
Helper lipid structure influences protein adsorption and delivery of lipid nanoparticles to spleen and liver†
Rui
Zhang‡
a,
Rakan
El-Mayta‡
a,
Timothy J.
Murdoch
 b,
Claude C.
Warzecha
c,
Margaret M.
Billingsley
a,
Sarah J.
Shepherd
a,
Ningqiang
Gong
a,
Lili
Wang
c,
James M.
Wilson
c,
Daeyeon
Lee
b and
Michael J.
Mitchell
*adefg
b,
Claude C.
Warzecha
c,
Margaret M.
Billingsley
a,
Sarah J.
Shepherd
a,
Ningqiang
Gong
a,
Lili
Wang
c,
James M.
Wilson
c,
Daeyeon
Lee
b and
Michael J.
Mitchell
*adefg
aDepartment of Bioengineering, University of Pennsylvania, Philadelphia, PA 19104, USA. E-mail: mjmitch@seas.upenn.edu
bDepartment of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, PA 19104, USA
cGene Therapy Program, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
dAbramson Cancer Center, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
eInstitute for Immunology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
fCardiovascular Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
gInstitute for Regenerative Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104, USA
First published on 15th December 2020
Abstract
Nucleic acids, such as messenger RNAs, antisense oligonucleotides, and short interfering RNAs, hold great promise for treating previously ‘undruggable’ diseases. However, there are numerous biological barriers that hinder nucleic acid delivery to target cells and tissues. While lipid nanoparticles (LNPs) have been developed to protect nucleic acids from degradation and mediate their intracellular delivery, it is challenging to predict how alterations in LNP formulation parameters influence delivery to different organs. In this study, we utilized high-throughput in vivo screening to probe for structure–function relationships of intravenously administered LNPs along with quartz crystal microbalance with dissipation monitoring (QCM-D) to measure the binding affinity of LNPs to apolipoprotein E (ApoE), a protein implicated in the clearance and uptake of lipoproteins by the liver. High-throughput in vivo screening of a library consisting of 96 LNPs identified several formulations containing the helper lipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) that preferentially accumulated in the liver, while identical LNPs that substituted DOPE with the helper lipid 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) preferentially accumulated in the spleen. Using QCM-D, it was found that one DOPE-containing LNP formulation (LNP 42) had stronger interactions with ApoE than an identical LNP formulation that substituted DOPE with DSPC (LNP 90). In order to further validate our findings, we formulated LNP 42 and LNP 90 to encapsulate Cy3-siRNA or mRNA encoding for firefly luciferase. The DSPC-containing LNP (LNP 90) was found to increase delivery to the spleen for both siRNA (two-fold) and mRNA (five-fold). In terms of liver delivery, the DOPE-containing LNP (LNP 42) enhanced mRNA delivery to the liver by two-fold and improved liver transfection by three-fold. Understanding the role of the helper lipid in LNP biodistribution and ApoE adsorption may aid in the future design of LNPs for nucleic acid therapeutics.
1. Introduction
Nucleic acid therapeutics represent a promising modality for treating diseases that have been considered ‘undruggable’ by existing small molecule medicines.1,2 Specifically, messenger RNAs (mRNAs) have been used in protein replacement therapies, short interfering RNAs (siRNAs) and antisense oligonucleotides (ASOs) function to silence specific genes of interest, and ribonucleoproteins (RNPs) and mRNAs encoding for Cas9 protein have been developed for gene editing applications.3–7 However, nucleic acids are large, hydrophilic, and highly negatively charged molecules; this prevents them from crossing cell membranes and reaching their intracellular targets.1,8 Additionally, they are unstable in circulation and prone to degradation by nucleases.8,57 Thus, in order to achieve therapeutic efficacy, safe and effective delivery vehicles are needed to overcome these obstacles to delivery.9,10Lipid nanoparticles (LNPs) have been developed as a non-viral delivery system for nucleic acids – functioning to protect cargo from degradation and mediate their intracellular delivery.7,11 LNPs typically contain an ionizable lipid component, which is unique in that it holds a neutral charge under physiological pH but becomes positively charged in the acidic endosomal compartment.12,13 This allows LNPs to undergo endosomal escape and subsequently release their nucleic acid cargo into the cytosol for gene regulation.14,15 In addition to the ionizable lipid, LNPs are commonly formulated with three additional excipients: a cholesterol component to improve LNP stability and membrane fusion, a helper phospholipid to aid in encapsulation efficiency and endosomal escape, and a lipid-anchored polyethylene glycol (PEG) conjugate (lipid-PEG) to minimize aggregation and immune cell opsonization.13,15–17 The potential for LNP clinical translation was emphasized recently when the United States Food and Drug Administration approved Onpattro, the first RNA interference (RNAi) LNP therapy for transthyretin-mediated amyloidosis.18,58 Recent investigations into altering and modifying LNP excipients, rather than the ionizable lipid, have revealed the importance of combinatorial effects among all LNP components.5,19,20 Certain changes in the tail structure of cholesterol, for instance, have been demonstrated to significantly boost transfection over similar formulations that modified the body or head of the cholesterol component.19 With the large number of formulation possibilities and relatively limited data observing the impacts of specific changes to LNP formulations, it remains challenging to predict how alterations in LNP formulation parameters may confer advantages for targeting different tissues.
Part of the reason why LNPs have been successful in the delivery of nucleic acids to the liver is due to the well-perfused nature of the organ and the fenestrations in the liver endothelium.21 Additionally, LNPs are known to interact with and bind proteins in circulation, leading to the formation of a protein corona, that can influence endogenous targeting to the liver.22 Specifically, apolipoprotein E (ApoE) is implicated in LNP uptake by hepatocytes through the low-density lipoprotein receptor (LDLR).21,23 Following systemic infusion, LNPs have also been observed to substantially accumulate in the spleen.24–26 This has been attributed to uptake by splenic macrophages of the mononuclear phagocytic system following surface opsonization and protein adsorption on the LNP surface.27–29 While LNP delivery to the spleen is a promising strategy for oncology and the development of vaccines,30 nucleic acid and lipid accumulation may trigger unwanted immune responses, such as cytokine release syndrome by massive IL-6 production in the spleen.31 In order to further improve the design of LNPs for nucleic acid therapeutics, it is important to understand how LNP formulation parameters are related to biodistribution, ApoE adsorption, and subsequent protein corona formation.
Here, we utilized both high-throughput in vivo LNP screening and quartz crystal microbalance with dissipation monitoring (QCM-D) to understand how LNP formulation parameters affect protein adsorption to LNPs and their delivery to the spleen and liver. For high-throughput screening, we used DNA barcoding to formulate and screen a library of 96 LNPs as a means of probing for LNP structure–function relationships. This approach allowed us to pool together the library of LNPs, inject them intravenously into mice, isolate tissues of interest, and quantify LNP biodistribution via deep sequencing. We then performed quantitative polymerase chain reaction (qPCR) to normalize the deep sequencing data and compare the delivery of LNPs across tissues. QCM-D was then used to investigate the extent to which different LNPs bound ApoE in order to validate the accumulation differences seen. Data was further validated by imaging the spleens of mice treated with these LNPs encapsulating fluorescent siRNA. Finally, luciferase mRNA-LNPs were synthesized to investigate the relationship between biodistribution and efficacy. Understanding the role of the helper lipid in directing LNP accumulation to the spleen or liver following ApoE adsorption may aid in the future design of LNPs for nucleic acid therapeutics.
2. Results and discussion
A high-throughput in vivo screening assay was used to probe structure–function relationships of intravenously administered LNPs.32,33 A library of 96 LNP formulations was synthesized for this screening assay. Each LNP was formulated by pipette mixing and encapsulated a unique DNA barcode (b-DNA). The LNPs within the library consisted of one ionizable lipid (C12-200),34 two different helper phospholipids (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) or 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC)), four different lipid-PEGs (C14-PEG1000, C14-PEG2000, C14-PEG3000, or C14-PEG5000), and two different ionizable lipid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :b-DNA weight ratios (5:1 or 10:1) – combined at six different molar ratios of cholesterol to lipid-PEG (varying from 48.5:1.5 to 1.5:48.5) (Table S1†). LNPs were characterized by dynamic light scattering (DLS) for hydrodynamic diameter and polydispersity (PDI) (Table S2†). 94 out of the 96 formulations formed LNPs based on DLS analysis of peaks and autocorrelation curves. The LNP library was then injected intravenously via the tail vein as a single pool, along with a naked b-DNA without an LNP carrier, into C57BL/6 mice. Six hours post-administration, tissues of interest (liver, spleen, kidney, small intestine, colon, stomach, cecum, feces) were isolated and DNA from these tissues was extracted. The b-DNA from these extracted DNA samples was amplified by polymerase chain reaction (PCR) and deep sequenced to determine the relative accumulation of LNPs in different tissues (Fig. 1A).
:b-DNA weight ratios (5:1 or 10:1) – combined at six different molar ratios of cholesterol to lipid-PEG (varying from 48.5:1.5 to 1.5:48.5) (Table S1†). LNPs were characterized by dynamic light scattering (DLS) for hydrodynamic diameter and polydispersity (PDI) (Table S2†). 94 out of the 96 formulations formed LNPs based on DLS analysis of peaks and autocorrelation curves. The LNP library was then injected intravenously via the tail vein as a single pool, along with a naked b-DNA without an LNP carrier, into C57BL/6 mice. Six hours post-administration, tissues of interest (liver, spleen, kidney, small intestine, colon, stomach, cecum, feces) were isolated and DNA from these tissues was extracted. The b-DNA from these extracted DNA samples was amplified by polymerase chain reaction (PCR) and deep sequenced to determine the relative accumulation of LNPs in different tissues (Fig. 1A).
