
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAzobenzene isomerization in condensed matter: lessons for the design of efficient light-responsive soft-matter systems
Luke W.
Giles
 a,
Charl F. J.
Faul
b and
Rico F.
Tabor
*a
a,
Charl F. J.
Faul
b and
Rico F.
Tabor
*a
aSchool of Chemistry, Monash University, Clayton 3800, Victoria, Australia. E-mail: rico.tabor@monash.edu; Tel: +61 3 9905 4558
bSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK
First published on 3rd June 2021
Abstract
Azobenzene-based materials have found applications in a wide variety of systems as a convenient method to induce photo-responsive behaviour through facile trans (E) to cis (Z) isomerization. Of particular interest are condensed matter systems, for which diverse assemblies have been applied in myriad ways to produce optical materials, controllable assemblies and functional surfaces. However, isomerization of azobenzene faces unique challenges in condensed systems when compared with dilute states (molecular solution and gas phase) where isomerization is best understood. In the present work, we explore recent studies looking at these challenges experienced in condensed matter phases, some solutions proposed to improve isomerization efficiency, and further give these issues wider context for the future development of light-responsive soft-matter systems.
1 Introduction
Stimuli-responsive materials are in high demand for the development of ‘smart' materials able to change or modify their physical and/or chemical properties upon application of a stimulus.1–3 This can be achieved through inclusion of molecules that respond to internal or external stimuli such as changes in pH, temperature, magnetism or light. Of these stimuli, light is particularly appealing as it is comparatively low energy and can be externally applied in a highly spatially and temporally resolved manner, and photochemical processes (particularly isomerizations) are generally selective, fast, and high yielding.4 There are a number of known light-responsive, or photochromic groups that exhibit conformational changes or breakage/formation of bonds upon irradiation of specific wavelengths of light. Examples of these groups include azobenzenes,5,6 stilbenes,7,8 diarylethenes,9–12 and spyropyrans12,13 (selected examples shown in Scheme 1).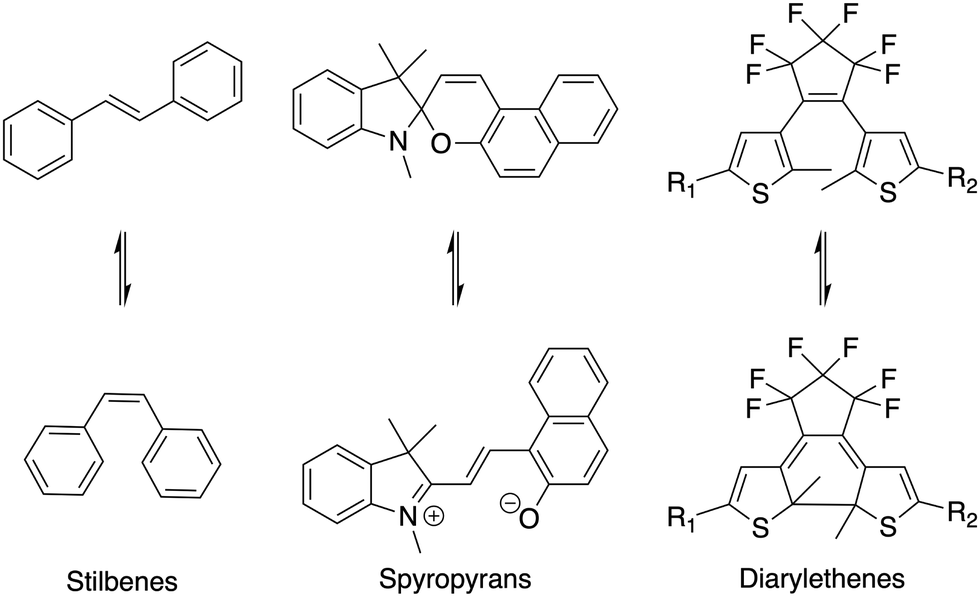 | ||
| Scheme 1 Examples of some known photochromic functional groups and their photo-induced structural changes. | ||
The azobenzene functional group is of particular interest for incorporation into ‘smart’ materials because it has many facile synthetic routes and the readily accessible π to π* transition can be used to induce isomerization.14,15 Azobenzenes have been used for the development of light-responsive materials including molecular electronics,16 photonics,17–21 sensors and actuators.22 They have been used to invoke photoswtiching in host–guest self-assembled structures,23–25 selective molecular capture26 and separations.27 Central to the effective application of the azobenzene group is an understanding of its E/Z isomerization, including the energy required for electronic excitation, kinetic rates of the isomerization process, and the mechanism of the switching process. The switching behaviour of azobenzene molecules is well described in the gas phase28 and in solution,29 however, additional complications arise in condensed and self-assembled systems such as liquid crystals (LCs), photo-surfactant assemblies, and surface adsorbed monolayers (SAMs), which will all be discussed later, particularly in Section 6.
While liquid crystal polymers also fit the criteria of being condensed matter phases of particular interest for the incorporation of light-responsive azobenzene units, they are, by and large, omitted from this review. They are omitted here because of additional complexities involved when considering liquid crystal polymers including reduced freedom of mixing of the azobenzene units when chemically bonded to a polymeric backbone, and the volume of studies investigating the topic. These reasons make azobenzene-containing liquid crystal polymers deserving, and indeed already the subject of reviews in their own right. Existing reviews cover aspects of azobenzene-containing liquid crystal polymer behaviour from targeted applications,30 surface and material structure control,31,32 and design principles.33
Condensed systems, which are generally of more use for application in ‘smart’ devices, incur additional complexity with potential for electronic quenching, exciton formation and steric considerations. This creates a multi-scale or many-body problem where behaviour of a condensed system cannot be assumed to be an additive summation of its constituent monomeric units.34 Surface adsorbed monolayers (SAMs) distinctly show all of these behaviours; when unaccounted for, these many body effects can completely quench or severely inhibit the photoisomerization behaviour of the monomeric units, as was seen in many of the early attempts at photoresponsive SAM formation.35–39 This has inspired a number of computational and experimental studies identifying mechanisms of deactivation and methods of reinstating isomerization behaviour. While SAMs offer an idealized case to study these interactions, the impacts of this area of study are more widely applicable for creation of efficient photo-switching devices based on a number of condensed matter systems.
This review focuses on the current state of the literature, examining the isomerization of azobenzene in condensed states. The example of SAMs is highlighted for the depth of insight these systems can offer on the underlying mechanisms behind isomerization quenching and tools that can be used to rectify these problems. However, the studies are put into the context of other condensed systems. The implications of increasing intermolecular interactions in these systems are examined, and the consequences of these interactions for ‘smart’ device formation are explored.
2 Switching mechanism
For realization of switchable materials and devices, the azobenzene core unit needs to be stimulated to isomerize between trans (E) and cis (Z) states. The trans isomer is more thermodynamically stable40,41 and adopts a planar structure,42,43 whereas the cis state is non-planar.44 Consequently, the trans state is generally the main focus of stimulated isomerization and the trans-dominant photostationary state is more close packed and susceptible to excimer formation due to the proximity of neighbouring azobenzene units.The azobenzene functional group undergoes a facile trans (E) to cis (Z) photo-induced isomerization. There is general agreement that trans to cis isomerization proceeds predominantly through excitation from from S2 ← S0 (π to π*) electronic states,45,46 with a smaller contribution from direct excitation through the symmetry forbidden S1 ← S0 (n to π*) transition.46,47 Isomerization then proceeds through one of four proposed mechanisms: rotation,43 inversion,40 concerted inversion48 or inversion-assisted rotation (Scheme 2). Multiple relaxation pathways are often called upon to explain various experimental observations.49 While the focus of this review is not specifically on the isomerization mechanism, it is important to note that this process is sensitive to changes in electronic states and hindrance of ring movement from the localized environment. A more general review on azobenzene switching can be found here.15
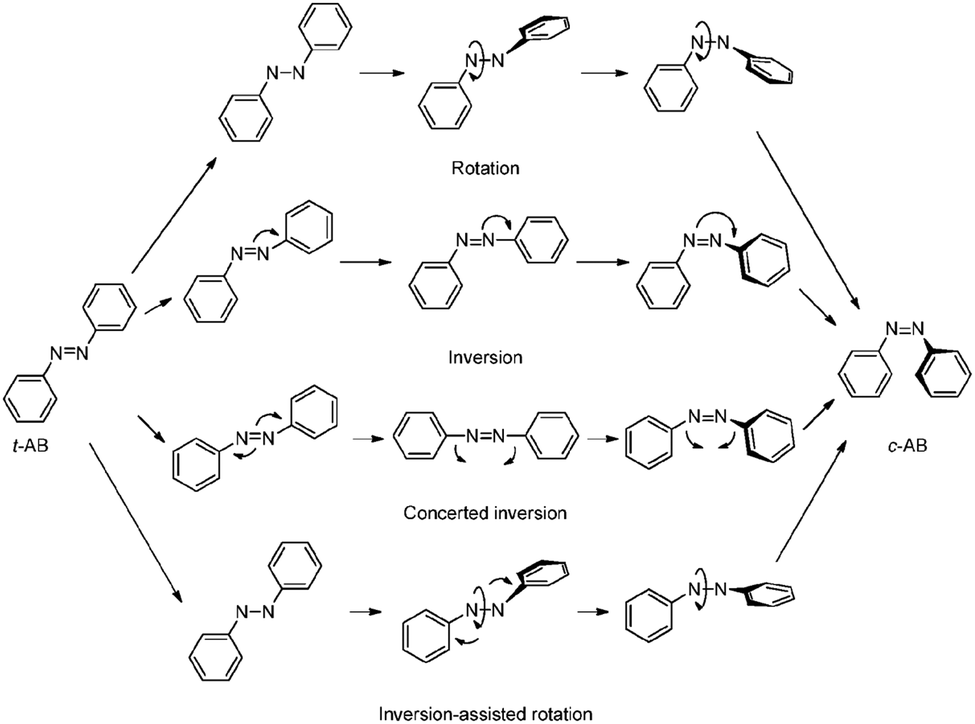 | ||
| Scheme 2 Proposed mechanisms for the trans to cis isomerization of azobenzene. Reproduced from ref. 15 with permission from The Royal Society of Chemistry.15 | ||
There are other design considerations that should be taken into account when designing azobenzene-containing materials for responsive materials. Since the S2 excitation is of the most importance for trans to cis isomerization, it is important to understand the influence that substitutions can have on the absorption spectrum of azobenzene. Particular effort has been made to employ substitutions to red-shift the absorption band of azobenzenes.50,51 Similarly, even the polarity of the solvent can have an effect on shifting the S2 peak.52 Care should be taken for each system to target the excitation wavelength used for greatest efficiency.
Substitutions can also have a stark effect on the stability of the cis isomer, therefore also having an effect on the rate of cis to trans reverse isomerization.53 One key example of this is so-called ‘push–pull’ azobenzenes, which have electron donating and electron withdrawing substitutions in opposing para positions (4- and 4′-positions) on the azobenzene rings. This “push–pull” arrangement can lead to the quantum yield of trans to cis isomerization becoming significantly less than that of the reverse cis to trans process.54
Cis to trans reverse isomerization may occur either from light stimulation or through thermal relaxation, which has an activation energy of around ∼95 kJ mol−1.55–57 Temperature does not affect the quantum yield of the trans to cis isomerization, however, the rate of thermal cis to trans isomerization increases with elevated temperature. Consequently, the trans isomer becomes more dominant at high temperatures. A more nuanced discussion of the effects of substitutions on the stability of the cis isomer and the rate of thermal cis to trans isomerization can be found here.15
While design aspects including substitutions, solvent choice, and temperature are known to have an effect on key aspects of the photo-response of azobenzene, these are not the focus of this review. Throughout this review, effects of steric hindrance, excimer formation and, briefly, inter-system quenching through surface interactions on trans to cis isomerization will be explored.
3 Surface interactions
Surface interactions are most relevant to SAMs, where azobenzene units are held in close proximity and at defined angles to the substrate, or systems where azobenzenes are adsorbed to a surface ‘face on’. While SAMs represent systems where azobenzene-containing molecules are bound strongly enough to be considered permanently in contact with the surface, surface interactions are also important to completely understand systems where transient adsorption is present.58 Most commonly, the Au(111) surface has been used for formation of SAM structures,12,59–66 however, other metallic surfaces such as Ag(111)67,68 have been used as well as silica,69,70 mica,71,72 and graphene.73In general, there are two common orientations in which azobenzene molecules associate with surfaces, dominated by forces of interaction with surface structures. First, as often demonstrated by unfunctionalized azobenzene, molecules may take a planar orientation to the surface.74–76 The second orientation is the typical SAM morphology where the azobenzene groups are grafted to the surface through a functional group, such as an alkane thiol, and distance from the surface is regulated by the length of the linking group. In alkane thiol SAMs, azobenzene rings typically orient themselves at an angle from the normal to the surface (Fig. 1).61,77
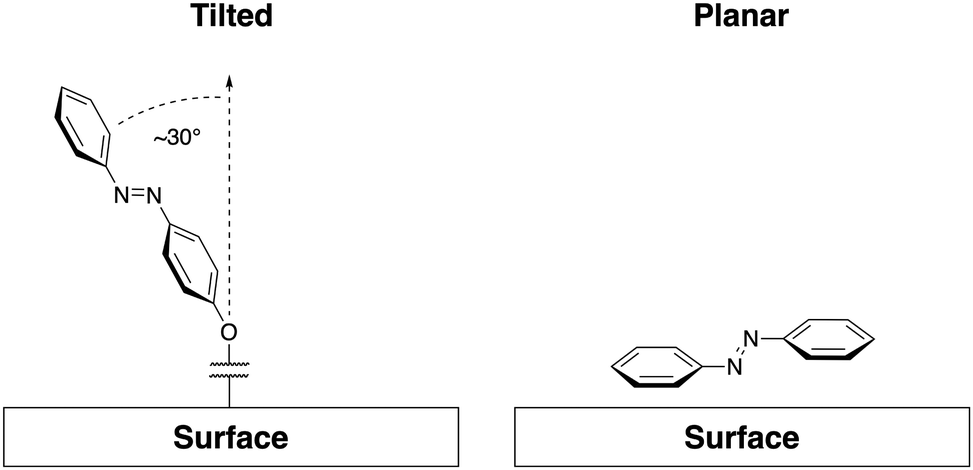 | ||
| Fig. 1 A representation of common surface interactions. Angled SAM arrangement with an example angle to the normal of ∼30° (left-hand side)61 and planar or “face on” adsorption common for unfunctionalized azobenzene (right-hand side).74,75 | ||
For both adsorption orientations, steric bulk and spacer groups have been the method of choice to electronically decouple azobenzene molecules from the surface. For the planar orientation, the difference is apparent when comparing azobenzene to tetra-tert-butylazobenzene.60,74–76,78,79 The effect of adsorption to surfaces can also in part be explained by changes in ground-state stability of the cis and trans isomers and their transition state energy.78,79 Similarly, azobenzene adsorbed in a planar orientation on a graphene substrate experiences changes in excitation energy via two competing mechanisms: substrate polarization which reduces optical band-gap, and dielectric screening weakening attractive interactions between electrons and holes. This causes an overall blueshift despite only weak hybridization with the substrate.73 An important outcome of this is that the effect on switching is, therefore, surface specific, as is shown by switching of tetra-tert-butylazobenzene on Au(111) surfaces but deactivation on a chemically similar Ag(111) surface.79
Similarly to the approach taken to azobenzene in a planar orientation to the surface, the switching behaviour of azobenzene SAMs can be controlled by changing the spacer length linking the molecule to the surface. Coupling to metal Au(111) is much greater than to Pt(111) surfaces, which can be explained by comparison of adsorption distance to the two surfaces; and coupling for the S2 state is greater than coupling of the S1 state for both Au(111) and Pt(111) surfaces.80
For spacer lengths of around three methylene (CH2) and one oxygen unit, this leads to charge-transfer times of around 80 ± 20 fs, much faster than the rate of azobenzene isomerization in solution which is in the order of ∼1 ps.61 This effect diminishes exponentially with spacer length to eleven methylene units, and diluting with decanethiolate can return switching functionality almost to that observed in solution.61
So, while surface interactions may cause deactivation of trans to cis isomerization, the extent of this effect is both specific to the surface present and is relatively easily mitigated through the use of bulky spacer groups. Surface interactions with azobenzene should be considered in functional materials including Au(111), Ag(111) and graphene surfaces that have been shown to cause deactivation of trans to cis isomerization.61,73,79 Pt(111) may offer an alternative metal surface less susceptible to disrupting the isomerization process.80
4 Steric effects
The influence of sterics on isomerization of azobenzene molecules can be isolated through the study of dilute systems with increasing hindrance from the surrounding medium. Moving from the gas phase to solution state, the dominant isomerization mechanism becomes the concerted inversion pathway.29 While Z to E isomerization is barely affected by this change, E to Z isomerization is significantly slowed in liquid compared with the gas phase.81When this theory is extended to isomerization in bulk liquid azobebenzene, the action of photoswitching is similar, with a change in rate of rotation of the benzene rings throughout the isomerization process. While this process occurs in less than a picosecond in the gas phase, the motion of the rings is significantly slowed by the increased steric hindrance of the melt occuring on the picosecond timescale and beyond.82
Titov et al. investigated the dynamics of azobenzene switching of a monomer, isolated dimer, and dimer in a SAM-like arrangement using the AM1/FOMO-CI method to account for differences in electronic spectra.83 It should be noted that the simulations did not include any surface interactions therefore simulations also resemble non-bound systems such as lamellar or smectic liquid-crystalline layers.84 Unit cell parameters were set to give a face to face distance of 3.5 Å between aromatic rings, typical for π–π interactions. This corresponded to a blue shift of around 0.1–0.2 eV for the π to π* transition. However, it should be noted that this shift is significantly less than has been noted in similar experimental systems.77 Importantly, despite the electronic shift in the system, the monomer and isolated dimer systems were found to have similar trans to cis quantum yields of Φ = 0.21 ± 0.05 and Φ = 0.19 ± 0.05, respectively. It was only in the packed systems where a large decrease in quantum yield to Φ = 0.11 ± 0.3 was noted. The similarity between quantum yields of monomeric and dimeric states may indicate that switching of electronically isolated dimers is not severely hindered and sterics play a larger part in switching quenching.
In general, increases in steric hindrance slow isomerization, which can be shown to decrease the quantum yield of the isomerization processes. However, short of complete conformational locking as has been noted in some crystalline solids, isomerization should remain feasible in condensed phases and improvements should be a measure of increasing quantum yield efficiency.
5 Excimers
Excimer formation occurs from close proximity of multiple azobenzene units through formation of H- and J-aggregates, arrangements where aggregated molecules have parallel or in-line transition dipoles, respectively.85,86 This translates to a face to face (H-aggregated) or top to tail (J-aggregated) arrangement for azobenzene-containing molecules (Fig. 2). In particular, we will consider H-aggregates of azobenzene which cause a hypsochomic shift (blue shift) in the S2 ← S0 excitation which has implications on the energy and efficiency of excitation for isomerization. Aggregation has been identified in a range of systems including photosurfactants,87 SAMs,88 and liquid crystals.89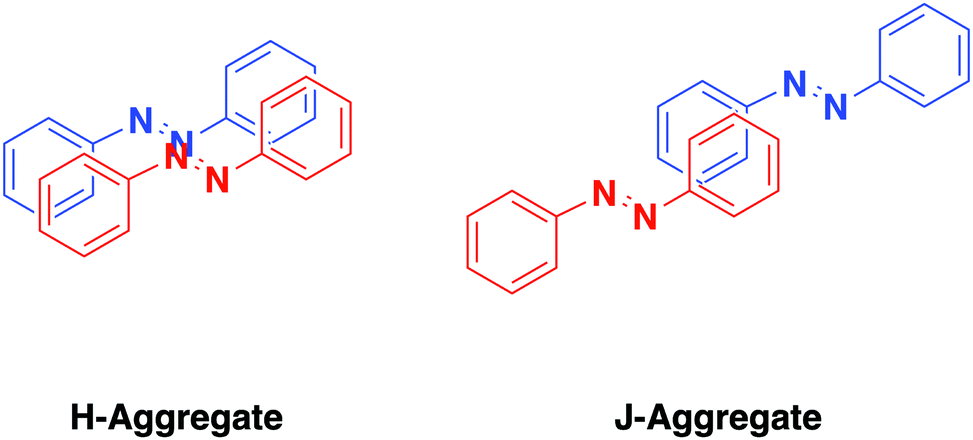 | ||
| Fig. 2 A representation of the arrangement of two azobenzene molecules into an H-aggregate (left-hand side) and a J-aggregate (right-hand side). | ||
Since aggregate formation causes changes in the spectral properties of azobenzene chromophores, it is reasonable to consider whether E/Z isomerization can be stimulated by excitation of aggregate structures. The possibility of using excitation of aggregate structure to stimulate isomerization was explored by Bronsh et al. by comparing photoisomerization yields from stimulation of different parts of the broadened S2 ← S0 excitation in an azobenzene-functionalized alkanethiolate SAM.62 Comparison of the effect of stimulation by light at a wavelength of 365 nm and 310 nm indicated a decrease in the fraction of cis isomers when stimulated with 310 nm irradiation. Therefore, it appears that the predominant pathway for photo-isomerization in aggregated structures remains via localized excitations, and strongly coupled, delocalized states do not contribute significantly and cannot be used for light harvesting or increasing trans to cis isomerization.77 For the design of efficient photo-switching systems, the focus must remain on minimization of aggregation and its effects.
So while aggregates cannot be efficiently stimulated to cause isomerization, it is also important to establish whether they will deactivate isomerization through electron delocalization processes. Utecht et al. considered the electronics of dimers, linear N-mers and two dimensionally packed linear N-mers using density functional theory on a (TD-) B3LYP/6-31G* level.90 Extension of calculations to consider N-mers was used to approximate Rabi transfer lifetimes for N-mer systems. For typical undiluted SAM spacings,77 this gave lifetimes orders of magnitude faster than trans to cis isomerization. Furthermore this was the case for enlarged spacings up to around 20 Å.
Contrary to the above result, Benassi et al. considered exciton transfer phenomena in azobenzene and bis[(1,1′)-biphen-4-yl]diazine in SAM-like arrangements and found that intermolecular exciton transfer should not represent a significant deactivation channel.91 This conclusion was reached by considering transfers taking on more Förster-type character, and calculation of exciton walk diffusion lengths, which were found to be sub-Å and subnanometer for azobenzene and the diphenyl analogue, respectively. The discrepancy with the result from Utecht et al. can be explained through differences in character of the transfers used to calculate timescales and diffusion lengths, as well as the predominant excitation used for the calculation. While Utecht et al. considered the S2 excited state, Benassi et al. considered the S1 excited state of more importance because isomerization can occur from either the S1 or S2 excited states28 and relaxation from the S2 to S1 excited state should occur faster than electronic excition transfer in the S2 state.
One method by which excimer formation has been dealt with from a practical aspect in SAMs is by controlling packing density of grafted azobenzene layers. Decreasing azobenzene density has been achieved by dilution of thiol-grafted azobenzene units with alkane thiols,92,94 substitution of bulky functional groups onto the azobenzene core-unit,95 the use of large bases for the azobenzene units68,96,97 and by increasing surface curvature.64,92
Systems that rely on a large steric ‘foot’ intrinsically hold a set spacing between the azobenzene core units when the feet are in the densest packing configuration possible.68,96,97 Similarly, addition of bulky groups to the azobenzene core group offsets packing between every azobenzene unit. Both use of large bases for adsorption onto metal surfaces68,96,97 and addition of meta and ortho substituted methyl and ethyl functional groups on the azobenzene aromatic ring95,98 have been shown to increase the fraction of azobenzene molecules switched during photo-isomerization, with the bulkier functional groups having a more pronounced effect. However, dilution with alkane thiols offers, by contrast, a statistical increase in spacing between azobenzene units because of the competitive adsorption of alkane thiols on the metal surface. Alkane thiol dilution has been shown to re-establish photo-isomerization behaviour even with minimal dilutions, from around 92 mol% azobenzene chromophore in the azobenzene/alkane thiol mix.92 These diluted systems still experience hypsochromic shifts in the π to π* transition (see Fig. 3) lending credence to the idea that in general it is the combination of steric hinderance and large, unbroken aggregates that is most detrimental to photoisomerization yield.
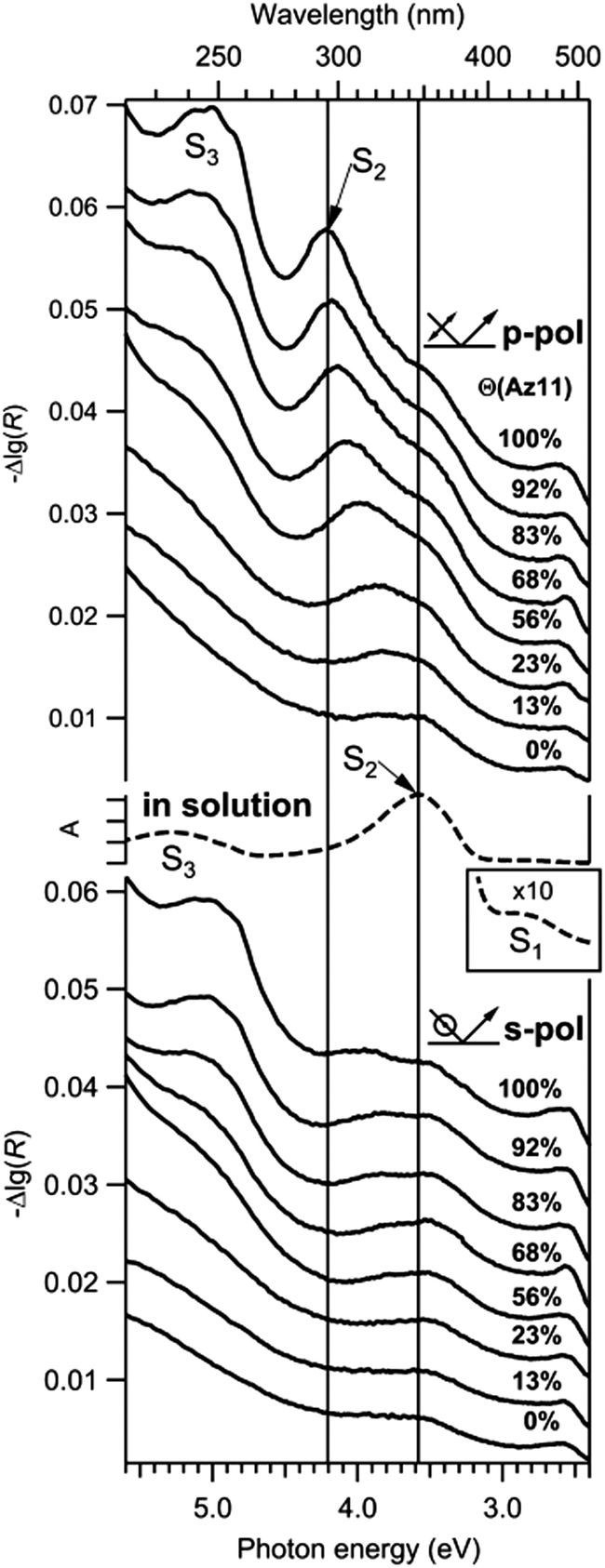 | ||
| Fig. 3 Series of p- and s-polarized differential reflectance (DR) spectra of SAMs for different Az11 coverages Θ (angle of incidence 45°, offsets were added). Schematic drawings indicate the orientation of the electric field vector of the incident light. An absorbance spectrum of trans-Az11 in methanol (middle, dashed) is plotted for comparison. The inset shows the upscaled S1 absorption band in solution. Reprinted (adapted) with permission from T. Moldt, D. Brete, D. Przyrembel, S. Das, J. R. Goldman, P. K. Kundu, C. Gahl, R. Klajn and M. Weinelt, Langmuir, 2015, 31, 1048–1057. Copyright (2015) American Chemical Society.92 | ||
A similar effect can be achieved by mixing azobenzenes with different functionalizations, which partially inhibits coupling of the chromophores, therefore preserving some of the spectral features of the constituent monomers.93Fig. 4 displays the calculated optical absorption spectra of dimers and densely packed SAM (p-SAM) structures with mixed –H, –CN and –CF3para-functionalizations on the azobenzene core. Optical spectra show improved S2 absorbance in comparison to analogous systems with a single functionalization (dotted lines), which is the main excitation known to cause E/Z isomerization. Hence, the presence of mixed functionalization partially counteracts the intermolecular coupling promoted by high molecular density.93
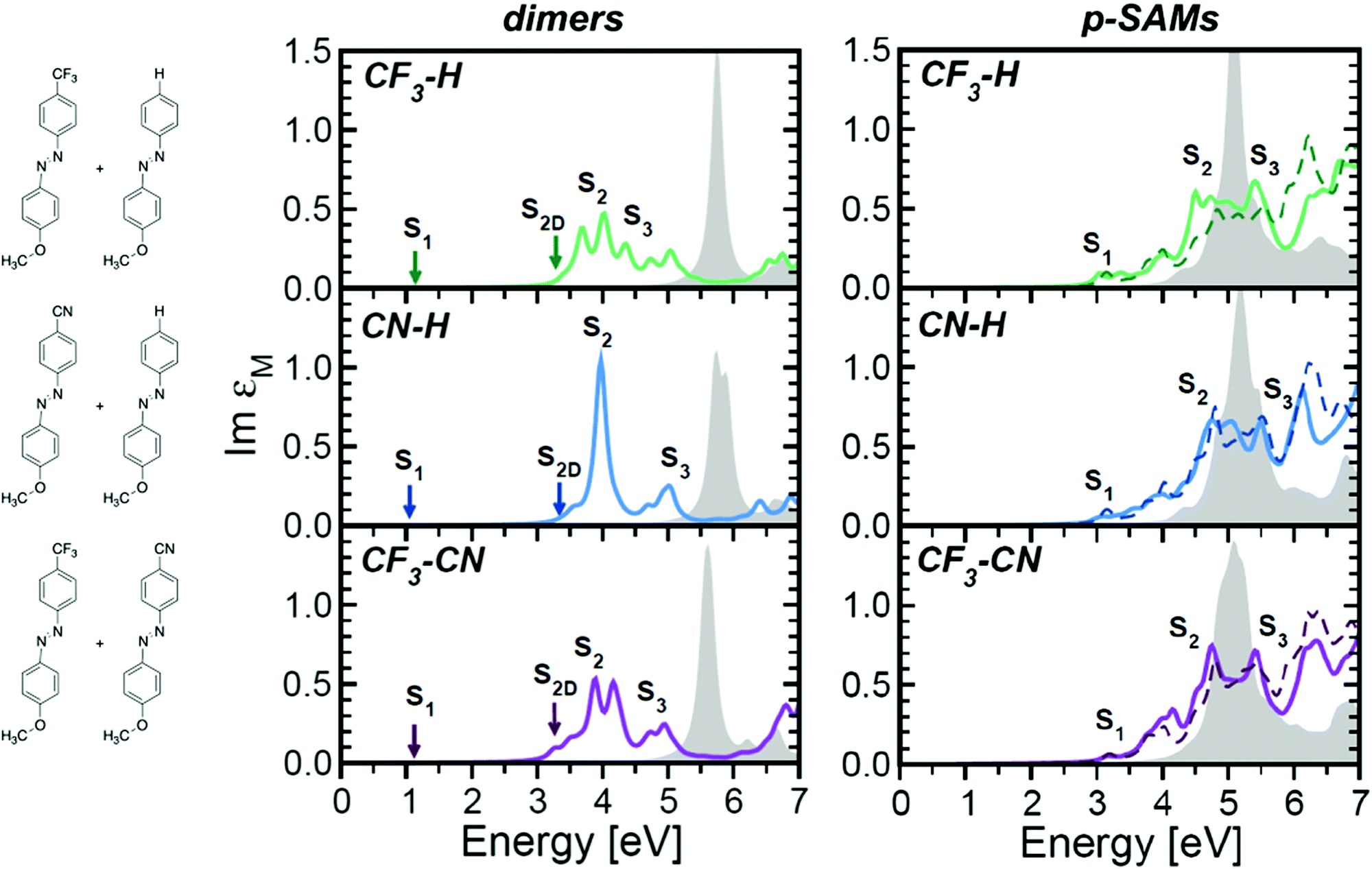 | ||
Fig. 4 Optical absorption spectra of dimers (left) and p-SAMs (right) with mixed functionalizations, expressed by Im![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) εM averaged over the three Cartesian components. Solid lines indicate the solutions of the BSE while filled gray areas correspond to the independent-particle approximation. Singlet excitons with vanishing oscillator strength are indicated by an arrow. For the p-SAMs, the averaged spectra of the homogenously functionalized SAMs with the corresponding terminations are also plotted (dashed lines). A Lorentzian broadening of 100 meV is applied to all spectra to mimic the excitation lifetime. Republished from Cocchi (2017) IOP Publishing. Reproduced with permission. All rights reserved.93 εM averaged over the three Cartesian components. Solid lines indicate the solutions of the BSE while filled gray areas correspond to the independent-particle approximation. Singlet excitons with vanishing oscillator strength are indicated by an arrow. For the p-SAMs, the averaged spectra of the homogenously functionalized SAMs with the corresponding terminations are also plotted (dashed lines). A Lorentzian broadening of 100 meV is applied to all spectra to mimic the excitation lifetime. Republished from Cocchi (2017) IOP Publishing. Reproduced with permission. All rights reserved.93 | ||
Another useful method is the use of curvature to moderate azobenzene packing. This approach has been explored through adsorption of azobenzene thiols to planar Au(111) and curved Au nanoparticle surfaces. In these systems, the same chromophore density gives ∼0.2 eV lower shift in the S2 ← S0 electronic transition92 (see Fig. 5). Similar systems have also be considered using computational methods, reconfirming the effect of decreased packing density due to curvature and finding that flexible backbones are necessary to take the most advantage from this additional spatial freedom.64
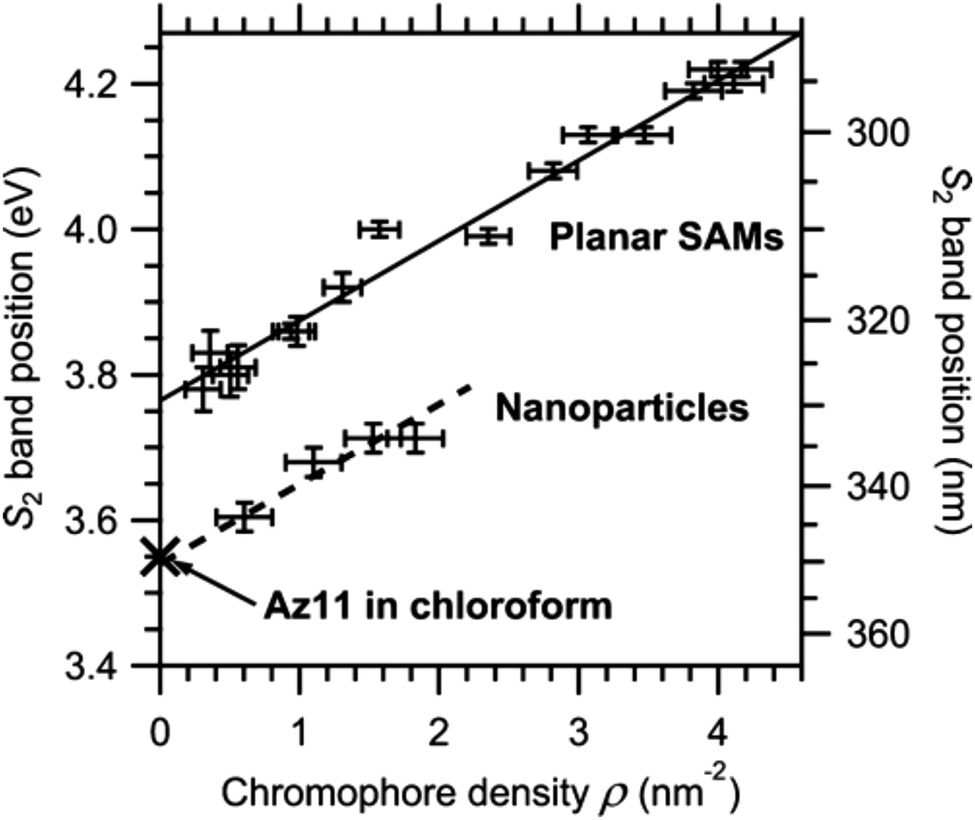 | ||
