
Discovery, synthesis and biological characterization of a series of N-(1-(1,1-dioxidotetrahydrothiophen-3-yl)-3-methyl-1H-pyrazol-5-yl)acetamide ethers as novel GIRK1/2 potassium channel activators†
Swagat
Sharma
a,
Lauren
Lesiak
a,
Christopher D.
Aretz
a,
Yu
Du
b,
Sushil
Kumar
a,
Nagsen
Gautam
a,
Yazen
Alnouti
a,
Nikilesh V.
Dhuria
c,
Yashpal S.
Chhonker
c,
C. David
Weaver
 b and
Corey R.
Hopkins
*a
b and
Corey R.
Hopkins
*a
aDepartment of Pharmaceutical Sciences, College of Pharmacy, University of Nebraska Medical Center, Omaha, NE, 68198 USA. E-mail: corey.hopkins@unmc.edu
bDepartment of Pharmacology, Vanderbilt University School of Medicine, Nashville, TN, 37232 USA
cDepartment of Pharmacy Practice and Science, College of Pharmacy, University of Nebraska Medical Center, Omaha, NE, 68198 USA
First published on 21st June 2021
Abstract
The present study describes the discovery and characterization of a series of N-(1-(1,1-dioxidotetrahydrothiophen-3-yl)-3-methyl-1H-pyrazol-5-yl)acetamide ethers as G protein-gated inwardly-rectifying potassium (GIRK) channel activators. From our previous lead optimization efforts, we have identified a new ether-based scaffold and paired this with a novel sulfone-based head group to identify a potent and selective GIRK1/2 activator. In addition, we evaluated the compounds in tier 1 DMPK assays and have identified compounds that display nanomolar potency as GIRK1/2 activators with improved metabolic stability over the prototypical urea-based compounds.
Introduction
The G protein-gated inwardly rectifying potassium channels (GIRK, Kir3) are a family of inward-rectifying potassium channels that are encoded by the genes KCNJ3, KCNJ6, KCNJ9, and KCNJ5.1,2 These channels are tetrameric complexes formed by homo- and heteroassembly among four related subunits (GIRK1–4 or Kir3.1–3.4).3–6 GIRK channels are key effectors in GPCR signaling pathways that modulate excitability in cells with GIRK1 being broadly expressed in the brain as well as numerous locations in the periphery including the heart, and endocrine tissues. Comparatively, the expression of GIRK2 and GIRK3 is highly localized to the brain, whereas GIRK4 is more prevalent in peripheral tissues including cardiac atrial myocytes, where it plays a key role in regulating heart rate.7–9 The overwhelming majority of GIRK channels exist as heteromeric complexes comprised of GIRK1 and one of the other GIRK subunits. The GIRK1/2 channel subtype is the most common within the brain, while other subunit combinations have more limited brain distribution.10–12 There is substantial preliminary evidence supporting the roles of GIRK channels in a number of physiological processes and as potential targets for numerous indications,13,14 such as pain perception,15–18 epilepsy,4,19,20 reward/addiction21–23 and anxiety.20 Unfortunately, there remains a lack of selective and brain-penetrant pharmacological tool compounds and as such, the role of GIRK channels as potential therapeutic targets is still not well understood.Previous reports of GIRK1/2 channel activators from our laboratories20,24–27 started from a high-throughput screen leading to the discovery of urea-containing compounds with in vivo activity in antiepileptic,19,20 anxiolytic20 and nociception models18,28 in mice. However, these compounds suffer from significant pharmacokinetic (PK) liabilities, namely, poor brain penetration as well as only modest selectivity against the GIRK1/4 channel subtype. Recent reports have identified additional activators from virtual screening efforts; however, these compounds are also based on the urea scaffold.29 We were interested in exploring compounds that were void of the urea moiety, and we have published on the discovery of amide-containing compounds.30,31 Unfortunately, these compounds suffer from significant metabolic stability issues (Fig. 1). One interesting observation that was made during these efforts was that the incorporation of a 1,1-dioxidotetrahydrothiophen-3-yl moiety on the privileged pyrazole (1vs.2 and 3vs.4) group imparted significant metabolic stability but at the expense of potency (Fig. 1). The reason for the stability imparted by the cyclic sulfone moiety is not known, but potentially could be due to circumventing cyclic oxidation of the cyclohexyl moiety. We then reinvestigated our initial high-throughput screening results and identified a new, ether containing scaffold that was intriguing to pair with the cyclic sulfone. Herein, we report the synthesis, biological characterization, and in vitro PK properties of a new ether series of compounds that incorporate the sulfone group while maintaining potency and stability.
