
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMacrocycle based dinuclear dysprosium(III) single molecule magnets with local D5h coordination geometry†
Jianfeng
Wu
ab,
Serhiy
Demeshko
a,
Sebastian
Dechert
 a and
Franc
Meyer
*a
a and
Franc
Meyer
*a
aInstitut für Anorganische Chemie, Universität Göttingen, Tammannstr. 4, D-37077 Göttingen, Germany. E-mail: franc.meyer@chemie.uni-goettingen.de
bSchool of Chemistry and Chemical Engineering, Northwestern Polytechnical University, Xi'an 710072, P. R. China
First published on 19th November 2021
Abstract
Targeted approaches for manipulating the coordination geometry of lanthanide ions are a promising way to synthesize high-performance single-molecule magnets (SMMs), but most of the successful examples reported to date focus on mononuclear complexes. Herein, we describe a strategy to assemble dinuclear SMMs with DyIII ions in approximate D5h coordination geometry based on pyrazolate-based macrocyclic ligands with two binding sites. A Dy4 complex with a rhomb-like arrangement of four DyIII as well as two dinuclear complexes having axial chlorido ligands (Dy2·Cl and Dy2*·Cl) were obtained; in the latter case, substituting Cl− by SCN− gave Dy2·SCN. Magneto-structural studies revealed that the μ-OH bridges with short Dy–O bonds dominate the magnetic anisotropy of the DyIII ions in centrosymmetric Dy4 to give a vortex type diamagnetic ground state. Dynamic magnetic studies of Dy4 identified two relaxation processes under zero field, one of which is suppressed after applying a dc field. For complexes Dy2·Cl and Dy2*·Cl, the DyIII ions feature almost perfect D5h environment, but both complexes only behave as field-induced SMMs (Ueff = 19 and 25 K) due to the weak axial Cl− donors. In Dy2·SCN additional MeOH coordination leads to a distorted D2d geometry of the DyIII ions, yet SMMs properties at zero field are observed due to the relatively strong axial ligand field provided by SCN− (Ueff = 43 K). Further elaboration of preorganizing macrocyclic ligands appears to be a promising strategy for imposing a desired coordination geometry with parallel orientation of the anisotropy axes of proximate DyIII ions in a targeted approach.
Introduction
Lanthanides have attracted increasing attention in the field of single-molecule magnetism during the past two decades,1 mostly due to their large magnetic moments and their often highly anisotropic character, which favors large energy barriers and high blocking temperatures for the magnetization relaxation.2 According to crystal field theory and electrostatic models,3 DyIII ions in an axial ligand field with D4d,4D5h,5D6h,6 and C∞v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 symmetry are likely to feature particularly large energy barriers and a low probability for quantum tunnelling of the magnetization (QTM). In light of these conceptual considerations, numerous dysprosium-based single-molecule magnets (SMMs) with excellent magnetic properties and hysteretic behaviour have been synthesized, reaching blocking temperatures up to 80 K.8 However, these high-performance Dy-based SMMs are mostly limited to mononuclear systems.
7 symmetry are likely to feature particularly large energy barriers and a low probability for quantum tunnelling of the magnetization (QTM). In light of these conceptual considerations, numerous dysprosium-based single-molecule magnets (SMMs) with excellent magnetic properties and hysteretic behaviour have been synthesized, reaching blocking temperatures up to 80 K.8 However, these high-performance Dy-based SMMs are mostly limited to mononuclear systems.
An increasingly pursued strategy for enhancing the SMM properties aims at promoting magnetic interactions in complexes with several proximate DyIII ions.9 Due to the usually high coordination numbers and flexibility of the coordination spheres of lanthanide ions,10 however, it is quite challenging to simultaneously control the geometry and ligand field in such oligonuclear complexes. Dinuclear systems represent the simplest case of oliogo- and polymetallic 4f element SMMs, and manipulating their ligand field and magnetic interactions appears to be comparatively facile.11 In many reported dinuclear DyIII complexes the two DyIII ions are ferromagnetically coupled,12 which reduces the QTM probability. In most of these complexes, however, the DyIII ions are surrounded by more than two ligands and reside in rather distorted coordination geometries of low symmetry, often approximating D2d13 and C2v,14 which is unfavourable for establishing an axial ligand field of the DyIII ions and usually does not result in a large anisotropy or high blocking temperature.
To realize axial ligand fields in dinuclear complexes in a targeted approach, careful ligand design is essential. On the one hand, the ligand should allow for high coordination numbers and should not be in conflict with the flexible nature of the 4f metal ions’ coordination spheres. On the other hand, the coordinating atoms of the ligand scaffold should be precisely arranged to enforce a specific coordination symmetry. To meet the two demands simultaneously, one efficient way is to place two strong donor ligands at the axial directions to establish the dominating anisotropy axis while surrounding the metal ion with weak field ligands in the equatorial plane to provide the high coordination number that is favored by DyIII ions. To that end, planar macrocycle-based scaffolds with weak N or O donors within the equatorial plane, which direct the binding of stronger exogenous donor ligands to the axial positions, show great promise for DyIII SMMs with large anisotropy. In this context, a few mononuclear dysprosium SMMs based on crownether15 or metallacrown ligands have been reported.16 Recently, Tong et al. achieved an approximately collinear arrangement of the DyIII–DyIII Ising moments of two DyIII monomers that are based on Cu5 metallacrowns with local D5h geometry; their magnetic signatures are a result of both antiferromagnetic coupling within the Cu5 metallacrowns as well as ferromagnetic Dy⋯Cu and Dy⋯Dy interactions.17 In order to exclude magnetic contributions from an open-shell metallacrown, the construction of dinuclear DyIII SMMs from planar macrocyclic ligands providing two pentadentate binding sites, combined with variation of axial coligands, appears to be a promising strategy to establish a proper ligand field of the DyIII ions in D5h coordination geometry.
With these thoughts in mind, and in view of our group's long history in the development of dinucleating pyrazolate ligands,18,19 we set out to synthesize dinuclear DyIII SMMs using bis(pyrazolato)-bridged macrocycles with two {N5} donor compartments that provide the metal ions’ equatorial coordination sphere. To that end, we conducted metal ion templated [2 + 2] ring formation reactions between pyrazole 3,5-dialdehyde and linear triamines. Unexpectedly, however, macrocycle formation did not occur and a Dy4 complex was obtained from these reactions (Scheme 1). We then turned to the corresponding pyrazole 3,5-diketone building block, which indeed led to the anticipated macrocycle-based dinuclear DyIII complexes (Scheme 2). All new complexes have been comprehensively characterized structurally and magnetically, and magneto-structural considerations have been used to rationalize the findings. The results are presented and further prospects of the approach are discussed in this paper.
