
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of push–pull triarylamine dyes containing 5,6-difluoro-2,1,3-benzothiadiazole units by direct arylation and their evaluation as active material for organic photovoltaics†
Tatiana
Ghanem
a,
Tony
Vincendeau
a,
Pablo Simón
Marqués
a,
Amir Hossein
Habibi
a,
Sana
Abidi
ab,
Ali
Yassin
a,
Sylvie
Dabos-Seignon
a,
Jean
Roncali
c,
Philippe
Blanchard
 a and
Clément
Cabanetos
*ad
a and
Clément
Cabanetos
*ad
aUniv Angers, CNRS, MOLTECH-ANJOU, SFR MATRIX, F-49000 Angers, France. E-mail: clement.cabanetos@cnrs.fr
bÉcole Nationale Supérieure d’Ingénieurs de Tunis (ENSIT), 13 Ave Taha Hussein, Tunis 1008, Tunisia
cBabes-Bolyai University, Faculty of Chemistry and Chemical Engineering, Department of Chemistry and SOOMCC, Cluj-Napoca, 11 Arany Janos Str., 400028, Cluj-Napoca, Romania
dBuilding Blocks for FUture Electronics Laboratory (2BFUEL), IRL2002, CNRS-Yonsei University, Seoul, South Korea
First published on 29th September 2021
Abstract
Two push–pull molecules involving a triarylamine donor unit connected to a dicyanovinyl acceptor moiety by a difluorobenzothiadiazole-thienyl block have been synthesized. In order to simplify the synthesis, avoid intermediate halogenation reactions and the formation of toxic organometallic by-products, building blocks were connected by direct (hetero)-arylation. The optimization of the experimental conditions of the coupling reactions is thus reported herein as well as the preliminary evaluation of the potential of the target compounds as active material in simple air-processed organic solar cells.
1. Introduction
During the past two decades, the synthesis of active materials for organic solar cells (OSCs) has generated a considerable research effort.1–3 The development of specifically designed conjugated polymers combined with the most recent non-fullerene acceptors has progressively led to OSCs with power conversion efficiencies (PCE) approaching 20%, i.e., close to those of silicon solar cells.4 Besides stability, the synthetic complexity, cost and scalability of active materials still remains a major problem for chemists involved in the development of active OPV materials.1,5 The introduction of molecules with unequivocal chemical structure as a possible alternative to polydisperse conjugated polymers around 2005 has represented a first attempt to address the problems of the cost and scalability of donor materials for OSCs.6–8 However, with a few exceptions, the synthesis of most of these molecular donors (MDs) resorts to transition-metal-catalyzed C–C cross-coupling reactions.9–11 This strategy implies the halogenation and functionalization of complementary building blocks by organometallic functional groups. In recent years the direct arylation of C–H activated aromatic or heteroaromatic moieties has emerged, as an appealing alternative to conventional organometallic coupling reactions.3,11,12 Although high conversion yields can be reached by means of careful optimizations of the reaction conditions, this method has been proven to be perfectly geared for the preparation of polymer and molecular donor materials.13–15 For instance, the synthesis of the triarylamine based push–pull TPA-T-DCV, (Fig. 1) flagship of our group, was recently simplified by the use of the C–H activation of the commercially available 2-thiophenecarboxaldehyde.16 Obtained at gram-scale in only two steps from cheap commercially available starting materials, TPA-T-DCV presents interesting potentialities as donor material in planar and co-evaporated bulk heterojunction OSCs.15–17 These promising first results paved the way to a myriad of structural variations on both the donor (D) and acceptor (A) constitutive blocks with the double objective of a better structure-properties rationalization and optimization of the photovoltaic performances.8,14–21 As shown by Wong and coworkers, the insertion of electron deficient (hetero)aromatic moiety such as benzothiadiazole (BzT) between the arylamine (D) and the dicyanovinyl group (A) represents a particularly interesting approach to finetune the ground- and excited-state characteristics of the D–A system.22 However, the use of the BzT building block generally requires harsh electrophilic halogenation conditions leading to moderate yields, mixtures of products and/or unstable intermediates.23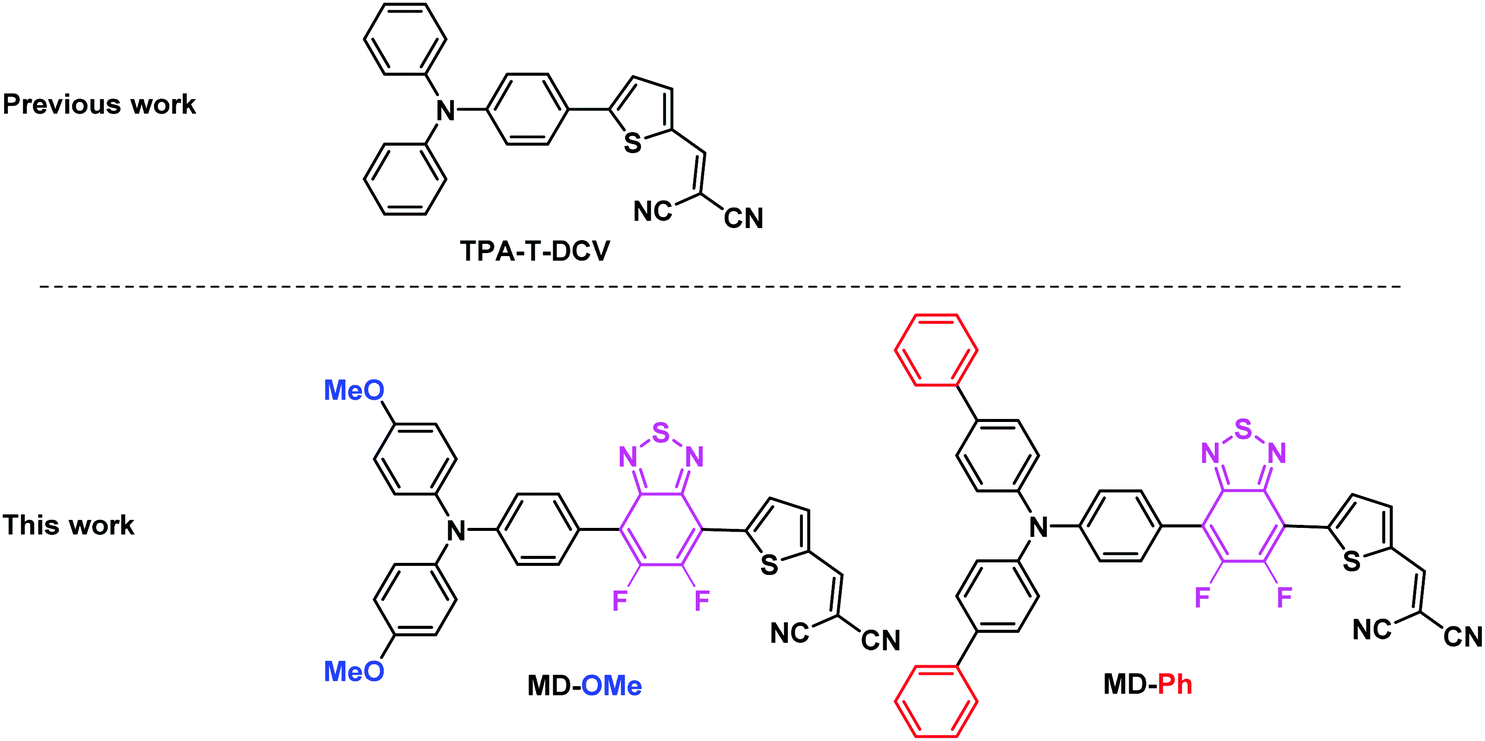 | ||
| Fig. 1 Structure of the early reported TPA-T-DCV and the two arylamine based push–pull studied herein, namely MD-OMe and MD-Ph. | ||
In this context, direct (hetero)-arylation appears to be a method of choice to overcome these synthetic problems.24,25 However, while Marder and co-workers have already reported the synthesis of symmetrical and unsymmetrical diaryl flanked 5,6-difluoro-2,1,3-benzothiadiazole (BT2F) derivatives using this methodology, the latter were not embedded in devices.19,26 Moreover, considering the very limited examples of reported BT2F based molecular systems used as active materials for organic photovoltaics, we report herein the synthesis, characterization and evaluation of the two molecular push–pull derivatives illustrated in Fig. 1. Hence, two different triphenylamine blocks were used to assess their impact on the electronic and charge transport properties of the molecules and therefore their photovoltaic efficiencies. While the methoxy groups of MD-OMe are expected to increase the donor strength of the triphenylamine block, the two biphenyl side groups of MD-Ph were recently found to significantly increase the hole-mobility of some arylamine based D–A materials.27
2. Results and discussion
Both push–pull molecules were initially prepared according to the synthetic route depicted in Scheme 1.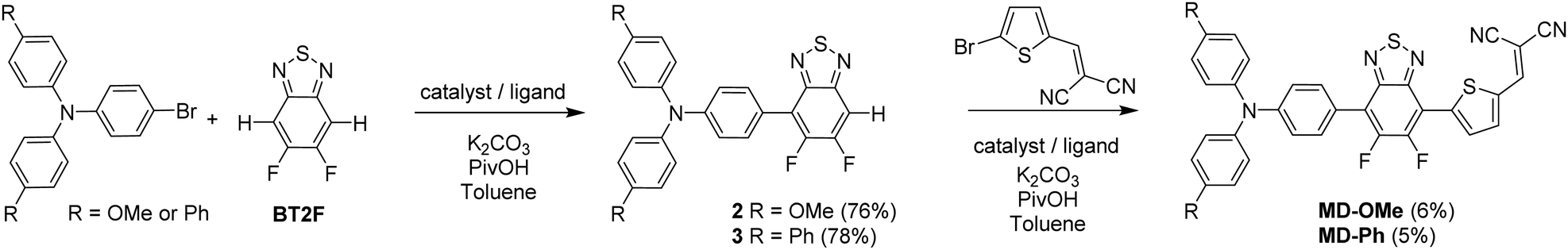 | ||
| Scheme 1 Synthetic route to MD-OMe and MD-Ph. | ||
Coupling conditions to afford arylamine functionalized unsymmetrical 5,6-difluoro-2,1,3-benzothiadiazole derivatives 2 and 3, were first carried out and optimized with the 4-bromo-N,N-bis(4-methoxyphenyl)aniline. As shown in Table 1, the first attempt was performed in presence of palladium(II) acetate and tri-tert-butylphosphonium tetrafluoroborate (PtBu3·HBF4) under conventional oil bath heating (entry 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | OMe-Br (eq) | Catalyst | Ligand | Heat source, temp./time | 2 (% yield) |
| 1 | 1.0 | Pd(OAc)2 | PtBu3·HBF4 | Oil bath 120 °C; 16h | 20 |
| 2 | 0.5 | Pd(OAc)2 | PtBu3·HBF4 | Oil bath (120 °C; 16h) | 33 |
| 3 | 0.5 | Pd(OAc)2 | PtBu3·HBF4 | μ-wave (150 °C; 1 h) | 36 |
| 4 | 0.5 | Pd(OAc)2 | P(o-MePh)3 | μ-wave (150 °C; 1 h) | 40 |
| 5 | 0.5 | Pd(OAc)2 | PCy3·HBF4 | μ-wave (150 °C; 1 h) | 76 |
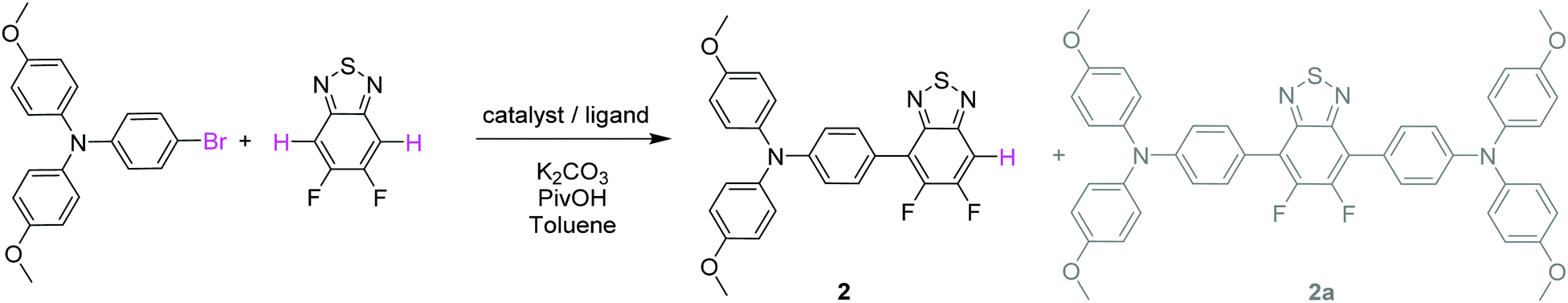
Despite operating in relatively diluted conditions, only moderate yields were obtained mainly because of the major formation of the symmetrical arylamine end capped derivative (2a). The reduction of the number of equivalents of bromo derivative by a factor of two results in a slight improvement of the conversion yield (entry 2). In parallel, the replacement of the conventional oil bath by microwave irradiation (entry 3) leads to a further slight improvement of the yield but considerably shortens the reaction time. One hour of irradiation appears as a good compromise between the consumption of the starting material and the formation of the symmetrical by-product (2a). Afterwards, the