
Quantitative proteomics of epigenetic histone modifications in MCF-7 cells under estradiol stimulation†
Yechen
Hu‡
ab,
Hao
Jiang‡
a,
Baofeng
Zhao
*a,
Kaiguang
Yang
 a,
Zhen
Liang
a,
Lihua
Zhang
*a and
Yukui
Zhang
a
a,
Zhen
Liang
a,
Lihua
Zhang
*a and
Yukui
Zhang
a
aKey Laboratory of Separation Sciences for Analytical Chemistry, National Chromatographic R&A Center, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian 116023, China. E-mail: lihuazhang@dicp.ac.cn; baofzhao@dicp.ac.cn
bNanjing Medical University, Nanjing 211166, China
First published on 18th December 2020
Abstract
Estrogen exposure has already been considered to be associated with tumorigenesis and breast cancer progression. To study the epigenetic regulation mechanism in MCF-7 cells under estrogen exposure, which normally results in cell proliferation and malignancy, a stable isotope labeling of amino acid (SILAC) based quantitative proteomics strategy was used to analyse histone post-translational modifications (PTMs) and protein differential expressions. In total, we have unambiguously identified 49 histone variants and quantified 42 of them, in which two differentially expressed proteins were found to be associated with breast cancers. Through the quantitative analysis of 470 histone peptides with a combination of different PTM types, including methylation (mono-, di-, and tri-), acetylation and phosphorylation, 150 of them were found to be differentially expressed. Through the biological analysis of the quantification results of both histone PTMs and proteins in MCF-7 cells, we found that (1) the histone variants H10 and H2AV have an effect on the adjustment of the nucleosome or chromatin structure and activate target genes; (2) after estrogen receptor (ER) activation by estrogen, the recruitment of histone acetyltransferase KAT7 might affect the acetylation at the N terminal of H4 (K5, K8 and K12) and also result in cross-talk between different acetylation sites; (3) different expression of histone deacetylase HDAC2 and its nucleo-cytoplasmic transportation process is important in the regulation of histone acetylation in MCF-7 cells under estrogen exposure.
Introduction
Breast cancer, one of the most common cancers in women with high mortality, could result from many risk factors, including not only the mutation of specific oncogenes, but also the epigenetic changes caused by the environment. Among them, estrogen exposure has already been considered to be associated with tumorigenesis and breast cancer progression.1 Generally, after binding with estrogens, estrogens receptors (ERs) could recruit various co-regulatory proteins in the nucleus including histone acetylases (HATs), histone acetyl enzymes (HDACs), histone methyltransferases (HMTs) and Polycomb group proteins.2 These co-regulatory proteins could change the post-translational modifications (PTMs) of histones and subsequently regulate transcription and expression of genes with the estrogen response element (ERE).3 So far, a lot of studies have focused on relevant impact factors of ER activity,4 quantitative changes of global proteins5,6 and the epigenetic changes of breast cancer under estrogen exposure,7 such as the evaluation of global DNA methylation, long non-coding RNA, and histone PTMs.Histones play important roles in ordering the DNA into structural units and gene regulation. More than 20 types of PTMs have been found on different histone variants, which play key roles in epigenetic regulation of gene expression.8,9 Traditional strategies for histone PTM research, like the molecular biology technique, have already been used to reveal plenty of important epigenetic regulation mechanisms in the process of estrogen induced mammary carcinogenesis. Pogribny10et al. analysed the morphological changes and alterations of DNA methylation, histone methylation and microRNA expression after six weeks of 17β-estradiol (E2) treatment and found that loss of tri-methylation on histone H3K9 and H4K20 played a crucial role in E(2)-induced mammary carcinogenesis in ACI rats, especially in the tumour initiation process. Through chromatin immune-precipitation, Walker11 and Spink12 proved that the tri-methylation level of histone H3K27 decreased after E2 activated the ER pathway in breast cancer carcinogenesis as well as tissue development. Also, histone acetylation on both proteins13 and the modification site level,14 like H4K8 (ref. 15) and H4K12,16 has been found to be associated with epigenetic deregulations and gene activation.
However, antibody-based analysis tools are limited by antibody specificity and preparation difficulty and are not suitable for large scale profiling of different histone PTM sites. With the development of mass spectrometry (MS), proteomics solutions have gained much attention in the study of the histone code.17 Recently, Freitas18et al. combined MS based proteomics and chromatin immune-precipitation technologies to evaluate the overall phosphorylation level of histone H1 variants on different modification sites. They demonstrated the up-regulated phosphorylation level of H1.2 and H1.4 during estrogen induced MCF-7 cell proliferation and found T146pho to be a potential biomarker for breast cancer. However, few studies have tried to keep an eye on global changes of histone PTMs in breast cancer under estrogen exposure.
In this study, we focused on the quantitative proteomics of histone PTMs in MCF-7 cells under estrogen exposure, integrating SILAC labelling, C8 reversed-phase fractionation and high mass accuracy LC-MS/MS identification. Through the relative quantification of total proteins, histone variants and PTMs, including acetylation, methylation and phosphorylation, the estradiol stimulation related epigenetic mechanism and co-regulated protein pathway have been discussed.
