
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CRISPR/Cas12a-mediated gold nanoparticle aggregation for colorimetric detection of SARS-CoV-2†
Yiren
Cao
,
Jinjun
Wu
 ,
Bo
Pang
,
Hongquan
Zhang
* and
X. Chris
Le
*
,
Bo
Pang
,
Hongquan
Zhang
* and
X. Chris
Le
*
Department of Laboratory Medicine and Pathology, University of Alberta, Edmonton, Alberta, Canada. E-mail: hongquan@ualberta.ca; xc.le@ualberta.ca
First published on 18th June 2021
Abstract
The trans-cleavage activity of the target-activated CRISPR/Cas12a liberated an RNA crosslinker from a molecular transducer, which facilitated the assembly of gold nanoparticles. Integration of the molecular transducer with isothermal amplification and CRISPR/Cas12a resulted in visual detection of the N gene and E gene of SARS-CoV-2 in 45 min.
CRISPR/Cas systems have revolutionized genome editing and are increasingly used for improving specificity and sensitivity of molecular assays. Various CRISPR-based assays have coupled CRISPR/Cas systems with nucleic acid amplification techniques.1 CRISPR/Cas systems have been incorporated within amplification strategies to improve the isothermal amplification,2 after the amplification to enhance the specificity of assays,3 or before the amplification to enrich rare and low-abundance targets.4
In the global fight against the COVID-19 pandemic, CRISPR/Cas systems have been playing a special role in the detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). CRISPR/Cas systems specifically recognized nucleic acid amplification products (amplicons), generated from the viral RNA of SARS-CoV-2, to create detectable readout signals and improve the specificity of the detection.5 In contrast to pH indicators and nonspecific DNA intercalating dyes, CRISPR/Cas systems are activated only by the specific sequences of amplicons complementary to pre-designed guide RNA (gRNA) sequences. Taking advantage of the trans-cleavage activity of CRISPR/Cas systems, researchers have incorporated fluorescence detection and lateral flow platforms into amplified detection of SARS-CoV-2.6,7
Colorimetric detection is advantageous for point-of-care applications because it produces visible signals without the need for an excitation light source or sophisticated equipment. Based on color changes due to aggregation of gold nanoparticles (AuNPs), colorimetric assays have been recently developed for the detection of SARS-CoV-2.8 However, these assays either have low sensitivity8a or require a long detection time.8b Here, we report a colorimetric assay that incorporates reverse transcription loop-mediated isothermal amplification (RT-LAMP) and the trans-cleavage activity of CRISPR/Cas12a to facilitate sequence-dependent aggregation of AuNPs, thus achieving isothermal and visualized detection of nucleic acids (Scheme 1). Because of the exponential amplification of LAMP and the target-specific trans-cleavage of CRISPR/Cas12a, our assay provides sensitive and specific detection of SARS-CoV-2 RNA. Unlike other colorimetric assays that use AuNPs, our assay incorporates a rationally designed hairpin transducer to facilitate spinning-accelerated aggregation of AuNPs, resulting in a color change recognizable within 1 min.