 | ||
| Scheme 1 Schematic drawing of the assembly of Dy4. | ||
 | ||
| Scheme 2 Schematic drawing of the assemblies of Dy2·Cl, Dy2*·Cl, and Dy2·SCN. | ||
Results and discussion
Synthesis and structural characterization of Dy4
Our initial objective was to assemble dinuclear DyIII complexes with macrocyclic ligands that are derived from the condensation of pyrazole 3,5-dialdehydes with diethylentriamine via a templated [2 + 2] ring formation reaction (Scheme 1). When using 4-phenyl-3,5-dicarbaldehyde-1H-pyrazole, however, the unexpected product [Dy4(μ3-OH)2L12(μ2-OH)2(DMF)4]·4ClO4 (Dy4; H2L1 = bis((5-dimethoxymethyl-4-phenyl-1H-pyrazol-methylene-amino)ethyl)-amine) was isolated. Herein, the ligand H2L1 was formed from the [1 + 2] condensation of one diethylenetriamine with two pyrazole dialdehyde building blocks and acetal formation with methanol at the remaining aldehyde functions (Scheme S1†). Such acetal formation is common for aldimine condensation reactions when alcohols are present.20 The pyrazole-based ligand is deprotonated during the complexation with DyIII ions.Complex Dy4 was isolated as yellow crystals by slow diffusion of diethyl ether into a DMF solution of the crude product. Single-crystal X-ray diffraction revealed that complex Dy4 crystallizes in the monoclinic space group P21/n (Table S1†) and shows centrosymmetry with the asymmetric unit containing half of the complex: one ligand, two DyIII ions, two coordinated DMF, two μ-OH bridges, and two ClO4−. The presence of OH groups is corroborated by a band at 3582 cm−1 for the O–H stretch in the IR spectrum of solid Dy4 (Fig. S1†). Each [L1]2− ligand strand in Dy4 is bent to bind two DyIII ions, one in the inner {N5} and one in the outer {N2O2} pocket, forming a Dy2 synthon with two additional DMF molecules coordinated to the DyIII ions. Two Dy2 subunits are connected by two μ3-OH and two μ-OH groups, forming Dy4 with the four DyIII ions arranged in a rhomb-like topology (Fig. 1). Four ClO4− are present in the lattice to balance the positive charges (Fig. S6†). Due to the centrosymmetric nature of the molecule, the four DyIII ions are in one plane. The two μ3-OH are located above and below this plane of the Dy4 rhomb, and the μ-OH each connect two DyIII ions at the edges of the rhomb. Selected atom distances and angles are listed in Tables S2 and S3.† Comparing the bond lengths, we find that the Dy–O6 (μ-OH) bonds (2.26 and 2.27 Å) are much shorter than the other coordination bonds. The anisotropy axes of DyIII ions are usually close to the shortest coordination bonds, which provides an efficient single/double atomic axial crystal field.21 Therefore, in our case, the magnetic anisotropies of the DyIII ions are probably related to the μ-OH bridges.
 | ||
| Fig. 1 Molecular structure of the cation of Dy4. Hydrogen atoms and counteranions have been omitted for clarity. | ||
Apart from the bond lengths, the individual coordination geometry also influences the anisotropy of DyIII ions. In complex Dy4, both Dy1 and Dy2 are eight coordinate but in different coordination environments (Fig. S10†). We used SHAPE22 to analyze the coordination geometry of the DyIII ions. As shown in Table S6,† the geometry of Dy1 is best described as a distorted square antiprism (D4d) with a CShM value of 1.36, while the coordination geometry for Dy2 is close to a biaugmented trigonal prism (C2v) with a CShM value of 1.90. Another factor that influences the magnetic properties of the complex is the magnetic coupling between DyIII ions.23 In Dy4 the Dy⋯Dy distances are in the range of 3.72–3.81 Å, which is expected to give rise to weak intramolecular magnetic interactions.
Synthesis and structural characterization of Dy2·Cl and Dy2*·Cl
Complexes [Dy2L2Cl4] (Dy2·Cl; H2L2 = hexamethyl-hexaaza-dipyrazolacycloicosaphane-2,9,12,19-tetraene) and [Dy2L3Cl4] (Dy2*·Cl; H2L3 = (octamethyl-hexaaza-dipyrazolacycloicosaphane-2,9,12,19-tetraene) were obtained from the in situ reaction of 3,5-diacetyl-4-methyl-1H-pyrazole with diethylenetriamine (for Dy2·Cl) or N,N-bis(2-aminoethyl)methylamine (for Dy2*·Cl) and DyCl3·6H2O (Scheme 2). Herein, the reaction of the diketone with a diamine in methanol forms the macrocyclic ligands rather than an open chain ligand bearing acetal groups as was observed for Dy4, which we attribute to the different propensity of ketones vs. aldehydes to react with alcohols. X-ray diffraction (XRD) quality crystalline material of both complexes was obtained upon recrystallization from methanol. The purity of the bulk samples was confirmed by powder XRD analyses (Fig. S3 and S4†). Thermogravimetric analyses evidenced that the compounds are stable up to 350 °C (Fig. S5†), which is beneficial for potential applications. Dy2·Cl and Dy2*·Cl both crystallize in the monoclinic space group P21/c with similar cell parameters (Table S1†). As shown in Fig. 2a and b, structural features of the two complexes are also very similar. Their molecular entities are centrosymmetry, containing one macrocyclic ligand, two DyIII ions, and four coordinated Cl−. The central pyrazolate bridges of the ligand are coplanar and span two {N5} pockets that each hosts a DyIII ion, as was anticipated. The Cl− anions coordinate to DyIII in axial positions above and below the ligand plane, forming an overall pentagonal bipyramidal (D5h) coordination polyhedron (Fig. 2). In the crystal packing, the Cl− ligands are in contact with the nearest methyl groups from neighboring molecules, presumably via weak hydrogen bonds (C–H⋯Cl distances in the range of 2.50–2.87 Å) to constitute 2D (Dy2·Cl, Fig. S7†) and 3D frameworks (Dy2*·Cl, Fig. S8†) in the lattice. The Dy–N bond lengths in the molecular structures are in the range of 2.44–2.48 Å and 2.45–2.55 Å for Dy2·Cl and Dy2*·Cl (Tables S4 and S5†), respectively. The Dy–Cl bond lengths in the two complexes are close to 2.61 Å, which is much longer than the Dy–N distances, indicating axially elongated pentagonal bipyramidal coordination spheres. The Cl–Dy–Cl angles are 169.18 and 174.17° for Dy2·Cl and Dy2*·Cl, respectively. The almost linear Cl–Dy–Cl arrangement reflects the rather small distortion of the D5h coordination geometry.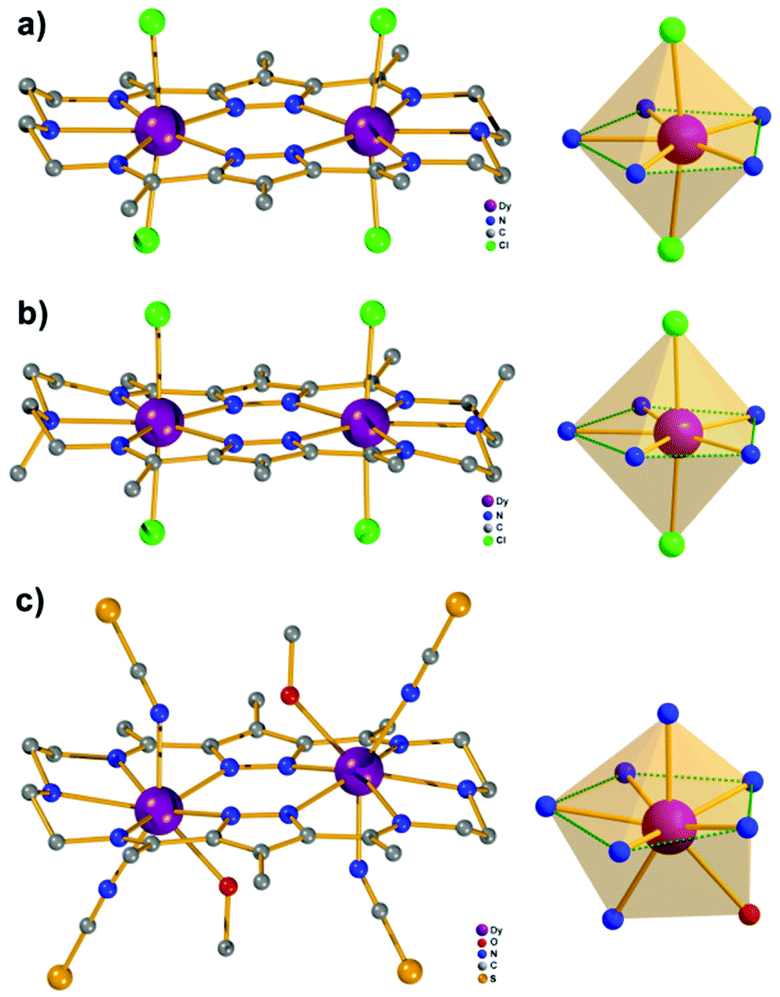 | ||
| Fig. 2 Structure views of complexes Dy2·Cl (a), Dy2*·Cl (b), and Dy2·NCS (c). The orange spheres represent the coordination polyhedron of the relevant DyIII ion in the complexes. Hydrogen atoms are omitted for clarity. | ||
To quantify the distortion of the coordination geometry, SHAPE analyses were carried out for the two complexes. The results are listed in Table S7.† As expected, the coordination polyhedra of the DyIII ions are close to a pentagonal bipyramid (D5h) with CShM values of 0.80 and 0.69 for Dy2·Cl and Dy2*·Cl, respectively. The small deviations indicate a successful control of the coordination geometry by these bis(pyrazolato) based macrocycle ligands, in accordance with the strategy outlined in the Introduction. Particularly, this is the first example of hosting two D5h coordinated DyIII ions within one macrocyclic ligand at rather short Dy⋯Dy distances (4.73 and 4.77 Å for Dy2·Cl and Dy2*·Cl, respectively). However, the weak Cl− donors at the axial positions result in a stretched D5h coordination environment, which is unfavorable for the anisotropy of the oblate DyIII ion. To modulate the ligand field and optimize potential SMMs properties (see below), substitution of the Cl− anions with stronger anionic N-donors was pursued in the case of complex Dy2·Cl.