Results and discussion
SILAC based quantitative proteomics is used for profiling of histone PTMs in MCF-7 cells under estradiol stimulation (Fig. 1), to find possible epigenetic changes and co-regulatory proteins and attempt to reveal the regulation mechanism.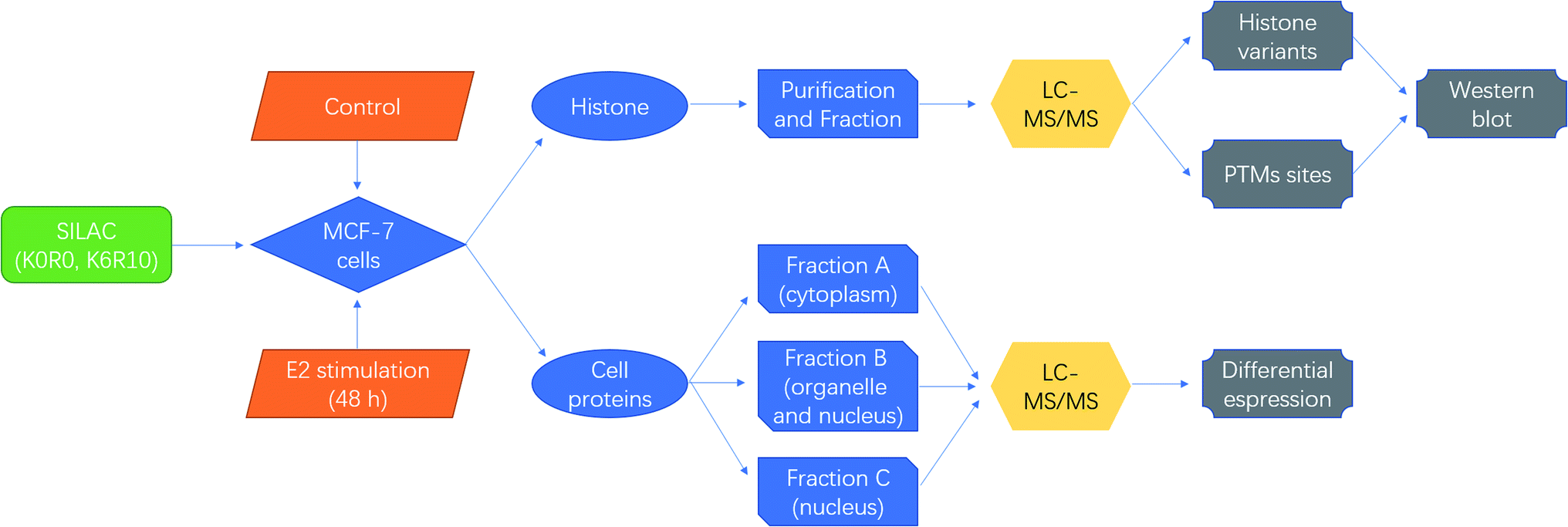 | ||
| Fig. 1 Schematic diagrams for quantitative proteomics of histone modifications in MCF-7 cells under estradiol stimulation. | ||
Quantification of histone variants
To increase the identification efficiency, histones were separated and purified into five fractions and further treated with lysine propionylation to improve LC performance and encourage MS detection. Through LC-MS/MS analysis, 49 histone variants were identified with high sequence coverage (ESI Table S1†). Among them, 42 histone variants were quantified with high confidence (Fig. 2). Consistent with previous reports,19 most histone variants remained unchanged during estrogen exposure except for histone H10 and H2AV which were up-regulated. This result was in accordance with a previous study where the H2A variant was up-regulated in the progression of certain breast cancer and involved in the process of ER related transcription activation.20 A follow-up study also found that the spliceosome of H2A helped to change the structure of chromatin and reduced the stability of nucleosomes.21 Hence, H2AV might also take part in such transcription activation in estradiol stimulated MCF-7 cells, resulting in proliferation and progression. Meanwhile, different variants of histone H1 have already been confirmed with high correlation with gene expression.22,23 As the most abundant chromatin-binding protein, the linker histone H1 binds to the nucleosomal core particle near the DNA entry and exit sites to protect the free linker DNA.24 The function of histone H1 has been underestimated for a very long time because it has been considered to be merely one of the structural chromatin components. However, it has become clear more recently that H1 has multiple functions in regulating development and disease progression by affecting heterochromatin formation, transcriptional regulation and epigenetics.25,26 There are 11 different H1 variants in mammals, including seven somatic-cell-specific variants and four germ-line-specific variants. These H1 variants act as specific gene regulators to differently affect gene expression rather than global regulators of gene expression, among which the H10 variant was reported to regulate more genes than the other linker histone H1 variants.27 Besides the known function in regulating cell differentiation, the H10 variant was reported to affect generation of epigenetic and functional intra-tumour heterogeneity in breast cancer recently.28 In this study, we found that the linker histone variant H10 was dramatically increased to be responsible for different gene expression in estradiol treated breast cancer MCF-7 cells. Thus, both histone variants H10 and H2AV could be considered potential clinical biomarkers or therapy targets for breast cancer progression.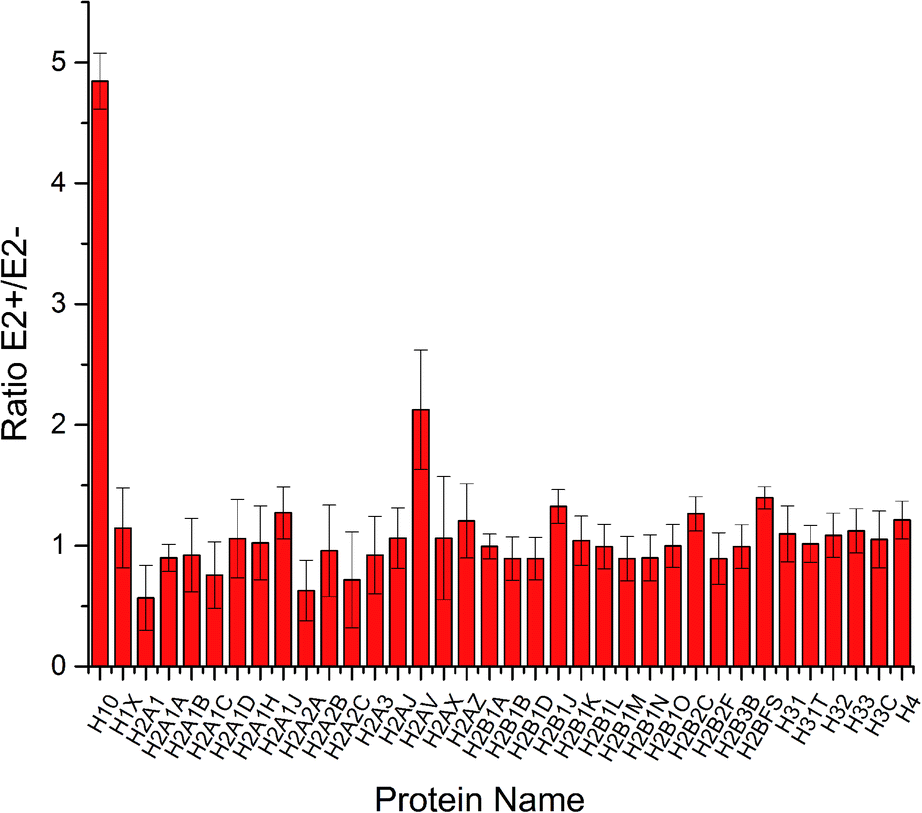 | ||
