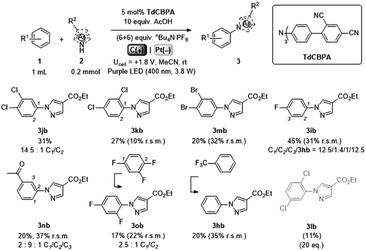
|
Mechanistic studies
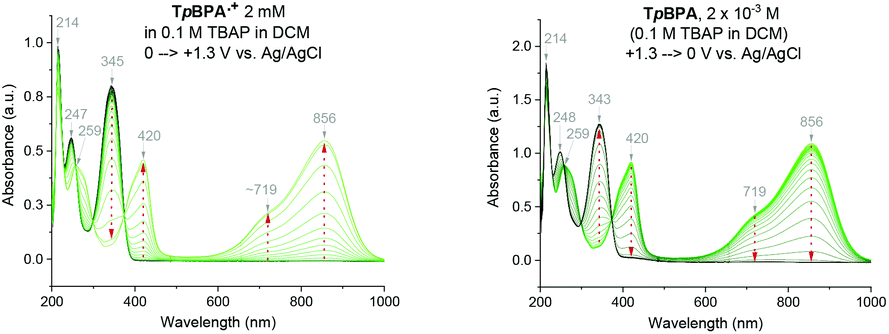
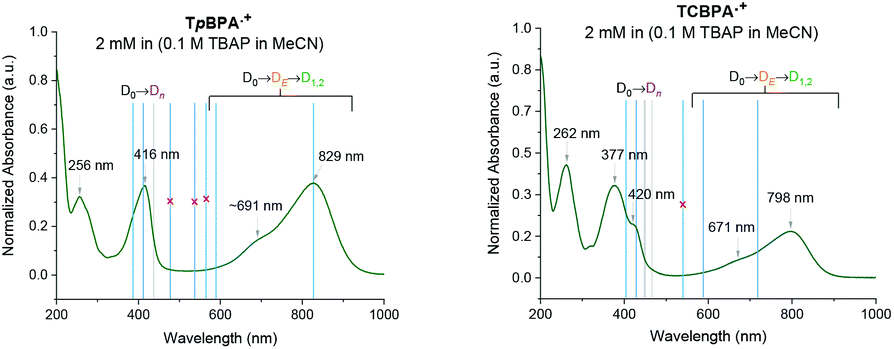
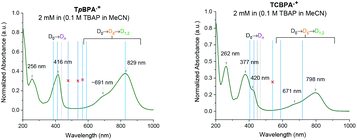
Spectroelectrochemical analysis of TpBPA (Fig. 5) TCBPA and TdCBPA (see ESI†) revealed the formation of their respective TPA˙+s by the disappearance of the TPA band (λmax = ca. 345 nm) and appearance of two broad absorption bands between 360–430 nm (λmax = ca. 420 nm) and between 600–900 nm (λmax = ca. 719, 856 nm), when the potential was increased from 0 to +1.3 V. Excellent reversibility was observed upon returning the potential to +0.0 V, indicating high stability of the TPA˙+s as e-PRCats.
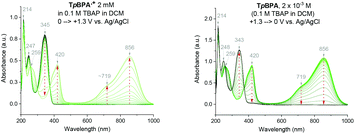 |
| | Fig. 5 Spectroelectrochemistry of TpBPA. | |
Neutral TpBPA possesses a strong absorption at λmax = 365 nm, where TpBPA˙+ absorbs poorly. Performing our optimized synthetic procedure for 3aa with 365 nm LEDs afforded only 11%, suggesting photoexcitation of neutral TpBPA is not a dominant mechanistic factor. The aforementioned control reaction without applied potential afforded only 4% of 3aa. In contrast to TpBPA, TCBPA does absorb appreciably at 395 nm. Nevertheless, in the absence of an applied potential for the optimized synthesis of 3gb, only a 12% yield of 3gb was observed. The very substantial yield differences when applied potential is present or absent confirm the pivotal role of the TPA˙+s and e-PRC as the main product-forming pathway. Inspecting the UV-visible absorption spectra of TpBPA˙+ and TCBPA˙+, we reasoned the longest wavelength bands (λmax at ca. 719 and 856 nm, respectively) must contain their D0 → D1 transitions. Based on the photochemical interpretation of Kasha's rule that prohibits photochemistry from higher order excited states, we irradiated the reactions forming 3aa and 3gb with 740 nm and 850 nm and were mystified to observe no reaction in either case (Fig. 6). That successful reaction was only observed at 395 nm implicated anti-Kasha behavior; a higher-order excited state participating in SET photooxidation. Such behavior is as surprising as it is intriguing, since (i) not only are examples of photochemistry violating Kasha's rule rarely reported in organic synthesis3b,c,28,29 but (ii) the reported lifetimes of photoexcited radical ion species are already ultrashort.30 In fact, despite a number of conPET/e-PRC reports invoking their photochemistry in super-oxidations/reductions, the question of how photoexcited radical ion species could ever participate in photochemical processes has eluded chemists. The ultrashort lifetimes of these species lie beneath timescales of diffusion-controlled bimolecular quenching.10