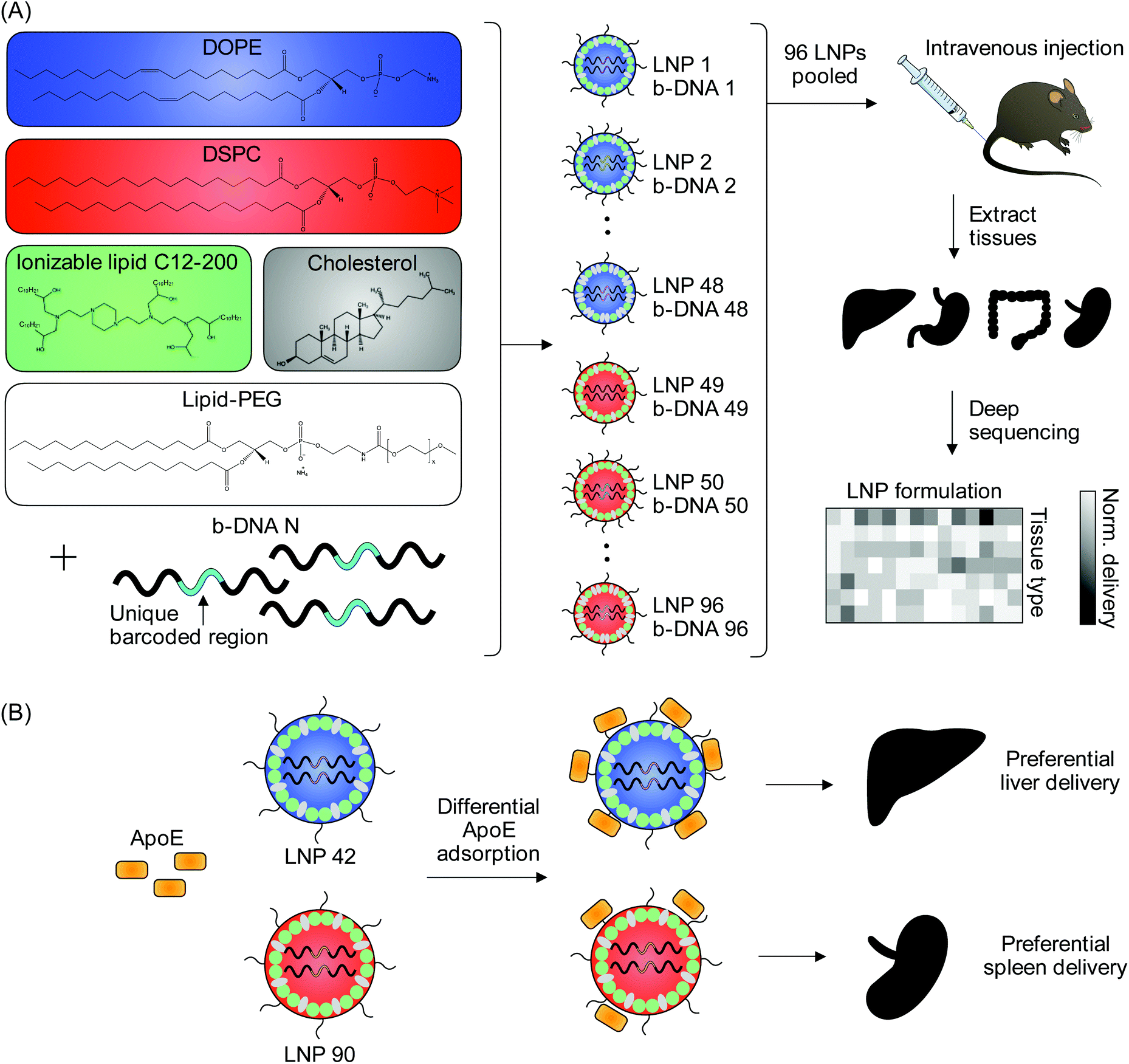 | ||
| Fig. 1 High-throughput in vivo screening and quartz crystal microbalance with dissipation monitoring (QCM-D) to quantify LNP delivery and protein adsorption properties. (A) LNPs were formulated with 1 ionizable lipid (C12-200), 6 different excipient molar ratios, 2 different ionizable lipid:b-DNA weight ratios, 2 different helper lipids, and 4 different lipid-anchored polyethylene glycol (PEG) conjugates (lipid-PEG) for a total of 96 LNP formulations. Details regarding the specific excipient molar ratios for each LNP are provided in Table S1.† LNPs were formulated by pipette mixing to encapsulate barcoded DNA (b-DNA). LNPs were pooled together and injected to C57BL/6 mice via tail vein (N = 4). Tissues were isolated six hours post-injection and accumulation of b-DNAs was quantified by deep sequencing. (B) Differences in LNP accumulation in the liver and spleen were identified and QCM-D was used as a means of quantifying the interactions between LNPs and apolipoprotein E (ApoE). | ||
Deep sequencing results demonstrated that b-DNAs delivered by LNPs, formulated with a cholesterol to lipid-PEG molar ratio of 48.5:1.5, accumulated in higher amounts in most tissues relative to the other molar ratios screened (Fig. S1†). Higher cholesterol content may improve LNP stability in circulation and subsequently increase the accumulation of these LNPs in most tissues.35 Additionally, LNPs formulated with a PEG molecular weight of 1000 Da, the lowest PEG molecular weight screened in this study, accumulated in higher amounts relative to the higher molecular weights screened (Fig. S2†). This is consistent with previous reports where higher molecular weight PEGs reduced cellular uptake by inhibiting interactions with target cell surfaces.36,37
Since the delivery of specific LNP formulations across different tissues cannot be compared using deep sequencing, we used quantitative polymerase chain reaction (qPCR) as a method of normalizing deep sequencing counts. qPCR was used to measure the total amount of b-DNAs in 5 nanograms of extracted DNA from all tissues analyzed. Sequencing counts were normalized by multiplying the existing ratio of counts across tissues for a specific LNP formulation by the ratio of total b-DNAs across all tissues as determined by qPCR (Fig. 2). A large majority of LNPs accumulated in either the liver or spleen, which is consistent with several previous reports.24–26,38,39
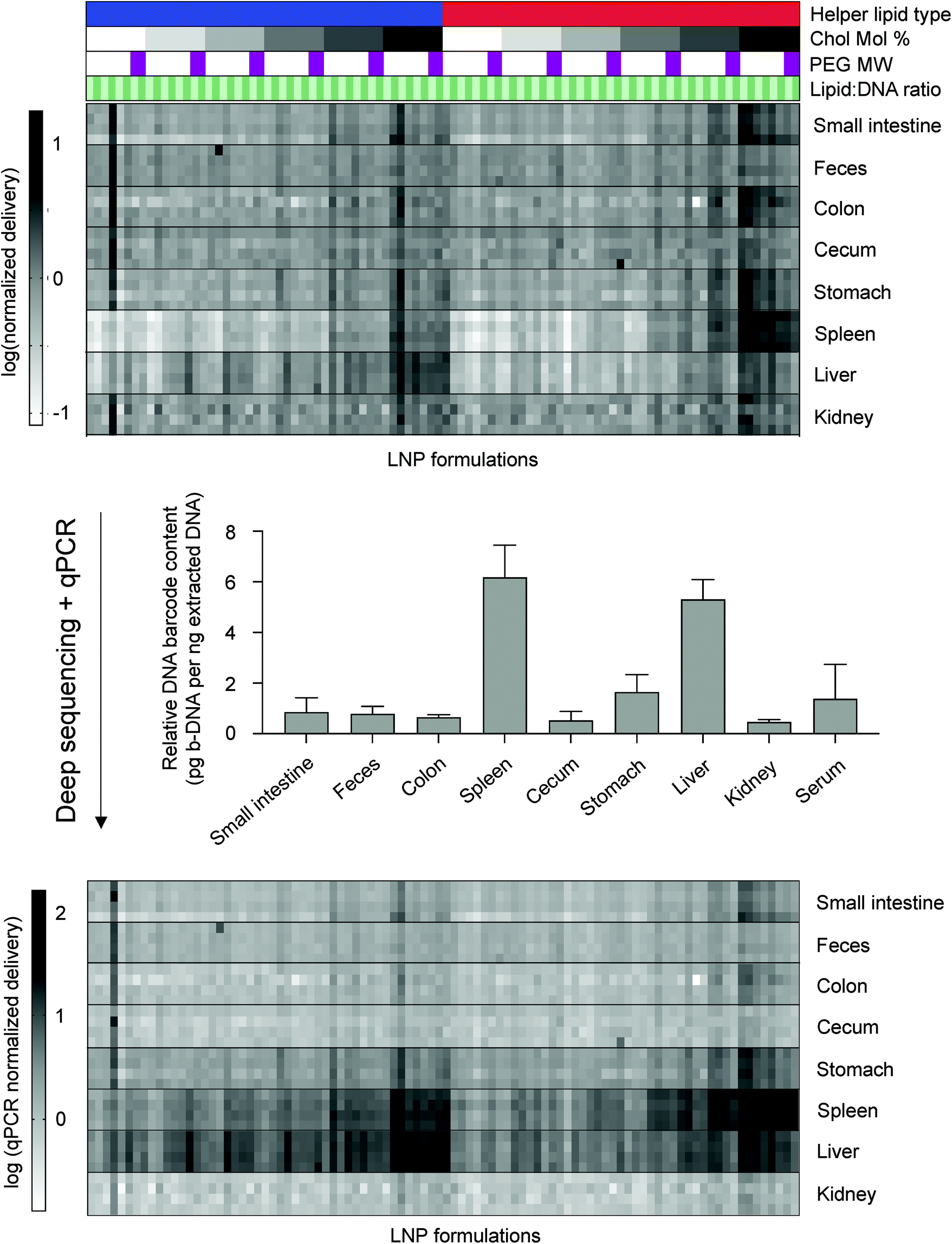 | ||
| Fig. 2 Combining deep sequencing-based accumulation measurements with quantitative polymerase chain reaction (qPCR) enables LNP delivery to be directly compared between different tissues. Along with a naked b-DNA, 96 LNP formulations were pooled together and injected via tail vein into C57BL/6 mice (N = 4). Tissues were isolated six hours post injection and extracted DNA was deep sequenced. A heatmap representing accumulation of LNPs to different tissues was generated. Darker clusters represent higher accumulation of a b-DNA in a particular tissue sample. LNP formulations are described above the heatmap. Helper lipid: Blue = DOPE, Red = DSPC. Ionizable lipid:b-DNA weight ratio: Light green = 5:1, Dark green = 10:1. PEG molecular weight: Light purple to dark purple represents increasing values from 1000, 2000, 3000, to 5000 Da. Cholesterol to lipid-PEG molar ratio: White to black represents increases in the molar percentage of cholesterol incorporated in LNPs from 1.5%, 10%, 20%, 30%, 40%, to 48.5%. Within the first heatmap, the accumulation of different LNPs within the same organ can be compared, but this comparison is not possible across organs. 5 nanograms of extracted DNA from various tissue samples was measured and total b-DNA in each tissue sample was amplified by qPCR. Relative amounts of b-DNA across tissue samples was then used to construct a second heatmap that allows for comparison across tissue samples. | ||
We then sought to further analyze the differences in accumulation in the liver and spleen of LNPs formulated with the two helper lipids used in this study. All LNPs formulated with DOPE were then compared to all LNPs formulated with DSPC, collectively, by performing a paired t-test. In the spleen, DSPC-containing LNPs accumulated in higher amounts than the DOPE-containing LNPs (**P = 0.0027) (Fig. 3A). The opposite was true for LNP accumulation in the liver, as DOPE-containing LNPs accumulated in higher amounts compared to DSPC-containing LNPs (****P < 0.0001) (Fig. 3B). To further explore individual differences, delivery to the liver and spleen of individual LNP pairs that only differed in the identity of the helper lipid were compared. In the spleen, LNPs formulated with DSPC accumulated in increased amounts over LNPs formulated with DOPE (Fig. 3C). In the liver, most of the LNP pairs demonstrated that incorporation of DOPE into the LNP formulation improved accumulation over LNPs formulated with DSPC (Fig. 3D). Although LNPs are widely used for nucleic acid delivery to the liver, several research reports have demonstrated the effectiveness of DSPC-containing LNPs for delivery to the spleen.26,40 This is likely, in part, due to LNP uptake by multiple myeloid cell types in the spleen, including macrophages, splenic reservoir monocytes, and dendritic cells.41 Delivery to cells in the spleen is especially important for the development of vaccines and immunotherapies.26,30,42,43 Further, a recent report observed that LNPs formulated with DOPE improved mRNA transfection in the liver over LNPs formulated with DSPC.24 Collectively, these data support our findings regarding the accumulation differences between DSPC-containing LNPs and DOPE-containing LNPs.