initial catalytic coupled was modified by replacing the tri-tert-butylphosphonium tetrafluoroborate either by the tri(o-tolyl)phosphine (entry 4) or the tricyclohexylphosphine tetrafluoroborate (entry 5). While similar conversion rates were monitored with the neutral ligand (entry 4), a descent synthetic yield reaching almost 80% was achieved with the bulkier charged analogue (entry 5). Applied to the biphenyl derivative, these conditions led to the target unsymmetrical 5,6-difluoro-2,1,3-benzothiadiazole based analogue 3 in similar good yields (78%). Each monoarylated products were then subsequently engaged in another cross-coupling reaction with the 2-bromo-5-(2,2-dicyanovinyl)thiophene.28 Even if the unsymmetrical functionalization of the BT2F core by direct arylation was successfully operated with electron rich moieties, low yields were achieved mainly due to the mild reactivity of the halogenated thiophene that appeared to be deactivated by the dicyanovinyl group (Scheme 1). Consequently, and as compromise between reactivity and extra synthetic steps, carbaldehyde functionalized unsymmetrical 5,6-difluoro-2,1,3 benzothiadiazole derivatives 4 and 5 were prepared by coupling 2 and 3 with the 5-bromothiophene-2-carbaldehyde respectively (Scheme 2).
 | ||
| Scheme 2 Second synthetic route considered to MD-OMe and MD-Ph through a Knoevenagel reaction. | ||
Though still modest, synthetic yields achieved were nonetheless significantly improved and are in consistency with early reported results.26 As a further step toward simplification, attempts to prepare these carbaldehyde derivatives by a one-pot like method were thus carried out. To do so, BT2F was first reacted with bromo arylamine before adding the 5-bromothiophene-2-carbaldehyde to the reaction mixture. After a second round of microwave irradiation, carbaldehyde 4 and 5 were successfully isolated with synthetic yields similar to those achieved with the above described two-step procedure. Finally, the later compounds were engaged in a Knoevenagel reaction with malononitrile to afford the corresponding final push–pull molecules. Despite full conversion confirmed in both cases by TLC, the biphenyl derivative was isolated in lower yield due to a limited solubility at room temperature.
Identity and purity of the final compound was confirmed by NMR spectroscopy and mass spectrometry (see ESI†). While thermal gravimetric analyses (TGAs) confirmed that both compounds can meet requirements of device fabrication, MD-OMe exhibits a significantly improved thermal stability compared to π-extended MD-Ph with a 5% weight loss recorded at 354 °C vs. 248 °C respectively (see ESI†). Fig. 2 shows the UV-vis absorption spectra of the two compounds in dichloromethane (DCM) and as thin film on glass.
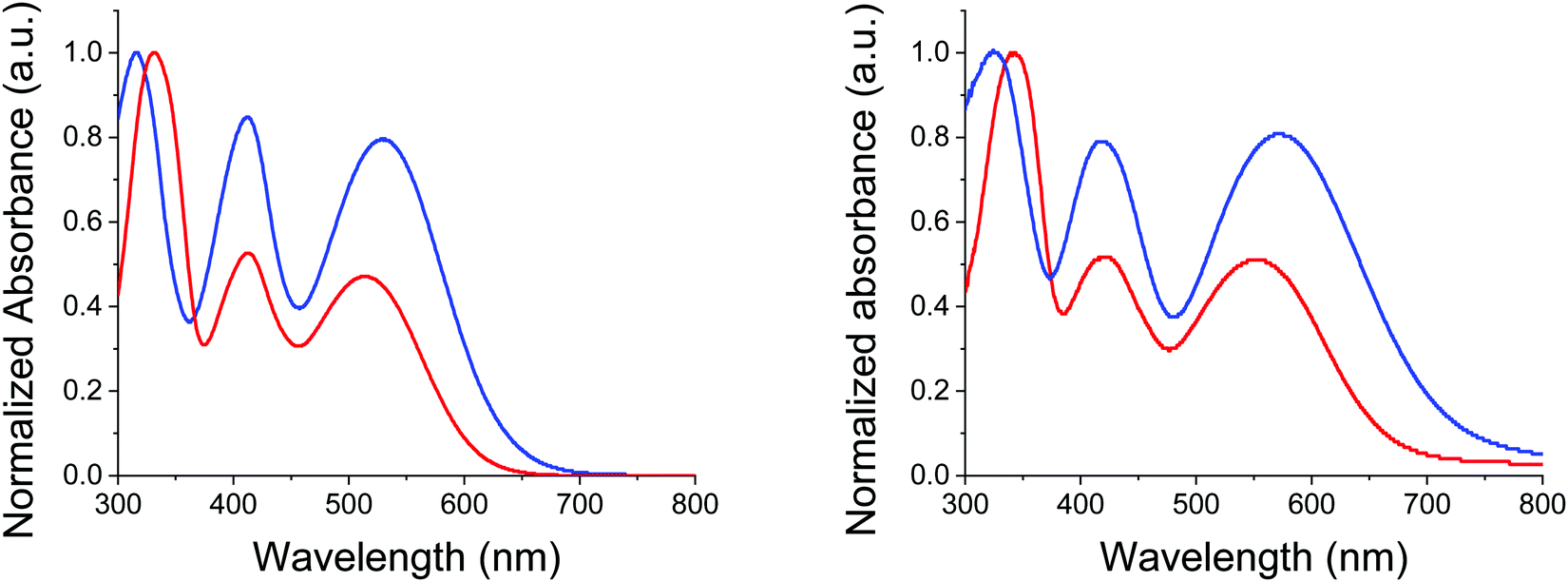 | ||
| Fig. 2 UV-Vis absorption spectra of MD-OMe (blue) and MD-Ph (red) in DCM (left) and as thin films on glass (right). | ||
The spectrum of both compounds shows three successive absorption bands with maxima at 332, 413 and 513 nm for the biphenyl compound MD-Ph in solution. Replacement of the biphenyl by a methoxy group produces, as expected, a small bathochromic shift of the maxima at longer wavelength (ca 14 nm) reflecting the higher electron donor effect of the methoxy group compared to a phenyl ring. This behavior results in a reduction of the optical bandgap estimated from the low-energy absorption onset of thin films from 1.86 to 1.68 eV. The comparison between the spectra of the two compounds shows that the introduction of the methoxy groups on the TPA block also increases the molar absorptivity of the visible absorption bands by almost a factor of two (Table 2).
| Molecule | λ maxabs solution (nm) | ε (M−1 cm−1) | λ maxabs thin film (nm) | E optg (eV)a | HOMO (eV)b | LUMO (eV)c |
|---|---|---|---|---|---|---|
| a Estimated from the low-energy absorption onset of thin films. b Determined by photoelectron spectroscopy in air (PESA). c LUMO = HOMO − Eoptg. | ||||||
| MD-OMe | 315 | 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 800 |
325 | 1.68 | −5.43 | −3.75 |
| 412 | 44100 |
421 | ||||
| 527 | 41100 |
571 | ||||
| MD-Phe | 332 | 42900 |
343 | 1.86 | −5.64 | −3.78 |
| 413 | 21900 |
423 | ||||
| 513 | 20200 |
552 | ||||
While the UV-vis absorption spectrum of TPA-T-DCV and many of its parent systems usually exhibits a single transition in the visible region, attributed to an internal charge transfer (ICT) from the TPA moiety to the DCV group, the spectra of MD-OMe and MD-Ph shows two distinct intense absorption bands in the 400–600 nm region. In order to get further insights on this question, ground-state density-functional theory (DFT) geometry optimizations and density-functional theory (TD-DFT) calculations were carried out using B3LYP/6-311G* basis sets (see ESI†).
As expected, these experiments first confirmed the ICT nature of the lowest energy contribution assigned to a HOMO → LUMO transition. Calculations indeed showed that the electronic density of the HOMO levels is mainly localized on the arylamine moiety while the LUMOs are on the benzothiadiazole/DCV blocks (Fig. 3).
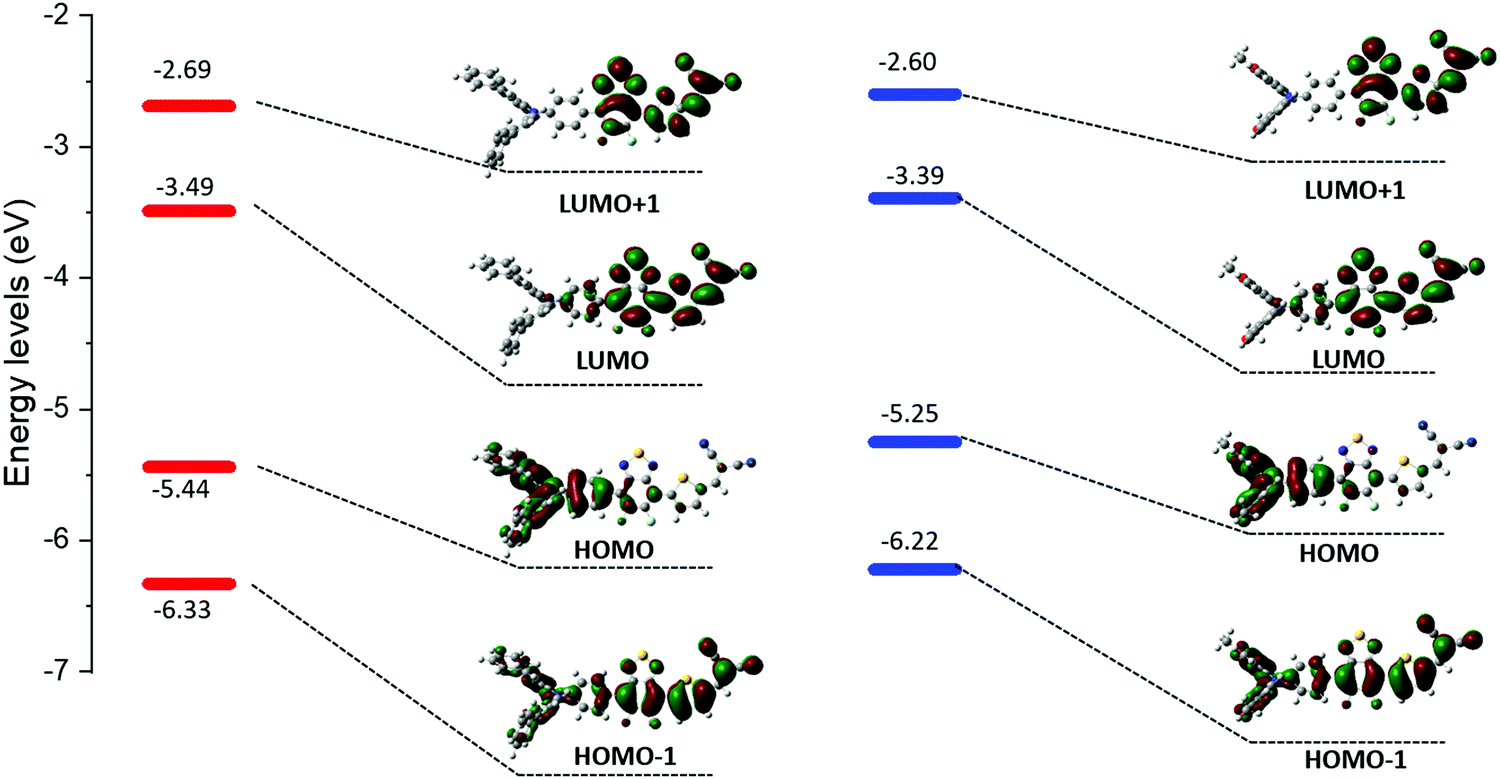 | ||
| Fig. 3 Optimized geometries and computed energy levels of MD-Ph (left) and MD-OMe (right). | ||
The absorption bands at ca 400 nm, were also found to have a charge-transfer character since they predominantly arise from a HOMO−1 → LUMO transition (see ESI†). Moreover, the band gap reduction induced by the electron-donating methoxy groups (MD-OMe) compared to simple phenyl rings was found to originate from the significant destabilization of the HOMO level exceeding by far that of the LUMO (ΔHOMO = 0.19 eV vs. ΔLUMO = 0.10 eV). Hence, photoelectron spectroscopy in air (PESA) measurements were subsequently performed and HOMO levels of −5.43 eV and −5.64 eV were calculated for MD-OMe and MD-Ph respectively. Once cross-tabulated with UV-visible experiments, the corresponding LUMO levels were simply calculated by adding the optical band gap (film spectra) to the HOMO values thus confirming, as depicted in the Fig. 4, the computed predictions.
