
Merging radical-polar crossover/cycloisomerization processes: access to polyfunctional furans enabled by metallaphotoredox catalysis†‡
Yongjun
Liu
b,
Wenping
Luo
b,
Tingting
Xia
a,
Yewen
Fang
 *ad,
Chan
Du
a,
Xiaoping
Jin
*c,
Yan
Li
*b,
Li
Zhang
c,
Wan
Lei
b and
Hao
Wu
a
*ad,
Chan
Du
a,
Xiaoping
Jin
*c,
Yan
Li
*b,
Li
Zhang
c,
Wan
Lei
b and
Hao
Wu
a
aSchool of Materials and Chemical Engineering, Ningbo University of Technology, No. 201 Fenghua Road, Ningbo 315211, China. E-mail: fang@nbut.edu.cn
bHubei Collaborative Innovation Center for Advanced Organic Chemical Materials and Ministry-of-Education Key Laboratory for Synthesis and Application of Organic Functional Molecules, Hubei University, No. 368 Youyi Dadao, Wuhan 430062, China. E-mail: liyanok@hubu.edu.cn
cDepartment of Pharmaceutical Engineering, Zhejiang Pharmaceutical College, No. 888 Yinxian Avenue East, Ningbo 315100, China. E-mail: jinxp@mail.zjpc.net.cn
dKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, No.345 Lingling Road, Shanghai 200032, China
First published on 1st February 2021
Abstract
With the radical derived from alkyl silicates or 4-alkyl-1,4-dihydropyridines as the surrogate for the nucleophile, the cyclisation of 2-(1-alkynyl)-2-alken-1-ones proceeds smoothly via consecutive reductive radical-polar crossover and cycloisomerization processes enabled by dual photoredox–copper catalysis. Both single-electron oxidation and reduction occur between the photocatalyst and radical precursor/adduct radical, generating the enolate ion without the need for a base and an exogenous oxidant–reductant. In contrast to the reported transition-metal catalysed cyclisation with the oxonium ion as the key intermediate, the nucleophilic attack of enolate-oxygen on the copper coordinated alkyne was proposed for this dual catalysis. This new methodology for the preparation of polyfunctional furans features mild conditions, a broad substrate scope, and good functional group tolerance.
Since Murphy and co-workers’ seminal contribution to radical-polar crossover (RPC) reactions,1 many organic transformations have been successfully developed via radical and ionic species in one pot.2 Due to the easily available radical precursors and the generation of radicals in a sustainable way, the modern RPC process by means of photoredox catalysis3 has emerged as an attractive synthetic strategy. In many reported reductive RPC process-based transformations,4,5 the ionic species was generated by two consecutive reductions of the electrophile6 or reduction of the radical derived from hydrogen atom transfer (HAT) catalysis (Scheme 1a).7 Specifically, the radical doesn't engage in the new bond formation of reaction components. However, with radical addition-polar termination (RAPT) as the strategy,8 a carbanion was generated from single-electron transfer (SET) reduction of the radical derived from radical addition toward a radical acceptor (Scheme 1b). Clearly, both radical and ionic species could be involved in the bond formations. Consequently, taking advantage of the high reactivity of radicals toward alkenes and tremendous well-known terminations of the ionic species, many interesting and useful reductive RAPT reactions could be designed and realized. Intrigued by the recent advances in the field of photocatalysed redox-neutral RPC reactions, the chemistry community now has a great new desire to extend the reductive RAPT strategy to more novel synthetic approaches.9
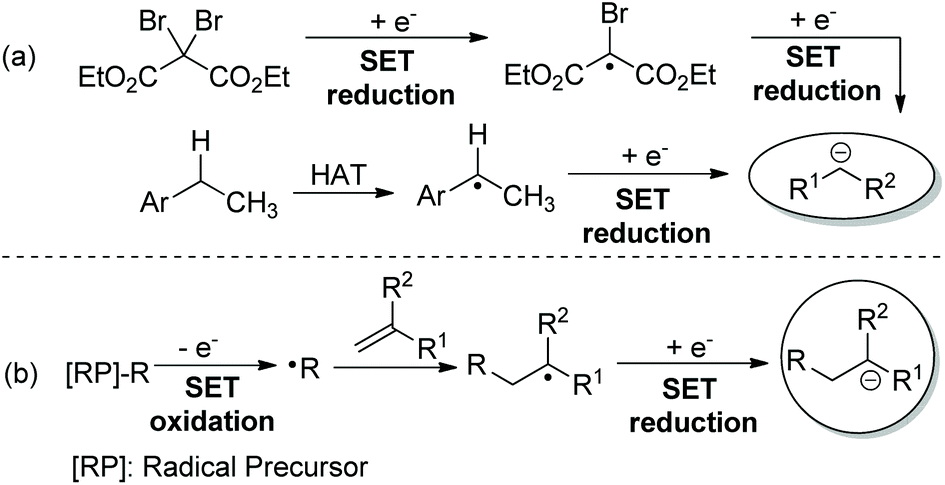 | ||
| Scheme 1 Generation of anionic species via the reductive RPC process. | ||
In the case of the base-mediated Michael reaction of α,β-unsaturated ketones, sometimes the expected 1,4-addition would be contaminated by 1,2-addition of the nucleophile to Michael acceptors (Scheme 2a). However, with RAPT as the strategy, it would be feasible to selectively and catalytically access ketone enolate via radical addition to alkene followed by SET reduction of the adduct radical (Scheme 2b). However, to the best of our knowledge, there is still no report on the synthetically useful termination of enolate-oxygen with reductive RAPT as the strategy for the formation of ketone enolate.