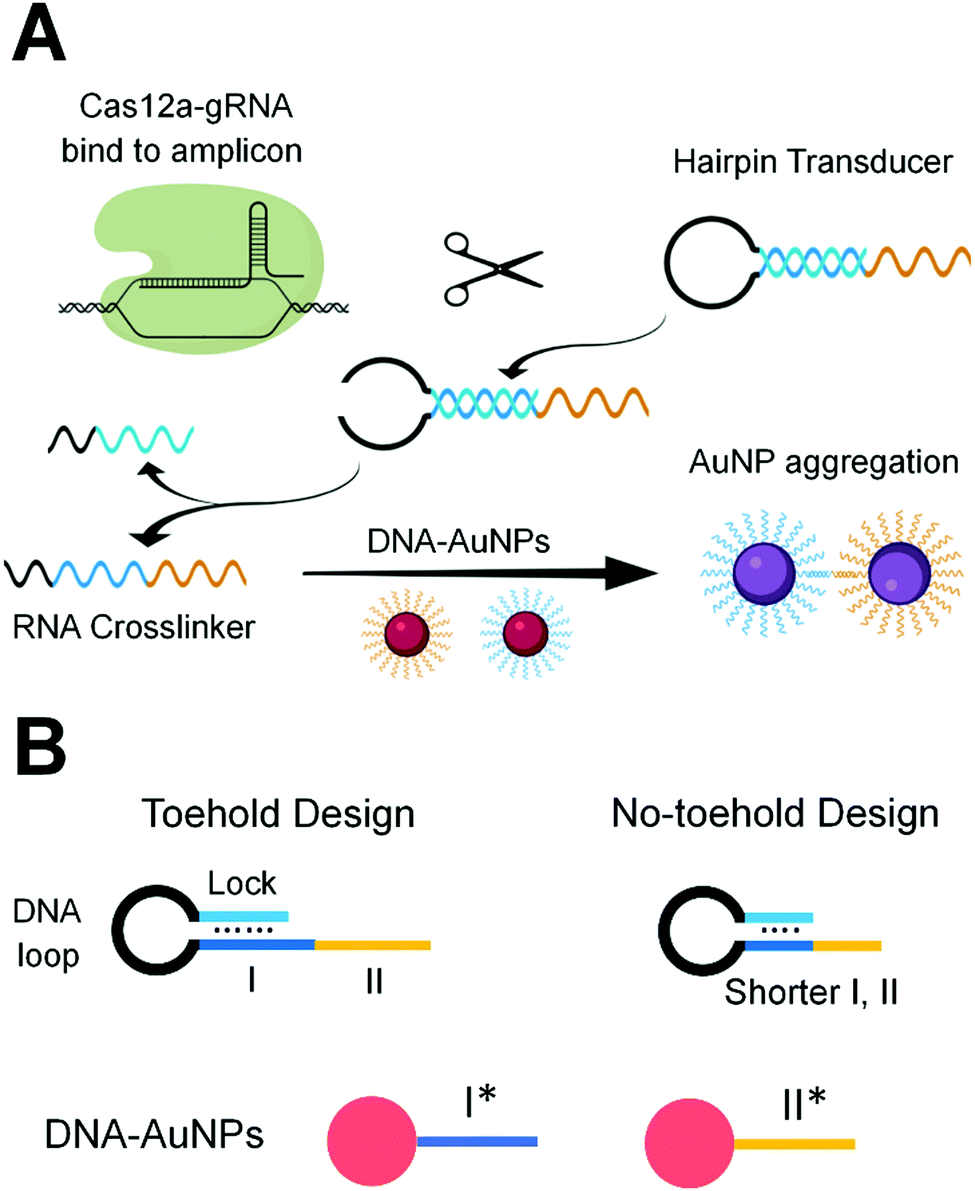 | ||
| Scheme 1 (A) The principle of Cas12a-mediated AuNP aggregation. The Cas12a-gRNA ribonucleoprotein specifically binds to the target sequence of the double-stranded DNA amplicon, which activates the Cas12a-gRNA ribonucleoprotein. This active enzyme trans-cleaves the DNA loop of the hairpin transducer, which destabilizes the hairpin and releases the RNA crosslinker from its hybrid. Hybridization of the RNA crosslinker with the ssDNAs on AuNPs results in the aggregation of AuNPs and the corresponding change in color from red to purple. (B) Hairpin transducers composed of an RNA crosslinker, a DNA loop, and a lock sequence. The RNA crosslinker sequence (subdomains I and II) within the hairpin transducer are complementary to the ssDNA sequences (I* and II*) that are conjugated on two separate AuNPs. In the toehold design, subdomain I is partially blocked by the lock sequence, leaving the unblocked portion to function as the toehold for toehold-mediated strand displacement of the lock. | ||
We designed gRNA sequences (Table S2, ESI†) that are complementary to specific sequences of the RT-LAMP amplicons of the N gene and E gene of SARS-CoV-2 (Table S1, ESI†). Binding of the Cas12a-gRNA ribonucleoprotein to the specific amplicon initiated the enzyme activity of Cas12a. The active Cas12a nuclease trans-cleaved the single-stranded (ss)DNA loop region of the hairpin transducer (HT). Cleavage of the hairpin changed the nature of the hybridization in the stem region from intra- to inter-molecular interaction, destabilized the hybrid, and liberated the RNA crosslinker. The free RNA crosslinker hybridized to two ssDNA sequences on separate AuNPs, resulting in aggregation of AuNPs and color change of the solution from red to purple (Scheme 1A).