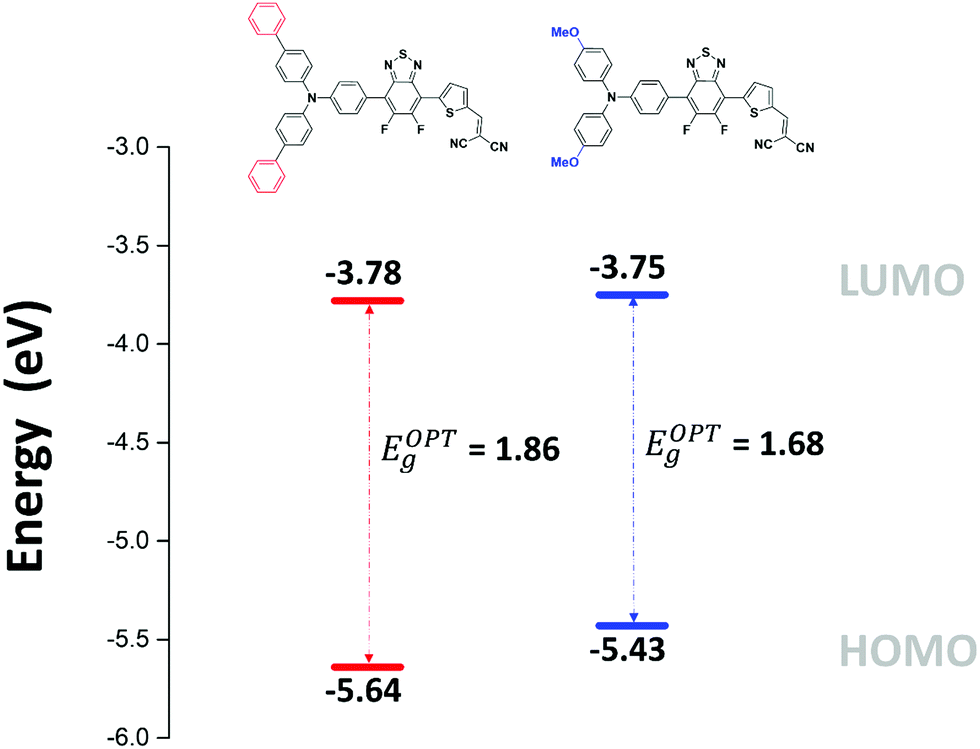 | ||
| Fig. 4 Energy level diagram estimated in solid state from PESA and UV-visible experiments. | ||
Designed as potential donor molecular systems for organic solar cells, such difference in HOMO level might have a significant impact on the photovoltaic parameters, in particular on the open circuit voltage (Voc). Consequently, simple air-processed bulk heterojunction (BHJ) solar cells using [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) as electron acceptor material were fabricated. Active layers with different donor: acceptor (D/A) weight to weight (w/w) ratios were processed in air from chloroform solutions, and embedded in devices of architectures ITO/PEDOT:PSS/active layer/Ca/Al. The photovoltaic data measured under AM 1.5 simulated solar light illumination (100 mW cm−2) are gathered in Table 3.
| Molecular Donor | D:A ratio (w/w) |
V oc (V) | J sc (mA cm−2) | FF | PCE (%) |
|---|---|---|---|---|---|
| MD-OMe | 1:1 |
0.70 ± 0.01 (0.71) | −3.15 ± 0.04 (−3.18) | 0.33 ± 0.02 (0.33) | 0.70 ± 0.04 (0.74) |
| MD-OMe | 1:2 |
0.84 ± 0.01 (0.85) | −4.53 ± 0.09 (−4.55) | 0.33 ± 0.01 (0.35) | 1.28 ± 0.05 (1.35) |
| MD-OMe | 1:3 |
0.76 ± 0.02 (0.77) | −4.72 ± 0.04 (−4.73) | 0.32 ± 0.02 (0.33) | 1.14 ± 0.06 (1.20) |
| MD-Ph | 1:1 |
1.00 ± 0.01 (1.01) | −4.50 ± 0.08 (−4.56) | 0.31 ± 0.02 (0.33) | 1.41 ± 0.08 (1.52) |
| MD-Ph | 1:2 |
1.00 ± 0.01 (1.01) | −5.43 ± 0.07(−5.47) | 0.33 ± 0.01 (0.34) | 1.81 ± 0.07 (1.88) |
| MD-Ph | 1:3 |
0.96 ± 0.02 (0.96) | −6.34 ± 0.09 (−6.43) | 0.32 ± 0.02 (0.34) | 1.95 ± 0.14 (2.09) |
The best power conversion efficiencies, of 1.35% and 2.09%, were reached with a 1:2 and 1:3 D/A ratio for MD-OMe and MD-Ph based devices, respectively. Comparison of their corresponding current density to voltage (J–V) curves revealed that the better efficiencies recorded in MD-Ph based OSCs mainly arise from their higher short circuit current (Jsc) and open circuit voltage (Voc) values (Fig. 5).