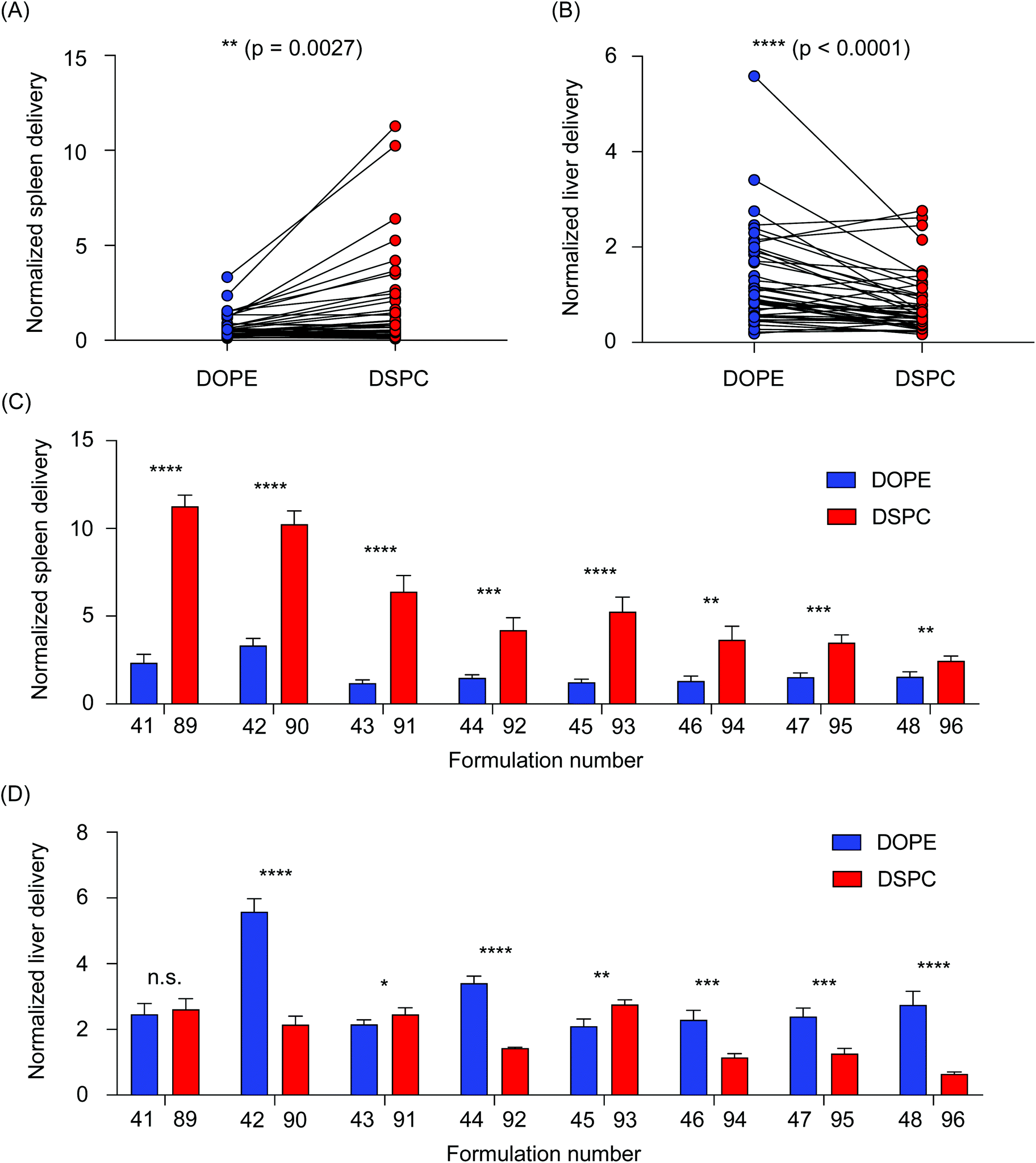 | ||
| Fig. 3 Helper lipid structure affects LNP delivery to the liver and spleen. (A and B) Accumulation of b-DNAs in the liver and spleen was plotted based on helper lipid incorporated into the LNPs. Connected data points represent LNPs with identical formulation parameters, with the exception of the type of helper lipid (DOPE or DSPC) incorporated into the formulation. (A) Overall, LNPs formulated with DSPC preferentially accumulated in the spleen (**p = 0.0027), (B) while LNPs formulated with DOPE preferentially accumulated in the liver (****p < 0.0001). (C and D) LNP pairings were identical in all parameters except for the type of the helper lipid (DOPE or DSPC) incorporated into the formulation. Bars in blue indicated LNPs that were formulated with DOPE. Bars in red indicated LNPs that were formulated with DSPC. (C) In the spleen, LNPs formulated with DSPC accumulated to a larger degree than their DOPE-containing counterparts. (D) In the liver, many LNPs formulated with DOPE accumulated to a larger degree than their DSPC-containing counterparts. Data was plotted as mean ± SD. N.S. denotes not significant, *P < 0.05, **P < 0.005, ***P < 0.001, ****P < 0.0001 by (A and B) Wilcoxon signed-rank test and (C and D) t-test. | ||
Given the differences in liver and spleen accumulation determined as a function of the helper lipid utilized in LNP formulations, we then assessed whether these helper lipids affect protein adsorption to LNPs. Systemically administered LNPs adsorb a variety of serum proteins onto their surface in vivo, which lends to the formation of the protein corona that influences cellular uptake and biodistribution.44,45 Of these serum proteins, ApoE has been largely implicated in the clearance and endogenous targeting of LNPs to the liver.46 A study demonstrated that pretreatment of mice with an siRNA knockdown of the LDLR impaired hepatic gene silencing by LNPs and that LNP-mediated gene silencing was impaired in ApoE−/− mice.46 Therefore, we hypothesized that differences existed in ApoE binding to LNPs selected from the in vivo screen that differed in terms of (i) their delivery to the liver and spleen, (ii) their helper lipid identity (DSPC vs. DOPE).
LNP 42, a top performing LNP that highly accumulated in the liver, was formulated using DOPE. LNP 90, a top performing LNP that highly accumulated in the spleen, was formulated using DSPC. The composition of these LNPs was identical aside from the type of helper lipid used (see Table S1†) and were therefore chosen for QCM-D experiments probing for LNP-ApoE interactions (Fig. 1B). QCM-D was chosen as it can monitor changes in the interfacial viscoelastic properties and mass-uptake in situ at a sufficiently high temporal resolution (∼1 s) detecting shifts in the frequency and dissipation of an oscillating quartz crystal.47 The technique is highly sensitive to changes in the mass and viscoelastic properties of materials adjacent to the sensor which cause shifts in the frequency and dissipation of an oscillating quartz crystal.47 Larger negative frequency shifts are typically associated with a higher coupled mass. The procedure is outlined schematically in Fig. 4A. For clarity, the frequency shifts for the third overtone were chosen for presentation (Fig. 4B); identical conclusions can be drawn from the frequency and dissipation shifts of all measured overtones. ApoE was adsorbed onto the Au-coated sensor by flowing ApoE solution for approximately 2 hours followed by rinsing in PBS buffer until the signal plateaued at a value of −57.5 ± 1.8 Hz. This shows that a nearly identical amount of ApoE is irreversibly adsorbed in both experiments.