Synthesis and structural characterization of Dy2·NCS
Complex [Dy2L2NCS4(MeOH)2] (Dy2·NCS) was obtained from Dy2·Cl by ligand substitution upon addition of NH4SCN, replacing Cl− by NCS− (Scheme 2). X-ray diffraction on single crystals obtained from methanol revealed that Dy2·NCS crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Table S1†); the molecular structure is shown in Fig. 2c. It features one dianionic macrocyclic ligand [L2]2−, two DyIII ions residing in the {N5} pockets, four SCN−, and two coordinated methanol solvent molecules (Fig. 2 and S9†). The {L2Dy2} core found in Dy2·Cl is preserved in Dy2·NCS, but two SCN− are now N-bound above and below the roughly planar core with Dy–N bond lengths of 2.40 and 2.42 Å. These axial bonds are shorter than the equatorial Dy–N bonds involving the macrocyclic ligand [L2]2− (2.45–2.55 Å). The two additional methanol ligands give rise to eight coordinate DyIII ions and move the DyIII 0.4 Å out of the equatorial {N5} plane towards the side of the methanol. Hence the NSCN–Dy–NSCN angles of 149.5° reflect strong deviation from a linear arrangement (Table S4†). SHAPE analysis suggests that the DyIII ions exhibit a distorted triangular dodecahedron geometry (D2d) with a CShM value of 2.6. In comparison with Dy2·Cl and Dy2*·Cl, the DyIII ions in Dy2·NCS reside in a lower symmetry coordination environment, yet the SCN− at the axial positions give rise to short Dy–N distances and a relatively strong ligand field; the latter was expected to prove beneficial in terms of SMM properties.
(Table S1†); the molecular structure is shown in Fig. 2c. It features one dianionic macrocyclic ligand [L2]2−, two DyIII ions residing in the {N5} pockets, four SCN−, and two coordinated methanol solvent molecules (Fig. 2 and S9†). The {L2Dy2} core found in Dy2·Cl is preserved in Dy2·NCS, but two SCN− are now N-bound above and below the roughly planar core with Dy–N bond lengths of 2.40 and 2.42 Å. These axial bonds are shorter than the equatorial Dy–N bonds involving the macrocyclic ligand [L2]2− (2.45–2.55 Å). The two additional methanol ligands give rise to eight coordinate DyIII ions and move the DyIII 0.4 Å out of the equatorial {N5} plane towards the side of the methanol. Hence the NSCN–Dy–NSCN angles of 149.5° reflect strong deviation from a linear arrangement (Table S4†). SHAPE analysis suggests that the DyIII ions exhibit a distorted triangular dodecahedron geometry (D2d) with a CShM value of 2.6. In comparison with Dy2·Cl and Dy2*·Cl, the DyIII ions in Dy2·NCS reside in a lower symmetry coordination environment, yet the SCN− at the axial positions give rise to short Dy–N distances and a relatively strong ligand field; the latter was expected to prove beneficial in terms of SMM properties.
Static magnetic properties
To explore the magnetic properties of Dy4, direct current (dc) magnetic susceptibility measurements were carried out on polycrystalline samples under an applied field of 1000 Oe in the temperature range 2–210 K. As shown in Fig. 3, the χMT product (χM is molar magnetic susceptibility) at 210 K is 52.97 cm3 K mol−1, which is slightly smaller than the expected value for four DyIII ions in the free-ion approximation (56.68 cm3 K mol−1, 6H15/2, g = 4/3 for DyIII ion). Upon cooling, the χMT product decreases gradually before dropping more quickly below 15 K, and it reaches 22.18 cm3 K mol−1 at 2 K. The drop of χMT at low temperatures can be attributed to the depopulation of the excited mJ sublevels of the ground J states of the DyIII ions, the onset of magnetic blocking, and a dominating diamagnetic ground state, which might result from antiferromagnetic interactions or from the toroidal arrangement of the magnetic axes of the individual DyIII ions.24 Under a 5000 Oe dc field, the temperature-dependent χMT data shows a similar profile. A plot of the molar magnetization (M) vs. H for Dy4 at 2 K (Fig. S13†) shows a relatively slow increase near zero field and a fast increase when rising the temperature (Fig. S13†), which is indicative of STM behavior. This is followed by a linear increase of M at high fields, reaching the value of 19.38 μB at 5 T, which is close to the expected value of 20 μB for four DyIII ions in a pure mJ = |±15/2> ground state. | ||
| Fig. 3 Temperature dependence of the product χMT for Dy4 at indicated fields between 2 and 210 K. | ||
To gain more insight into the magnetic properties of Dy4, the anisotropy axes of the DyIII ions were calculated by the Magellan program25 based on the structure determined crystallographically. As shown in Fig. 4, the magnetic anisotropy axes of all DyIII ions are close to Dy–O6 directions. This is mainly because of the short Dy–O bonds giving a relatively stronger ligand field that dominates the orientation of the local magnetic moments. Overall, the anisotropy axes of the four DyIII ions are arranged in a toroidal fashion, suggesting the potential existence of single-molecule toroic (SMT) behavior.24b,26 However, this calculation is based on an electrostatic model that does not include any intramolecular interaction and excited states, therefore future quantitative evaluation of the ground states and of the magnetic interaction J through ab initio calculations is desirable.26a,27
 | ||
| Fig. 4 Orientations of the main magnetic axes of the local magnetizations in the ground state of Dy4. | ||
For the dinuclear complexes Dy2·Cl, Dy2*·Cl, and Dy2·NCS, the variable temperature χMT data show values of 27.63, 27.67, and 27.30 cm3 K mol−1, respectively, at 210 K (Fig. 5). These values are close to the expected one for two DyIII ions in the free-ion approximation (28.34 cm3 K mol−1). Upon cooling, χMT decreases gradually before dropping more quickly below 50 K to reach 13.70 (Dy2·Cl), 14.09 (Dy2*·Cl), and 13.45 cm3 K mol−1 (Dy2·NCS) at 2 K. The drop of the χMT product at low temperatures can be ascribed to dominating antiferromagnetic interactions between the two DyIII ions. Plots of the molar magnetization (M) vs. H at 2 K (Fig. S14 and S15†) show a sharp increase at low fields followed by a linear increase at high fields, reaching the values of 10.24 (Dy2·Cl), 10.57 (Dy2*·Cl), and 11.51 μB (Dy2·NCS) at 5 T; these values are close to the expected value 10 μB for two DyIII ions in a pure mJ = |±15/2> ground state.
