
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ambruticins: tetrahydropyran ring formation and total synthesis†
James I.
Bowen
 a,
Luoyi
Wang
b,
Matthew P.
Crump
a and
Christine L.
Willis
*a
a,
Luoyi
Wang
b,
Matthew P.
Crump
a and
Christine L.
Willis
*a
aSchool of Chemistry, University of Bristol, Bristol, BS8 1TS, UK. E-mail: chris.willis@bristol.ac.uk
bInstitute of Microbiology, Chinese Academy of Sciences, NO. 1 Beichen West Road, Chaoyang District, Beijing, 100101, China
First published on 28th June 2021
Abstract
The ambruticins are a family of polyketide natural products which exhibit potent antifungal activity. Gene knockout experiments are in accord with the proposal that the tetrahydropyran ring of the ambruticins is formed via the AmbJ catalysed epoxidation of the unsaturated 3,5-dihydroxy acid, ambruticin J, followed by regioselective cyclisation to ambruticin F. Herein, a convergent approach to the total synthesis of ambruticin J is described as well as model studies involving epoxidation and cyclisations of unsaturated hydroxy esters to give tetrahydropyrans and tetrahydrofurans. The total synthesis involves preparation of three key fragments which were united via a Suzuki–Miyaura cross-coupling and Julia–Kocienski olefination to generate the required carbon framework. Global deprotection to a triol and selective oxidation of the primary alcohol gave, after hydrolysis of the lactone, ambruticin J.
Introduction
Oxygen heterocycles, including dihydropyrans (DHPs) and tetrahydropyrans (THPs), are common structural features of biologically active natural products and FDA approved small molecule drugs.1 The ambruticins (e.g. ambruticins F and S) are an interesting family of myxobacterial polyketide-derived natural products, which contain both these heterocycles (Scheme 1).2,3 Ambruticin S exhibits potent antifungal activity against a variety of fungal pathogens including Coccidioides immitis, Histoplasma capsulatum and Blastomyces dermatitidis.4–6 The antimycotic activity originates from interaction with the high-osmolarity glycerol (HOG) protein kinase signalling pathway, and no toxicity was observed in mice dosed with ambruticin S.7,8 Due to this promising antifungal activity, and fascinating structure, ambruticin S has been a target for several total syntheses.9–13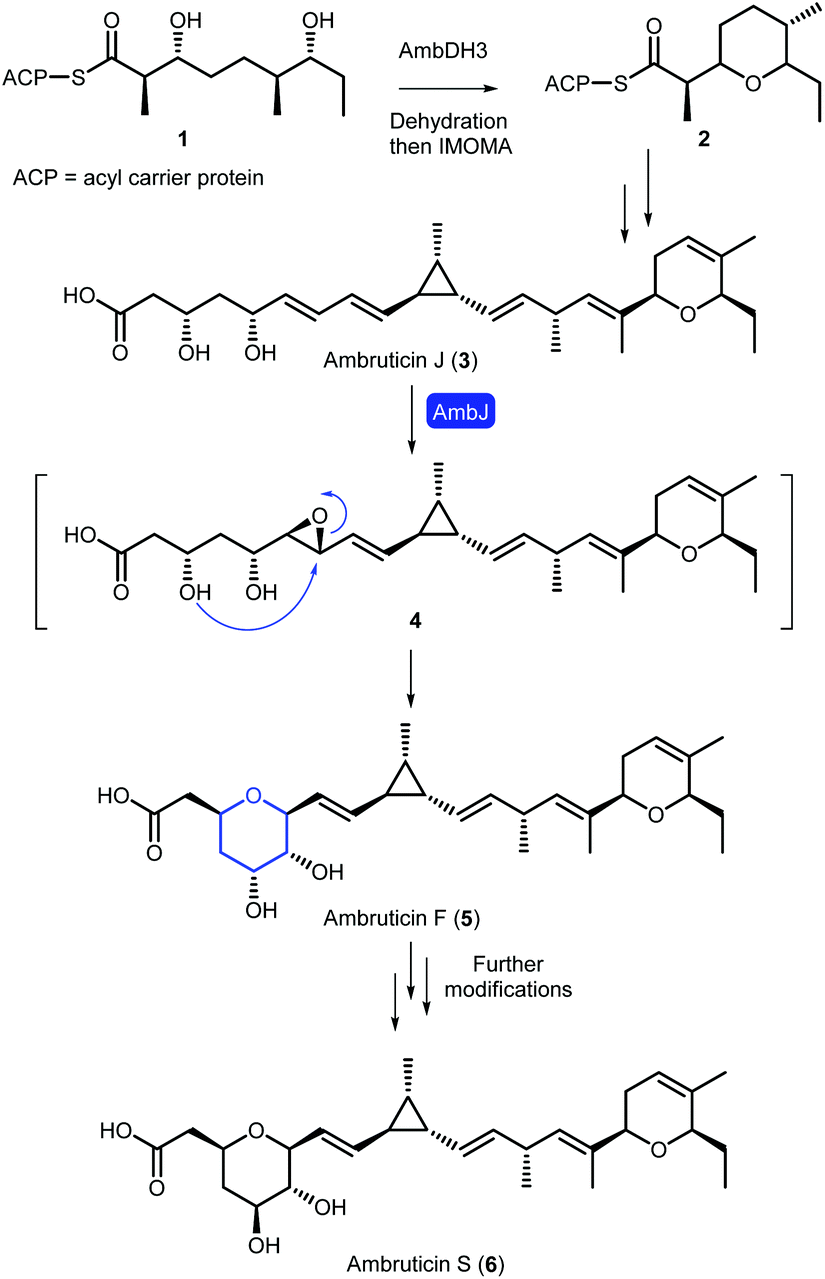 | ||
| Scheme 1 Proposed biosynthesis of ambruticin S. | ||