The hairpin transducer has two important functions: it serves as a substrate of Cas12a and releases a crosslinker to assemble DNA-functionalized AuNPs. We designed the hairpin transducer to contain three functional domains: an RNA crosslinker, a DNA loop, and a DNA lock (Scheme 1B). The DNA lock sequence hybridized to a portion of the RNA crosslinker, caging the crosslinker in the hairpin structure and keeping it unavailable for the assembly of DNA-functionalized AuNPs. We designed the crosslinker using the RNA sequence, which cannot be trans-cleaved by Cas12a. Because the trans-cleavage activity of Cas12a only cleaves ssDNA, not RNA, the activated Cas12a cleaves the ssDNA loop region of the hairpin transducer. We added an inverted thymine to the 3′-end of the hairpin to inhibit its extension by the polymerase present in the reaction mixture. Incorporating these design considerations into isothermal amplification, Cas12a recognition, and colorimetric detection, we were able to minimize background and achieve sensitive and specific detection of target nucleic acid sequences.
As an example, the detection of SARS-CoV-2 viral RNA was completed within 45 min, including 30 min for RT-LAMP, 10 min for Cas12a-mediated trans-cleavage reaction, and instantaneous aggregation of AuNPs (Scheme S1, ESI†).
Achieving high sensitivity and rapid signal response requires efficient liberation of the RNA crosslinker from the DNA lock upon the target-initiated trans-cleavage of the hairpin transducer. In the absence of the target, the RNA crosslinker must be firmly blocked by the DNA lock to ensure low background. For these two reasons, we designed and tested several hairpin transducers (Scheme 1B and Table S3, ESI†). In the toehold design (Scheme 1B, left), the DNA lock only partially blocked subdomain I of the RNA crosslinker, leaving the unblocked portion to serve as a toehold for the rapid displacement of the lock. We intended to use a long DNA lock to stably block the RNA crosslinker (thus, low background), and at the same time, to achieve the fast interaction of subdomain I with I* on the AuNP immediately after the target-initiated cleavage of the DNA loop (thus, high sensitivity and fast response). This design took advantage of the toehold-mediated strand displacement reaction to accelerate the hybridization between I and I* without the need for spontaneous dissociation of the DNA lock from the RNA crosslinker.
We tested four hairpin transducers of the toehold design, HT-t1, HT-t2, HT-t3, and HT-t4, in the detection of a model DNA sequence, a DNA activator recognizable by the gRNA-Cas12a ribonucleoprotein (Table S2, ESI†). The length of the DNA lock in these hairpin transducers varied from 14 to 11 nucleotides (nt), leaving the unblock toehold on subdomain I as 1 to 4 nt. The results (Fig. S1A, ESI†) from the analysis of the DNA activator (+) show clear color changes when comparing no hairpin transducer with any of the four hairpin transducers. The changes in color and absorbance were more significant with the use of hairpin transducers of increasing length of toehold. These results are consistent with the expected consequence of toehold-mediated strand displacement reactions. We also concurrently measured the background from reaction mixtures containing the identical reagents but no DNA activator (−). In all cases, background was observable (Fig. S1, ESI†). These results suggest dynamic dissociation (or leakage) of the lock strand from subdomain I of the crosslinker strand, resulting in background assembly of DNA-functionalized AuNPs. We estimated the melting temperature of the hybrids in the toehold hairpin design, which was over 54.5 °C. The toehold might have induced the “leakage” from otherwise stable hybrids at room temperature.