| Fig. 2 Quantification results of histone variants in MCF-7 cells under E2 stimulation. | ||
Quantification of proteins in MCF-7 cells under estradiol stimulation
To profile the comprehensive pathway map in such a process, proteins from different fractions in MCF-7 cells were quantified and analysed together (Fig. 1). In total, 1589, 1303 and 588 proteins were quantified successfully from fraction A, B and C, respectively. The differentially expressed proteins are listed in ESI Table S2,† and the related pathway is shown in Table S3.† Through KEGG analysis, it was found that most cytoplasmic proteins take part in the citrate cycle and glycolysis, which have already been reported to be related to the accommodation of histone deacetylases (HDACs) in both MCF-7 and MCF-10 cells.29Furthermore, through cluster and pathway analysis of up- and down-regulated proteins in different fractions (Fig. 3A), cluster 2 and 6 showed a significantly higher expression level in fraction B and C, which means certain proteins tend to be “enriched” in the organelles or nucleus. GO analysis demonstrated that most higher expressed proteins in cluster 2 are located in the nucleosome and are responsible for conjunction with DNA and construction of chromatin. For cluster 6, they prefer to play roles in the synthesis of nucleotides and ATP, located on the mitochondrial membrane. The KEGG pathway results showed that proteins in both clusters were involved in the process of neurodegenerative disorders like Huntington's disease30 and systemic lupus erythematosus31,32 (Fig. 3B). The common feature of these two diseases is the pathogenesis of epigenetic regulation of gene expression in the process, such as histone PTMs and DNA methylation, especially histone acetylation levels. Previous studies also demonstrated that HDAC inhibitors could be potential drugs for neurodegenerative disorders.33–35 These results suggested that estradiol induced breast cancer progression shared the same pathway with the pathogenesis of neurodegenerative disorders and lupus erythematosus, mainly related to changes in histone PTMs, which also gave us a hint that the histone acetylation level and HDACs played a key role in estrogen induced breast cancer progression.
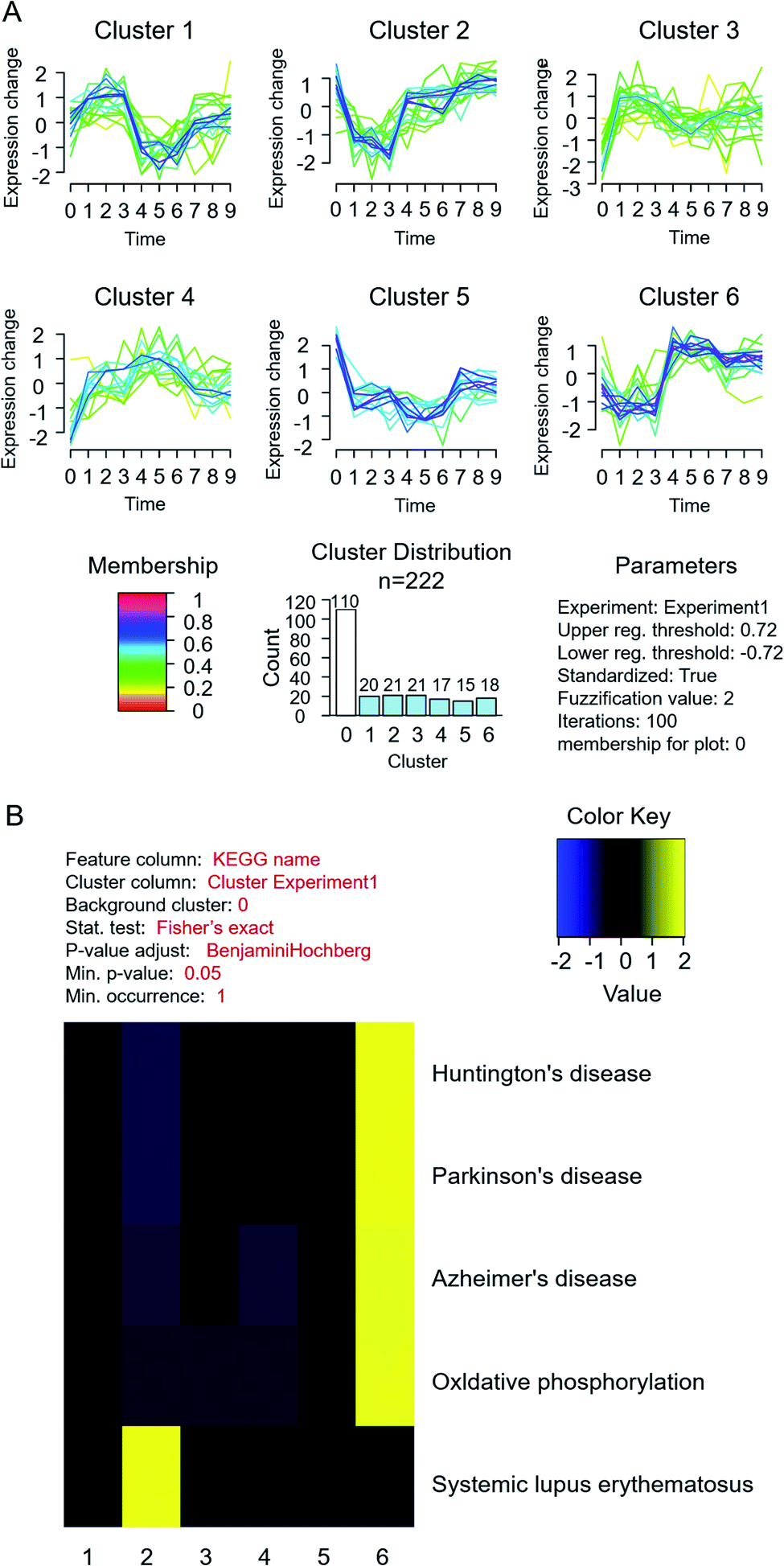 | ||
| Fig. 3 Cluster and pathway analysis of quantified differential proteins in MCF-7 cells under estradiol stimulation. (A) Cluster analysis; (B) KEGG pathway analysis. | ||
Quantification of histone PTMs
In total, 470 histone peptides with different PTMs were quantified, including mono- and di-methylation on lysine and arginine, acetylation and trimethylation on lysine, and phosphorylation on serine, threonine and tyrosine. As shown in ESI Fig. S2,† 150 histone PTM sites were found to be up- or down-regulated and distributed in different histone variants. The detailed quantification results for different histone PTM sites on H1, H2A, H2B, H3 and H4 are listed in ESI Tables S4, S5, S6, S7 and S8, respectively.