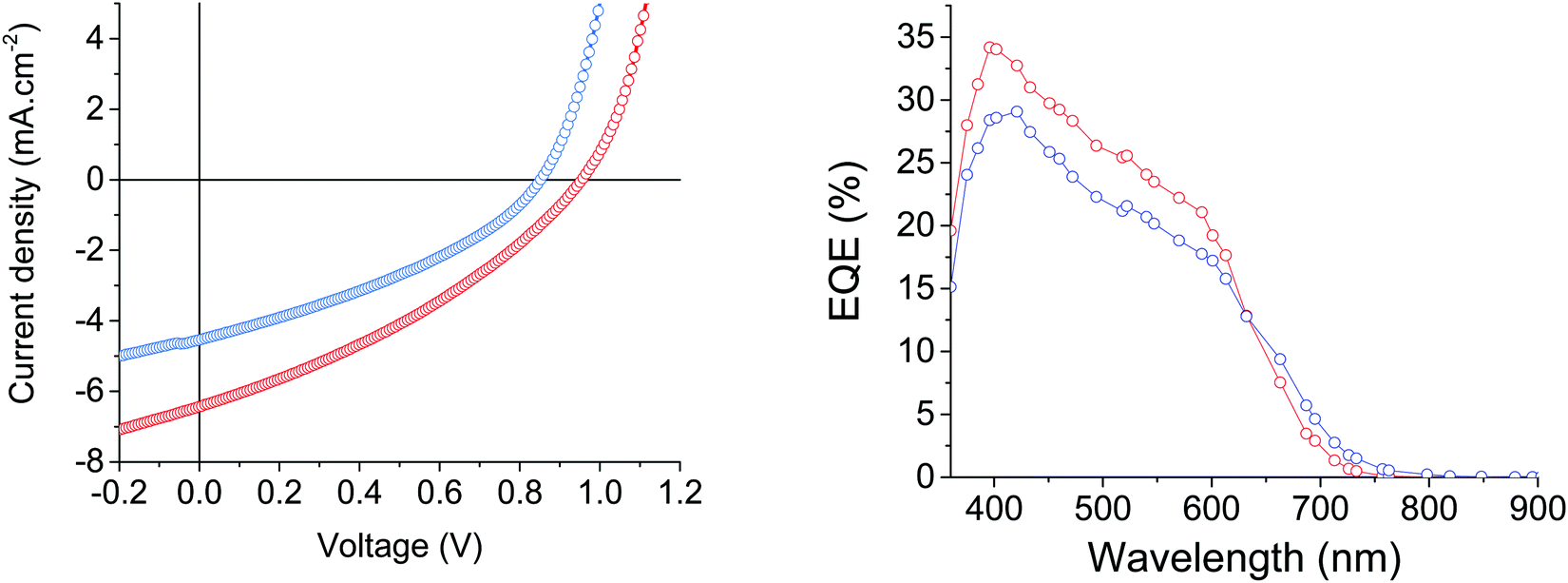 | ||
| Fig. 5 Current density–voltage characteristics and EQE spectra of the best MD-OMe (blue) and MD-Ph (red) based organic solar cells. | ||
While the latter parameter (Voc) can be correlated to the deeper HOMO level of the biphenyl derivative, MD-OMe was nonetheless characterized by better absorbing properties in the visible range which was expected to lead to higher Jsc. Interestingly and in agreement with the recorded Jsc values, external quantum efficiency measurements performed on the best devices also highlighted an improved contribution of the biphenyl derivative to the photocurrent compared to its methoxy counterpart (Fig. 5). Hence, in an attempt to rationalize these results, morphologies of the active layers were first probed by atomic force microscopy (AFM). As illustrated in Fig. 6, smooth surface topographies with similar roughness (RMS) parameters of ca 0.6 nm were observed in both cases (Fig. 6).
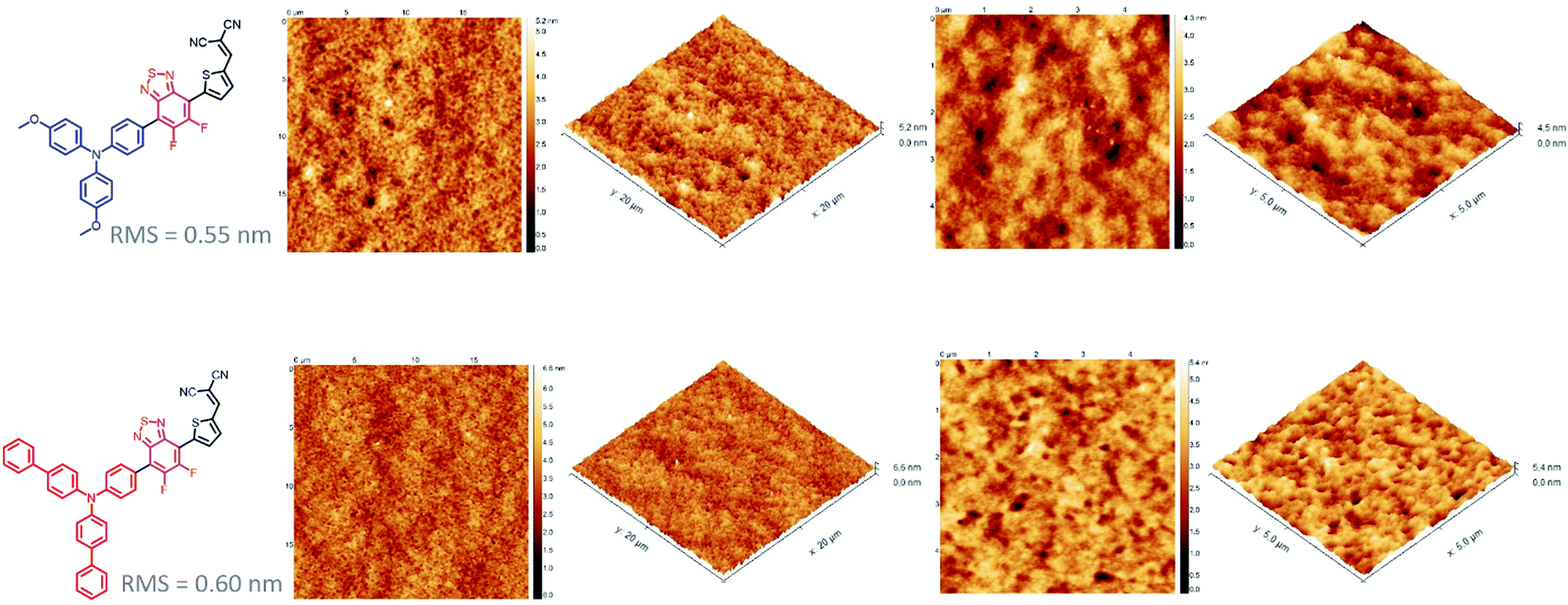 | ||
| Fig. 6 AFM phase images (20 × 20 and 5 × 5 μm) of optimized MD-OMe and MD-Ph based active layers. | ||
Nonetheless, MD-Ph/PC71BM blend exhibits nano domains of smaller size than those observed with the methoxy derivative which can be the harbinger of a better biphenyl induced self-organization of the donor molecule which can, in turn, be beneficial for the charge transport properties and therefore the Jsc parameter.
Consequently, both optimized active layers were subsequently embedded in hole-only devices of architecture ITO/PEDOT:PSS/donor/Au. Through the use of the space charge limited current (SCLCs) method, hole mobilities of ca 7.3 × 10−5 and 8.3 × 10−4 cm2 V−1 s−1 were measured for MD-OMe and MD-Ph respectively (see ESI†). Consequently, this order-of-magnitude difference might compensate the lower absorbing properties of the active layer and contribute to the higher Jsc value delivered by the MD-Ph based devices.
3. Conclusion
In summary, two new triphenylamine based push–pull molecules containing 5,6-difluoro-2,1,3-benzothiadiazole units have been synthesized. In order to simplify the synthesis procedure and limit the formation of potentially toxic by-products, two major steps of the synthesis were performed using optimized direct (hetero)-arylation conditions. As a step towards simplification, the synthetic route was further shortened by the use of a “one-pot” procedure without modification of the catalytic system nor purification of intermediates. The analysis of the electronic properties of the two compounds shows, in agreement with theoretical results, that the methoxy derivative presents a red-shifted absorption, a higher molecular absorption coefficient and leads to a material of smaller band gap. The evaluation of the photovoltaic properties of the two compounds as donor material in BHJ cells shows that in spite of less favorable optical properties, the biphenyl derivative leads to better performances due to the conjunction of a higher open-circuit voltage and better charge transport properties. Evaluated in simple air processed devices, these preliminary results pave the way to further optimization and device engineering to reveal the true potential of these accessible and greener materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The French ministry of Research is acknowledged for the PhD grant of T. G. (project MOSA). P. S. M. thanks the European Union's Horizon 2020 research and innovation program under Marie Sklodowska Curie Grant agreement No. 722651 (SEPOMO). A. H. A and S. A. thank the University of Isphan and the University of Tunis for their grants, respectively.References
- M. Riede, D. Spoltore and K. Leo, Organic Solar Cells—The Path to Commercial Success, Adv. Energy Mater., 2021, 11(1), 2002653, DOI:10.1002/aenm.202002653.
- L. Duan and A. Uddin, Progress in Stability of Organic Solar Cells, Adv. Sci., 2020, 7(11), 1903259, DOI:10.1002/advs.201903259.
- L. Hong, H. Yao, Y. Cui, Z. Ge and J. Hou, Recent Advances in High-Efficiency Organic Solar Cells Fabricated by Eco-Compatible Solvents at Relatively Large-Area Scale, APL Mater., 2020, 8(12), 120901, DOI:10.1063/5.0027948.
- Y. Lin, A. Magomedov, Y. Firdaus, D. Kaltsas, A. El-Labban, H. Faber, D. R. Naphade, E. Yengel, X. Zheng, E. Yarali, N. Chaturvedi, K. Loganathan, D. Gkeka, S. H. AlShammari, O. M. Bakr, F. Laquai, L. Tsetseris, V. Getautis and T. D. Anthopoulos, 18.4% Organic Solar Cells Using a High Ionization Energy Self-Assembled Monolayer as Hole-Extraction Interlayer, ChemSusChem, 2021, 14, 3569–3578, DOI:10.1002/cssc.202100707.
- R. Po and J. Roncali, Beyond Efficiency: Scalability of Molecular Donor Materials for Organic Photovoltaics, J. Mater. Chem. C, 2016, 4(17), 3677–3685, 10.1039/C5TC03740A.
- J. Roncali, P. Leriche and P. Blanchard, Molecular Materials for Organic Photovoltaics: Small Is Beautiful, Adv. Mater., 2014, 26(23), 3821–3838, DOI:10.1002/adma.201305999.
- A. Venkateswararao and K.-T. Wong, Small Molecules for Vacuum-Processed Organic Photovoltaics: Past, Current Status, and Prospect, Bull. Chem. Soc. Jpn., 2021, 94(3), 812–838, DOI:10.1246/bcsj.20200330.
- C. Cabanetos, P. Blanchard and J. Roncali, Arylamine Based Photoactive Push-Pull Molecular Systems: A Brief Overview of the Chemistry “Made in Angers, Chem. Rec., 2019, 19(6), 1123–1130, DOI:10.1002/tcr.201800184.
- J. Kudrjasova, J. Kesters, P. Verstappen, J. Brebels, T. Vangerven, I. Cardinaletti, J. Drijkoningen, H. Penxten, J. Manca, L. Lutsen, D. Vanderzande and W. Maes, A Direct Arylation Approach towards Efficient Small Molecule Organic Solar Cells, J. Mater. Chem. A, 2016, 4(3), 791–795, 10.1039/C5TA09023G.
- K. Okamoto, J. Zhang, J. B. Housekeeper, S. R. Marder and C. K. Luscombe, C–H Arylation Reaction: Atom Efficient and Greener Syntheses of π-Conjugated Small Molecules and Macromolecules for Organic Electronic Materials, Macromolecules, 2013, 46(20), 8059–8078, DOI:10.1021/ma401190r.
- J. He, Y. J. Jo, X. Sun, W. Qiao, J. Ok, T. Kim and Z. Li, Squaraine Dyes for Photovoltaic and Biomedical Applications, Adv. Funct. Mater., 2021, 31(12), 2008201, DOI:10.1002/adfm.202008201.
- T. Bura, J. T. Blaskovits and M. Leclerc, Direct (Hetero)Arylation Polymerization: Trends and Perspectives, J. Am. Chem. Soc., 2016, 138(32), 10056–10071, DOI:10.1021/jacs.6b06237.
- M. Mainville and M. Leclerc, Direct (Hetero)Arylation: A Tool for Low-Cost and Eco-Friendly Organic Photovoltaics, ACS Appl. Polym. Mater., 2021, 3(1), 2–13, DOI:10.1021/acsapm.0c00883.
- P. Josse, S. Dayneko, Y. Zhang, S. Dabos-Seignon, S. Zhang, P. Blanchard, G. C. Welch and C. Cabanetos, Direct (Hetero)Arylation Polymerization of a Spirobifluorene and a Dithienyl-Diketopyrrolopyrrole Derivative: New Donor Polymers for Organic Solar Cells, Molecules, 2018, 23(4), 962, DOI:10.3390/molecules23040962.
- Z. Ni, H. Wang, H. Dong, Y. Dang, Q. Zhao, X. Zhang and W. Hu, Mesopolymer Synthesis by Ligand-Modulated Direct Arylation Polycondensation towards n-Type and Ambipolar Conjugated Systems, Nat. Chem., 2019, 11(3), 271–277, DOI:10.1038/s41557-018-0200-y.
- J. Grolleau, F. Gohier, M. Allain, S. Legoupy, C. Cabanetos and P. Frère, Rapid and Green Synthesis of Complementary D–A Small Molecules for Organic Photovoltaics, Org. Electron., 2017, 42, 322–328, DOI:10.1016/j.orgel.2016.12.046.
- A. Leliège, C.-H. L. Régent, M. Allain, P. Blanchard and J. Roncali, Structural Modulation of Internal Charge Transfer in Small Molecular Donors for Organic Solar Cells, Chem. Commun., 2012, 48(71), 8907–8909, 10.1039/C2CC33921H.
- A. Labrunie, Y. Jiang, F. Baert, A. Leliège, J. Roncali, C. Cabanetos and P. Blanchard, Small Molecular Push–Pull Donors for Organic Photovoltaics: Effect of the Heterocyclic π-Spacer, RSC Adv., 2015, 5(124), 102550–102554, 10.1039/C5RA21958B.
- J. W. Choi, C.-H. Kim, J. Pison, A. Oyedele, D. Tondelier, A. Leliège, E. Kirchner, P. Blanchard, J. Roncali and B. Geffroy, Exploiting the Potential of 2-((5-(4-(Diphenylamino)Phenyl)Thiophen-2-Yl)Methylene)Malononitrile as an Efficient Donor Molecule in Vacuum-Processed Bulk-Heterojunction Organic Solar Cells, RSC Adv., 2014, 4(10), 5236–5242, 10.1039/C3RA47059H.
- Y. Jiang, C. Cabanetos, M. Allain, P. Liu and J. Roncali, Manipulation of the Band Gap and Efficiency of a Minimalist Push–Pull Molecular Donor for Organic Solar Cells, J. Mater. Chem. C, 2015, 3(20), 5145–5151, 10.1039/C5TC00913H.
- A. Labrunie, P. Josse, S. Dabos-Seignon, P. Blanchard and C. Cabanetos, Pentaerythritol Based Push–Pull Tetramers for Organic Photovoltaics, Sustainable Energy Fuels, 2017, 1(9), 1921–1927, 10.1039/C7SE00345E.
- L.-Y. Lin, Y.-H. Chen, Z.-Y. Huang, H.-W. Lin, S.-H. Chou, F. Lin, C.-W. Chen, Y.-H. Liu and K.-T. Wong, A Low-Energy-Gap Organic Dye for High-Performance Small-Molecule Organic Solar Cells, J. Am. Chem. Soc., 2011, 133(40), 15822–15825, DOI:10.1021/ja205126t.
- J. Zhang, T. C. Parker, W. Chen, L. Williams, V. N. Khrustalev, E. V. Jucov, S. Barlow, T. V. Timofeeva and S. R. Marder, C–H-Activated Direct Arylation of Strong Benzothiadiazole and Quinoxaline-Based Electron Acceptors, J. Org. Chem., 2016, 81(2), 360–370, DOI:10.1021/acs.joc.5b02551.
- Y. Jiang, C. Cabanetos, M. Allain, S. Jungsuttiwong and J. Roncali, Manipulation of the Electronic and Photovoltaic Properties of Materials Based on Small Push-Pull Molecules by Substitution of the Arylamine Donor Block by Aliphatic Groups, Org. Electron., 2016, 37, 294–304, DOI:10.1016/j.orgel.2016.06.035.
- S. Yu, F. Liu, J. Yu, S. Zhang, C. Cabanetos, Y. Gao and W. Huang, Eco-Friendly Direct (Hetero)-Arylation Polymerization: Scope and Limitation, J. Mater. Chem. C, 2017, 5(1), 29–40, 10.1039/C6TC04240F.
- J. Zhang, W. Chen, A. J. Rojas, E. V. Jucov, T. V. Timofeeva, T. C. Parker, S. Barlow and S. R. Marder, Controllable Direct Arylation: Fast Route to Symmetrical and Unsymmetrical 4,7-Diaryl-5,6-Difluoro-2,1,3-Benzothiadiazole Derivatives for Organic Optoelectronic Materials, J. Am. Chem. Soc., 2013, 135(44), 16376–16379, DOI:10.1021/ja4095878.
- P. S. Marqués, J. M. Andrés Castán, P. Josse, M. Blais, A. H. Habibi, I. Ramirez, K. Walzer, J. Roncali, P. Blanchard and C. Cabanetos, Effect of 4-Biphenyl Groups on the Charge Transport and Photovoltaic Properties of Arylamine Based Push–Pull Systems, New J. Chem., 2020, 44(27), 11441–11447, 10.1039/D0NJ02019B.
- R. Fitzner, E. Reinold, A. Mishra, E. Mena-Osteritz, H. Ziehlke, C. Körner, K. Leo, M. Riede, M. Weil, O. Tsaryova, A. Weiß, C. Uhrich, M. Pfeiffer and P. Bäuerle, Dicyanovinyl–Substituted Oligothiophenes: Structure-Property Relationships and Application in Vacuum-Processed Small Molecule Organic Solar Cells, Adv. Funct. Mater., 2011, 21(5), 897–910, DOI:10.1002/adfm.201001639.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00798j |
| This journal is © The Royal Society of Chemistry 2021 |