
Injectable nanofibrillar hydrogels based on charge-complementary peptide co-assemblies†
Bethsymarie
Soto Morales
a,
Renjie
Liu
 a,
Juanpablo
Olguin
a,
Abigail M.
Ziegler
a,
Stephanie M.
Herrera
a,
Kimberly L.
Backer-Kelley
b,
Karen L.
Kelley
b and
Gregory A.
Hudalla
*a
a,
Juanpablo
Olguin
a,
Abigail M.
Ziegler
a,
Stephanie M.
Herrera
a,
Kimberly L.
Backer-Kelley
b,
Karen L.
Kelley
b and
Gregory A.
Hudalla
*a
aJ. Crayton Pruitt Family Department of Biomedical Engineering, University of Florida, Gainesville, Florida 32611, USA. E-mail: ghudalla@bme.ufl.edu; Tel: +1 (352) 273-9326
bUF Interdisciplinary Center for Biotechnology Research – 2033 Mowry Road University of Florida, Gainesville, Florida 32610, USA
First published on 5th January 2021
Abstract
Injectable hydrogels are attractive for therapeutic delivery because they can be locally administered through minimally-invasive routes. Charge-complementary peptide nanofibers provide hydrogels that are suitable for encapsulation of biotherapeutics, such as cells and proteins, because they assemble under physiological temperature, pH, and ionic strength. However, relationships between the sequences of charge-complementary peptides and the physical properties of the hydrogels that they form are not well understood. Here we show that hydrogel viscoelasticity, pore size, and pore structure depend on the pairing of charge-complementary “CATCH(+/−)” peptides. Oscillatory rheology demonstrated that co-assemblies of CATCH(4+/4−), CATCH(4+/6−), CATCH(6+/4−), and CATCH(6+/6−) formed viscoelastic gels that can recover after high-shear and high-strain disruption, although the extent of recovery depends on the peptide pairing. Cryogenic scanning electron microscopy demonstrated that hydrogel pore size and pore wall also depend on peptide pairing, and that these properties change to different extents after injection. In contrast, no obvious correlation was observed between nanofiber charge state, measured with ζ-potential, and hydrogel physical properties. CATCH(4+/6−) hydrogels injected into the subcutaneous space elicited weak, transient inflammation whereas CATCH(6+/4−) hydrogels induced stronger inflammation. No antibodies were raised against the CATCH(4+) or CATCH(6−) peptides following multiple challenges in vehicle or when co-administered with an adjuvant. These results demonstrate that CATCH(+/−) peptides form biocompatible injectable hydrogels with viscoelastic properties that can be tuned by varying peptide sequence, establishing their potential as carriers for localized delivery of therapeutic cargoes.
Introduction
Hydrogels formed from peptides that self-assemble into nanofibers are widely used in biomedical applications, such as drug delivery, tissue engineering, regenerative medicine, and immune engineering.1–8 Synthetic peptides are attractive as building blocks for hydrogels because they can be made from natural amino acids that are well-tolerated metabolites, as well as non-natural amino acids that provide unique chemical features. Peptide-based hydrogels have been shown to be biocompatible with various cells and tissues.9–11 Often, they elicit little to no inflammation,12–15 with the peptides being weakly immunogenic despite being foreign to the host.9,11,16 The amino acid sequence can be tailored to create peptides that spontaneously assemble into fibrillar hydrogels in aqueous conditions. Alternatively, peptides can be designed to form hydrogels in response to a specific stimulus, such as a change in temperature, salt concentration, or pH,7,17–21 which is advantageous for encapsulating sensitive biologic cargoes. Hydrogel mechanical properties can be tailored by varying peptide concentration or amino acid sequence. Furthermore, because the hydrogels form through physical crosslinking of nanofibers, they often undergo shear-thinning and recovery, which enables their delivery via minimally-invasive injection.22–25 Finally, bioactive moieties, such as peptides, small molecule drugs, proteins, or carbohydrates can be appended onto a fibrillizing peptide domain to create hydrogels endowed with specific functional properties, such as cell adhesion, antigen presentation, molecular recognition, controlled drug release, and enzymatic degradation, among others.12,13,15Co-assembly, in which two different peptide molecules associate to form a single fibrillar architecture, is a simple way to expand the range of structural, mechanical, and functional properties of peptide-based hydrogels.13,15,26 Co-assembly can take many forms, such as cooperative co-assembly, random co-assembly, destructive co-assembly, or self-sorting.27 Changing the amino acid sequence of either peptide can alter their co-assembly propensity, nanofiber morphology, and hydrogel mechanical properties,12,20,28–30 such as stiffness, pore size, and molecular transport.29,31 Selective co-assembly is a special case in which two different molecules, A and B, cannot assemble when alone, but when combined associate to form two-component (-ABABAB-) β-sheet nanofibers. Replacing neutral residues with charged residues in known synthetic self-assembling peptide sequences is an effective way to encode selective co-assembly because like-charged molecules repel each other, while opposites attract.12,32,33 For example, the pair referred to as Co-Assembly Tags based on Charge complementarity, or “CATCH(+/−)” are 11 amino acid long variants of the self-assembling peptide Q11,12,34,35 P11–13 and P11–14 were derived from P11–2,32 and KVW10 and EVW10 are variants of MAX1.36 Selective co-assembly of charge-complementary peptides can be triggered at physiologic temperature, pH, and ionic strength, which makes these systems ideal for encapsulating cells or creating hydrogels with immobilized protein domains.12 Despite an increasing number of charge-complementary co-assembling peptide pairs reported in the literature, though, their use as biomaterials remains limited by a lack of understanding of the mechanisms of nanostructure formation, the mechanical properties of the hydrogels that they form, and their biocompatibility.