† Many differential histone PTMs were consistent with previous research related to breast cancer proliferation and malignancy, such as K36 multi-methylation on H3 and T4 phosphorylation on H11 and H14, which have been considered to be cyclin-dependent kinases, and participated in the cell cycle and transcription.36 Besides, rarely reported modification sites were quantified with different expression after estradiol stimulation in MCF-7 cells, like H3Y99 phosphorylation and H4R23 mono-methylation. All these results indicated that changes of histone PTMs play crucial roles in estradiol stimulation in MCF-7 cells.Among all these different histone PTMs, most acetylation sites stayed at a high expression level after estrogen exposure in MCF-7 cells, especially for the N-terminal amino acids (AAs) of histone variants. In the N-terminal of histones H2AV and H2AZ, which share the most similar sequence (AGGKAGKDSGKAKAKAVSR and AGGKAGKDSGKAKTKAVSR), the acetylation levels of K5, K8 and K12 were all found to be up-regulated (Fig. 4). It is the same with the acetylation sites on histone H4, which possesses 4 available sites K5, K8, K12 and K16. H2AK5 acetylation is well-known for its transcription activation function in many biological processes.37 Different combinations of acetylation sites were found to be up-regulated inordinately (Fig. 5). The overexpression of acetylation on K5, K8 and K12 has already been reported in Kristy's study.38 However, their data didn't show the exact combination of acetylation sites, because the immuno-histochemistry technique is not adequate to give detailed information. In our quantitative proteomics results, besides the differentially expressed histone modification sites, multiple histone acetylation sites also presented competitive inhibition or synergistic action. Even though the acetylation occurred on a single site or a combination of multiple sites (2 or 3), the estrogen exposure led to a high level of acetylation expression in the H2 N-terminal peptide (Fig. 4A, blue line chart). However, the intensity of the H2 N-terminal peptide with acetylation sites was decreased (Fig. 4A, column chart), which might be caused by the stereo-hindrance effect of certain histone acetyltransferases (HATs) or HDACs. In addition, the abundance of H2 N terminal peptides with acetylation sites increased more than 5 times when di-methylation at K12 or tri-methylation at K16 was found (Fig. 4B). Similarly, two or more acetylation site combination H4 N-terminal peptide expression was increased in breast cancer cells under estrogen treatment (Fig. 5A, blue line chart). The overexpression of acetylation on H4K8 was further validated by western blotting (Fig. 5C). However, the intensity of the H4 N-terminal peptide with multiple acetylation sites was decreased compared with peptides without acetylation and with a single acetylation site (Fig. 5A, column chart). Besides, mono-methylation at R17 in the same peptide suppressed the estradiol-induced acetylation at H4 N-terminal peptide levels (Fig. 5B). All these results suggested that multiple histone acetylation sites as well as methylation sites in the same peptide presented competitive inhibition or synergistic action in estradiol induced breast cancer progression.