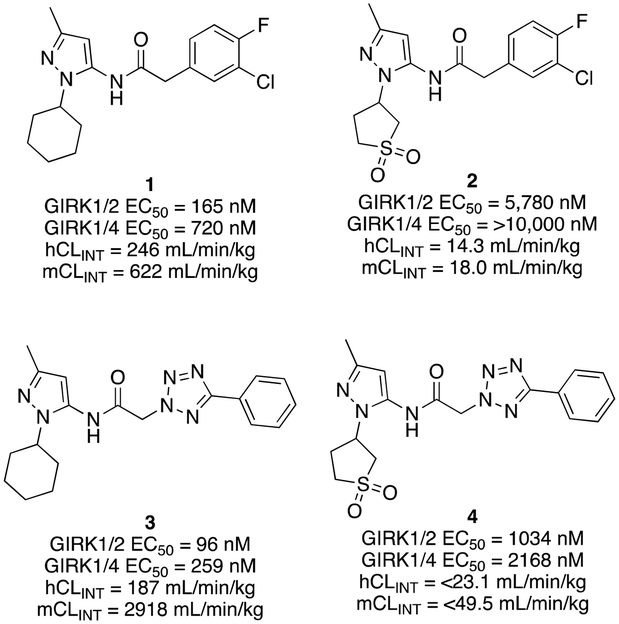 | ||
| Fig. 1 Previously disclosed phenylacetamide and 2H-tetrazole-2-ylacetamide GIRK1/2 activators. | ||
Results and discussion
Chemistry
The compounds were synthesized as outlined in Scheme 1 (additional synthetic schemes are outlined in the ESI† procedures).31 The procedure was designed to be a modular approach that facilitates a variety of substitution patterns on both the ether (amine) and the pyrazole head group. The β-keto nitrile, 5, was cyclized with 3-hydrazineyltetrahydrothiophene-1,1-dioxide, 6, with AcOH/EtOH under reflux to yield the aminopyrazole 7. The pyrazole, 7, was then reacted with chloroacetyl chloride to yield the α-chloro coupling partner, 10a. Alternatively, the phenol or aniline, 8, was added to ethyl 2-bromoacetate under basic conditions leading to the ester, 9, which was saponified (LiOH, rt) yielding the acid, 10b. To access the final targets, 11a–z, either the appropriately substituted aminopyrazole, 7, was condensed with the acid, 10b, using propylphosphonic anhydride (T3P, CH2Cl2), or the phenol or amine, 8, was reacted with the α-chloro compound 10a to yield the desired GIRK1/2 activators.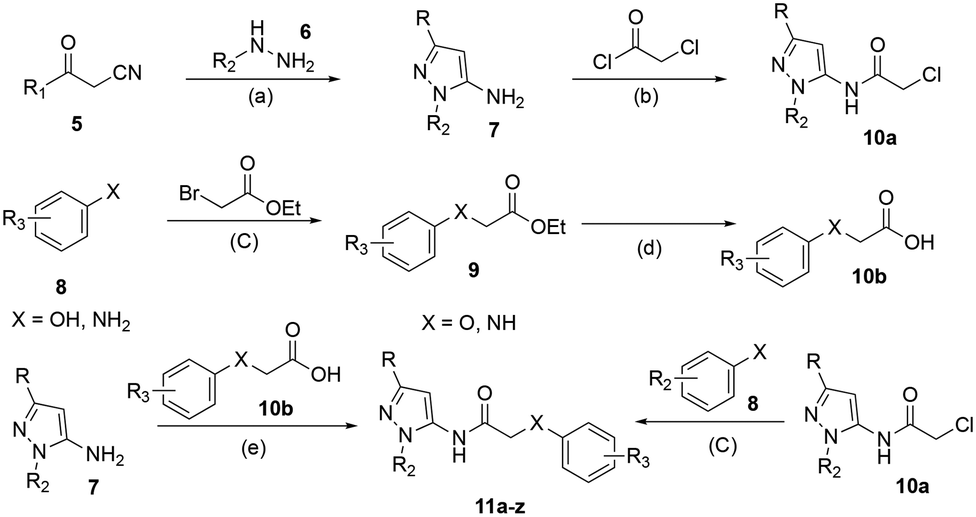 | ||
Scheme 1 Synthesis of ether containing GIRK1/2 activators. Reagents and condition: (a) AcOH, EtOH, reflux, 72 h; (b) Et3N, CH2Cl2, rt, 12 h; (c) K2CO3, DMF, 60 °C, 16 h; (d) LiOH, H2O/MeOH/THF (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5:7.5), rt, 16 h; (e) T3P, Et3N, CH2Cl2, rt, 12 h. :1.5:7.5), rt, 16 h; (e) T3P, Et3N, CH2Cl2, rt, 12 h. | ||
Evaluation of biological activity
The initial structure–activity relationship (SAR), which was centered around the right-hand portion of the molecule, is detailed in Table 1. The SAR data was obtained by testing on HEK293 cells expressing GIRK channels using thallium flux assays, as previously described.30,31 The initial molecule, 11a, based on a similar compound obtained from a high-throughput screening effort contained the 2,4-dichlorophenoxy moiety combined with the 1,1-dioxidotetrahydrothiophen-3-yl that was identified previously. 11a was a potent GIRK1/2 activator and was selective against the closely related GIRK1/4 (11a, GIRK1/2 EC50 = 137 nM; GIRK1/4 EC50 = 702 nM); this selectivity versus GIRK1/4 constitutes one of the most selective compounds reported. Selectively eliminating one of the chlorine atoms led to reduced GIRK1/2 activity (11b–c); however, the activity was recouped by moving the dichloro substitution pattern around the molecule (2,3-dichloro, 11d, EC50 = 239 nM; 2,5-dicholoro, 11e, EC50 = 150 nM). Replacing the 4-chlorine with a bromine or fluorine retained activity (11f, g), but substituting a 2-fluorine led to a 10-fold loss of activity (11h). The same activity pattern was observed with the di- and monobromine compounds (11i–k), and the difluorine (11l) was less active. Further substitutions around the phenyl ring system did not produce a more potent compound (11m–p) with the exception of the 2,4-di(trifluoromethyl) (11o). Adding more bulk with the napthyl/quinoline (11p, q) or indole/indazole (11u, v) was not a productive change, nor was the addition of a nitrogen atom in the form of a pyridine ring (11s, t). The ether linkage could be replaced with an amine (11w, EC50 = 592 nM) with only a ∼4-fold loss of activity; however, if the amine was alkylated with a methyl group (11x) the activity loss was ∼22-fold. Lastly, we investigated the 3-position of the pyrazole group (R1) as this area had been previously explored. Moving from the methyl to the ethyl group (11y) led to an equipotent compound, but, adding more bulk in the form of the cyclobutyl group (11z) led to a loss of activity. Previous scaffolds tolerated the cyclobutyl group, although the SAR was minimal. In addition, the previous amide scaffold did not tolerate an additional methylene spacer group, a two-carbon spacer between the amide and phenyl group whereas the ether compounds tolerate a two-atom (carbon–oxygen) spacer.30|
|
||||
|---|---|---|---|---|
| Cmpd | R1 | R | GIRK1/2 (nM ± SEM; % ± SEM)a,b | GIRK1/4 (nM ± SEM; % ± SEM)a,b |
| a Mean potency values (±SEM) were obtained from a minimum of triplicate determinations; values are average of n = 3. b Mean efficacy values (±SEM) shown are obtained from a minimum of triplicate determinations; values are normalized to standard compound, VU0466551. | ||||
| 11a | Me |
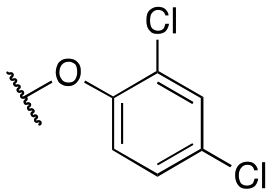
|
137 ± 23; 95 ± 2 | 702 ± 211; 76 ± 4 |
| 11b |
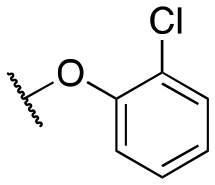
|
962 ± 170; 92 ± 5 | 3133 ± 289; 59 ± 5 | |
| 11c |
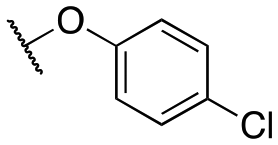
|
4659 ±18; 91 ± 3 | >10000; >77 |
|
| 11d |
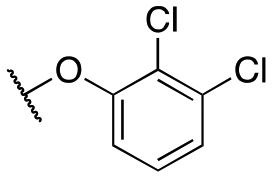
|
239 ± 50; 84 ± 5 | 601 ± 137; 44 ± 4 | |
| 11e |

|
150 ± 38; 88 ± 4 | 563 ± 73; 68 ± 3 | |
| 11f |
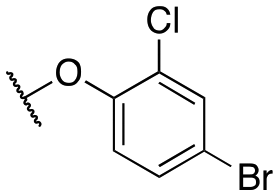
|
132 ± 24; 93 ± 4 | 335 ± 65; 76 ± 5 | |
| 11g |
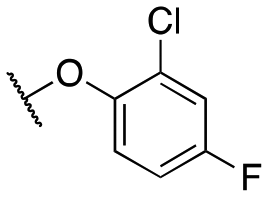
|
448 ± 56; 90 ± 4 | 1536 ± 73; 61 ± 3 | |
| 11h |
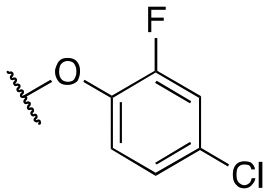
|
1454 ± 12; 91 ± 2 | 3919 ± 54; 52 ± 2 | |
| 11i |
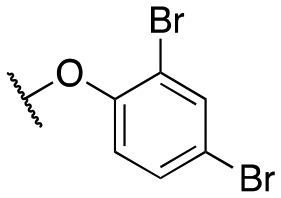
|
64 ± 7; 94 ± 2 | 229 ± 28; 77 ± 3 | |
| 11j |
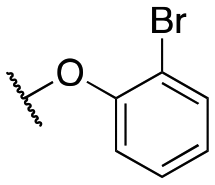
|
493 ± 80; 86 ± 4 | 1277 ± 155; 48 ± 2 | |
| 11k |
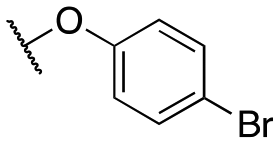
|
4480 ± 404; 96 ± 11 | >10000; >54 |
|
| 11l |
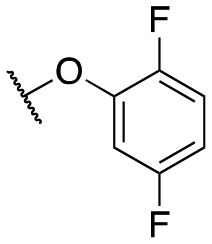
|
6021 ± 1480; 100 ± 7 | >10000; >40 |
|
| 11m |
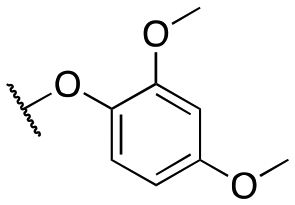
|
>10000; >40 |
Inactive | |
| 11n |
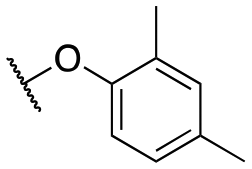
|
1739 ± 154; 99 ± 2 | 5541 ± 882; 57 ± 4 | |
| 11o |
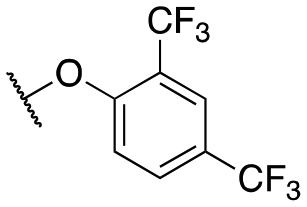
|
146 ± 21; 88 ± 2 | 730 ± 97; 52 ± 2 | |
| 11p |
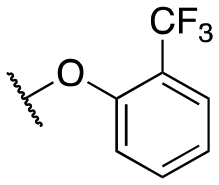
|
484 ± 93; 81 ± 4 | 1286 ± 257; 48 ± 4 | |
| 11q |
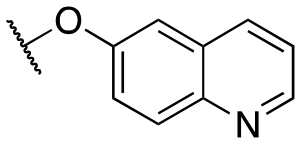
|
Inactive | Inactive | |
| 11r |
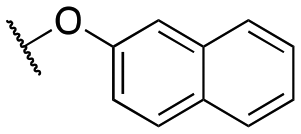
|
4200 ± 570; 98 ± 4 | >10000; >41 |
|
| 11s |
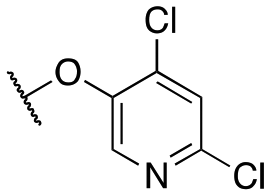
|
>10000; >61 |
>10000; >61 |
|
| 11t |
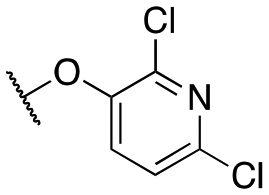
|
3472 ± 174; 70 ± 4 | 5378 ± 270; 42 ± 2 | |
| 11u |
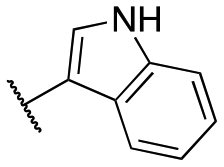
|
Inactive | Inactive | |
| 11v |
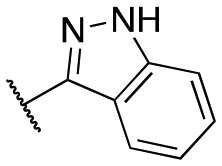
|
Inactive | Inactive | |
| 11w |
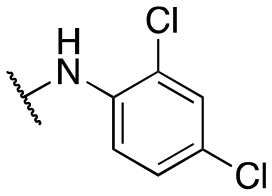
|
592 ± 51; 98 ± 2 | >3000; >60 | |
| 11x |
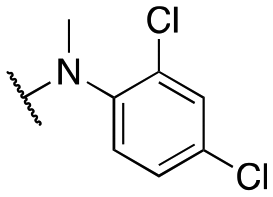
|
2955 ± 235; 77 ± 9 | 3839 ± 368; 24 ± 3 | |
| 11y | Et |
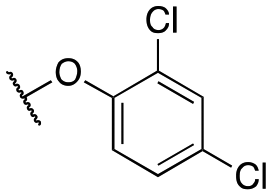
|
187 ± 34; 79 ± 4 | 360 ± 98; 36 ± 4 |
| 11z |
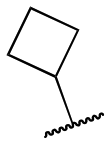
|
1400 ± 449; 21 ± 1 | Inactive | |
Having explored the right-hand ether linked portion of the molecule, we next wanted to turn our attention to the pyrazole moiety (Table 2). As with our previous attempts to replace the pyrazole moiety, all compounds synthesized were devoid of most activity (12a–d), with the exceptions being the benzoisoxazole (12c, EC50 = 609 nM) and pyridine moiety (12d, EC50 = 782 nM). In addition to replacing the pyrazole, we investigated amide replacements or alkylation of the amide as a way to improve the metabolic stability which was mainly caused by amidolysis (see below). Unfortunately, the compounds were not active (carbamate, 12e; N-methyl amide, 12g). The thioamide (12f) did retain some activity, however.
| Cmpd | Structure | GIRK1/2 (nM ± SEM; % ± SEM)a,b | GIRK1/4 (nM ± SEM; % ± SEM)a,b |
|---|---|---|---|
| a Mean potency values (±SEM) were obtained from a minimum of triplicate determinations; values are average of n = 3. b Mean efficacy values (±SEM) shown are obtained from a minimum of triplicate determinations; values are normalized to standard compound, VU0466551. | |||
| 12a |
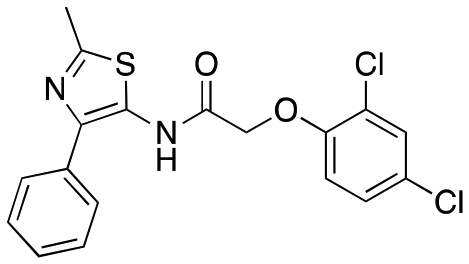
|
Inactive | Inactive |
| 12b |
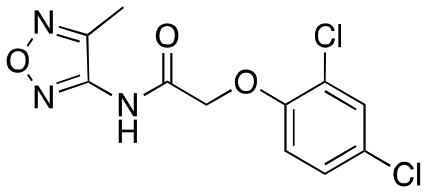
|
>6000; >50 | >8000; >52 |
| 12c |
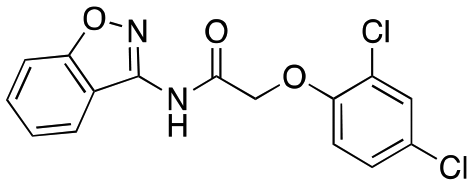
|
609 ± 169; 20 ± 1 | Inactive |
| 12d |
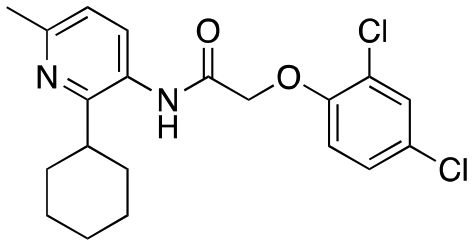
|
782 ± 155; 44 ± 3% | Inactive |
| 12e |
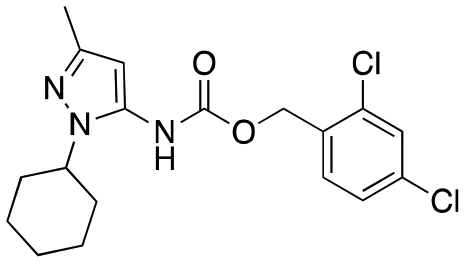
|
Inactive | Inactive |
| 12f |
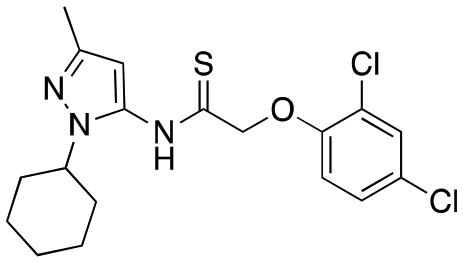
|
>4000; >46 | Inactive |
| 12g |
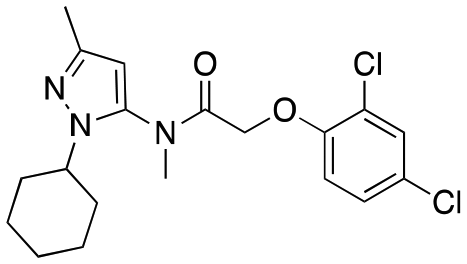
|
723 ± 88; 103 ± 4 | 2869 ± 282; 99 ± 6 |
| 12h |
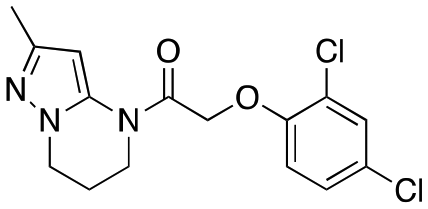
|
Inactive | Inactive |
Finally, we investigated the substitution of the nitrogen of the privileged pyrazole group (Table 3). As stated previously, we identified the 1,1-dioxidotetrahydrothiophen-3-yl group as one that imparts significant stability in the liver microsome assay; however, we wanted to confirm that this group was still the best in terms of both potency and stability. As we have seen previously, substituted alkyl groups were active GIRK1/2 activators when added to the pyrazole group. Namely, the isopropyl (13a), cyclohexyl (13c), 4,4-difluorocyclohexyl (13e) and pyran (13f) were all equipotent or more potent than 11a against GIRK1/2, with varying selectivity versus GIRK1/4. Notably, 13e (4,4-difluorocyclohexyl) was one of the most potent GIRK1/2 activators identified (EC50 = 33 nM). Interestingly, when the 5-membered sulfone ring in 11a was expanded to the 6-membered ring (13b) there was an ∼4-fold loss of activity, and the 4,4-dimethylcyclohexyl derivative (13d) was also much less active.