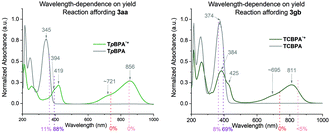 |
| | Fig. 6 Wavelength dependence on product yields. | |
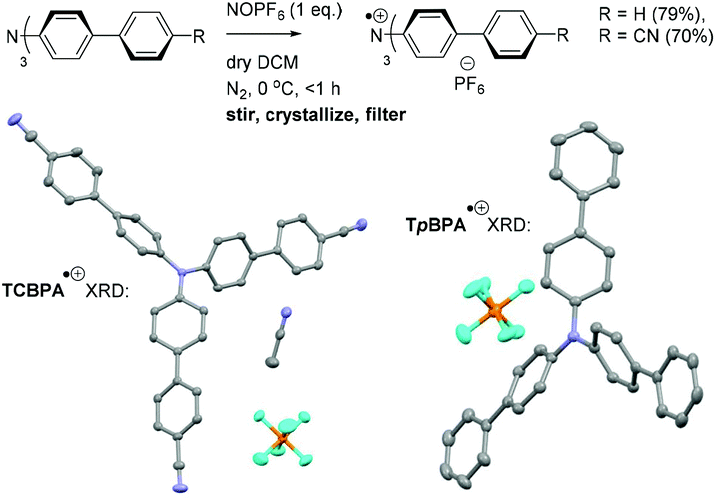
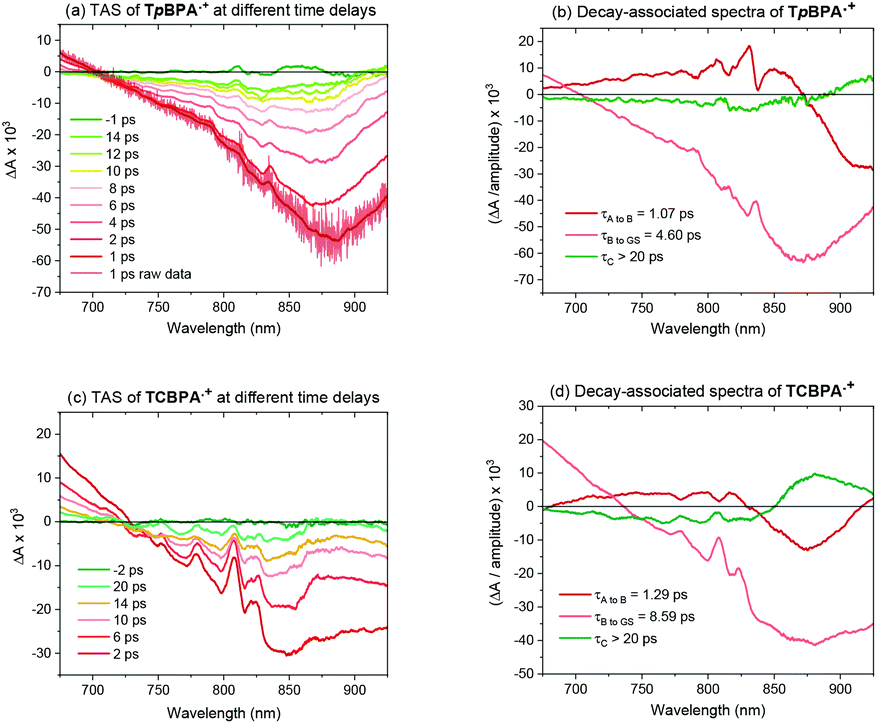
Greatly aiding our mechanistic study was the fact that TPA˙+s can be isolated as bench stable PF6 salts.23 Their crystal structures revealed a common propeller-type structure consistent with parent TPAs (Fig. 7). However, attempts to investigate quenching of photoexcited TPA˙+s were thwarted by the fact that they do not exhibit steady-state fluorescence (see ESI†). Consistent with this observation, reported lifetimes of related excited N radical cation species31 lie within the femto- to picosecond timeframe. Transient absorption spectroscopy (TAS) was therefore employed to determine the lifetimes of excited TpBPA˙+ and TCBPA˙+. Pumping with a broadband visible light source (490–900 nm)32 revealed a ground state bleach in the 600–850 nm bands (Fig. 8) and an excited state absorption between 490–570 nm, indicative of absorption by the D1 (or D2) excited state. The lifetimes of the D1 excited states of TpBPA˙+ and TCBPA˙+ were 4.6 ps and 8.6 ps, respectively, clearly ruling out diffusion-controlled bimolecular quenching. It is reasonable to assume higher order excited states possessing even shorter lifetimes.
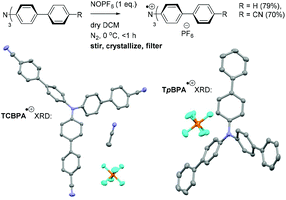 |
| | Fig. 7 Synthesis of TpBPA˙+, TCBPA˙+ and their XRD crystal structures. Thermal ellipsoids are set at the 50% probability level. H atoms omitted for clarity; C atoms (grey), F atoms (cyan), N atoms (blue), P atoms (orange). | |
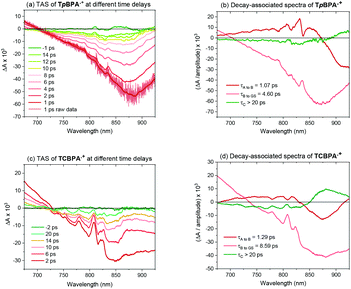 |
| | Fig. 8 Transient absorption and decay-associated spectra of TPA˙+s. | |
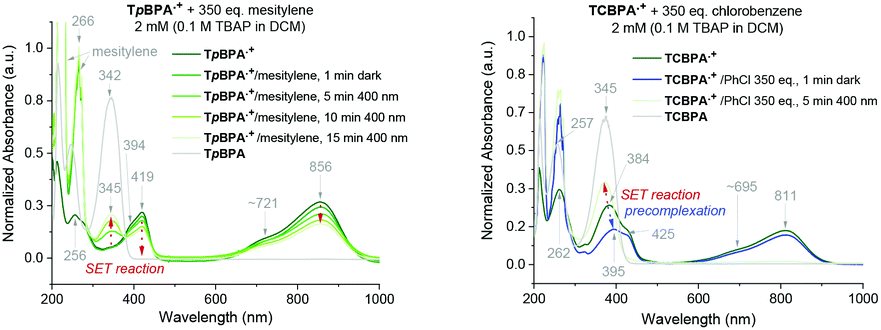
Precomplexation could rationalize productive, unimolecular SET and the aforementioned anti-Kasha behaviour. Presuming that precomplexes may possess different UV-vis absorptions than their parent TPA˙+s, we investigated quenching of the absorption of TPA˙+s in the presence of representative concentrations of arenes (Fig. 9). In the presence of mesitylene however, the spectrum of TpBPA˙+ was unchanged. Gratifyingly, irradiation with 395 nm light effected gradual conversion of TpBPA˙+ to TpBPA, corroborating the expected SET from mesitylene to the photoexcited TPA˙+. Interestingly and in contrast, the spectrum of TCBPA˙+was altered by PhCl; a small bathochromic perturbation of the peak at 384 nm to 395 nm occurred. Irradiation with 395 nm completely converted TCBPA˙+ to TCBPA after minutes.