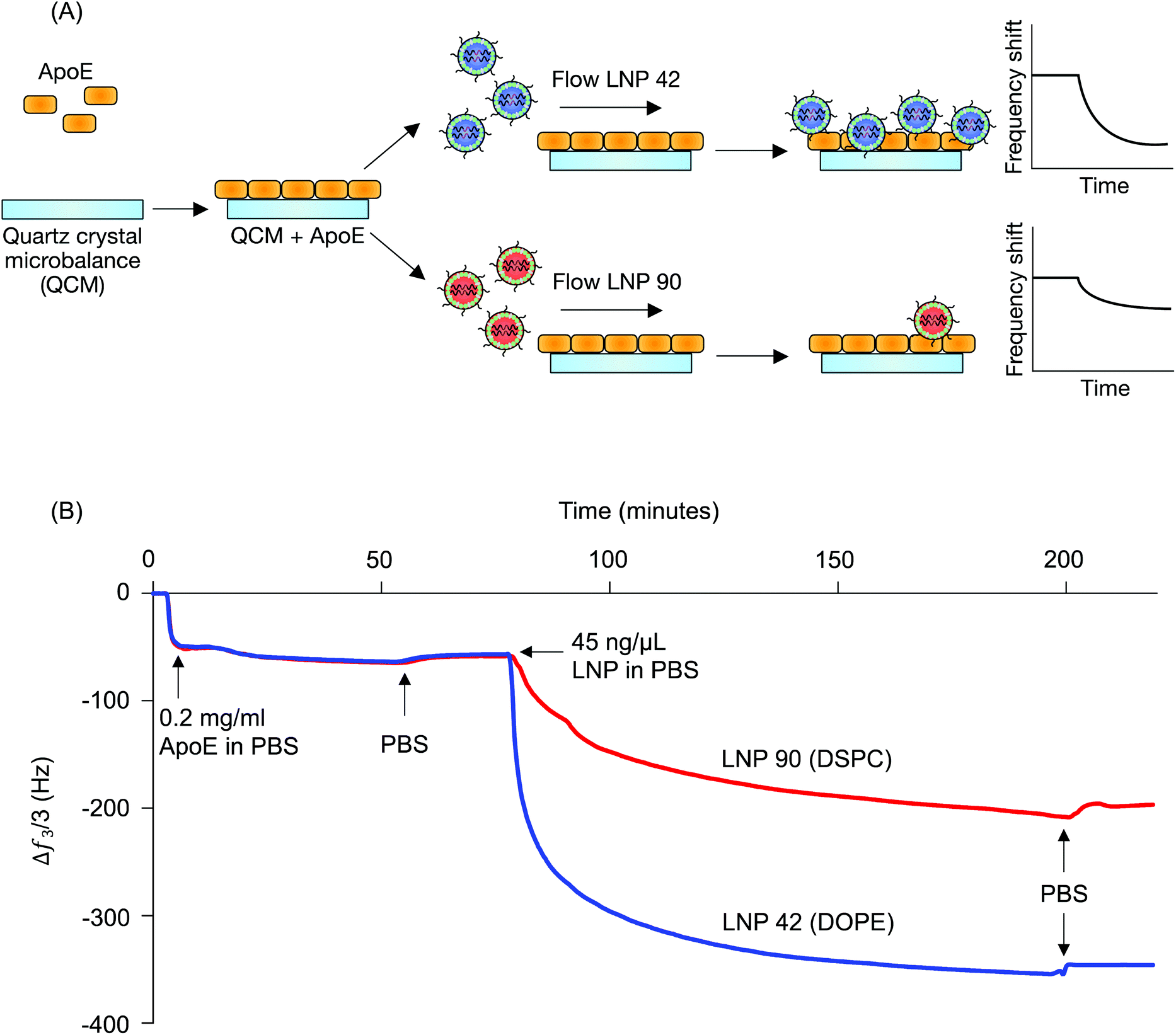 | ||
| Fig. 4 Helper lipid structure affects LNP interactions with apolipoprotein E. (A) Quartz crystal microbalance with dissipation monitoring was used to investigate interactions between ApoE and LNP formulations that differed only in terms of their helper lipid composition (DOPE vs. DSPC). ApoE layers were irreversibly adsorbed on Au-coated quartz crystals, followed by the target LNP solution (45 ng μl−1 b-DNA in LNPs). Adsorption of particles results in a negative frequency shift of the quartz crystal. (B) The frequency shift of the third overtone versus time shows a larger decrease in frequency when the DOPE-containing LNP (LNP 42) is flowed over the quartz crystal than the DSPC-containing LNP (LNP 90), indicating greater adsorption onto the ApoE layer. PBS rinses indicate that the particles irreversibly adsorbed to the ApoE layer. | ||
LNPs were then flowed over the adsorbed ApoE layers. Both LNPs adsorbed irreversibly on the ApoE layer with frequency shifts of −350 Hz and −200 Hz for LNP 42 and LNP 90, respectively, when rinsed with PBS. The larger magnitude of the frequency shift for LNP corresponds to increased adsorption of ApoE to the LNP formulated with DOPE over the LNP formulated with DSPC. Furthermore, the dissipation shift for a given frequency shift is smaller for LNP 42 than for LNP 90, which is qualitatively consistent with a more rigid coupling between ApoE and LNP 42 (Fig. S3A†). Control experiments for LNPs formulated with C12-200 interacting with the bare Au sensor show a larger negative frequency shift for the DSPC formulation (Fig. S4†). This is the reverse of the magnitudes observed in the presence of ApoE and is further evidence of stronger interactions between the DOPE-containing LNPs and ApoE.
To provide further support for the role of the helper lipid in LNP-ApoE interactions, additional experiments were performed with a different ionizable lipid (C14-4) previously used for mRNA delivery.48 In these experiments, the final frequency and dissipation shifts for both LNPs formulated with DOPE and DSPC were similar (Fig. S3B†). However, the peak rate of adsorption (i.e. dF/dt) was approximately 3 times larger for the DOPE-containing LNP (Fig. S5†). Both the DSPC and DOPE LNPs formulated with C14-4 displayed no difference in the rate and degree of adsorption when interacting with a bare Au sensor (Fig. S4†). This suggests that the high peak rate of LNPs on the ApoE-coated sensor results from specific interactions between ApoE and the LNP formulated with DOPE. Thus, LNPs formulated with DOPE show preferential interaction with ApoE independent of the choice of ionizable lipid. Increased membrane fluidity has been previously shown to enhance protein adsorption.49,50 The helper lipid DOPE is an unsaturated lipid and may therefore be expected to increase the membrane fluidity of LNPs relative to the saturated DSPC, which could account for the observed stronger interactions with ApoE. Given ApoE is implicated in LNP uptake by the liver, the QCM-D results coincide with our previous observation concerning higher accumulation of LNPs formulated with DOPE in the liver over LNPs formulated with DSPC.21,51
Next, we sought to validate the observed biodistribution differences between LNPs 42 and 90 by formulating these LNPs to encapsulate a fluorescently labeled Cy3-siRNA. We intravenously injected these LNPs into two separate groups of mice (N = 3 mice per group) and isolated spleens six hours post-injection. We quantified the fluorescence intensity of dissected spleens by rinsing with PBS and imaging with an in vivo imaging system (IVIS) (Fig. 5A). Following our previous analysis, the DSPC-containing LNP accumulated to a larger degree in the spleens of these mice, demonstrated by higher radiant efficiency, compared to the spleens isolated from mice treated with the DOPE-containing LNP (**P = 0.0019) (Fig. 5B and C). Due to the strong autofluorescence of liver tissue, liver images for Cy3-siRNA accumulation were difficult to acquire and were not included. These experiments do not inform functional delivery of nucleic acids, so we formulated LNPs 42 and 90 to encapsulate an mRNA encoding for firefly luciferase to better evaluate the relationship between biodistribution and efficacy.
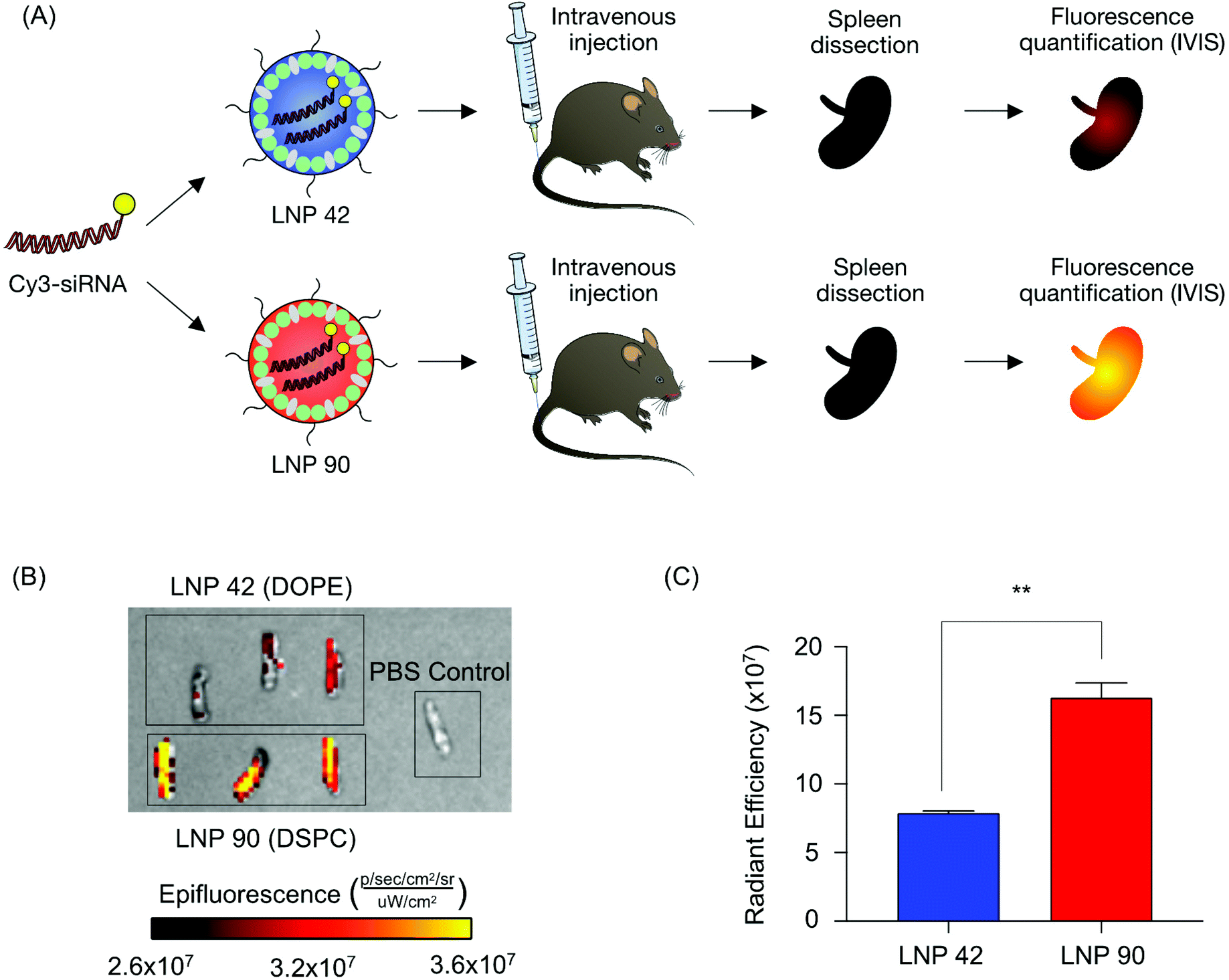 | ||
| Fig. 5 Validation of helper lipid effects on LNP-mediated delivery of fluorescently labeled siRNA to the spleen. (A) LNP 42 and LNP 90 were formulated by pipette mixing to encapsulate a fluorescent Cy3-siRNA. Both LNPs were formulated at molar percentages of 48.5% cholesterol, 1.5% lipid-PEG1000, 40% ionizable lipid, and 10% helper lipid at an ionizable lipid:b-DNA weight ratio of 10:1. These LNPs only differed in the identity of their helper lipid; LNP 42 was formulated with DOPE and LNP 90 was formulated with DSPC. C57BL/6 mice were intravenously injected with either LNP 42 or LNP 90 (5 μg siRNA per injection). Six hours post injection, spleens were isolated from mice, and their mean fluorescence intensity was measured by IVIS imaging. N = 3 mice per group. (B) Representative images of fluorescence detection in spleens from mice treated with either LNP 42 or LNP 90. (C) Fluorescence intensity in the spleens of injected mice was quantified. (**P = 0.0019). Data was plotted as mean ± SD. **P < 0.005 by t-test. | ||
LNP 42 and LNP 90 were formulated with a microfluidic device to encapsulate mRNA encoding for firefly luciferase. LNPs were intravenously administered to two separate groups of C57BL/6 mice (N = 4) via tail vein injection. Total luminescent flux was quantified 6 hours post-injection using an IVIS Spectrum imaging system. Data was acquired from live whole mice before the livers and spleens were isolated and imaged (Fig. S6† and Fig. 6). Representative images for N = 2 mice suggest a higher luminescence signal from the livers of mice injected with LNP 42 and LNP 90 (Fig. 6A). Quantification of the bioluminescence data demonstrated a three-fold increase in liver transfection when switching from LNP 90 (containing DSPC) to LNP 42 (containing DOPE) (***P = 0.0001) (Fig. 6B). However, differences between the LNPs delivering cargo to the spleen was less apparent, in part, due to the overall weak luminescent signal in the spleen (P = 0.1493) (Fig. 6C). We sought to quantify total luciferase mRNA abundance in the liver and spleen by reverse transcriptase qPCR (RT-qPCR) for comparison with our previous biodistribution data. Relative mRNA abundance was normalized to the housekeeping gene, GAPDH. In the liver, switching the helper phospholipid from DSPC to DOPE resulted in a two-fold increase in luciferase mRNA abundance (*P = 0.0263) (Fig. 6D). In the spleen, switching the helper phospholipid from DOPE to DSPC resulted in a five-fold increase in luciferase mRNA abundance (***P = 0.0003) (Fig. 6E).
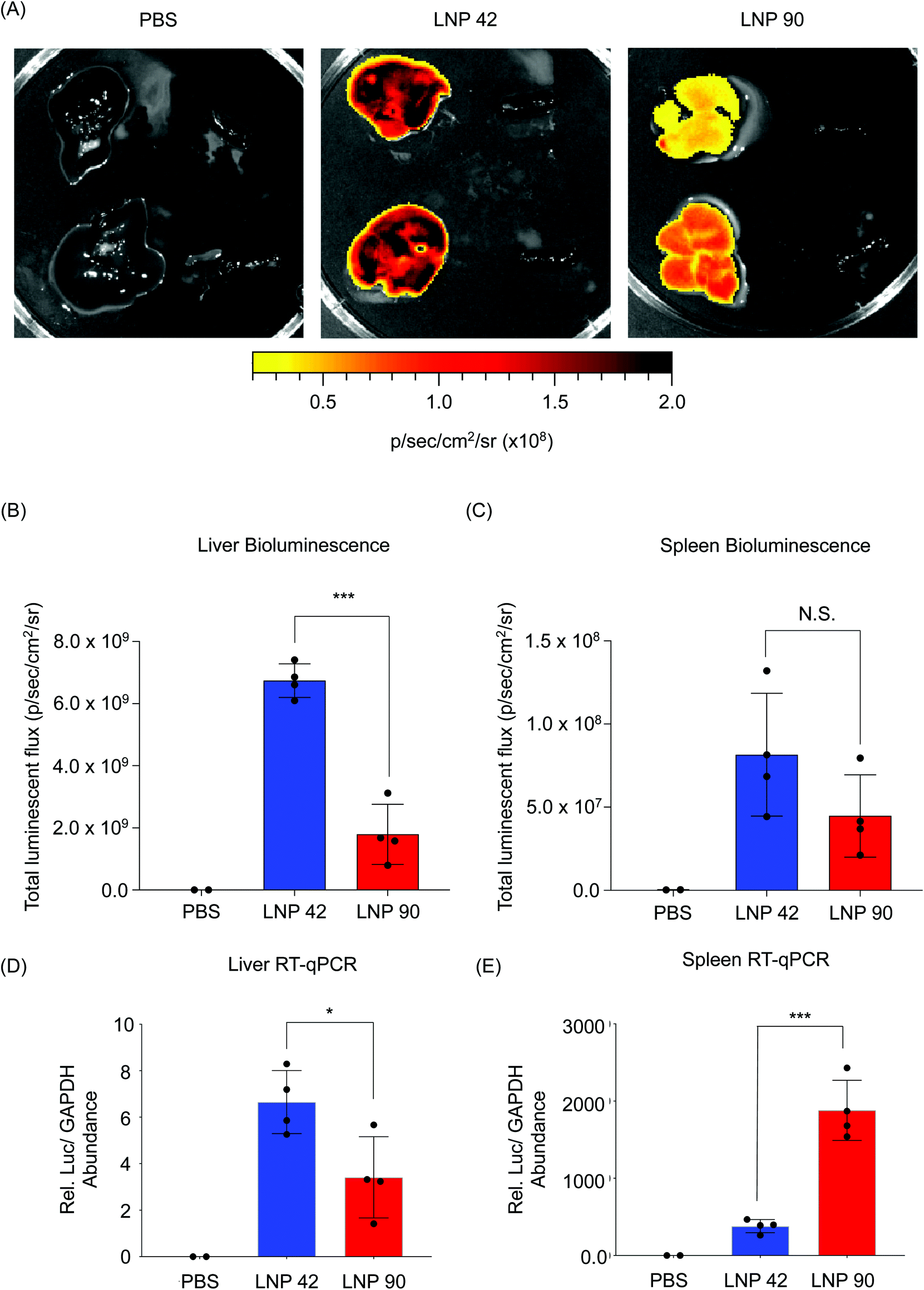 | ||
| Fig. 6 LNPs formulated with mRNA encoding for firefly luciferase provides insight into the relationship between biodistribution and efficacy. (A) LNP 42 and LNP 90 were formulated by microfluidic mixing to encapsulate an mRNA encoding for firefly luciferase. C57BL/6 mice were intravenously injected with either LNP 42 or LNP 90 at 0.2 mg kg−1 mRNA via tail vein. Total luminescent flux was quantified 6 hours post-injection. N = 4 mice per group. N = 2 mice per PBS control group. Representative images for N = 2 mice are shown. Within each image, isolated livers (left) were imaged next to isolated spleens (right). (B and C) Total luminescent flux from the isolated (B) livers (***P = 0.0001) and (C) spleens (P = 0.1493) was quantified and plotted. (D and E) Reverse transcriptase qPCR (RT-qPCR) was performed to quantify the amounts of luciferase mRNA present in each group. Relative luciferase mRNA abundance was calculated by CT values that were normalized to the housekeeping gene, GAPDH. (D) In the liver, mice injected with LNP 42 had approximately 2 times the amount of mRNA as mice injected with LNP 90. (*P = 0.0263). (E) In the spleen, mice injected with LNP 90 had approximately 5 times the amount of mRNA as mice injected with LNP 42. (***P = 0.0003). Data was plotted as mean ± SD. *P < 0.05 and ***P < 0.001 by t-test. | ||
Improving organ-specific delivery to certain tissues of interest, while decreasing delivery to off-target tissues, has been challenging in the design of LNPs. Overall, DOPE and DSPC are two commonly used helper phospholipids for LNP formulation.26,52,53 After intravenous injection, many LNPs reach their target liver tissue, but a large number of LNPs also accumulate in the spleen.26,40 Our results suggest that by switching the helper phospholipid from DSPC to DOPE, LNP delivery to the spleen can be decreased two-fold for siRNA-LNPs and five-fold for mRNA-LNPs (Fig. 5B and 6E). Additionally, DOPE-containing LNPs improved mRNA transfection in the liver by over three-fold with a two-fold improvement in overall mRNA delivery to the liver (Fig. 6B and D). mRNA delivery to the spleen is considered to be an attractive strategy for the development of vaccines and immunotherapies due to the high number of immune cells that reside in the spleen.30 Although LNP 90 accumulated to a very high degree in the spleen, this did not translate to functional mRNA transfection. This may be attributed to the fact that immune cells are notably difficult to transfect.54,55 Therefore, future efforts aim to improve spleen-specific functional nucleic acid delivery.
3. Conclusion
Overall, our work aimed to probe structure–function relationships of intravenously administered LNPs delivered to the liver and spleen and investigate their protein adsorption properties, using high-throughput in vivo screening and QCM-D, respectively. Through high-throughput screening, several LNPs formulated with a low molecular weight lipid-PEG and a high cholesterol to lipid-PEG molar ratio accumulated in higher amounts in the analyzed tissues. Further, we identified LNP formulations that differed only in their helper lipid composition, but accumulated in different amounts in the liver and spleen. Because ApoE adsorption has been implicated in endogenous targeting of LNPs to the liver, we investigated how altering the helper lipid component in LNPs influenced interactions with ApoE.21 Using QCM-D, it was shown that LNPs formulated with DOPE had stronger interactions with ApoE and accumulated in increased amounts in the livers of injected mice compared to LNPs formulated with DSPC, which had weaker interactions with ApoE and accumulated to a greater degree in the spleens of injected mice. Improving our understanding of how key LNP components interact with ApoE and other serum proteins will offer insight into future design of new delivery materials for nucleic acid therapeutics.4. Materials and methods
4.1. DNA barcode (b-DNA) design
b-DNA design parameters followed a previous report.33 b-DNAs were single-stranded and consisted of 61 nucleotides with five consecutive phosphorothioate bonds at each end. The barcode region comprised of 10 nucleotides in the center of the oligonucleotide. An additional 10 random nucleotides were included at the 3′ region of the barcode. Both the 5′ and 3′ ends of each b-DNA were conserved and contained priming sites for Illumina adapters. A full list of b-DNA sequences can be found in Table S3.† All oligonucleotides were purchased from Integrated DNA Technologies and were purified via standard desalting procedures.4.2. Ionizable lipid synthesis and characterization
C12-200 and C14-4 were synthesized as previously described.34,48 Epoxide-terminated alkyl chains (Sigma Aldrich) were reacted with polyamine cores by Michael addition chemistry. The reagents were reacted at a 7-molar excess of alkyl chains at 80 °C for 48 hours. The resulting lipids were mixed with Celite® 545 (Sigma Aldrich) and all solvent was evaporated using a Rotavapor R-300 (BÜCHI). The ionizable lipids were purified by using a CombiFlash Nextgen 300+ chromatography system (Teledyne ISCO) and target fractions were identified by liquid chromatography-mass spectrometry.4.3. Lipid nanoparticle (LNP) formulation and characterization
b-DNA LNPs and Cy3-siRNA LNPs were formulated by pipette mixing at an aqueous:organic ratio of 3:1. The lipid-containing organic phase was mixed into the nucleic acid-containing aqueous phase. The organic phase was prepared by dissolving an ionizable lipid, helper lipid, cholesterol, and lipid-PEG in ethanol at the molar ratios found in Table S1.† The aqueous phase was prepared using 10 mM pH 4.0 citrate buffer containing either a unique b-DNA, Cy3-siRNA (Sigma Aldrich Universal neg control), or luciferase mRNA at a weight ratio of 10:1 or 5:1 (ionizable lipid:nucleic acid). mRNA-LNPs were formulated by microfluidic mixing at an aqueous:organic flow rate ratio of 3:1. The formulated LNPs were then dialyzed against PBS in a 96-well microdialysis plate (10000 MWCO, Thermo Fisher Scientific, 88260) at room temperature for 2 hours. C12-200 and C14-4 were synthesized in-house. Cholesterol was purchased from Sigma Aldrich (C8667). The helper lipids 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE, 850725P) and 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC, 850365P) were purchased from Avanti Polar Lipids. The lipid-PEGs used in this study, 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-1000] (ammonium salt) (C14-PEG1000, 880710P), 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (C14-PEG2000, 880150P), 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-3000] (ammonium salt) (C14-PEG3000, 880310P), and 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-5000] (ammonium salt) (C14-PEG5000, 880210P), were also purchased from Avanti Polar Lipids. DNA, siRNA, and mRNA concentration in LNP formulations was determined using a NanoDrop Spectrophotometer (Thermo Fisher Scientific). Mean hydrodynamic diameter and polydispersity (PDI) of LNPs were measured using a Zetasizer Nano ZS instrument (Malvern Panalytical).