|
|
|||
|---|---|---|---|
| Cmpd | R | GIRK1/2 (nM ± SEM; % ± SEM)a,b | GIRK1/4 (nM ± SEM; % ± SEM)a,b |
| a Mean potency values (±SEM) were obtained from a minimum of triplicate determinations; values are average of n = 3. b Mean efficacy values (±SEM) shown are obtained from a minimum of triplicate determinations; values are normalized to standard compound, VU0466551. | |||
| 13a |
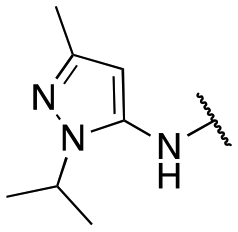
|
133 ± 7; 98 ± 1 | 607 ± 50; 78 ± 2 |
| 13b |
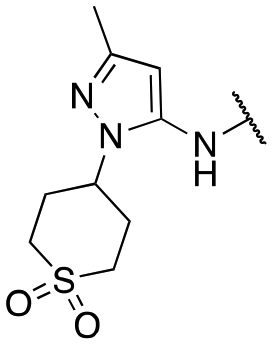
|
577 ± 58; 65 ± 2 | 1171 ± 89; 44 ± 1 |
| 13c |
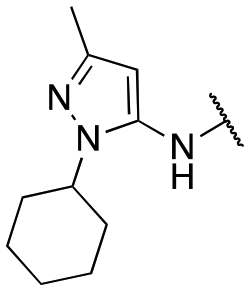
|
91 ± 8; 98 ± 2 | 430 ± 43; 94 ± 3 |
| 13d |
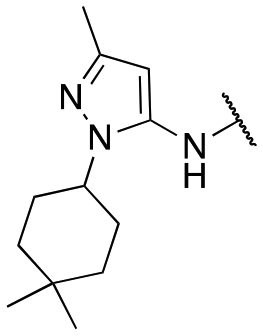
|
>677; >28 | Inactive |
| 13e |
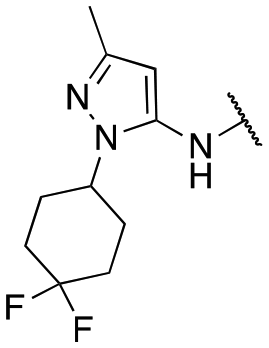
|
33 ± 3; 84 ± 2 | 145 ± 21; 71 ± 3 |
| 13f |
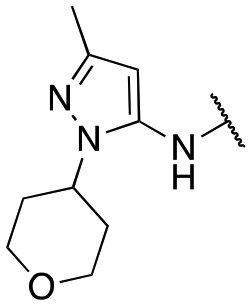
|
88 ± 7; 108 ± 2 | 371 ± 27; 86 ± 3 |
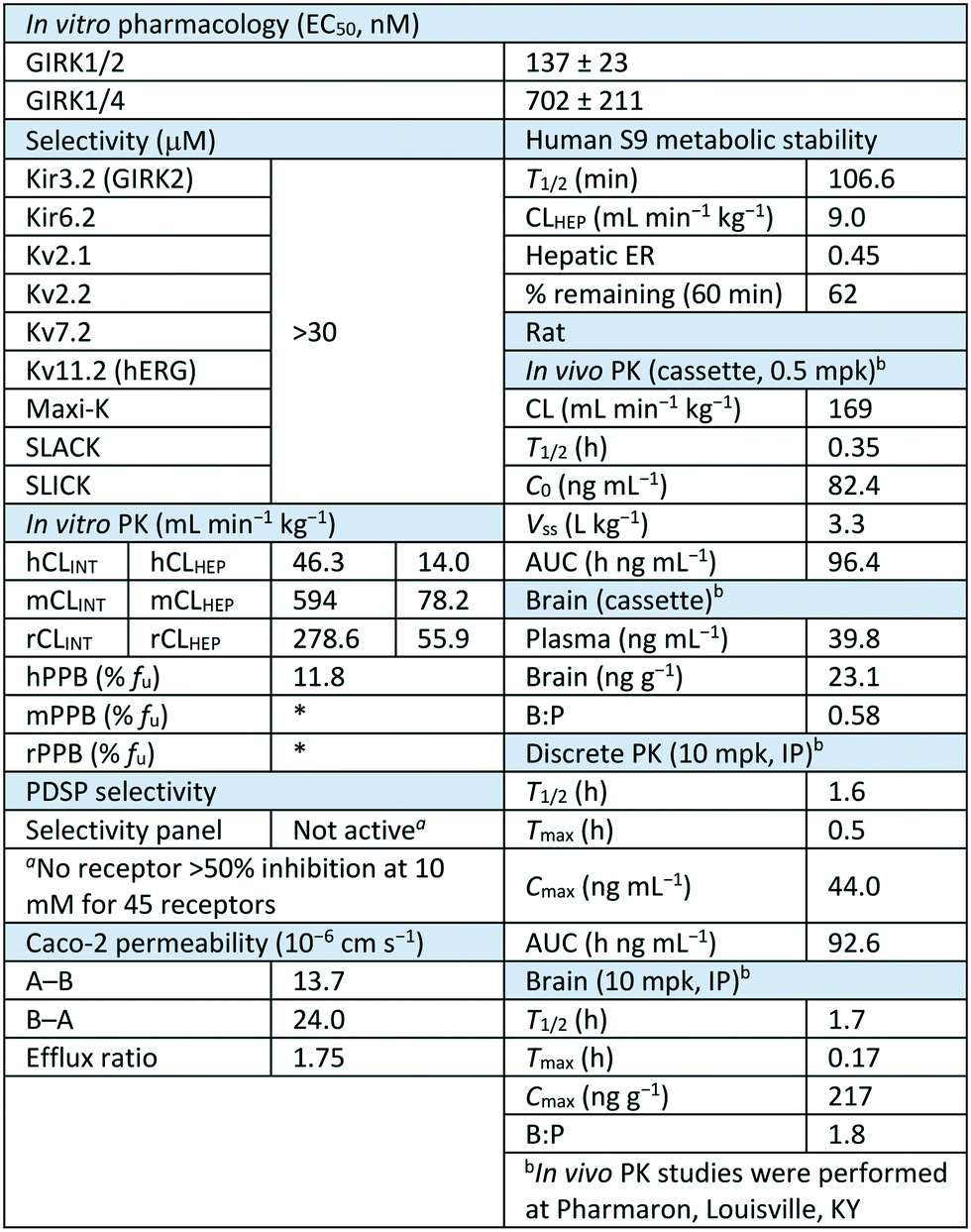
Next, we further evaluated selected compounds based on liver microsome stability and plasma protein binding (Table 4).32,33 As we had seen previously, the incorporation of the 1,1-dioxidotetrahydrothiophen-3-yl moiety imparted significant improvement in the metabolic stability. Interestingly, there appears to be some divergence in the stability in human liver microsomes with 11a and 13b being the most stable (13b contains the six-membered sulfone). However, the compounds with better mouse liver microsome stability are 11o (2,4-trifluomethyl) and 11w (the amino analog of 11a). All of the compounds that contain alkyl or cyclic alkyl southern fragments were all metabolically unstable which is consistent with our previous observations. A metabolite ID study showed the presence of the amide hydrolysis of the aminopyrazole as the major metabolite in both human and mouse liver microsomes (see ESI† Fig. S1). All of the compounds showed excellent human free fraction (with a few exceptions) with% fu values above 5%. However, nearly all of the compounds were deemed to be unstable in mouse plasma and thus the free fraction was unable to be obtained. The lone exceptions were 11w and 12e, even though both of these compounds showed mouse liver microsome instability. Next, we chose two compounds to evaluate in rat liver microsomes and protein binding to see if there was a species difference between mouse and rat. Both 11a and 13b showed high clearance in rat microsomes (similar to that in mouse) and were unstable in rat plasma protein binding assay (not shown). Although these compounds showed high clearance in the rat, we also evaluated the compounds in a tissue distribution study to obtain plasma and brain concentrations in order to inform any future animal studies. We dosed 11a and 11w in an IV cassette study, and 11a showed appreciable levels in both plasma and brain with a ratio of 0.56 (Kp).34,3511w had similar levels in the plasma but did not have any detectable levels in the brain – potentially due to the presence of the amine (NH); which the additional hydrogen bond donor could negatively impact the brain penetration.