To eliminate the background that might have been caused by the toehold-mediated strand displacement reaction, we designed and tested several hairpin transducers not containing any toehold but having the entire subdomain I fully blocked by the lock DNA (Scheme 1B, right). The success of this design hinges on fast spontaneous dissociation of the DNA lock sequence from the RNA crosslinker and efficient hybridization of the crosslinker with the ssDNA sequences on AuNPs. A shorter crosslinker and lock are desirable for spontaneous dissociation of the DNA lock, but too short an RNA crosslinker can result in slow hybridization between I and I* and between II and II*. We designed five crosslinkers (Table S4, ESI†) with subdomains I + II being 10 + 10, 9 + 9, 8 + 8, 7 + 7, and 6 + 6 nt. We determined the shortest length of the crosslinker needed for sufficient assembly of DNA-functionalized AuNPs. We mixed 100 nM of each crosslinker with 1 nM of two DNA-functionalized AuNPs and determined the aggregation of AuNPs by measuring the absorbance (Fig. S3, ESI†). Changes in absorbance due to AuNP aggregation were the highest with the use of crosslinkers 10 + 10, 9 + 9, and 8 + 8 nt. Thus, we chose 20, 18, and 16 nt as the lengths of the crosslinkers to construct hairpin transducers HT20, HT18, and HT16. Application of these hairpin transducers substantially reduced the background (Fig. 1). The highest difference in absorbance between the positive sample and negative control was observed with the use of HT18. This was a consequence of efficient blocking of the crosslinker, resulting in negligible background. Thus, HT18 was selected as the hairpin transducer for the detection of SARS-CoV-2.
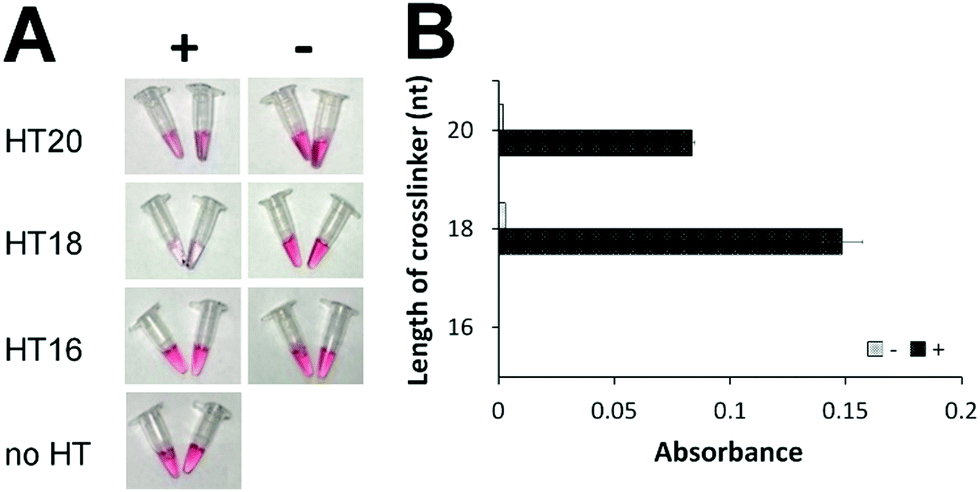 | ||
| Fig. 1 Comparison of hairpin transducers of different crosslinker lengths without a toehold by colorimetric assay (A) and UV-Vis absorbance (B). ‘+’ indicates samples containing 20 nM model DNA sequence (DNA activator for Cas12a-gRNA). ‘−’ indicates samples containing all reagents but no DNA activator. ‘no HT’ indicates the negative control, containing all reagents except hairpin transducer. ‘Absorbance’ is the difference of UV-Vis absorbance values at 530 nm (Fig. S4, ESI†) between the samples and the negative control. | ||
We aimed to achieve fast AuNP aggregation and obvious color change which is desirable for point-of-care testing. We optimized the concentration of Na+ to accelerate the assembly of DNA-functionalized AuNPs and generate fast AuNP aggregation (Fig. 2A). We prepared a series of AuNP solutions each containing 1 nM of two types of DNA-functionalized AuNPs, and 0 or 200 nM crosslinker of 18 nt (L18, Table S4, ESI†) to induce AuNP aggregation. The increase of Na+ concentration efficiently accelerated AuNP aggregation. A color change was observed immediately after the addition of 0.8 M NaCl, while the color remained unchanged in the blank sample (negative control) after 20 min (Fig. 2B).