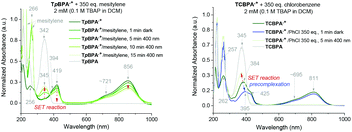 |
| | Fig. 9 UV-vis spectra of TpBPA˙+ (top) and TCBPA˙+ (bottom) in the presence of mesitylene and PhCl, respectively and after irradiation at 395 nm. | |
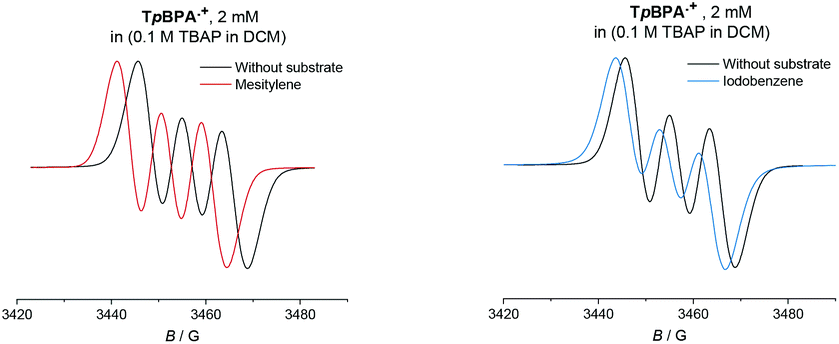
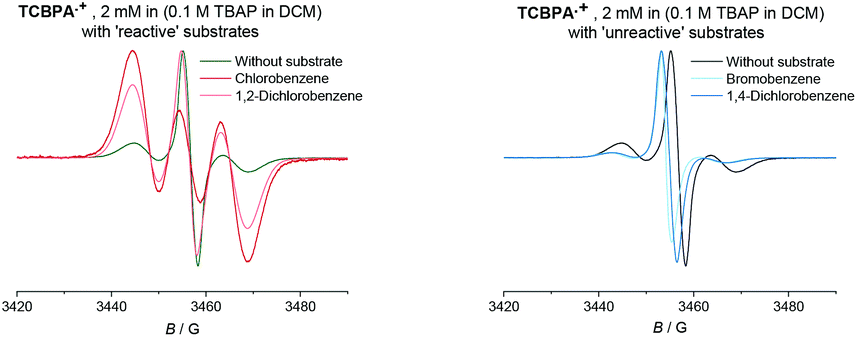
Given the paramagnetic nature of TPA˙+s, we reasoned that a change in their EPR spectra in the presence of representative arene concentrations would be more conclusive in corroborating precomplexation. The EPR signal of TpBPA˙+ showed a triplet (aN = 8.9 G). Addition of mesitylene to TpBPA˙+ caused its EPR signal to shift (ΔB = 4.5 G) to lower G values (Fig. 10), but the signal shape was largely unchanged. This indicates that the spin density of the TpBPA˙+ is largely unaffected when it undergoes precomplexation with mesitylene. A less pronounced shift in G value occurred in the presence of iodobenzene (ΔB = 2.0 G) but flattening of the triplet shoulders was observed (see ESI†). Based on these spectroscopic differences, we hypothesize that a different type of precomplex occurs in this case that is ‘unreactive’, to rationalize the inability to engage this substrate under the reaction conditions despite its more accessible redox potential (Epox = +2.07 vs. SCE) than toluene (Epox = +2.28 V vs. SCE).33 In contrast to TpBPA˙+, simulated fitting of the EPR signal from TCBPA˙+ revealed two radical species, one triplet and one superimposed singlet appearing as a large broad central feature. We presume TCBPA˙+ exists as a mixture of rotamers in solution, one propeller-type as observed in the solid state, and one in which a biphenyl unit falls into conjugation with the N radical cation, consistent with reported behaviour for similar compounds.34,35 Addition of PhCl caused a notable change in the signal shape, giving exclusively the triplet signal (Fig. 11), consistent with the change in UV-vis (Fig. 9) and indicating that the spin density of TCBPA˙+ is notably affected by precomplexation with PhCl. A similar change was detected upon addition of 1,2-dichlorobenzene, but not for its 1,4-congenor (17% yield of 3jbvs. 6% yield of 3lb under TCPBA e-PRC) and not for PhBr (22% of 3fb from TCPBA e-PRC) which instead all gave spectra clearly favoring the ‘unreactive’ complex (singlet signal).36
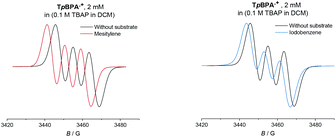 |
| | Fig. 10 EPR spectra of TpBPA˙+ with reactive mesitylene (left, 350 eq.) and unreactive iodobenzene (right, 350 eq.). | |
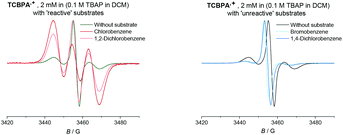 |
| | Fig. 11 EPR spectra of the TCBPA˙+ with reactive (left) and unreactive (right) substrates (350 eq.). | |
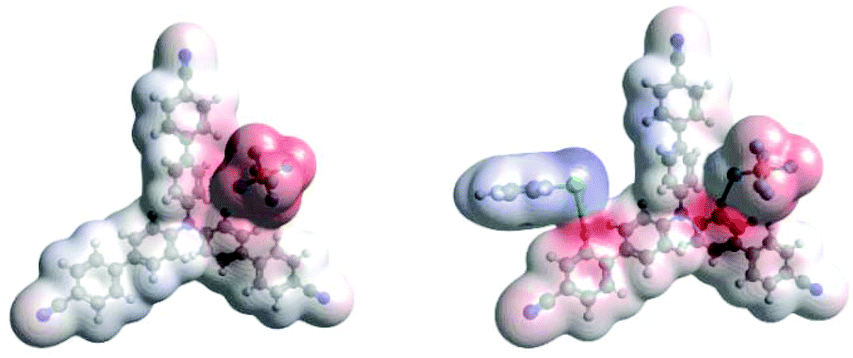
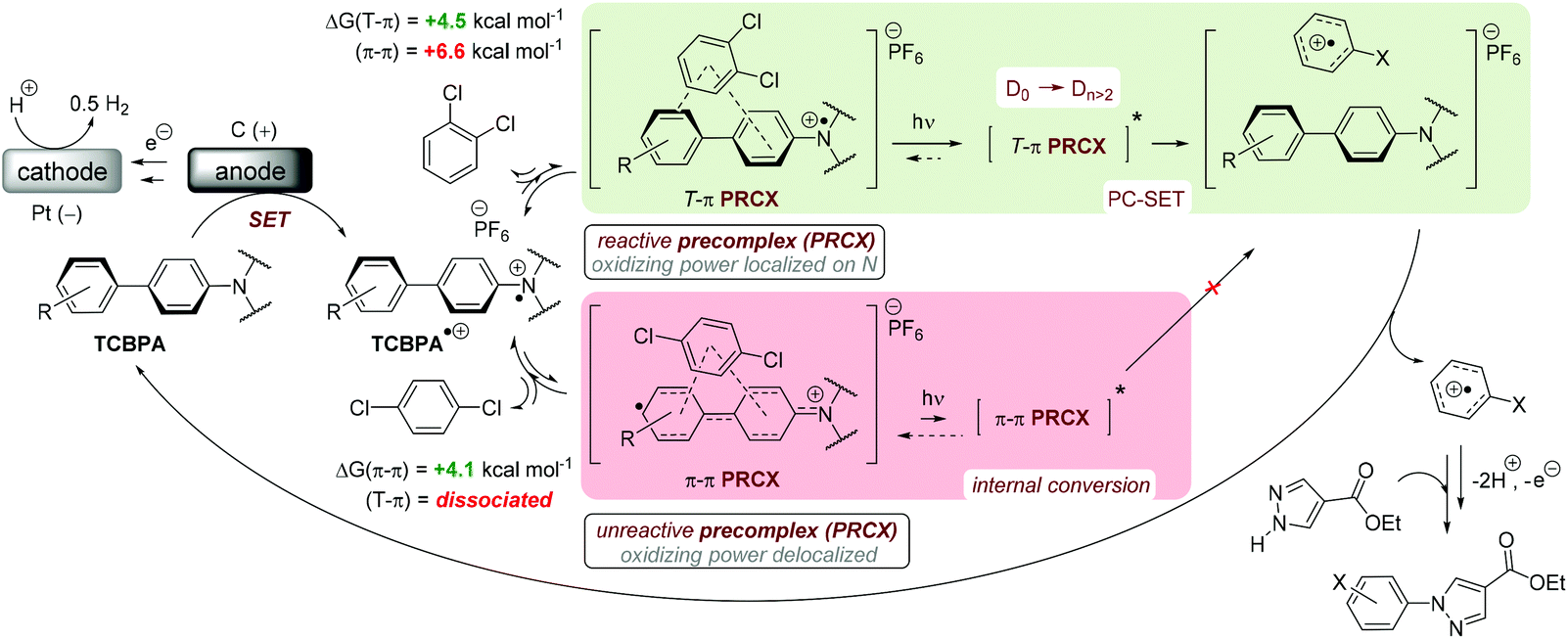
Density Functional Theory (DFT) calculations modelled precomplexation of various TPA˙+/arene combinations (Table 6). For unsymmetrical (halo)arene substrates, orientations of the complex with halogen facing both ‘in’ to the N radical cation and ‘out’ were explored (see ESI† for full investigations). We assumed that π-stacking interactions37 at the TPA˙+'s biphenyl unit could be responsible for precomplexation. Attempts to position PhCl or mesitylene substrates in a sandwich or parallel-displaced π–π stacking interaction (“π–π” complex) around the inner N-bearing ring of their respective TPA˙+s led to dissociation, whereas positioning of the substrates around the terminal aromatic ring identified local minima resembling