In 2006, Reeves proposed a biosynthetic pathway to the ambruticins, based on results from analysis of the biosynthetic gene cluster alongside gene knockout experiments.14 More recently, Hahn and co-workers have revealed that the DHP is formed via dehydration followed by an intramolecular oxa-Michael addition (IMOMA) catalysed by AmbDH3 of linear polyketide 1 to give THP 2 (Scheme 1).15 Oxidation to the DHP is proposed to occur later in the pathway. The value of AmbDH3 as a biocatalyst for use in synthesis has been reported recently.16 However, a detailed understanding of the formation of the THP in ambruticin biosynthesis remains elusive. The gene ambJ encodes for a flavin-dependent monooxygenase and cultures of the ΔambJ mutant of Sorangium cellulosum yielded syn-diol ambruticin J (3).14 Hence it was proposed that THP formation occurs through epoxidation of ambruticin J to 4, catalysed by AmbJ, followed by selective 6-endo epoxide ring opening.17
Creation of THPs via regioselective cyclisation of epoxy-alcohols has been investigated in the biosynthesis of several natural products. For example, we have shown that in mupirocin biosynthesis the Rieske oxidase MupW catalyses an initial oxidation of acyclic precursor 7 to epoxide 8, and the epoxide hydrolase MupZ is required for regioselective THP formation. Without MupZ, tetrahydrofuran 9 is exclusively formed via a 5-exo cyclisation (Scheme 2).18 Similar cyclisations have been reported for lasalocid biosynthesis in which Lsd18 catalyses alkene epoxidation and the epoxide hydrolase (Lsd19) is required for THP formation.19
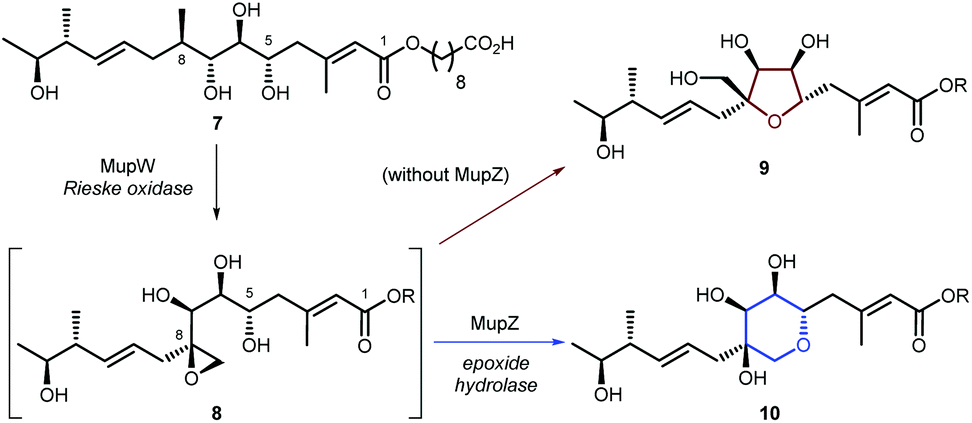 | ||
| Scheme 2 THP ring formation in mupirocin biosynthesis. | ||
In accord with these biosynthetic studies, it has been shown that in the absence of structural features which stabilise a 6-endo transition state or destabilise the 5-exo transition state, cyclisation of 4,5-epoxy alcohols predominantly form the favoured smaller tetrahydrofuran ring.20–23 No epoxide hydrolase has been identified in the ambruticin biosynthetic gene cluster. Hence, it raises the intriguing question as to whether AmbJ catalyses both formation and cyclisation of epoxide 4 or if THP formation occurs spontaneously with the adjacent alkene in 6,7-epoxy-ambruticin J (4) stabilising the 6-endo transition state leading to ambruticin F (5).24,25
With our ongoing interest in the synthesis and biosynthesis of THP containing natural products, we aim to elucidate the mechanism of THP formation in ambruticin biosynthesis.26,27 Herein, we describe our approach for the total synthesis of ambruticin J required as a substrate for AmbJ as well as cyclisation studies on model unsaturated alcohols. Whilst this manuscript was in preparation, Taylor and co-workers reported the first total synthesis of ambruticin J which uses a similar strategy to that which we have developed.28
Results and discussion
Total synthesis of ambruticin J
Our approach for the total synthesis of ambruticin J needed to be flexible to enable the preparation of analogues for later substrate specificity studies with AmbJ. Due to the reported instability of ambruticin J to conjugate elimination and cyclopropane ring opening, we opted to introduce the carboxylic acid in the final synthetic step.14 Our strategy took inspiration from a number of natural product syntheses which led to a modular approach, whereby three fragments could be united at a late stage of the synthesis (Scheme 3). The framework of ambruticin J was to be constructed through a Julia–Kocienski olefination between aldehyde 11 and the known sulfone 1211 (prepared using a phosphonamide anion olefination previously developed by Hanessian in the total synthesis of ambruticin S).13 Aldehyde 11 was to be prepared via a Suzuki–Miyaura cross-coupling of vinyl iodide 13 and vinyl boronic ester 14 with the latter being constructed using cationic cyclopropane methodology developed by Taylor.29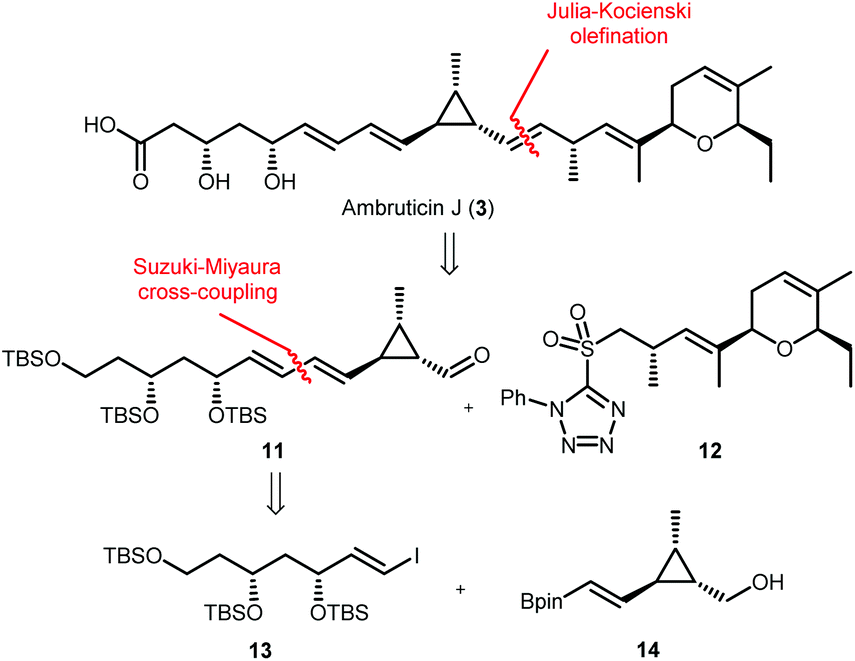 | ||
| Scheme 3 Retrosynthetic analysis of ambruticin J. | ||
To begin, vinyl iodide 13 was prepared via an aldol reaction of aldehyde 16 with acylated thiazolidinethione auxiliary 15 followed by a decarboxylative Claisen condensation with ethyl potassium malonate 18 to afford β-ketoester 19 (Scheme 4).30 Selective reduction of 19 gave syn-diol 20 in 71% yield over the three steps.31,32 Attempted reduction of ester 20 with DIBAL-H resulted in unwanted lactol formation. Hence, a sequence of silyl protection, reduction and further protection was used giving protected triol 22 in 73% over three steps. Conversion of vinyl silane 22 to vinyl iodide 13 proved problematic, with classical conditions such as NIS in MeCN providing the required vinyl iodide in <50% yield and as a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of E/Z isomers.33 Adopting Zakarian's conditions with hexafluoroisopropanol as the solvent increased the yield to 55% but with a similar E/Z ratio.34 However, excellent (E)-selectivity was achieved using a three step sequence developed by Sato.35 Initial epoxidation of vinyl silane 22 with mCPBA afforded a diastereomeric mixture of epoxides 23 in 73% yield. Regioselective epoxide ring opening with LiSnBu3 and subsequent Peterson olefination provided the trans-vinyl stannane which was readily converted to (E)-vinyl iodide 13 in 34% overall yield from simple starting materials on a multigram scale.36