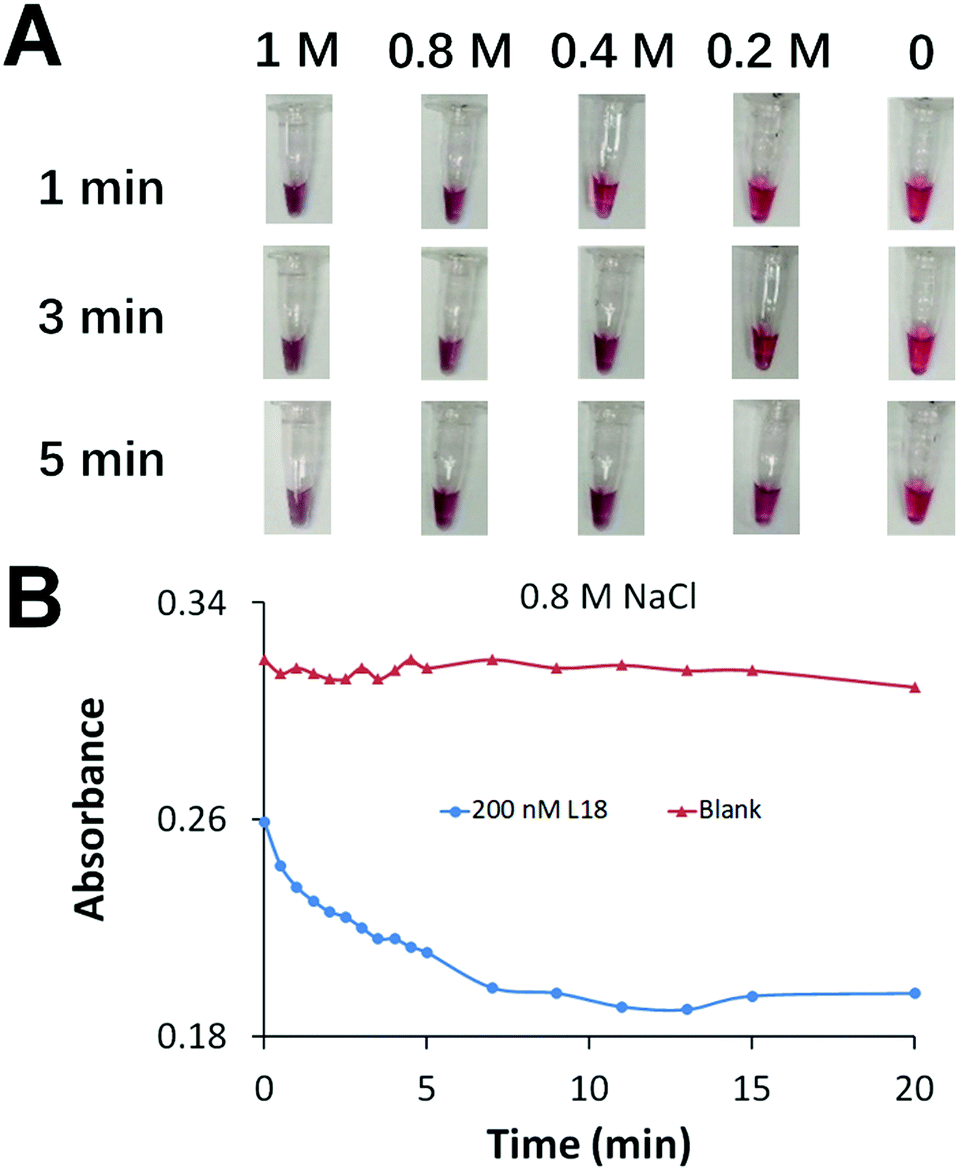 | ||
| Fig. 2 (A) Rapid aggregation of DNA-functionalized AuNPs assisted by the crosslinker sequence that was derived from the hairpin transducer. The samples contained 200 nM crosslinker L18, 1 nM each of two DNA-functionalized AuNPs, and different concentrations of NaCl. The photographs were taken at 1, 3, and 5 min after the addition of NaCl. (B) Absorbance measured at 530 nm for 20 min. Time 0 was when 0.8 M NaCl was added to the solution that contained 200 nM crosslinker L18 and 1 nM each of two DNA-functionalized AuNPs. | ||
Although the color change within 1 min was suitable for rapid detection, it was difficult to reliably differentiate the weak color change from red to purple. Full AuNP aggregation and an obvious color change required 10 min of incubation (Fig. 2B). To achieve rapid detection, we used a portable spinner to centrifuge the AuNP solutions for only 10 s. Precipitation of the aggregated AuNPs in positive samples reduced the purple color of the solution to nearly colorless (Fig. 3). The dispersed AuNPs in the negative sample remained the red color. The brief 10 s centrifugation of the AuNP solutions also improved the sensitivity of the colorimetric detection (Fig. S5, ESI†).
 | ||
| Fig. 3 Representative images from triplicate analysis of samples containing varying concentrations of the SARS-CoV-2 viral RNA. The N gene was detected. 5 μL of the viral RNA sample was used. The tubes labelled blank contained all reagents but no RNA sample (negative control). | ||
We examined the applicability of the assay for the detection of the E gene and N gene of SARS-CoV-2 viral RNA. We chose to detect these two genes because the E gene is highly conserved among all beta coronaviruses, such as SARS-CoV-2, SARS-CoV, and bat SARS-like coronavirus, whereas the N gene enables the differentiation of SARS-CoV-2 from other coronaviruses.
We determined the reaction time needed for RT-LAMP by testing a series of solutions containing varying amounts of the viral RNA, from 375 to 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 750000 copies (Fig. S6, ESI†). We monitored the RT-LAMP reaction in real time using SYBR Green detection. Amplification curves show that 30 min was sufficient for RT-LAMP to amplify low copies of the viral RNA. We then used the CRISPR/Cas12a-mediated AuNP aggregation assay to test these solutions (Fig. 3). The color of the negative control solution that contained all the reagents but no viral RNA remained red, whereas the color of all positive sample solutions changed from red to purple and then to nearly colorless after centrifugation for 10 s. Aggregated AuNP pellets were observed from all positive samples, the cause of the disappearance of the purple color.
750000 copies (Fig. S6, ESI†). We monitored the RT-LAMP reaction in real time using SYBR Green detection. Amplification curves show that 30 min was sufficient for RT-LAMP to amplify low copies of the viral RNA. We then used the CRISPR/Cas12a-mediated AuNP aggregation assay to test these solutions (Fig. 3). The color of the negative control solution that contained all the reagents but no viral RNA remained red, whereas the color of all positive sample solutions changed from red to purple and then to nearly colorless after centrifugation for 10 s. Aggregated AuNP pellets were observed from all positive samples, the cause of the disappearance of the purple color.
We determined the sensitivity of the assay by testing low copies of the viral RNA of SARS-CoV-2. For the detection of the N gene, the color change was observed for all triplicate tests of 225, 300, and 375 copies of the viral RNA (Fig. S8, ESI†). One of the three samples containing 75 copies of the viral RNA also showed visible color change. For the detection of the E gene, as few as 300 copies of the viral RNA were consistently detectable (Fig. S10, ESI†). These results of reproducibly detecting 225 copies of the N gene and 300 copies of the E gene are consistent with those of real-time RT-LAMP, suggesting that the sensitivity of the assay is governed by RT-LAMP. Thus, our colorimetric assay using the transducer-mediated AuNP aggregation maintained the sensitivity of the RT-LAMP assay (Fig. S7 and S9, ESI†). Integration of the Cas12a system for recognition of the specific target sequence improved the specificity of the assay.
We further applied the colorimetric assay to the analysis of clinical samples for diagnosis and screening of COVID-19 (Table S6, ESI†). We tested 54 clinical respiratory swab samples, half of which were SARS-CoV-2 positive as determined by RT-qPCR. Using the colorimetric assay for detection of the N gene, we reported 25 samples as SARS-CoV-2 positive. We were not able to detect very low concentrations of SARS-CoV-2 RNA in two samples that required 35.4 and 37 threshold cycles of RT-qPCR to achieve positive detection. Our colorimetric assay reported negative results for all 27 SARS-CoV-2 negative clinical samples. Therefore, the overall clinical sensitivity and specificity of the assay were 92.6% and 100%, respectively, for the detection of these 54 clinical samples (Table S5, ESI†). Similar results were obtained for the detection of the E gene (Table S8, ESI†).
We have developed a colorimetric assay that integrates isothermal RT-LAMP amplification, target-initiated trans-cleavage activity of CRISPR-Cas12a, and transducer-directed aggregation of AuNPs. We successfully applied the assay to the detection of SARS-CoV-2 RNA, which was completed within 45 min at a single controlled temperature. Detection with the naked eye and elimination of sophisticated equipment make this assay promising for point-of-care testing of COVID-19. The assay can be easily modified, by adjusting the LAMP primers and gRNA, for the detection of other pathogens.
YC, JW, and BP contributed to conceptualization, investigation, data curation, formal analysis, visualization, validation, writing, review, and editing. HZ and XCL contributed to conceptualization, formal analysis, funding acquisition, project administration, resources, supervision, writing, review, and editing.
We thank the Canadian Institutes of Health Research, the New Frontiers in Research Fund, the Natural Sciences and Engineering Research Council of Canada, and the Social Sciences and Humanities Research Council of Canada for their support. We thank Dr Kanti Pabbaraju, Dr Graham Tipples, and LeeAnn Turnbull (Provincial Public Health Laboratory, Alberta Precision Laboratories, Edmonton, Canada) for providing RNA extracts of the clinical specimens and the RT-qPCR results.
Conflicts of interest
There are no conflicts to declare.References
- (a) M. J. Kellner, J. G. Koob, J. S. Gootenberg, O. O. Abudayyeh and F. Zhang, Nat. Protoc., 2019, 14, 2986 CrossRef CAS PubMed; (b) R. Zhou, Y. Li, T. Dong, Y. Tang and F. Li, Chem. Commun., 2020, 56, 3536 RSC; (c) W. M. Tian, X. L. Liu, G. T. Wang and C. H. Liu, Chem. Commun., 2020, 56, 13445 RSC; (d) Y. Xiong, J. J. Zhang, Z. L. Yang, Q. B. Mou, Y. Ma, Y. H. Xiong and Y. Lu, J. Am. Chem. Soc., 2020, 142, 207 CrossRef CAS PubMed; (e) W. Feng, A. M. Newbigging, J. Tao, Y. Cao, H. Peng, C. Le, J. Wu, B. Pang, J. Li, D. L. Tyrrell, H. Zhang and X. C. Le, Chem. Sci., 2021, 12, 4683 RSC.
- (a) T. Wang, Y. Liu, H. H. Sun, B. C. Yin and B. C. Ye, Angew. Chem., Int. Ed., 2019, 58, 5382 CrossRef CAS PubMed; (b) W. Zhou, L. Hu, L. Ying, Z. Zhao, P. K. Chu and X. F. Yu, Nat. Commun., 2018, 9, 1 CrossRef PubMed; (c) K. Zhang, R. Deng, X. Teng, Y. Li, Y. Sun, X. Ren and J. Li, J. Am. Chem. Soc., 2018, 140, 11293 CrossRef CAS PubMed.
- (a) J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438 CrossRef CAS PubMed; (b) J. S. Chen, E. Ma, L. B. Harrington, M. D. Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 27, 436 CrossRef PubMed; (c) K. Pardee, A. A. Green, M. K. Takahashi, D. Braff, G. Lambert, J. W. Lee, T. Ferrante, D. Ma, N. Donghia, M. Fan, N. M. Daringer, I. Bosch, D. M. Dudley, D. H. O’Connor, L. Gehrke and J. J. Collins, Cell, 2016, 165, 1255 CrossRef CAS PubMed; (d) J. S. Gootenberg, O. O. Abudayyeh, M. J. Kellner, J. Joung, J. J. Collins and F. Zhang, Science, 2018, 360, 439 CrossRef CAS PubMed.
- (a) S. H. Lee, J. Yu, G. H. Hwang, S. Kim, H. S. Kim, S. Ye, K. Kim, J. Park, D. Y. Park, Y. K. Cho, J. S. Kim and S. Bae, Oncogene, 2017, 36, 6823 CrossRef CAS PubMed; (b) W. Gu, E. D. Crawford, B. D. O’Donovan, M. R. Wilson, E. D. Chow, H. Retallack and J. L. DeRisi, Genome Biol., 2016, 17, 41 CrossRef CAS PubMed; (c) A. Aalipour, J. C. Dudley, S. Park, S. Murty, J. J. Chabon, E. A. Boyle, M. Diehn and S. S. Gambhir, Clin. Chem., 2018, 64, 307 CrossRef CAS PubMed; (d) J. Quan, C. Langelier, A. Kuchta, J. Batson, N. Teyssier, A. Lyden, S. Caldera, A. McGeever, B. Dimitrov, R. King, J. Wilheim, M. Murphy, L. P. Ares, K. A. Travisano, R. Sit, R. Amato, D. R. Mumbengegwi, J. L. Smith, A. Bennett, R. Gosling, P. M. Mourani, C. S. Calfee, N. F. Neff, E. D. Chow, P. S. Kim, B. Greenhouse, J. L. DeRisi and E. D. Crawford, Nucleic Acids Res., 2019, 47, 83 CrossRef PubMed.
- (a) F. Zhang, O. O. Abudayyeh and J. S. Gootenberg, A protocol for detection of COVID-19 using CRISPR diagnostics, 2020, https://www.broadinstitute.org/files/publications/special/COVID-19%20detection%20(updated).pdf; (b) J. P. Broughton, X. Deng, G. Yu, C. L. Fasching, V. Servellita, J. Singh, X. Miao, J. A. Streithorst, A. Granados, A. Sotomayor-Gonzalez, K. Zorn, A. Gopez, E. Hsu, W. Gu, S. Miller, C. Y. Pan, H. Guevara, D. A. Wadford, J. S. Chen and C. Y. Chiu, Nat. Biotechnol., 2020, 38, 870 CrossRef CAS; (c) S. K. Kailasa, V. N. Mehta, J. R. Koduru, H. Basu, R. K. Singhal, Z. V. P. Murthy and T. J. Park, Analyst, 2021, 146, 1489 RSC.
- (a) X. Ding, K. Yin, Z. Li, R. V. Lalla, E. Ballesteros, M. M. Sfeir and C. Liu, Nat. Commun., 2020, 11, 4711 CrossRef CAS PubMed; (b) M. Patchsung, K. Jantarug, A. Pattama, K. Aphicho, S. Suraritdechachai, P. Meesawat, K. Sappakhaw, N. Leelahakorn, T. Ruenkam, T. Wongsatit, N. Athipanyasilp, B. Eiamthong, B. Lakkanasirorat, T. Phoodokmai, N. Niljianskul, D. Pakotipraph, S. Chanarat, A. Homchan, R. Tinikul, P. Kamutira, K. Phiwkaow, S. Soithongcharoen, C. Kantiwiriyawanitch, V. Pongsupasa, D. Trisrivirat, J. Jaroensuk, T. Wongnate, S. Maenpuen, P. Chaiyen, S. Kamnerdnakta, J. Swangsri, S. Chuthapisith, Y. Sirivatanauksorn, C. Chaimayo, R. Sutthent, W. Kantakamalakul, J. Joung, A. Ladha, X. Jin, J. S. Gootenberg, O. O. Abudayyeh, F. Zhang, N. Horthongkham and C. Uttamapinant, Nat. Biomed. Eng., 2020, 4, 1140 CrossRef PubMed.
- B. Pang, J. Xu, Y. Liu, H. Peng, W. Feng, Y. Cao, J. Wu, H. Xiao, K. Pabbaraju, G. Tipples, M. A. Joyce, H. A. Saffran, D. L. Tyrrell, H. Zhang and X. C. Le, Anal. Chem., 2020, 92, 16204 CrossRef CAS PubMed.
- (a) P. Moitra, M. Alafeef, K. Dighe, M. B. Frieman and D. Pan, ACS Nano, 2020, 14, 7617 CrossRef CAS PubMed; (b) W. S. Zhang, J. Pan, F. Li, M. Zhu, M. Xu, H. Zhu, Y. Yu and G. Su, Anal. Chem., 2021, 93, 4126 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc02546e |
| This journal is © The Royal Society of Chemistry 2021 |