4.4. Animal experiments
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals at the University of Pennsylvania and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Pennsylvania. To evaluate LNP biodistribution by deep sequencing, 8-week-old female C57BL/6 mice (Jackson Laboratory, 18–21 g) were intravenously injected with a pool of different b-DNA LNP formulations via the tail vein, along with a naked b-DNA as a negative control. The mice were injected at a dose of 0.4 μg per each b-DNA. For the initial b-DNA delivery screen, tissue samples were isolated 6 hours post-injection, frozen in liquid nitrogen, mechanically ground into powder using a Geno/Grinder (SPEX Sample Prep) and stored at −80 °C until further analysis. In the Cy3-siRNA fluorescence experiment, 8-week-old female C57BL/6 mice (Jackson Laboratory, 18–21 g) were intravenously injected with 5 μg of fluorescently labeled siRNA formulated into LNPs. Spleens were isolated 6 hours post-injection, rinsed in 1× PBS, and imaged by IVIS. To evaluate functional luciferase-mRNA delivery by LNPs, male C57BL/6 mice aged 6–8 weeks (Jackson Laboratory, 23–30 g) were administered a single 0.1 mL intravenous dose of formulated LNPs encapsulating luciferase mRNA in PBS pH 7.4 (Gibco) via tail vein injection. Bioluminescence imaging was performed with an IVIS Spectrum Imaging system (Caliper Life Sciences). Mice were administered D-luciferin (PerkinElmer) at a dose of 150 mg kg−1 (IP) and anesthetized 5 minutes later by ketamine/xylazine (IP). Live animal and organ image acquisition occurred 20 minutes and 30 minutes after D-luciferin administration, respectively. Exposure times for whole animal and organs were 30 seconds and 1 second, respectively. Bioluminescence values were quantified by measuring photon flux in the region of interest where bioluminescence signal emanated using the Living IMAGE Software provided by Caliper.4.5. Luciferase RT-qPCR
RNA was extracted from the liver and spleen using Trizol (Invitrogen) and treated with TURBO DNA-free Kit (ThermoFisher). cDNA was synthesized using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) with random hexamer priming. qPCR was performed on an ABI7500 thermocycler using Taqman assays for rodent GAPDH (Applied Biosystems cat# 4308313) and firefly Luciferase (F: 5′-ACCATCGCCCTGATCATGA; R: 5′-GCTACGCCCTTGGGCAAT; Probe: FAM-CAGTAGTGGCAGTACCG-MGB). Relative luciferase mRNA abundance was calculated by CT values that were normalized to the housekeeping gene, GAPDH.4.6. b-DNA extraction and library preparation
In order to extract DNA from frozen tissues, approximately 30 mg of ground tissue was resuspended in a lysis buffer that contained 100 mM Tris-HCl (Fisher Scientific, 50155887), 5 mM EDTA (Fisher Scientific, 50997738), 0.2% SDS (Fisher Scientific, 507513793), 200 mM NaCl (Fisher Scientific, S318100), and 0.2 mg mL−1 proteinase K (Thermo Fisher Scientific, PI17916).56 Extracted DNA was further purified by Zymo Oligo Clean and Concentrator columns (Zymo Research, D4060) according to the manufacturer's protocol. b-DNA amplification was conducted by PCR using the following reagents: 5 μL 5× HF Phusion buffer, 0.5 μL 10 mM dNTPs, 0.25 μL Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific, F530S), 1.18 μL extracted oligonucleotides, 1 μL 5 μM forward primer, 1 μL 5 μM full length reverse primer, 2 μL DMSO, and 15.25 μL H2O. The following PCR cycling conditions were used: 98 °C for 12 seconds, 67 °C for 22 seconds, and 72 °C for 28 seconds, for a total of 35 cycles. Primer sequences shown below were added in the above mixture:Forward primer (Universal):
5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT
Full length reverse primer:
5′-CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT
XXXXXXXX: denotes 8 nucleotide organ barcode. The full list of reverse primers can be found in Table S4.†
PCR products were run by gel electrophoresis on 3% agarose (Universal Medical, IB70060) in Tris-acetate-EDTA buffer (Fisher Scientific, 24710030). Amplified b-DNA (144 bp) was excised from the gel, pooled, and purified by Zymo Gel Extraction columns (Zymo Research, D4001) according to the manufacturer's protocol. The purified products were stored at −20 °C until deep sequencing.
4.7. Deep sequencing and barcode delivery quantification
All deep-sequencing runs were performed using multiplexed runs on Illumina MiSeq (Illumina). PCR product pools were quantitated using the KAPA Library Quantification Kit for next-generation sequencing. PCR product pools were loaded onto flow cells at 4 nM concentration.b-DNA delivery of a specific barcode to a certain tissue was calculated following these 3 steps: (i) dividing the number of sequencing reads of one barcode delivered by a single LNP formulation by the total amount of reads from all barcodes delivered by all LNPs in a specific tissue; (ii) dividing the number of sequencing reads of the same barcode (utilized in (i)) by the total amount of reads from all barcodes of all LNPs in the non-injected LNP pool and (iii) dividing the results from (i) by the results from (ii).
4.8. Quantitative-polymerase chain reaction (qPCR)
Primer sequences were designed using Primer-BLAST (National Institute of Health). Both forward primer and reverse primer sequences were designed to bind the conserved region of b-DNA. Primer sequences are shown below:Forward primer:
5′-AGACGTGTGCTCTTCCGAT
Reverse primer:
5′-ACACGACGCTCTTCCGAT
qPCR was performed using Fast SYBR™ Green Master Mix (Thermo Fisher Scientific, 4385612). qPCR master solution was prepared as follows: 10 μl Fast SYBR Green Master Mix (2×), 1 μL 5 μM forward primer, 1 μL 5 μM reverse primer, 3 μL molecular biology grade water. Subsequently, 5 μL of 1 ng μL−1 extracted DNA from each tissue sample were mixed with 15 μL qPCR master solution, reaching a final volume of 20 μL. To make qPCR reaction solution for a standard curve, 5 μL b-DNA of known concentrations were made by serial dilution and mixed with 15 μL of the same qPCR master solution. The cycling conditions were carried out following the manufacturer's protocol: after 20 seconds of denaturation at 95 °C, 50 cycles with 2-segment amplification were performed consisting of 3 seconds at 95 °C for denaturation and 30 seconds at 60 °C for polymerase elongation.
After the qPCR reaction, a calibration curve was obtained by plotting the logarithm of the concentrations of b-DNA standard and their corresponding value of cycle threshold (Ct). This calibration curve was then used to determine the b-DNA concentration of tissue samples. For all the tissue samples, 5 ng of total extracted DNA was added for each reaction, which allows the amount of b-DNA (based on qPCR results) from different tissue samples to be directly compared. Therefore, the amount of b-DNA in each tissue sample was normalized to picogram b-DNA per ng total extracted DNA.
4.9. Quartz crystal microbalance with dissipation monitoring (QCM-D)
Au coated QCM-D sensors (QSX301, Biolin Scientific) with a resonance frequency of 4.95 MHz were cleaned via 10 min UV-Ozone, followed by 5 min base piranha treatment (TL1 protocol). The sensors were then rinsed thoroughly in MilliQ water, dried with dry N2, and followed by a final 10 min of UV-Ozone treatment. Shifts in the resonance frequency (ΔF) and dissipation (ΔD) for odd overtones (n = 1,3,…,13) were monitored using an E4 QCM-D (Q-Sense Inc., Gothenburg, Sweden). All shifts are reported relative values for the sensors equilibrated in PBS buffer. All solutions were introduced using via a peristaltic pump (IPC N-4, Isamatec) at a nominal flowrate of 50 μL min−1.ApoE was deposited on the sensor by flowing 0.2 mg mL−1 ApoE in PBS over the crystals. After ∼10 min, intermittent pumping (50 μL or ∼1.25 cell volumes of continuous pumping followed by a 4–9 minutes pause) was used to preserve material. After 2 h of adsorption, the ApoE layers were rinsed with PBS buffer at 50 μL min−1 until equilibrated (∼20 min). Measured shifts were nearly identical for each experiment (e.g. ΔF3/3 = −57.5 ± 1.8 Hz), indicating that comparisons of subsequent measurements of ApoE binding are valid.
Interactions between LNPs selected from the library screen and ApoE were tested by flowing 45 ng μL−1 LNP in PBS over the PBS-rinsed ApoE layer. Again, intermittent pumping was performed after ∼10 min of continuous pumping. Here, 50 μL was pumped over one minute, followed by a 4 minutes pause. PBS buffer was introduced using continuous pumping after 2 h of adsorption of the nanoparticles onto the ApoE layer.
Control measurements were performed by flowing nanoparticle solutions over clean Au sensors equilibrated in PBS buffer until the frequency change had plateaued before subsequent rinsing in PBS buffer. The above procedure was repeated with LNPs formulated with C14-4, but a 9 minutes pause was used instead after every 50 μL that was pumped.
Conflicts of interest
J. M. W. is a paid advisor to and holds equity in Scout Bio and Passage Bio; he holds equity in Surmount Bio; he also has sponsored research agreements with Amicus Therapeutics, Biogen, Elaaj Bio, Janssen, Moderna, Passage Bio, Scout Bio, Surmount Bio, and Ultragenyx, which are licensees of Penn technology. M. J. M. is an inventor on a patent related to this work filed by the Trustees of the University of Pennsylvania (PCT/US20/56252). J. M. W. and L. W. are inventors on patents that have been licensed to various biopharmaceutical companies and for which they may receive payments. The remaining authors declare that they have no conflicts of interest.Acknowledgements
M. M. B. is supported by a Tau Beta Pi Graduate Research Fellowship and an NIH Training in HIV Pathogenesis T32 Program (T32 AI007632). S. J. S. is supported by a National Science Foundation (NSF) Graduate Research Fellowship (DGE 1845298). M. J. M. acknowledges support from a Burroughs Wellcome Fund Career Award at the Scientific Interface (CASI), a US National Institutes of Health (NIH) Director's New Innovator Award (DP2 TR002776), a grant from the American Cancer Society (129784-IRG-16-188-38-IRG), the National Institutes of Health (NCI R01 CA241661, NCI R37 CA244911 and NIDDK R01 DK123049), an Abramson Cancer Center (ACC)–School of Engineering and Applied Sciences (SEAS) Discovery Grant (P30 CA016520), and a 2018 AACR-Bayer Innovation and Discovery Grant, Grant Number 18-80-44-MITC. C. C. W., L. W. and J. M. W. acknowledge support from a private funding source.References
- X. Shen and D. R. Corey, Chemistry, Mechanism and Clinical Status of Antisense Oligonucleotides and Duplex RNAs, Nucleic Acids Res., 2018, 46(4), 1584–1600, DOI:10.1093/nar/gkx1239.
- K. A. Whitehead, R. Langer and D. G. Anderson, Knocking down Barriers: Advances in SiRNA Delivery, Nat. Rev. Drug Discovery, 2009, 8(2), 129–138, DOI:10.1038/nrd2742.
- R. L. Ball, P. Bajaj and K. A. Whitehead, Oral Delivery of SiRNA Lipid Nanoparticles: Fate in the GI Tract, Sci. Rep., 2018, 8(1), 1–12, DOI:10.1038/s41598-018-20632-6.
- S. Ramaswamy, N. Tonnu, K. Tachikawa, P. Limphong, J. B. Vega, P. P. Karmali, P. Chivukula and I. M. Verma, Systemic Delivery of Factor IX Messenger RNA for Protein Replacement Therapy, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(10), E1941–E1950, DOI:10.1073/pnas.1619653114.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Selective Organ Targeting (SORT) Nanoparticles for Tissue-Specific MRNA Delivery and CRISPR–Cas Gene Editing, Nat. Nanotechnol., 2020, 15(4), 313–320, DOI:10.1038/s41565-020-0669-6.
- J. Watts and D. Corey, Gene Silencing by SiRNAs and Antisense Oligonucleotides in the Laboratory and the Clinic, J. Pathol., 2012, 226(2), 365–379, DOI:10.1002/path.2993.Gene.
- K. A. Hajj and K. A. Whitehead, Tools for Translation: Non-Viral Materials for Therapeutic MRNA Delivery, Nat. Rev. Mater., 2017, 2, 1–17, DOI:10.1038/natrevmats.2017.56.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Delivery Materials for siRNA Therapeutics, Nat. Mater., 2013, 12(11), 967–977, DOI:10.1038/nmat3765.
- I. Lostalé-Seijo and J. Montenegro, Synthetic Materials at the Forefront of Gene Delivery, Nat. Rev. Chem., 2018, 2(10), 258–277, DOI:10.1038/s41570-018-0039-1.
- O. S. Fenton, K. N. Olafson, P. S. Pillai, M. J. Mitchell and R. Langer, Advances in Biomaterials for Drug Delivery, Adv. Mater., 2018, 30(29), 1–29, DOI:10.1002/adma.201705328.
- A. Schroeder, C. G. Levins, C. Cortez, R. Langer and D. G. Anderson, Lipid-Based Nanotherapeutics for SiRNA Delivery, J. Intern. Med., 2010, 267(1), 9–21 CrossRef CAS.
- K. A. Whitehead, J. R. Dorkin, A. J. Vegas, P. H. Chang, J. Matthews, O. S. Fenton, Y. Zhang, K. T. Olejnik, V. Yesilyurt, D. Chen, S. Barros, B. Klebanov, T. Novobrantseva, R. Langer and D. G. Anderson, Degradable Lipid Nanoparticles with Predictable In Vivo SiRNA Delivery Activity, Nat. Commun., 2014, 5(1), 4277, DOI:10.1038/ncomms5277.
- Y. Granot and D. Peer, Delivering the Right Message: Challenges and Opportunities in Lipid Nanoparticles-Mediated Modified MRNA Therapeutics—An Innate Immune System Standpoint, Semin. Immunol., 2017, 34(July), 68–77, DOI:10.1016/j.smim.2017.08.015.
- M. J. Mitchell, R. K. Jain and R. Langer, Engineering and Physical Sciences in Oncology: Challenges and Opportunities, Nat. Rev. Cancer, 2017, 17(11), 659–675 CrossRef CAS.
- A. J. Mukalel, R. S. Riley, R. Zhang and M. J. Mitchell, Nanoparticles for Nucleic Acid Delivery: Applications in Cancer Immunotherapy, Cancer Lett., 2019, 458(February), 102–112, DOI:10.1016/j.canlet.2019.04.040.
- S. J. Allison and J. Milner, RNA Interference by Single- and Double-Stranded SiRNA with a DNA Extension Containing a 3′ Nuclease-Resistant Mini-Hairpin Structure, Mol. Ther. – Nucleic Acids, 2013, 2(12), 1–8, DOI:10.1038/mtna.2013.66.
- X. Cheng and R. J. Lee, The Role of Helper Lipids in Lipid Nanoparticles (LNPs) Designed for Oligonucleotide Delivery, Adv. Drug Delivery Rev., 2016, 99, 129–137, DOI:10.1016/j.addr.2016.01.022.
- K. Garber, Alnylam Launches Era of RNAi Drugs, Nat. Biotechnol., 2018, 36(9), 777–778, DOI:10.1038/nbt0918-777.
- S. Patel, N. Ashwanikumar, E. Robinson, Y. Xia, C. Mihai, J. P. Griffith, S. Hou, A. A. Esposito, T. Ketova, K. Welsher, J. L. Joyal, Ö. Almarsson and G. Sahay, Naturally-Occurring Cholesterol Analogues in Lipid Nanoparticles Induce Polymorphic Shape and Enhance Intracellular Delivery of MRNA, Nat. Commun., 2020, 11(1), 1–13, DOI:10.1038/s41467-020-14527-2.
- L. Miao, J. Lin, Y. Huang, L. Li, D. Delcassian, Y. Ge, Y. Shi and D. G. Anderson, Synergistic Lipid Compositions for Albumin Receptor Mediated Delivery of MRNA to the Liver, Nat. Commun., 2020, 11(1), 2424, DOI:10.1038/s41467-020-16248-y.
- A. Akinc, W. Querbes, S. De, J. Qin, M. Frank-Kamenetsky, K. N. Jayaprakash, M. Jayaraman, K. G. Rajeev, W. L. Cantley, J. R. Dorkin, J. S. Butler, L. Qin, T. Racie, A. Sprague, E. Fava, A. Zeigerer, M. J. Hope, M. Zerial, D. W. Sah, K. Fitzgerald, M. A. Tracy, M. Manoharan, V. Koteliansky, A. De Fougerolles and M. A. Maier, Targeted Delivery of RNAi Therapeutics with Endogenous and Exogenous Ligand-Based Mechanisms, Mol. Ther., 2010, 18(7), 1357–1364, DOI:10.1038/mt.2010.85.
- A. Chonn, S. C. Semple and P. R. Cullis, Association of Blood Proteins with Large Unilamellar Liposomes in Vivo, J. Biol. Chem., 1992, 267(26), 18759–18765 CrossRef CAS.
- J. A. Kulkarni, P. R. Cullis and R. Van Der Meel, Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility, Nucleic Acid Ther., 2018, 28(3), 146–157, DOI:10.1089/nat.2018.0721.
- K. J. Kauffman, J. R. Dorkin, J. H. Yang, M. W. Heartlein, F. Derosa, F. F. Mir, O. S. Fenton and D. G. Anderson, Optimization of Lipid Nanoparticle Formulations for MRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs, Nano Lett., 2015, 15(11), 7300–7306, DOI:10.1021/acs.nanolett.5b02497.
- B. Shi, E. Keough, A. Matter, K. Leander, S. Young, E. Carlini, A. B. Sachs, W. Tao, M. Abrams, B. Howell and L. Sepp-Lorenzino, Biodistribution of Small Interfering RNA at the Organ and Cellular Levels after Lipid Nanoparticle-Mediated Delivery, J. Histochem. Cytochem., 2011, 59(8), 727–740, DOI:10.1369/0022155411410885.
- T. I. Novobrantseva, A. Borodovsky, J. Wong, B. Klebanov, M. Zafari, K. Yucius, W. Querbes, P. Ge, V. M. Ruda, S. Milstein, L. Speciner, R. Duncan, S. Barros, G. Basha, P. Cullis, A. Akinc, J. S. Donahoe, K. N. Jayaprakash, M. Jayaraman, R. L. Bogorad, K. Love, K. Whitehead, C. Levins, M. Manoharan, F. K. Swirski, R. Weissleder, R. Langer, D. G. Anderson, A. De Fougerolles, M. Nahrendorf and V. Koteliansky, Systemic RNAi-Mediated Gene Silencing in Nonhuman Primate and Rodent Myeloid Cells, Mol. Ther. – Nucleic Acids, 2012, 1(1), e4, DOI:10.1038/mtna.2011.3.
- S. Nie, Understanding and Overcoming Major Barriers in Cancer Nanomedicine, Leadersh. Equ. Strateg. Individ. who are champions Child. youth, Fam, 2010, vol. 5(4), pp. 155–166, DOI:10.2217/nnm.10.23.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nanoparticle Uptake: The Phagocyte Problem, Nano Today, 2015, 10(4), 487–510, DOI:10.1016/j.nantod.2015.06.006.
- P. W. Bisso, S. Gaglione, P. P. G. Guimarães, M. J. Mitchell and R. Langer, Nanomaterial Interactions with Human Neutrophils, ACS Biomater. Sci. Eng., 2018, 4(12), 4255–4265, DOI:10.1021/acsbiomaterials.8b01062.
- L. M. Kranz, M. Diken, H. Haas, S. Kreiter, C. Loquai, K. C. Reuter, M. Meng, D. Fritz, F. Vascotto, H. Hefesha, C. Grunwitz, M. Vormehr, Y. Hüsemann, A. Selmi, A. N. Kuhn, J. Buck, E. Derhovanessian, R. Rae, S. Attig, J. Diekmann, R. A. Jabulowsky, S. Heesch, J. Hassel, P. Langguth, S. Grabbe, C. Huber, Ö. Türeci and U. Sahin, Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy, Nature, 2016, 534(7607), 396–401, DOI:10.1038/nature18300.
- M. T. Abrams, M. L. Koser, J. Seitzer, S. C. Williams, M. A. Dipietro, W. Wang, A. W. Shaw, X. Mao, V. Jadhav, J. P. Davide, P. A. Burke, A. B. Sachs, S. M. Stirdivant and L. Sepp-Lorenzino, Evaluation of Efficacy, Biodistribution, and Inflammation for a Potent SiRNA Nanoparticle: Effect of Dexamethasone Co-Treatment, Mol. Ther., 2010, 18(1), 171–180, DOI:10.1038/mt.2009.208.
- J. E. Dahlman, K. J. Kauffman, Y. Xing, T. E. Shaw, F. F. Mir, C. C. Dlott, R. Langer, D. G. Anderson and E. T. Wang, Barcoded Nanoparticles for High Throughput in Vivo Discovery of Targeted Therapeutics, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(8), 2060–2065, DOI:10.1073/pnas.1620874114.
- P. P. G. Guimaraes, R. Zhang, R. Spektor, M. Tan, A. Chung, M. M. Billingsley, R. El-Mayta, R. S. Riley, L. Wang, J. M. Wilson and M. J. Mitchell, Ionizable Lipid Nanoparticles Encapsulating Barcoded MRNA for Accelerated in Vivo Delivery Screening, J. Controlled Release, 2019, 316, 404–417, DOI:10.1016/j.jconrel.2019.10.028.
- K. T. Love, K. P. Mahon, C. G. Levins, K. A. Whitehead, W. Querbes, J. R. Dorkin, J. Qin, W. Cantley, L. L. Qin, T. Racie, M. Frank-Kamenetsky, K. N. Yip, R. Alvarez, D. W. Y. Sah, A. De Fougerolles, K. Fitzgerald, V. Koteliansky, A. Akinc, R. Langer and D. G. Anderson, Lipid-like Materials for Low-Dose, in Vivo Gene Silencing, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(5), 1864–1869, DOI:10.1073/pnas.0910603106.
- M. Ramezanpour, M. L. Schmidt, I. Bodnariuc, J. A. Kulkarni, S. S. W. Leung, P. R. Cullis, J. L. Thewalt and D. P. Tieleman, Ionizable Amino Lipid Interactions with POPC: Implications for Lipid Nanoparticle Function, Nanoscale, 2019, 11(30), 14141–14146, 10.1039/c9nr02297j.
- H. Hatakeyama, H. Akita and H. Harashima, The Polyethyleneglycol Dilemma: Advantage and Disadvantage of PEGylation of Liposomes for Systemic Genes and Nucleic Acids Delivery to Tumors, Biol. Pharm. Bull., 2013, 36(6), 892–899, DOI:10.1248/bpb.b13-00059.
- D. Pozzi, V. Colapicchioni, G. Caracciolo, S. Piovesana, A. L. Capriotti, S. Palchetti, S. De Grossi, A. Riccioli, H. Amenitsch and A. Laganà, Effect of Polyethyleneglycol (PEG) Chain Length on the Bio-Nano- Interactions between PEGylated Lipid Nanoparticles and Biological Fluids: From Nanostructure to Uptake in Cancer Cells, Nanoscale, 2014, 6(5), 2782–2792, 10.1039/c3nr05559k.
- O. S. Fenton, K. J. Kauffman, R. L. McClellan, E. A. Appel, J. R. Dorkin, M. W. Tibbitt, M. W. Heartlein, F. De Rosa, R. Langer and D. G. Anderson, Bioinspired Alkenyl Amino Alcohol Ionizable Lipid Materials for Highly Potent in Vivo MRNA Delivery, Adv. Mater., 2016, 28(15), 2939–2943, DOI:10.1002/adma.201505822.
- O. S. Fenton, K. J. Kauffman, J. C. Kaczmarek, R. L. McClellan, S. Jhunjhunwala, M. W. Tibbitt, M. D. Zeng, E. A. Appel, J. R. Dorkin, F. F. Mir, J. H. Yang, M. A. Oberli, M. W. Heartlein, F. DeRosa, R. Langer and D. G. Anderson, Synthesis and Biological Evaluation of Ionizable Lipid Materials for the In Vivo Delivery of Messenger RNA to B Lymphocytes, Adv. Mater., 2017, 29(33), 1–7, DOI:10.1002/adma.201606944.
- S. Sabnis, E. S. Kumarasinghe, T. Salerno, C. Mihai, T. Ketova, J. J. Senn, A. Lynn, A. Bulychev, I. McFadyen, J. Chan, Ö. Almarsson, M. G. Stanton and K. E. Benenato, A Novel Amino Lipid Series for MRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-Human Primates, Mol. Ther., 2018, 26(6), 1509–1519, DOI:10.1016/j.ymthe.2018.03.010.
- F. Leuschner, P. Dutta, R. Gorbatov, T. I. Novobrantseva, J. S. Donahoe, G. Courties, K. M. Lee, J. I. Kim, J. F. Markmann, B. Marinelli, P. Panizzi, W. W. Lee, Y. Iwamoto, S. Milstein, H. Epstein-Barash, W. Cantley, J. Wong, V. Cortez-Retamozo, A. Newton, K. Love, P. Libby, M. J. Pittet, F. K. Swirski, V. Koteliansky, R. Langer, R. Weissleder, D. G. Anderson and M. Nahrendorf, Therapeutic SiRNA Silencing in Inflammatory Monocytes in Mice, Nat. Biotechnol., 2011, 29(11), 1005–1010, DOI:10.1038/nbt.1989.
- H. Matsuo, N. Yoshimoto, M. Iijima, T. Niimi, J. Jung, S.-Y. Jeong, E. K. Choi, T. Sewaki, T. Arakawa and S. Kuroda, Engineered Hepatitis B Virus Surface Antigen L Protein Particles for in Vivo Active Targeting of Splenic Dendritic Cells, Int. J. Nanomed., 2012, 3341 CAS.
- D. Schmid, C. G. Park, C. A. Hartl, N. Subedi, A. N. Cartwright, R. B. Puerto, Y. Zheng, J. Maiarana, G. J. Freeman, K. W. Wucherpfennig, D. J. Irvine and M. S. Goldberg, T Cell-Targeting Nanoparticles Focus Delivery of Immunotherapy to Improve Antitumor Immunity, Nat. Commun., 2017, 8(1), 1–11, DOI:10.1038/s41467-017-01830-8.
- D. Chen, S. Ganesh, W. Wang and M. Amiji, Plasma Protein Adsorption and Biological Identity of Systemically Administered Nanoparticles, Nanomedicine, 2017, 12(17), 2113–2135, DOI:10.2217/nnm-2017-0178.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Biomolecular Coronas Provide the Biological Identity of Nanosized Materials, Nat. Nanotechnol., 2012, 7(12), 779–786, DOI:10.1038/nnano.2012.207.
- Y. Suzuki and H. Ishihara, Structure, Activity and Uptake Mechanism of SiRNA-Lipid Nanoparticles with an Asymmetric Ionizable Lipid, Int. J. Pharm., 2016, 510(1), 350–358, DOI:10.1016/j.ijpharm.2016.06.124.
- I. Reviakine, D. Johannsmann and R. P. Richter, Hearing What You Cannot See and Visualizing What You Hear: Interpreting Quartz Crystal Microbalance Data from Solvated Interfaces, Anal. Chem., 2011, 83(23), 8838–8848, DOI:10.1021/ac201778h.
- M. M. Billingsley, N. Singh, P. Ravikumar, R. Zhang, C. H. June and M. J. Mitchell, Ionizable Lipid Nanoparticle-Mediated MRNA Delivery for Human CAR T Cell Engineering, Nano Lett., 2020, 20(3), 1578–1589, DOI:10.1021/acs.nanolett.9b04246.
- D. W. Lee, X. Banquy, K. Kristiansen, Y. Kaufman, J. M. Boggs and J. N. Israelachvili, Lipid Domains Control Myelin Basic Protein Adsorption and Membrane Interactions between Model Myelin Lipid Bilayers, Proc. Natl. Acad. Sci., 2014, 111(8), E768 LP–E775, DOI:10.1073/pnas.1401165111.
- A. H. Pande, S. Qin and S. A. Tatulian, Membrane Fluidity Is a Key Modulator of Membrane Binding, Insertion, and Activity of 5-Lipoxygenase, Biophys. J., 2005, 88(6), 4084–4094, DOI:10.1529/biophysj.104.056788.
- Y. Dong, K. T. Love, J. R. Dorkin, S. Sirirungruang, Y. Zhang, D. Chen, R. L. Bogorad, H. Yin, Y. Chen, A. J. Vegas, C. A. Alabi, G. Sahay, K. T. Olejnik, W. Wang, A. Schroeder, A. K. R. Lytton-Jean, D. J. Siegwart, A. Akinc, C. Barnes, S. A. Barros, M. Carioto, K. Fitzgerald, J. Hettinger, V. Kumar, T. I. Novobrantseva, J. Qin, W. Querbes, V. Koteliansky, R. Langer and D. G. Anderson, Lipopeptide Nanoparticles for Potent and Selective SiRNA Delivery in Rodents and Nonhuman Primates, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(11), 3955–3960, DOI:10.1073/pnas.1322937111.
- F. DeRosa, B. Guild, S. Karve, L. Smith, K. Love, J. R. Dorkin, K. J. Kauffman, J. Zhang, B. Yahalom, D. G. Anderson and M. W. Heartlein, Therapeutic Efficacy in a Hemophilia B Model Using a Biosynthetic MRNA Liver Depot System, Gene Ther., 2016, 23(10), 699–707, DOI:10.1038/gt.2016.46.
- F. DeRosa, L. Smith, Y. Shen, Y. Huang, J. Pan, H. Xie, B. Yahalom and M. W. Heartlein, Improved Efficacy in a Fabry Disease Model Using a Systemic MRNA Liver Depot System as Compared to Enzyme Replacement Therapy, Mol. Ther., 2019, 27(4), 878–889, DOI:10.1016/j.ymthe.2019.03.001.
- J. J. Drabick, J. Glasspool-Malone, S. Somiari, A. King and R. W. Malone, Cutaneous Transfection and Immune Responses to Intradermal Nucleic Acid Vaccination Are Significantly Enhanced by in Vivo Electropermeabilization, Mol. Ther., 2001, 3(2), 249–255, DOI:10.1006/mthe.2000.0257.
- S. Przybylski, M. Gasch, A. Marschner, M. Ebert, A. Ewe, G. Helmig, N. Hilger, S. Fricke, S. Rudzok, A. Aigner and J. Burkhardt, Influence of Nanoparticle-Mediated Transfection on Proliferation of Primary Immune Cells in Vitro and in Vivo, PLoS One, 2017, 12(5), 1–16, DOI:10.1371/journal.pone.0176517.
- P. W. Laird, A. Zijderveld, K. Linders, M. A. Rudnicki, R. Jaenisch and A. Berns, Simplified Mammalian DNA Isolation Procedure, Nucleic Acids Res., 1991, 19(15), 4293, DOI:10.1093/nar/19.15.4293.
- X. Han, M. J. Mitchell and G. Nie, Nanomaterials for Therapeutic RNA Delivery, Matter, 2020, vol. 3, Issue 6, pp. 1948–1975, ISSN 2590–2385, DOI:10.1016/j.matt.2020.09.020.
- M. J. Mitchell, M. M. Billingsley and R. M. Haley, et al., Engineering precision nanoparticles for drug delivery, Nat Rev Drug Discov, 2020 DOI:10.1038/s41573-020-0090-8.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01609h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |