
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving detection capability for single particle inductively coupled plasma mass spectrometry with microdroplet sample introduction†
Jovana
Kocic
 ,
Detlef
Günther
and
Bodo
Hattendorf
*
,
Detlef
Günther
and
Bodo
Hattendorf
*
Department of Chemistry and Applied Biosciences, ETH Zurich, Zurich, Switzerland. E-mail: bodo@inorg.chem.ethz.ch
First published on 26th November 2020
Abstract
Single particle inductively coupled plasma mass spectrometry (sp-ICPMS) is an attractive technique for fast measurement of elemental composition, mass and particle number concentration (PNC) of metal containing nanoparticles (NPs). In order to investigate NPs <10 nm using ICPMS, low instrumental background and high detection efficiency are primary requirements. This study evaluated the performance of a sector-field ICPMS with standard and enhanced sensitivity (“Jet”) vacuum interfaces with different sample introduction setups: conventional pneumatic nebulization with (DSN) and without aerosol desolvation (PN) and microdroplet generation (MDG). Additionally, the influence of nitrogen gas as an addition to a dry aerosol was studied. In this study, transport efficiencies (TEs) and detection efficiencies (DEs) are determined for the different instrumental setups. Gold NP suspensions were analysed and evaluated for PNC and size. Applying counting statistics, the size limit of detection (LODsize) of gold nanoparticles (Au NPs) was estimated to be 6.1 nm and 4.7 nm for PN and MDG with the standard interface, and 3.6 nm and 3.1 nm for DSN and MDG with the “Jet” interface and nitrogen addition, respectively. Additionally, DEs for various elements were determined. 11 isotopes (27Al, 47Ti, 63Cu, 107Ag, 111Cd, 115In, 133Cs, 140Ce, 193Ir, 197Au, and 238U) were measured at a mass resolving power (MRP) of 300 while an MRP of 4000 was used for 56Fe and 66Zn. DEs obtained for the conventional nebulization system with a spray chamber (PN) were in the range of 10−4 to 10−2 counts per atom (low resolution) and 10−6 to 10−5 counts per atom (medium resolution), while significant improvement in DE was obtained for the MDG setup with the “Jet” interface and nitrogen addition resulting in the range of 10−2 to 10−1 counts per atom (low resolution) and 10−4 to 10−3 counts per atom (medium resolution). The enhancement in DE was most pronounced for isotopes of lower m/z indicating reduced mass discrimination of the “Jet” interface with nitrogen gas added to the sample aerosol. The corresponding LODSIZE could thus be decreased by 10 or 2 times for example for Al- and Au-containing NPs, respectively. At the same time the use of an MDG for sample introduction allowed for 98.5% TE in the analyses of NP suspensions, while a TE of 10% (PN) or 23% (DSN) was obtained with pneumatic nebulizers.
Introduction
The number of commercial products containing engineered nanoparticles (NPs) is constantly expanding.1,2 Thus, the development of validated methods and protocols is required in order to ensure reproducible properties of NP characteristics and its relation to environmental safety.2–5 In this respect, various international projects are going on, with the aim of developing and evaluating different analytical techniques for NP assessment (e.g. AceNano within Horizon2020 (ref. 6)). NP characterization includes the determination of mass, size, shape, particle number concentration (PNC), morphology, surface characterization, elemental/chemical composition, and exposure.7 Single particle inductively coupled plasma mass spectrometry (sp-ICPMS) offers fast analysis of different parameters needed for characterization of inorganic NPs: elemental composition and mass as well as possibly size, PNC, and distinction from dissolved fractions.8–11 The concept was introduced in 2003 by Degueldre and Favarger, who analysed colloidal particle fractions in suspension by ICPMS, using time-resolved data acquisition.12–16 By 2019, the total number of reports about sp-ICPMS increased to over 300, as recently highlighted in a review by Mozhayeva et al.17Different sample introduction systems are being coupled to ICPMS for NP analysis.18–21 Most frequently, pneumatic nebulizers are used as they are most readily available. One of the major limitations of this configuration however is low transport efficiency (TE). For conventional concentric nebulizers with Scott-type or cyclonic spray chambers the TE is in the range of 5–10%, with a sample flow rate between 100 and 1000 μL min−1.22 Other, high efficiency nebulizers have been developed, whose reported TEs reach >90% at <10 μL min−1 sample flow rate23 but such systems require precise control of the flow rate and are less robust against clogging. Pneumatic nebulizers with aerosol desolvation operate by the same principle, but the solvent is evaporated in a heated spray chamber and removed by membrane desolvation. TEs however were reported to span across a wide range, depending on operating conditions. Todoli et al.,24 reported a TE of up to 90% when applying 0.6 mL min−1 as the sample flow rate, while 18.7% TE at 0.11 mL min−1 sample flow rate was reported by Hadioui et al.21 using a different system. The incomplete and variable sample transfer of the conventional nebulizers allows an accurate determination of TE crucial for assessing the true PNC of the analysed sample.25 Pace et al.22 described different approaches for TE determination termed: “waste collection”, “particle size”, and “particle frequency” methods. While the waste collection method does not require NP reference materials for TE assessment, it was shown that it may overestimate TE due to solvent evaporation inside the spray chamber. Therefore, the “particle size” and “particle frequency” are considered more reliable but they rely on reference materials, with known PNC and/or size need.
The requirement of precise TE determination can in principle be circumvented using total consumption sample introduction systems like microdroplet generators. This method was already used by Hieftje and co-workers back in 1968 to study fundamental aspects of evaporation of droplets and particles in flames.26 In 1997, Olesik et al.27 used a microdroplet generator (MDG) and introduced individual droplets into an ICP for optical emission and mass spectrometry, and, in 2010, for the first time diluted particle suspensions were introduced into the ICPOES by the group of K. Niemax at ISAS Dortmund.28 With the motivation to increase the sensitivity and accuracy MDGs were considered an attractive alternative for discrete low-volume sample introduction systems for ICPMS, and investigated further for example by Gschwind et al.,20,29,30 Koch et al.31 and Shigeta et al.32
The use of an MDG for NP analyses provides several advantages over conventional nebulization:
- TE of practically 100% for sufficiently small droplets and verification on a per-sample basis. By adding an internal standard and counting the number of signal spikes caused by its isotope in the transient signal, this number can be related to the number of droplets injected and the TE determined. Once stable droplet trajectories have been established, any drift in TE can be readily verified. Quasi-simultaneous spectrum acquisition by ICP-TOFMS can achieve this in parallel to the particle analysis, while using sequential MS detection requires these measurements to be interspersed in the particle analyses.
- The low sample flux reaching the ICP per event (i.e. 8.2 pL for droplets of 25 μm) allows for a higher PNC to be analysed while keeping the occurrence of multiple events per acquisition to a minimum. To allow multiple events to occur at a rate of less than 1%, it follows from Poisson statistics that a particle should be detected in less than 2% of all acquisitions (i.e. droplets or dwell times) on average. For the conditions mentioned before, this amounts to a max PNC of >2 × 106 mL−1, while the conventional sp-ICPMS with the pneumatic nebulizer (e.g. 20 μL min−1 sample flux into the ICP and 1 ms dwell time) may reach this limit at a PNC of less than 105 mL−1. MDG sample introduction thus reduces sample preparation and possible artefacts caused by changing the solvent composition.
- Droplet desolvation before the ICP reduces the instantaneous heat required to evaporate the droplet residue and/or NP. Together with more localized vaporization inside the ICP this should also translate into a higher DE.21,33
Today, various technically interesting nanomaterials appear as quantum dots (QDs), which may contain just between several 100 and a few 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms of a particular element1,34–43 where DEs in the permil-range are required. Recent studies using the high sensitivity sector-field ICPMS with the conventional nebulization system however reported DEs for gold in the range of 0.01–0.06% (ion counts per atom).44–47 Additionally, Hadioui et al.21 have compared wet and dry plasma conditions and reported DEs of 0.08 and 0.3%, respectively.
000 atoms of a particular element1,34–43 where DEs in the permil-range are required. Recent studies using the high sensitivity sector-field ICPMS with the conventional nebulization system however reported DEs for gold in the range of 0.01–0.06% (ion counts per atom).44–47 Additionally, Hadioui et al.21 have compared wet and dry plasma conditions and reported DEs of 0.08 and 0.3%, respectively.
In this study we used a sector field ICPMS with an optimized vacuum interface to achieve maximum sensitivity for NP detection. The modified interface consists of a specific “Jet” sampler cone, an “X” skimmer cone and a high capacity dry interface pump. Earlier reports indicate that a more than 5-fold increase in sensitivity could be obtained for dry plasma conditions by laser ablation sampling and with the addition of mL min−1 amounts of nitrogen to the carrier gas.48
The performance of the instrument for NP detection was evaluated for conventional pneumatic nebulization with the cyclonic spray chamber (PN) or a desolvation system (DSN) and an MDG for sample introduction. The performance of the “Jet” interface was evaluated by comparing DEs and LODsize49 for various elements to the standard interface configuration. Additionally, TEs for PN, DSN and MDG operation were investigated.
Experimental
Materials
Au NP standards in citrate buffer with nominal sizes of 10 nm (diameter of 8.8 ± 0.4 nm, and Au mass concentration of 0.053 mg mL−1, ≈7.7 × 1012 NP mL−1) and 30 nm (diameter of 31 ± 3 nm, and Au mass concentration of 0.052 mg mL−1, ≈1.7 × 1011 NP mL−1) from NanoComposix, San Diego, California, USA were used for the studies. Nanoparticle suspensions were stored in a fridge at around 5 °C, and were allowed to warm up to room temperature for 15–30 minutes prior to use. Before each dilution step, suspensions were vortexed (Vortex Genie 2, Scientific Industries, USA) for 1 min, and then sonicated (Faust Laborbedarf AG, Schaffhausen, Switzerland) for 2 min. Suspensions were diluted gravimetrically with different concentrations of sodium citrate (0.1 mM and 1 mM), which had been filtered through 0.22 μm filters. Gold nanoparticle suspensions were doped with 0.05–1 μg kg−1 of dissolved Ir (Merck AG, Darmstadt, Germany) which served as a droplet tracer for MDG experiments, or as a tracer for instrumental drift for experiments with spray chamber and DSN sample introduction setups. To minimize sedimentation of nanoparticles, all suspensions were sonicated prior to the measurements. Each nanoparticle suspension was freshly diluted from the stock suspension prior to the measurements.Single-element standards were prepared from stock solutions from Inorganic Ventures, Christiansburg, Virginia, USA (1000 mg L−1: Al, Fe, Cu, Cs, Ce), Merck AG, Darmstadt, Germany (1000 mg L−1: Zn, Ag, Cd, In, Ir, Au, and 10 mg L−1: U), and Alfa Aesar, Thermo Fisher Scientific, UK (1000 mg L−1: Ti) in 1% HNO3 (subboiled) or in 3% HCl (optima grade, Fisher Scientific, United Kingdom). Concentrations of the standard solutions were in the range of 1–10 μg kg−1 for the spray chamber and MDG with the standard interface, and 0.01–100 μg kg−1 for DSN and MDG sample introduction systems with the “Jet” interface and nitrogen addition (see Table S1†).
All sample dilutions were prepared gravimetrically using an electronic balance (scale reading of 0.1 mg, Mettler AT400, Mettler-Toledo, Greifensee, Switzerland).
Instrumentation and data acquisition