| Fig. 5 S2 band position on NPs and in mixed SAMs versus chromophore density, ρ. The S2 band position of Az11 in solution is added for comparison. All data points correspond to absorption maxima directly read from the spectra. The solid line represents a fit to the SAM data points, the dashed line has the same slope, and an offset was added to compare with results from NPs. Reprinted (adapted) with permission from T. Moldt, D. Brete, D. Przyrembel, S. Das, J. R. Goldman, P. K. Kundu, C. Gahl, R. Klajn and M. Weinelt, Langmuir, 2015, 31, 1048–1057. Copyright (2015) American Chemical Society.92 | ||
While excimer formation is a significant pathway for deactivation of azobenzene isomerization, predominantly through formation of excitation modes that do not lead to isomerization,62 breaking up the aggregate structure is effective in restoring the isomerization process. This method of restoring isomerization ability has been demonstrated through dilution with non-azobenzene containing species,92 addition of bulky spacers,95,98 and increases in curvature.92 Even dilution with azobezene species with different functionalization may improve isomerization efficiency.93
6 Application to functional soft-matter systems
So far we have discussed a number of lessons learnt from routes for the reactivation of concentrated and densely packed azobenzene systems. This section will focus on other functional soft-matter systems from the myriad ways azobenzene has been applied, such as azosurfactants99 and liquid crystals.100,101 Here it will be contended that these same principles are of immense value for the design of efficient photo-switching systems based off azobenzene and what explore questions remain to be answered. However, SAM systems will not be further discussed because the importance of reactivation of azobenzene switching in these systems is held to be self-evident.6.1 Liquid crystals (LCs)
Azobenzene-based materials have been applied to liquid-crystalline (LC) materials as thermotropic,102–106 lyotropic107–110 and polymeric systems111–118 for a diverse array of applications. Some recent reviews of various applications of azobenzene containing liquid crystals can be found here.119–123 Azobenzene LC formation can be achieved both with the azobenzene-containing molecule as the major component, or as a dopant into other systems. The trans to cis isomerization of azobenzene is generally employed to either induce a phase change (most commonly an order to disorder transition) or to induce changes in alignment of the LC phase.124 For this reason, using azobenzene's geometry change to transition between mesophases or to change mesophase parameters remains a fertile avenue of investigation.Since LC materials possess anisotropic ordering, the angle of incident light is also an important consideration for calculation of the efficiency of azobenzene isomerization. The dipole of the azobenzene chromophore should align with the polarization of the incident photon for excitation to occur.125 This is the underlying mechanism via which LC alignment is controlled using azobenzene dopants. For this reason, it is simpler to here compare UV-visible spectra to look for H-aggregation rather than only looking for studies on isomerization efficiency, however, care should be taken because often it is only a dilute solution-state absorbance spectrum of the azobenzene chromophore that is reported.103,126–128
UV-visible spectra of films of azobenzene-containing liquid crystals, where the azobenzene component is the main constituent,129 where it is a minor dopant130,131 and liquid crystal polymers,132 often display spectral shifts in the π to π* excitation that are consistent with those observed for partially diluted SAMs.92 The extent of hypsochromic shifting observed appear to trend as expected, with greater aggregate formation arising from increases in azobenzene content. Trends dependent on liquid-crystalline mesophase, therefore local molecular arrangement, are less clear.131 Thought the temperature dependence of the optical spectra, especially in thermotropic systems, is fairly well established.133 Since photo-responsive behaviour is retained, the effect of azobenzene chromophore aggregation will be most measurable in isomerization kinetic rates and photo-isomerization efficiency. This is an area which could obviously benefit from a systematic study to compare the extent of aggregation and its effects on isomerization either in a single-component system with diverse phase behaviour or through doping into various mesophases of comparable host systems, such as a series of cyanobiphenyl (nCB) thermotropic liquid crystals, from which both nematic (N) and smectic (Sm) phases can be accessed.
Since dilute systems are less affected by H-aggregation induced deactivation and are sufficient in ability to cause order to disorder changes and photo-induced allignment,124 what advantages can increased azobenzene content bring, and when is it appropriate to consider? While doping into thermotropic liquid-crystalline mesophases is often advantageous, for many lyotropic systems, the simplicity of using a single main component can offer greater robustness in the face of small perturbations to the system, such as salt concentration in aqueous systems. Additionally, while order-to-disorder transitions are common and relatively easy to induce, order-to-order or mesophase-to-mesophase transitions can be more difficult to achieve. This kind of order-to-order transition can be achieved through fine control of molecular structure, which has been demonstrated in lyotropic and thermotropic systems where the azobenzene component is the main component.107,134
The obvious exception to complete order-to-disorder transitions for azobenzene-doped systems comes from cholesteric liquid crystals (CLCs) and the so called blue-phase (BP), which is a frustrated “double-twisted” form of the cholesteric phase.135 In these systems, azobenzene dopants have been employed to induce mesophase changes from the frustrated BP to cholesteric phases,136,137 as well as partial disorder transitions.138 Additionally they can be employed to change only mesophase parameters such as selective control of wavelength reflection in BP liquid crystals and controlled changes to cholesteric pitch length in CLCs.136,137 While such concentrated systems may be difficult photo-isomerize,89 the chirality of self-assembled structures and addition of bulky substituent groups may offset molecular packing effects on isomerization efficiency.
While lower concentrations of azobenzene content in liquid-crystalline media offer the same advantages in decreasing, though not always eliminating, H-aggregate formation to retain isomerization efficiency, the structure transitions accessible to high azobenzene content systems can offer advantages for targeted mesophase-to-mesophase transitions. Therefore, concentration and avoiding H-aggregate formation should not be the only considerations in photo-responsive liquid crystal design, but should be secondary to the targeted system transition the application calls for. However, in situ UV-visible analysis is important for quantification and identification of intermolecular interactions, and should be done in addition to dilute solution state analysis of the azobenzene chromophore. The design principles of dilution and including sterically bulky functional groups can be employed where necessary to fine tune system response.