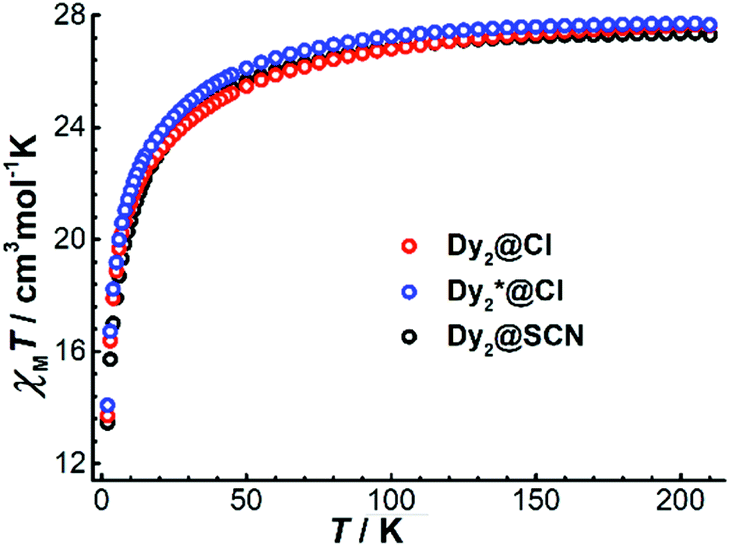 | ||
| Fig. 5 Temperature dependence of the product χMT for Dy2·Cl, Dy2*·Cl, and Dy2·NCS at 5000 Oe between 2 and 210 K. | ||
Dynamic magnetic properties
Alternating current (ac) susceptibility measurements were performed on Dy4 under an oscillating field of 3.0 Oe to study the dynamic magnetic properties. Two overlapped out-of-phase (χ′′) peaks at 9 and 14 K are detected in the temperature-dependent ac susceptibility at zero dc field (Fig. S17†), suggesting the presence of two relaxation processes. This behavior usually occurs in polynuclear SMMs that contain more than one asymmetric DyIII ion.28 In Dy4, the DyIII ions are in two different coordination environments, viz. a distorted square antiprism (D4d) and a biaugmented trigonal prism (C2v) as described above, which might translate into two relaxation processes. To further explore these relaxation processes, field-dependent ac measurements were carried out at 9 and 14 K (Fig. S18†). A maximum of the χ′′ signal is observed around 800 Oe for the measurement at 9 K, while a minimum is detected at the same field for the measurement at 14 K, suggesting that the application of a dc field can efficiently regulate the relaxation process. As shown in Fig. S18,† only one temperature-dependent χ′′ peak at 9 K is observed under 800 Oe field. Compared with the measurement without field, the χ′′ signal at 14 K is suppressed. Temperature- and frequency-dependent χ′′ signals show broad peaks under zero dc field (Fig. 6 and Fig. S19†). After applying an 800 Oe dc field, the broad χ′′ peaks become sharper, indicating only one relaxation process involved in the magnetic moment reversal. | ||
| Fig. 6 Frequency-dependent ac susceptibility of Dy4 under zero (left) and 800 Oe (right) dc field. | ||
The Cole–Cole plots represented as χ′′ versus χ′ at zero applied field (Fig. 7, top) show broad semi-circular profiles, suggesting multiple relaxation processes. After applying an 800 Oe dc field, however, the Cole–Cole plots evolve into single semi-circular curves, indicative of a single relaxation process. Fitting the Cole–Cole plots at zero-field with the CC-FIT program29 by assuming a double relaxation Debye model confirms the presence of two relaxation processes. Fitting of the data obtained under an 800 Oe field gives one relaxation process. The relevant fitting parameters are listed in Tables S9 and S10.†
 | ||
| Fig. 7 The Cole–Cole diagrams (top) and τ vs. T−1 plots (bottom) under zero (left) and 800 Oe (right) dc field for Dy4. The solid lines indicate the best fits. | ||
The relaxation time (τ) for magnetic moment reversal was extracted from the best fits of the Cole–Cole plots. We fitted the relevant τ versus 1/T plots with the following equation:30
 | (1) |
exp(−Ueff/T) represent quantum tunneling, direct, Raman and Orbach relaxation processes,31 respectively. The best fit gave the anisotropy barriers Ueff = 59.2 K with τ0 = 9.3 × 10−6 s for the slow relaxation phase (SR) and Ueff = 48.7 K and τ0 = 1.1 × 10−5 s for the fast relaxation phase (FR). Other parameters obtained from the fitting are listed in Table S11.† For the relaxation under 800 Oe dc field, a similar fitting using the above equation was carried out and gave the anisotropy barrier Ueff = 46 K and τ0 = 1.2 × 10−5 s (Fig. 7). The energy barrier, especially the relaxation times in the low-temperature region, is found to be comparable with the relevant values of the FR phase under zero dc field (Tables S10 and S11†). Generally, the application of a dc field can suppress the quantum tunneling of the magnetization (QTM).32 In the case of Dy4, however, the dc field seems to suppress one of the thermal relaxation processes. We hypothesize that the two relaxation processes are from isolated and coupled relaxation pathways.33 The coupled relaxation process is probably related to the intramolecular interactions, which could be suppressed by a proper dc field. Further investigation of the magnetic interactions and coupled magnetic states will require ab initio calculations.23a,34
Alternating current (ac) susceptibility measurements were also performed on the dinuclear complexes. Both Dy2·Cl and Dy2*·Cl show an out of phase ac susceptibility signal (χ′′) without peak under zero dc field (S20†), indicative of slow relaxation. We then performed field-dependent ac measurements at 2 K with a frequency of 1488 Hz to determine the optimal dc field. As shown in Fig. S21,† the ac plots are similar for the two complexes, the χ′′ signal showing broad peaks around 300 and 1800 Oe as well as a trough at 1200 Oe. However, when ac measurements of the two complexes were performed at 300 Oe (Fig. S22†), the χ′′ signal did not show any frequency-dependent peaks, and the relaxation time could not be extracted from the data. We suspect that the peaks at 300 and 1800 Oe originate from fast quantum tunneling of the magnetization (QTM), which is related to the cross of the energy gap. To suppress this relaxation process, a 1200 Oe dc field was then chosen and applied in all following ac susceptibility measurements. Indeed, the χ′′ signal showed temperature- and frequency-dependent out of phase peaks under 1200 Oe dc field (Fig. 8), indicative of field-induced single molecule magnetic behavior. The profiles of the ac plots of the two complexes are similar because of the essentially identical structural features and coordination geometries. In contrast, complex Dy2·NCS shows temperature and frequency-dependent χ′′ peaks under zero dc field. Extracting the relaxation time (τ) for magnetic moment reversals from the Cole–Cole plots by using the CC-FIT software (Tables S12–S14†) and fitting the relevant τ versus 1/T plots yields the anisotropy barriers Ueff of 19.1, 25.1, and 43.1 K for Dy2·Cl, Dy2*·Cl, and Dy2·NCS (Fig. S23†), respectively. Other parameters obtained from the fitting are listed in Table S15.†
 | ||
| Fig. 8 Frequency-dependent ac susceptibility of Dy2·Cl (left), Dy2*·Cl (middle), and Dy2·NCS (right) under indicated fields and temperatures. | ||
In Dy2·Cl and Dy2*·Cl, the DyIII ions are in almost perfect D5h geometry. However, the energy barriers are not particularly high, and only field-induced SMM properties are observed. This can be ascribed to the weak Cl− donors that lead to an axially elongated ligand field, which is unfavorable for the anisotropy of oblate DyIII ions. When replacing the Cl− with N-bound SCN−, the resulting complex Dy2·SCN shows SMMs properties even under zero field, as the SCN− donors give relatively short Dy–N bonds and provide a relatively stronger axial ligand field. Although the coordination geometry of the DyIII ions in Dy2·SCN deviates from D5h, the axial ligand field still plays an important role for dominating the anisotropy.
To further analyze the anisotropy of the dinuclear complexes, the anisotropy axes of the DyIII ions were calculated using the Magellan program.25 Assignments of the ligand charges are shown in Fig. S24.† The magnetic anisotropy axes of the DyIII ions in complexes Dy2·Cl and Dy2*·Cl are close to the Cl–Dy–Cl vectors (Fig. 9, Fig. S24†). In both complexes the Dy–N distances are in the range of 2.44–2.55 Å and the Dy–Cl distances are 2.61 Å, as described above. The long axial coordination bonds result in a weak ligand field, in which large anisotropy could not be generated. However, the anisotropy of the DyIII ions is still along the axial directions. In complex Dy2·NCS, the anisotropy axes of the DyIII ions are close to the Dy–NSCN directions, specifically those involving the linearly bound SCN− ligands, because of the relatively short Dy–NSCN bonds (compared with the Dy–N bonds involving the equatorial ligand) that give rise to a roughly axial ligand field (Fig. 9).
 | ||
| Fig. 9 Orientations of the main magnetic axes and local magnetizations of the ground state of the dinuclear complexes Dy2·Cl (a), Dy2*·Cl (b), and Dy2·NCS (c). | ||
Calculation of the anisotropy axes with the Magellan program uses an electrostatic model based on the crystallographically determined structures, where the magnetic anisotropy axes of the DyIII ions are related to the negatively charged donors. To investigate the influence of the charges on the orientations of the anisotropy axes, we reduced the charge on Cl and N (in SCN−) to half (Fig. S25†). As shown in Fig. S26,† the resulting anisotropy axes are still close to the directions mentioned above (with deviation angles of 1.0, 0.9, and 4.5° for Dy2·Cl, Dy2*·Cl, and Dy2·NCS, respectively, Table S16†). Overall, the anisotropy axes are directed roughly along the pseudo-C5 axis defined by the equatorial plane that is constituted by the {N5} pockets of the macrocyclic ligand, and by the axial (pseudo) halide ligands; as a result, the anisotropy of the DyIII ions appears to be dominated by the approximate local D5h geometry, which is predetermined by the planar dinucleating macrocyclic scaffold.
Conclusions
In summary, the template condensation of pyrazole-3,5-dicarbonyl synthons with linear triamines in the presence of dysprosium salts gave the expected bis(pyrazolato) bridged dinuclear DyIII complexes Dy2·Cl and Dy2*·Cl when using 3,5-diacetyl-4-methyl-1H-pyrazole and DyCl3·6H2O; the products feature a macrocyclic ligand providing two {N5} binding pockets and axial chlorides. However, the combination of 4-phenyl-3,5-dicarbaldehyde-1H-pyrazole and Dy(ClO4)3·6H2O resulted in a tetranuclear complex Dy4 with an open-chain ligand strand due to incomplete imine condensation and the presence of peripheral acetal groups. Studies of the magnetic properties and their analysis in view of the molecular structures suggested a toroidal orientation of the anisotropy axes and a diamagnetic ground state for Dy4 because of its centrosymmetric structure and the strongly coordinating μ-OH bridges in the plane of the rhomb-like Dy4 arrangement. The dynamic magnetic properties of complex Dy4 revealed two relaxation processes under zero field, one of which can be suppressed after applying a weak dc field. The DyIII ions in dinuclear complexes Dy2·Cl and Dy2*·Cl exhibit almost perfect D5h coordination geometry. Due to the weak Cl− donors on the axial directions, however, both complexes only behave as field-induced SMMs. Replacement of the axial Cl− donors by stronger SCN− donors produced Dy2·SCN that shows SMM signatures even at zero dc field, though additional MeOH ligands increase the DyIII coordination number to 8 and decrease the local symmetry. Magnetostructural studies indicate that although the DyIII ions are exposed to a weak ligand field, their more or less pronounced local D5h geometry in the dinuclear complexes still dominates the orientation of the anisotropy axes.Achieving high magnetic anisotropies that translate into favorable performances of SMMs is a consistent pursuit for chemists working in the field of molecular magnetism. A recent focus in DyIII chemistry has been on the synthesis of mononuclear complexes with pronounced axial ligand field. Herein, a complementary strategy for enhancing the SMM properties has been pursued that targets preorganized dinuclear complexes in which a local D5h geometry of the two DyIII ions and a parallel orientation of their anisotropy axes are enforced by planar macrocyclic scaffolds with two pentadentate binding compartments. Although the SMM properties found for the present first examples of such DyIII2 complexes are not yet particularly impressive, the beneficial use of bis (pentadentate) macrocyclic scaffolds for controlling the DyIII coordination chemistry has been demonstrated, and further modifications of the axial donors for modulating the ligand field appear to be promising. Work in the latter direction is underway in our laboratory.
Experimental
General synthetic considerations
All chemicals and solvents were commercially obtained and used as received without any further purification. 4-Phenyl-3,5-dicarbaldehyde-1H-pyrazole and 3,5-diacetyl-4-methyl-1-H-pyrazole were synthesized under ambient conditions following a previously reported method.35 IR measurements of solid samples were performed with a Cary 630 FTIR spectrometer equipped with Dial Path and Diamond ATR accessory and analyzed by FTIR MicroLab software. IR bands (Fig. S1 and S2†) were labeled according to their relative intensities with vs. (very strong), s (strong), m (medium), and w (weak). Powder X-ray diffraction measurements were recorded on Bruker D8 advance X-Ray diffractometer using Cu-Kα radiation. Thermogravimetric analyses were performed on a Netzsch STA449F3 TG-DSC instrument in the range of 25–1000 °C with a heating rate of 10 K min−1 under N2 atmosphere. Elemental analyses were carried out using an Elementar Vario EL III instrument by the analytical laboratory of the Institute of Inorganic Chemistry at the Georg-August-University Göttingen.Synthesis of Dy4
The ligand and complex were synthesized via a one-pot procedure: a mixture of Dy(ClO4)3·6H2O (1 mmol) and 4-phenyl-3,5-dicarbaldehyde-1H-pyrazole (1 mmol) in 50 mL methanol was stirred and heated at 70 °C, then a solution of diethylenetriamine (1 mmol) in 20 mL methanol was added dropwise. After heating overnight at 70 °C, the solution was allowed to cool to room temperature. A yellow precipitate formed, which was separated by filtration, washed with methanol and dried under vacuum. Yellow crystals of complex Dy4 suitable for X-ray diffraction were obtained by slow diffusion of diethyl ether into a DMF solution of the crude product. Yield: 90 mg, (20%, based on metal salt). Elemental analysis (%) calcd for [Dy4(μ3-OH)2L12(μ2-OH)2(DMF)4]·4ClO4 (C72H102Cl4Dy4N18O32, MW = 2523.51): C, 34.27, H, 4.07, N, 9.99; found C, 33.57, H, 4.11, N, 9.71. IR (solid, ATR)![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) [cm−1] = 3582 (w), 3287 (w), 2933 (br), 1647 (s), 1529 (w), 1497 (w), 1435 (m), 1383 (m), 1308 (m), 1207 (w), 1077 (s), 1013 (m), 937 (m), 890 (w), 811 (w), 773 (s), 702 (m), 670 (m), 656 (m), 621 (s), 537 (w), 520 (w), 403 (w).
[cm−1] = 3582 (w), 3287 (w), 2933 (br), 1647 (s), 1529 (w), 1497 (w), 1435 (m), 1383 (m), 1308 (m), 1207 (w), 1077 (s), 1013 (m), 937 (m), 890 (w), 811 (w), 773 (s), 702 (m), 670 (m), 656 (m), 621 (s), 537 (w), 520 (w), 403 (w).
Synthesis of Dy2·Cl
The complex was isolated from an in situ reaction (Scheme 2). A mixture of DyCl3·6H2O (1 mmol) and 3,5-diacetyl-4-methyl-1H-pyrazole (1 mmol) in 50 mL methanol was stirred and heated at 70 °C, then a solution of diethylenetriamine (1 mmol) in 20 mL methanol was added dropwise. The reaction was heated overnight at 70 °C. After the solution was allowed to cool to room temperature, the solvent was removed under vacuum and the residue was washed with ether to remove unreacted substrates. Pure colorless crystals of the product Dy2·Cl were obtained by recrystallization of the crude product from methanol. Yield: 70 mg, (15%, based on metal salt). Elemental analysis (%) calcd for [Dy2L2Cl4] (C24H36Cl4Dy2N10, MW = 931.43): C, 30.95, H, 3.90, N, 15.04; found C, 29.88, H, 4.08, N, 14.31. IR (solid, ATR) [cm−1] = 2922 (w), 2875 (w), 1610 (vs), 1504 (w), 1466 (w), 1455 (w), 1447 (w), 1418 (m), 1348 (m), 1292 (s), 1266 (w), 1208 (s), 1124 (m), 1096 (m), 1078 (m), 1050 (m), 1034 (s), 1012 (m), 967 (m), 933 (m), 827 (s), 749 (w), 721 (m), 553 (w), 532 (w), 479 (m), 426 (w).
Synthesis of Dy2*·Cl
The complex was isolated from an in situ reaction (Scheme 2). A mixture of DyCl3·6H2O (1 mmol), and 3,5-diacetyl-4-methyl-1H-pyrazole (1 mmol) in 50 mL methanol was stirred and heated at 70 °C, then a solution of N,N-bis(2-aminoethyl)methylamine (1 mmol) in 20 mL methanol was added dropwise. The reaction was heated overnight at 70 °C. After the solution was allowed to cool to room temperature, the solvent was removed under vacuum and the residue was washed with ether to remove unreacted substrates. Pure colorless crystals of the product Dy2*·Cl were obtained by recrystallization of the crude product from methanol. Yield: 58 mg, (12%, based on metal salt). Elemental analysis (%) calcd for [Dy2L3Cl4] (C26H40Cl4Dy2N10, MW = 959.48): C, 32.55, H, 4.20, N, 14.60; found C, 31.39, H, 4.13, N, 13.95. IR (solid, ATR) [cm−1] = 2906 (w), 1607 (vs), 1463 (w), 1433 (w), 1384 (w), 1348 (m), 1307 (w), 1294 (s), 1259 (w), 1211 (s), 1125 (m), 1094 (m), 1072 (w), 1049 (w), 966 (w), 927 (w), 779 (m), 724 (w), 554 (w), 539 (w), 479 (w), 427 (w).
Synthesis of Dy2·NCS
Complex Dy2·NCS was obtained from Dy2·Cl by ligand substitution (Scheme 2). A mixture of Dy2·Cl (0.1 mmol) and NH4NCS (0.4 mmol) in 30 mL methanol was heated to 70 °C overnight without stirring, then the solution was slowly cooled to room temperature. Yellow crystals of complex Dy2·NCS suitable for X-ray diffraction were obtained. Yield: 70 mg, (65%, based on Dy2·Cl). Elemental analysis (%) calcd for [Dy2L2(NCS)4(MeOH)2] (C30H44Dy2N14O2S4, MW = 1086.03): C, 33.18, H, 4.08, N, 18.06, S, 11.81; found C, 31.80, H, 3.94, N, 18.16, S, 11.94. IR (solid, ATR) [cm−1] = 2867 (w), 2931 (w), 2048 (vs), 1608 (s), 1465 (w), 1425 (s), 1352 (m), 1301 (s), 1210 (m), 1127 (m), 1100 (m), 1075 (s), 994 (w), 968 (s), 935 (s), 908 (m), 831 (m), 810 (m), 741 (w), 719 (m), 711 (m), 530 (w), 478 (m), 427 (w).
Crystallography
Crystal data and details of the data collections are given in Table S1.† X-ray data were collected on a STOE IPDS II diffractometer (graphite monochromated Mo-Kα radiation, λ = 0.71073 Å) by use of ω scans at −140 °C. The structures were solved with SHELXT and refined on F2 using all reflections with SHELXL-2018.36 Non-hydrogen atoms were refined anisotropically. Most hydrogen atoms were placed in calculated positions and assigned to an isotropic displacement parameter of 1.2/1.5 Ueq(C) and 1.2 Ueq(N) in case of Dy2·Cl. The oxygen or nitrogen bound hydrogen atoms in Dy4 and Dy2·SCN were refined using DFIX restraints (dO–H = 0.82 Å, dN–H = 1 Å (only for Dy4)) and applying a fixed isotropic displacement parameter of 0.08 Å2 in case of Dy4. The counterions in Dy4 (ClO4−) were found to disordered (occupancy factors: 0.879(5)/0.121(5) and 0.318(3)/0.682(3)). SADI (dCl–O & dO⋯O) restraints and EADP constraints were applied to model the disordered parts. The CH3-N(C2H4)2-moiety of the ligand in Dy2*·Cl was found to be disordered (occupancy factors: 0.698(8)/0.302(8)). SADI (dN–C & dC–C) and RIGU restraints were applied to model the disordered part. The unit cell of Dy4 contains highly disordered solvent molecules (DMF and H2O) for which no satisfactory model for a disorder could be found. The solvent contribution to the structure factors was calculated with PLATON SQUEEZE37 and the resulting .fab file was processed with SHELXL using the ABIN instruction. The empirical formula and derived values are in accordance with the calculated cell content. Face-indexed absorption corrections were performed numerically with the program X-RED. CCDC 2103370–2103373† contains the supplementary crystallographic data for this paper.Magnetic measurements
Magnetic susceptibility measurements were recorded on a Quantum-Design MPMS XL-5 SQUID magnetometer equipped with a 5 T magnet. The fresh sample was transferred into a capsule and covered with perfluoropolyether based inert oil Fomblin Y45 to prevent any loss of solvent molecules. Direct current (dc) magnetic susceptibility measurements were performed on polycrystalline samples of Dy4, Dy2·Cl, Dy2*·Cl, and Dy2·SCN in the temperature range 2–210 K (below the pour point of the oil Fomblin Y45), under an applied field of 5000 Oe (1000 and 5000 Oe for Dy4). The field-dependent magnetizations for all complexes were measured in the field range 0–5 T at 2 K. The dynamics of the magnetization were investigated by measuring the ac susceptibility under zero static field and a 3.0 Oe ac oscillating field. Diamagnetic corrections were made with the Pascal's constants38 for all the constituent atoms as well as the experimentally determined contributions of the sample holder.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
J. W. thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship.Notes and references
- (a) N. Ishikawa, M. Sugita, T. Ishikawa, S.-y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed; (b) P. Zhang and J. Tang, Lanthanide Single Molecule Magnets, Springer-Verlag, 2015 Search PubMed; (c) A. Dey, P. Kalita and V. Chandrasekhar, ACS Omega, 2018, 3, 9462–9475 CrossRef CAS PubMed.