In this report, we characterized hydrogels fabricated via co-assembly of four pairs of CATCH peptide variants: CATCH(4+) [Ac-QQKFKFKFKQQ-Am] & CATCH(4−) [Ac-QQEFEFEFEQQ-Am] (“CATCH4+/4−”); CATCH(4+) and CATCH(6−) [Ac-EQEFEFEFEQE-Am] (“CATCH4+/6−”); CATCH(6+) [Ac-KQKFKFKFKQK-Am] & CATCH(4−) (“CATCH6+/4−”); and CATCH(6+) and CATCH(6−) (“CATCH6+/6−”) (Fig. 1), where the number denotes the number of lysine or glutamic acid residues in each molecule. Prior reports suggest that hydrophobicity, length of the peptide sequence, and complementary ionic interactions can influence the formation of hydrogels, their physical properties, and their response to mechanical stresses.31,37 Further, peptide sequence and nanofiber charge can influence biocompatibility and immunogenicity.38 Although all complementary CATCH pairs can co-assemble into β-sheet nanofibers,35 how the peptide sequence and peptide charge contribute to the biophysical properties of CATCH hydrogel networks is not well understood. Using a combination of oscillatory rheology, transmission electron microscopy, cryogenic-scanning electron microscopy, and ζ-potential measurements, we studied relationships between CATCH peptide pairings, hydrogel rheology, and biocompatibility. All of the CATCH peptide pairs form viscoelastic hydrogels that undergo shear-thinning and recovery; however, the extent of recovery depends on which two peptides are co-assembled together. Shear-thinning and recovery enables minimally-invasive injection directly into a tissue site of interest. Following injection, CATCH hydrogels elicit varying degrees of inflammation. Weakly inflammatory CATCH peptides are not immunogenic, despite being foreign to the host. This study advances our understanding of the physical properties of co-assembled peptide hydrogels and establishes their potential as injectable biomaterials.
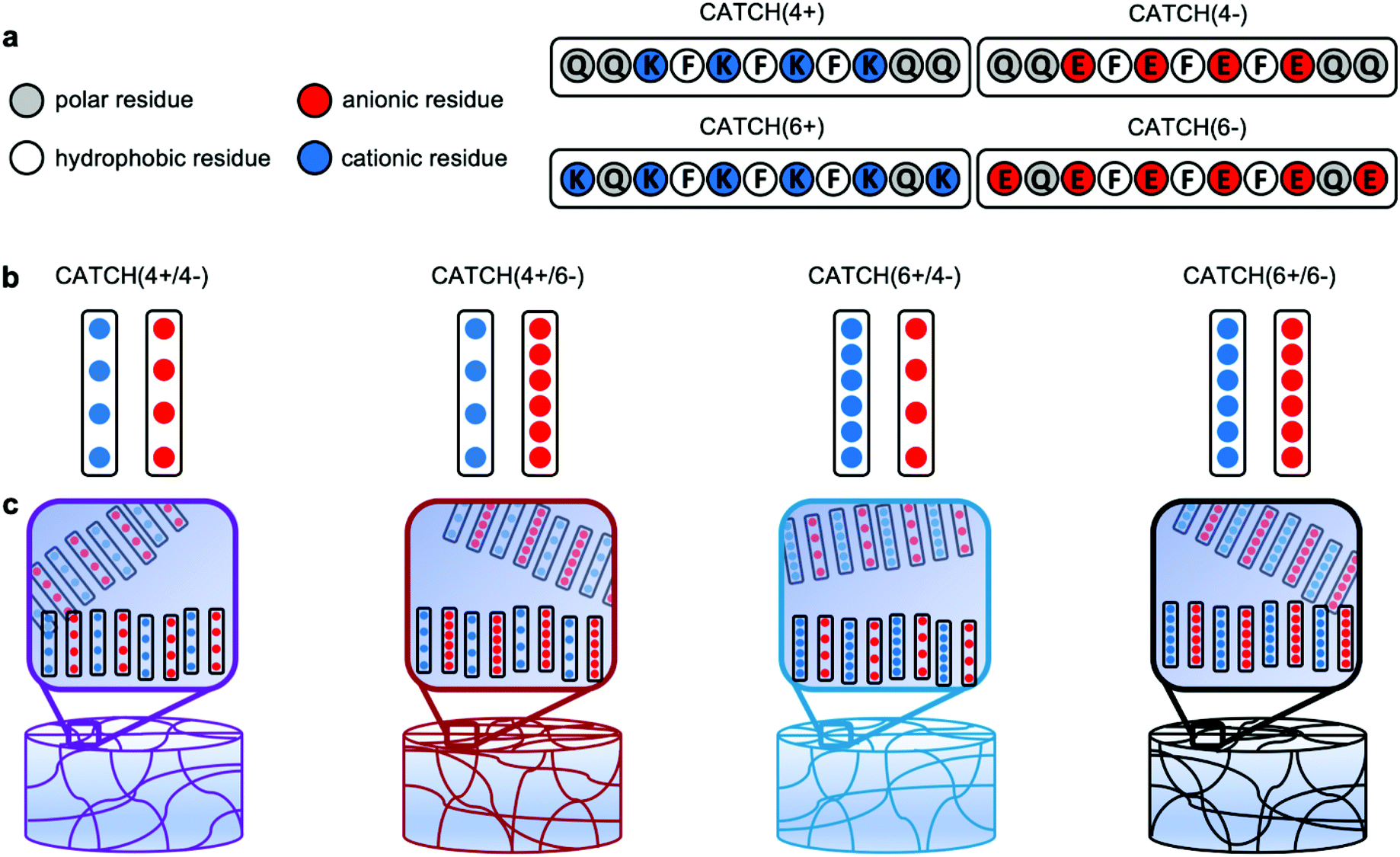 | ||
| Fig. 1 Schematic representation of CATCH(+/−) nanofibrillar hydrogels. (a) Molecular design and primary sequence of the CATCH(4+), CATCH(6+), CATCH(6−), and CATCH(4−) peptides. (b) CATCH(+/−) peptides co-assemble into nanofibrillar hydrogels when combined at an equimolar ratio in aqueous media. (c) Here we studied hydrogels formed from the CATCH(+/−) pairs: CATCH(4+/4−), CATCH(4+/6−), CATCH(6+/4−) and CATCH(6+/6−). | ||
Results
CATCH peptides co-assemble into viscoelastic hydrogels
At a total peptide concentration of 12 mM, equimolar mixtures of CATCH(4+/4−), CATCH(4+/6−), CATCH(6+/6−), and CATCH(6+/4−) formed self-supporting hydrogels in 1× phosphate-buffered saline (PBS) (Fig. 2). When subjected to oscillating rheology, all CATCH hydrogels had a ratio of storage modulus (G′) to loss modulus (G′′) that was greater than 1 at different angular frequencies (Fig. 2a–d), indicative of viscoelastic behavior at 0.5% strain. CATCH(4+/4−) formed hydrogels with G′ of 3.84 ± 0.70 kPa (Fig. 2a). In contrast, CATCH(6+/6−) formed weaker hydrogels with G′ close to 0.50 ± 0.08 kPa (Fig. 2d). CATCH(4+/6−) and CATCH(6+/4−) formed hydrogels of intermediate stiffness with storage moduli of 0.97 ± 0.27 kPa and 0.78 ± 0.06 kPa, respectively (Fig. 2b and c).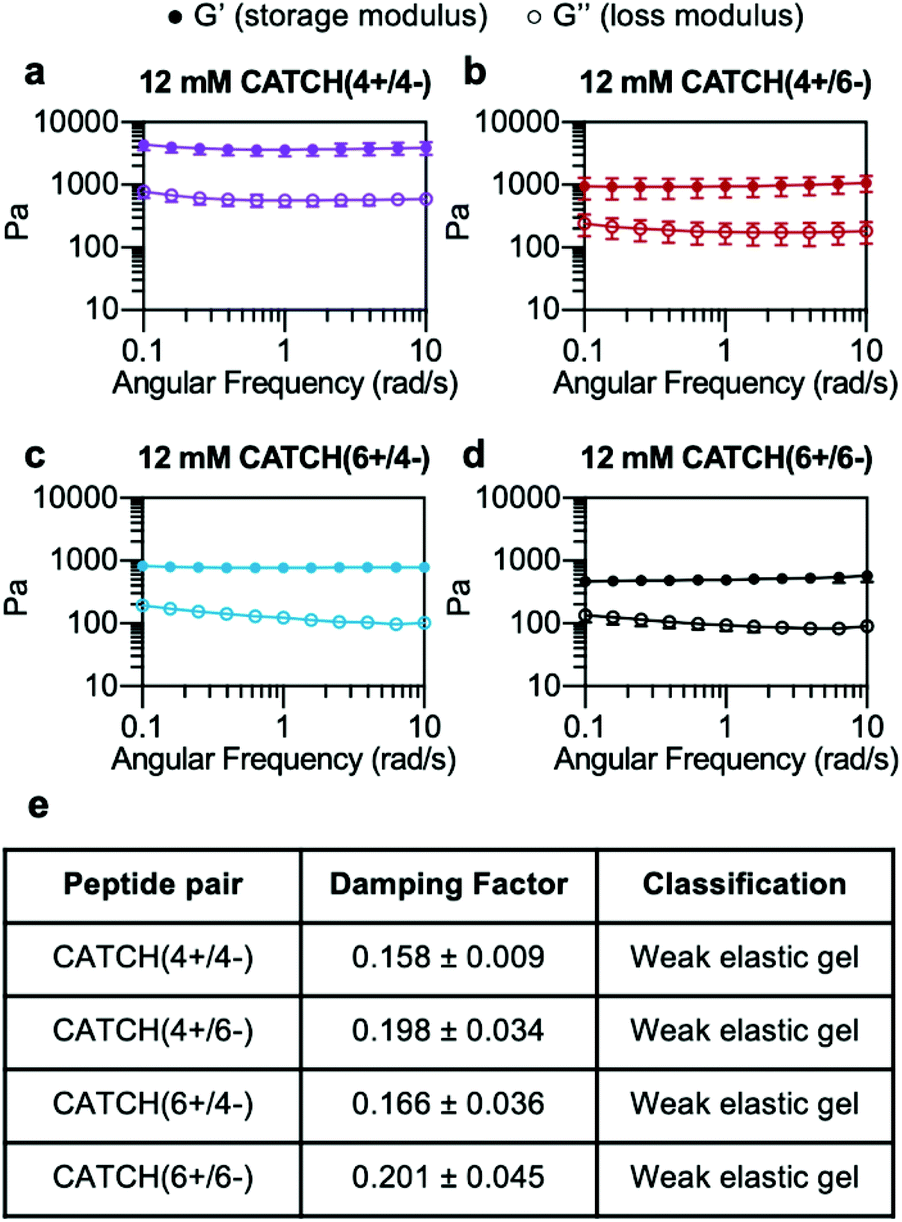 | ||
| Fig. 2 Oscillating rheology of CATCH(+/−) peptide hydrogels. (a–d) G′ and G′′ of 12 mM CATCH(4+/4−), CATCH(4+/6−), CATCH(6+/4−), and CATCH (6+/6−) hydrogels at different angular frequencies. (e) Average damping factor of 12 mM CATCH(+/−) hydrogels, calculated as the average of the G′′/G′ ratio at each angular frequency. Data are presented as the mean ± standard deviation (n = 3). | ||
The damping factor (G′′/G′) of all 12 mM CATCH hydrogels ranged from 0.1 to 0.25, characteristic of weak elastic gels39,40 (Fig. 2e). Over the range of 2–16 mM, CATCH(4+/6−) hydrogel storage modulus increased with total CATCH peptide concentration (Fig. 3a). The damping factor at each of these concentrations was between 0.1 and 1, indicating that all of the hydrogels were weakly elastic regardless of their storage modulus (Fig. 3b). Moreover, it was observed that at high frequency the 2 mM hydrogel had a damping factor greater than 1, indicating the material was near its gelation point at this concentration (Fig. 3b).
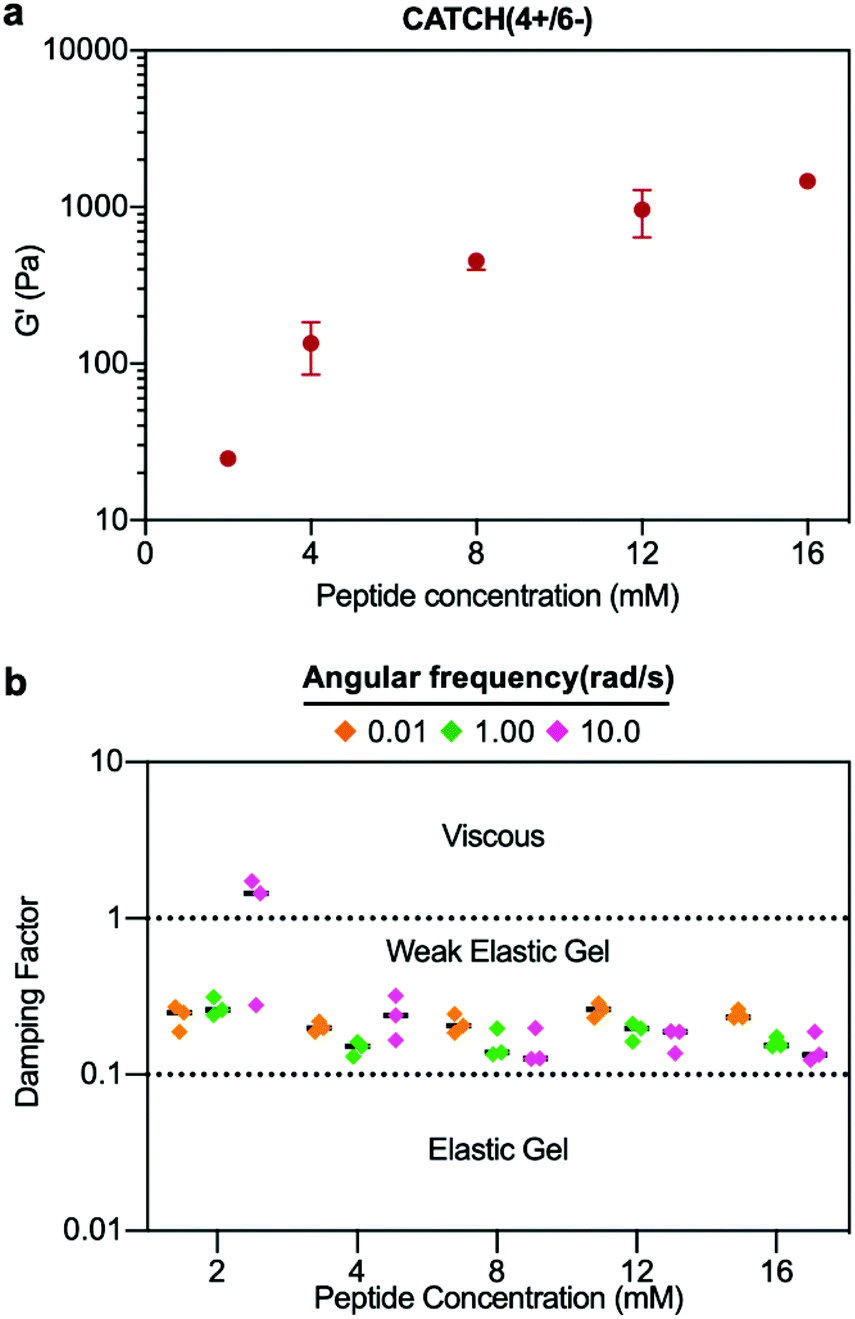 | ||
| Fig. 3 Concentration-dependence of CATCH(4+/6−) hydrogel viscoelasticity. (a) Storage modulus of CATCH(4+/6−) hydrogels assembled at different peptide concentrations. (b) Damping factor of CATCH(4+/6−) hydrogels assembled at different peptide concentrations at angular frequencies of 0.1, 1 and 10 rad s−1. [CATCH(4+)] = [CATCH(6−)] in all samples. Data are presented as the mean ± standard deviation (n = 3). | ||
Based on these observations, the rheological properties of the CATCH (4+/4−), CATCH(6+/4−), and CATCH(6+/6−) pairs were evaluated at 2 mM total peptide concentration (Fig. S1†). CATCH(4+/4−) formed a self-supporting hydrogel at 2 mM, with a storage modulus close to 1 kPa over the frequency range of 0.1–10 rad s−1, indicating that this formulation was well above its gelation point (Fig. S1a†). In contrast, CATCH(6+/6−) had the properties of a viscoelastic solid at 0.1–4 rad s−1, but those of a liquid at higher frequencies (Fig. S1d†). Similar to CATCH(4+/6−), CATCH(6+/4−) hydrogels were near their gel point at 2 mM, as indicated by a decrease in G′ and an increase in G′′ at high frequency (Fig. S1b and c†). Together, these observations demonstrated that CATCH(4+/4−) formed the stiffest hydrogels, while CATCH(6+/6−) formed the softest hydrogels.
CATCH(+/−) hydrogel shear thinning and recovery varies with peptide pairing
Step-shear flow measurements were used to evaluate the injectability of CATCH hydrogels, which requires that they flow under high shear and then recover viscoelasticity when the applied forces are removed.12,25,41 Shear thinning was observed for all of the CATCH pairs, and viscosity recovered upon transition from high to low shear rate (Fig. 4). CATCH(4+/6−), CATCH(6+/4−), and CATCH(6+/6−) hydrogels demonstrated similar shear-dependent changes in viscosity over three low-high shear rate cycles (Fig. 4b–d). In contrast, CATCH(4+/4−) demonstrated shear-thinning at both low and high shear rates over the first two cycles, but only at a high shear rate in the third cycle (Fig. 4a).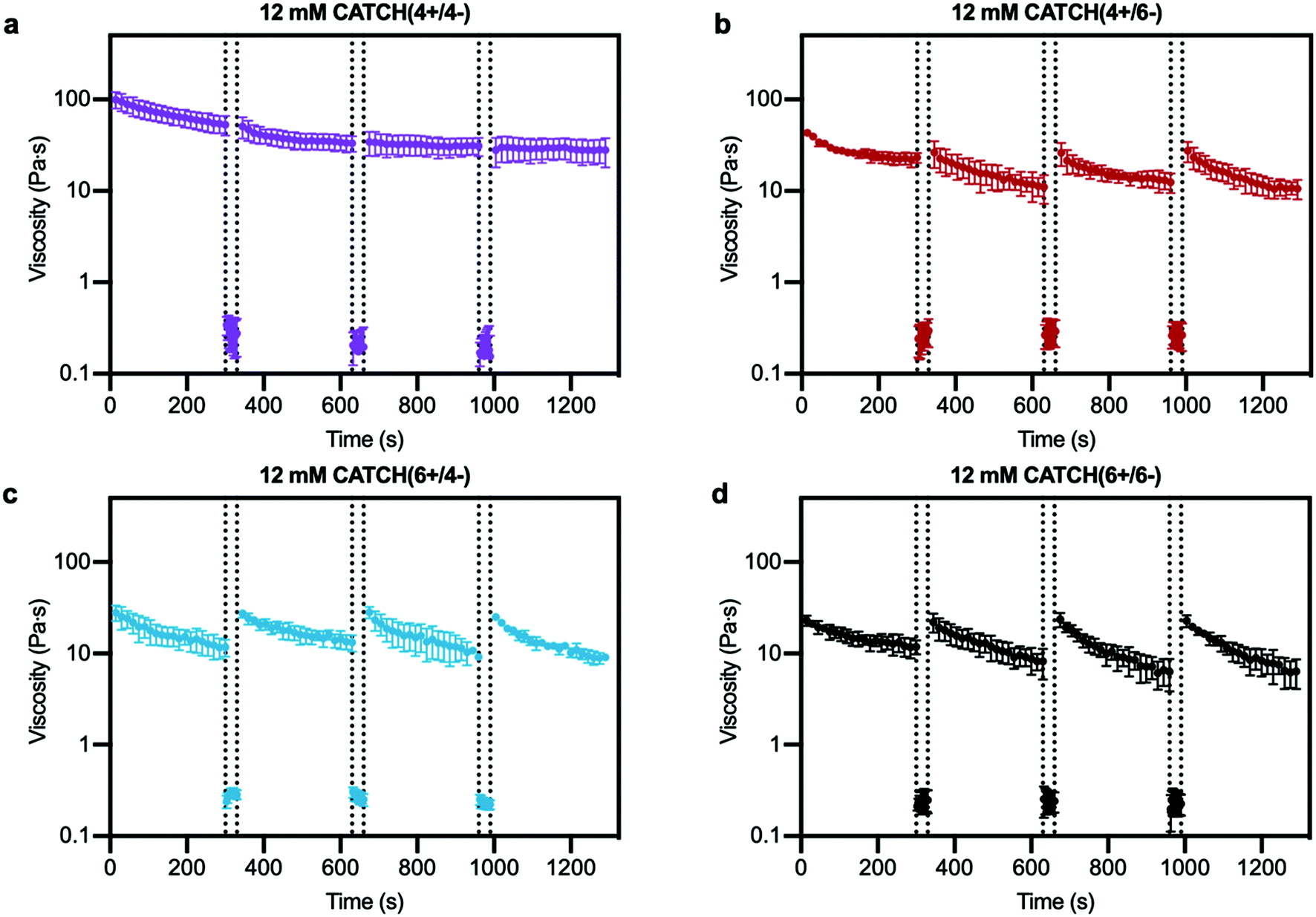 | ||
| Fig. 4 Step-shear flow rate of CATCH(+/−) hydrogels. (a–d) Viscosity at 0.5 s−1 shear rate before and after flow of CATCH(+/−) hydrogels at 100 s−1 shear rate (between dashed lines). Data are presented as the mean ± standard deviation (n = 3). | ||
Oscillating rheology was used to determine if CATCH hydrogels undergo viscoelastic recovery, indicative of network self-healing and restored elasticity, following high-strain disruption.42 Within 30 s after 1000% strain disruption, all four CATCH pairs had a G′/G′′ ratio > 1 indicative of a viscoelastic solid; however, differences in the percentage and rate of recovery were observed (Fig. 5). CATCH(4+/4−) recovered 100% of its initial stiffness within 132 seconds (Fig. 5a). In contrast, CATCH(6+/6−), CATCH(4+/6−) and CATCH(6+/4−) recovered 63%, 60% and 95% of their initial stiffnesses, respectively, after 10 minutes (Fig. 5b–d). Taken together, these data demonstrate that all CATCH hydrogels can recover after both shear and strain disruption, but the extent of recovery depends on the peptide combination.
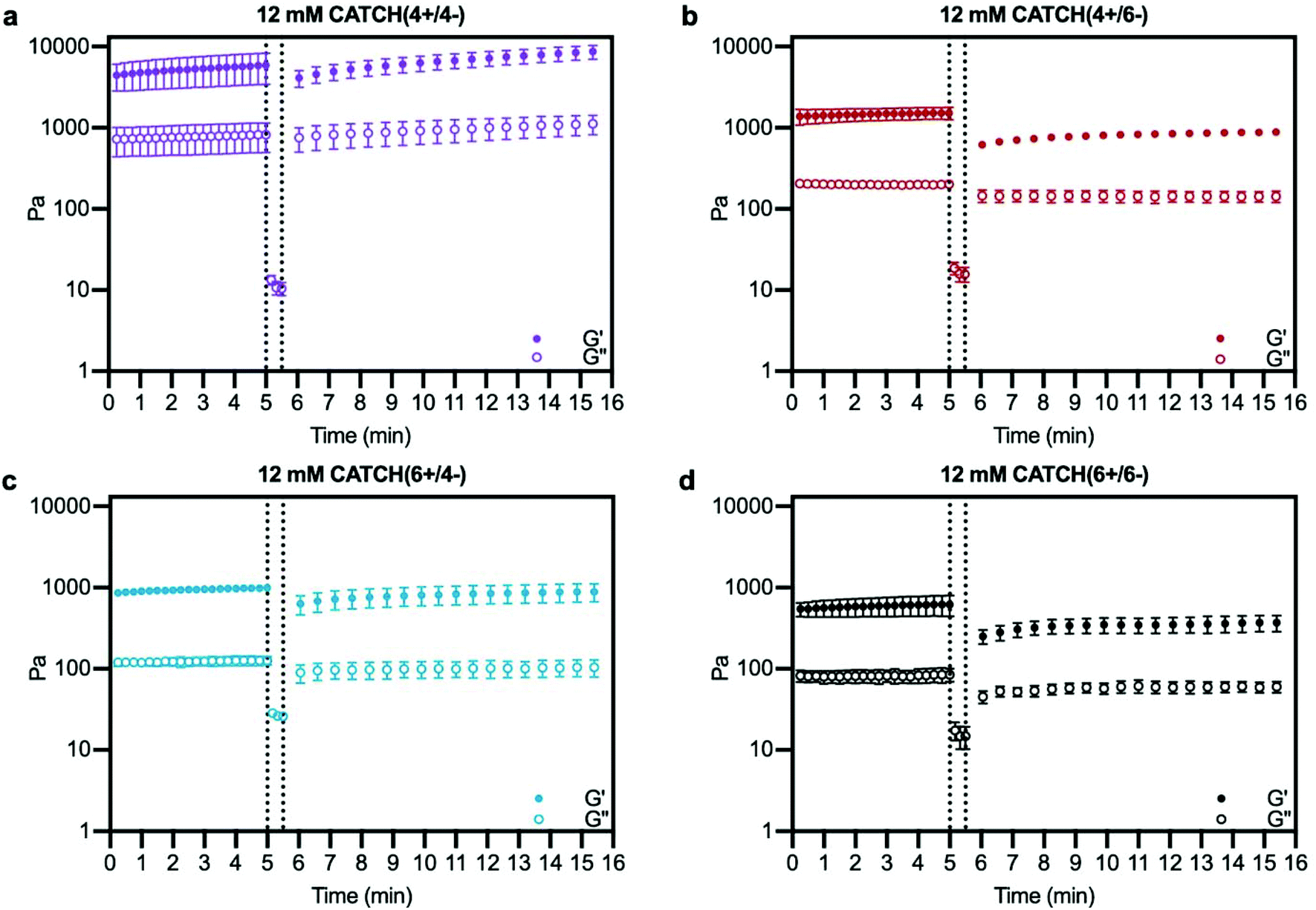 | ||
| Fig. 5 Viscoelasticity restoration kinetics of CATCH(+/−) hydrogels after high-strain disruption. (a–d) Storage modulus and loss modulus before (left of dashed line) and after (right of dashed line) disruption of CATCH(+/−) hydrogels at 1000% strain. Data are presented as the mean ± standard deviation (n = 3). | ||
Porosity of CATCH hydrogels varies with peptide pairing
Cryogenic-scanning electron microscopy (Cryo-SEM), which enables observation of CATCH hydrogel structure in the hydrated state, demonstrated that network porosity differed with different peptide pairings (Fig. 6a). Cryo-SEM images suggested that CATCH(4+/4−) hydrogels had pores with relatively small diameters, as well as regions of mats that lacked any observable porosity (Fig. 6a). In contrast, hydrogels formed from the other CATCH combinations had no obvious organization, less uniform pore structures, and fewer if any regions of dense mats (Fig. 6a). Pore size measurements indicated that 12 mM CATCH(4+/4−) hydrogels had pores with a mean diameter of 0.87 ± 0.32 μm. 12 mM CATCH(6+/6−) hydrogels had a slightly larger pore size, with a mean diameter of 1.41 ± 0.81 μm. CATCH(4+/6−) hydrogels had an even larger pore size, with a mean diameter of 2.26 ± 0.62 μm, while CATCH(6+/4−) hydrogels had the largest pore size, with a mean diameter of 2.84 ± 1.03 μm (Fig. 6c and Fig. S2†). The thickness of the hydrogel pore walls also differed with peptide pairing (Fig. 6d). CATCH(4+/4−) and CATCH(6+/4−) hydrogel pores had thicker walls, with average thicknesses of 0.18 ± 0.06 μm and 0.24 ± 0.12 μm, respectively. In contrast, CATCH(6+/6−) had thinner walls, with an average thickness of 0.10 ± 0.04 μm. CATCH(4+/6−) hydrogel pores had the thinnest walls, with an average thicknesses of 0.05 ± 0.02 μm.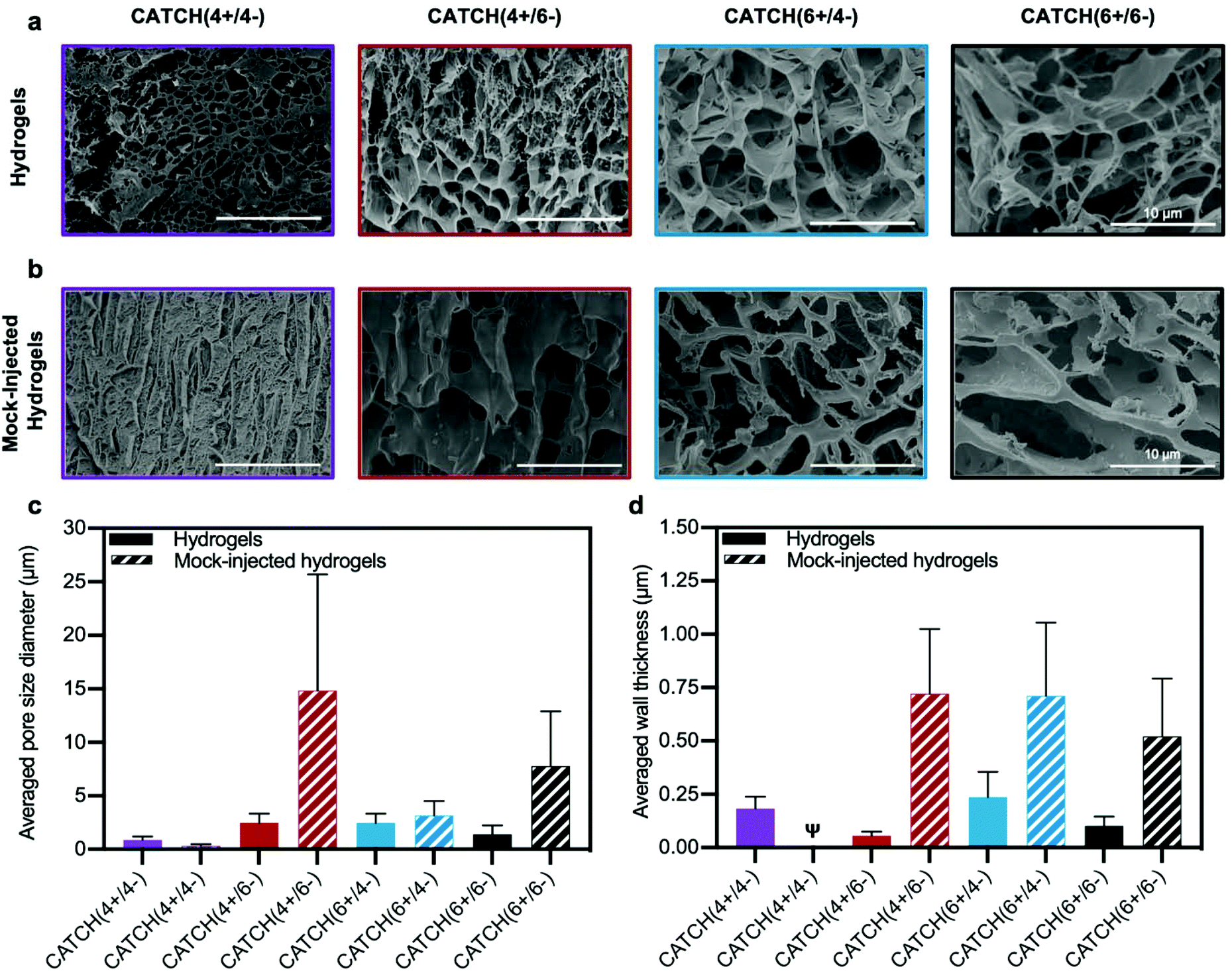 | ||
| Fig. 6 Cryogenic electron microscopy analysis of CATCH(+/−) hydrogel morphology. (a) Cryo-SEM micrographs of 12 mM CATCH(4+/4−), CATCH(4+/6−), CATCH(6+/4−), and CATCH(6+/6−) hydrogels in the hydrated state. (b) Cryo-SEM micrographs of 12 mM CATCH(4+/4−), CATCH(4+/6−), CATCH(6+/4−), and CATCH(6+/6−) mock-injected hydrogels. (c) Average pore diameter measured from Cryo-SEM micrographs of CATCH(+/−) hydrogels and mock-injected hydrogels (Fig. 5a and b) (n = 30 measurements). (d) Average wall thickness measured from Cryo-SEM micrographs of CATCH(+/−) hydrogels and mock-injected hydrogels (Fig. 5a and b) (n = 30 measurements). ψ indicates that wall thickness could not be reliably quantified from the Cryo-SEM micrographs. | ||