6.2 Photo-surfactants
Photo-surfactants represent an area of research that has displayed continual development over a number of decades,99 with many applications of interest including modulation of wetting on surfaces, variation of interfacial tension,140 controlled delivery and uptake141–143 and exploration of many biological processes.144 These systems often deal with self-assembled structures of photo-surfactants and more complex mixtures. This means that the distribution and local structures formed, through processes such as membrane rafting, and changes in local curvature, can add layers of complexity to these systems and the way in which they respond to light stimuli. H-aggregation, indicative of areas of intermolecular interaction between azobenzene functional groups, has been identified in a range of self-assembled photo-surfactant structures including Langmuir–Blodgett films,145 worm-like micelles,146 bilayers and vesicles.139,147 It is of note that small spherical micelles are significantly less affected by H-aggregation, which can be inferred to be a result of their consistently greater curvature.148Kuiper et al. provide an example of the impact of H-aggregation on the efficiency of photo-surfactant isomerization within vesicles.139 Through the investigation of three azobenzene-based photo-surfactants in 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) vesicles, varying degrees of hypsochromic shifting was observed in UV-visible spectra of the composite vesicles, which directly correlated with isomerization efficiency and response of the structures. It has been suggested that the amount of shifting in the S2 excitation peak is indicative of various types of azo-surfactant aggregate structures within the vesicle bilayers (Fig. 6). Whilst photo-isomerization was achieved in samples with only a moderate hypsochromic shift of the S2 excitation peak, it seems likely that this is largely influenced by mobility within the surfactant bilayer and an equilibrium with the isolated monomeric state. Clearly, despite this being a multicomponent system where the azobenzene component is effectively diluted, there are cases in which the photo-isomerization could be vastly improved with additional measures to facilitate decoupling of the excimer complexes. Since membrane rafting is well established in many systems, methods for structure interrogation via small-angle scattering have been developed.149 This presents well as a method by which the structure–function relationships between aggregate structures and system photo-response could be identified.
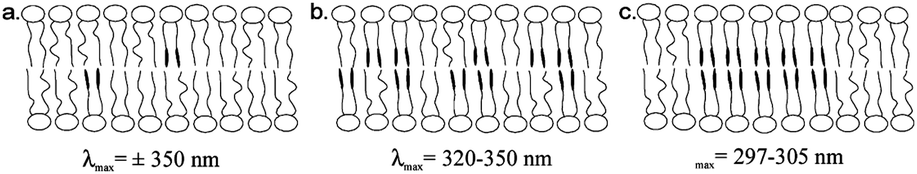 | ||
| Fig. 6 Schematic presentations of the type of aggregates of azobenzenes and corresponding wavelengths of maximal absorbance (λmax): (a) low concentration of azobenzene amphiphile, no H-aggregation; (b) H-aggregation but no microdomain formation; (c) H-aggregation and microdomain formation. Reprinted (adapted) with permission from J. M. Kuiper and J. B. F. N. Engberts, Langmuir, 2004, 20, 1152–1160. Copyright (2004) American Chemical Society.139 | ||
Aggregation of azobenzene into raft structures could be of particular interest in systems where there is a steric- or curvature-induced constraint on the system. This may include cubic surfactant mesophases where there exist regions of greater and lower curvature. Indeed, bicontinuous cubic mesophases may be a way in which curvature relationships can be interrogated, since various Bragg peaks represent these sub-regions of greater or lesser curvature, as can be inferred from inverse small-angle scattering methods.150 Such an investigation into the relationship between azobenzene aggregation in cubic mesophases and the influence of photo-isomerization in single-component and doped systems may offer a pathway by which the relationship between curvature and H-aggregate formation can be understood. Such an investigation would provide insights to the mechanistic action of azobenzene control in these systems as well as parallels to processes such as in meso protein crystallization.151
The influence of curvature on the aggregation and action of azobenzene is also evident in a variety of other systems including single-component photo-rheological fluids.146 Formulation of a photo-rheological fluid from a system of a single photo-surfactant in water confers benefits of simplicity and robustness in comparison to more complex formulations. However, in single-component formulations of worm-like micelles as photo-rheological fluids, an elliptical cross-section in trans-dominant aggregates has been proposed to be caused by a combination of π–π interaction-dominated curvature and steric interaction-dominated curvature (Fig. 7). It is of note that Kelly et al. also achieved the formation of a single-component photo-rheological fluid from another photo-surfactant in a similar fashion.152 While the worm-like micelles formed in their investigation were adequately fit with a simple cylindrical cross-section, these systems displayed UV-visible absorbance as a combination of hypsochromic shifted and monomeric species. While exposure times required for isomerization will be largely dominated by optical density, future iterative improvements on the design of photo-rheological fluids may include investigation into additional aromatic substitutions to moderate π–π interactions. Doing so may offer a facile method for increasing the relatively small difference in isomer ratios between trans and cis-dominant photo-stationary states that have been noted in similar systems.153
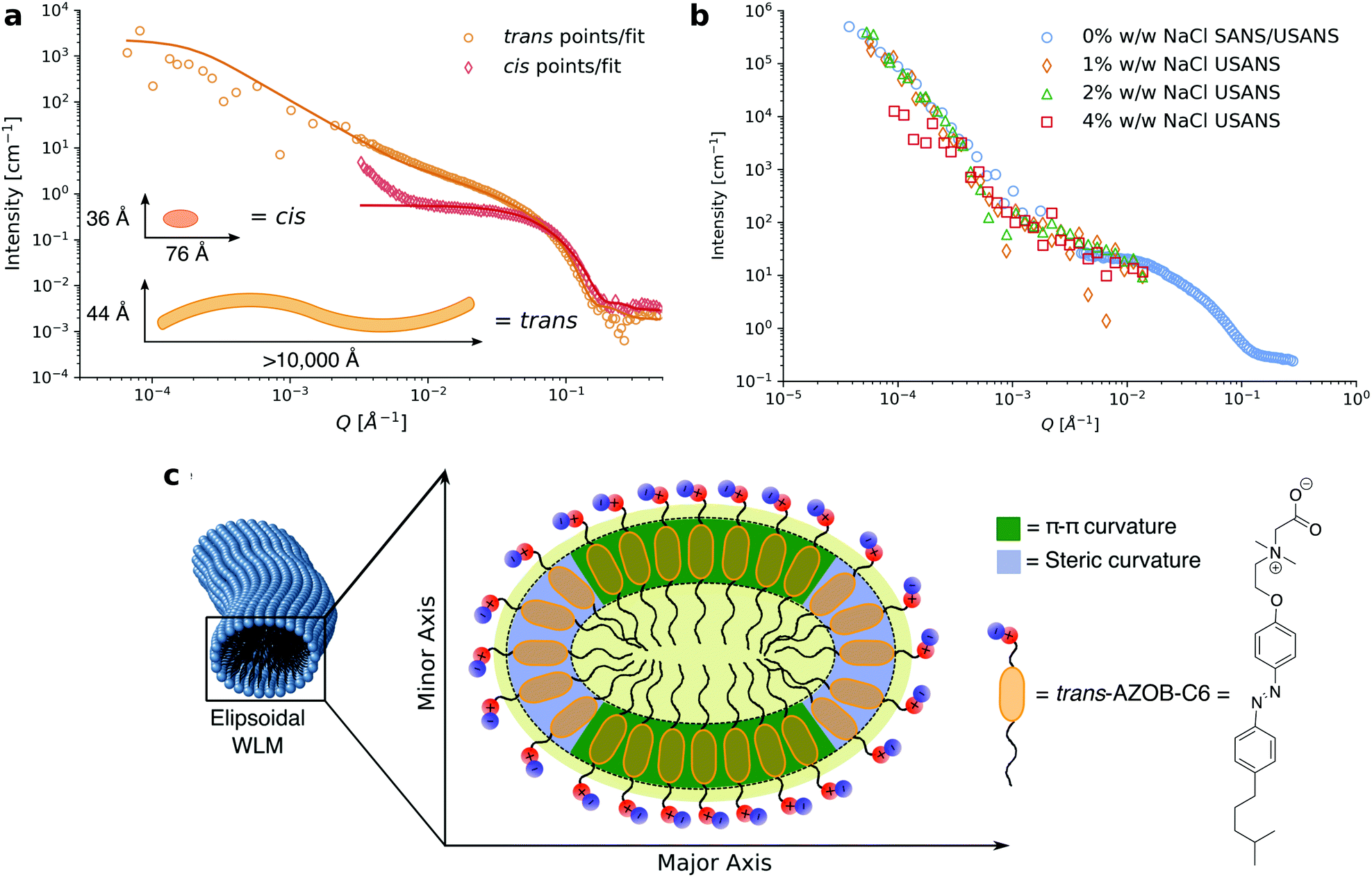 | ||
| Fig. 7 (a) Reduced 1D neutron scattering data for 15 mM solutions of 2-(dimethyl(2-(4-((4-(4-methylpentyl)phenyl)diazenyl)phenoxy)ethyl)ammonio)acetate (named AZOB-C6 in the original paper) in its cis (SANS only) and trans (SANS and USANS) isomers in D2O (hollow points) with model fits and fitting parameters generated from the fits (solid lines). (b) USANS patterns for 1% w/w surfactant AZOB-C6 in D2O with increasing concentrations of NaCl. Note that the scattering pattern for the 1% w/w formulation without salt appears different to that of the 15 mM solution as inter-micellar interactions contribute to the scattering pattern at this concentration. (c) Schematic cross section of ellipsoidal WLMs formed by AZOB-C6 showing sections of steric and sections of π–π driven curvature that drive the ellipsoidal cross section.146 Reprinted (adapted) from C. S. G. Butler, J. P. King, L. W. Giles, J. B. Marlow, M. L. P. Vidallon, A. Sokolova, L. de Campo, K. L. Tuck and R. F. Tabor, J. Colloid Interface Sci., 2021, 594, 669–680. Copyright (2021), with permission from Elsevier. | ||
Azobenzene-containing surfactants show a preference for flattened regions with elliptical micellar aggregates.154 This trend is likely to be influenced by the propensity of the azobenzene moiety to π-stack into H-aggregate structures. Upon trans to cis isomerization of photo-surfactant micelles, a number of structural effects have been noted. These changes include decreases in the number of aggregated molecules, decreases in the length and size of the photo-surfactant aggregates and, if close enough to the critical micelle concentration (CMC), complete disaggregation into monomeric units.155 It is also interesting to consider how the rate of isomerization changes beyond the CMC. This question was explored by Arya et al. who followed the isomerization of several azbenzene-containing surfactants before and after their CMC via UV-visible spectroscopy.156 By considering the rate constant for the monomeric component to be distinct from the rate constant for components in the micellar aggregates it was found that the isomerization rate within micelles was around 80% slower compared with single molecules. While this difference was considered to be mostly attributed to steric effects, questions remain around elucidating the effect that H-aggregation plays within this relationship.
Photo-surfactants are also frequently brought into contact with a variety of substrates as a way to introduce a photo-response without the need for direct chemical conjugation of the azobenzene moiety and substrate. A review of this method of photo control with a specific focus on microgels, DNA molecules, polymer brushes and colloidal particles can be found here.157 Azobenzene derivatives have also been considered in many other mixed systems with substrates including graphene158,159 and graphene oxide,160,161 polyelectrolytes,162,163 and metal surfaces.164–167 Conveniently, some metal nanocrystal systems are well positioned for in situ monitoring using surface enhanced Raman spectroscopy (SERS).166,167 Photo-isomerization can be observed these systems, which in contrast to the deactivation observed for surface interactions with some SAMS, can be speculated to be largely because of the presence of photo-surfactants both on the surface and in bulk solution. Therefore, similar to the conclusion made by Santer et al., the design of the azobenzene chromophores can be largely independent of the target substrate.157 However, interactions cannot be completely discounted as the example systems used for SERS experiments indicate that surface interactions can have implications for the isomerization kinetics and photo-stationary states.166,167
Surface interactions, to some extent, inhibiting or influencing the rate of trans to cis isomerization, set up a system with interplay between two isomerization rates (Ksurf and Ksol) and rates of adsorption and desorption from the surface (Kad and Kde respectively in Fig. 8). It is of note that similar two-rate kinetic systems have previously been modelled for more complete understanding of the system.168 Multi-rate systems may also share further similarities to micellar systems in which isomerization rates within micelles and bulk solutions may differ. If the difference between regular micellar structures and bulk solution is not stark enough, then regions of H-aggregations identified within some micellar structures such as the worm like micelles presented here,146 may display more noticeable multi-rate kinetics.
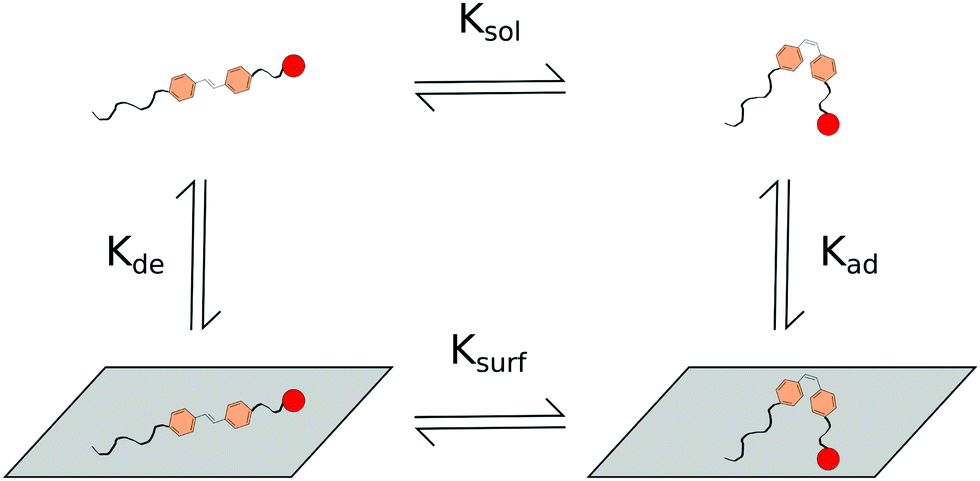 | ||
| Fig. 8 Scheme for the proposed interactions of photo-responsive components interacting with surfaces. | ||
In the complex assemblies observed from photo-surfactant systems, it is apparent that these molecules show a preference for low curvature in many of their micellar aggregate structures. This is likely to be influenced by π–π interactions and, often, H-aggregate formation. This effect can even extend to heterogeneous structure formation in mixed systems, whereby photo-surfactant aggregate formation occurs over homogeneous mixing with the azobenzene-free component. While micellar aggregate formation alone has been shown to have an effect on trans (E) to cis (Z) isomerization rate, more work is required to understand the full effect of internalization of H-aggregate structures within these self-assemblies. By contrast, where photo-surfactants in solution state are brought into contact with heterogeneous surfaces, azobenzene chromophore design can generally be independent of the target substrate with minimal drawbacks. A large contributing factor to this observation may be the equilibrium of photo-surfactants existing between the target surface and bulk solution. However, considering their importance to SAMs, surface interactions should still be considered as a potential explanation for any unanticipated results, and surface enhanced Raman spectroscopy (SERS) is particularly well placed to continue investigation into these relationships.
7 Conclusions and outlook
Photo-responsive systems are continually finding more practical applications in a variety of system arrangements, from relatively dilute solutions to condensed phases including liquid crystals and monolayer films, with ultimate uses from photonic materials to controllable electronics. Inactivation of trans (E) to cis (Z) isomerization seen in many early studies into surface adsorbed monolayer (SAM) films has lead to subsequent investigation that has revealed the mechanisms by which the photo-isomerization process may be quenched. These deactivation pathways include surface energy transfers and excimer formation. While steric effects have also been shown to have an effect on the kinetic rate of isomerization, the effect of steric hindrance is generally limited to a kinetic effect rather than complete quenching.While investigations into SAMs and the mechanisms of deactivation in this specific set of condensed matter systems have yielded promising results for the fine control of azobenzene isomerization in condensed phases, these same deactivation phenomena are relevant and indeed present in many other condensed matter systems, including liquid crystals, Langmuir–Blodgett films, and photo-surfactant assemblies. While deactivation phenomena are often overlooked in the study of these condensed systems, further investigation is warranted to completely understand their effects. Exploration of these phenomena may begin with the in situ analysis of the UV-visible spectra and H-aggregation present in condensed systems, as well as more extensive studies to understand the heterogeneous distribution of azobenzene chromophores in mixed systems as has been observed in many photo-surfactant systems. Greater isomerization efficiency should be achievable by employing design principles gained from investigations into SAM systems including dilutions, bulky spacers to control surface interactions, conjugation of steric spacers to the aromatic rings, and mixtures including azobenzene chromophores with varied substitutions. Better understanding of isomerization deactivation pathways and employing system design principle to manage them will help ensure the continued development of functional light-responsive systems with fast, high-yielding isomerization into the future.
Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors would like to thank AINSE Limited for providing financial assistance (Award: PGRA). This research was supported by an Australian Government Research Training Program (RTP) Scholarship. Finally, the authors would also like to acknowledge that the COVID-19 lock-down period was a strong influence for the initiation of writing this review and hope this finds the readers in good health.Notes and references
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 Search PubMed.
- R. Yerushalmi, A. Scherz, M. E. Van Der Boom and H. B. Kraatz, J. Mater. Chem., 2005, 15, 4480–4487 Search PubMed.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 Search PubMed.
- H. Zhang, X. Meng and P. Li, Prog. Chem., 2008, 20, 657–672 Search PubMed.
- L. Dong, Y. Feng, L. Wang and W. Feng, Chem. Soc. Rev., 2018, 47, 7339–7368 Search PubMed.
- Y. Luo and Q. Zhang, Adv. Condens. Matter Mater. Res., 2010, 7, 1–97 Search PubMed.
- T. Sako and M. Sato, Petrotech, 1997, 20, 373–378 Search PubMed.
- D. H. Waldeck, Chem. Rev., 1991, 91, 415–436 Search PubMed.
- P. Bauer, M. Sommer, J. Thurn, M. Pärs, J. Köhler and M. Thelakkat, Chem. Commun., 2013, 49, 4637–4639 Search PubMed.
- S. J. Lim, B. K. An, D. J. Sang, M. A. Chung and Y. P. Soo, Angew. Chem., Int. Ed., 2004, 43, 6346–6350 Search PubMed.
- S. A. Díaz, L. Giordano, J. C. Azcárate, T. M. Jovin and E. A. Jares-Erijman, J. Am. Chem. Soc., 2013, 135, 3208–3217 Search PubMed.
- W. R. Browne and B. L. Feringa, Annu. Rev. Phys. Chem., 2009, 60, 407–428 Search PubMed.
- J. E. Stumpel, D. Liu, D. J. Broer and A. P. H. J. Schenning, Chem. – Eur. J., 2013, 19, 10922–10927 Search PubMed.
- E. Merino, Chem. Soc. Rev., 2011, 40, 3835–3853 Search PubMed.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 Search PubMed.
- X. Huang and T. Li, J. Mater. Chem. C, 2020, 8, 821–848 Search PubMed.
- P. Zhou, Y. Li, X. Li, S. Liu and Y. Su, Liq. Cryst. Rev., 2016, 4, 83–100 Search PubMed.
- F. Zhao, Z. Pan, C. Wang, Y. Zhou and M. Qin, Opt. Quantum Electron., 2014, 46, 1491–1498 Search PubMed.
- X. Wang, Y. Mi, D. Wang, W. He, H. Cao and H. Yang, Mol. Cryst. Liq. Cryst., 2016, 630, 1–5 Search PubMed.
- U. Hrozhyk, S. Serak, N. Tabiryan, L. Hoke, D. M. Steeves, G. Kedziora and B. Kimball, Proc. SPIE, 2008, 7050, Liquid Crystals XII, 705007 Search PubMed.
- I. A. Budagovsky, V. N. Ochkin, S. A. Shvetsov, A. S. Zolot’ko, A. Y. Bobrovsky, N. I. Boiko and V. P. Shibaev, Mol. Cryst. Liq. Cryst., 2017, 647, 100–106 Search PubMed.
- L. Dong and Y. Zhao, Mater. Chem. Front., 2018, 2, 1932–1943 Search PubMed.
- D. Xia, G. Yu, J. Li and F. Huang, Chem. Commun., 2014, 50, 3606–3608 Search PubMed.
- M. Liu, X. Yan, M. Hu, X. Chen, M. Zhang, B. Zheng, X. Hu, S. Shao and F. Huang, Org. Lett., 2010, 12, 2558–2561 Search PubMed.
- X. Chi, X. Ji, D. Xia and F. Huang, J. Am. Chem. Soc., 2015, 137, 1440–1443 Search PubMed.
- Y. Liu, H. Wang, P. Liu, H. Zhu, B. Shi, X. Hong and F. Huang, Angew. Chem., 2021, 133, 5830–5834 Search PubMed.
- Y. Liu, H. Wang, L. Shangguan, P. Liu, B. Shi, X. Hong and F. Huang, J. Am. Chem. Soc., 2021, 143, 3081–3085 Search PubMed.
- P. Cattaneo and M. Persico, Phys. Chem. Chem. Phys., 1999, 1, 4739–4743 Search PubMed.
- G. Tiberio, L. Muccioli, R. Berardi and C. Zannoni, ChemPhysChem, 2010, 11, 1018–1028 Search PubMed.
- H. Yu, J. Mater. Chem. C, 2014, 2, 3047–3054 Search PubMed.
- C. Zhu, Y. Lu, J. Sun and Y. Yu, Langmuir, 2020, 36, 6611–6625 Search PubMed.
- X. Pang, J.-a. Lv, C. Zhu, L. Qin and Y. Yu, Adv. Mater., 2019, 31, 1904224 Search PubMed.
- J. Vapaavuori, C. G. Bazuin and A. Priimagi, J. Mater. Chem. C, 2018, 6, 2168–2188 Search PubMed.
- L. D. Site, Entropy, 2014, 16, 23 Search PubMed.
- R. Wang, T. Iyoda, L. Jiang, D. A. Tryk, K. Hashimoto and A. Fujishima, J. Electroanal. Chem., 1997, 438, 213–219 Search PubMed.
- S. D. Evans, S. R. Johnson, H. Ringsdorf, L. M. Williams and H. Wolf, Langmuir, 1998, 14, 6436–6440 Search PubMed.
- J. Zhang, J. K. Whitesell and M. A. Fox, Chem. Mater., 2001, 13, 2323–2331 Search PubMed.
- R. Micheletto, M. Yokokawa, M. Schroeder, D. Hobara, Y. Ding and T. Kakiuchi, Appl. Surf. Sci., 2004, 228, 265–270 Search PubMed.
- I. Willner, V. Pardo-Yissar, E. Katz and K. T. Ranjit, J. Electroanal. Chem., 2001, 497, 172–177 Search PubMed.
- H. Rau and E. Lueddecke, J. Am. Chem. Soc., 1982, 104, 1616–1620 Search PubMed.
- W. M. Horspool and F. Lenci, CRC handbook of organic photochemistry and photobiology, CRC Press, Boca Raton, 2004 Search PubMed.
- C. J. Brown, Acta Crystallogr., 1966, 21, 146–152 Search PubMed.
- C. R. Crecca and A. E. Roitberg, J. Phys. Chem. A, 2006, 110, 8188–8203 Search PubMed.
- L. Gagliardi, G. Orlandi, F. Bernardi, A. Cembran and M. Garavelli, Theor. Chem. Acc., 2004, 111, 363–372 Search PubMed.
- C. L. Forber, E. C. Kelusky, N. J. Bunce and M. C. Zerner, J. Am. Chem. Soc., 1985, 107, 5884–5890 Search PubMed.
- P. Bortolus and S. Monti, J. Phys. Chem., 1979, 83, 648–652 Search PubMed.
- T. Cusati, G. Granucci, M. Persico and G. Spighi, J. Chem. Phys., 2008, 128, 194312 Search PubMed.
- H. M. D. Bandara, T. R. Friss, M. M. Enriquez, W. Isley, C. Incarvito, H. A. Frank, J. Gascon and S. C. Burdette, J. Org. Chem., 2010, 75, 4817–4827 Search PubMed.
- C. Xu, L. Yu, F. L. Gu and C. Zhu, Phys. Chem. Chem. Phys., 2018, 20, 23885–23897 Search PubMed.
- M. J. Hansen, M. M. Lerch, W. Szymanski and B. L. Feringa, Angew. Chem., Int. Ed., 2016, 55, 13514–13518 Search PubMed.
- D. B. Konrad, G. Savasci, L. Allmendinger, D. Trauner, C. Ochsenfeld and A. M. Ali, J. Am. Chem. Soc., 2020, 142, 6538–6547 Search PubMed.
- S. Kobayashi, H. Yokoyama and H. Kamei, Chem. Phys. Lett., 1987, 138, 333–338 Search PubMed.
- M. Han and T. Honda, Sci. China: Chem., 2011, 54, 1955–1961 Search PubMed.
- G. Gabor and E. Fischer, J. Phys. Chem., 1971, 75, 581–583 Search PubMed.
- H. Rau and E. Lueddecke, J. Am. Chem. Soc., 1982, 104, 1616–1620 Search PubMed.
- A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, J. Am. Chem. Soc., 2004, 126, 3234–3243 Search PubMed.
- J. Garcia-Amorós, G. Stopa, G. Stochel, R. van Eldik, M. Martnez and D. Velasco, Phys. Chem. Chem. Phys., 2018, 20, 1286–1292 Search PubMed.
- T. M. McCoy, S. A. Holt, A. M. Rozario, T. D. M. Bell and R. F. Tabor, Adv. Mater. Interfaces, 2017, 4, 1700803 Search PubMed.
- E. Benassi, G. Granucci, M. Persico and S. Corni, J. Phys. Chem. C, 2015, 119, 5962–5974 Search PubMed.
- C. Bronner, M. Schulze, S. Hagen and P. Tegeder, New J. Phys., 2012, 14, 043023 Search PubMed.
- C. Gahl, R. Schmidt, D. Brete, S. Paarmann and M. Weinelt, Surf. Sci., 2016, 643, 183–189 Search PubMed.
- W. Bronsch, T. Moldt, L. Boie, C. Gahl and M. Weinelt, J. Phys.: Condens. Matter, 2017, 29, 484002 Search PubMed.
- W. Bronsch, D. Przyrembel, L. Boie, C. Gahl and M. Weinelt, Appl. Phys. Lett., 2017, 111, 081601 Search PubMed.
- C. Liu, D. Zheng, W. Hu, Q. Zhu, Z. Tian, J. Zhao, Y. Zhu and J. Ma, Nanoscale, 2017, 9, 16700–16710 Search PubMed.
- D. Bedrov, J. B. Hooper, M. A. Glaser and N. A. Clark, Langmuir, 2016, 32, 4004–4015 Search PubMed.
- Z. Tian, J. Wen and J. Ma, Mol. Simul., 2015, 41, 28–42 Search PubMed.
- R. J. Maurer, W. Liu, I. Poltavsky, T. Stecher, H. Oberhofer, K. Reuter and A. Tkatchenko, Phys. Rev. Lett., 2016, 116, 146101 Search PubMed.
- K. Scheil, T. G. Gopakumar, J. Bahrenburg, F. Temps, R. J. Maurer, K. Reuter and R. Berndt, J. Phys. Chem. Lett., 2016, 7, 2080–2084 Search PubMed.
- N. Delorme, J. F. Bardeau, A. Bulou and F. Poncin-Epaillard, Langmuir, 2005, 21, 12278–12282 Search PubMed.
- M. Min, G. S. Bang, H. Lee and B.-C. Yu, Chem. Commun., 2010, 46, 5232–5234 Search PubMed.
- D. Bleger, A. Ciesielski, P. Samori and S. Hecht, Chem. – Eur. J., 2010, 16, 14256–14260 Search PubMed.
- M. El Garah, F. Palmino and F. Cherioux, Langmuir, 2010, 26, 943–949 Search PubMed.
- Q. Fu, C. Cocchi, D. Nabok, A. Gulans and C. Draxl, Phys. Chem. Chem. Phys., 2017, 19, 6196–6205 Search PubMed.
- S. Hagen, F. Leyssner, D. Nandi, M. Wolf and P. Tegeder, Chem. Phys. Lett., 2007, 444, 85–90 Search PubMed.
- M. Wolf and P. Tegeder, Surf. Sci., 2009, 603, 1506–1517 Search PubMed.
- J. Cho, L. Berbil-Bautista, N. Levy, D. Poulsen, J. M. J. Frechet and M. F. Crommie, J. Chem. Phys., 2010, 133, 234707 Search PubMed.
- C. Gahl, R. Schmidt, D. Brete, E. R. McNellis, W. Freyer, R. Carley, K. Reuter and M. Weinelt, J. Am. Chem. Soc., 2010, 132, 1831–1838 Search PubMed.
- E. R. McNellis, J. Meyer and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 205414 Search PubMed.
- R. J. Maurer and K. Reuter, Angew. Chem., Int. Ed., 2012, 51, 12009–12011 Search PubMed.
- E. Benassi and S. Corni, J. Phys. Chem. C, 2014, 118, 25906–25917 Search PubMed.
- M. Böckmann, N. L. Doltsinis and D. Marx, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 78, 036101 Search PubMed.
- M. Böckmann, N. L. Doltsinis and D. Marx, J. Phys. Chem. A, 2010, 114, 745–754 Search PubMed.
- E. Titov, G. Granucci, J. P. Götze, M. Persico and P. Saalfrank, J. Phys. Chem. Lett., 2016, 7, 3591–3596 Search PubMed.
- J. W. Goodby, R. J. Mandle, E. J. Davis, T. Zhong and S. J. Cowling, Liq. Cryst., 2015, 42, 593–622 Search PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 Search PubMed.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 Search PubMed.
- G. Xu, K. Okuyama and M. Shimomura, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1992, 213, 105–115 Search PubMed.
- C. L. Yeung, S. Charlesworth, P. Iqbal, J. Bowen, J. A. Preece and P. M. Mendes, Phys. Chem. Chem. Phys., 2013, 15, 11014–11024 Search PubMed.
- M. Gupta, V. Pal and S. K. Pal, New J. Chem., 2018, 42, 8765–8772 Search PubMed.
- M. Utecht, T. Klamroth and P. Saalfrank, Phys. Chem. Chem. Phys., 2011, 13, 21608–21614 Search PubMed.
- E. Benassi and S. Corni, J. Phys. Chem. C, 2013, 117, 25026–25041 Search PubMed.
- T. Moldt, D. Brete, D. Przyrembel, S. Das, J. R. Goldman, P. K. Kundu, C. Gahl, R. Klajn and M. Weinelt, Langmuir, 2015, 31, 1048–1057 Search PubMed.
- C. Cocchi and C. Draxl, J. Phys.: Condens. Matter, 2017, 29, 394005 Search PubMed.
- D. T. Valley, M. Onstott, S. Malyk and A. V. Benderskii, Langmuir, 2013, 29, 11623–11631 Search PubMed.
- M. Han, T. Honda, D. Ishikawa, E. Ito, M. Hara and Y. Norikane, J. Mater. Chem., 2011, 21, 4696–4702 Search PubMed.
- N. R. Krekiehn, M. Müller, U. Jung, S. Ulrich, R. Herges and O. M. Magnussen, Langmuir, 2015, 31, 8362–8370 Search PubMed.
- U. Jung, C. Schütt, O. Filinova, J. Kubitschke, R. Herges and O. Magnussen, J. Phys. Chem. C, 2012, 116, 25943–25948 Search PubMed.
- D. Ishikawa, E. Ito, M. Han and M. Hara, Langmuir, 2013, 29, 4622–4631 Search PubMed.
- R. F. Tabor, T. M. McCoy, Y. Hu and B. L. Wilkinson, Bull. Chem. Soc. Jpn., 2018, 91, 932–939 Search PubMed.
- A. R. Yuvaraj, M. Y. Mashitah and R. Lutfor, Mol. Cryst. Liq. Cryst., 2016, 631, 21–30 Search PubMed.
- E. Wuckert, M. D. Harjung, N. Kapernaum, C. Mueller, W. Frey, A. Baro, F. Giesselmann and S. Laschat, Phys. Chem. Chem. Phys., 2015, 17, 8382–8392 Search PubMed.
- S. A. A. Santos, W. C. Costa, I. H. Bechtold, R. A. P. Halfen, A. A. Merlo and L. F. Campo, Liq. Cryst., 2019, 46, 655–665 Search PubMed.
- Z. Miao, Y. Zhang, Y. Zhao, Z. Wang and D. Wang, Mol. Cryst. Liq. Cryst., 2014, 591, 10–18 Search PubMed.
- D. Wang, Y. Huang, J.-M. Lv, Y. Min, Y.-Y. Quan, L.-N. Ge, M. Tian and D.-S. Yao, Liq. Cryst., 2018, 1–12 Search PubMed.
- R. Yang, D. Zhao, G. Dong, Y. Liu and D. Wang, Crystals, 2018, 8, 147 Search PubMed.
- Y. Zakrevskyy, J. Stumpe and C. F. J. Faul, Adv. Mater., 2006, 18, 2133–2136 Search PubMed.
- S. Peng, Q. Guo, T. C. Hughes and P. G. Hartley, Langmuir, 2014, 30, 866–872 Search PubMed.
- S. Peng, Q. Guo, P. G. Hartley and T. C. Hughes, J. Mater. Chem. C, 2014, 2, 8303–8312 Search PubMed.
- J. E. Houston, E. A. Kelly, M. Kruteva, K. Chrissopoulou, N. Cowieson and R. C. Evans, J. Mater. Chem. C, 2019, 7, 10945–10952 Search PubMed.
- L. W. Giles, J. B. Marlow, C. S. G. Butler, G. A. Turpin, L. de Campo, S. T. Mudie, C. F. J. Faul and R. F. Tabor, Phys. Chem. Chem. Phys., 2020, 22, 4086–4095 Search PubMed.
- X. Pang, J.-a. Lv, C. Zhu, L. Qin and Y. Yu, Adv. Mater., 2019, 31, 1904224 Search PubMed.
- X. Pang, B. Xu, X. Qing, J. Wei and Y. Yu, Macromol. Rapid Commun., 2018, 39, 1700237 Search PubMed.
- P. Van Der Asdonk, H. C. Hendrikse, A. C. Sauli, S. P. M. Kraaijkamp and P. H. J. Kouwer, J. Mater. Chem. C, 2016, 4, 8263–8269 Search PubMed.
- C. Weber, T. Liebig, M. Gensler, L. Pithan, S. Bommel, D. Bléger, J. P. Rabe, S. Hecht and S. Kowarik, Macromolecules, 2015, 48, 1531–1537 Search PubMed.
- R. Weglowski, A. Kozanecka-Szmigiel, W. Piecek, J. Konieczkowska and E. Schab-Balcerzak, Opt. Commun., 2017, 400, 144–149 Search PubMed.
- J. E. Koskela, J. Vapaavuori, J. Hautala, A. Priimagi, C. F. J. Faul, M. Kaivola and R. H. A. Ras, J. Phys. Chem. C, 2012, 116, 2363–2370 Search PubMed.
- J. J. Xiong, D. Shen, Z. G. Zheng and X. Q. Wang, Chin. Phys. B, 2016, 25, 096401 Search PubMed.
- H. Yamada, M. Kukino, Z. A. Wang, R. Miyabara, N. Fujimoto, J. Kuwabara, K. Matsuishi and T. Kanbara, J. Appl. Polym. Sci., 2015, 132, 41275 Search PubMed.
- D.-Y. Kim and K.-U. Jeong, Liq. Cryst. Today, 2019, 28, 34–45 Search PubMed.
- L. Wang and Q. Li, Photoact. Funct. Soft Mater., 2019, 91–123 Search PubMed.
- T. Seki, K. Fukuhara, D. Tanaka, T. Nakai, M. Hara and S. Nagano, Mol. Cryst. Liq. Cryst., 2017, 653, 1–8 Search PubMed.
- P. Zhou, Y. Li, X. Li, S. Liu and Y. Su, Liq. Cryst. Rev., 2016, 4, 83–100 Search PubMed.
- M. Alaasar, Liq. Cryst., 2016, 43, 2208–2243 Search PubMed.
- Y. Yu and T. Ikeda, J. Photochem. Photobiol., C, 2004, 5, 247–265 Search PubMed.
- D.-Y. Kim, D.-G. Kang, M.-H. Lee, J.-S. Kim, C.-R. Lee and K.-U. Jeong, Chem. Commun., 2016, 52, 12821–12824 Search PubMed.
- Z. C. Miao, Y. M. Zhang, Y. Z. Zhao and D. Wang, Mol. Cryst. Liq. Cryst., 2013, 582, 98–106 Search PubMed.
- X. Wang, Z. Li, H. Zhao and S. Chen, R. Soc. Open Sci., 2020, 7, 200474 Search PubMed.
- I. Niezgoda, J. Jaworska, D. Pociecha and Z. Galewski, Liq. Cryst., 2015, 42, 1148–1158 Search PubMed.
- W. Li, S. Nagano, K. Yonetake and T. Seki, Mol. Cryst. Liq. Cryst., 2012, 563, 112–120 Search PubMed.
- G. Wang, M. Zhang, T. Zhang, J. Guan and H. Yang, RSC Adv., 2012, 2, 487–493 Search PubMed.
- J. G. Fernsler, M. A. Glaser, R. Shao, D. A. Coleman, J. E. Maclennan, D. R. Link, C. Chang, K. Lanham, D. Walba, C. Boyer, J. A. Zasadzinski and N. A. Clark, Liq. Cryst., 2017, 44, 769–783 Search PubMed.
- S. M. Alauddin, N. F. K. Aripin, T. S. Velayutham and A. Martinez-Felipe, Molecules, 2020, 25, 2579 Search PubMed.
- Y. Zakrevskyy, J. Stumpe, B. Smarsly and C. F. J. Faul, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2007, 75, 031703 Search PubMed.
- M. Alaasar, S. Poppe, Y. Cao, C. Chen, F. Liu, C. Zhu and C. Tschierske, J. Mater. Chem. C, 2020, 8, 12902–12916 Search PubMed.
- J. Yang, W. Zhao, W. He, Z. Yang, D. Wang and H. Cao, J. Mater. Chem. C, 2019, 7, 13352–13366 Search PubMed.
- W. L. He, M. Li, S. Q. Liu, M. J. Wei, C. Liu, L. L. Li, Z. Yang, D. Wang and H. Cao, Liq. Cryst., 2018, 45, 370–380 Search PubMed.
- X. Chen, L. Wang, C. Li, J. Xiao, H. Ding, X. Liu, X. Zhang, W. He and H. Yang, Chem. Commun., 2013, 49, 10097–10099 Search PubMed.
- L. Yin, Y. Wu, J. Gao, J. Ma, Z. Hu, G. Zou and Q. Zhang, Soft Matter, 2015, 11, 6145–6151 Search PubMed.
- J. M. Kuiper and J. B. F. N. Engberts, Langmuir, 2004, 20, 1152–1160 Search PubMed.
- C. Herdes, E. E. Santiso, C. James, J. Eastoe and E. A. Müller, J. Colloid Interface Sci., 2015, 445, 16–23 Search PubMed.
- Y. Orihara, A. Matsumura, Y. Saito, N. Ogawa, T. Saji, A. Yamaguchi, H. Sakai and M. Abe, Langmuir, 2001, 17, 6072–6076 Search PubMed.
- M. Akamatsu, P. A. FitzGerald, M. Shiina, T. Misono, K. Tsuchiya, K. Sakai, M. Abe, G. G. Warr and H. Sakai, J. Phys. Chem. B, 2015, 119, 5904–5910 Search PubMed.
- S. Aleandri, C. Speziale, R. Mezzenga and E. M. Landau, Langmuir, 2015, 31, 6981–6987 Search PubMed.
- S. H. Donaldson, C. T. Lee, B. F. Chmelka and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 15699–15704 Search PubMed.
- E. Piosik, M. Kotkowiak, I. Korbecka, Z. Galewski and T. Martyński, Phys. Chem. Chem. Phys., 2017, 19, 23386–23396 Search PubMed.
- C. S. G. Butler, J. P. King, L. W. Giles, J. B. Marlow, M. L. P. Vidallon, A. Sokolova, L. de Campo, K. L. Tuck and R. F. Tabor, J. Colloid Interface Sci., 2021, 594, 669–680 Search PubMed.
- M. Shimomura and T. Kunitake, J. Am. Chem. Soc., 1987, 109, 5175–5183 Search PubMed.
- R. F. Tabor, R. J. Oakley, J. Eastoe, C. F. J. Faul, I. Grillo and R. K. Heenan, Soft Matter, 2009, 5, 78–80 Search PubMed.
- D. Marquardt, F. A. Heberle, J. D. Nickels, G. Pabst and J. Katsaras, Soft Matter, 2015, 11, 9055–9072 Search PubMed.
- M. Impéror-Clerc, Interface Focus, 2012, 2, 589–601 Search PubMed.
- L. van’t Hag, L. de Campo, N. Tran, A. Sokolova, R. Trenker, M. E. Call, M. J. Call, C. J. Garvey, A. E. Leung, T. A. Darwish, A. Krause-Heuer, R. Knott, T. G. Meikle, C. J. Drummond, R. Mezzenga and C. E. Conn, Langmuir, 2019, 35, 8344–8356 Search PubMed.
- E. A. Kelly, N. Willis-Fox, J. E. Houston, C. Blayo, G. Divitini, N. Cowieson, R. Daly and R. C. Evans, Nanoscale, 2020, 12, 6300–6306 Search PubMed.
- E. A. Kelly, J. E. Houston and R. C. Evans, Soft Matter, 2019, 15, 1253–1259 Search PubMed.
- C. Blayo, J. E. Houston, S. M. King and R. C. Evans, Langmuir, 2018, 34, 10123–10134 Search PubMed.
- R. F. Tabor, M. J. Pottage, C. J. Garvey and B. L. Wilkinson, Chem. Commun., 2015, 51, 5509–5512 Search PubMed.
- P. Arya, J. Jelken, N. Lomadze, S. Santer and M. Bekir, J. Chem. Phys., 2020, 152, 024904 Search PubMed.
- S. Santer, J. Phys. D: Appl. Phys., 2017, 51, 013002 Search PubMed.
- M. Döbbelin, A. Ciesielski, S. Haar, S. Osella, M. Bruna, A. Minoia, L. Grisanti, T. Mosciatti, F. Richard, E. A. Prasetyanto, L. De Cola, V. Palermo, R. Mazzaro, V. Morandi, R. Lazzaroni, A. C. Ferrari, D. Beljonne and P. Samorì, Nat. Commun., 2016, 7, 11090 Search PubMed.
- F. Cardano, M. Frasconi and S. Giordani, Front. Chem., 2018, 6, 102 Search PubMed.
- K. M, H. Chi, T. Li, H. Wang and S. Thomas, Composites, Part B, 2019, 173, 106978 Search PubMed.
- T. M. McCoy, A. C. Y. Liu and R. F. Tabor, Nanoscale, 2016, 8, 6969–6974 Search PubMed.
- J. Liu, K. Kobayashi, L. Yang, N. Takisawa and K. Shirahama, J. Colloid Interface Sci., 1999, 213, 412–416 Search PubMed.
- C. T. Lee, K. A. Smith and T. A. Hatton, Langmuir, 2009, 25, 13784–13794 Search PubMed.
- A. Mat Lazim, M. Bradley and J. Eastoe, Langmuir, 2010, 26, 11779–11783 Search PubMed.
- L. Lysyakova, N. Lomadze, D. Neher, K. Maximova, A. V. Kabashin and S. Santer, J. Phys. Chem. C, 2015, 119, 3762–3770 Search PubMed.
- G. Angelini, L. Scotti, A. Aceto and C. Gasbarri, J. Mol. Liq., 2019, 284, 592–598 Search PubMed.
- L. Sun, C. Wang, Y. Pan, T. Chen and Z. Lv, J. Raman Spectrosc., 2020, 51, 756–763 Search PubMed.
- S. Tripathi and R. F. Tabor, J. Colloid Interface Sci., 2016, 476, 119–131 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |