
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalable electrochemical synthesis of diaryliodonium salts†
Mohamed
Elsherbini
and
Wesley J.
Moran
 *
*
Department of Chemistry, University of Huddersfield, Queensgate, Huddersfield HD1 3DH, UK. E-mail: w.j.moran@hud.ac.uk
First published on 4th May 2021
Abstract
Cyclic and acyclic diaryliodonium are synthesised by anodic oxidation of iodobiaryls and iodoarene/arene mixtures, respectively, in a simple undivided electrolysis cell in MeCN–HFIP–TfOH without any added electrolyte salts. This atom efficient process does not require chemical oxidants and generates no chemical waste. More than 30 cyclic and acyclic diaryliodonium salts with different substitution patterns were prepared in very good to excellent yields. The reaction was scaled-up to 10 mmol scale giving more than four grams of dibenzo[b,d]iodol-5-ium trifluoromethanesulfonate (>95%) in less than three hours. The solvent mixture of the large-scale experiment was recovered (>97%) and recycled several times without significant reduction in yield.
Introduction
Owing to their ready availability, low toxicity, unique chemical properties and structural diversity, hypervalent iodine reagents have found tremendous applications in organic synthesis.1 Iodonium salts,2 a class of hypervalent iodine reagents with two carbon ligands, have practical synthetic applications as oxidants,3 photosensitisers in photopolymerisation4 and electrophilic group transfer reagents in a wide range of metal-catalysed and metal-free cross coupling reactions,5 in addition to their medical and pharmaceutical applications.6 Diaryliodonium salts Ar2I+X− are, typically, highly stable compounds that have been extensively studied for over a century. Their high electrophilicity and the superior leaving group ability of iodoarenes stand behind their versatility as arylating agents for a wide range of nucleophiles.7 In addition, they serve as efficient aryne synthons.8 Furthermore, cyclic diaryliodonium salts provide a handle for the rapid construction of polycyclic heterocyclic and carbocyclic compounds of various classes.9Over the long history of diaryliodonium salts, various synthetic routes have been developed and improved over time to achieve high levels of efficiency and selectivity.1a,7 Typically, the synthesis of diaryliodonium salts involves oxidation of iodoarenes to the corresponding λ3-iodanes followed by ligand exchange with arenes or organometallic arenes and anion exchange if necessary. Extended reaction cascades that involve initial iodination of arenes with molecular iodine are also known.10 The reaction sequence can be achieved stepwise or in one-pot.11 Most of these synthetic approaches to iodonium salts are under acidic conditions, although some neutral or basic conditions have been developed. In addition, synthesis under flow conditions has been achieved.12
Hypervalent iodine compounds are often lauded in the literature as mild, “green” reagents and are considered eco-friendly alternatives to various chemical oxidants, especially heavy metal oxidising agents. Unfortunately, syntheses typically require the utilisation of toxic and/or hazardous, and sometimes expensive, chemical oxidants, usually in large excess. This is accompanied by the generation of waste, which increases their ecological footprint and reduces the “greenness” of the overall process. This problem can, in theory, be alleviated by the development of electrochemical syntheses of hypervalent iodine reagents.13 Unsurprisingly, the electrochemical synthesis14 of hypervalent iodine reagents is a current hot topic,13,15 however the published methods for the electrochemical synthesis of diaryliodonium salts are scarce and suffer from limited substrate scopes and low current efficiencies. In addition, published procedures suffer from the formation of side products and complex mixtures as a result of the reaction medium; usually, a mixture of H2SO4/AcOH/Ac2O, that leads to acylation of electron-rich aromatic substrates.16 Herein, we report a simple, scalable “green” electrochemical synthesis of cyclic and acyclic diaryliodonium salts with wide scope. A very recent report on the electrochemical synthesis of aryliodine(III) reagents including some diaryliodonium salts emerged during the final preparation of this work.17
Results and discussion