Measurements were performed using a sector-field ICPMS (Element XR, Thermo Scientific, Bremen, Germany), employing a mass resolving power (MRP, m/Δm) of 300 (Al, Ti, Cu, Ag, Cd, In, Cs, Ce, Ir, Au, and U) or 4000 (Fe, and Zn). We are aware that 47Ti and 63Cu may be affected by spectral overlap, depending on the matrix composition. However in order to compare the performance for a wide range of elements we used pure standard solutions where we would not expect substantial interference. Fe and Zn were measured at higher MRP because of the presence of the plasma-based background ions ArO+ and ArCN+. A wet aerosol was produced using a concentric nebulizer (MicroMist™) with the conventional cyclonic spray chamber, while a DSN with the same nebulizer (Nu Instruments, DSN-100, Wrexham, United Kingdom), and the microdroplet generator, MDG (Microdrop Technologies, GmbH, Norderstedt, Germany) were used for the introduction of dry aerosols. Solutions were self-aspirated with pneumatic nebulization, and the nebulizer uptake (nominal 200 μL min−1) was determined for the optimized nebulizer gas flow rate by recording the consumed sample mass gravimetrically. Using the MDG, droplets were introduced to the ICP using a dispenser head with a nozzle of 50 μm inner diameter. The triple pulse mode (a sequence of three individually programmable voltage pulses applied to the piezo-actuator) was used for producing droplet diameters of 22–25 μm, at a frequency of 100 Hz.32 The MDG was inserted into an in-house built adapter,30 containing 4 symmetrically arranged gas inlets. Two windows in the adapter, placed downstream near the end of the MDG nozzle, allow monitoring droplets, via a CCD camera. Argon and nitrogen gas were introduced via 4 inlets at the adapter, while helium was introduced through the dispenser head to accelerate desolvation of the droplets.50 The droplets were captured using Virtual Dub (V. 1.9.11, Avery Lee, Free Software Foundation Inc., Cambridge, USA) and their size was determined during post-processing using ImageJ (National of Health, Bethesda, MA, USA).51 The MDG-adapter setup was mounted at the ICP torch holder using a custom-made tray, allowing the entire setup to move together with the torch (see Fig. S1†). A schematic overview of different setups used is presented in Fig. 1. Additionally, standard nickel sampler and skimmer cones were used for wet (spray chamber) and dry (MDG) plasma conditions, and compared to the “Jet” configuration, when nitrogen gas was mixed to the dry aerosols prior to the ICP (DSN and MDG).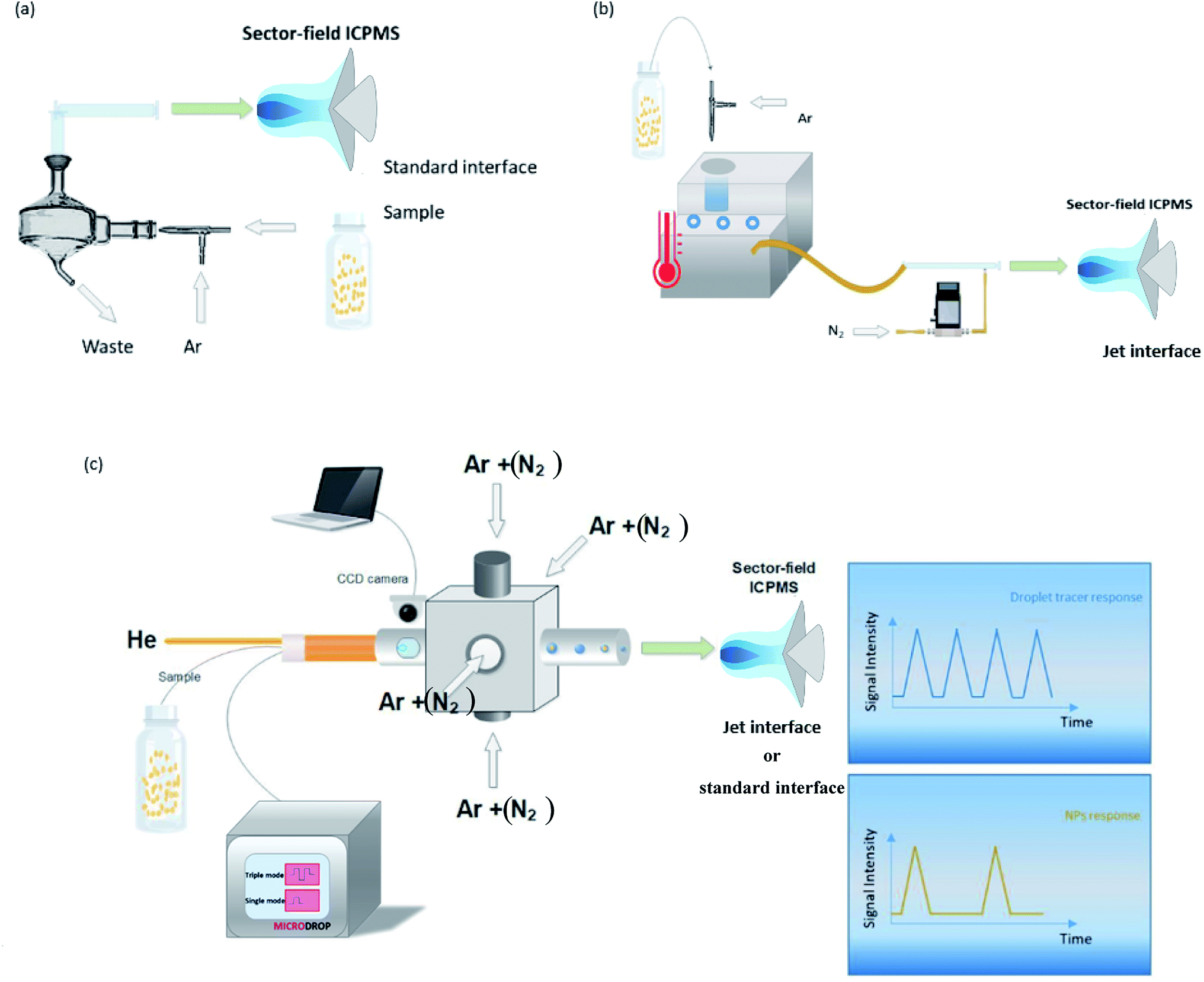 | ||
| Fig. 1 Pneumatic nebulization with the conventional cyclonic spray chamber (a), and the desolvation nebulization system, DSN (b), and the microdroplet generator, MDG (c) are presented. | ||
The instrument's data acquisition system was used to record the transient signals by repeated rapid mass scans at the plateau of the mass peak (so called E-scan of 5% of the total peak width) at dwell times of 1 ms as described previously.20 Shorter dwell times are principally of advantage when recording NP events which typically last for <0.5 ms because less noise is integrated per data point and lower limits of detection can be obtained.44–47 The main objective of this study was however the comparison of DE and TE for the different sample introduction methods and 1 ms was used to reduce data volume and processing times.
The instrument software however requires a minimum settling time of 1 ms to be included before each scan where no data are recorded. This reduces the duty cycle of the measurement and has to be considered when for example determining TE and PNC. For the conditions chosen in these experiments the duty cycle was 98.5% which was considered acceptable for the purpose of the study. The instrument also uses an SEM detector with two-stage signal acquisition. The ion counting mode was exclusively used in these experiments to avoid cross-calibration errors. Ion counting provides a linear range of approximately 3–5 106 cps with detector dead time correction. A transient signal shorter than the 1 ms dwell time and yielding a total of 3000 counts may however exhibit peak intensities where the common dead time correction, assuming a non-paralyzable detector, may fail. To avoid dead time-related artifacts, we adjusted the concentrations of ionic solutions and particle sizes whenever possible to yield less than 106 cps or 1000 counts per ms.
The “size method”22 was used for TE determination with the pneumatic nebulizer systems (PN and DSN) using a suspension of 30 nm Au NPs and a gold ionic solution of 0.0014 and 0.013 μg L−1, respectively. In order to avoid ion counting errors when using the DSN, the instrument's sensitivity was reduced to <1000 counts per NP by reducing the “focus lens” settings.
The TE of the MDG setup was determined by counting the number of events from dissolved Ir in the NP suspensions and relating it to the number of droplets produced by the MDG at the beginning and the end of each NP analysis.
The acquisition parameters of ICPMS for all three setups are given in Table 1.
| Parameter | Value |
|---|---|
| Samples per peak | 1000 |
| Sample (dwell) time, ms | 1 |
| Settling time, ms | 1 |
| Mass window, % | 5 |
| Runs (NPs) | 5000 |
| Runs (dissolved analytes) | 1000 |
| Passes | 1 |
| Detection mode | Counting |
| # replicates (NPs) | 5 |
| # replicates (Ir) | 1 |
| # replicates (dissolved analytes) | 2 |
| Total acquisition time, s (NPs) | 1250 |
| Total acquisition time, s (dissolved analytes) | 1320 |
Operating conditions used for the different sample introduction setups are listed in Table 2. Optimization was performed on a daily basis for the highest sensitivity. MDG settings are listed in Table S2.†
| PN standard interface | MDG standard interface | DSN “Jet” interface | MDG “Jet” interface | |
|---|---|---|---|---|
| a 25 μm droplet diameter, 100 Hz droplet frequency. b Al, Ce, Cs, Ti. c Ag, Cd, Fe, In, U, Au, Ir, Cu, Zn. | ||||
| Sampling depth, mm | −1.1b, 0c | −4.8 | −4.8 | −4.8 |
| Carrier gas, L min−1 | 1.2 | 1.3 | 1.2–1.8 | 1.2 |
| Sample uptake, μL min−1 | 120 | 0.049a | 90–160 | 0.049a |
| Auxiliary gas, L min−1 | 1.0 | 1.2 | 1.1 | 1.2 |
| RF-power, W | 1350 | 1330 | 1350 | 1330 |
| Nitrogen gas, mL min−1 | — | 0–14 | 16.5–21 | 0–20 |
| Membrane gas, L min−1 | — | — | 4.1 | — |
| Hot gas flow, L min−1 | — | — | 0.4 | — |
| Helium gas, L min−1 | — | 0.450 | — | 0.3 |
| Remarks | UO+/U+ = 6% | — | — | — |
Data evaluation
All measurements were carried out in time resolved mode and raw data were processed using the custom written code for Matlab ® (R2017b, MathWorks, Inc.) and further data evaluation was done by using Excel ® (Microsoft Corp.). Background subtraction was done by selecting a threshold value, where the instrumental background could be separated from the sample signals. Further, a split correction was applied to all signal intensities which were equal or higher than this threshold value.In order to obtain statistically representative data, at least 500 NP events were recorded per analysis. NP size calibration was carried out via 30 nm Au NP reference materials for PN and DSN sample introduction. When using the MDG system, an ionic Au standard was introduced to determine the absolute sensitivity. Hence, the measured signals of Au NPs were divided by the absolute sensitivity (ion counts per mass) and DE (ion counts per atoms introduced). DEs for 13 elements were calculated using eqn (1) for pneumatic nebulization with or without desolvation and eqn (2) for the MDG setup:
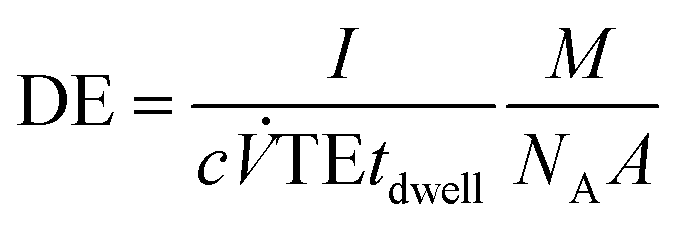 | (1) |
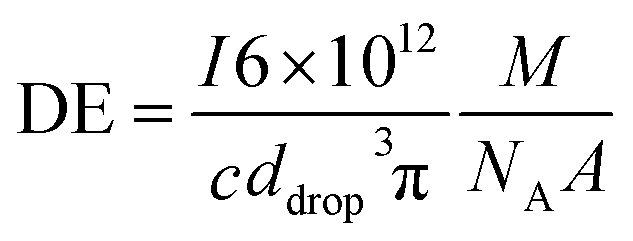 | (2) |
![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) (mL s−1) is the sample uptake, c (g mL−1) is the concentration of the element in solution, A is the relative abundance of the isotope measured, TE is the transport efficiency of the sample introduction system, tdwell (s) is the used dwell (integration) time, and ddrop (μm) is the diameter of the droplet generated with the MDG. The sample uptake for pneumatic nebulization was determined gravimetrically and the droplet diameter was captured continuously via the CCD camera.
(mL s−1) is the sample uptake, c (g mL−1) is the concentration of the element in solution, A is the relative abundance of the isotope measured, TE is the transport efficiency of the sample introduction system, tdwell (s) is the used dwell (integration) time, and ddrop (μm) is the diameter of the droplet generated with the MDG. The sample uptake for pneumatic nebulization was determined gravimetrically and the droplet diameter was captured continuously via the CCD camera.
ICPMS in principle can only yield the mass of an element in an NP. Accordingly, we report NP limits of detection (LODs) generally based on mass (eqn (3)) and assuming Poisson-noise dominated background signals.25,52 For NPs with known stoichiometry and assuming a spherical shape they were transformed into a size LOD (eqn (4)) as well.
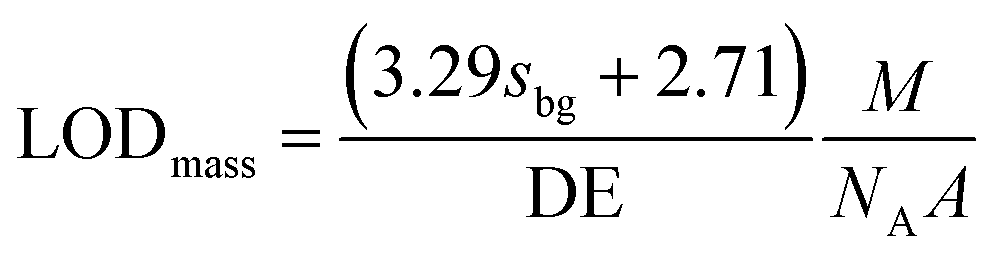 | (3) |
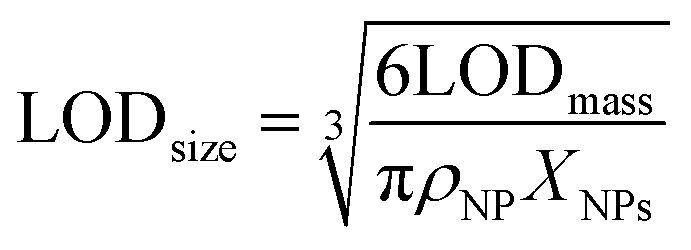 | (4) |
Results and discussion
Transport efficiency (TE)
TEs for PN and DSN were determined using the size method, while the TE for the MDG sample introduction setup was established via the number of events recorded for droplets containing dissolved Ir in an Au suspension with 0.1 mM citrate buffer (see Fig. S2†). With conventional nebulization without desolvation a transport efficiency of 10.5 ± 0.6% was reached in the best case. With desolvation the actual sample volume reaching the ICP could be doubled (23 ± 7%) because a substantial fraction of the aerosol droplets were at least partly evaporated to allow for more efficient transfer than with the conventional spray chamber arrangement. The MDG on the other hand provided almost quantitative (98.5 ± 0.6%) TE. The transient signals for the droplet tracer Ir show a highly reproducible occurrence at the dispensing frequency (Fig. S2†), which lets us assume that in fact all droplets reached the ICP but a fraction of them were not detected because they arrived during the “blind time” of the acquisition system.Detection efficiencies
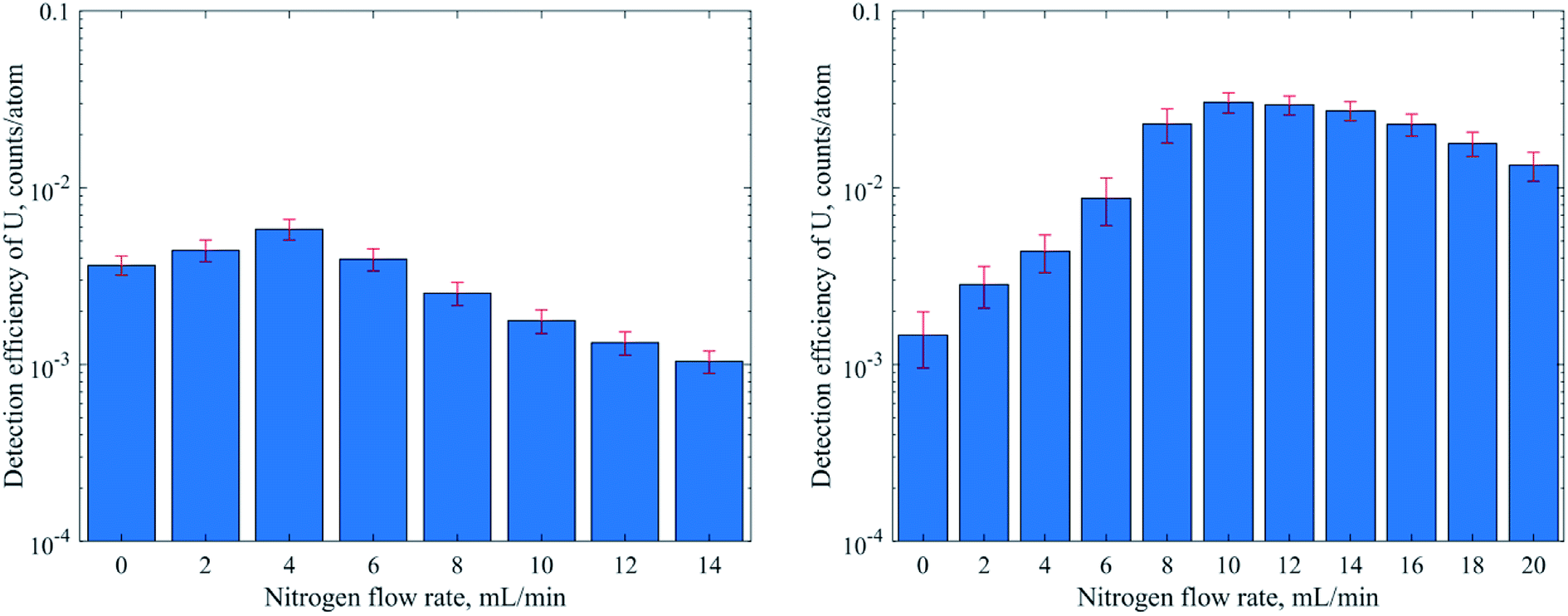 | ||
| Fig. 2 Effect of nitrogen addition to the carrier gas on the detection efficiency of dissolved uranium, when the MDG setup with the standard (left) or “Jet” (right) interface was used. | ||
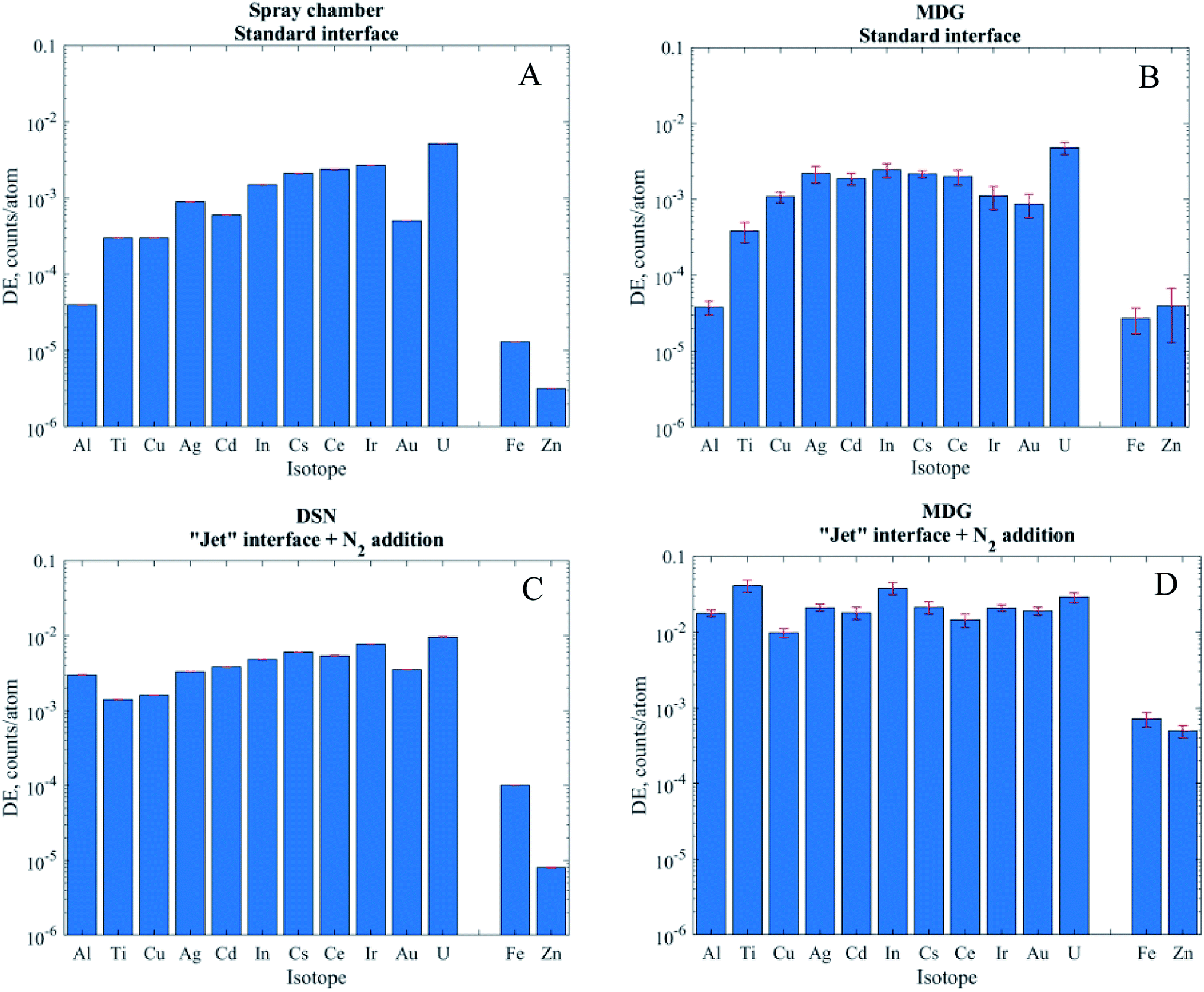 | ||
| Fig. 3 DEs of different elements when using the spray chamber (A) and MDG (B) with the standard interface, and the DSN (C) and MDG (D) with the “Jet” interface. Measurements are performed in low resolution. Error bars in A and C are hardly visible. As Fe and Zn are measured in medium resolution, they are presented separated from the rest of the isotopes which are measured in low resolution. | ||
When using pneumatic nebulization without desolvation and the MDG with the standard interface the DEs obtained were between 10−4 and 10−2 for isotopes measured in low resolution (MRP = 300). In line with the commonly observed mass discrimination, the DEs generally increased with m/z of the isotopes apart from elements with high ionization energies like Au. Operation in medium resolution (MRP of 3000, Fe and Zn) typically reduces ion transmission by a factor of 10 and, accordingly, DEs of 10−5 (Fe) and 3 × 10−6 (Zn) were obtained. For operation with the “Jet” interface and nitrogen addition on the other hand not only substantially higher DEs could be achieved but also the instrument's mass discrimination was reduced substantially if not eliminated across the range of isotopes investigated. Al or Ti for example showed almost three orders of magnitude higher DE with the “Jet” interface, while the DE of U only increased by only a factor of 2 (DSN) or 6 (MDG). Overall, DEs were slightly higher with the MDG when compared to PN and DSN methods. In particular using the “Jet” interface resulted in 3–8 times higher DE. Ti and Zn on the other hand reproducibly showed an exceptional increase by more than 20 times, which is not fully understood at this stage.
Characterization of NPs and LODs
For the comparison of NP analyses with the different instrumental configurations two suspensions of Au NPs were used. Due to their age the sizes were verified by transmission electron microscopy and found to be in good agreement with those originally specified by the manufacturers. Ionic Au standards and 30 nm Au NPs (size reference value: 31 ± 3 nm, measured 32 ± 4 nm) were used to either determine the transport efficiency in PN and DSN measurements via the “size” and “frequency” methods. They were also used to determine the DE for the calibration of the mass/size of the 10 nm Au NPs (size reference value: 8.8 ± 0.4 nm, measured 8.8 ± 0.4 nm) and PNC with all setups. For MDG experiments also ionic standards containing a known concentration of Au were used to determine the DE. When using the “Jet” configuration however the 30 nm NPs' ion signal intensities were at the upper limit of the linear dynamic range of the ion counting detection mode. Therefore, a set of experiments was carried out using the DSN where the signal intensity was artificially reduced by about 10 times by de-focusing the ion beam in the ion optics of the ICPMS.Results for the NP size and PNC are compiled in Table 3. When using the 31 nm Au NPs for calibration of the DE, only the conventional PN method and the DSN with the de-focused ion beam yielded acceptable size values for the 8.8 nm NPs. Both methods using the higher sensitivity of the “Jet” interface resulted in particles sizes that were approximately 50% too high. This is a direct result of the ion signals being near the upper end of the linear detection range. As can be seen in the histograms (Fig. S3C†) the ion counts recorded for 30 nm Au NPs with the “Jet” interface appear to be truncated as the signal exceeds 3000 counts. In fact, when extrapolating the signal for ionic standards or the 10 nm NPs (Fig. S4†), the 30 nm Au NPs should yield a mean value near 10000 counts or 10 Mcps, which is outside the linear range for ion counting with this secondary electron multiplier. Thus, the sensitivity or DE is underestimated by approximately 3 times, yielding a proportionally higher mass or about 50% larger size. The deviation in size obtained with the MDG and standard interface can however not readily be explained by this effect, because the signal intensities for the 31 nm Au NPs should have been well within the linear detection range (Fig. S3A and B†).
| Method | Size, nm | PNC, 1012 mL−1 | ||
|---|---|---|---|---|
| NPa | Ionicb | Sizec | Freq.d | |
| a Based on DE calibration using 31 nm Au NPs. b Based on DE calibration by a dissolved Au standard. c Based on TE determination using the “size” method. d Based on TE determination using the “frequency” method. e Based on TE determination via the droplet tracer. | ||||
| PN Std | 8.4 ± 1.6 | 5.2 ± 0.9 | 7.3 ± 0.4 | |
| DSN Jet | 12.5 ± 1.1 | 2.5 ± 0.5 | 8.5 ± 0.6 | |
| DSN Jet de-focused | 8.9 ± 1.2 | 5.1 ± 1.6 | 6.9 ± 0.4 | |
| MDG Std | 12.2 ± 1.5 | 8.9 ± 1.1 | 4.70 ± 0.06e | 5.7 ± 0.1 |
| MDG Jet | 12.9 ± 1.4 | 8.9 ± 0.9 | 4.38 ± 0.07e | 5.8 ± 0.1 |
When using dissolved standards with suitable concentrations on the other hand, the size of the 8.8 nm NPs could be determined with good agreement. From the results discussed before, it appears that calibration of the DE via NP suspensions requires a critical selection of an appropriate particle size. Larger particles often allow for better distinction between NP and background signals but too intense signals can cause underestimation of the DE and at the same time overestimation of the TE (see below). DE calibration using ionic standards however provides greater flexibility to match the calibrants' signal intensities to the mass spectrometer's linear range and mitigates the bias in size determination.
The PNCs determined with either approach were generally lower than the reference values (7.7 × 1012 NP mL−1) given by the manufacturer. When using pneumatic nebulization (PN or DSN) and the “size” method to determine TE, PNCs of 5.1 × 1012 mL−1 and 5.2 × 1012 mL−1 were obtained unless the 30 nm Au NPs used for calibration exceeded the linear range. In the latter case about 50% lower PNC was obtained, which is a direct result of underestimating the DE. Considering the age of the suspensions these deviations are considered to be caused by particles that may have settled during storage and were eventually not re-suspended before dilution. This assumption would be supported by the fact that using the 30 nm Au suspension to calibrate the TE via the “frequency” method22 yielded a PNC closer to the expected value.
Using the MDG on the other hand, the PNC determined with the standard interface configuration was slightly below the 5 × 1012 mL−1 obtained using PN and DSN, while the experiments with the “Jet” interface yielded a 15% lower value. The latter again indicates that the detection electronics might have “missed” events that exceeded the linear detection range. Another possible explanation for the lower PNC results for the MDG setup might be an inaccurate sizing of the droplets, but the fact that the NP size could be accurately reproduced using ionic standards does not support this.
Limits of detection
The use of the MDG in combination with the “Jet” interface yielded substantially higher DE for all elements investigated when compared to the conventional nebulization methods. Of particular interest for NP analyses is however that the mass discrimination of the ICPMS was substantially reduced. Due to the fact that most natural and many engineered and natural NPs consist of the more abundant lighter elements their analysis will benefit the most from the enhanced detection capability using the combination of the “Jet” interface and MDG for sample introduction. To highlight this feature, Table 4 compares the LODs in terms of mass and corresponding size of selected elements for conventional sp-ICP-MS operation and MDG sample introduction with the “Jet” interface. It needs to be kept in mind that any size/mass LOD ultimately depends on the instrumental background and the dissolved fraction of an element in the suspension. The data thus merely indicate that the higher DE for low mass isotopes can allow for reducing the lowest detectable NP sizes by 10 times in the best case (e.g. Al), while the only moderately higher DE for the heaviest isotopes does not yield appreciably lower size/mass LODs. In particular, in the latter case the concurrent increase of the instrumental background with DE appears to be the limiting factor.| PN | MDG “Jet” | PN | MDG “Jet” | |||||
|---|---|---|---|---|---|---|---|---|
| Sensitivity, cts g−1 | Bckg, cts | Sensitivity, cts g−1 | Bckg, cts | LODmass, ag | LODsize, nm | LODmass, ag | LODsize, nm | |
| Al | 9.37 × 1017 | 449 | 3.96 × 1020 | 485 | 283 | 58.5 | 0.354 | 6.3 |
| Al2O3 | 535 | 63.5 | 0.669 | 6.8 | ||||
| Ti | 2.95 × 1017 | 26 | 3.77 × 1019 | 29 | 91.3 | 33.8 | 0.765 | 6.9 |
| TiO2 | 152 | 41.0 | 1.28 | 8.3 | ||||
| Fe | 1.27 × 1017 | 4 | 7.01 × 1018 | 1 | 88.6 | 27.8 | 1.27 | 6.8 |
| Fe3O4 | 122 | 35.6 | 1.76 | 8.7 | ||||
| Cu | 2.29 × 1018 | 41 | 6.40 × 1019 | 15 | 13.4 | 14.2 | 0.60 | 5.0 |
| CuO | 16.8 | 17.2 | 0.75 | 6.1 | ||||
| Zn | 8.24 × 1015 | 1 | 1.27 × 1018 | 1 | 869 | 61.5 | 7.84 | 12.8 |
| ZnO | 1080 | 71.4 | 9.76 | 14.9 | ||||
| ZnS | 1290 | 84.6 | 11.7 | 17.6 | ||||
| Ag | 2.67 × 1018 | 8 | 6.10 × 1019 | 212 | 5.73 | 10.1 | 1.70 | 6.8 |
| Ce | 8.96 × 1018 | 1 | 5.50 × 1019 | 0.1 | 0.864 | 5.9 | 0.120 | 3.0 |
| CeO2 | 1.06 | 6.4 | 0.147 | 3.3 | ||||
| Au | 1.44 × 1018 | 0.5 | 5.79 × 1019 | 2 | 4.06 | 7.3 | 0.271 | 3.0 |
| U | 1.31 × 1019 | 1 | 7.23 × 1019 | 1 | 0.494 | 3.7 | 0.354 | 3.3 |
| Cd | 8.91 × 1017 | 4 | 2.77 × 1019 | 17 | 12.8 | 14.2 | 0.94 | 5.9 |
| CdS | 16.5 | 18.7 | 1.2 | 7.8 | ||||
Conclusions
In this work, the capabilities of different instrumental configurations for sp-ICPMS analysis were compared with respect to detection and transport efficiency, DE and TE, and the achievable LODsize for nanoparticle detection. The conventional pneumatic nebulizer spray chamber combination yielded a TE of 10.5% and DE was in the range of 10−5 to 10−3 ion counts per atom present in the ICP. Isotopes detected using a mass resolving power of 4000 instead of 300 had correspondingly about 10 times lower DEs. A higher TE (23%) could be obtained by using a desolvating nebulizer system (DSN), while microdroplet introduction could provide quantitative sample transfer to the ICP. Determination of the transport efficiency was straightforward with the MDG setup but required adequately sized NPs when the pneumatic nebulizers were used. If the available NPs for TE calibration approach or exceed the linear detection range, TE will be overestimated. This has a direct impact on DE calibration and thus determination of NPs size and PNC. Here again MDG sampling proved beneficial because adjusting the element's concentration in the droplets provides a highly flexible method to ensure signal intensities with the mass spectrometer's linear range. The most significant impact for nanomaterial analyses is however provided through the use of the “Jet” interface. Substantially higher sensitivities can be realized, in particular in combination with MDG sampling, and specifically for isotopes of low m/z. The mass spectrometer's mass discrimination is substantially reduced and results in almost 3 orders of magnitude enhancement in sensitivity for 27Al for example. The LODsize for Al-containing NPs can thus be lowered by an order of magnitude. Unless isotopes of poor sensitivity like 66Zn have to be analysed the size limit of detection was in the single digit nm range for mono-elemental NPs of all elements investigated. Because of the flexibility with respect to calibration of the instrument's sensitivity and percentage detection efficiency, compared to conventional methods of NP analysis, we consider the combination of microdroplet generation and the Jet interface as a highly valuable method for accurate and sensitive determination of NPs in the nm range. It paves the way to be able to also detect and quantify quantum dots containing just a few 1000 of detectable atoms.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work has received funding from European Union Horizon 2020 Programme (H2020) under grant agreement no 720952 via SBFI Ref-1131-52302. A special thanks goes to Ralf Kägi for support and the TEM measurements. We also would like to thank the reviewers of this manuscript for their helpful and constructive comments.References
- W. J. Stark, P. R. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- M. E. Vance, T. Kuiken, E. P. Vejerano, S. P. McGinnis, M. F. Hochella and D. R. Hull, Beilstein J. Nanotechnol., 2015, 6, 1769–1780 CrossRef CAS.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS.
- F. Gottschalk and B. Nowack, J. Environ. Monit., 2011, 13, 1145–1155 RSC.
- M. F. Hochella, D. W. Mogk, J. Ranville, I. C. Allen, G. W. Luther, L. C. Marr, B. P. McGrail, M. Murayama, N. P. Qafoku, K. M. Rosso, N. Sahai, P. A. Schroeder, P. Vikesland, P. Westerhoff and Y. Yang, Science, 2019, 363, eaau8299 CrossRef.
- Acenano, http://acenano-project.eu, accessed 1 December 2018 Search PubMed.
- F. Laborda, E. Bolea, G. Cepriá, M. T. Gómez, M. S. Jiménez, J. Pérez-Arantegui and J. R. Castillo, Anal. Chim. Acta, 2016, 904, 10–32 CrossRef CAS.
- F. von der Kammer, P. L. Ferguson, P. A. Holden, A. Masion, K. R. Rogers, S. J. Klaine, A. A. Koelmans, N. Horne and J. M. Unrine, Environ. Toxicol. Chem., 2012, 31, 32–49 CrossRef CAS.
- L. Hendriks, B. Ramkorun-Schmidt, A. Gundlach-Graham, J. Koch, R. N. Grass, N. Jakubowski and D. Günther, J. Anal. At. Spectrom., 2019, 34, 716–728 RSC.
- K. Mehrabi, A. Gundlach-Graham, D. Günther and A. Gundlach-Graham, Environ. Sci.: Nano, 2019, 6, 3349–3358 RSC.
- M. D. Montaño, F. Von Der Kammer, C. W. Cuss and J. F. Ranville, J. Anal. At. Spectrom., 2019, 34, 1768–1772 RSC.
- C. Degueldre and P. Y. Favarger, Colloids Surf., A, 2003, 217, 137–142 CrossRef CAS.
- C. Degueldre, P. Y. Favarger and C. Bitea, Anal. Chim. Acta, 2004, 518, 137–142 CrossRef CAS.
- C. Degueldre and P. Y. Favarger, Talanta, 2004, 62, 1051–1054 CrossRef CAS.
- C. Degueldre, P. Y. Favarger, R. Rossé and S. Wold, Talanta, 2006, 68, 623–628 CrossRef CAS.
- C. Degueldre, P. Y. Favarger and S. Wold, Anal. Chim. Acta, 2006, 555, 263–268 CrossRef CAS.
- D. Mozhayeva and C. Engelhard, J. Anal. At. Spectrom., 2020, 35(9), 1740–1783 RSC.
- D. M. Mitrano, A. Barber, A. Bednar, P. Westerhoff, C. P. Higgins and J. F. Ranville, J. Anal. At. Spectrom., 2012, 27, 1131 RSC.
- J. Tuoriniemi, T. Holbrook, G. Cornelis, M. Schmitt, H.-J. Stärk and S. Wagner, J. Anal. At. Spectrom., 2020, 1678–1686 RSC.
- S. Gschwind, M. D. L. Aja Montes and D. Günther, Anal. Bioanal. Chem., 2015, 407, 4035–4044 CrossRef CAS.
- M. Hadioui, G. Knapp, A. Azimzada, I. Jreije, L. Frechette-Viens and K. J. Wilkinson, Anal. Chem., 2019, 91, 13275–13284 CrossRef CAS.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361–9369 CrossRef CAS.
- IAS Inc., MSAG, http://iasinc.jp/english/product5.html, accessed 15 July 2020.
- J.-L. Todoli and J.-M. Mermet, Liq. Sample Introd. ICP Spectrom., 2008, pp. 119–146 Search PubMed.
- F. Laborda, J. Jiménez-Lamana, E. Bolea and J. R. Castillo, J. Anal. At. Spectrom., 2013, 28, 1220–1232 RSC.
- G. M. Hieftje and H. V. Malmstadt, Anal. Chem., 1968, 40, 1860–1867 CrossRef CAS.
- J. W. Olesik, Appl. Spectrosc., 1997, 51(5), 158A–175A CrossRef CAS.
- C. C. Garcia, A. Murtazin, S. Groh, V. Horvatic and K. Niemax, J. Anal. At. Spectrom., 2010, 25, 645–653 RSC.
- S. Gschwind, L. Flamigni, J. Koch, O. Borovinskaya, S. Groh, K. Niemax and D. Günther, J. Anal. At. Spectrom., 2011, 26, 1166 RSC.
- S. Gschwind, H. Hagendorfer, D. A. Frick and D. Günther, Anal. Chem., 2013, 85, 5875–5883 CrossRef CAS.
- J. Koch, L. Flamigni, S. Gschwind, S. Allner, H. Longerich and D. Günther, J. Anal. At. Spectrom., 2013, 28, 1707–1717 RSC.
- K. Shigeta, H. Traub, U. Panne, A. Okino, L. Rottmann and N. Jakubowski, J. Anal. At. Spectrom., 2013, 28, 646 RSC.
- J. H. D. Hartley, S. J. Hill and L. Ebdon, Spectrochim. Acta, Part B, 1993, 48, 1421–1433 CrossRef.
- E. Roduner, Chem. Soc. Rev., 2006, 35, 583–592 RSC.
- T. Verano-Braga, R. Miethling-Graff, K. Wojdyla, A. Rogowska-Wrzesinska, J. R. Brewer, H. Erdmann and F. Kjeldsen, ACS Nano, 2014, 8, 2161–2175 CrossRef CAS.
- L. Shang, K. Nienhaus and G. Nienhaus, J. Nanobiotechnol., 2014, 12, 5 CrossRef.
- M. Yao, L. He, D. J. McClements and H. Xiao, J. Agric. Food Chem., 2015, 63, 8044–8049 CrossRef CAS.
- F. Krieg, Q. K. Ong, M. Burian, G. Raino, D. Naumenko, H. Amenitsch, A. Süess, M. Grotevent, F. Krumeich, I. Maryna, I. Shorubalko, F. Stellacci, M. V. Kovalenko, F. Krieg, Q. K. Ong, M. Burian, G. Rainò and D. Naumenko, J. Am. Chem. Soc., 2019, 141(50), 19839–19849 CrossRef CAS.
- S. T. Ochsenbein, F. Krieg, Y. Shynkarenko, G. Rainò and M. V. Kovalenko, ACS Appl. Mater. Interfaces, 2019, 11, 21655–21660 CrossRef CAS.
- Y. Kelestemur, Y. Shynkarenko, M. Anni, S. Yakunin, M. L. De Giorgi and M. V. Kovalenko, ACS Nano, 2019, 13(12), 13899–13909 CrossRef CAS.
- T. J. Brunner, P. Wick, P. Manser, P. Spohn, R. N. Grass, L. K. Limbach, A. Bruinink and W. J. Stark, Environ. Sci. Technol., 2006, 40, 4374–4381 CrossRef CAS.
- M. Auffan, J. Rose, J. Y. Bottero, G. V. Lowry, J. P. Jolivet and M. R. Wiesner, Nat. Nanotechnol., 2009, 4, 634–641 CrossRef CAS.
- E. Oh, J. B. Delehanty, K. E. Sapsford, K. Susumu, R. Goswami, J. B. Blanco-Canosa, P. E. Dawson, J. Granek, M. Shoff, Q. Zhang, P. L. Goering, A. Huston and I. L. Medintz, ACS Nano, 2011, 5, 6434–6448 CrossRef CAS.
- P. Shaw and A. Donard, J. Anal. At. Spectrom., 2016, 31, 1234–1242 RSC.
- J. Tuoriniemi, G. Cornelis and M. Hassellöv, J. Anal. At. Spectrom., 2015, 30, 1723–1729 RSC.
- G. Cornelis and M. Hassellöv, J. Anal. At. Spectrom., 2014, 29, 134–144 RSC.
- R. Peters, Z. Herrera-Rivera, A. Undas, M. Van Der Lee, H. Marvin, H. Bouwmeester and S. Weigel, J. Anal. At. Spectrom., 2015, 30, 1274–1285 RSC.
- Z. Hu, Y. Liu, S. Gao, W. Liu, W. Zhang, X. Tong, L. Lin, K. Zong, M. Li, H. Chen, L. Zhou and L. Yang, J. Anal. At. Spectrom., 2012, 27, 1391–1399 RSC.
- L. A. Currie, Anal. Chim. Acta, 1999, 391, 105–126 CrossRef CAS.
- J. Koch, L. Flamigni, S. Gschwind, S. Allner, H. Longerich and D. Günther, J. Anal. At. Spectrom., 2013, 28, 1707–1717 RSC.
- U. W. S. Rasband, ImageJ, U.S. National of Health, Bethesda, MA, http://imagej.nih.gov/ij/ Search PubMed.
- S. Lee, X. Bi, R. B. Reed, J. F. Ranville, P. Herckes and P. Westerhoff, Environ. Sci. Technol., 2014, 48, 10291–10300 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ja00421a |
| This journal is © The Royal Society of Chemistry 2021 |