- Z. Zhu, M. Guo, X.-L. Li and J. Tang, Coord. Chem. Rev., 2019, 378, 350–364 CrossRef CAS.
- (a) J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC; (b) J.-L. Liu, Y.-C. Chen and M.-L. Tong, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC.
- (a) J. Wu, J. Jung, P. Zhang, H. Zhang, J. Tang and B. Le Guennic, Chem. Sci., 2016, 7, 3632–3639 RSC; (b) P. Cen, M. Wang, X. Ma, L. Chen, Y.-Q. Zhang, Y. Li, D. Tian and X. Liu, CrystEngComm, 2020, 22, 6856–6863 RSC.
- (a) Y.-C. Chen, J.-L. Liu, L. Ungur, J. Liu, Q.-W. Li, L.-F. Wang, Z.-P. Ni, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 2829–2837 CrossRef CAS PubMed; (b) K.-X. Yu, J. G. C. Kragskow, Y.-S. Ding, Y.-Q. Zhai, D. Reta, N. F. Chilton and Y.-Z. Zheng, Chem, 2020, 6, 1777–1793 CrossRef CAS.
- (a) A. Canaj, S. Dey, E. Regincós Marti, C. Wilson, G. Rajaraman and M. Murrie, Angew. Chem., Int. Ed., 2019, 58, 14146–14151 CrossRef CAS PubMed; (b) Z.-H. Li, Y.-Q. Zhai, W.-P. Chen, Y.-S. Ding and Y.-Z. Zheng, Chem. – Eur. J., 2019, 25, 16219–16224 CrossRef CAS PubMed.
- (a) C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439 CrossRef CAS PubMed; (b) F. S. Guo, B. M. Day, Y. C. Chen, M. L. Tong, A. Mansikkamaki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed.
- (a) H. L. C. Feltham and S. Brooker, Coord. Chem. Rev., 2014, 276, 1–33 CrossRef CAS; (b) J. Hamacek and A. Vuillamy, Eur. J. Inorg. Chem., 2018, 1155–1166 CrossRef CAS.
- (a) J. Acharya, N. Ahmed, J. Flores-Gonzalez, P. Kumar, F. Pointillart, O. Cador, S. K. Singh and V. Chandrasekhar, Dalton Trans., 2020, 49, 13110–13122 RSC; (b) H. Ke, Y. Yang, W. Wei, Y. Jiang, Y.-Q. Zhang, G. Xie and S. Chen, Dalton Trans., 2020, 49, 10594–10602 RSC.
- (a) C. Jin, X.-L. Li, Z. Liu, A. Mansikkamäki and J. Tang, Dalton Trans., 2020, 49, 10477–10485 RSC; (b) Y. Huang, J.-X. Li, Y. Ge, X.-M. Zhang, Y. Xu, Y. Li, Y.-Q. Zhang and J.-L. Yao, Dalton Trans., 2020, 49, 8976–8984 RSC; (c) P. Cen, X. Liu, Y.-Q. Zhang, J. Ferrando-Soria, G. Xie, S. Chen and E. Pardo, Dalton Trans., 2020, 49, 808–816 RSC.
- (a) D. Li, M.-M. Ding, Y. Ge, D. F. Tello Yepes, M. Sun, M. S. Najib, Y. Li, Y.-Q. Zhang and J.-l. Yao, New J. Chem., 2020, 44, 20634–20642 RSC; (b) F.-X. Shen, K. Pramanik, P. Brandão, Y.-Q. Zhang, N. C. Jana, X.-Y. Wang and A. Panja, Dalton Trans., 2020, 49, 14169–14179 RSC; (c) M. Kong, X. Feng, J. Wang, Y.-Q. Zhang and Y. Song, Dalton Trans., 2020, 50, 568–577 RSC.
- (a) M. Fondo, J. Corredoira-Vázquez, A. M. Garcia-Deibe, J. Sanmartin Matalobos, S. Gómez-Coca, E. Ruiz and E. Colacio, Inorg. Chem. Front., 2021, 8, 2532–2541 RSC; (b) Y. Ge, D. Li, G. Wang, Y. Cui, M. S. Najib, Y. Li and B.-L. Wang, Chem. – Asian J., 2019, 14, 2846–2852 CAS.
- (a) J. Wu, D. Liu, Q. Yang, Y. Ge, J. Tang and Z. Qi, New J. Chem., 2021, 45, 2200–2207 RSC; (b) S. Liu, J. Lu, X.-L. Li, Z. Zhu and J. Tang, Dalton Trans., 2020, 49, 12372–12379 RSC.
- (a) H. Wada, S. Ooka, T. Yamamura and T. Kajiwara, Inorg. Chem., 2017, 56, 147–155 CrossRef CAS PubMed; (b) Y.-S. Ding, T. Han, Y.-Q. Hu, M. Xu, S. Yang and Y.-Z. Zheng, Inorg. Chem. Front., 2016, 3, 798–807 RSC.
- (a) J. Wang, Z.-Y. Ruan, Q.-W. Li, Y.-C. Chen, G.-Z. Huang, J.-L. Liu, D. Reta, N. F. Chilton, Z.-X. Wang and M.-L. Tong, Dalton Trans., 2019, 48, 1686–1692 RSC; (b) H. L. C. Feltham, R. Clérac, A. K. Powell and S. Brooker, Inorg. Chem., 2011, 50, 4232–4234 CrossRef CAS PubMed; (c) S.-G. Wu, Z.-Y. Ruan, G.-Z. Huang, J.-Y. Zheng, V. Vieru, G. Taran, J. Wang, Y.-C. Chen, J.-L. Liu, L. T. A. Ho, L. F. Chibotaru, W. Wernsdorfer, X.-M. Chen and M.-L. Tong, Chem, 2021, 7, 982–992 CrossRef CAS; (d) W. Rui-Chen, Wu Si-Guo, L. Jun-Liang, J. Jian-Hua, H. Guo-Zhang, Li Quan-Wen and T. Ming-Liang, Acta Chim. Sin., 2020, 78, 412–418 CrossRef.
- J. Wang, Q.-W. Li, S.-G. Wu, Y.-C. Chen, R.-C. Wan, G.-Z. Huang, Y. Liu, J.-L. Liu, D. Reta, M. J. Giansiracusa, Z.-X. Wang, N. F. Chilton and M.-L. Tong, Angew. Chem., Int. Ed., 2021, 60, 5299–5306 CrossRef CAS PubMed.
- (a) B. Schneider, S. Demeshko, S. Dechert and F. Meyer, Inorg. Chem., 2012, 51, 4912–4914 CrossRef CAS PubMed; (b) M. Steinert, B. Schneider, S. Dechert, S. Demeshko and F. Meyer, Angew. Chem., Int. Ed., 2014, 53, 6135–6139 CrossRef CAS PubMed; (c) M. Steinert, B. Schneider, S. Dechert, S. Demeshko and F. Meyer, Inorg. Chem., 2016, 55, 2363–2373 CrossRef CAS PubMed.
- (a) G. Leibeling, S. Demeshko, S. Dechert and F. Meyer, Angew. Chem., Int. Ed., 2005, 44, 7111–7114 CrossRef CAS PubMed; (b) D.-H. Manz, P.-C. Duan, S. Dechert, S. Demeshko, R. Oswald, M. John, R. A. Mata and F. Meyer, J. Am. Chem. Soc., 2017, 139, 16720–16731 CrossRef CAS PubMed; (c) E. Ferretti, S. Dechert, S. Demeshko, M. C. Holthausen and F. Meyer, Angew. Chem., Int. Ed., 2019, 58, 1705–1709 CrossRef CAS PubMed.
- (a) S. J. Archibald, A. J. Blake, S. Parsons, M. Schröder and R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 1997, 173–180 RSC; (b) S. J. Archibald, A. J. Blake, M. Schröder and R. E. P. Winpenny, J. Chem. Soc., Chem. Commun., 1994, 1669–1670 RSC; (c) B. T. Gregg, K. C. Golden and J. F. Quinn, Tetrahedron, 2008, 64, 3287–3295 CrossRef CAS.
- (a) Y.-N. Guo, L. Ungur, G. E. Granroth, A. K. Powell, C. Wu, S. E. Nagler, J. Tang, L. F. Chibotaru and D. Cui, Sci. Rep., 2014, 4, 5471 CrossRef CAS PubMed; (b) J. Xiong, H.-Y. Ding, Y.-S. Meng, C. Gao, X.-J. Zhang, Z.-S. Meng, Y.-Q. Zhang, W. Shi, B.-W. Wang and S. Gao, Chem. Sci., 2017, 8, 1288–1294 RSC; (c) M. Guo, J. Wu, O. Cador, J. Lu, B. Yin, B. Le Guennic and J. Tang, Inorg. Chem., 2018, 57, 4534–4542 CrossRef CAS PubMed; (d) M. Guo, Y.-Q. Zhang, Z. Zhu and J. Tang, Inorg. Chem., 2018, 57, 12213–12221 CrossRef CAS PubMed.
- (a) D. Casanova, P. Alemany, J. M. Bofill and S. Alvarez, Chem. – Eur. J., 2003, 9, 1281–1295 CrossRef CAS PubMed; (b) S. Alvarez and M. Llunell, J. Chem. Soc., Dalton Trans., 2000, 3288–3303 RSC.
- (a) Y.-N. Guo, G.-F. Xu, W. Wernsdorfer, L. Ungur, Y. Guo, J. Tang, H.-J. Zhang, L. F. Chibotaru and A. K. Powell, J. Am. Chem. Soc., 2011, 133, 11948–11951 CrossRef CAS PubMed; (b) S. K. Langley, C. Le, L. Ungur, B. Moubaraki, B. F. Abrahams, L. F. Chibotaru and K. S. Murray, Inorg. Chem., 2015, 54, 3631–3642 CrossRef CAS PubMed; (c) S. K. Langley, L. Ungur, N. F. Chilton, B. Moubaraki, L. F. Chibotaru and K. S. Murray, Inorg. Chem., 2014, 53, 4303–4315 CrossRef CAS PubMed.
- (a) J. Tang, I. Hewitt, N. T. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729–1733 CrossRef CAS PubMed; (b) L. Ungur, S.-Y. Lin, J. Tang and L. F. Chibotaru, Chem. Soc. Rev., 2014, 43, 6894–6905 RSC; (c) G. Fernandez Garcia, D. Guettas, V. Montigaud, P. Larini, R. Sessoli, F. Totti, O. Cador, G. Pilet and B. Le Guennic, Angew. Chem., Int. Ed., 2018, 57, 17089–17093 CrossRef CAS PubMed.
- N. F. Chilton, D. Collison, E. J. L. McInnes, R. E. P. Winpenny and A. Soncini, Nat. Commun., 2013, 4, 2551 CrossRef PubMed.
- (a) L. F. Chibotaru, L. Ungur and A. Soncini, Angew. Chem., Int. Ed., 2008, 47, 4126–4129 CrossRef CAS PubMed; (b) P.-H. Guo, J.-L. Liu, Z.-M. Zhang, L. Ungur, L. F. Chibotaru, J.-D. Leng, F.-S. Guo and M.-L. Tong, Inorg. Chem., 2012, 51, 1233–1235 CrossRef CAS PubMed; (c) J. Wu, S.-Y. Lin, S. Shen, X.-L. Li, L. Zhao, L. Zhang and J. Tang, Dalton Trans., 2017, 46, 1577–1584 RSC.
- (a) S.-Y. Lin, W. Wernsdorfer, L. Ungur, A. K. Powell, Y.-N. Guo, J. Tang, L. Zhao, L. F. Chibotaru and H.-J. Zhang, Angew. Chem., Int. Ed., 2012, 51, 12767–12771 CrossRef CAS PubMed; (b) L. Ungur, S. K. Langley, T. N. Hooper, B. Moubaraki, E. K. Brechin, K. S. Murray and L. F. Chibotaru, J. Am. Chem. Soc., 2012, 134, 18554–18557 CrossRef CAS PubMed; (c) K. R. Vignesh, S. K. Langley, A. Swain, B. Moubaraki, M. Damjanović, W. Wernsdorfer, G. Rajaraman and K. S. Murray, Angew. Chem., Int. Ed., 2018, 57, 779–784 CrossRef CAS PubMed.
- (a) I. F. Díaz-Ortega, J. M. Herrera, D. Aravena, E. Ruiz, T. Gupta, G. Rajaraman, H. Nojiri and E. Colacio, Inorg. Chem., 2018, 57, 6362–6375 CrossRef PubMed; (b) Y.-N. Guo, G.-F. Xu, P. Gamez, L. Zhao, S.-Y. Lin, R. Deng, J. Tang and H.-J. Zhang, J. Am. Chem. Soc., 2010, 132, 8538–8539 CrossRef CAS PubMed; (c) L. Zhang, J. Jung, P. Zhang, M. Guo, L. Zhao, J. Tang and B. Le Guennic, Chem. – Eur. J., 2016, 22, 1392–1398 CrossRef CAS PubMed.
- D. Reta and N. F. Chilton, Phys. Chem. Chem. Phys., 2019, 21, 23567–23575 RSC.
- (a) X.-L. Li, H. Li, D.-M. Chen, C. Wang, J. Wu, J. Tang, W. Shi and P. Cheng, Dalton Trans., 2015, 44, 20316–20320 RSC; (b) J. Wu, S. Demeshko, S. Dechert and F. Meyer, Chem. Commun., 2020, 56, 3887–3890 RSC.
- (a) R. Orbach, Proc. R. Soc. London, Ser. A, 1961, 264, 485–495 CrossRef CAS; (b) R. Orbach, Proc. R. Soc. London, Ser. A, 1961, 264, 458–484 CrossRef CAS.
- F.-S. Guo, A. Mansikkamaki, M.-L. Tong, Y.-C. Chen and R. Layfield, Angew. Chem., Int. Ed., 2019, 58, 10163–10167 CrossRef CAS PubMed.
- T. Han, Y. Ding, Z.-H. Li, K.-X. Yu, Y.-Q. Zhai, N. F. Chilton and Y.-Z. Zheng, Chem. Commun., 2019, 55, 7930–7933 RSC.
- L. Ungur and L. F. Chibotaru, Phys. Chem. Chem. Phys., 2011, 13, 20086–20090 RSC.
- A. Sachse, L. Penkova, G. Noël, S. Dechert, O. A. Varzatskii, I. O. Fritsky and F. Meyer, Synthesis, 2008, 800–806 CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
- E. A. Boudreaux and L. N. Mulay, Theory and Applications of Molecular Paramagnetism, John Wiley & Sons, New York, 1976 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR spectra, XRD, and TGA are depicted in Fig. S1–S5. Crystallographic data are listed in Table S1 and Fig. S6–S9. Selected bond lengths and angles are listed in Tables S2–S5. SHAPE calculations are given in Tables S6–S8 and Fig. S10–S12. The M vs. H plots are presented in Fig. S13–S16. Ac susceptibilities are presented in Fig. S17–S23. Magellan calculations are given in Tables S16 and Fig. S24–S26. CCDC 2103370–2103373. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt02815d |
| This journal is © The Royal Society of Chemistry 2021 |