Diaryliodonium salts are air and moisture stable compounds, but their physical properties and synthetic utility are strongly affected by the nature of the anionic part of the molecule. Previously published electrochemical methods for their preparation utilised H2SO4/AcOH/Ac2O as the reaction medium but these suffered from low yields and side product formation. In addition, isolation was problematic as the initially formed hydrogen sulfate salts required an additional ion exchange step. Usually this was achieved with halide salts such that the iodonium salts were isolated and characterised as halides, mainly iodides. Unfortunately, the synthetic utility of diaryliodonium salts with halide counterions is limited due to their extremely poor solubility in organic solvents and the nucleophilicity of the halide anions. On the other hand, diaryliodonium triflates are easily soluble in organic solvents and are widely used in organic synthesis.9,18 Thereby, this work focuses on the direct electrochemical synthesis of diaryliodonium triflates and hence triflic acid (TfOH) was the acid of choice for the process optimisation.The oxidation of 2-iodobiphenyl (1a) to the corresponding cyclic diaryliodonium triflate 2a was attempted using the Electrasyn device with an undivided cell setup. In accordance with several previous reports on the anodic oxidation of iodoarenes into the corresponding hypervalent iodine reagents, we initiated our studies using a glassy carbon (GC) anode and a platinum (Pt) cathode.13 The initial attempt at electrolysis of a solution of 1a (0.15 mmol) in 2,2,2-trifluoroethanol (TFE, 5 mL) containing TfOH (0.75 mmol, 5 equiv.) applying a constant current of 5 mA (Table 1, entry 1) was unsuccessful as the cell voltage increased rapidly to reach the cell limit (30 V) within a few minutes. Replacing TFE with 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) enhanced the solution conductivity, and the electrolysis could be completed leading to the formation of the iodonium salt 2a in 70% yield after passing a charge of 2.5 F (entry 2). Despite this encouraging result, the cell voltage was still high, 12 V at the beginning of the electrolysis which dropped gradually to 5 V at the end of the electrolysis. This problem was alleviated by using a solvent mixture of HFIP and MeCN (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) leading to the formation of 2a in excellent yield (95%, entry 3).
:1) leading to the formation of 2a in excellent yield (95%, entry 3).
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | 1a [mol L−1] | Current [mA] | Charge [F] | 2a yield [%] |
| a Electrolyses were carried out with an Electrasyn 2.0, using a 5 mL glass vial equipped with a glassy carbon (GC) anode and a Pt cathode; electrode immersed area: 2.8 cm2. Reaction conditions: 2-iodobiphenyl (1a) dissolved in 5 mL solvent, TfOH (5 equiv.), constant current electrolysis. b TfOH (3 or 4 equiv.). TFE: 2,2,2-trifluoroethanol; HFIP: 1,1,1,3,3,3-hexafluoro-2-propanol. | |||||
| 1 | TFE | 0.03 | 5 | — | — |
| 2 | HFIP | 0.03 | 5 | 2.5 | 70 |
| 3 | HFIP–MeCN (4:1) |
0.03 | 5 | 2.5 | 95 |
| 4 | MeCN | 0.03 | 5 | 2.5 | 80 |
| 5 | HFIP–MeCN (1:1) |
0.03 | 5 | 2.5 | 83 |
| 6 | HFIP–MeCN (4:1) |
0.03 | 7.5 | 2.5 | 88 |
| 7 | HFIP–MeCN (4:1) |
0.03 | 10 | 2.5 | 89 |
| 8 | HFIP–MeCN (4:1) |
0.03 | 5 | 2.0 | 78 |
| 9 | HFIP–MeCN (4:1) |
0.03 | 5 | 2.25 | 86 |
| 10 | HFIP–MeCN (4:1) |
0.06 | 5 | 2.5 | 96 |
| 11 | HFIP–MeCN (4:1) |
0.1 | 5 | 2.5 | 85 |
| 12b | HFIP–MeCN (4:1) |
0.06 | 5 | 2.5 | 95 |
Interestingly, running the electrolysis in acetonitrile without fluorinated alcohol (entry 4) led to the isolation of salt 2a in 80%, which is not significantly different from using a 1:1 mixture of HFIP and MeCN (entry 5, see the ESI† for more results of solvent screening). Increasing the current density from 1.8 mA cm−2 to 2.7 and 3.6 mA cm−2 whilst maintaining the same total charge transfer (entries 6 and 7) led to a decrease of the reaction yield to 88 and 89%, respectively. Reducing the charge passed to the theoretical amount (2.0 F) led to a significant decrease of the reaction yield (78%, entry 8) while passing a charge of 2.25 F (entry 9) led to the formation of 2a in 86% yield. Doubling the concentration of 1a (entry 10) did not negatively affect the reaction yield, as 2a was isolated in 96% yield, while increasing the concentration to 0.1 M (entry 11) was accompanied by a gradual increase in the cell voltage due to the precipitation of the product during the electrolysis which led to a drop in the yield to 85% (see the ESI† for more experiments). Applying the optimum conditions (entry 10) but using 3 or 4 equivalents of TfOH did not lead to any reduction in the yield (entry 12) but was accompanied by a significant increase in the cell voltage.
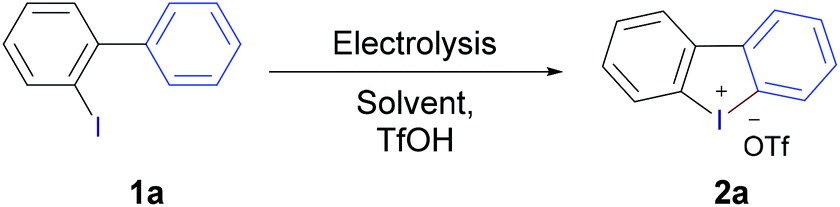
To explore the scope of substrates, a wide range of iodobiaryls with various substitution patterns in both rings were synthesised and subjected to the optimised reaction conditions (Table 1, entry 10). The results (Scheme 1) showed that the reaction proceeded smoothly in most cases giving the desired cyclic diaryliodonium salts 2 in very good to excellent yields. Under the standard reaction conditions 2-iodobiphenyl afforded the corresponding iodonium triflate 2a in 96% yield, replacing triflic acid with tosylic acid and trifluoroacetic acid afforded the tosylate and trifluoroacetate analogues in 93% and 70%, respectively, showing the flexibility of the method for producing the desired iodonium salts with different counter anions. The relatively lower yield in the case of trifluoroacetic acid could be attributed to the poor conductivity of the reaction solution; the addition of water (150 μL) was required to improve the conductivity and complete the electrolysis. Substrates without substituents on the ArI moiety furnished the desired products (2b–k) in very good to excellent yields regardless of the nature of the substituent in the arene part, with excellent tolerance of cyano, ketone, halogen, alkyl, and aryl substituents. 2-Fluorodibenzo[b,d]iodol-5-ium trifluoro-methanesulfonate (2b) was obtained in 88% using 3′-fluoro-2-iodo-1,1′-biphenyl (1b), containing the fluorine in the arene moiety, while the analogous substrate 5-fluoro-2-iodo-1,1′-biphenyl (1b′) containing the fluorine in the ArI moiety led to a slight decrease in the yield (82%). Highly electron-rich substrates were problematic. The methoxy-substituted product 2l was obtained in just 44% yield, as the experiment could not be completed. Electrolysis was stopped after passing only 1.25 F due to unwanted polymerisation and electrode passivation that led to the gradual increase of the cell voltage till the cell limit; a problem that has been reported previously for electrolysis of electron-rich aryliodides.13a,19 The same problem was observed in the case of 2m and 2o. Fortunately, this problem was alleviated by replacing the glassy carbon anode with platinum and alternating the polarity of the electrolysis each 20 seconds. Under these modified conditions, the electrolysis could be completed without problems and product 2l was obtained in a very good yield (87%). Also, product 2o was obtained in very good yield (81%) under the modified conditions, however product 2m was not obtained and decomposition/polymerisation was observed under both conditions. Substrates with one methyl group in the ArI moiety led to products 2n and 2p–r in excellent yields, higher than 90% in all cases regardless of the nature of the substituent in the arene moiety. In addition, electron-deficient substrates with electron-withdrawing groups in both parts of the molecule performed very well and gave the corresponding iodonium salts (2u–2x) in high yields (79%–91%). All substrates containing chlorine substituents gave the desired iodonium salts in excellent yields (>90%). The lowest yields obtained (59% and 75%) for products 2k and 2j having Ph and tBu substituent, respectively maybe attributed to the low solubility of the corresponding substrates in the reaction medium.
 | ||
| Scheme 1 Reaction scope of the electrochemical synthesis of cyclic diaryliodonium salts 2. aTfOH replaced with TsOH·H2O (2 equiv.). bTfOH replaced with CF3CO2H (5 equiv.). cUsing substrate 1b′. dObtained using Pt–Pt with alternating polarity. | ||
Electron-rich heterocyclic substrates proved challenging. Compounds S1y, S1z, and S1aa for example underwent polymerisation/decomposition and the corresponding heterocyclic iodonium salts were not obtained under either the standard or the modified reaction conditions. The anodic oxidation of pyrrole typically leads to deposition to pyrrole black and other polymeric materials.20 In addition, these electron-rich heterocyclic systems are prone to polymerisation under acidic conditions.21
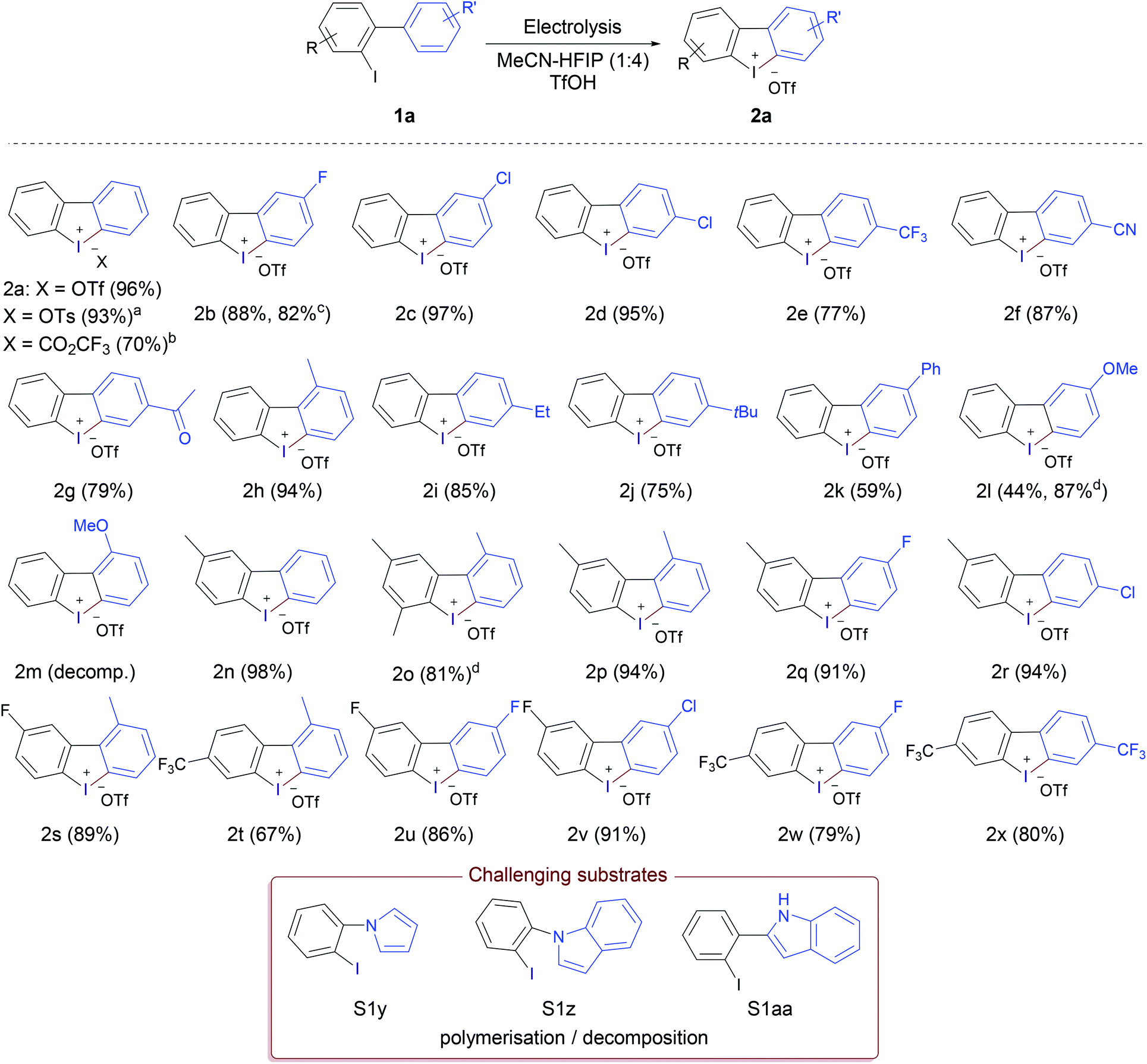
Although the original focus of this work was the electrochemical synthesis of cyclic diaryliodonium salts, the excellent results achieved encouraged us to explore the general applicability of the developed method and extending the reaction scope to acyclic diaryliodonium salts (Scheme 2). Under the same reaction conditions (Table 1, entry 10), diphenyliodonium trifluoromethanesulfonate (5a) was obtained in 55% yield from the reaction of iodobenzene and 1.0 equivalent of benzene. Increasing the amount of benzene to 1.2, 1.5 and 2.0 equivalents led to a gradual improvement of the reaction outcome, forming 5a in 67%, 80% and 88% yield, respectively. The reaction scope was studied using 1.5 equivalents of the arene. Electrolysis of iodobenzene without addition of any other arene component led to the isolation of (4-Iodophenyl)(phenyl)iodonium trifluoromethanesulfonate (5b) in excellent yield (91%).
 | ||
| Scheme 2 Substrate scope of acyclic diaryliodonium triflates 5. aUsing iodomesitylene and bromobenzene. bProduct 2a was obtained instead. | ||
Anodic oxidation of the relatively electron-deficient 4-fluoroiodobenzene in the presence of benzene, toluene and mesitylene furnished the corresponding diaryliodonium salts 5c,d,f in high yields, while using electron-deficient arene counterpart, namely, benzotrifluoride did not lead to successful formation of the desired iodonium salt 5e. The electrochemical oxidation of 1-bromo-4-iodobenzene in the presence of toluene gave 5g in a relatively lower yield (70%). Replacing toluene with mesitylene achieved the corresponding iodonium salt 5h in higher yield (81%), which was formed in lower yield (58%) when using iodomesitylene and bromobenzene. Using a more electron rich aryliodide (4-iodotoluene) and electron-rich arene (mesitylene) led to the formation of 5j in excellent yield (97%). Like the methoxy-containing cyclic product 2m, the acyclic product 5k was not obtained and decomposition/polymerisation was observed upon electrolysis of 2-methoxyiodobenzene in the presence of benzene as the arene counterpart. The competition between oxidation/intramolecular coupling and oxidation/intermolecular coupling was studied by anodic oxidation of 2-iodobiphenyl (1a) in the presence of one equivalent of each of benzene and mesitylene. The experiment using benzene showed the exclusive formation of the cyclic iodonium salt 2a in 95% yield without any trace of the acyclic product 5l. While the reaction using mesitylene led to the formation of the acyclic product 5m as a major product along with a small percentage of the cyclic product 2a in a 10:1 ratio.
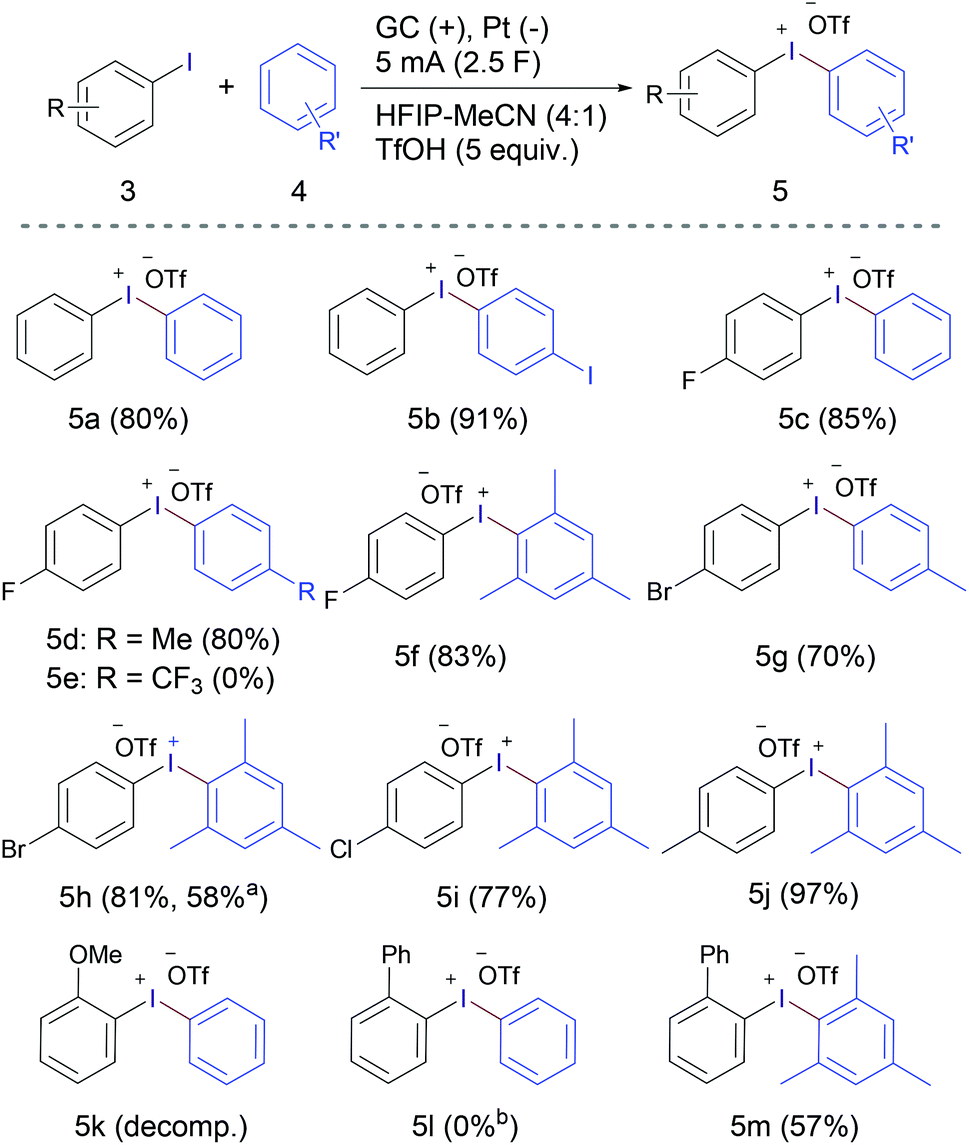
The scalability of the developed method was demonstrated using a beaker-type cell equipped with three parallel 5 × 5 cm electrodes [anode (GC)-cathode (Pt)-anode (GC), see the ESI† for details]. Electrolysis of 10 mmol of 2-iodobiaryl (1a) applying a total current of 92 mA (46 mA on each anode, 1.84 mA cm−2) led to the isolation of 4.17 g of product 2a (97%) in 5.8 hours (2.5 F) with excellent purity (99.9%) (Fig. 1, experiment 1). Clearly demonstrating the practicality of the developed method at the gram-scale. Performing the same experiment using m-chloroperbenzoic acid (mCPBA) on the same scale requires 3.5 g mCPBA (15.0 mmol) and produces equivalent amount of benzoic acid as chemical waste.22 To improve the “greenness” of the whole process and to reduce its ecological footprint more, the solvent system of the first large-scale experiment was collected using a rotary evaporator and recycled several times. The solvent recovery efficiency was very high (97–99%) and the recovered solvent was used for three more cycles. In these cases, the formation of 2a was observed in excellent yield and purity (Fig. 1, experiments 2–4).
 | ||
| Fig. 1 Scale-up/solvent recycling data. Purity of product 2a was determined by 1H NMR using 1,4-dibromobenzene as an internal standard; experiment 6: the recovered solvent was dried over MS before use; experiment 7 was performed applying 92 mA on each anode (double current density: 3.68 mA cm−2) and fresh solvent mixture. | ||
A subsequent fifth cycle showed a significant reduction of the product purity (85%), but without significant reduction of the actual yield (yield x purity %). We supposed that this reduction of purity could be due to the increase of the water content of the solvent with recycling and collection. To test this hypothesis, the solvent recovered from experiment number five was dried using molecular sieves prior to use, which led to a regain of the high purity and yield of the product 2a (Fig. 1, experiment 6). In addition, doubling the current density from 1.84 mA cm−2 to 3.68 mA cm−2, applying a total current of 184 mA (92 mA on each anode), did not have a significant negative impact on the reaction outcome and furnished 2a in high purity and yield, while cutting the reaction time in half to 2.9 hours (Fig. 1, experiment 7).
Conclusions
In conclusion, the synthesis of a wide range of cyclic and acyclic diaryliodonium salts was achieved in high to excellent yields via anodic oxidation of iodobiaryls and iodoarene/arene mixtures using a simple undivided cell. The reaction proceeds smoothly in a mixture of HFIP, MeCN and TfOH under constant current electrolysis, avoiding the use of added salts such as Bu4NBF4, Bu4NClO4 or LiClO4 typically used in batch-type electrolyses as supporting electrolytes. Using triflic acid as a volatile added electrolyte in addition to its role as a reactant rendered the isolation of the formed iodonium salts very simple and efficient. The flexibility of the method was proven by obtaining iodonium salts with other counterions by simply replacing triflic acid with the desired acid. Electrolysis of electron-rich heterocyclic substrates was unsuccessful with this setup and led to polymerisation/decomposition of the starting materials without the isolation of the desired heterocyclic iodonium salts. The easy scalability of the developed method was demonstrated by running a large-scale experiment (10 mmol, 33 folds) leading to the isolation of more than four grams of product 2a (>97%) with excellent purity (>99%). In addition, the solvent system of the large-scale experiment was easily recovered (>97%) using a rotary evaporator and recycled several times without significant reduction of the reaction yield, improving the overall efficiency, economy, and the ecological impact of the developed method.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Leverhulme Trust for their generous funding (Grant No: RPG-2019-058). We also thank all the Moran group members for helpful discussions and assistance. Many thanks to Dr S. Ward for mass spectrometric analysis and Dr N. McLay for NMR analysis.Notes and references
- (a) V. V. Zhdankin, Hypervalent Iodine Chem, John Wiley & Sons Ltd, Chichester, UK, 2013, pp. 21–143 CrossRef; (b) Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis, ed. T. Wirth, [in: Top. Curr. Chem., 2016, 373]., Springer-Verlag, 2016 Search PubMed; (c) A. Parra, Chem. Rev., 2019, 119, 12033–12088 CrossRef CAS PubMed.
- (a) M. S. Yusubov, A. V. Maskaev and V. V. Zhdankin, ARKIVOC, 2011, 2011, 370–409 Search PubMed; (b) K. Kepski, C. R. Rice and W. J. Moran, Org. Lett., 2019, 21, 6936–6939 CrossRef CAS PubMed.
- (a) J. Qurban, M. Elsherbini, H. Alharbi and T. Wirth, Chem. Commun., 2019, 55, 7998–8000 RSC; (b) A. Yoshimura, K. C. Nguyen, S. C. Klasen, A. Saito, V. N. Nemykin and V. V. Zhdankin, Chem. Commun., 2015, 51, 7835–7838 RSC.
- (a) A. Baralle, P. Garra, F. Morlet-Savary, C. Dietlin, J. Fouassier and J. Lalevée, Macromol. Rapid Commun., 2020, 41, 1900644 CrossRef CAS PubMed; (b) J. Kabatc, J. Ortyl and K. Kostrzewska, RSC Adv., 2017, 7, 41619–41629 RSC; (c) T. Brömme, D. Oprych, J. Horst, P. S. Pinto and B. Strehmel, RSC Adv., 2015, 5, 69915–69924 RSC.
- (a) N. R. Deprez and M. S. Sanford, Inorg. Chem., 2007, 46, 1924–1935 CrossRef CAS PubMed; (b) D. R. Stuart, Chem. – Eur. J., 2017, 23, 15852–15863 CrossRef CAS PubMed; (c) D. P. Hari, S. Nicolai and J. Waser, in PATAI'S Chem. Funct. Groups, John Wiley & Sons, Ltd, Chichester, UK, 2018, pp. 1–58 Search PubMed; (d) E. Zawia, D. J. Hamnett and W. J. Moran, J. Org. Chem., 2017, 82, 3960–3964 CrossRef CAS PubMed; (e) A. Rodríguez and W. J. Moran, J. Org. Chem., 2016, 81, 2543–2548 CrossRef PubMed.
- (a) V. W. Pike, J. Labelled Compd. Radiopharm., 2017, 1–32 Search PubMed; (b) P. Das, E. Tokunaga, H. Akiyama, H. Doi, N. Saito and N. Shibata, Beilstein J. Org. Chem., 2018, 14, 364–372 CrossRef CAS PubMed; (c) N. Nguyen, D. W. Wilson, G. Nagalingam, J. A. Triccas, E. K. Schneider, J. Li, T. Velkov and J. Baell, Eur. J. Med. Chem., 2018, 148, 507–518 CrossRef CAS PubMed.
- E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052–9070 CrossRef CAS PubMed.
- D. R. Stuart, Synlett, 2017, 28, 275–279 CrossRef CAS.
- (a) D. Zhu, Z. Wu, L. Liang, Y. Sun, B. Luo, P. Huang and S. Wen, RSC Adv., 2019, 9, 33170–33179 RSC; (b) A. Boelke, P. Finkbeiner and B. J. Nachtsheim, Beilstein J. Org. Chem., 2018, 14, 1263–1280 CrossRef CAS PubMed; (c) M. Wang, Q. Fan and X. Jiang, Org. Lett., 2018, 20, 216–219 CrossRef CAS PubMed.
- E. Lindstedt, M. Reitti and B. Olofsson, J. Org. Chem., 2017, 82, 11909–11914 CrossRef CAS PubMed.
- N. Soldatova, P. Postnikov, O. Kukurina, V. V. Zhdankin, A. Yoshimura, T. Wirth and M. S. Yusubov, Beilstein J. Org. Chem., 2018, 14, 849–855 CrossRef CAS PubMed.
- (a) G. Laudadio, H. P. L. Gemoets, V. Hessel and T. Noël, J. Org. Chem., 2017, 82, 11735–11741 CrossRef CAS PubMed; (b) N. S. Soldatova, P. S. Postnikov, M. S. Yusubov and T. Wirth, Eur. J. Org. Chem., 2019, 2081–2088 CrossRef CAS.
- (a) M. Elsherbini, B. Winterson, H. Alharbi, A. A. Folgueiras-Amador, C. Génot and T. Wirth, Angew. Chem., Int. Ed., 2019, 58, 9811–9815 ( Angew. Chem. , 2019 , 131 , 9916–9920 ) CrossRef CAS PubMed; (b) M. Elsherbini and T. Wirth, Chem. – Eur. J., 2018, 24, 13399–13407 CrossRef CAS PubMed; (c) T. Broese and R. Francke, Org. Lett., 2016, 18, 5896–5899 CrossRef CAS PubMed.
- (a) D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–123400 RSC; (b) A. L. Rauen, F. Weinelt and S. R. Waldvogel, Green Chem., 2020, 22, 5956–5960 RSC; (c) D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573–4591 CrossRef CAS PubMed; (d) D. E. Collin, A. A. Folgueiras-Amador, D. Pletcher, M. E. Light, B. Linclau and R. C. D. Brown, Chem. – Eur. J., 2020, 26, 374–378 CrossRef CAS PubMed; (e) T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CrossRef CAS; (f) M. C. Leech and K. Lam, Acc. Chem. Res., 2020, 53, 121–134 CrossRef CAS PubMed; (g) F. J. Holzhäuser, G. Creusen, G. Moos, M. Dahmen, A. König, J. Artz, S. Palkovits and R. Palkovits, Green Chem., 2019, 21, 2334–2344 RSC.
- (a) S. R. Waldvogel, S. Arndt, D. Weis and K. Donsbach, Angew. Chem., Int. Ed., 2020, 59, 8036–8041 CrossRef PubMed; (b) A. F. Roesel, T. Broese, M. Májek and R. Francke, ChemElectroChem, 2019, 6, 4229–4237 CrossRef CAS; (c) R. Francke, T. Broese and A. F. Roesel, in PATAI'S Chem. Funct. Groups, John Wiley & Sons, Ltd, Chichester, UK, 2018, pp. 1–22 Search PubMed; (d) L. Massignan, X. Tan, T. H. Meyer, R. Kuniyil, A. M. Messinis and L. Ackermann, Angew. Chem., Int. Ed., 2020, 59, 3184–3189 CrossRef CAS PubMed; (e) S. Doobary, A. T. Sedikides, H. P. Caldora, D. L. Poole and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 1155–1160 CrossRef CAS PubMed.
- (a) L. L. Miller and A. K. Hoffmann, J. Am. Chem. Soc., 1967, 89, 593–597 CrossRef CAS; (b) M. J. Peacock and D. Pletcher, Tetrahedron Lett., 2000, 41, 8995–8998 CrossRef CAS; (c) K. Watts, W. Gattrell and T. Wirth, Beilstein J. Org. Chem., 2011, 7, 1108–1114 CrossRef CAS PubMed.
- B. Zu, J. Ke, Y. Guo and C. He, Chin. J. Chem., 2021, 39, 627–632 CrossRef CAS.
- (a) K. Zhu, Y. Wang, Q. Fang, Z. Song and F. Zhang, Org. Lett., 2020, 22, 1709–1713 CrossRef CAS PubMed; (b) G. Kervefors, L. Kersting and B. Olofsson, Chem. – Eur. J., 2021, 27, 5790–5795 CrossRef CAS PubMed; (c) S. N. Yunusova, A. S. Novikov, N. S. Soldatova, M. A. Vovk and D. S. Bolotin, RSC Adv., 2021, 11, 4574–4583 RSC.
- Y. Amano and S. Nishiyama, Heterocycles, 2008, 75, 1997–2003 CrossRef CAS.
- J. K. Howard, K. J. Rihak, A. C. Bissember and J. A. Smith, Chem. – Asian J., 2016, 11, 155–167 CrossRef CAS PubMed.
- J. K. Laha, M. K. Hunjan, S. Hegde and A. Gupta, Org. Lett., 2020, 22, 1442–1447 CrossRef CAS PubMed.
- (a) D. Zhu, Q. Liu, B. Luo, M. Chen, R. Pi, P. Huang and S. Wen, Adv. Synth. Catal., 2013, 355, 2172–2178 CrossRef CAS; (b) M. Jiang, J. Guo, B. Liu, Q. Tan and B. Xu, Org. Lett., 2019, 21, 8328–8333 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ob00457c |
| This journal is © The Royal Society of Chemistry 2021 |