To assess the effect of shear-thinning on hydrogel network architecture, CATCH(+/−) hydrogels were subjected to a mock injection through the same fine-gauge needle used for in vivo studies and then observed in the hydrated state using cryo-SEM (Fig. 6b). The micrographs suggested that the network porosity and thickness of the hydrogel pore walls were affected in all formulations by the shear forces experienced in the needle. In particular, an increase in regions of dense mats were observed in CATCH(4+/4−) hydrogels after injection, which corresponded with a smaller pore size and qualitatively thicker walls. In contrast, CATCH(6+/6−) and CATCH(4+/6−) hydrogels had larger pore sizes and thicker walls after injection, while CATCH(6+/4−) hydrogels had a similar pore size after injection, but thicker pore walls (Fig. 6b–d). Coupled with the rheology data, these observations suggest that pore size, pore structure, and pore wall thickness likely all contribute to the stiffness, shear-thinning, strain deformation, and recovery of CATCH hydrogels.
Nanofiber aggregation potential varies with peptide pairing
The morphology of CATCH nanofibers under non-gelling conditions was viewed with transmission electron microscopy (Fig. S3†). CATCH(4+/4−) nanofibers appeared as mats of large aggregates, whereas CATCH(6+/6−) nanofibers were dispersed, consistent with a prior report.35 Nanofibers formed from the CATCH(4+/6−) pair also had a dispersed morphology, consistent with a prior report.12 The CATCH(6+/4−) pair formed nanofibers with a dispersed morphology, which was similar to that of the CATCH(6+/6−) and CATCH(4+/6−) pairs. The observed nanofiber morphologies were generally consistent with the hydrogel pore structures observed with cryo-SEM. Collectively, these observations suggest that the greater initial stiffness, lower gel point, and improved recovery of CATCH(4+/4−) hydrogels may result from the tendency of CATCH(4+/4−) nanofibers to aggregate; however, the complex rheological properties of CATCH hydrogels likely also depend on other molecular-level aspects of the system that govern nanofiber entanglement and lateral association, such as β-sheet morphology and β-strand organization.ζ-Potential is a poor predictor of CATCH hydrogel physical properties
Based on the different morphologies observed with Cryo-SEM and TEM, ζ-potential of the different CATCH peptides alone and in combination was measured to determine if aggregation potential correlated with nanofiber charge state (Fig. S4a–f†). The sign of the ζ-potential of the individual CATCH peptides correlated with the expected charge based on their amino acid content over the concentration range of 0.2–1 mM (Fig. S4a–c†). At a concentration of 1 mM total peptide, the CATCH(4+/6−) and CATCH(6+/4−) pairs had opposing ζ-potentials of −23.88 ± 0.34 mV and 24.18 ± 1.01 mV, respectively (Fig. S4d†), as expected based on the imbalance of charged amino acid residues in each peptide strand. Likewise, at a concentration of 1 mM total peptide, the CATCH(4+/4−) and CATCH(6+/6−) pairs had near-neutral ζ-potentials of −0.95 ± 0.59 mV and 0.08 ± 0.52 mV, respectively (Fig. S4d†), consistent with the equal number of charged amino acid residues in each strand. The measured ζ-potentials of the CATCH peptide pairs were not concentration-dependent over the range of 0.2–1 mM (Fig. S4d–f†), which is near or above the critical concentration of fibrillization.35 While these measurements could explain the aggregation prone behaviour of the CATCH(4+/4−) pair, they do not explain the behaviour of the CATCH(6+/6−) pair, which formed dispersed nanofibers at low concentrations and weaker porous gels at higher concentrations. Likewise, these measurements cannot explain the differences in pore size and wall thickness of CATCH(4+/6−) and CATCH(6+/4−) hydrogel networks. Thus, in general, ζ-potential is a poor predictor of the aggregation potential of CATCH(+/−) nanofibers, and in turn the porosity and rheological properties of CATCH(+/−) hydrogels.CATCH(4+/6−) hydrogels are biocompatible
The rheological properties of CATCH hydrogels suggested that they could find use as vehicles for minimally-invasive therapeutic delivery. However, the CATCH peptides are non-natural, “designer” sequences that are not found in mammalian systems. Thus, the recognition of CATCH peptides or hydrogels as foreign matter by the host immune system could limit their usefulness as delivery vehicles. The biocompatibility of CATCH hydrogels likely depends on the peptide sequences, the charge state of the nanofibers, and the physical properties of the hydrogel. To assess the biocompatibility of CATCH hydrogels, we evaluated the cytotoxicity of CATCH nanofibers, the inflammatory response following local injection of CATCH hydrogels in subcutaneous tissue, and adaptive immunity against the CATCH peptides following multiple exposures.We first evaluated the cytotoxicity of nanofibers formed from each CATCH(+/−) combination using an NIH3T3 fibroblast in vitro assay.43,44 Cell metabolic activity, an indicator of fibroblast viability, was greater than 80% relative to untreated cells at 24 h, indicating that none of the nanofibers were cytotoxic at any of the concentrations tested (Fig. S5†). These observations were consistent with the low cytotoxicity reported for the Q11 peptide,11,14 which is the parent sequence of the CATCH peptides.
Next, we characterized the onset and duration of inflammation at the site of injection of a CATCH hydrogel. For this, we chose to evaluate inflammation after an injection of CATCH(4+/6−) or CATCH(6+/4−) hydrogels. These hydrogel formulations were chosen based on their shear-thinning and recovery properties, strain-deformation recovery properties, and porous architecture after a mock injection. Further, we sought to test whether anionic CATCH(4+/6−) hydrogels would be better tolerated than cationic CATCH(6+/4−) hydrogels, a hypothesis that was informed by a prior report demonstrating charge-dependent differences in immunogenicity of Q11 variants.38
CATCH(4+/6−) hydrogels injected into the subcutaneous space of female C57BL/6J mice elicited weak inflammation, measured as change in paw diameter, which peaked at 6 h and resolved within a few days (Fig. 7). In contrast, mice that received an injection of CATCH(6+/4−) hydrogel had a large change in paw diameter, reaching a maximum of 1.32 ± 0.06 fold increase relative to the contralateral vehicle-injected paw at 24 hours (Fig. 7a). The fold change observed at 24 hours after injection of the CATCH(6+/4−) hydrogel was comparable to that of mice that received an injection of aqueous λ-carrageenan,45,46 a viscous polysaccharide solution used as a positive control for sterile inflammation. Over the next 96 hours, the paw diameter of animals that received a CATCH(6+/4−) hydrogel injection decreased, yet remained slightly elevated relative to baseline; the paw diameter of animals that received a λ-carrageenan injection remained significantly elevated (Fig. 7a). Collectively, these observations suggest that nanofiber charge indeed influences the inflammatory response to CATCH peptide hydrogels, and that the anionic CATCH(4+/6−) hydrogel is the more appropriate formulation for in vivo applications because it elicits weak local inflammation that resolves quickly.
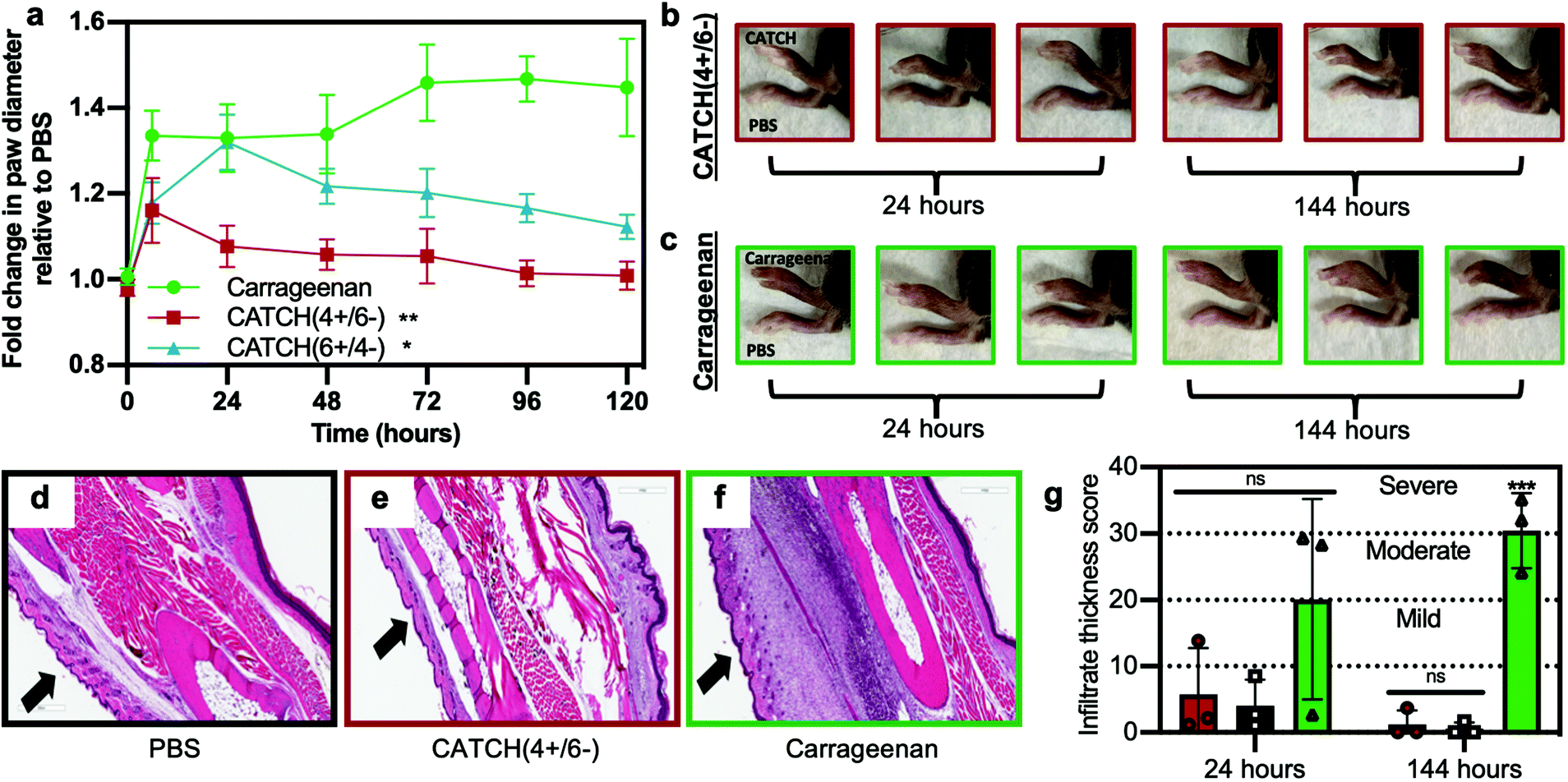 | ||
| Fig. 7 Analysis of inflammation induced following subcutaneous injection of CATCH(+/−) hydrogels into C57BL/6J mice. (a) Fold change in paw thickness after injection of 12 mM CATCH(4+/6−) hydrogel (red square), 12 mM CATCH(6+/4−) hydrogel (blue triangle), λ-carrageenan (green circle, positive control), relative to PBS vehicle injected into contralateral paw (vehicle control). (b and c) Representative digital photographs of mouse paws at t = 24 or 144 h after injection of 12 mM CATCH(4+/6−) or λ-carrageenan. (d–f) Representative histology sections taken from tissues at t = 144 h after injection of PBS vehicle, 12 mM CATCH(4+/6−) hydrogel, or λ-carrageenan. (g) Infiltrate thickness score at t = 24 and 144 h after injection of PBS vehicle, 12 mM CATCH(4+/6−) hydrogel, or λ-carrageenan determined from histology sections. Data presented as mean ± standard deviation (n = 5) in (a). ** represents p < 0.01, * represents p < 0.05, repeated measures ANOVA with Dunnett's multiple comparison. Data presented as mean ± standard deviation (n = 3) in (g). *** represents p < 0.001, ANOVA with Tukey's post-hoc. | ||
To assess sex-related differences in the inflammatory response to CATCH hydrogels, we injected a CATCH(4+/6−) hydrogel into the subcutaneous space of male C57BL/6J mice. We observed a similar trajectory of paw diameter changes when compared to female mice that received a CATCH(4+/6−) hydrogel (Fig. S6†). Although the measured differences in paw diameter were statistically different than those of paws into which only PBS vehicle was injected, the change in paw diameter following injection of CATCH(4+/6−) hydrogels into male mice was significantly less than the change following injection of λ-carrageenan. These observations generally suggested that there were no sex-related differences in the innate immune response to CATCH(4+/6−) hydrogels.
We used histology to assess changes in cellular infiltration at the injection site over time. Injection of a CATCH(4+/6−) hydrogel led to minimal cell infiltration when compared to injection of 1× PBS vehicle or λ-carrageenan (Fig. 7d–g and Fig. S7†). At 144 hours, cell infiltration decreased in tissues that received a CATCH(4+/6−) hydrogel, whereas cell infiltration significantly increased in tissues that received a λ-carrageenan injection (Fig. 7d–g). In vivo imaging of a CATCH hydrogel labeled with IRdye-Cy5.5 demonstrated that some of the material was present at the injection site for more than 12 days, which indicated that inflammation resolved before the gel was cleared (Fig. S8†).
To determine if anti-CATCH antibodies were raised, serum was collected from each animal on day 28, following subcutaneous injection of a CATCH(4+/6−) hydrogel into the paw on day 0 and injection of either CATCH(4+) or CATCH(6−) in PBS on day 14 (Fig. 8a). No measurable serum IgGs were detected against either CATCH(4+) or CATCH(6−) (Fig. 8b and Fig. S9†). These animals received a second subcutaneous injection of either CATCH(4+) or CATCH(6−) in PBS on day 42, and serum was collected at day 56. No measurable serum IgGs were detected against either CATCH(4+) or CATCH(6−) after this secondary challenge (Fig. 8b and Fig. S9†). An independent cohort of mice received two injections (day 0 and 28) of CATCH(4+) or CATCH(6−) emulsified in TiterMax®, a polymer adjuvant that is effective for raising antibodies against peptide and protein antigens47–49 (Fig. 8c). No IgGs against either CATCH(4+) or CATCH(6−) were detected in serum collected on days 35 or 42 (Fig. 8d). Collectively, these observations demonstrate that CATCH(4+) and CATCH(6−) are poor immunogens in C57BL/6J mice. These observations are supported by predictions from the Immune Epitope Database (IEDB), which estimated that CATCH(4+) and CATCH(6−) peptides would be poor MHC II binders, and in turn, poor immunogens, in C57BL/6J mice (Fig. S10†). These observations were also consistent with Q11 which has previously been reported as non-immunogenic in C57BL/6J mice,11,16 and was predicted to be a poor binder of C57BL/6J MHC II by the IEDB.
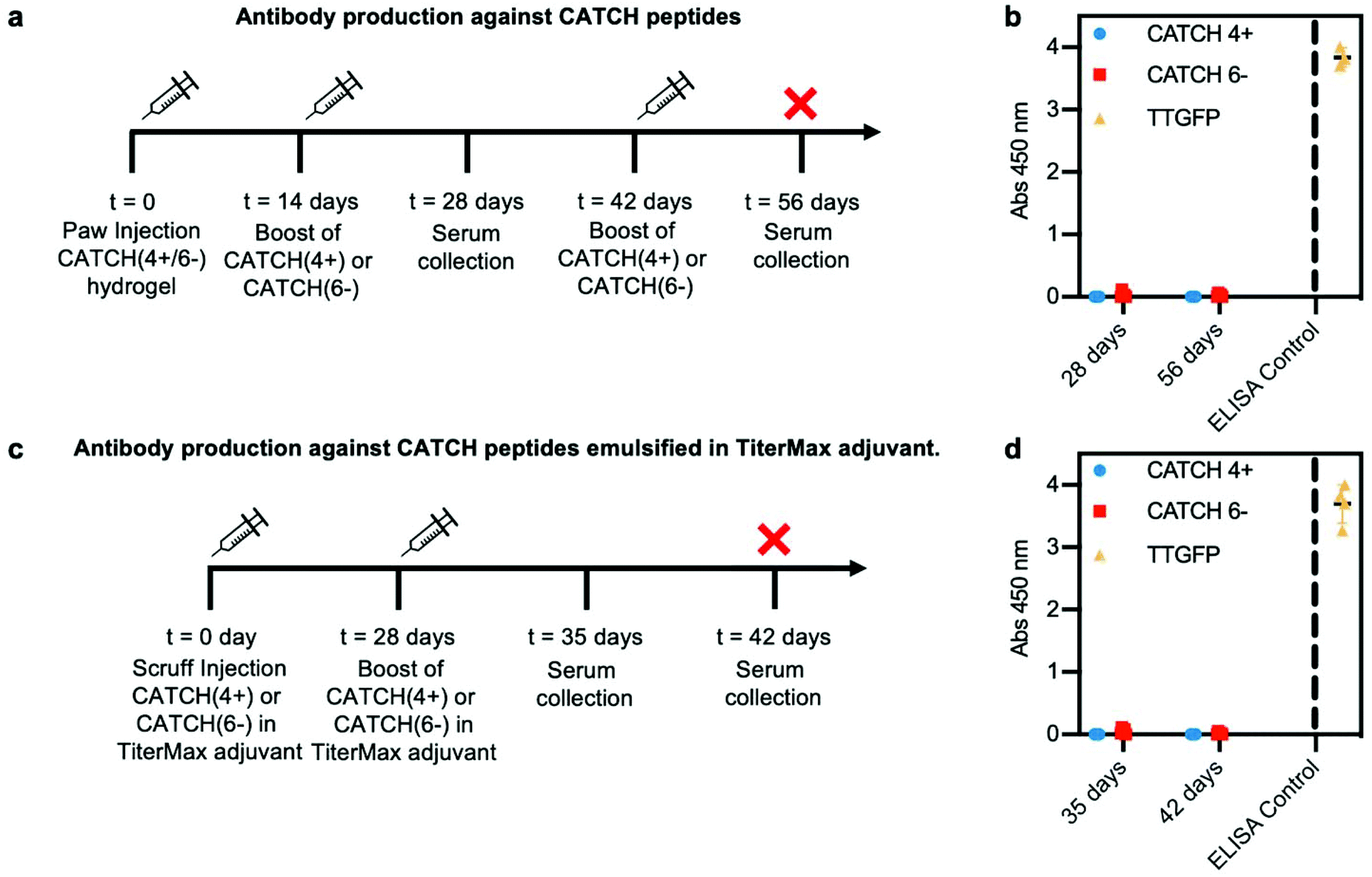 | ||
| Fig. 8 Analysis of serum immunoglobulins raised against CATCH(4+) and CATCH(6−) by C57BL/6 mice. (a) Injection and sample collection schedule for challenge with CATCH(4+/6−) hydrogel followed by CATCH(4+) or CATCH(6−) in vehicle. (b) Total serum IgG reactive against CATCH(4+), CATCH(6−), and TT-GFP (positive control). (c) Injection and sample collection schedule for challenge with CATCH(4+) or CATCH(6−) emulsified in TiterMax® adjuvant. (d) Total serum IgG reactive against CATCH(4+), CATCH(6−), and TT-GFP (positive control). Data presented as mean ± standard deviation (n = 5). | ||
Discussion
This report demonstrates that charge-complementary CATCH peptide pairs form viscoelastic gels that can undergo shear-thinning and recovery, but the extent of recovery depends on the peptide pair. Physical properties of the CATCH hydrogels, such as their pore size and pore structure, depend on the peptide pairing. No obvious correlation was observed between nanofiber charge state and the hydrogel rheological properties or pore structure. CATCH nanofibers were not toxic to fibroblasts in vitro. Further, CATCH(4+/6−) hydrogels injected into the subcutaneous space elicited weak inflammation, which resolved in a few days, and the peptides were not immunogenic.The rheological properties of hydrogels are important to consider when assessing their potential for in vivo use. The mechanical properties of a biomaterial should closely match that of the tissue into which they will be placed. Toward this end, viscoelastic solids, such as hydrogels, are attractive for use in compliant tissues and non-load bearing applications.50–52 Furthermore, viscoelastic solids that undergo shear-thinning and recovery are advantageous for minimally-invasive delivery via injection. All CATCH(+/−) combinations formed viscoelastic solids with damping factors characteristic of weak elastic gels. However, the storage modulus of the hydrogels varied with peptide pairing. It was observed that the charge-matched pairs, CATCH(4+/4−) and CATCH(6+/6−), formed the stiffest and the weakest hydrogels, respectively, while the charge-mismatched pairs formed gels with intermediate moduli. These data suggested that the total number of charged amino acid residues influences the mechanical properties of CATCH hydrogels to a greater extent than the ratio of cationic to anionic residues.
The hydrogel network architecture, which is governed by nanofiber entanglement and lateral association, could provide additional insight into the rheological properties of CATCH hydrogels. We assessed the network architecture by viewing the hydrogels in the hydrated state using cryogenic SEM. We observed that hydrogels formed from the CATCH(4+/4−) pair had the smallest pore diameters, as well as regions of mats with no observable porosity. Further, the walls of the pores in CATCH(4+/4−) hydrogels were thicker than those in the CATCH(6+/6−) hydrogels. Collectively, these data suggested that the combination of small pores and thick pore walls contributed to the increased stiffness of CATCH hydrogels. These observations were consistent with prior reports of other hydrogels systems where small pore sizes and thick pore walls were associated with stiffer networks.53,54 However, these correlations are complicated by the observation that the CATCH(4+/6−), CATCH(6+/4−), and CATCH(6+/6−) hydrogels had similar pore sizes and pore wall thicknesses, yet the charge-mismatched pairs formed gels with an ∼2-fold higher G′. Thus, network architecture alone cannot explain the rheological properties of CATCH hydrogels.
We observed that CATCH hydrogel shear thinning and recovery also vary with CATCH peptide pairing in a complex way. All of the CATCH hydrogels recovered viscoelasticity within 30 seconds after high strain disruption. However, only the CATCH(4+/4−) pair demonstrated full recovery of G′ after high-strain disruption. This suggested that the small pores and relatively thick pore walls of the CATCH(4+/4−) pair collectively dissipated the energy of applied forces more effectively than networks with a looser pore architecture. The viscosity of all of the CATCH hydrogels also recovered after high-shear. However, after three step-shear cycles, the CATCH(4+/4−) pair viscosity remained constant at a low shear rate, while viscosity changed over time at low shear for all other CATCH pairs. All CATCH hydrogels also demonstrated a significant change in network architecture after mock injection. Notably, the CATCH(4+/6−) hydrogel demonstrated the greatest change in pore diameter after mock injection and the lowest extent of recovery after high-strain disruption. In contrast, CATCH(6+/4), CATCH(6+/6−), and CATCH(4+/6−) hydrogels demonstrated comparable changes in pore wall thickness. Collectively, these data suggested that susceptibility to changes in pore diameter may inform rheological recovery of CATCH hydrogels, although this is again complicated by the observation that the CATCH(6+/6−) hydrogel recovered its initial G′ faster and to a greater extent than the CATCH(6+/4−) hydrogel, yet it also had a greater change in pore diameter after mock injection.
We hypothesized that nanofiber charge state may govern network architecture and hydrogel rheological properties by dictating the propensity for inter-nanofiber interactions, such as entanglement and lateral association. The charge state of the CATCH(+/−) nanofibers aligned with predictions based on the amino acid sequences of the peptides, with charge-matched pairs forming near-neutral assemblies and charge-imbalanced pairs forming charged assemblies. In light of the observed differences in the architecture and rheological properties of the CATCH(4+/4−) and CATCH(6+/6−) hydrogels, these ζ-potential measurements suggested that there was no direct correlation between nanofiber charge state and hydrogel properties.
From the observed differences in ζ-potential of the individual peptides one would likely predict that the nanofibers assembled from the CATCH(4+/4−) and CATCH(6+/6−) pairs would be net negative; however, this did not align with experimental observations in which the nanofibers had a net neutral ζ-potential. The observed net neutral ζ-potential for CATCH(4+/4−) and CATCH(6+/6−) nanofibers may be explained by recently published NMR measurements and computational simulations, which suggest that the cationic peptides are present in stoichiometric excess of the anionic peptides in CATCH nanofibers.34,35 Here, ζ-potential measurements indicated that lysine-rich CATCH(+) peptides have a relatively low absolute charge when alone, especially when compared to the CATCH(−) peptides. We propose that this increases the probability that CATCH(+) can couple with CATCH(+), whereas CATCH(−):CATCH(−) interactions are unfavorable, which leads to more cationic strands in the nanofibers. Indeed, recent reports have shown that the cationic CATCH peptides have greater propensity for self-association than their anionic counterparts, especially at high and low ionic strength.35 Collectively, these observations demonstrate that the interplay between CATCH peptide charge, β-sheet assembly, and nanofiber composition is highly complex, and suggest that advancing our understanding of the rheological behaviour of CATCH peptide hydrogels will require greater knowledge of these molecular-level aspects of the system.
Based on their similar rheological properties, network architectures, and lack of cytotoxicity, we characterized the host response to CATCH(4+/6−) and CATCH(6+/4−) hydrogels after injection into the subcutaneous space. Consistent with a prior report that characterized immunogenicity of charged peptide nanofibers,38 we observed that the anionic CATCH(4+/6−) hydrogel elicited weaker inflammation than the cationic CATCH(6+/4−) hydrogel. In vivo imaging with a labeled peptide indicated that the CATCH(4+/6−) hydrogel persisted at the injection site for a considerably longer duration than the inflammation observed after injection (Fig. S8†), suggesting that the hydrogels do not induce chronic inflammation. Prior reports demonstrated that soluble nanofibers of Q11, the parent peptide of the CATCH family, did not induce inflammation at a subcutaneous injection site.14 Here, we attribute the weak inflammation of the CATCH(4+/6−) hydrogel to the increase in density of material that was injected. Finally, no antibodies were raised against CATCH(4+) or CATCH(6−), despite being synthetic sequences that are foreign to the host, even when co-delivered with a potent adjuvant. Predictions made using the IEDB database estimated that CATCH(+) and CATCH(−) would bind poorly to MHC II of C57BL/6J mice, which supported the lack of an immune response observed in our studies. Further, prior reports demonstrated that Q11, the parent peptide of the CATCH family, was not immunogenic in C57BL/6J mice,11,16 which was again supported by IEDB predictions. Collectively, these data demonstrate that CATCH(4+/6−) hydrogels are relatively well-tolerated by both the innate and adaptive immune systems of the host, despite being non-native, “designer” sequences.
Conclusions
CATCH hydrogels are viscoelastic solids that undergo shear-thinning and recovery. The stiffness and extent of recovery depend on the pairs of peptides that are co-assembled. A CATCH(4+/6−) hydrogel injected into the subcutaneous space elicits weak inflammation, and humoral immunity is not raised against the CATCH peptides despite them being foreign to the host. Collectively, these data suggest that CATCH(4+/6−) hydrogels may be suitable as carriers for cells, proteins, or other therapeutic cargoes in vivo.Materials and methods
Preparation of peptide stock solutions
All peptides were synthesized and purified by Genscript. Cationic CATCH peptides (CATCH(4+) and CATCH(6+)) were added to deionized water at a working concentration of 20 mM determined by weight and dissolved with sonication. The anionic (CATCH(4−) and CATCH(6−)) peptides were dissolved by adding deionized water to yield a concentration of 20 mM determined by weight and adjusting the pH to ∼6.8 with sodium hydroxide. The concentration was verified using Phenylalanine absorbance (λ = 258 nm). Stock solutions were diluted to 2× final peptide concentration (2–16 mM based on experiment, described below) using 10× phosphate buffered saline (PBS) for a final concentration of 1× PBS.Hydrogel and nanofiber preparation
Equal volumes of each stock solution were mixed together to yield an equimolar peptide mixture at the final concentration. Mixtures were prepared at 0.5–1 mM and incubated 18 h at room temperature to allow nanofiber assembly.For oscillatory rheology, stock peptide solutions were heated for 5 min at 68 °C and 70 μL of each peptide solution was pipetted onto a single spot on a glass slide to create a 140 μL peptide mixture. These equimolar peptide mixtures were prepared over the range of 2–16 mM. A second glass slide was placed on top of a 2 mm spacer to form peptide mixtures into a cylindrical shape. Glass slides were coated with Sigmacote (Sigma-Aldrich) to render them hydrophobic. Peptide solutions between glass slides were cured for 1 h at room temperature to allow hydrogel formation.
For injections, stock peptide solutions were heated for 5 min at 68 °C, to facilitate mixing, and then sequentially drawn into the syringe barrel.
Oscillatory rheology
Cryo-scanning electron microscopy
Cryo-scanning electron microscopy (cryo-SEM), combines the high-performance imaging of scanning electron microscopy and cryogenic sample preparation techniques to investigate structures and materials in their native, hydrated state. Plastic polymers, hydrogels, and emulsified products are often either damaged by the electron beam or are not stable in the vacuum environment of an SEM. Such samples can be cryo-stabilized by freezing and transferred to a cryo-preparation chamber under vacuum.The cryo-SEM experiments were performed using a Quorum PP3010 T cryotransfer system (Quorum Technologies, Electron Microscopy Sciences) attached to a Hitachi SU5000FE VP-SEM (Hitachi High Technologies, America). Samples of 12 mM CATCH hydrogels were prepared between glass slides as described above. After curing, hydrogels were mounted by gentle transfer using a small spatula from the glass slide onto a carbon adhesive tab on a 10 mm copper stub (Electron Microscopy Science). Mock-injected samples of 12 mM CATCH hydrogels were prepared in syringes as described above. After curing, the hydrogels were injected onto glass slides, and then transferred to the carbon adhesive tab on a copper stub. Extra water was removed with a filter paper wedge, and then a small amount of colloidal graphite-OCT low temperature adhesive mixture was applied to the edge of the hydrogel.
After attaching the sample stub to the transfer shuttle, the hydrogel was vitrified in liquid ethane within a metal crucible surrounded by a liquid nitrogen reservoir. The ethane frozen sample was then rapidly plunged into the PrepDek® workstation liquid nitrogen slush at −210 °C under vacuum then immediately transferred to the cryo-preparation chamber. To reveal the hydrogel internal structure and to provide a clean surface uncontaminated by atmospheric water, the frozen hydrogel was fractured transverse along the top of the sample with a top mounted, micrometer cooled fracturing knife and longitudinal fracture along the hydrogel side using the side-mounted knife.
Specimen sublimation was initiated by increasing the temperature to −90 °C for 40 minutes. To further reveal pore wall details, the sublimation settings were optimized to −60 °C for 15 minutes without introduction of dehydration artifacts. The prep chamber temperature returned to −145 °C and the hydrogel was rendered conductive with sputter coat of platinum for 60 seconds at 10 mA in an argon atmosphere, approximately 3 nm thickness, then loaded into the nitrogen gas-cooled cold stage inside the SEM chamber at −140 °C. The hydrogel remained frozen during imaging at −140 °C, under high vacuum conditions, accelerating voltage 5 keV, emission current 187 μA with a working distance between 5 and 10 mm. At least three images were taken at random locations across each sample. Pore size and wall thickness were characterized by measuring 30 random locations using Image J (ImageJ software, NIH Image, MD, USA.). Average pore size is presented as mean ± standard deviation. Wall thickness is presented as mean ± standard deviation for each hydrogel.
Transmission electron microscopy
To view nanofibers, 1 mM equimolar peptide mixtures were prepared as described above. After 18 h incubation at room temperature, solutions of nanofibers were adsorbed onto Formvar/carbon grids (FCF400-CU-UB, Electron Microscopy Sciences) by placing grids on top of 10 μL sample solution for 5 min. Grids were dried by tilting onto a Kimwipe (Kimberly-Clark) and samples were negatively stained with a 2% aqueous solution of uranyl acetate for 30 s. All samples were imaged with a FEI Tecnai Spirit transmission electron microscope (FEI, The Netherlands) housed in the University of Florida Interdisciplinary Center for Biotechnology Research.ζ-Potential measurements
For ζ-potential measurements, 1 mM, 500 μM, and 200 μM solutions of CATCH peptides alone or in combination were prepared as described above, except 20 mM ammonium bicarbonate was used in place of 1 × PBS. Peptide samples, as well as a buffer blank, were analyzed using a Dynamic Light Scattering system (DLS; Particle Sizing Systems, Port Richey, FL, USA). Each sample was measured in triplicate with three runs in each cycle. It is worth noting that for samples of CATCH peptides alone, the hydrodynamic radius of an unfolded 1.5 kDa peptide should be about 1 nm, which is the above the detection limit of the machine (0.1 nm).Cytotoxicity studies
For cytotoxicity studies, equimolar mixtures of CATCH peptides at 0.5, 0.75, and 1 mM were prepared as described above. NIH 3T3 fibroblasts (ATCC) were seeded in a 96-well plate at 2 × 105 cells per well in 50 μL of cell culture media (Dulbecco's Modified Eagle Medium (Gibco), 10% fetal bovine serum (HyClone), 1% penicillin–streptomycin (Gibco)). Cells were incubated at 37 °C and 5% CO2. After 2 hours, 50 μL of each CATCH peptide mixture or 1× PBS was added to each well. Cells were incubated for 24 h. The solution in each well was removed and replaced with fresh medium, followed by adding 20 μL of CellTiter-Blue (Promega). Fluorescence was measured after 2 hours using a SpectraMax M3 plate reader (ex: 560 nm/em: 590 nm).In vivo studies
All animal procedures were performed in accordance with USDA guidelines, as well as the Guidelines for Care and Use of Laboratory of the University of Florida, according to protocols approved by the University of Florida Institutional Animal Care and Use Committee. Cohorts of 15–17 weeks old male and female C57BL/6J mice were purchased from Jackson Laboratories. For subcutaneous injections, mice were anesthetized using 2% isoflurane.To assess inflammation in male mice, cohorts (n = 5) received a subcutaneous injection of 40 μL of 12 mM CATCH(4+/6−) hydrogel, or λ-carrageenan (positive control) into the top of the foot. Swelling of the paws was measured using calipers on day 0 (before injection), 6 hours after injection, and on days 1, 2, 3, 4, 5 and 6 after injection.
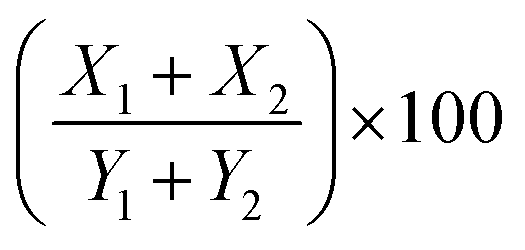 ; where (X1) is the thickness of infiltrate relative to the thickness of the right section of the paw (Y1), and (X2) is the thickness of infiltrate relative to the thickness of the left section of the paw (Y2). Finally, the scores were binned using the following scale: absent (0–9%), mild (10–19%), moderate (20–29%), and severe (>30%).
; where (X1) is the thickness of infiltrate relative to the thickness of the right section of the paw (Y1), and (X2) is the thickness of infiltrate relative to the thickness of the left section of the paw (Y2). Finally, the scores were binned using the following scale: absent (0–9%), mild (10–19%), moderate (20–29%), and severe (>30%).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100, 1:1000, 1:10000). Diluted serum was added to the blocked wells, and incubated for 1 h at room temperature. Serum was removed and plates were washed three times with PBST. 100 μL of peroxidase-conjugated goat anti-mouse IgG was added to each well (1:5000 in PBS with 1% BSA) and then incubated for 1 h at room temperature. Secondary antibody solution was removed and plates were washed five times with PBST. Then, plates were developed with 100 μL of TMB substrate for 30 min at room temperature. Finally, 100 μL of stop solution (0.16 M sulfuric acid) was added, and absorbance was measured at 450 nm using a SpectraMax M3 plate reader.
:100, 1:1000, 1:10000). Diluted serum was added to the blocked wells, and incubated for 1 h at room temperature. Serum was removed and plates were washed three times with PBST. 100 μL of peroxidase-conjugated goat anti-mouse IgG was added to each well (1:5000 in PBS with 1% BSA) and then incubated for 1 h at room temperature. Secondary antibody solution was removed and plates were washed five times with PBST. Then, plates were developed with 100 μL of TMB substrate for 30 min at room temperature. Finally, 100 μL of stop solution (0.16 M sulfuric acid) was added, and absorbance was measured at 450 nm using a SpectraMax M3 plate reader.
To further evaluate the immunogenicity of CATCH peptides, female mice received a 100 μL scruff injection of 1 mM CATCH(4+) or 1 mM CATCH(6−) peptide emulsified in TiterMax® (Sigma-Aldrich) (n = 5 per peptide) on day 0, and a 50 μL injection on day 28. Blood was drawn weekly from the facial vein, and the animals were euthanized on day 42. Sera was analyzed for anti-peptide total IgG antibodies via ELISA according to methods described above.
TT-GFP protein was used as positive control for the ELISA assay. Historical data showed this protein raised antibodies in C57BL/6J mice when given with an adjuvant.55
Peptide immunogenicity prediction
A bioinformatics method was used to estimate the immunogenicity of CATCH(4+) and CATCH(6−) in the C57BL/6J mouse. Peptide sequences were submitted to http://www.iedb.org and MHC binding predictions (H2-IAb background) were made on 07/06/2020 using the IEDB analysis resource consensus tool.56,57Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the National Institute of Health (5 R21 EB024762) and the National Science Foundation (CBET-1743432). We would like to thank Dr Blanka Sharma (Dept of Biomedical Engineering, University of Florida) for access to the DLS system, Dr Christine Schmidt (Dept of Biomedical Engineering, University of Florida) for access to the rheometer, and Dr Thomas Angelini (Dept of Mechanical and Space Engineering, University of Florida) for access to the rheometer.References
- P. J. S. King, M. Giovanna Lizio, A. Booth, R. F. Collins, J. E. Gough, A. F. Miller and S. J. Webb, Soft Matter, 2016, 6, 1915–1923 RSC.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS.
- S. Young, M. Wong, Y. Tabata and A. G. Mikos, J. Controlled Release, 2005, 109, 256–274 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS.
- Y. Li, J. Rodrigues and H. Tomás, Chem. Soc. Rev., 2012, 41, 2193–2221 RSC.
- J. D. Tang, C. Mura and K. J. Lampe, J. Am. Chem. Soc., 2019, 141, 4886–4899 CrossRef CAS.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS.
- M. E. Davis, J. P. M. Motion, D. A. Narmoneva, T. Takahashi, D. Hakuno, R. D. Kamm, S. Zhang and R. T. Lee, Circulation, 2005, 111, 442–450 CrossRef CAS.
- L. A. Haines-Butterick, D. A. Salick, D. J. Pochan and J. P. Schneider, Biomaterials, 2008, 29, 4164–4169 CrossRef CAS.
- J. P. Jung, A. K. Nagaraj, E. K. Fox, J. S. Rudra, J. M. Devgun and J. H. Collier, Biomaterials, 2009, 30, 2400–2410 CrossRef CAS.
- D. T. Seroski, A. Restuccia, A. D. Sorrentino, K. R. Knox, S. J. Hagen and G. A. Hudalla, Cell. Mol. Bioeng., 2016, 9, 335–350 CrossRef CAS.
- R. Liu and G. A. Hudalla, Molecules, 2019, 24, 1450 CrossRef CAS.
- J. Chen, R. R. Pompano, F. W. Santiago, L. Maillat, R. Sciammas, T. Sun, H. Han, D. J. Topham, A. S. Chong and J. H. Collier, Biomaterials, 2013, 34, 8776–8785 CrossRef CAS.
- G. A. Hudalla, T. Sun, J. Z. Gasiorowski, H. Han, Y. F. Tian, A. S. Chong and J. H. Collier, Nat. Mater., 2014, 13, 829–836 CrossRef CAS.
- J. S. Rudra, Y. F. Tian, J. P. Jung and J. H. Collier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 622–627 CrossRef CAS.
- J. H. Collier, B. H. Hu, J. W. Ruberti, J. Zhang, P. Shum, D. H. Thompson and P. B. Messersmith, J. Am. Chem. Soc., 2001, 123, 9463–9464 CrossRef CAS.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS.
- M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086–7098 RSC.
- A. Lampel, S. A. McPhee, H. A. Park, G. G. Scott, S. Humagain, D. R. Hekstra, B. Yoo, P. W. J. M. Frederix, T. De Li, R. R. Abzalimov, S. G. Greenbaum, T. Tuttle, C. Hu, C. J. Bettinger and R. V. Ulijn, Science, 2017, 356, 1064–1068 CrossRef CAS.
- Y. Tian, F. B. Polzer, H. V. Zhang, K. L. Kiick, J. G. Saven and D. J. Pochan, Biomacromolecules, 2018, 19, 4286–4298 CrossRef CAS.
- B. D. Ratner and S. J. Bryant, Annu. Rev. Biomed. Eng., 2014, 6, 41–75 CrossRef.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- H. D. Lu, M. B. Charati, I. L. Kim and J. A. Burdick, Biomaterials, 2012, 33, 2145–2153 CrossRef CAS.
- C. Yan, A. Altunbas, T. Yucel, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Soft Matter, 2010, 6, 5143–5156 RSC.
- S. Bera, S. Mondal, Y. Tang, G. Jacoby, E. Arad, T. Guterman, R. Jelinek, R. Beck, G. Wei and E. Gazit, ACS Nano, 2019, 13, 1703–1712 CrossRef CAS.
- P. Makam and E. Gazit, Chem. Soc. Rev., 2018, 47, 3406–3420 RSC.
- C. J. Bowerman and B. L. Nilsson, Biopolymers, 2012, 98, 169–184 CrossRef CAS.
- J. P. Jung, J. Z. Gasiorowski and J. H. Collier, Biopolymers, 2010, 94, 49–59 CrossRef CAS.
- L. W. Chang, T. K. Lytle, M. Radhakrishna, J. J. Madinya, J. Vélez, C. E. Sing and S. L. Perry, Nat. Commun., 2017, 8, 1–8 CrossRef.
- E. Prince and E. Kumacheva, Nat. Rev. Mater., 2019, 4, 99–115 CrossRef.
- S. Kyle, S. H. Felton, M. J. McPherson, A. Aggeli and E. Ingham, Adv. Healthcare Mater., 2012, 1, 640–645 CrossRef CAS.
- K. M. Wong, Y. Wang, D. T. Seroski, G. E. Larkin, A. K. Mehta, G. A. Hudalla, C. K. Hall and A. K. Paravastu, Nanoscale, 2020, 12, 4506–4518 RSC.
- Q. Shao, K. M. Wong, D. T. Seroski, Y. Wang, R. Liu, A. K. Paravastu, G. A. Hudalla and C. K. Hall, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4710–4717 CrossRef CAS.
- D. T. Seroski, X. Dong, K. M. Wong, R. Liu, Q. Shao, A. K. Paravastu, C. K. Hall and G. A. Hudalla, Commun. Chem., 2020, 3, 1–11 CrossRef.
- S. Ramachandran, Y. Tseng and Y. B. Yu, Biomacromolecules, 2005, 6, 1316–1321 CrossRef CAS.
- S. Zhang, F. Gelain and X. Zhao, Semin. Cancer Biol., 2005, 15, 413–420 CrossRef.
- Y. Wen, A. Waltman, H. Han and J. H. Collier, ACS Nano, 2016, 10, 9274–9286 CrossRef CAS.
- D. Sun and B. Yoo, LWT–Food Sci. Technol., 2015, 64, 205–211 CrossRef CAS.
- Z. A. Syahariza and H. Y. Yong, J. Food Meas. Charact., 2017, 11, 1586–1591 CrossRef.
- M. H. Chen, L. L. Wang, J. J. Chung, Y. H. Kim, P. Atluri and J. A. Burdick, ACS Biomater. Sci. Eng., 2017, 3, 3146–3160 CrossRef CAS.
- J. L. Mann, A. C. Yu, G. Agmon and E. A. Appel, Biomater. Sci., 2018, 6, 10–37 RSC.
- C. Diaferia, E. Gianolio, P. Palladino, F. Arena, C. Boffa, G. Morelli and A. Accardo, Adv. Funct. Mater., 2015, 25, 7003–7016 CrossRef CAS.
- J. A. Luckanagul, C. Pitakchatwong, P. Ratnatilaka Na Bhuket, C. Muangnoi, P. Rojsitthisak, S. Chirachanchai, Q. Wang and P. Rojsitthisak, Carbohydr. Polym., 2018, 181, 1119–1127 CrossRef CAS.
- A. J. Iqbal, A. L. F. Sampaio, F. Maione, K. V. Greco, T. Niki, M. Hirashima, M. Perretti and D. Cooper, Am. J. Pathol., 2011, 178, 1201–1209 CrossRef.
- C. J. Morris, Methods Mol. Biol., 2003, 225, 115–121 Search PubMed.
- O. Haçariz, G. Sayers, M. McCullough, M. Garrett, J. O'Donovan and G. Mulcahy, Vaccine, 2009, 27, 45–50 CrossRef.
- X. Deng, S. A. Meyers, T. L. Tollner, A. I. Yudin, P. D. Primakoff, D. N. He and J. W. Overstreet, J. Reprod. Immunol., 2002, 54, 93–115 CrossRef CAS.
- M. Yang, A. Clavijo, M. Li, K. Hole, H. Holland, H. Wang and M. Y. Deng, J. Immunol. Methods, 2007, 321, 174–181 CrossRef CAS.
- E. Caló and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267 CrossRef.
- M. C. Catoira, L. Fusaro, D. Di Francesco, M. Ramella and F. Boccafoschi, J. Mater. Sci. Mater. Med., 2019, 30, 115 CrossRef.
- A. K. Means and M. A. Grunlan, ACS Macro Lett., 2019, 8, 705–713 CrossRef.
- L. Yue, S. Wang, V. Wulf and I. Willner, Nat. Commun., 2019, 10, 4774 CrossRef.
- Q. Zhang, H. Lu, N. Kawazoe and G. Chen, Acta Biomater., 2014, 10, 2005–2013 CrossRef CAS.
- S. A. Farhadi, E. Bracho-Sanchez, M. M. Fettis, D. T. Seroski, S. L. Freeman, A. Restuccia, B. G. Keselowsky and G. A. Hudalla, Nat. Commun., 2018, 9, 4943 CrossRef.
- P. Wang, J. Sidney, C. Dow, B. Mothé, A. Sette and B. Peters, PLoS Comput. Biol., 2008, 4, e1000048 CrossRef.
- P. Wang, J. Sidney, Y. Kim, A. Sette, O. Lund, M. Nielsen and B. Peters, BMC Bioinf., 2010, 11, 568 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm01372b |
| This journal is © The Royal Society of Chemistry 2021 |