:1 mixture of E/Z isomers.33 Adopting Zakarian's conditions with hexafluoroisopropanol as the solvent increased the yield to 55% but with a similar E/Z ratio.34 However, excellent (E)-selectivity was achieved using a three step sequence developed by Sato.35 Initial epoxidation of vinyl silane 22 with mCPBA afforded a diastereomeric mixture of epoxides 23 in 73% yield. Regioselective epoxide ring opening with LiSnBu3 and subsequent Peterson olefination provided the trans-vinyl stannane which was readily converted to (E)-vinyl iodide 13 in 34% overall yield from simple starting materials on a multigram scale.36
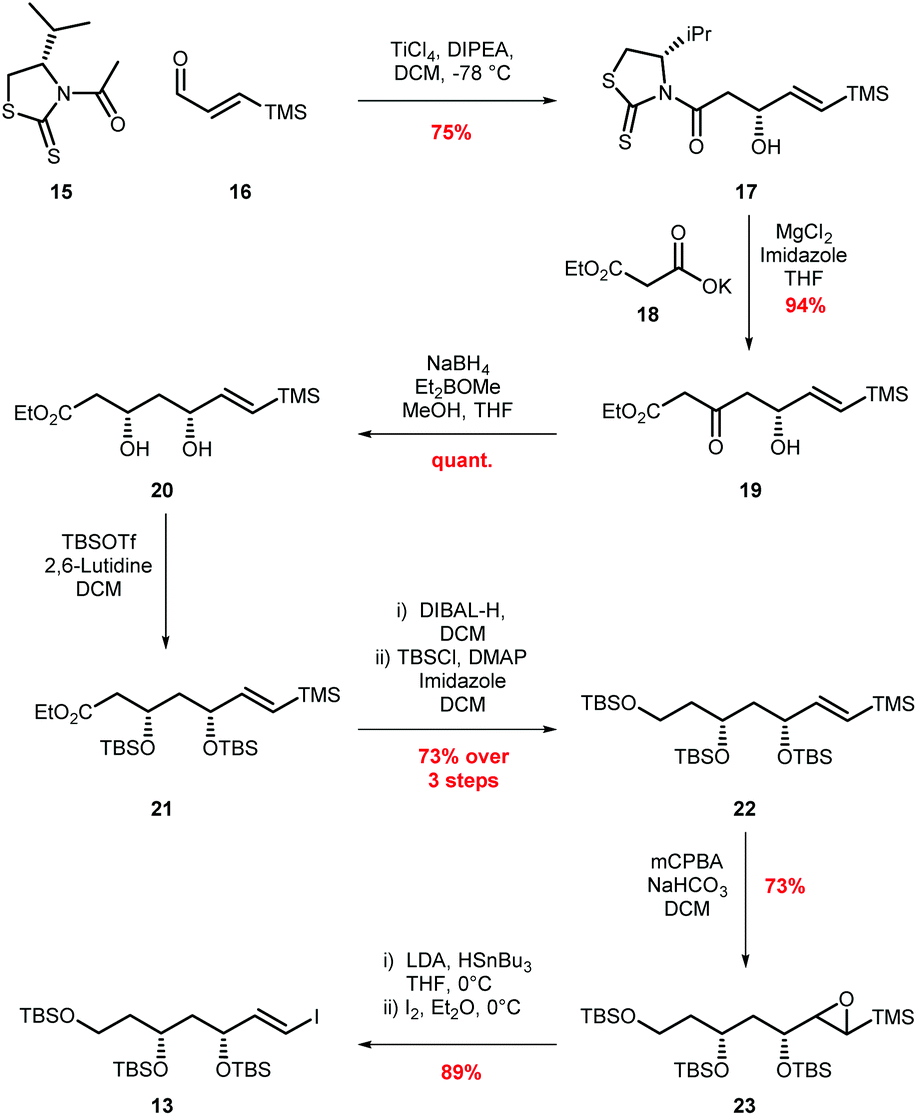 | ||
| Scheme 4 Synthesis of vinyl iodide 13. | ||
Our attention then turned to the synthesis of the vinyl boronic ester coupling partner 14 (Scheme 5) and our approach to this fragment proved to be similar to that described in the recently released paper by Taylor.28 Homoallylic alcohol 27 was prepared from boronic ester 24 in two steps utilising Hall's protocol,37 beginning with a Matteson homologation of boronic ester 24, followed by in situ Grignard addition to give allyl boronic ester 25. Lewis acid catalysed allylation with aldehyde 26 (prepared in two steps from ethylene glycol) provided the required homoallylic alcohol 27 in 75% yield over the two steps. Formation of vinyl cyclopropane 28 proceeded in 96% yield by treatment of alcohol 27 with triflic anhydride.29 The rate of addition of triflic anhydride proved key to the success of this reaction. If addition was too slow, competitive silylation of the starting material by TMSOTf, generated in the reaction, was observed. Conversely, faster rates of addition led to an exotherm and reduced the stereocontrol. Dihydroxylation of alkene 28 followed by cleavage with NaIO4 gave aldehyde 29 required for the Seyferth–Gilbert homologation with Ohira–Bestmann reagent followed by silyl deprotection to alcohol 32.38,39 Finally, hydroboration with Schwartz's reagent provided vinyl boronic ester 14 in 97% yield.40 Overall, this eight step synthetic route gave boronic ester 14 in 51% yield on a multi-gram scale.
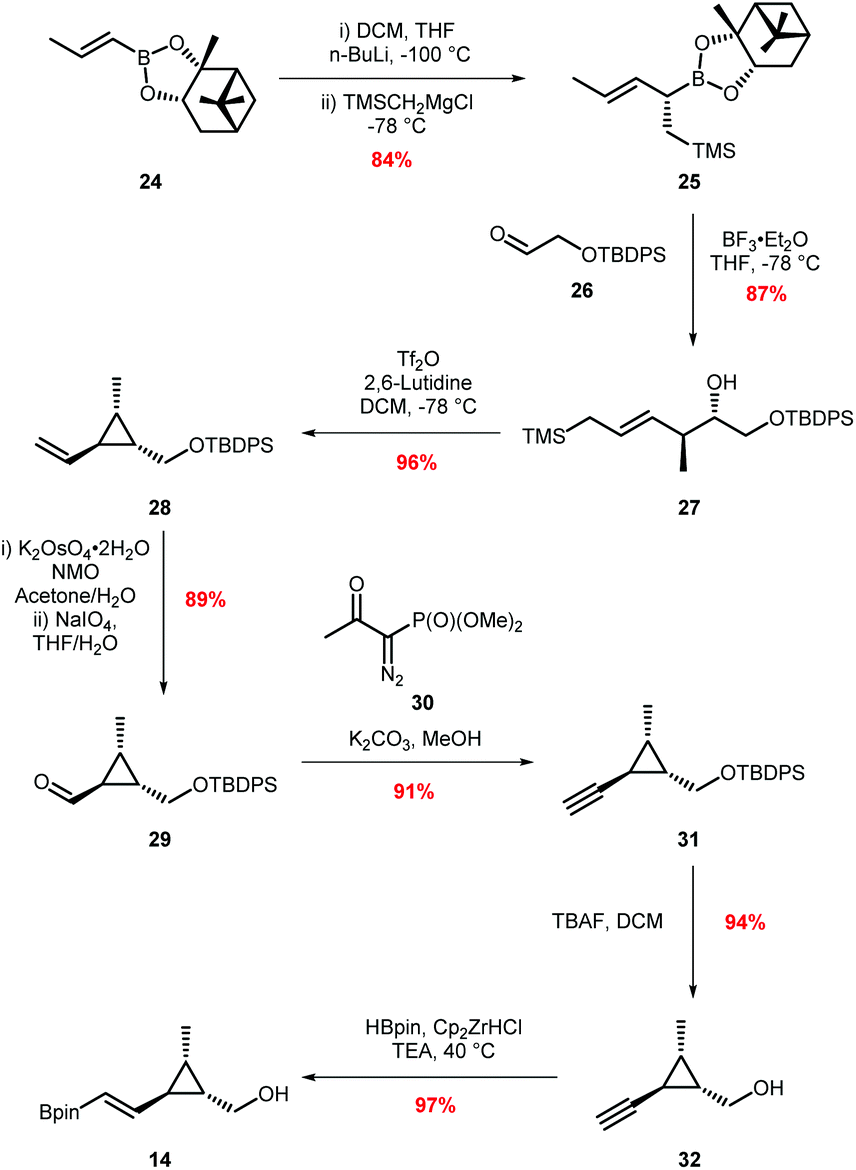 | ||
| Scheme 5 Synthesis of vinyl boronic ester 14. | ||
Sulfone 12 (Scheme 3) was prepared as previously described by Hanessian and Markó.41,42 However some of the steps were optimised and the modified procedures are reported in the ESI.† With all three fragments 12, 13 and 14 in hand the key carbon–carbon bond forming reactions were investigated (Scheme 6).
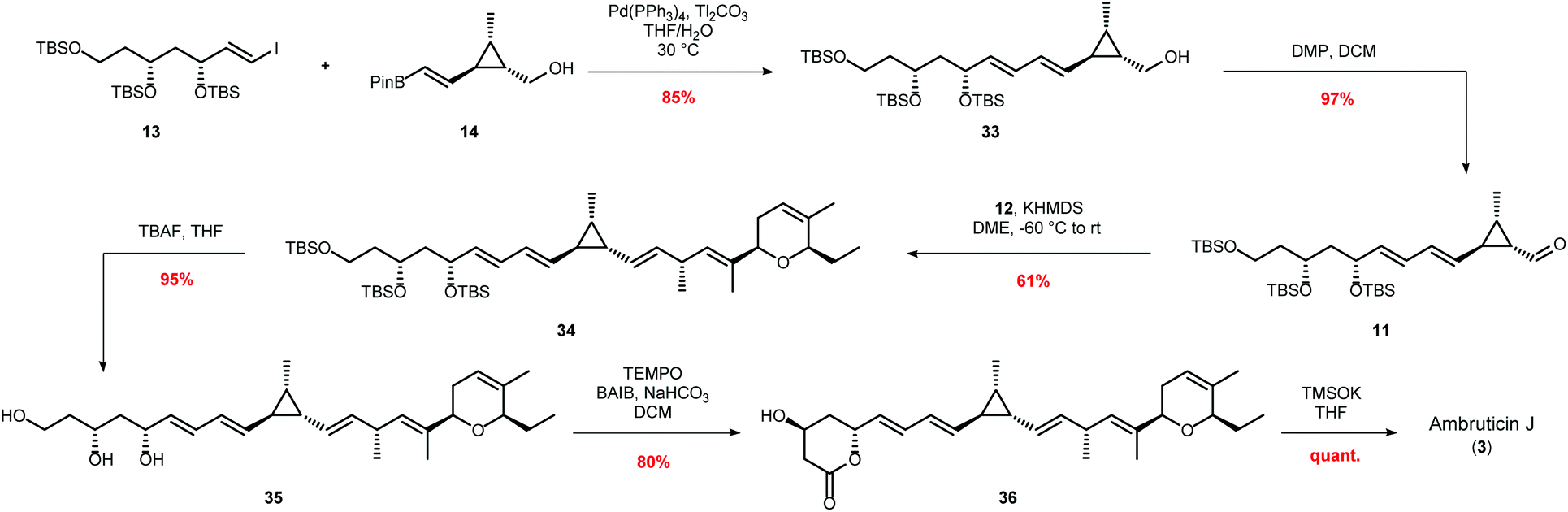 | ||
| Scheme 6 Completing the synthesis of ambruticin J via key thallium-accelerated Suzuki–Miyaura cross-coupling and Julia–Kocienski olefination. | ||
Initial attempts to couple vinyl iodide 13 and vinyl boronic ester 14via a Pd(PPh3)4 catalysed Suzuki–Miyaura cross-coupling with various potassium and sodium bases were low yielding. However, using thallium carbonate as a base gave the required E,E-diene in 85% yield.43 Oxidation of alcohol 33 with DMP to aldehyde 11 followed by a Julia–Kocienski olefination using equimolar amounts of sulfone 12 and aldehyde 11 with KHMDS in DME gave E-alkene 34 in only 44% isolated yield.44 However, using a slight excess (1.3 eq.) of sulfone 12 improved the yield to 61%. With the complete skeleton of ambruticin J now assembled, 34 was deprotected with TBAF to liberate triol 35 and selective oxidation of the primary alcohol was achieved using TEMPO-BAIB to give lactone 36.45 Careful monitoring of this oxidation was required to prevent formation of unwanted products. Finally, lactone 36 was hydrolysed under mild conditions to give ambruticin J (3). Potassium trimethylsilanolate was selected for this final transformation as Reeves et al. had reported that ambruticin J is unstable even to weak acid and base.14,46 The spectroscopic data for ambruticin J were in accord with that reported by Taylor.28
Model cyclisation studies
Factors which control cyclisation of 4,5-epoxy alcohols to selectively give either tetrahydropyrans or tetrahydrofurans are of widespread interest, stimulated in part by a fascination in the biosynthetic origin of polyether natural products such as the monensins and brevotoxins.47 In the absence of structural features which either stabilise a 6-endo transition state or destabilise a 5-exo transition state, cyclisations of 4,5-epoxy alcohols tend to favour the smaller THF rather than the THP.20–23 As no epoxide hydrolase has been identified in the ambruticin biosynthetic gene cluster, cyclisation of putative trans epoxide 4 to ambruticin F may be catalysed by either AmbJ or a further as yet unknown enzyme; alternatively an enzyme may not be required (Scheme 1). Indeed, it is possible that THP ring formation may be favoured over the more typical smaller heterocycle (THF) due to stabilisation of the 6-endo transition state by the 8,9-double bond, in accord with early studies by Nicolaou.24,25 Such cyclisations have been used to good effect in synthesis as exemplified in the recent total synthesis of the THP containing natural product meayamycin B.48 Thus, model studies were undertaken to gain an insight into the preferred mode of cyclisation of simple 4,5-epoxy alcohols.First 3,5-dihydroxy ester 40 required as a short chain analogue of ambruticin J was prepared in 4 steps by an analogous route to that used for the synthesis of vinyl iodide 13 (Scheme 7A). Asymmetric aldol condensation of unsaturated aldehyde 37 with acylated thiazolidinethione 15 followed by a decarboxylative Claisen condensation cleaved the auxiliary to give β-hydroxy-ketone 39. Stereoselective Narasaka-Prasad reduction of 39 delivered the required dihydroxy-ester in 54% overall yield.31,32 Next a selective epoxidation of diene 40 was required. Using the free alcohol to direct Sharpless epoxidation, allylic alcohol 40 was assumed to give epoxide 41, although it could not be detected even in the NMR spectrum of the crude product as cyclisation had occurred giving THP 42.49,50 It is well established that such epoxy alcohols are unstable and readily cyclise, for example as described by Oishi in studies towards the synthesis of yessotoxin using Sharpless epoxidation conditions.51 The structure of 42 was confirmed by acetylation to 43 and subsequent NMR analysis (see ESI†). No further products, including THF 44, were detected. This result is in accord with the proposal that THP formation is favoured by the presence of the adjacent π-system stabilising the 6-endo transition state in conjunction with destabilisation of the 5-exo transition state by inductive effects of the C-5 alcohol (Fig. S2†).25
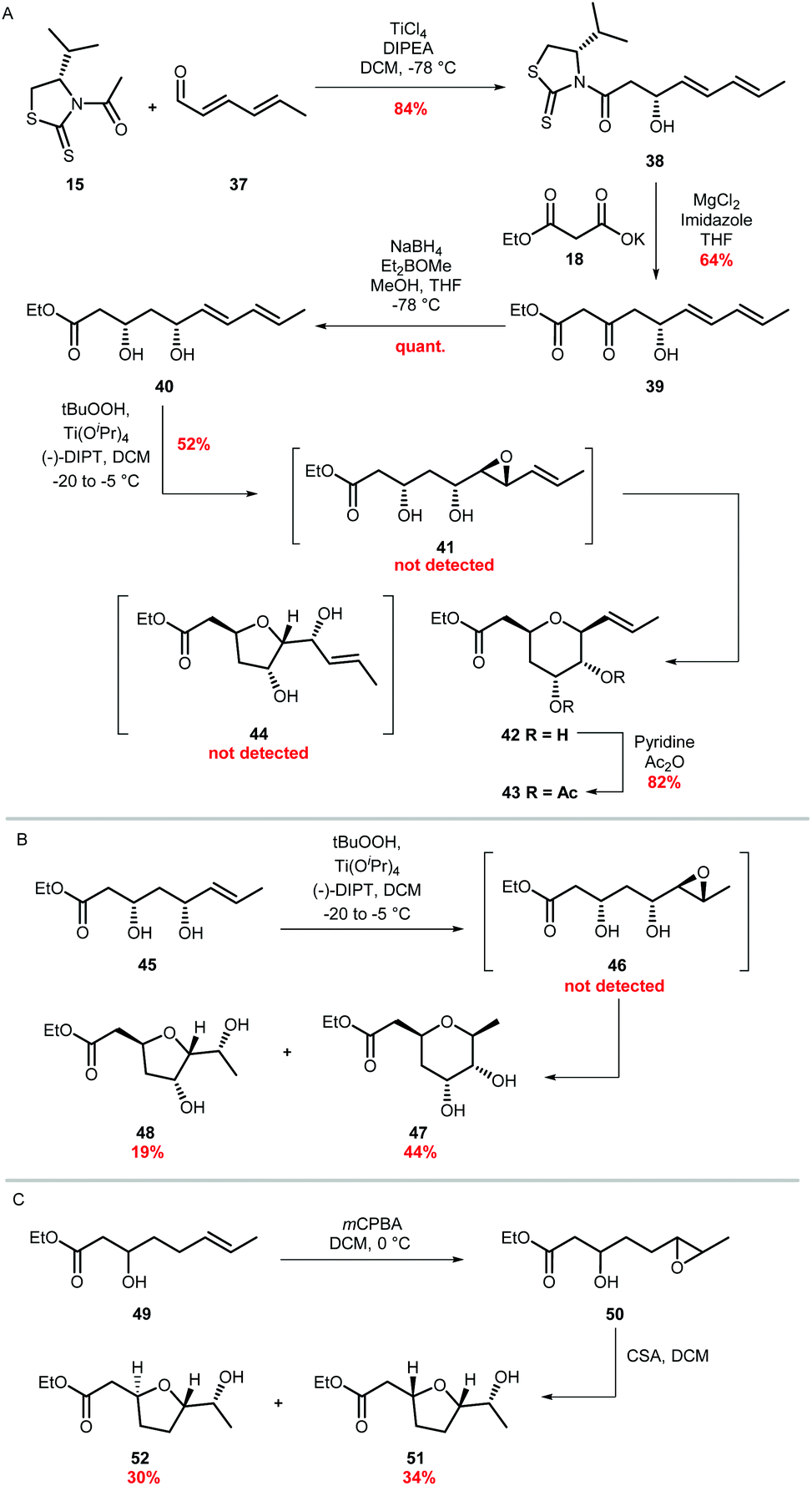 | ||
| Scheme 7 (A) Synthesis and epoxidation of diene 40 (B) Epoxidation of ester 45 and (C) Epoxidation of ester 49. | ||
To gain further support for this proposal two further model unsaturated alcohols were prepared. The first, dihydroxy ester 45 lacking the second double bond, was synthesised by an analogous route to that used for 40 but starting from crotonal (Scheme 7B). Sharpless epoxidation of 45 gave a mixture of THP and THF products 47 and 48 in 44% and 19% isolated yield respectively, with no further products detected (Scheme 7B). Finally, unsaturated alcohol 49 was prepared and treated with mCPBA to give a mixture of epoxides 50 which were not purified but on addition of CSA gave THFs 51 and 52 arising from the expected 5-exo cyclisation (Scheme 7C). These results taken together are in accord with the proposal that both the C-5 alcohol and 8,9-alkene of ambruticin J may play a role in directing intramolecular epoxide ring opening to form ambruticin F. Hence, if a non-enzymatic cyclisation of epoxide 4 occurs then, based on these simple model studies, formation of the THP would be reasonable.
Conclusions
AmbJ is a flavin-dependent monooxygenase proposed to catalyse oxidation of ambruticin J to epoxide 4 (Scheme 1).14 However, as no epoxide hydrolase has been identified in the ambruticin biosynthetic gene cluster, it is not clear if AmbJ also catalyses a 6-endo epoxide ring opening to give ambruticin F or if THP formation occurs spontaneously. The results of model studies described herein on the chemical epoxidation and cyclisation of unsaturated alcohols (40, 45 and 49) are in accord with the proposal that both the C-5 alcohol and 8,9-alkene of ambruticin J may play a role in directing 6-endo intramolecular ring opening to form ambruticin F. However, studies with AmbJ are essential to fully understand the biosynthesis of ambruticin F. To facilitate these biochemical investigations, we have completed a convergent total synthesis of ambruticin J. The approach involved construction of the carbon framework via the synthesis of three fragments 12, 13 and 14 which were united via a thallium-accelerated Suzuki–Miyaura cross-coupling and Julia–Kocienski olefination. A global deprotection of 34, selective oxidation to lactone 36 and finally hydrolysis under mild conditions gave ambruticin J. This modular route may be readily adapted to generate ambruticin J analogues to investigate the substrate specificity of AmbJ and these studies are currently underway in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Bristol Chemical Synthesis Centre for Doctoral Training, funded by EPSRC (EP/G036764/1), and the University of Bristol, for a PhD studentship to JIB.Notes and references
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed.
- S. M. Ringel, R. C. Greenough, S. Roemer, D. Connor, A. L. Gutt, B. Blair, G. Kanter and M. von Strandtmann, J. Antibiot., 1977, 30, 371 CrossRef CAS PubMed.
- V. Michelet and J.-P. Genet, Curr. Org. Chem., 2005, 9, 405 CrossRef CAS.
- G. Höfle, H. Steinmetz, K. Gerth and H. Reichenbach, Liebigs Ann. Chem., 1991, 1991, 941 CrossRef.
- F. Hahn and F. M. Guth, Nat. Prod. Rep., 2020, 37, 1300 RSC.
- S. Shadomy, D. M. Dixon, A. Espinel-Ingroff, G. E. Wagner, H. P. Yu and H. J. Shadomy, Antimicrob. Agents Chemother., 1978, 14, 99 CrossRef CAS PubMed.
- H. B. Levine, S. M. Ringel and J. M. Cobb, Chest, 1978, 73, 202 CrossRef CAS PubMed.
- P. Knauth and H. Reichenbach, J. Antibiot., 2000, 53, 1182 CrossRef CAS PubMed.
- A. S. Kende, Y. Fujii and J. S. Mendoza, J. Am. Chem. Soc., 1990, 112, 9645 CrossRef CAS.
- T. A. Kirkland, J. Colucci, L. S. Geraci, M. A. Marx, M. Schneider, D. E. Kaelin and S. F. Martin, J. Am. Chem. Soc., 2001, 123, 12432 CrossRef CAS PubMed.
- P. Liu and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 10772 CrossRef CAS PubMed.
- E. Lee, S. J. Choi, H. Kim, H. O. Han, Y. K. Kim, S. J. Min, S. H. Son, S. M. Lim and W. S. Jang, Angew. Chem., Int. Ed., 2002, 41, 176 CrossRef CAS PubMed.
- S. Hanessian, T. Focken, X. Mi, R. Oza, B. Chen, D. Ritson and R. Beaudegnies, J. Org. Chem., 2010, 75, 5601 CrossRef CAS PubMed.
- B. Julien, Z.-Q. Tian, R. Reid and C. D. Reeves, Chem. Biol., 2006, 13, 1277 CrossRef CAS PubMed.
- G. Berkhan and F. Hahn, Angew. Chem., Int. Ed., 2014, 53, 14240 CrossRef CAS PubMed.
- T. Hollmann, G. Berkhan, L. Wagner, K. H. Sung, S. Kolb, H. Geise and F. Hahn, ACS Catal., 2020, 10, 4973 CrossRef CAS.
- In this paper, 6-endo refers to the cyclisation of an epoxy-alcohol in which a 6-membered ring (THP) is formed, and the parent epoxide C–C bond is located inside the newly formed oxygen heterocycle. In contrast, 5-exo refers to cyclisation of an epoxy-alcohol where the epoxide C–C bond is outside the newly formed THF ring.
- L. Wang, A. Parnell, C. Williams, N. A. Bakar, M. R. Challand, M. W. van der Kamp, T. J. Simpson, P. R. Race, M. P. Crump and C. L. Willis, Nat. Catal., 2018, 1, 968 CrossRef CAS.
- Y. Shichijo, A. Migita, H. Oguri, M. Watanabe, T. Tokiwano, K. Watanabe and H. Oikawa, J. Am. Chem. Soc., 2008, 130, 12230 CrossRef CAS PubMed.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734 RSC.
- K. Gilmore, R. K. Mohamed and I. V. Alabugin, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 487 CAS.
- J. Na, K. N. Houk, C. G. Shevlin, K. D. Janda and R. A. Lerner, J. Am. Chem. Soc., 1993, 115, 8453 CrossRef CAS.
- I. Vilotijevic and T. F. Jamison, Science, 2007, 317, 1189 CrossRef CAS PubMed.
- K. C. Nicolaou, M. E. Duggan, C.-K. Hwang and P. K. Somers, J. Chem. Soc., Chem. Commun., 1985, 111, 1359 RSC.
- K. C. Nicolaou, C. V. C. Prasad, P. K. Somers and C. K. Hwang, J. Am. Chem. Soc., 1989, 111, 5330 CrossRef CAS.
- E. R. Venegas and C. L. Willis, Org. Lett., 2020, 22, 2548 CrossRef CAS PubMed.
- P. T. Seden, J. P. H. Charmant and C. L. Willis, Org. Lett., 2008, 10, 1637 CrossRef CAS PubMed.
- R. E. Taylor, K. Trentadue, C. Chang and A. Nalin, Chem. – Eur. J., 2021 DOI:10.1002/chem.202100975.
- R. E. Taylor, F. C. Engelhardt, M. J. Schmitt and H. Yuan, J. Am. Chem. Soc., 2001, 123, 2964 CrossRef CAS.
- M. B. Hodge and H. F. Olivo, Tetrahedron, 2004, 60, 9397 CrossRef CAS.
- T. E. Smith, M. Djang, A. J. Velander, C. W. Downey, K. A. Carroll and S. van Alphen, Org. Lett., 2004, 6, 2317 CrossRef CAS PubMed.
- K.-M. Chen, G. E. Hardtmann, K. Prasad, O. Repič and M. J. Shapiro, Tetrahedron Lett., 1987, 28, 155 CrossRef CAS.
- D. P. Stamos, A. G. Taylor and Y. Kishi, Tetrahedron Lett., 1996, 37, 8647 CrossRef CAS.
- E. A. Ilardi, C. E. Stivala and A. Zakarian, Org. Lett., 2008, 10, 1727 CrossRef CAS PubMed.
- S. Okamoto, T. Shimazaki, Y. Kobayashi and F. Sato, Tetrahedron Lett., 1987, 28, 2033 CrossRef CAS.
- P. F. Hudrlik, J. P. Arcoleo, R. H. Schwartz, R. N. Misra and R. J. Rona, Tetrahedron Lett., 1977, 18, 591 CrossRef.
- F. Peng and D. G. Hall, J. Am. Chem. Soc., 2007, 129, 3070 CrossRef CAS PubMed.
- G. Roth, B. Liepold, S. Müller and H. Bestmann, Synthesis, 2003, 59 CrossRef.
- J. Pietruszka and A. Witt, Synthesis, 2006, 4266 CrossRef CAS.
- Y. D. Wang, G. Kimball, A. S. Prashad and Y. Wang, Tetrahedron Lett., 2005, 46, 8777 CrossRef CAS.
- S. Hanessian, T. Focken and R. Oza, Org. Lett., 2010, 12, 3172 CrossRef CAS PubMed.
- J. Pospíšil and I. E. Markó, J. Am. Chem. Soc., 2007, 129, 3516 CrossRef PubMed.
- S. A. Frank, H. Chen, R. K. Kunz, M. J. Schnaderbeck and W. R. Roush, Org. Lett., 2000, 2, 2691 CrossRef CAS PubMed.
- P. R. Blakemore, J. Chem. Soc., Perkin Trans. 1, 2002, 2, 2563 RSC.
- T. M. Hansen, G. J. Florence, P. Lugo-Mas, J. Chen, J. N. Abrams and C. J. Forsyth, Tetrahedron Lett., 2003, 44, 57 CrossRef CAS.
- E. D. Laganis and B. L. Chenard, Tetrahedron Lett., 1984, 25, 5831 CrossRef CAS.
- I. Vilotijevic and T. F. Jamison, Angew. Chem., Int. Ed., 2009, 48, 5250 CrossRef CAS.
- R. K. Bressin, S. Osman, I. Pohorilets, U. Basu and K. Koide, J. Org. Chem., 2020, 85, 4637 CrossRef CAS.
- Y. Gao, J. M. Klunder, R. M. Hanson, H. Masamune, S. Y. Ko and K. B. Sharpless, J. Am. Chem. Soc., 1987, 109, 5765 CrossRef CAS.
- B. Schmidt and O. Kunz, Beilstein J. Org. Chem., 2013, 9, 2544 CrossRef PubMed.
- T. Oishi, M. Suzuki, K. Watanabe and M. Murata, Tetrahedron Lett., 2006, 47, 3975 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and NMR spectra. See DOI: 10.1039/d1ob00883h |
| This journal is © The Royal Society of Chemistry 2021 |