
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational engineering of Luminiphilus syltensis (R)-selective amine transaminase for the acceptance of bulky substrates†
Eleni
Konia
a,
Konstantinos
Chatzicharalampous
a,
Athina
Drakonaki
 a,
Cornelia
Muenke
b,
Ulrich
Ermler
b,
Georgios
Tsiotis
a and
Ioannis V.
Pavlidis
*a
a,
Cornelia
Muenke
b,
Ulrich
Ermler
b,
Georgios
Tsiotis
a and
Ioannis V.
Pavlidis
*a
aDepartment of Chemistry, University of Crete, Voutes University Campus, 70013, Heraklion, Greece. E-mail: ipavlidis@uoc.gr
bMax Planck Institute of Biophysics, Max-von-Laue-Straße 3, 60438, Frankfurt am Main, Germany
First published on 11th November 2021
Abstract
Despite the plethora of information on (S)-selective amine transaminases, the (R)-selective ones are still not well-studied; only a few structures are known to date, and their substrate scope is limited, apart from a few stellar works in the field. Herein, the structure of Luminiphilus syltensis (R)-selective amine transaminase is elucidated to facilitate engineering towards variants active on bulkier substrates. The V37A variant exhibited increased activity towards 1-phenylpropylamine and to activity against 1-butylamine. In contrast, the S248 and T249 positions, located on the β-turn in the P-pocket, seem crucial for maintaining the activity of the enzyme.
Transaminases (TAs, E.C. 2.6.1.x) have attracted the scientific interest of the field due to their ability to easily synthesize optically pure amines.1 The structure of the substrate-binding site, typically composed of a small and a large binding pocket over pyridoxal 5′-phosphate (PLP),2 leads to excellent enantioselectivity in most cases. To expand the synthetic potential of these enzymes, several research groups have been working on the enlargement of the small binding pocket, which typically accommodates only a methyl group. There have been several cases of successful engineering of (S)-selective amine transaminases (ATAs), which belong to fold class I of PLP-dependent enzymes.3 The transfer of knowledge among (S)-ATAs has been proven feasible, and methodologies to identify (S)-selective ATAs active against bulky substrates from the gene pool have been proposed.4 However, the (R)-selective ATAs, which belong to fold-class IV, are less studied. The first methodology to guide (R)-ATA identification from the sequence pool was published in 2010 by Höhne and his coworkers.2 In the same year, Savile and coworkers published stellar protein engineering work to construct (R)-ATAs for the production of sitagliptin,5 highlighting their synthetic potential and evolvability. Since then, knowledge about (R)-ATAs is sparse; only a few structures are available, and we are still far from predicting the substrate scope of an enzyme solely from the sequence. Very recently, an interesting meta-analysis of the available structures of fold type IV TAs and their sequences shed some light on their substrate specificity.6
To contribute to this field, we decided to work with the (R)-selective ATA from Luminiphilus syltensis (LS_ATA), which was identified by Höhne et al., an enzyme previously mentioned as Gamma proteobacterium ATA.2 This TA was selected, as it exhibited the highest activity against the benchmark substrate, 1-phenylethylamine (PEA), among several (R)-ATAs (data not shown), and its sequence is quite unique compared with the known fold IV enzymes. In public databases, LS_ATA is annotated as a “branched-chain amino acid transaminase” (BCAT). The closest resolved structure is the BCAT from the thermophilic archaea Geoglobus acetivorans (PDB code: 5cm0),7 which presents only 41% identity to LS_ATA. However, Höhne et al. already observed that the activity for valine synthesis was lower than that for PEA,2 while BCATs exhibit the opposite catalytic profile.8 Höhne and coworkers suggested two motifs to distinguish BCATs from (R)-ATAs,2 but LS_ATA does not have any of them. The βX- and βY-strands of LS_ATA (GVFDVVSAW and ASIRFIVT, respectively) are a mix of the motifs found in BCATs and (R)-ATAs. However, the β-turn of the P-pocket of LS_ATA has the sequence STAG, which resembles more the motif of (R)-ATAs.6 Thus, as LS_ATA is structurally and functionally closer to the (R)-ATA, we will refer to it as such.
To guide our engineering efforts, we crystallized LS_ATA and determined its X-ray structure. The L. syltensis (R)-ATA was structurally characterized at a resolution of 1.6 Å, in the orthorhombic space group P212121, in the internal aldimine state between the PLP and catalytic lysine, K154. LS_ATA was found as a homohexamer built up of a trimer of dimers (Fig. 1). Gel filtration and blue native polyacrylamide gel electrophoresis experiments (Fig. S1, ESI†) confirmed the homohexameric state. Although the dimeric structure is the most common for (S)-ATAs, several (R)-ATAs are organized in higher multimers. The TAs from Geoglobus acetivorans and Archeoglobus fulgidus also form hexamers,7 while the (R)-ATA from Thermomyces stellatus was mainly found as the tetramer (∼90%).9 A higher order of the self-organization of (R)-ATAs seems to be a common feature for stabilizing the functional dimer. PLP is bound in a canonical manner into the interdomain cleft, covalently linked with K154. The substrate binding site positioned between two monomers of the dimer is in an open conformation as the O-pocket loop (100–107) is highly flexible and directed into the bulk solvent. This finding is in contrast to that of G. acetivorans ATA in the complex with α-ketoglutarate (PDB code: 5e25), in which the O-pocket loop points towards PLP, and largely shields the substrate.7 Peisach and coworkers suggested that a “carboxylate trap” (a Tyr, an Arg, and a His residue) is responsible for the specificity of D-amino acid aminotransferases (DAATs).10 However, these residues are not found on the respective positions in LS_ATA.10
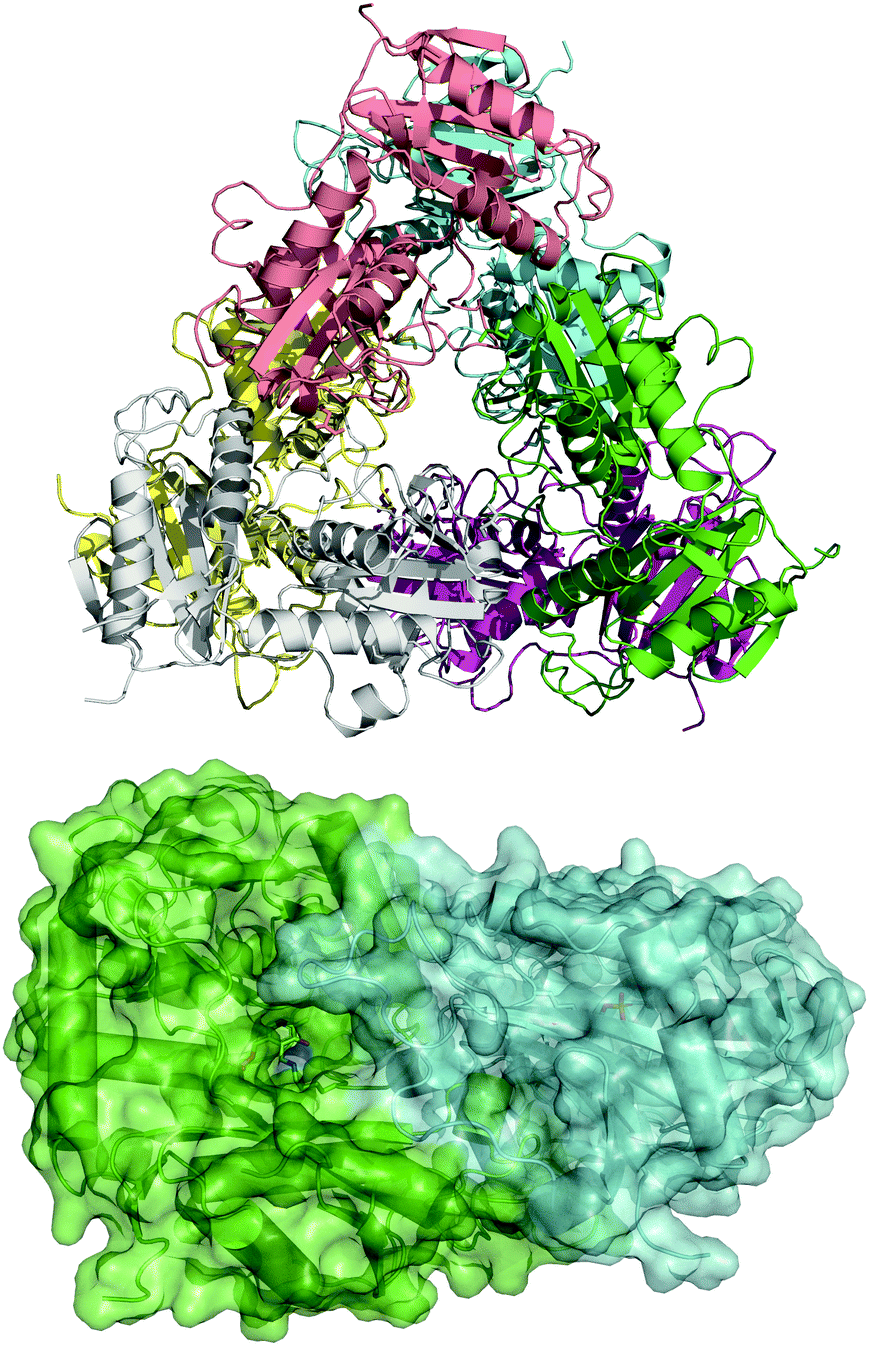 | ||
| Fig. 1 The hexameric form of LS_ATA (upper), where each chain is presented with another color, and the dimer (lower) where the internal aldimine (sticks) is formed in the interface. PDB code: 7p3t. | ||
When LS_ATA is characterized with the two optically pure amines separately via a photometric assay,11 it exhibits minor activity with (S)-PEA, especially at higher pH values (Table S1, ESI†). However, its preference for the (R)-amine was higher (Eapp ∼ 70), as expected.2 To explain the enantioselectivity of LS_ATA, the structure was refined in water, and the lowest energy structure was selected for further analysis. The dimer of chains A and C had the best quality, according to Molprobity.12 The quinonoids of (R)- and (S)-PEA were built in this dimer, as this is the most demanding intermediate of the catalytic mechanism.
As shown in Fig. 2, the quinonoid can be formed in both conformations. However, the Re-face quinonoid has a lower formation energy compared with the Si-face equivalent (−1521.65 kJ mol−1 and −1497.861 kJ mol−1, respectively), as the small binding pocket does not provide enough space for an aryl group. The selectivity for the (R)-enantiomer could also be attributed to the specific residues forming the binding pockets. The large binding pocket is formed from the polar residues F35, R92, and R158. These residues might be involved in stabilizing the aromatic ring of the Re-face quinonoid. F35 is at the correct distance and orientation to establish π–π stacking with the Re-face quinonoid (Fig. 2A). However, they cannot adequately interact with the much smaller methyl group of the Si-face quinonoid (Fig. 2B).
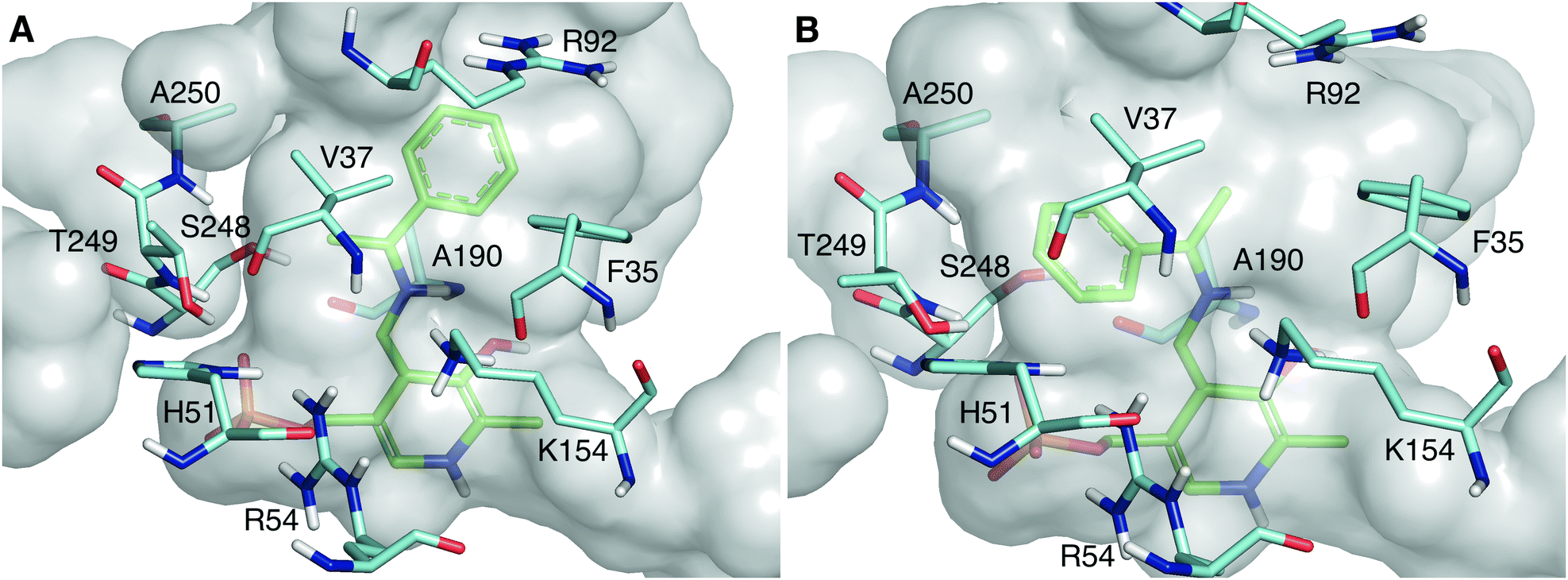 | ||
| Fig. 2 The active site pocket of LS_ATA bearing the Re-face quinonoid (A) and Si-face quinonoid (B). The quinonoid intermediates are shown in green, and residues within 4 Å from the intermediate are shown in cyan (all belong to the A chain). The catalytic lysine, K154, is in front of the quinonoid. All non-polar hydrogens are removed for clarity. | ||
The wild type LS_ATA did not exhibit any detectable activity with (R)-1-phenyl propylamine ((R)-PPA) and (R)-1-phenyl butylamine ((R)-PBA) via the photometric assay. Despite the small size difference of the small substituent, bulkier substrates seem not to fit in. This was also a challenging target for (S)-ATAs, that many research groups have worked upon.13 In order to rationally design an LS_ATA variant able to accept bulkier substrates, we first checked in silico all possible clashes. As shown in Fig. 3A, the methyl group is nicely accommodated in the small binding pocket. However, the propyl group of PBA causes clashes with several amino acids in the small binding pocket, namely V37, S248, T249 and A250, despite its flexibility and the energy minimization of the complex (Fig. 3B). We decided to proceed with alanine screening, as alanine is the smaller amino acid able to maintain the structural integrity of proteins. Obviously, A250 was omitted from the screening, and V37, S248, and T249 were selected for mutagenesis. V37 is part of the βX-strand, and is present both in DAATs and (R)-ATAs,6 while S248 and T249 are found on the β-turn in the P-pocket of LS_ATA. S248 is present more often in DAATs, while T249 is present in (R)-ATAs, showcasing that LS_ATA is not fitting into the identified motifs.
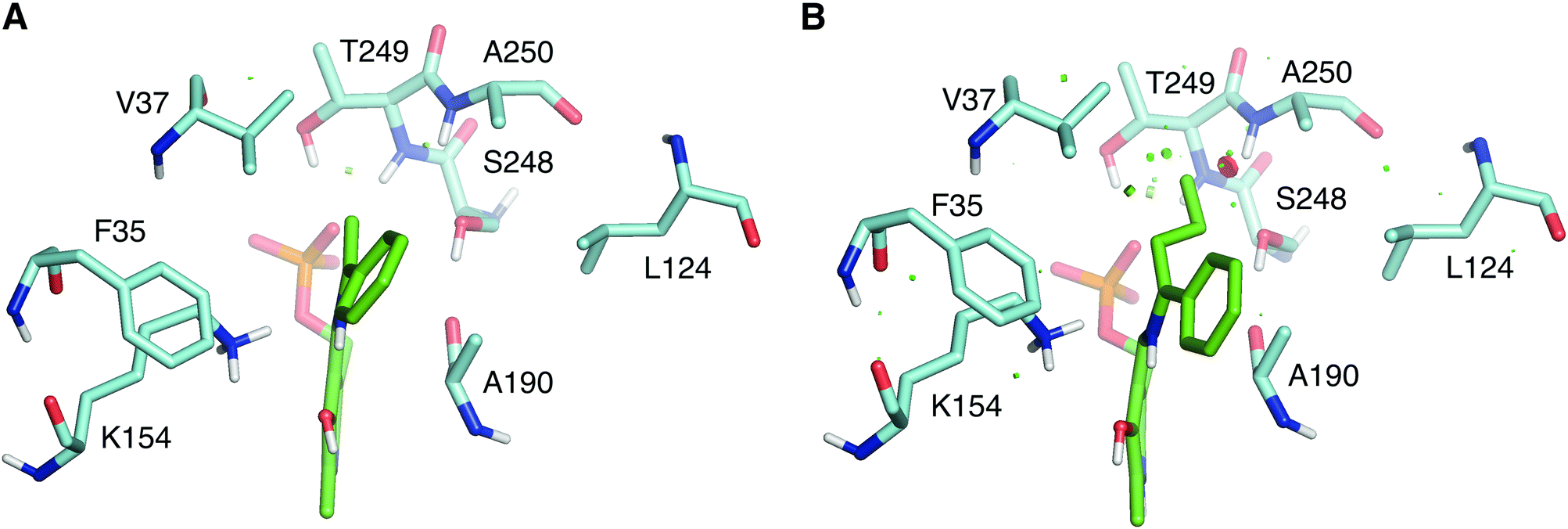 | ||
| Fig. 3 Comparison of (A) (R)-PEA and (B) (R)-PBA quinonoids (green) in the small binding pocket. Residues within 4 Å from the small substituent are shown in cyan. | ||
The activity of all variants was screened with the photometric assay by Schätzle et al.11 As shown in Table S2 (ESI†), the V37A variant exhibits significant activity towards (R)-PPA, while the wild-type has no detectable activity. When mutation S248A is introduced, the variants are inactive, while T249A mutation has a negative effect on the activity of LS_ATA. V37A also decreases by 66% the activity of LS_ATA against the benchmark substrate (PEA); nevertheless, it exhibits broader substrate scope. The double mutant V37A/T249A also exhibits detectable activity against (R)-PPA, showcasing that V37A is the mutation responsible for the expansion of the substrate scope.
To further investigate the negative effect of S248A and T249A mutations, we monitored the formation of pyridoxamine phosphate (PMP), which is the first half-reaction.14 We used the wild-type as the positive control and we investigated all the variants found inactive against (R)-PEA using the photometric assay. As shown in Fig. S2 (ESI†), the internal aldimine, found in the resting state of the enzyme, has an absorption peak at about 400–410 nm. The addition of (R)-PEA to a solution with the active enzyme, such as wild-type LS_ATA, leads to the formation of PMP, which is observed at about 340 nm. Among all mutants that did not exhibit any detectable activity with (R)-PEA, only the S248A variant presents a peak at 340 nm. However, a peak at 410 nm is also observed, indicating that a fraction of PLP remains in the internal aldimine state, unable to form PMP. The V37A/S248A and V38A/T249A variants do not seem able to form PMP at all. These results indicate that the observed lack of activity is not due to the loss of the ability to coordinate pyruvate, but because PMP is not formed as efficiently (or not at all) from these variants. S248 and T249 establish two hydrogen bonds (with the amide and the hydroxyl group of the side chain, respectively) with the phosphate group of PLP, a significant group for the coordination of the cofactor in the active site of the enzyme. We hypothesize that these mutations cause minor structural changes in the active site of LS_ATA, leading to the loss of activity. Nevertheless, all mutants were soluble and had their characteristic yellow color, showing at least some ability to bind PLP in their active site.
To characterize the enantioselectivity of the enzyme, the kinetic resolutions of (R,S)-PEA and (R,S)-PPA were performed in a larger scale (8 mM) with the wild-type and the single mutant V37A. As shown in Fig. 4, the wild-type and V37A variant exhibit similar activity against (R,S)-PEA. Both enzymes convert all (R)-PEA in 3 h and no further conversion is observed (even after 24 h), indicating high enantioselectivity towards (R)-amines. However, in the photometric assay (Table S2, ESI†), a three times higher specific activity of the wild-type, compared with V37A, against (R)-PEA was observed. More striking is the fact that the wild-type is active with (R,S)-PPA, while no activity was detected in the photometric assay. However, the activity is low, and even after 24 h (R)-PPA is not fully converted. The V37A mutation increases this activity, and the (R)-amine is fully converted after 24 h. This discrepancy with the data from the photometric assay led us to test the activity of both variants against (R)-PBA (4 mM). The V37A variant could convert (R)-PBA, with full conversion achieved after 24 h, while the wild-type did not exhibit any significant activity. The divergence between the two assays may arise from the substrate concentration difference, as the photometric assay was performed with 1 mM amine, while the kinetic resolution assay with 8 mM of racemate. The initial velocity observed for the wild-type with PPA is over the detection limit of the photometric assay. We hypothesize that the affinity constants for the amines are in the low mM range; thus the initial velocity is heavily influenced by the concentration.
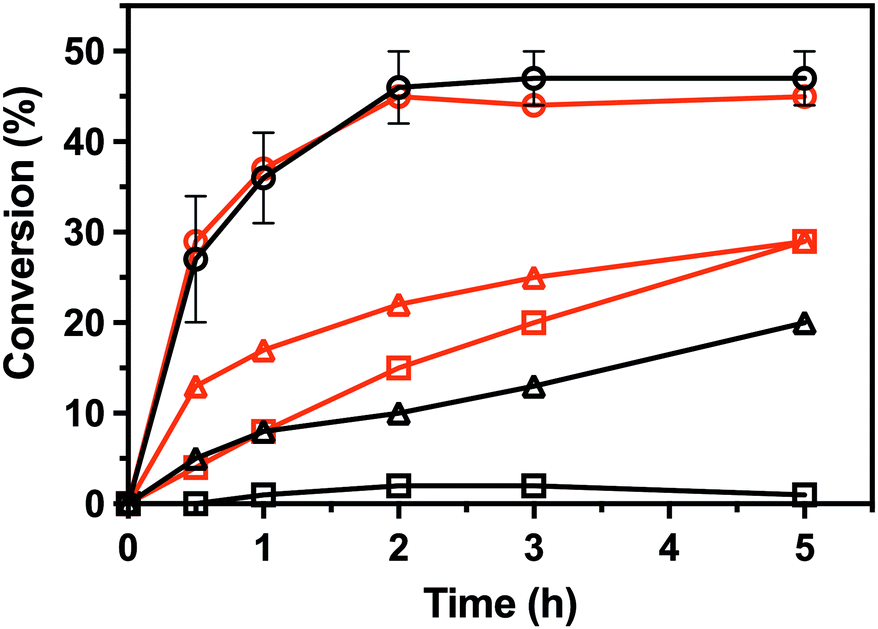 | ||
| Fig. 4 Conversion in the kinetic resolutions of (R,S)-PEA (circle) and (R,S)-PPA (triangle), as well as the deamination of (R)-PBA (square), using pyruvate as the amine acceptor from the LS_ATA wild-type (black) and the V37A variant (red). All experiments were performed at least in duplicate. Errors smaller than symbols are not presented. Experimental details are found in the ESI.† | ||
The kinetic resolution mode is not synthetically interesting for (R)-ATAs, as they provide the (S)-enantiomer. There are several (S)-ATAs that can efficiently produce (S)-PBA.4,13b Despite our efforts using several amine donors (isopropylamine, D-alanine, and (R)-PEA), we were not successful in asymmetric synthesis with propiophenone and butyrophenone (data not shown). It seems that the activity of the V37A variant is quite low to observe reasonable conversion in the asymmetric synthesis mode. We believe, however, that the V37A variant is a good template for protein engineering, to obtain efficient biocatalysts that can produce bulky (R)-amines.
Conclusively, the structure of LS_ATA, a unique enzyme – in terms of sequence – of fold class IV, will provide some insights into (R)-ATAs. We hope that our work will contribute to the understanding of the substrate scope of (R)-ATAs, and the guidance of protein engineering efforts. However, the mutations in the β-turn of the P-pocket showed that secondary interactions are crucial for the activity of (R)-ATAs and that further studies on the role of the amino acids in the small binding pocket are required.
We would like to thank Dr. M. Höhne (University of Greifswald) for providing us with the wild-type construct, and Prof. H. Michel (Max Planck Institute of Biophysics) for his support. The research project was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “1st Call for H.F.R.I. Research Projects to support Faculty Members & Researchers and the Procurement of High-Cost Research Equipment Grant” (Project Number: 664).
Conflicts of interest
There are no conflicts to declare.References
- (a) S. A. Kelly, S. Mix, T. S. Moody and B. F. Gilmore, Appl. Microbiol. Biotechnol., 2020, 104(11), 4781–4794 CrossRef CAS PubMed; (b) M. D. Patil, G. Grogan, A. Bommarius and H. Yun, Catalysts, 2018, 8(7), 254 CrossRef; (c) F. Guo and P. Berglund, Green Chem., 2017, 19(2), 333–360 RSC; (d) M. Fuchs, J. E. Farnberger and W. Kroutil, Eur. J. Org. Chem., 2015, 6965–6982 CrossRef CAS PubMed.
- M. Höhne, S. Schätzle, H. Jochens, K. Robins and U. T. Bornscheuer, Nat. Chem. Biol., 2010, 6, 807–813 CrossRef PubMed.
- (a) H. Land, F. Ruggieri, A. Szekrenyi, W.-D. Fessner and P. Berglund, Adv. Synth. Catal., 2020, 362, 812–821 CrossRef CAS; (b) H.-G. Kim, S.-W. Han and J.-S. Shin, Adv. Synth. Catal., 2019, 361, 2594–2606 CAS; (c) M. S. Weiß, I. V. Pavlidis, P. Spurr, S. P. Hanlon, B. Wirz, H. Iding and U. T. Bornscheuer, ChemBioChem, 2017, 18, 1022–1026 CrossRef PubMed; (d) M. Genz, O. Melse, S. Schmidt, C. Vickers, M. Dörr, T. van den Bergh, H.-J. Joosten and U. T. Bornscheuer, ChemCatChem, 2016, 8, 3199–3202 CrossRef; (e) S.-W. Han, E.-S. Park, J.-Y. Dong and J.-S. Shin, Appl. Environ. Microbiol., 2015, 81, 6994–7002 CrossRef CAS PubMed.
- I. V. Pavlidis, M. S. Weiß, M. Genz, P. Spurr, S. P. Hanlon, B. Wirz, H. Iding and U. T. Bornscheuer, Nat. Chem., 2016, 8(11), 1076–1082 CrossRef CAS PubMed.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- E. Y. Bezsudnova, V. O. Popov and K. M. Boyko, Appl. Microbiol. Biotechnol., 2020, 104, 2343–2357 CrossRef CAS PubMed.
- M. N. Isupov, K. M. Boyko, J. M. Sutter, P. James, C. Sayer, M. Schmidt, P. Schonheit, A. Y. Nikolaeva, T. N. Stekhanova, A. V. Mardanov, N. V. Ravin, E. Y. Bezsudnova, V. O. Popov and J. A. Littlechild, Front. Bioeng. Biotechnol., 2019, 7, 7 CrossRef PubMed.
- E. Y. Bezsudnova, K. M. Boyko, A. Y. Nikolaeva, Y. S. Zeifman, T. V. Rakitina, D. A. Suplatov and V. O. Popov, Biochimie, 2019, 158, 130–138 CrossRef CAS PubMed.
- C. M. Heckmann, L. J. Gourlay, B. Dominguez and F. Paradisi, Front. Bioeng. Biotechnol., 2020, 8, 707 CrossRef PubMed.
- D. Peisach, D. M. Chipman, P. W. van Ophem, J. M. Manning and D. Ringe, Biochemistry, 1998, 37, 4958–4967 CrossRef CAS PubMed.
- S. Schätzle, M. Höhne, E. Redestad, K. Robins and U. T. Bornscheuer, Anal. Chem., 2009, 81(19), 8244–8248 CrossRef PubMed.
- I. W. Davis, A. Leaver-Fay, V. B. Chen, J. N. Block, G. J. Kapral, X. Wang, L. W. Murray, W. B. Arendall 3rd, J. Snoeyink, J. S. Richardson and D. C. Richardson, Nucleic Acids Res., 2007, 35, W375–W383 CrossRef PubMed.
- (a) Y. Xie, F. Xu, L. Yang, H. Liu, X. Xu, H. Wang and D. Wei, Catal. Sci. Technol., 2021, 11, 2461–2470 RSC; (b) A. Nobili, F. Steffen-Munsberg, H. Kohls, I. Trentin, C. Schulzke, M. Höhne and U. T. Bornscheuer, ChemCatChem, 2015, 7(5), 757–760 CrossRef CAS.
- M. Voss, C. Xiang, J. Esque, A. Nobili, M. J. Menke, I. André, M. Höhne and U. T. Bornsheuer, ACS Chem. Biol., 2020, 15, 416–424 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and data, X-ray data, public depository codes and links. See DOI: 10.1039/d1cc04664k |
| This journal is © The Royal Society of Chemistry 2021 |