| Cmpd | Intrinsic clearance (mL min−1 kg−1)a,d | Plasma protein binding (% fu)b,d | ||||
|---|---|---|---|---|---|---|
| hCLINT | hCLHEP | mCLINT | mCLHEP | H | M | |
| 11a | 46.3 | 14.0 | 594 | 78.2 | 11.8 | |
| 11i | 93 | 16.4 | 672.2 | 79.4 | 4.8 | |
| 11o | 50.9 | 14.4 | 134.6 | 53.9 | 6.8 | |
| 11w | 44.9 | 13.8 | 174.2 | 59.3 | 8.5 | 13.1 |
| 11y | 165.1 | 17.8 | 1174.1 | 83.6 | 5.4 | |
| 12e | 341.4 | 19.8 | 2806.7 | 87.2 | 0.32 | 0.49 |
| 12f | 250.7 | 19.4 | >5930 | >88.7 | <0.06 | |
| 13a | 323.8 | 18.8 | 4950 | 88.4 | ||
| 13b | 25.4 | 11.5 | 294 | 68.9 | 16.0 | |
| 13e | 295.1 | 18.7 | >5930 | >88.7 | 6.6 | |
| 13f | 100.4 | 16.7 | 2970 | 87.4 | 16.3 | |
| rCLINT | rCLHEP | |||||
|---|---|---|---|---|---|---|
| 11a | 278.6 | 55.9 | ||||
| 13b | 216.7 | 52.9 |
| In vivo cassette (rat, IV)e | ||||||
|---|---|---|---|---|---|---|
| 11a | 11w | |||||
| a Predicted hepatic clearance based on intrinsic clearance in mouse and human liver microsomes using the well-stirred organ CL model (binding terms excluded). b f u = fraction unbound. c Unstable in human or mouse plasma. d In vitro DMPK studies performed at Q2 Solutions, Indianapolis, IN. e In vivo cassette studies performed at Pharmaron, Louisville, KY. | ||||||
| Plasma | 168 | 143 | ||||
| Brain | 87.1 | BLQ | ||||
| B:P |
0.56 | N/A | ||||
Next, we utilized thallium flux assays to functionally evaluate the selectivity of subset of compounds in a diverse panel of potassium channels (ESI† Table S2). The compounds were evaluated against Kir3.2 (homomeric GIRK2), Kir6.2/SUR1, Kv2.1, Kv2.2, Kv7.2, Kv11.1 (hERG), Maxi-K, SLACK, and SLICK. 11a showed complete selectivity against all ion channels tested. In fact, all of the compounds tested (11a, 11y, 13a, 13e, 13f) showed excellent selectivity, with only a select few compounds showed any activity, albeit it weak (>20 μM). As 11a has emerged as the lead compound based on potency, stability and ion channel selectivity, we finally profiled this compound against the Psychoactive Drug Screening Panel at the University of North Carolina, Chapel Hill.36 This panel consists of 45 receptors and transporters that are of importance in the CNS. Gratifyingly, 11a was clean against this larger panel of receptors, confirming 11a as a selective GIRK1/2 activator.
Lastly, 11a was evaluated in further assays in order to better assess the value of the compound as an in vivo tool compound (Table 5). In addition to the potency, in vitro PK, and selectivity assays, 11a was run in a Caco-2 experiment in order to assess permeability as well as efflux, and it was determined to be highly permeable and have a low efflux ration (ER = 1.75). 11a was also tested in a human liver S9 liver stability assay and was shown to be a low clearance molecule (supporting the liver microsome result) (CLHEP = 9.0 mL min−1 kg−1) with 62% remaining at 60 min. Next, we dosed 11a in an in vivo rat cassette, and it was shown to be highly cleared with a short half-life (T1/2 = 0.35 h). Finally, 11a was evaluated in a discrete in vivo PK experiment in order to assess the PK properties when dosed individually. Interestingly, when 11a was dosed IP, the compound had improved plasma and brain half-life (plasma, t1/2 = 1.6 h; brain, t1/2 = 1.7 h) and significantly improved brain:plasma ratio (Kp = 1.8).
|
Conclusions
In summary, an HTS campaign identified a new ether-based scaffold that we coupled with a previously identified N-(1-(1,1-dioxidotetrahydrothiophen-3-yl)-3-methyl-1H-pyrazol-5-yl) head group to afford a new class of GIRK1/2 activators. Chemical optimization improved potency, human liver microsome stability, and brain penetration in rat (Kp). Additional profiling showed the lead compound, 11a, to be selective against a wide panel of diverse potassium channels as well as against a group of important CNS receptors and transporters. Further separation of the enantiomers and evaluation in in vivo animal models of pain and epilepsy will be reported in due course.Author contributions
C. R. H. oversaw and designed the chemistry. S. S., L. L. and C. D. A. performed the synthetic chemistry work. C. R. H. and Y. S. C. designed and performed the drug metabolism and pharmacokinetic in vitro experiments and in vivo studies. N. V. D. performed the in vivo PK experiments as well as Pharmaron. Y. A. designed the S9 stability studies and N. G. and S. K. performed the studies. C. D. W. designed and analyzed the in vitro pharmacology experiments. Y. D. performed and analyzed the in vitro pharmacology experiments. C. R. H. wrote the manuscript with input from all authors.Conflicts of interest
C. D. W. is an owner of WaveFront Biosciences and ION Biosciences – sellers of the Panoptic kinetic imaging plate reader and the thallium-sensitive fluorescent indicators used in these studies.Acknowledgements
The authors would like to thank Q2 Solutions (Indianapolis, IN USA) for the in vitro DMPK experiments and Pharmaron (Louisville, KY) for the in vivo DMPK experiments. The study was supported by a grant from the NIH (NIMH: R01MH107399) to C. R. H. and C. D. W.Notes and references
- M. Yamada, A. Inanobe and Y. Kurachi, Pharmacol. Rev., 1998, 50, 723 CAS.
- N. Dascala, Cell. Signalling, 1997, 9, 551 CrossRef.
- C. Lüscher and P. A. Slesinger, Nat. Rev. Neurosci., 2010, 11, 301 CrossRef PubMed.
- R. Luján, E. M. Fernandez de Velasco, C. Aguado and K. Wickman, Trends Pharmacol. Sci., 2014, 37, 20 Search PubMed.
- P. Aryal, H. Dvir, S. Choe and P. A. Slesinger, Nat. Neurosci., 2009, 12, 988 CrossRef CAS PubMed.
- H. Hibino, A. Inanobe, K. Furutani, S. Murakami, I. Findlay and Y. Kurachi, Physiol. Rev., 2010, 90, 291 CrossRef CAS PubMed.
- G. Labouebe, M. Lomazzi, H. G. Cruz, C. Creton, R. Lujan, M. Li, Y. Yanagawa, K. Obata, M. Watanabe, K. Wickman, S. B. Boyer, P. A. Slesinger and C. Luscher, Nat. Neurosci., 2007, 10, 1559 CrossRef CAS PubMed.
- C. L. Huang, P. A. Slesinger, P. J. Casey, Y. N. Jan and L. Y. Jan, Neuron, 1995, 15, 1133 CrossRef CAS PubMed.
- Q. Lei, M. B. Jones, E. M. Talley, A. D. Schrier, W. E. McIntire, J. C. Garrison and D. A. Bayliss, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9771 CrossRef CAS PubMed.
- G. Krapivinsky, E. Gordon, K. Wickman, B. Velimirovic, L. Krapivinsky and D. Clapham, Nature, 1995, 374, 135 CrossRef CAS PubMed.
- N. Dascal, W. Schreibmayer, N. F. Lim, W. Wang, C. Chaykin, L. DiMagno, C. Labarca, B. L. Kieffer, C. Gaveriaux-Ruff and D. Trollinger, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 10235 CrossRef CAS PubMed.
- Y. J. Liao, Y. N. Jan and L. Y. Jan, J. Neurosci., 1996, 16, 7137 CrossRef CAS.
- D. Jeremic, I. Sanchez-Rodriguez, L. Jimenez-Diaz and J. D. Navarro-Lopez, Pharmacol. Ther., 2021, 223, 107808 CrossRef CAS PubMed.
- Y. Zhao, I. Gameiro-Ros, I. W. Glaaser and P. A. Slesinger, Trends Pharmacol. Sci., 2021, 42, 203 CrossRef CAS PubMed.
- C. Lyu, J. Mulder, S. Barde, K. Sahlholm, H. Zeberg, J. Nilsson, P. Århem, T. Hökfelt, K. Fried and T.-J. S. Shi, Mol. Pain, 2015, 11, 44 CrossRef PubMed.
- S. Bruehl, J. S. Denton, D. F. Lonergan, M. E. Koran, M. Chont, C. Sobey, S. Fernando, W. S. Bush, P. Mishra and T. A. Thornton-Wells, Pain, 2013, 154, 2853 CrossRef CAS PubMed.
- D. Nishizawa, K. Fukuda, S. Kasai, Y. Ogai, J. Hasegawa, N. Sato, H. Yamada, F. Tanioka, H. Sugimura, M. Hayashida and K. Ikeda, J. Pharmacol. Sci., 2014, 126, 253 CrossRef CAS PubMed.
- M. Kimura, H. Shiokawa, Y. Karashima, M. Sumie, S. Hoka and K. Yamaura, PLoS One, 2020, 15, e0239094 CrossRef CAS PubMed.
- K. Kaufmann, I. Romaine, E. Days, C. Pascual, A. Malik, L. Yang, B. Zou, Y. Du, G. Sliwoski, R. D. Morrison, J. Denton, C. M. Niswender, J. S. Daniels, G. A. Sulikowski, X. Xie, C. W. Lindsley and C. D. Weaver, ACS Chem. Neurosci., 2013, 4, 1278 CrossRef CAS PubMed.
- N. Wydeven, E. M. Fernandez de Velasco, Y. Du, M. A. Benneyworth, M. C. Hearing, R. A. Fischer, M. J. Thomas, C. D. Weaver and K. Wickman, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10755 CrossRef CAS PubMed.
- E. M. Fernandez de Velasco, N. McCall and K. Wickman, Int. Rev. Neurobiol., 2015, 123, 201 CrossRef.
- N. Sugaya, T. Kobayashi and K. Ikeda, J. Drug Alcohol Res., 2013, 2, 235823 CrossRef.
- R. A. Rifkin, S. J. Moss and P. A. Slesinger, Trends Pharmacol. Sci., 2017, 38, 378 CrossRef CAS PubMed.
- C. M. Niswender, K. A. Johnson, Q. Luo, J. E. Ayala, C. Kim, P. J. Conn and C. D. Weaver, Mol. Pharmacol., 2008, 73, 1213 CrossRef CAS PubMed.
- S. J. Ramos-Hunter, D. W. Engers, K. Kaufmann, Y. Du, C. W. Lindsley, C. D. Weaver and G. A. Sulikowski, Bioorg. Med. Chem. Lett., 2013, 23, 5195 CrossRef CAS PubMed.
- W. Wen, W. Wu, C. D. Weaver and C. W. Lindsley, Bioorg. Med. Chem. Lett., 2014, 24, 5102 CrossRef CAS PubMed.
- W. Wen, W. Wu, I. Romaine, K. Kaufman, Y. Du, G. A. Sulikowski, C. D. Weaver and C. W. Lindsley, Bioorg. Med. Chem. Lett., 2013, 23, 4562 CrossRef CAS PubMed.
- K. K. Abney, M. Bubser, Y. Du, K. A. Kozek, T. M. Bridges, C. W. Lindsley, J. S. Daniels, R. D. Morrison, K. Wickman, C. R. Hopkins, C. K. Jones and C. D. Weaver, ACS Chem. Neurosci., 2019, 10, 1294 CrossRef CAS.
- X. Yu, L. Cantwell, A. I. Molosh, L. D. Plant, D. Gazgalis, S. D. Fitz, E. T. Dustrude, Y. Yang, T. Kawano, S. Garai, S. F. Noujaim, A. Shekhar, D. E. Logothetis and G. A. Thakur, J. Biol. Chem., 2020, 295, 3614 CrossRef PubMed.
- J. M. Wieting, A. K. Vadukoot, S. Sharma, K. K. Abney, T. M. Bridges, J. S. Daniels, R. D. Morrison, K. Wickman, C. D. Weaver and C. R. Hopkins, ACS Chem. Neurosci., 2017, 8, 1873 CrossRef CAS PubMed.
- S. Sharma, K. A. Kozek, K. K. Abney, S. Kumar, N. Gautam, Y. Alnouti, C. D. Weaver and C. R. Hopkins, Bioorg. Med. Chem. Lett., 2019, 29, 791 CrossRef CAS PubMed.
- R. S. Obach, Drug Metab. Dispos., 1999, 27, 1350 CAS.
- G. Chang, S. J. Steyn, J. P. Umland and D. O. Scott, ACS Med. Chem. Lett., 2010, 1, 50 CrossRef CAS.
- T. M. Bridges, R. D. Morrison, F. W. Byers, S. Luo and J. S. Daniels, Pharmacol. Res. Perspect., 2014, 2, e00077 Search PubMed.
- N. F. Smith, F. I. Raynaud and P. Workman, Mol. Cancer Ther., 2007, 6, 428 CrossRef CAS PubMed.
- J. Besnard, G. F. Ruda, V. Setola, K. Abecassis, R. M. Rodriguez, X. P. Huang, S. Norval, M. F. Sassano, A. I. Shin, L. A. Webster, F. R. Simeons, L. Sojanovsky, A. Prat, N. G. Seidah, D. B. Constam, G. R. Bickerton, K. D. Read, W. C. Wetsel, I. H. Gilbert, B. L. Roth and A. L. Hopkins, Nature, 2012, 492, 220 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00129a |
| This journal is © The Royal Society of Chemistry 2021 |