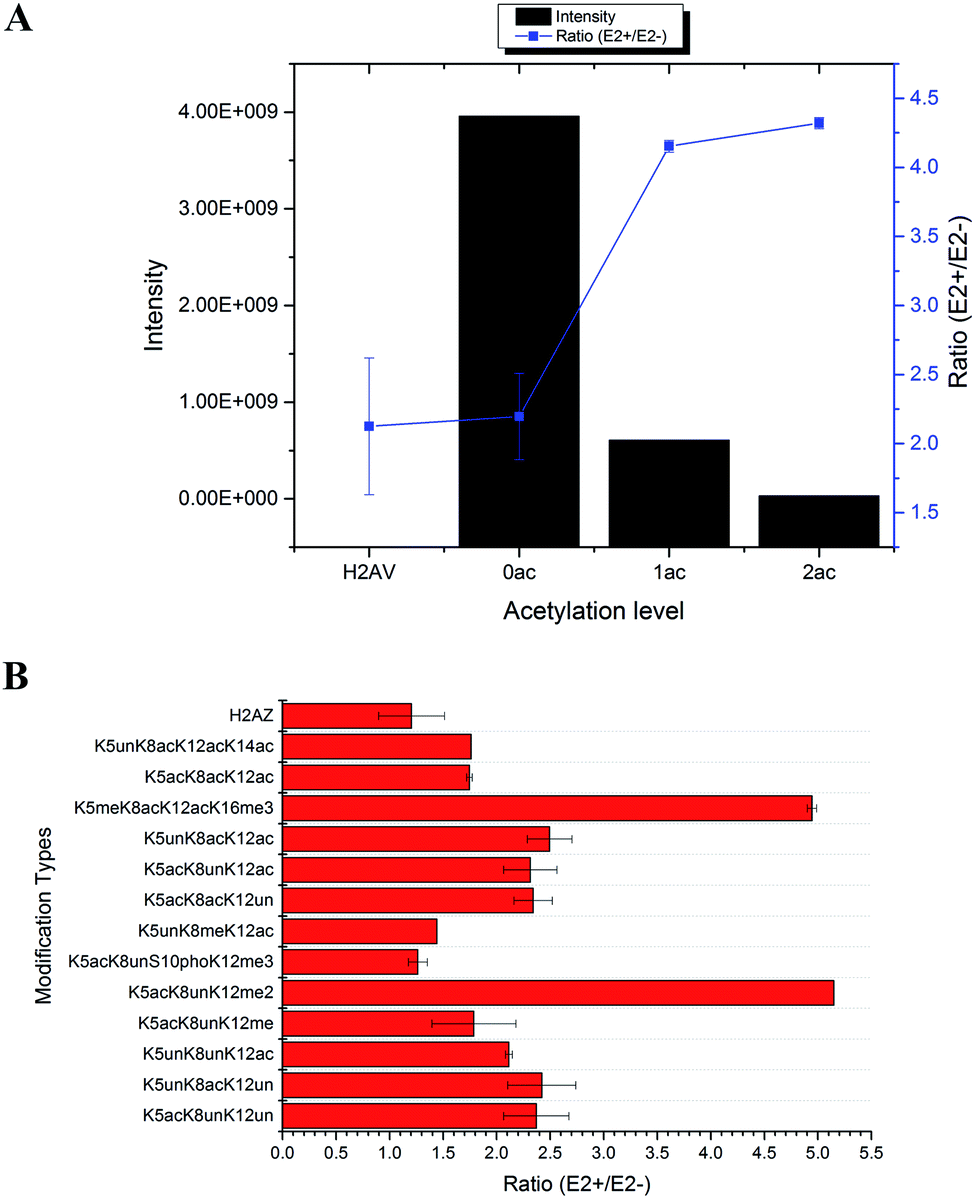 | ||
| Fig. 4 Quantification results of acetylation sites at the N-terminal of H2AV and H2AZ in MCF-7 cells under estradiol stimulation. (A) Intensity and ratio of peptides with different acetylation levels; (B) ratio of peptides with different combination sites of PTMs. | ||
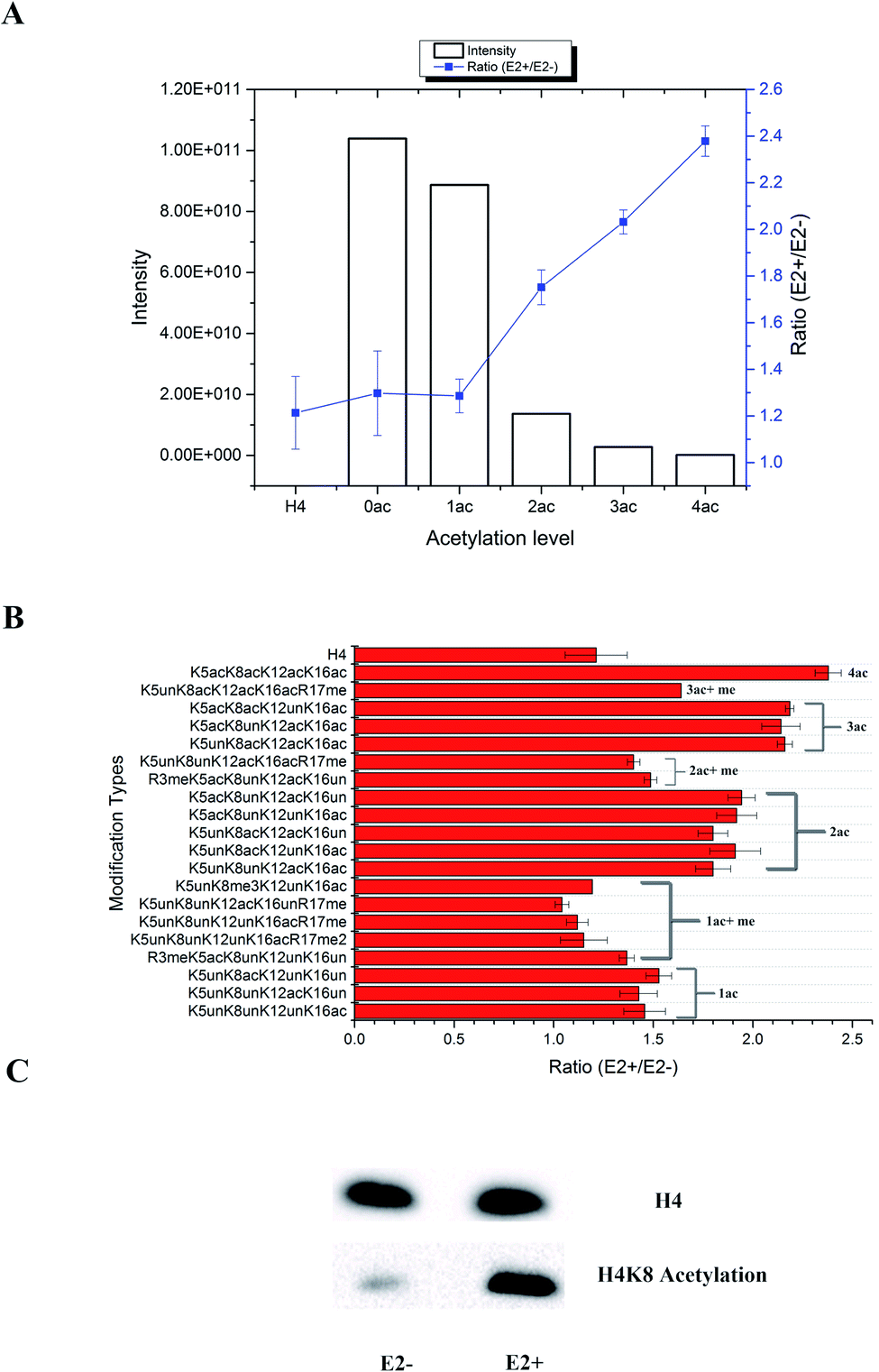 | ||
| Fig. 5 Quantification results of acetylation sites at the N-terminal of H4 in MCF-7 cells under estradiol stimulation. (A) Intensity and ratio of peptides with different acetylation levels; (B) ratio of peptides with different combination sites of PTMs; (C) western blotting validation of H4K8 acetylation. | ||
The acetylation level normally relies on the expression of certain HATs or HDACs. All the above results indicated that the estradiol stimulation might have an effect on different HDACs or HATs, resulting in different expression of acetylation site combinations. We summarize the quantification results of all histone PTM related enzymes in Table S9.† Two acetylation related enzymes, KAT7 and HDAC2, were found to be changed. KAT7 was already reported to have acetyltransferase activity for H4K5, H4K8 and H4K12, but not for H4K16, which might explain why peptides with H4K16 acetylation were expressed higher in treated MCF-7 cells. Meanwhile, the expression level of HDAC2 was different between the cytoplasm and nuclear fraction, indicating possible transportation of HDACs from the cytoplasm to the nucleus. It is noteworthy that HDAC inhibitors have already become a popular research topic for tumour targeting therapy, which has been proved to kill cancer cells,39 such as the FDA approved cancer treatment drugs SAHA and FK228. The HDAC inhibitor Vorinostat40 for treatment of drug resistant breast cancer is in Phase II clinical trials. Through quantitative proteomics experiments, it's obvious that multiple acetylation sites played more important and selective roles than single sites, at least during the estrogen related epigenetic regulation pathway. It is more likely that this unusual histone acetylation phenomenon was controlled by certain HDACs or HATs selectively. However, many factors including different ER/ER dimers as well as co-regulators in the ER protein complexes will selectively activate the different HATs/HDACs in breast cancer cells and the function of multiple HATs/HDACs on the ER protein complexes also remains unclear. Therefore, the detailed interactions between liganded ERs and epigenetic modifying proteins are essential to investigate, which would aid in the identification of new diagnostic and therapeutic targets for breast cancer.
Experimental
Materials and chemicals
Bovine serum albumin (BSA), phenylmethanesulfonyl fluoride (PMSF), urea, a protease inhibitor cocktail, 17β-estradiol (E2), sodium dodecyl sulfate (SDS), dithiothreitol (DTT), iodoacetamide (IAA), formic acid (FA), trifluoroacetic acid (TFA) and dextran coated charcoal were ordered from Sigma-Aldrich (St. Louis, MO, USA). A phosphatase inhibitor cocktail was bought from Thermo Fisher Scientific (Rockford, USA). Trypsin (bovine pancreas, TPCK treated) was bought from Promega (Madison, WI, USA). NP-40 and BCA assay kits were bought from Beyotime (Shanghai, China). Acetonitrile (ACN, HPLC grade) and methanol (HPLC grade) were bought from Merck (Darmstadt, Germany). Water was purified using a Milli-Q system (Millipore, Milford, MA, USA). All the other chemicals and solvents were analytical-grade from Kermel (Tianjin, China). All histone antibodies, including the antibodies of H4 and H4K8 acetylation were ordered from PTM Biolabs (Hangzhou, China).Fused-silica capillaries (250 μm i.d. × 375 μm o.d. and 150 μm i.d. × 375 μm o.d.) were purchased from Polymicro Technologies (Phoenix, AZ, USA). Reprosil-Pur C18-AQ particles (5 μm, 120 Å) were ordered from Dr. Maisch (Ammerbuch-Entringen, Germany). An ultrasonic cell disruptor was purchased from Cole-Parmer (Vernon Hills, IL, USA). A SpeedVac was purchased from Thermo-Fisher (San Jose, CA, USA). Polyvinylidene difluoride (PVDF) membranes, protein markers, a Trans-Blot Turbo and a ChemiDoc™ XRS+ system for western blotting were bought from BIO-RAD (Hercules, CA, USA).
MCF-7 cell culture and estrogen exposure
MCF-7 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). For SILAC analysis, MCF-7 cells were cultured in lysine- and arginine-deficient DMEM containing 10% dialyzed FBS and 1% penicillin/streptomycin, supplemented with 50 mg L−1 14N4/12C6-Argine and 12C6-Lysine for a light medium or 50 mg L−1 15N4/13C6-Argine and 13C6-Lysine for a heavy medium (Thermo-Fisher, San Jose, CA, USA). Cells were cultured for six passages to allow complete incorporation of heavy amino acids. After SILAC labelling, the culture medium was replaced with no phenol red DMEM containing 10% dialyzed FBS, which has already been treated with dextran coated charcoal to remove estrogen analogs. After 48 h of replacement, 17β-estradiol (E2) was added into the heavy medium for 48 h. MCF-7 cells were then digested with trypsin and rinsed with ice-cold 1× PBS 3 times. Equal numbers of light and heavy labelled cells were mixed together for further protein extraction.Sample preparation
Previously washed MCF-7 cell pellets (5 × 106) were lysed with 1 mL nuclei isolation buffer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NIB-250 buffer (15 mM Tris–HCl, 15 mM NaCl, 60 mM KCl, 5 mM MgCl2, 1 mM CaCl2, 250 mM sucrose, pH 7.5), containing 0.2% NP-40, 1% protease inhibitor cocktail, 1% phosphatase inhibitor cocktail, 1 mM PMSF and 10 mM sodium butyrate, and then homogenized with gentle pipetting for 10 min at 4 °C. The solution was then centrifuged at 1000 × g for 10 min at 4 °C. The nucleus pellet was washed with 500 μL NIB-250 buffer twice in an ice-bath. Then the supernatants were combined as cell protein fraction A, which contains mostly cytoplasmic components.
:NIB-250 buffer (15 mM Tris–HCl, 15 mM NaCl, 60 mM KCl, 5 mM MgCl2, 1 mM CaCl2, 250 mM sucrose, pH 7.5), containing 0.2% NP-40, 1% protease inhibitor cocktail, 1% phosphatase inhibitor cocktail, 1 mM PMSF and 10 mM sodium butyrate, and then homogenized with gentle pipetting for 10 min at 4 °C. The solution was then centrifuged at 1000 × g for 10 min at 4 °C. The nucleus pellet was washed with 500 μL NIB-250 buffer twice in an ice-bath. Then the supernatants were combined as cell protein fraction A, which contains mostly cytoplasmic components.
Slowly add 200 μL 0.2 M H2SO4 to the pellet, re-suspend by gentle pipetting and then incubate the sample with gentle shaking for 3 h at 4 °C. Then centrifuge the solution at 3400 × g for 10 min at 4 °C. Wash the pellet with 100 μL 0.2 M H2SO4. The pellet marked as cell protein fraction B contains mostly nuclear and organelle components.
Combine the supernatant and mix with a suitable volume of TCA to achieve a final concentration of 17%. Let the mixture precipitate on ice overnight. Then centrifuge at 3400 × g for 10 min at 4 °C. Discard the supernatant and rinse the tube with 100% ice-cold acetone. Air dry the pellet and dissolve the histones with 100 μL 20 mM NH4HCO3 (pH 8.0). Then centrifuge at 3400 × g for 30 min at 4 °C and transfer the supernatant to a new tube as histones. The pellet marked as cell protein fraction C contains mostly non-histone proteins in the nucleus. Measure the protein concentration by BCA assay.
According to a previous report,41 histones were purified and separated into different fractions by C8-RPLC, collected as histone H1, H2B, H2A, H4 and H3 (ESI Fig. S1†), and then derivatized with propionic anhydride, except for histone H1. All histone fractions were then dissolved in 100 μL 20 mM NH4HCO3 (pH 8.0). Trypsin was added into the protein solution with a substrate-to-enzyme ratio of 20:1 (m/m) and incubated at 37 °C overnight. Then, the reaction was terminated by adding FA to adjust the pH of the solution to 2–3.
For protein fraction A, the protein solution was denatured at 90 °C for 10 min. The samples were then reduced with 10 mM DTT at 56 °C for 1.5 h and alkylated with 20 mM IAA at room temperature for 40 min in the dark. Trypsin was then added at a substrate-to-enzyme ratio of 50:1 (m/m) and incubated at 37 °C overnight. For protein fraction B and C, the protein pellets were dissolved with 4% SDS, and then treated with the FASP process.
All the samples were stored at −80 °C before usage.
Nano-RPLC-ESI-MS/MS analysis
A nano-RPLC-ESI-MS/MS system was set up to realize quantitative proteomics research, consisting of Q Exactive mass spectrometer and Ultimate™ 3000 RSLC nano HPLC systems (Thermo Fisher, San Jose, CA, USA). The experimental conditions of nano-RPLC were as follows: the C18 separation column (75 μm i.d., 13 cm length) was home-made with Reprosil-Pur C18-AQ particles (5 μm, 120 Å); mobile phase, 2% (v/v) ACN containing 0.1% (v/v) FA (A), 98% (v/v) ACN containing 0.1% (v/v) FA (B); flow rate, 260 nL min−1; separation gradient: 0–2 min, 2–5% B; 2–72 min, 5–22% B; 72–102 min, 22–35% B; 102–115 min, 35–80% B; 115–125 min, 80% B.For Q Exactive analysis, the total ion chromatograms and mass spectra were recorded from m/z 300 to 1850 with Xcalibur software (version 2.3.0) in positive ion mode. The temperature of the ion transfer capillary and the spray voltage were set at 27.5 °C and 2.6 kV. MS/MS spectra were acquired using a data-dependent top 12 method and precursor selection was based on parent ion intensity from the survey scans. Higher-energy C-trap dissociation (HCD) was used for fragmentation. One full MS scan with a maximum injection time of 50 ms (resolution = 70000, at 200 m/z) was followed by 12 MS/MS scans in centroid mode (resolution = 17500, at 200 m/z), with a maximum injection time of 100 ms. The isolation window of the precursors was 2 Da. The normalized collision energy was set to 28. The dynamic exclusion duration was set to 25 s.
MS data analysis
For the analysis of proteins from fractions of the cell lysate, all MS/MS spectra raw files were searched against the human database from the UniProt repository using MaxQuant42 (version 1.5.2.8). Mass tolerances were set as 6 ppm for precursor ions and 20 ppm for fragments. All peptides were searched using fully tryptic cleavage constraints with up to 2 missed cleavages. Carbamidomethylation of cysteine was searched as a static modification (+57.02 Da). Oxidation on methionine (+15.99 Da) and acetylation on the protein N-terminal (+42.01 Da) was set as variable modifications. The results were filtered with FDR control (≤1% at both the spectral and protein level). For the quantification parameter settings, the SILAC label was selected, in which the heavy labels were marked as Lys6 and Arg10. Only unique and razor peptides were used for protein quantification. For evaluation of protein changes in different fractions, all raw files were searched together marked as a different “experiment” in the software setting.For the analysis of histone fractions, all MS/MS spectra raw files were searched against the human histone database (55 histone proteins) from the UniProt repository using MASCOT (version 2.4) and quantified using MASCOT Distiller43 (version 2.5). Mass tolerances were set as 7 ppm for precursor ions and 20 ppm for fragments. All peptides were searched using fully Arg-C cleavage constraints with up to 2 missed cleavages except that trypsin with up to 6 missed cleavages was used for the histone H1 fraction. Oxidation on methionine residues (+15.99 Da), deamidation on asparagine and glutamine (+0.98 Da), acetylation on the protein N-terminal (+42.01 Da) and lysine, methylation (mono-, di-, and tri-) on lysine, methylation (mono- and di-) on arginine, and phosphorylation on serine, threonine and tyrosine were considered variable modifications. The SILAC label was selected. All the quantification results were filtered as the following parameters: p value (0.5), expected cut-off (0.5), STD error (0.25), fraction (0.5), and correlation (0.7). And all peptides are shown in bold red. Peptide results with a quantification ratio higher than 10 or less than 0.1 were removed.