T-type stacking interaction (“T–π” complexes). For this complex, minimal change in the spin density was detected for TpBPA˙+ + mesitylene (Fig. 12), whereas a large shift in spin density occurred for TCBPA˙+ + PhCl where the Cl atom was facing inwards (Fig. 13). This is consistent with the changes in EPR and UV-vis spectra, and so we assigned this T–π complex as the one responsible for the triplet EPR signal and successful reactivity, since. On the other hand, for less successful substrate PhBr and unsuccessful PhI (no product, 60% recovered 2b), a π–π complex was presumed to be responsible for the broad singlet EPR signal. Delocalization of the N radical cation over the biphenyl aromatic system would lead to stabilization, presumably decreasing Epox of the photoexcited TPA˙+. Free energies of precomplexation were endergonic at the levels of theory employed,38 this was confirmed to be a result of the implicit solvation model (see ESI†). It is most important to consider relative trends. Intermolecular distances for T–π stacking and π–π stacking were close to previously-reported distances for simpler complexes/dimers.39–41
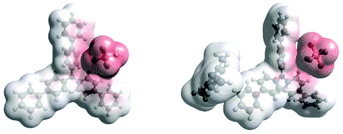 |
| | Fig. 12 DFT spin densities of TpBPA˙+ without (left) vs. with (right) Mesitylene. | |
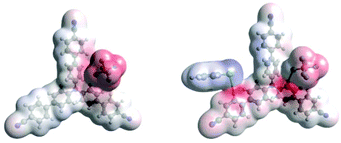 |
| | Fig. 13 DFT spin densities of TCBPA˙+ without (left) vs. with (right) PhCl, T–π complex, Cl atom facing “in”. | |
Table 6 Free energies and intermolecular distances for T–π or π–π precomplexes
| Complexa |
Complexation ΔG (kcal mol−1) |
Intermolecular distance (Å) |
| T–π |
π–π |
T–π |
π–π |
| N.D. not determined. In all cases, MeCN solvent was modelled implicitly. Pseudopotentials were applied to I and Br atoms (see ESI†). Intermolecular distances quoted are not centroid-to-centroid of the arene rings and are defined in the ESI.† Hypothesized orientation of the precomplex matching UV-vis/EPR data and reactivity patterns. uB3LYP/6-31+g(d,p). ωB97XD/6-31+g(d,p). Halogen atom(s) facing “out”. Halogen atom(s) facing “in”. Rearranged from the T–p complex. Dissociated. |
|
TpBPA˙+ + 1,3,5-TMB |
+7.2b |
+4.9c,f |
3.3–5.5b |
3.6–4.2c,f |
|
TpBPA˙+ + PhI |
+28.4b,d |
+28.3b,d |
4.7–6.5b,d |
5.2–5.5b,d |
| +28.1b,e |
+26.1b,e |
3.8–6.2b,e |
6.2–6.8b,e |
|
TCBPA˙+ + PhCl |
+4.5b,d |
+2.9c,e,f |
4.5–6.8b,d |
3.7–4.3c,e,f |
| +3.5c,d |
|
3.2–5.3c,d |
|
| +5.1b,e |
|
4.8–7.4b,e |
|
|
TCBPA˙+ + PhBr |
+30.8b,d |
29.7b,d |
3.4–6.2b,d |
4.9–5.4b,d |
| +31.4b,e |
N.D.b,e |
3.7–6.9b,e |
N.D.b,e |
|
TCBPA˙+ + 1,2PhClCl |
+4.5b,d |
+6.6b,d |
4.2–6.4b,d |
5.3–5.7b,d |
| +4.7b,e |
N.D.b,e |
4.6–6.9b,e |
N.D.b,e |
|
TpBPA˙+ + 1,3-PhClCl |
+4.9b,d |
N.D.b,d,g |
N.D.b,d |
N.D.b,d,g |
| +5.2b,e |
N.D.b,e,g |
4.9–7.4b,e |
N.D.b,e,g |
|
TCBPA˙+ + 1,4-PhClCl |
N.D.b,g |
+4.2b,d |
N.D.b,g |
5.5–5.8b,d |
Mesitylene, chlorobenzene, 1,2-dichlorobenzene and their respective TPA˙+s all had accessible ΔG values (+3.5–7.2 kcal mol−1) for the ‘reactive’ T–π complex. An accessible ΔG was also found for 1,3-dichlorobenzene (+4.9–5.2 kcal mol−1) consistent with its reactivity (20% 3 kb under the TCBPA e-PRC conditions of Table 4). Iodo- and bromobenzene as substrates gave very high ΔG values for T–π complexes and their π–π complexes were more accessible albeit still highly endergonic. Attempts to obtain a T–π complex for TCBPA˙+ with 1,4-dichlorobenzene led to dissociation, while its π–π complex was accessible.