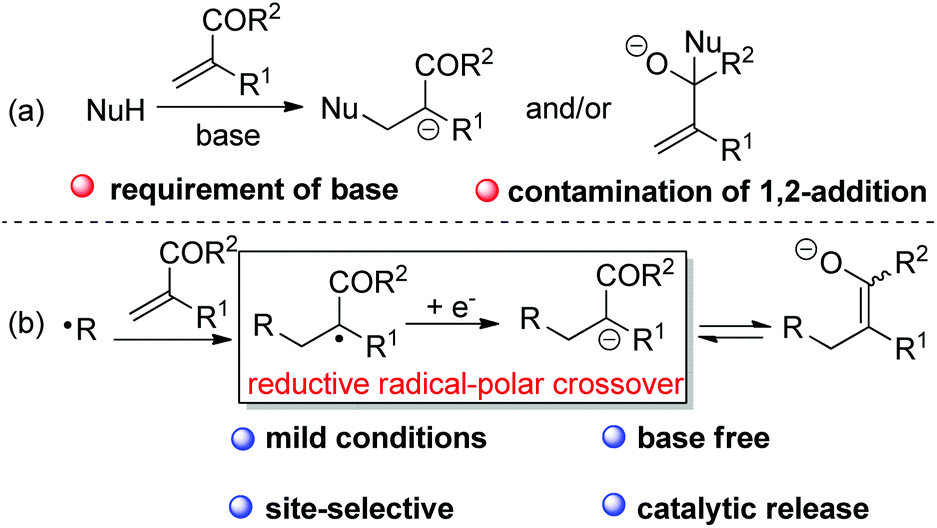 | ||
| Scheme 2 Ketone enolate formation via the Michael addition or reductive RPC process. | ||
Highly functionalized furans are key structural units in many natural products and important pharmaceuticals.10 They also serve as useful building blocks in synthetic chemistry. For these reasons, numerous efforts have been devoted to the construction of highly functionalized furans and their further transformations.11 Among the available methods to prepare furans, cyclisation of 2-(1-alkynyl)-2-alken-1-ones with various nucleophiles enabled by transition-metal catalysis is highly reliable.12 According to the investigation by Larock and other groups, the reaction was proposed to proceed via the formation of an oxonium ion that is trapped by various nucleophiles to afford the corresponding furans (Scheme 3a). However, compared to the broad substrate scope of O-nucleophiles, the carbon-based nucleophiles are only limited to electron-rich arenes and indoles. Inspired by our and other groups’ photoredox-catalysed cyclopropanations13,14 and Giese-type reactions,8,15 we envision that a carbanion could be generated via SET reduction of the radical derived from the Giese-type addition of the radical with 2-(1-alkynyl)-2-alken-1-one. After the generation of the enolate ion via the reductive RPC process, we hypothesize that a subsequent keto–enol tautomerism equilibrium would be established under base-free conditions. In the presence of a suitable catalyst, the following transition-metal-catalysed cycloisomerization of alk-3-yn-1-one would occur to furnish the expected furan (Scheme 3b). Herein, under mild and base–additive free conditions, we report that polyfunctional furans could be efficiently prepared via consecutive RPC and cycloisomerization processes enabled by dual photoredox–copper catalysis. With a radical as the surrogate for carbon-centered nucleophiles, this new method is nicely complementary to the well-developed transition-metal catalysed cyclisation of 2-(1-alkynyl)-2-alken-1-ones.
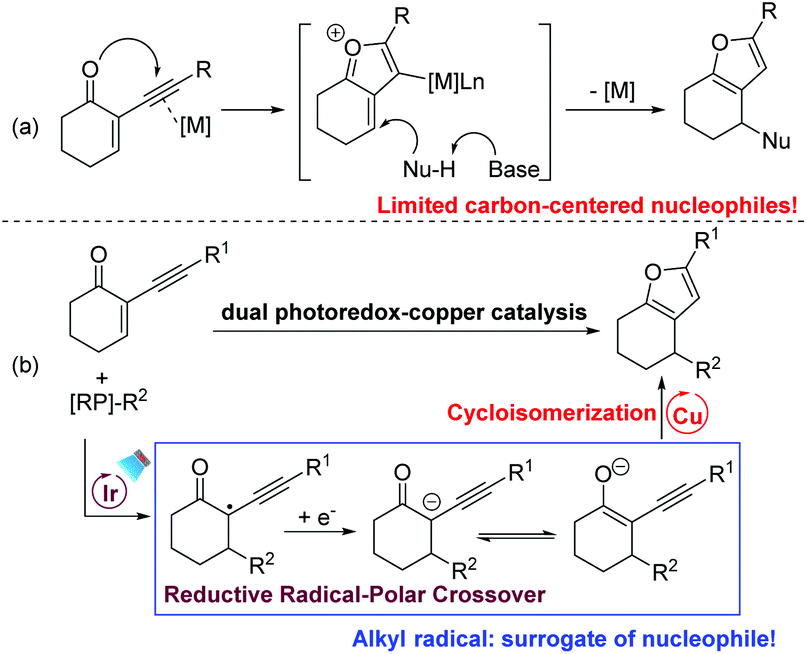 | ||
| Scheme 3 Two different pathways for accessing furans from yne-enones. | ||
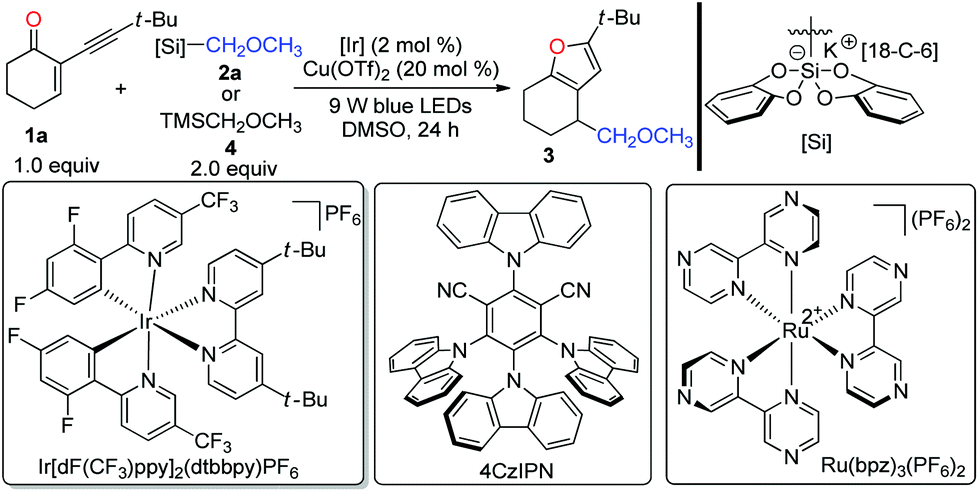
To determine whether our designed combined catalysis is feasible, we began to optimize the reaction conditions with 2-(3,3-dimethylbutynyl)-2-cyclohexen-1-one 1a and bis-catecholatosilicate 2a as the model substrates. According to the results presented in Table 1, the expected polysubstituted furan 3 could be nicely isolated in 87% yield in the presence of Ir[dF(CF3)ppy]2(dtbbpy)PF6 (2 mol%) and Cu(OTf)2 (20 mol%) in DMSO at room temperature upon irradiation with blue light-emitting diodes (LEDs) for 24 h (entry 1). The use of the commercially available (methoxymethyl)trimethylsilane 4 led to no formation of 3 (entry 2). Similar to our previous observation, DMSO is superior to DMF, DMA, and THF (entries 3–5). Using Ru(bpz)3(PF6)216 as the photocatalyst afforded the desired furan 3 in a 64% yield (entry 6). Interestingly, with the organic photocatalyst 4CzIPN,17 the reaction also worked smoothly with a 62% yield (entry 7). Further evaluation indicated that both Cu(II) (entries 1, 12 and 13) and Cu(I) catalysts (entries 8–11) were able to promote the desired transformation. Remarkably, we found that the copper catalyst plays a critical role in this reaction, since using other Lewis catalysts resulted in a severe decrease of the efficiency (see the ESI‡ for more optimizations). Control experiments confirmed that visible light, a photocatalyst, and a copper catalyst were all required for this cascade reaction (entries 14–16).
|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | Yield of 3 (%) |
| a Standard reaction conditions: A reaction mixture of 1a (0.2 mmol), 2a (0.4 mmol), [Ir] (2 mol%), and DMSO (6.0 mL) was irradiated using 9 W blue LEDs for 24 h at room temperature (cooling with a fan). [Ir]: Ir[dF(CF3)ppy]2(dtbbpy)PF6; [Ru]: Ru(bpz)3(PF6)2. b Yield of the isolated product 3. c NMR yield (500 MHz) was reported using p-nitroacetophenone as an internal standard. d The reaction was performed in the dark. | ||
| 1 | None | 87 |
| 2c | 4 instead of 2a | 0 |
| 3 | In DMF | 57 |
| 4 | In DMA | 16 |
| 5 | In THF | 47 |
| 6 | [Ru] instead of [Ir] | 64 |
| 7 | 4CzIPN instead of [Ir] | 62 |
| 8 | CuCl | 86 |
| 9 | CuI | 81 |
| 10 | Cu(CH3CN)4PF6 | 83 |
| 11 | CuOTf | 47 |
| 12 | CuBr2 | 18 |
| 13 | Cu(OAc)2 | 81 |
| 14c,d | Without light | 0 |
| 15c | No [Ir] | 0 |
| 16c | Without Cu(OTf)2 | 0 |
With the optimized reaction conditions in hand (entry 1, Table 1), we next turned our attention to investigating the general synthetic applicability as well as the potential limitations of this novel dual photoredox–copper catalysis. As highlighted in Table 2, with 2-(3,3-dimethylbutynyl)-2-cyclohexen-1-one 1a and 2-phenylethynyl-2-cyclohexene-1-one 1b as the radical acceptors, a range of alkyl bis(catecholato)silicates 2 could engage in this cascade reaction to produce various furans 5 and 6 in moderate to high yields. Taking advantage of the low oxidation potential, bis-catecholato silicon compounds (Eox between +0.34 and +0.87 V vs. SCE in DMF) are reliable radical precursors for the generation of reactive primary radicals.18,19 Pleasingly, a series of unstabilized primary radicals could be nicely engaged as shown by the formation of products 5a–d and 6a–e. Generally, the reactions between 1a and 2 provided better yields compared to the corresponding reactions of 1b with 2. Possibly, the tert-butyl group is much more beneficial for the stabilization of the organocopper intermediate derived from the cycloisomerization process. In addition to the linear alkyl radicals, the branched iso-butyl radical furnished the products 5e and 6f in 73% and 22% yields, respectively. Expectedly, the α-oxygenated radical reacted properly to produce 5f (87%) and 6g (45%). Furthermore, the secondary radical generated from bis(catecholato)cyclohexylsilicate was also converted into the corresponding products in good yields (5g and 6h).
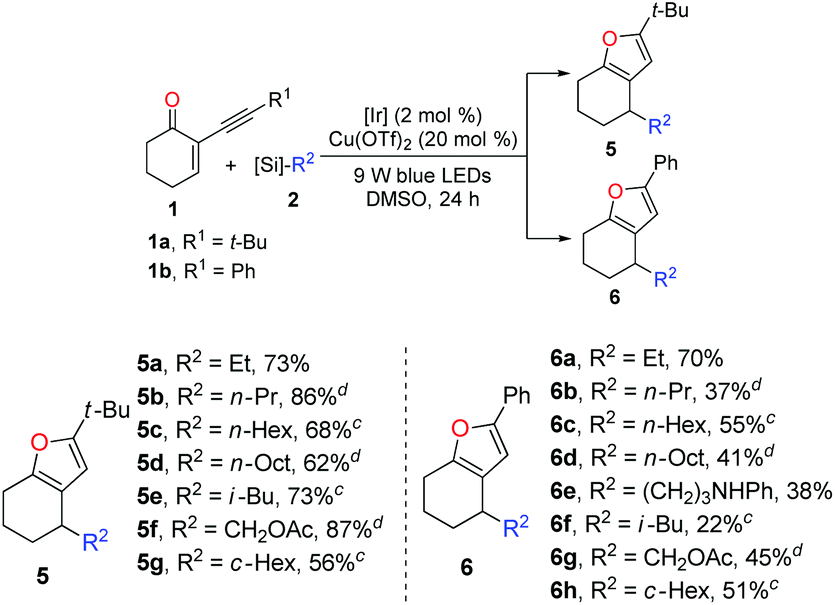
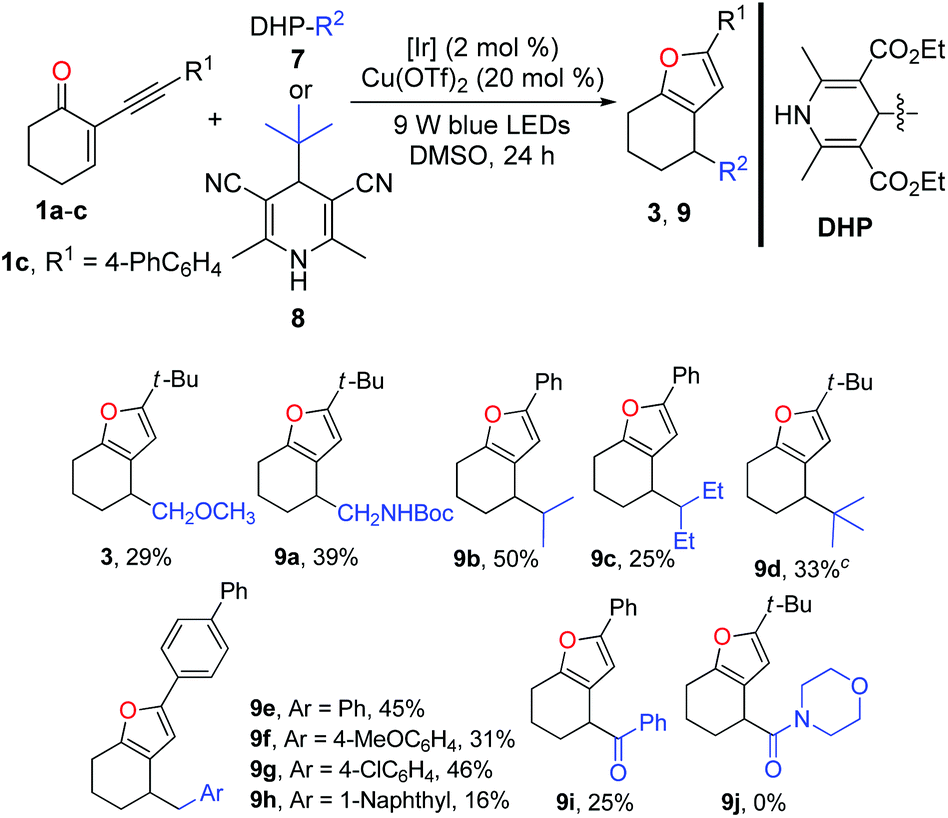
Inspired by the successful application of alkyl silicates as the alkyl radical precursors, various 4-alkyldihydropyridines (DHPs)20 were used to test the generality of the radical source for this dual catalysis. Generally, the success and efficiency of this cascade reaction were highly dependent on the radical precursors. In light of the yield, the results of the reactions with alkyl-DHPs 7 as the radical precursors are inferior to those of the reactions of 1 with alkylsilicates. As listed in Table 3, methoxymethyl and N-Boc protected aminomethyl radicals derived from the corresponding dihydropyridine precursors could engage in the cascade reaction to afford the corresponding furans 3 and 9a in 29% and 39% yields, respectively. However, the ethyl radical derived from 4-ethyldihydropyridine did not give the desired product. Interestingly, secondary alkyl-DHPs engaged in the cascade reactions smoothly, in which moderate yields of furans 9b and 9c were obtained. Pleasingly, the tert-butyl radical generated from Hantzsch nitrile 8 reacted well with 1a to give furan 9d in 33% yield. Of note, with 1a and 1b as the radical acceptors, it is difficult to purify the furans due to the contamination of the undesired homocoupling products of benzyl radicals. Fortunately, the purification problem could be circumvented using the reaction of enone 1c in combination with ArCH2-DHPs. Notably, benzylic DHPs containing not only an electron-donating group (–OMe) but also a weak electron-withdrawing group (–Cl) on the phenyl ring succeeded in delivering the target furans 9e–h in moderate to good yields. Interestingly, the benzoyl radical generated from the acyl-DHP21 also successfully underwent the cascade reaction, producing 9i in 25% yield. As a limitation, 4-carbamoyl-1,4-dihydropyridine22 failed to participate in the cascade reaction.
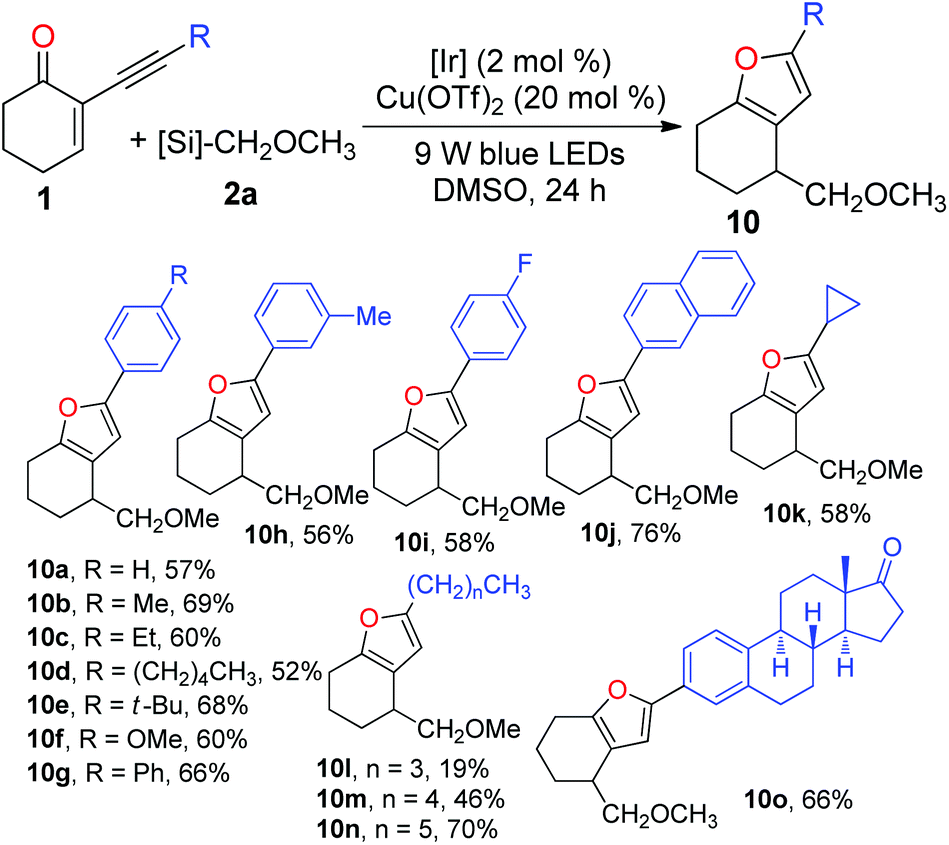
After investigating the scope of radical precursors, we next examined the influence of alkynes 1 using 2a as the radical source. Generally, both aromatic and aliphatic alkynes were suitable substrates for the tandem reaction and could be readily cyclised to give the desired furans in good to high yields (Table 4). Electronic variations in the aryl ring of the alkynes have no pronounced effect on the efficiency of the tandem addition–cyclisation processes, delivering furans 10a–j in 52–76% yields. Additionally, 5-cyclopropylfuran 10k and a set of 5-alkyl substituted furans 10l–n could be readily prepared using the current cascade reactions. Moreover, the reaction of the estrone derivative with 2a successfully generated the corresponding furan 10o in 66% yield. This demonstrates that this protocol dealing with the preparation of furans is viable in a complex molecular setting.
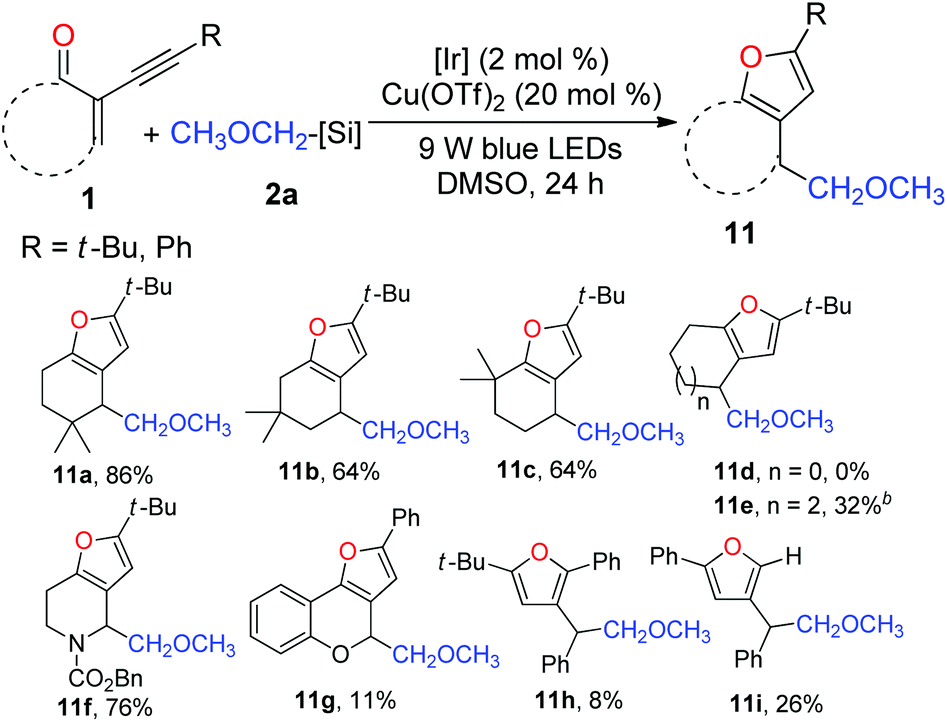
Subsequently, we focused our attention on the examination of the scope of 2-(1-alkynyl)-2-alken-1-ones 1. As presented in Table 5, varying the steric demand on the cyclohexene ring did not diminish the efficiency of the tandem reaction, affording furans 11a–c in 64–86% yields. Somewhat surprisingly, the reaction of the five-membered enynone with 2a failed to give the expected furan 11d. However, 11e could be prepared from the seven-membered enynone in a synthetically useful yield. Interestingly, the nitrogen-containing six-membered enynone was readily transformed into furan 11f in 76% yield. The benzopyran-derived enynone could likewise be used, and the resulting furan 11g was isolated in 11% yield. Delightfully, subjecting acyclic radical acceptors to the standard reaction conditions afforded furans 11h and 11i, albeit in low yields.
To have a better insight into the mechanism, we have conducted several preliminary mechanistic experiments. When 1b was subjected to the standard conditions in the presence of halomethyl silicates 12, cyclopropane 13 was obtained instead of the furan product in both reactions (Scheme 4a), thereby supporting the involvement of the photoredox-catalysed reductive RPC process.13,14a Clearly, the cyclopropanation process outcompeted the copper-catalysed cycloisomerization reaction. Moreover, in the presence of 30.0 equiv. of deuterated methanol, the furan product was obtained via dual catalysis instead of the copper-catalysed cyclisation of 1a with deuterated methanol as a nucleophile, suggesting that photoredox-catalysed radical addition to alkene is more facile than the copper-induced cyclisation of carbonyl oxygen onto the triple bond (Scheme 4b). According to the result of deuterium incorporation in 14, the protonation of the carbon–copper bond was involved.
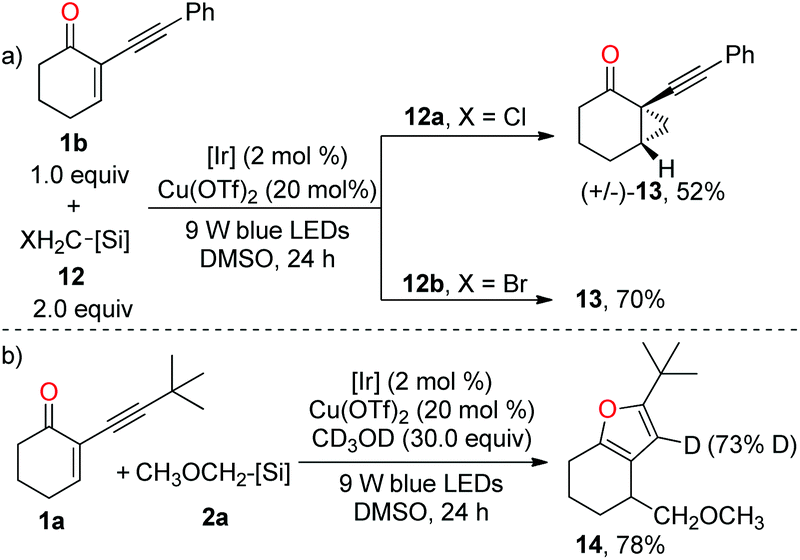 | ||
| Scheme 4 Mechanistic experiments. | ||
Based on these results, a prospective mechanism involving reductive RPC and copper-catalysed cycloisomerization processes is shown in Scheme 5. The initial SET oxidation of alkyl bis(catecholato)silicates 2 or 4-alkyl DHPs 7 by the photoexcited photocatalyst *[Ir(III)] leads to the formation of the reduced species [Ir(II)] and alkyl radical I.9a The alkyl radical reacts with the acceptor alkene 1 to provide adduct radical II. After SET reduction of II by the reduced photocatalyst, enolate ion III is formed and the photocatalyst is regenerated in its ground state, thus closing the redox-neutral photocatalytic cycle. After the generation of the enolate ion, subsequent keto–enol tautomerism equilibrium between III and IV would be established. Subsequent coordination of the alkynyl moiety of enolate to the copper catalyst induces a cyclisation of the enolate oxygen onto the triple bond. Protonation of the resulting organocopper intermediate VI produces furan 3 with simultaneous regeneration of the copper catalyst.
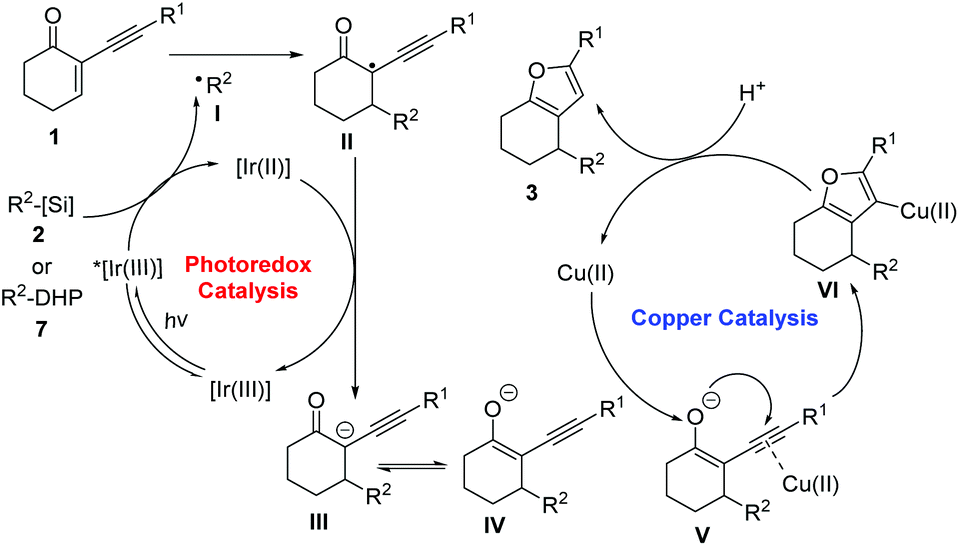 | ||
| Scheme 5 Proposed mechanistic pathway. | ||
Lastly, we briefly evaluated the further transformation of the furan product (Scheme 6). Furan 3 was treated with DDQ in toluene to yield benzofuran 15 in 42% yield, thus providing a regioselective method for the preparation of 4-substituted benzofuran.
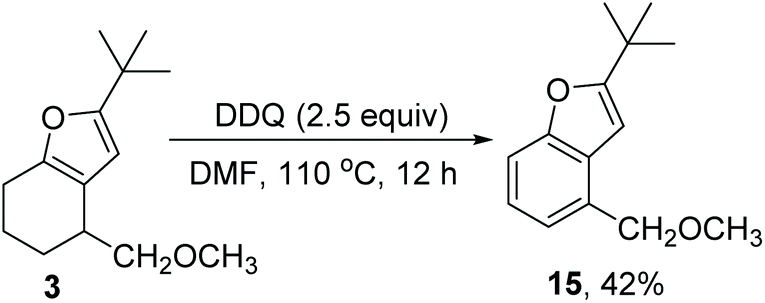 | ||
| Scheme 6 DDQ-induced dehydrogenation of the furan product. | ||
In summary, we have developed a mild visible-light-promoted photoredox/copper-catalysed cyclisation of 2-(1-alkynyl)-2-alken-1-ones with radicals for the preparation of polyfunctional furans. With alkyl silicates or 4-alkyl-1,4-dihydropyridines as the radical precursors, a range of alkyl radicals could be used as the surrogate for the carbon-centered nucleophile for this cascade reaction. In addition to a broad scope of radical precursors, a set of cyclic and acyclic 2-(1-alkynyl)-2-alken-1-ones are suitable radical acceptors for this dual catalysis. This protocol tolerates a wide range of functional groups and is viable in a complex molecular setting. This methodology also features the redox-neutral RPC process and the selective formation of ketone enolate under base-free conditions. A mechanistic pathway involving consecutive photoredox-catalysed reductive RPC and copper-catalysed cycloisomerization processes is proposed. The resulting furan can be readily transformed into 4-substituted benzofuran through DDQ-promoted dehydrogenation. We anticipate that this methodology will inspire synthetic chemists with new enthusiasm for the application of the reductive RPC process in combination with transition metal catalysis for the preparation of important and useful molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (no. LY20B020008), the Ningbo Natural Science Foundation (no. 2019A610130, 2019A610203, and 2015A610278), the Science and Technology Innovation 2025 Major Project of Ningbo (no. 2019B10112), and the National Training Programs for Innovation and Entrepreneurship for Undergraduates. We acknowledge Professor Chaozhong Li (SIOC) for generous support and valuable discussions.Notes and references
- C. Lampard, J. A. Murphy and N. Lewis, Tetrathiafulvalene as a catalyst for radical-polar crossover reactions, J. Chem. Soc., Chem. Commun., 1993, 295–297 RSC.
- For selected examples on radical-polar crossover process-based reactions, see: (a) K. P. Shing Cheung, D. Kurandina, T. Yata and V. Gevorgyan, Photoinduced Palladium-Catalyzed Carbofunctionalization of Conjugated Dienes Proceeding via Radical-Polar Crossover Scenario: 1,2-Aminoalkylation and Beyond, J. Am. Chem. Soc., 2020, 142, 9932–9937 CrossRef CAS; (b) H. Wang, Y. Gao, C. Zhou and G. Li, Visible-Light-Driven Reductive Carboarylation of Styrenes with CO2 and Aryl Halides, J. Am. Chem. Soc., 2020, 142, 8122–8129 CrossRef CAS; (c) M. Kischkewitz, K. Okamoto, C. Mück-Lichtenfeld and A. Studer, Radical-polar crossover reactions of vinylboron ate complexes, Science, 2017, 355, 936–938 CrossRef CAS; (d) C. A. Discolo, E. E. Touney and S. V. Pronin, Catalytic Asymmetric Radical-Polar Crossover Hydroalkoxylation, J. Am. Chem. Soc., 2019, 141, 17527–17533 CrossRef CAS; (e) R. Wang, M. Ma, X. Gong, X. Fan and P. J. Walsh, Reductive Cross-Coupling of Aldehydes and Imines Mediated by Visible Light Photoredox Catalysis, Org. Lett., 2019, 21, 27–31 CrossRef; (f) X. Zhang, Z. Zhang, J.-N. Song and Z. Wang, Reductive radical-initiated 1,2-C migration assisted by an azidyl group, Chem. Sci., 2020, 11, 7921–7926 RSC; (g) Q. Fu, Z.-Y. Bo, J.-H. Ye, T. Ju, H. Huang, L.-L. Liao and D.-G. Yu, Transition metal-free phosphonocarboxylation of alkenes with carbon dioxide via visible-light photoredox catalysis, Nat. Commun., 2019, 10, 3592 CrossRef; (h) W.-J. Zhou, Z.-H. Wang, L.-L. Liao, Y.-X. Jiang, K.-G. Cao, T. Ju, Y. Li, G.-M. Cao and D.-G. Yu, Reductive dearomative arylcarboxylation of indoles with CO2 via visible-light photoredox catalysis, Nat. Commun., 2020, 11, 3263 CrossRef CAS.
- For selected reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (b) J. W. Tucker and C. R. J. Stephenson, Shining Light on Photoredox Catalysis: Theory and Synthetic Applications, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS; (c) K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS; (d) Q.-Q. Zhou, Y.-Q. Zou, L.-Q. Lu and W.-J. Xiao, Visible-Light-Induced Organic Photochemical Reactions through Energy-Transfer Pathways, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS; (e) Q. Liu and L.-Z. Wu, Recent advances in visible-light-driven organic reactions, Natl. Sci. Rev., 2017, 4, 359–380 CrossRef CAS; (f) Y. Chen, L.-Q. Lu, D.-G. Yu, C.-J. Zhu and W.-J. Xiao, Visible light-driven organic photochemical synthesis in China, Sci. China: Chem., 2019, 62, 24–57 CrossRef CAS; (g) W. Zhou, Y. Jiang, L. Chen, K. Liu and D.-G. Yu, Visible-Light Photoredox and Palladium Dual Catalysis in Organic Synthesis, Chin. J. Org. Chem., 2020, 40, 3697–3713 CrossRef.