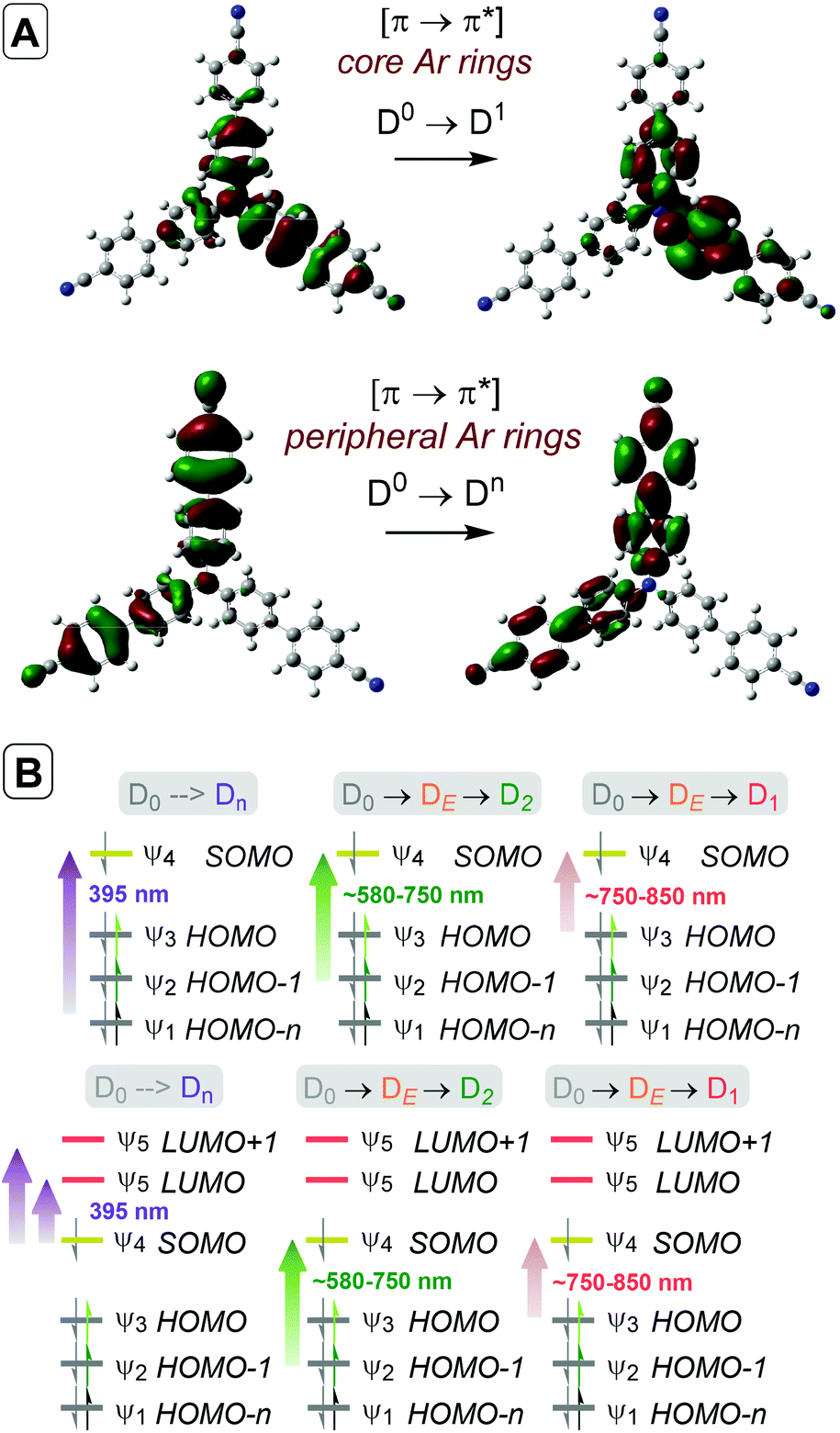
Time-dependent density functional theory (TD-DFT) calculations investigated the energies of higher order excited Dn states for TpBPA˙+, TCBPA˙+ and TdCBPA˙+, using both CAM-B3LYP/6-31G(d,p) and ωB97XD/6-31+G(d,p).31a,42 The calculated UV-visible spectra of TpBPA˙+ and TCBPA˙+ in MeCN reasonably agreed with experimental spectra (Fig. 14). The broad visible band (ca. 580–850 nm) is known to result from symmetry breaking35a of the first excited state DE in solution and in the excited state to give the D1 and D2 states, meaning TD-DFT transitions of the 580–850 nm region were less accurate than those of higher order excitations. Canonical molecular orbital (MO) calculations31a,43 revealed that excited states involved HOMO-‘n’ to SOMO transitions, typical of hole-particle excitations (Fig. 15A).16e,31a Due to the complexity in interpretation of Canonical MO transitions of higher excited state transitions, Natural Transition Orbitals (NTOs) were employed to visualize the changes in ‘hole density’.44 For all TPA˙+s studied, the first (and second) excited states (D0→DE→D1,2) involved π → π* transitions around the core aromatics, while the higher order excited states (D0→Dn) corresponding to 395 nm involved π → π* transitions around the peripheral aromatics (Fig. 15B). The concentration of ‘hole density’ at peripheral aromatics is exactly where it would be in closest proximity to substrate arenes in reactive T–π precomplexes. Ruling out participation of the first two excited states and states accessed at wavelengths <380 nm, we predict ‘effective maximum’ excited state potentials of TpBPA˙+ at +4.02 V, TCBPA˙+ at +4.19 V and TdCBPA˙+ at +4.41 V vs. SCE from the Rehm–Weller equation.45 A mechanism is proposed consistent with spectroscopic and computational studies herein (Fig. 16). Anodic SET oxidation generates the TPA˙+ from its TPA. Photoexcitation of the TPA˙+ to its D1/D2 or higher Dn states followed by bimolecular SET reductive quenching is prohibited by the TPA˙+'s picosecond lifetime. Instead, preassociation occurs to give a reactive T–π or an unreactive π–π precomplex (PRCX), depending on the sterics of the arene substrate.46,47 In the latter case, conjugative stabilization of the N radical cation decreases Epox of the *TPA˙+ below the threshold for productive unimolecular SET such that photoexcitation of the PRCX leads simply to non-radiative photophysical relaxation processes (such as internal conversion). In the former case, photoexcitation of the PRCX yields unimolecular SET reductive quenching of the *TPA˙+, regenerating the TPA and generating the arene radical cation to be intercepted by the N-heterocyclic nucleophile 2b followed by loss of protons and further SET (anodic or by the TPA˙+) to yield product 3. The prerequisite for precomplexation rationalizes the typical requirement for an excess of arene (3.5 eq. up to 1 mL, ∼40 eq. herein) to drive precomplexation equilibrium in arene amination reactions mediated by radical cations.15c,16e
 |
| | Fig. 14 Computed (CAM-B3LYP/6-31G(d,p), CPCM = MeCN, blue lines) vs. experimental UV-vis spectra of TpBPA˙+ and TCBPA˙+ in MeCN containing 0.1 M nBu4N·PF6. Negligible TD-DFT excitations (coefficient f < 0.0020) in grey. Assignments based on the optical transitions. | |
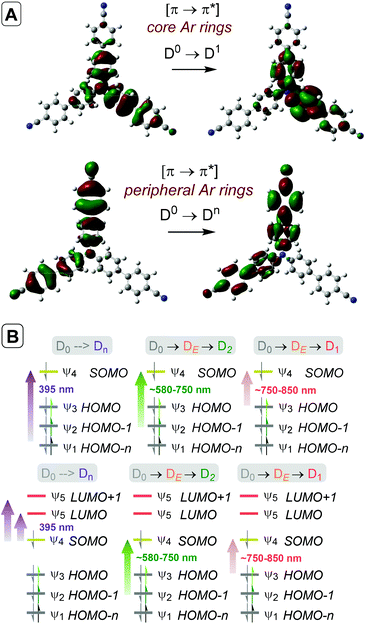 |
| | Fig. 15 (A) Natural Transition Orbitals (NTOs) depicting photoexcitation of TCBPA˙+ to D1 or Dn. (B) TD-DFT predicted orbital transitions of TpBPA˙+ (top) and TCBPA˙+ (bottom). Wavelengths are derived from optical transitions. | |
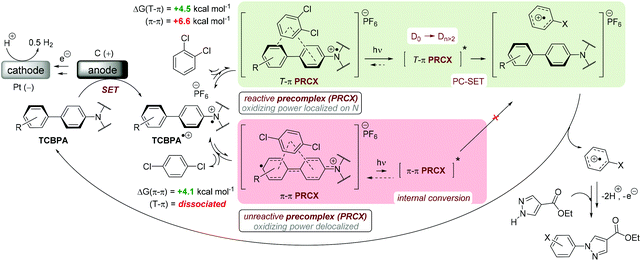 |
| | Fig. 16 Proposed e-PRC mechanism involving precomplexation modes for π-stacking and photochemistry from a higher order (than D1/D2) photoexcited state. | |
Conclusions
We report tri(p-substituted)biarylamines (TPAs) as a novel class of tunable, electroactivated photoredox catalyst. Photoexcited tris(p-substituted)biarylaminium radical cations (TPAs˙+) are demonstrated as highly potent SET oxidants. Straightforward customization of TPAs allows tuning of UV-vis absorptions, redox potentials and handles for precomplexation within the TPAs˙+. We report the first evidence of dispersion controlled (π-stacking) precomplexation in synthetic photoelectrochemistry and in the photochemistry of excited radical ions, which serves as a unique control element. Precomplexation enables remarkable photochemical phenomena: (i) circumvention of the ultrashort lifetimes of excited radical ion states for their use in photocatalysis, (ii) anti-Kasha fashion engagement of higher-order excited states in photocatalysis, (iii) overturning of thermodynamic selectivity dictated by redox potentials by virtue of steric/electronic factors involved in the precomplex. We are excited to witness future opportunities in reactivity and selectivity that dispersion precomplexation may provide to photocatalysis, in addition to recent developments in the control of photochemical outcomes through confined spaces and ordering of solvent.48–50
Author contributions
S. W. performed the optimization study, conducted all photoelectrochemical reactions, synthesized TPA˙+s and measured spectroelectrochemistry and UV-vis spectra of compounds. S. W., J. P. B. and J. Ž. designed TPAs while J. Ž. and M. D. synthesized second generation TPAs. Under the guidance of J. R., P. H. measured and analyzed all EPR spectra of TPA˙+s and precomplexes. V. B. and J. P. B. shared the DFT calculations of precomplexes, spin densities and TD-DFT calculations. D. J. S. performed XRD studies on TPA˙+s. Under the guidance of E. T. and J. H., A. K. measured TAS of TPA˙+s. E. T. measured fluorescence, EEM spectra and TCSPC of TPA˙+s. J. H. and E. T. analyzed and interpreted TAS and luminescence data. J. B. and S. W. together wrote the manuscript and analyzed all other spectroscopic data. J. B. conceived and guided the study, designed photoelectrochemical cells, conducted all CV measurements, guided the overall project and facilitated collaborations. All authors checked the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Alexander von Humboldt Foundation for funding, provided within the framework of the Sofja Kovalevskaja Award endowed by the German Federal Ministry of Education and Research. We thank Prof. John C. Walton for helpful discussions on the interpretation of EPR spectra. We thank Regina Hoheisel for assistance and training in spectroelectrochemistry. We thank Prof. Patrick Nuernberger and Dr Roger-Jan Kutta for insightful discussions on excited state photophysics. We thank Dr Peter R. Clark for insightful preliminary discussions on π-stacking template-controlled photochemistry of triarylamine radical cations. We thank Prof. Burkhard König for providing infrastructural support and Tobias Karl for preliminary advice on photoelectrochemical reaction setups. Computational work was supported by The Ministry of Education, Youth and Sports from the Large Infrastructures for Research, Experimental Development and Innovations Project “e-Infrastructure CZ – LM2018140”. This manuscript is dedicated to the memory of Dr Matthew P. John who first suggested the use of TPA˙+s as oxidizing photocatalysts.
Notes and references
- Selected reviews:
(a) K. D. Moeller, Tetrahedron, 2000, 56, 9527–9554 CrossRef;
(b) J.-I. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 7, 2265–2299 CrossRef PubMed;
(c) B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC;
(d) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef PubMed;
(e) A. Weibe, T. Geishoff, S. Mohle, E. Rodrigo, M. Zirbes and S. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef PubMed.
- Selected reviews:
(a) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef PubMed;
(b) J. M. R. Narayanam and C. R. J. Stepenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC;
(c) C. Prier, D. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef PubMed;
(d) S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561–574 RSC;
(e) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS;
(f) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS.
-
(a) I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS;
(b) I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, B. D. Duneitz, A. M. Moran and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS;
(c) J. P. Cole, D.-F. Chen, M. Kudisch, R. M. Pearson, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2020, 142, 13573–13581 CrossRef CAS.
-
(a) M. Majek, U. Faltermeier, B. Dick, R. Pérez-Ruiz and A. J. von Wangelin, Chem. – Eur. J., 2015, 21, 15496–15501 CrossRef CAS PubMed;
(b) C. G. López-Calixto, M. Liras, V. A. de la Peña O‘Shea and R. Pérez-Ruiz, Appl. Catal., B, 2018, 237, 18–23 CrossRef;
(c) C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 10, 11023–11029 RSC;
(d) B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Nature, 2019, 565, 343–346 CrossRef CAS.
- Electrode materials can be modified with chiral additives to perform enantioselective reductions or oxidations, see:
(a) M. Ghosh, V. S. Shinde and M. Rueping, Beilstein J. Org. Chem., 2019, 15, 2710–2746 CrossRef CAS PubMed;
(b) Q. Lin, L. Li and S. Luo, Chem. – Eur. J., 2019, 25, 10033–10044 CrossRef CAS PubMed.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC.
-
(a)
T. Fuchigami, M. Atobe and S. Inagi, Fundamentals and Applications of Organic Electrochemistry: Synthesis, Materials, Devices, Wiley-VCH, Weinheim, 1st edn, 2015, p. 217 Search PubMed;
(b) N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- C. Costentin and J.-M. Savéant, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11147–11152 CrossRef CAS.
-
(a) A. M. Couper, D. Pletcher and F. C. Walsh, Chem. Rev., 1990, 90, 857–865 CrossRef;
(b) D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed.
- For a dedicated review, see: J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed.
- For reviews comparing photo-, electo- and photoelectrochemical strategies, see:
(a) R. H. Verschueren and W. M. De Borggraeve, Molecules, 2019, 24, 2122 CrossRef;
(b) J. Liu, L. Lu, D. Wood and S. Lin, ACS Cent. Sci., 2020, 6, 1317–1340 CrossRef CAS.
- For highlights, see:
(a) L. Capaldo, L. L. Quadri and D. Ravelli, Angew. Chem., Int. Ed., 2019, 131, 17670–17672 CrossRef;
(b) Y. Yu, P. Guo, J.-S. Zhong, Y. Yuan and K.-Y. Ye, Org. Chem. Front., 2020, 7, 131–135 RSC.
-
(a) H. Tateno, Y. Miseki and K. Sayama, Chem. Commun., 2017, 53, 4378–4381 RSC;
(b) T. Li, T. Kasahara, J. He, K. E. Dettelbach and G. M. Sammis, Nat. Commun., 2017, 8, 390 CrossRef;
(c) H. Tateno, Y. Iguchi, Y. Miseki and K. Sayama, Angew. Chem., Int. Ed., 2018, 57, 11238–11241 CrossRef CAS PubMed;
(d) L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366–373 CrossRef CAS.
-
(a) R. Scheffold and R. Orlinski, J. Am. Chem. Soc., 1983, 105, 7200–7202 CrossRef CAS;
(b) F. Wang and S. Stahl, Angew. Chem., Int. Ed., 2019, 58, 6385–6390 CrossRef CAS.
-
(a) H. Yan, Z.-W. Hou and H.-C. Xu, Angew. Chem., 2019, 131, 4640–4643 CrossRef;
(b) X.-L. Lai, X.-M. Shu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS PubMed;
(c) H. Huang and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 59, 658–662 CrossRef;
(d) W. Zhang, K. L. Carpenter and S. Lin, Angew. Chem., Int. Ed., 2020, 59, 409–417 CrossRef CAS;
(e) Y. Qiu, A. Scheremetjew, L. H. Finger and L. Ackermann, Chem. – Eur. J., 2020, 26, 3241–3246 CrossRef CAS PubMed.
-
(a) J.-C. Moutet and G. Reverdy, Tetrahedron Lett., 1979, 20, 2389–2393 CrossRef;
(b) J.-C. Moutet and G. Reverdy, J. Chem. Soc., Chem. Commun., 1982, 654–655 RSC;
(c) S. S. Shukla and J. F. Rusling, J. Phys. Chem., 1985, 89, 3352–3358 CrossRef;
(d) B. R. Eggins and P. K. J. Robertson, J. Chem. Soc., Faraday Trans., 1994, 90, 2249–2256 RSC;
(e) H. Huang, Z. M. Strater, M. Rauch, J. Shee, T. J. Sisto, C. Nickolls and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 13318–13322 CrossRef CAS;
(f) H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS;
(g) N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS PubMed.
- R. I. Walter, J. Am. Chem. Soc., 1955, 77, 5999–6002 CrossRef CAS.
- For selected reviews, see:
(a) Z. Ning and N. Tian, Chem. Commun., 2009, 5483–5495 RSC;
(b) P. Cias, C. Slugovc and G. Gescheidt, J. Phys. Chem. A, 2011, 115, 14519–14525 CrossRef CAS PubMed.
- For selected reviews, see:
(a) E. Steckhan, Angew. Chem., Int. Ed. Engl., 1986, 25, 683–701 CrossRef;
(b) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- For selected examples, see:
(a) T. Fuchigami, M. Tetsu, T. Tajima and H. Ishii, Synlett, 2001, 1269–1271 CrossRef CAS;
(b) X. Wu, A. P. Davis and A. J. Fry, Org. Lett., 2007, 9, 5633–5636 CrossRef CAS;
(c) C.-Y. Cai and H.-C. Xu, Nat. Commun., 2018, 9, 3551 CrossRef PubMed.
-
(a) R. A. Pabon, D. J. Bellville and N. L. Bauld, J. Am. Chem. Soc., 1983, 105, 5158–5159 CrossRef CAS;
(b) D. W. Reynolds, K. T. Lorenz, H. S. Chiou, D. J. Bellville, R. A. Pabon and N. L. Bauld, J. Am. Chem. Soc., 1987, 109, 4960–4968 CrossRef CAS.
-
(a) P. S. Engel, A. K. M. M. Hoque, J. N. Scholz, H. J. Shine and K. H. Whitmire, J. Am. Chem. Soc., 1988, 110, 7880–7882 CrossRef CAS;
(b) L. Eberson and B. Olofsson, Acta Chem. Scand., 1989, 43, 698–701 CrossRef CAS;
(c) F. Ciminale, L. Lopez, A. Nacci, L. D'Accolti and F. Vitale, Eur. J. Org. Chem., 2005, 1597–1603 CrossRef CAS.
- J. P. Barham, M. P. John and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 15482–15487 CrossRef CAS.
- N. A. Romero, K. A. Margrey, E. N. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef.
- S. Das, P. Natarajan and B. König, Chem. – Eur. J., 2017, 23, 18161–18165 CrossRef PubMed.
- S. Fukuzumi, J. Yuasa, N. Satoh and T. Suenobu, J. Am. Chem. Soc., 2004, 126, 7585–7594 CrossRef CAS.
- For detailed mechanistic discussions on the preference of certain substrates to undergo C–H activation vs. C–F substitution, see: V. A. Pistritto, M. E. Schutzbach-Horton and D. A. Nicewicz, J. Am. Chem. Soc., 2020, 142, 17187–17194 CrossRef PubMed ; also see ref. 15c.
- M. Fujitsuka and T. Majima, J. Photochem. Photobiol., C, 2018, 35, 25–37 CrossRef.
- A. P. Demchenko, V. I. Tomin and P.-T. Chou, Chem. Rev., 2017, 117, 13353–13381 CrossRef CAS PubMed.
- Selected examples:
(a) D. Gosztola, M. P. Niemczyk, W. Svec, A. S. Lukas and M. R. Wasielewski, J. Phys. Chem. A, 2000, 104, 6545–6551 CrossRef CAS;
(b) C. Lu, M. Fujitsuka, A. Sugimoto and T. Majima, J. Phys. Chem. C, 2016, 120, 12734–12741 CrossRef CAS.
-
(a) J. A. Christensen, B. T. Phelan, S. Chaudhuri, A. Acharya, V. S. Batista and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 5290–5299 CrossRef CAS;
(b) J. Grilj, P. Buchgraber and E. Vauthey, J. Phys. Chem. A, 2012, 116, 7516–7522 CrossRef;
(c) J. Köhler, T. Quast, J. Buback, I. Fischer, T. Brixner, P. Nuernberger, B. Geiß, J. Mager and C. Lambert, Phys. Chem. Chem. Phys., 2012, 14, 11081–11089 RSC.
- Detailed TAS studies are ongoing to further elucidate the nature of higher order excited states and the influences of precomplexation.
- W. C. Neikam, G. M. Dimeler and M. M. Desmond, J. Electrochem. Soc., 1964, 111, 1190–1192 CrossRef.
- L. Fajarí, R. Papoular, M. Reig, E. Brillas, J. L. Jorda, O. Vallcorba, J. Rius, D. Velasco and L. Juliá, J. Org. Chem., 2014, 79, 1771–1777 CrossRef PubMed.
- Here ‘symmetry breaking’ refers to the rotation of one N-aryl unit into conjugation with the N radical cation, resulting in a symmetry change of C3 → C2 as proposed in the following references:
(a) S. Amthor, B. Noller and C. Lambert, Chem. Phys., 2005, 316, 141–152 CrossRef CAS;
(b) Y. Su, X. Wang, L. Wang, Z. Zhang, X. Wang, Y. Song and P. P. Power, Chem. Sci., 2016, 7, 6514–6518 RSC . In ref. 35bthe thermally-excited triplet state therein represents a dimeric analog of the TPA˙+s herein and, like TCBPA˙+, displays a broad central singlet-type signal.
- Drastic changes in the EPR spectra of TPA˙+s cannot be rationalized by solvent effects. Signals did not change in shape or shift when compared in DCM vs. MeCN vs. 0.1 M nBu4N·PF6 in MeCN. Arenes with similar viscosities give rise to different signals in the EPR of TCBPA˙+ and anisotropy typical of solvent effects is not observed, see ESI.†.
-
(a) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS;
(b) C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- Endergonic binding is reported for systems involving π–π dispersion interactions under a similar level of theory, see:
(a) L. M. da Costa, S. R. Stoyanov, S. Gusarov, X. Tan, M. R. Gray, J. M. Stryker, R. Tykwinski, J. W. de M. Carneiro and P. R. Seidl, Energy Fuels, 2012, 26, 2727–2735 CrossRef;
(b) A. Muraoka and M. Hayashi, Chem. Phys. Lett., 2020, 748, 137393 CrossRef . Reactions with a 20 kcal mol−1 barrier are spontaneous at room temperature, see ref. 23.
-
(a) D. E. Williams and Y. Xiao, Acta Crystallogr., 1993, 49, 1 CrossRef;
(b) M. O. Sinnokrot and C. D. Sherrill, J. Am. Chem. Soc., 2004, 126, 7690–7697 CrossRef PubMed;
(c) M. Hajji, H. Mtiraoui, N. Amiri, M. Msaddek and T. Guerfel, Int. J. Quantum Chem., 2019, 119, e26000 CrossRef.
- The arene centroid-centroid model (ref. 37a) was deemed lacking in the case of these complexes where arenes bridged both aromatics of the TPA˙+ biphenyl unit. For rationalization of distances, see ESI.†.
- Similar DFT levels were successfully used to model π-stacking interactions of similar or very large systems, see ref. 38. The long-range-corrected functional ωB97XD includes a dispersion model and provides comparable results to expensive calculations for dispersion-type interactions, see:
(a) T. Gao, T. Li, W. Li, L. Li, C. Fang, H. Li, L. Hu, Y. Lu and Z.-M. Su, J. Cheminf., 2016, 8, 24 CrossRef;
(b) K. Wang, J. Lv and J. Miao, Theor. Chem. Acc., 2015, 134, 5 Search PubMed.
- CAM-B3LYP and ωb97XD have been used for TD-DFT of similar carbazole radical cations. Agreements between calculated and experimental UV-vis spectra are reasonable. For example, see: Z. Zara, J. Iqbal, K. Ayub, M. Irfan, A. Mahmood, R. A. Khera and B. Eliasson, J. Mol. Struct., 2017, 1149, 282–298 CrossRef CAS.
- Calculated canonical MOs resembled similar reported compounds, see: B. C. Lin, C. P. Cheng and Z. P. M. Lao, J. Phys. Chem. A, 2003, 107, 5241–5251 CrossRef.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef.
- Calculated from the sum of the ground state redox potential (vs. SCE) and the transition energy (eV) closest to 395 nm predicted by computational methods (average of both methods), see ESI.†.
- Dispersive precomplexation in this way requires a precursor photocatalyst, and is fundamentally distinct from formation of EDA complexes where the chromophore is generated in situ from two colorless precursors. For a review, see:
(a) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef PubMed.
- Further refinements to our working mechanistic proposal of precomplexation, including explicit solvent molecules, the possibility of multiple substrate arene binding and the excited state dynamics of precomplexes are ongoing and will be reported in due course.
- N. Berg, S. Bergwinkl, P. Nuernberger, D. Horinek and R. M. Gshwind, Extended Hydrogen Bond Networks for Effective Proton-Coupled Electron Transfer (PCET) Reactions: The Unexpected Role of Thiophenol and Its Acidic Channel in Photocatalytic Hydroamidations, J. Am. Chem. Soc., 2021, 143, 724–735 CrossRef PubMed.
- M. Giedyk, R Narobe, S. Weiß, D. Touraud, W. Kunz and B. König, Photocatalytic activation of alkyl chlorides by assembly-promoted single electron transfer in microheterogeneous solutions, Nat. Catal., 2020, 3, 40–47 CrossRef CAS.
- J. Kaur, A. Shahin and J. P. Barham, Photocatalyst-Free, Visible-Light-Mediated C(sp3)-H Arylation of Amides via a Solvent-Caged EDA Complex, Org. Lett., 2021 DOI:10.1021/acs.orglett.1c00132.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopy, XRD, computational coordinates. CCDC 2035879, 2035880 and 2038665. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qo01609h |
| ‡ These authors contributed equally to this work. |
|
| This journal is © the Partner Organisations 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Jonas
Žurauskas‡
a,
Jonas
Žurauskas‡