- S. Tsunoi, M. Tanaka, S. Tsunoi, I. Ryu, S. Yamasaki, N. Sonoda and M. Komatsu, Tandem annulations: a one operation construction of bicyclo[3.3.0]octan-1-ol and bicyclo[3.2.1]octan-1-ol skeletons by a three-component coupling reaction of alk-4-enyl iodides with CO and alkenes in the presence of zinc, Chem. Commun., 1997, 1889–1890 RSC.
- For reviews, see: (a) L. Pitzer, J. L. Schwarz and F. Glorius, Reductive radical-polar crossover: traditional electrophiles in modern radical reactions, Chem. Sci., 2019, 10, 8285–8291 RSC; (b) R. J. Wiles and G. A. Molander, Photoredox-Mediated Net-Neutral Radical/Polar Crossover Reactions, Isr. J. Chem., 2020, 60, 281–293 CrossRef CAS; (c) Z. Zhang, J.-H. Ye, T. Ju, L.-L. Liao, H. Huang, Y.-Y. Gui, W.-J. Zhou and D.-G. Yu, Visible-Light-Driven Catalytic Reductive Carbonylation with CO2, ACS Catal., 2020, 10, 10871–10885 CrossRef CAS.
- Y. Zhang, R. Qian, X. Zheng, Y. Zeng, J. Sun, Y. Chen, A. Ding and H. Guo, Visible light induced cyclopropanation of dibromomalonates with alkenes via double-SET by photoredox catalysis, Chem. Commun., 2015, 51, 54–57 RSC.
- (a) Q.-Y. Meng, T. E. Schirmer, A. L. Berger, K. Donabauer and B. König, Photocarboxylation of Benzylic C−H Bonds, J. Am. Chem. Soc., 2019, 141, 11393–11397 CrossRef CAS; (b) A. L. Berger, K. Donabauer and B. König, Photocatalytic carbanion generation from C−H bonds-reductant free Barbier/Grignard-type reactions, Chem. Sci., 2019, 10, 10991–10996 RSC.
- Y. Liu, W. Luo, J. Wu, Y. Fang, Y. Li, X. Jin, L. Zhang, Z. Zhang, F. Xu and C. Du, Radical addition-polar termination cascade: efficient strategy for photoredox-neutral-catalysed cyclopropanation and Giese-type reactions of alkenyl N-methylminodiacetyl boronates, Org. Chem. Front., 2020, 7, 1588–1592 RSC.
- (a) A. Cartier, E. Levernier, V. Corcé, T. Fukuyama, A.-L. Dhimane, C. Ollivier, I. Ryu and L. Fensterbank, Carbonylation of Alkyl Radicals Derived from Organosilicates through Visible-Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2019, 58, 1789–1793 CrossRef CAS; (b) K. Donabauer, K. Murugesan, U. Rozman, S. Crespi and B. König, Chem. – Eur. J., 2020, 26, 12945–12950 CrossRef CAS.
- For selected reviews, see: (a) A. T. Merritt and S. V. Ley, Clerodane diterpenoids, Nat. Prod. Rep., 1992, 9, 243–287 RSC; (b) P. A. Roethle and D. Trauner, The chemistry of marine furanocembranoids, pseudopteranes, gersolanes, and related natural products, Nat. Prod. Rep., 2008, 25, 298–317 RSC.
- For selected examples, see: (a) T. Naveen, A. Deb and D. Maiti, Copper/P(tBu)3-Mediated Regiospecific Synthesis of Fused Furans and Naphthofurans, Angew. Chem., Int. Ed., 2017, 56, 1111–1115 CrossRef CAS; (b) D. Nitsch and T. Bach, Bismuth(III) Triflate-Catalyzed Synthesis of Substituted 2-Alkenylfurans, J. Org. Chem., 2014, 79, 6372–6379 CrossRef CAS; (c) T. J. Donohoe, J. F. Bower and J. A. Basutto, Olefin cross-metathesis-based approaches to furans: procedures for the preparation of di- and trisubstituted variants, Nat. Protoc., 2010, 5, 2005–2010 CrossRef CAS; (d) J. Bao, H. Tian, P. Yang, J. Deng and J. Gui, Moldular Synthesis of Functionalized Butenolides by Oxidative Furan Fragmentation, Eur. J. Org. Chem., 2020, 339–347 CrossRef CAS; (e) T. J. Donohoe, A. Raoof, G. C. Freestone, I. D. Linney, A. Cowley and M. Helliwell, Synthesis of Enantiopure Dihydropyranones: Aldol-Based Ring Expansion of Dihydrofurans, Org. Lett., 2002, 4, 3059–3062 CrossRef CAS; (f) T. Montagnon, M. Tofi and G. Vassilikogiannakis, Using singlet oxygen to synthesize polyoxygenated natural products from furans, Acc. Chem. Res., 2008, 41, 1001–1011 CrossRef CAS.
- (a) T. Yao, X. Zhang and R. C. Larock, J. Am. Chem. Soc., 2004, 126, 11164–11165 CrossRef CAS; (b) T. Yao, X. Zhang and R. C. Larock, Synthesis of Highly Substituted Furans by the Electrophile-Induced Coupling of 2-(1-Alkynyl)-2-alken-1-ones and Nucleophiles, J. Org. Chem., 2005, 70, 7679–7685 CrossRef CAS; (c) N. T. Patil, H. Wu and Y. Yamamoto, Cu(I) Catalyst in DMF: An Efficient Catalytic System for the Synthesis of Furans from 2-(1-Alkynyl)-2-alken-1-ones, J. Org. Chem., 2005, 70, 4531–4534 CrossRef CAS; (d) C. H. Oh, V. R. Reddy, A. Kim and C. Y. Rhim, Nucleophile-assisted Pt-catalyzed cyclization of enynones: an access to synthesis of highly substituted furans, Tetrahedron Lett., 2006, 47, 5307–5310 CrossRef CAS; (e) V. Rauniyar, Z. J. Wang, H. E. Burks and F. D. Toste, Enantioselective Synthesis of Highly Substituted Furans by a Copper(II)-Catalyzed Cycloisomerization-Indole Addition Reaction, J. Am. Chem. Soc., 2011, 133, 8486–8489 CrossRef CAS; (f) F. Hu, T. Chen, J. Yan, M. Cheng, L. Huang and Y. Hu, Au-catalyzed cascade addition/cyclization/H-transfer reactions of 3-(1-alkynyl)chromones to construct 4H-Furo[3,2-c]pyrans scaffold, RSC Adv., 2012, 2, 11238–11241 RSC; (g) Z. Zhang, V. Smal, P. Retailleau, A. Voituriez, G. Frison, A. Marinetti and X. Guinchard, Tethered Counterion-Directed Catalysis: Merging the Chiral Ion-Pairing and Bifunctional Ligand Strategies in Enantioselective Gold(I) Catalysis, J. Am. Chem. Soc., 2020, 142, 3797–3805 CrossRef CAS; (h) Z. Li, J. Peng, C. He, J. Xu and H. Ren, Silver(I)-Mediated Cascade Reaction of 2-(1-Alkynyl)-2-alken-1-ones with 2-Naphthols, Org. Lett., 2020, 22, 5768–5772 CrossRef CAS.
- (a) T. Guo, L. Zhang, X. Liu, Y. Fang, X. Jin, Y. Yang, Y. Li, B. Chen and M. Ouyang, Visible-Light-Promoted Redox-Neutral Cyclopropanation Reactions of α-Substituted Vinylphosphonates and Other Michael Acceptors with Chloromethyl Silicate as Methylene Transfer Reagent, Adv. Synth. Catal., 2018, 360, 4459–4463 CrossRef CAS; (b) W. Luo, Y. Yang, Y. Fang, X. Zhang, X. Jin, G. Zhao, L. Zhang, Y. Li, W. Zhou, T. Xia and B. Chen, Photoredox-Catalyzed Cyclopropanation of 1,1-Disubstituted Alkenes via Radical-Polar Crossover Process, Adv. Synth. Catal., 2019, 361, 4215–4221 CrossRef CAS; (c) W. Luo, Y. Fang, L. Zhang, T. Xu, Y. Liu, Y. Li, X. Jin, J. Bao, X. Wu and Z. Zhang, Bromomethyl Silicate: A Robust Methylene Transfer Reagent for Radical-Polar Crossover Cyclopropanation of Alkenes, Eur. J. Org. Chem., 2020, 1778–1781 CrossRef CAS.
- (a) J. P. Phelan, S. B. Lang, J. S. Compton, C. B. Kelly, R. Dykstra, O. Gutierrez and G. A. Molander, Redox-Neutral Photocatalytic Cyclopropanation via Radical/Polar Crossover, J. Am. Chem. Soc., 2018, 140, 8037–8047 CrossRef CAS; (b) C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Synthesis of Functionalized Cyclopropanes from Carboxylic Acids by a Radical Addition-Polar Cyclization Cascade, Angew. Chem., Int. Ed., 2018, 57, 15430–15434 CrossRef CAS.
- T. Guo, L. Zhang, Y. Fang, X. Jin, Y. Li, R. Li, X. Li, W. Cen, X. Liu and Z. Tian, Visible-Light-Promoted Decarboxylative Giese Reactions of α-Aryl Ethenylphosphonates and the Application in the Synthesis of Fosmidomycin Analogue, Adv. Synth. Catal., 2018, 360, 1352–1357 CrossRef CAS.
- D. M. Schultz, J. W. Sawicki and T. P. Yoon, An improved procedure for the preparation of Ru(bpz)3(PF6)2 via a high-yielding synthesis of 2,2′-bipyrazine, Beilstein J. Org. Chem., 2015, 11, 61–65 CrossRef.
- (a) J. Luo and J. Zhang, Donor-Acceptor Fluorophores for Visible-Light-Promoted Organic Synthesis: Photoredox/Ni Dual Catalytic C(sp3)-C(sp2) Cross-Coupling, ACS Catal., 2016, 6, 873–877 CrossRef CAS; (b) C. Lévêque, L. Chenneberg, V. Corcé, C. Ollivier and L. Fensterbank, Chem. Commun., 2016, 52, 9877–9880 RSC; (c) For a review, see: J. Chen, Y. Li, L. Mei and H. Wu, Application of Photosensitizer 2,4,5,6-Tetrakis(carbazol-9-yl)-1,3-dicyanobenzene in Photo-induced Transition-Metal-Free Organic Synthesis, Chin. J. Org. Chem., 2019, 39, 3040–3050 CrossRef CAS.
- (a) Y. Nishigaichi, A. Suzuki, T. Saito and A. Takuwa, First examples of hypervalent enhancement of photoallylation by allylsilicon compounds via photoinduced electron transfer, Tetrahedron Lett., 2005, 46, 5149–5151 CrossRef CAS; (b) V. Corcé, L.-M. Chamoreau, E. Derat, J.-P. Goddard, C. Ollivier and L. Fensterbank, Silicates as Latent Alkyl Radical Precursors: Visible-Light Photocatalytic Oxidation of Hypervalent Bis-Catecholato Silicon Compounds, Angew. Chem., Int. Ed., 2015, 54, 11414–11418 CrossRef; (c) M. Joufroy, D. N. Primer and G. A. Molander, Base-Free Photoredox/Nickel Dual-Catalytic Cross-Coupling of Ammonium Alkylsilicates, J. Am. Chem. Soc., 2016, 138, 475–478 CrossRef.
- For reviews, see: (a) C. Chuit, R. J. P. Corriu, C. Reye and J. C. Young, Reactivity of penta- and hexacoordinate silicon compounds and their role as reaction intermediates, Chem. Rev., 1993, 93, 1371–1448 CrossRef CAS; (b) J.-P. Goddard, C. Ollivier and L. Fensterbank, Photoredox Catalysis for the Generation of Carbon Centered Radicals, Acc. Chem. Res., 2016, 49, 1924–1936 CrossRef CAS; (c) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Recent Advances in Alkyl Carbon-Carbon Bond Formation by Nickel/Photoredox Cross-Coupling, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS.
- For reviews, see: (a) S. Ye and J. Wu, 4-Substituted Hantzsch Esters as Alkylation Reagents in Organic Synthesis, Acta Chim. Sin., 2019, 77, 814–831 CrossRef CAS; (b) P.-Z. Wang, J.-R. Chen and W.-J. Xiao, Hantzsch esters: an emerging versatile class of reagents in photoredox catalyzed organic synthesis, Org. Biomol. Chem., 2019, 17, 6936–6951 RSC.
- G. Goti, B. Bieszczad, A. Vega-Peñaloza and P. Melchiorre, Stereocontrolled Synthesis of 1,4-Dicarbonyl Compounds by Photochemical Organocatalytic Acyl Radical Addition to Enals, Angew. Chem., Int. Ed., 2019, 58, 1213–1217 CrossRef CAS.
- N. Alandini, L. Buzzetti, G. Favi, T. Schulte, L. Candish, K. D. Collins and P. Melchiorre, Amide Synthesis by Nickel/Photoredox-Catalyzed Direct Carbamoylation of (Hetero)Aryl Bromides, Angew. Chem., Int. Ed., 2020, 59, 5248–5253 CrossRef CAS.
Footnotes |
| † In memory of Prof. Kilian Muñiz. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and compound characterisation data. See DOI: 10.1039/d0qo01472a |
| This journal is © the Partner Organisations 2021 |