All the quantification results of proteins from cell lysate fractions were processed with Z-score normalization and one sample T-test using Perseus (http://coxdocs.org/doku.php?id=perseus:start). Ratios of histone PTM peptides were normalized using the median ratio of all histone unmodified peptides. A ratio higher than 1.65 or less than 0.606 was considered differential expression.
All the gene ontology (GO) information was obtained from the Uniprot database (http://www.uniprot.org/), including molecular function (GOMF), biological process (GOBP) and cellular component (GOCC). GproX44 was used for the cluster and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of quantified proteins. Enrichment evaluation was further realized using Fisher's exact test with a p-value cut-off of 0.05.
Western blotting
For western blotting of histone PTMs, equal amounts of histone proteins from different samples were resolved by 16% SDS-PAGE and transferred to PVDF membranes using a Trans-Blot Turbo semidry transfer system according to the manual (room temperature, 25 V, 1 A, 30 min). The membrane was then blocked for 1 h in Tris-buffered saline solution supplemented with 0.1% Tween-20 (TBST) and 5% skim milk powder and incubated with the primary antibody (1:1000) overnight at 4 °C. Membranes were then washed and incubated with goat anti-rabbit horseradish peroxidase conjugates (1:5000) for 30 min at room temperature. The blots were scanned with a ChemiDoc™ XRS+ system and digitalized with Image Lab™. The antibodies used in this study include the histone H4 mouse monoclonal antibody and the acetyl-histone H4 (Lys8) rabbit polyclonal antibody.
Conclusions
In summary, integrating SILAC labelling, C8 reversed-phase fractionation and high mass accuracy LC-MS/MS identification, the quantitative proteomics workflow has been used to reveal the histone PTM regulation mechanism in the process of estradiol stimulated MCF-7 cells. We have unambiguously quantified 42 core histone variants and more than 470 histone peptides with different PTM combinations. On this basis, we attempted to delineate the overall state of the histone PTM map containing some of the most important and usual modifications which are highly correlated with gene expression. Relative changes in histone variants (H10 and H2AV) have been found, elucidating that the estradiol stimulation could affect breast cancer cells possibly through the “inside” changes of the chromosome, leading to gene activation and further cancer cell proliferation. More importantly, histone PTMs play very significant regulatory roles in this process, especially for the acetylation at N-terminals of histone H4 and H2AV/H2AZ. Through the comparison results of MS analysis and western blotting, competitive inhibition or synergistic action of multiple histone acetylation sites has been presented on histone H4 (H4K5, H4K8, H4K12 and H4K16) and expression and translocation changes of histone PTM related enzymes (KAT7 and HDAC2) have also been found, which reveals that possible functional interaction exists among histone PTMs and co-regulatory proteins in the nuclear system of breast cancer. All these results provided a relatively comprehensive view of epigenetic histone PTMs of MCF-7 cells under estrogen exposure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Key Research and Development Program of China (2017YFA0505003 and 2017YFC1200401), National Natural Science Foundation (91543201, 21725506, 21874131 and 31770893), CAS Key Project in Frontier Science (QYZDY-SSW-SLH017), Cooperation agreement of the Innovation Academy for Precision Measurement Science and Technology and innovation program of science and research from the DICP, CAS.Notes and references
- A. Johansson, D. Palli, G. Masala, S. Grioni, C. Agnoli, R. Tumino, M. C. Giurdanella, F. Fasanelli, C. Sacerdote, S. Panico, A. Mattiello, S. Polidoro, M. E. Jones, M. J. Schoemaker, N. Orr, K. Tomczyk, N. Johnson, O. Fletcher, V. Perduca, L. Baglietto, P. A. Dugue, M. C. Southey, G. G. Giles, D. R. English, R. L. Milne, G. Severi, S. Ambatipudi, C. Cuenin, V. Chajes, I. Romieu, Z. Herceg, A. J. Swerdlow, P. Vineis and J. M. Flanagan, Clin. Epigenet., 2019, 11, 66 CrossRef.
- D. Chi, H. Singhal, L. Li, T. F. Xiao, W. H. Liu, M. Pun, R. Jeselsohn, H. S. He, E. Lim, R. Vadhi, P. Rao, H. Long, J. Garber and M. Brown, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11437–11443 CrossRef CAS.
- P. Y. Song, Y. R. Li, Y. Dong, Y. Y. Liang, H. N. Qu, D. Qi, Y. Lu, X. S. Jin, Y. T. Guo, Y. Y. Jia, X. Q. Wang, W. H. Xu and C. S. Quan, J. Exp. Clin. Cancer Res., 2019, 38, 534 Search PubMed.
- Y. Wei and J. Huang, J. Steroid Biochem. Mol. Biol., 2019, 191, 105380 CrossRef CAS.
- A. P. Drabovich, M. P. Pavlou, C. Schiza and E. P. Diamandis, Mol. Cell. Proteomics, 2016, 15, 2093–2107 CrossRef CAS.
- H. J. Johansson, F. Socciarelli, N. M. Vacanti, M. H. Haugen, Y. F. Zhu, I. Siavelis, A. Fernandez-Woodbridge, M. R. Aure, B. Sennblad, M. Vesterlund, R. M. Branca, L. M. Orre, M. Huss, E. Fredlund, E. Beraki, O. Garred, J. Boeke, T. Sauer, W. Zhao, S. Nord, E. K. Hoglander, D. C. Jans, H. Brismar, T. H. Haukaas, T. F. Bathen, E. Schlichting, B. Naume, T. Luders, E. Borgen, V. N. Kristensen, H. G. Russnes, O. C. Lingjaerde, G. B. Mills, K. K. Sahlberg, A. L. Børresen-Dale and J. Lehtiö, Nat Commun., 2019, 10, 1600 CrossRef.
- C. C. Faria, M. S. Peixoto, D. P. Carvalho and R. S. Fortunato, Oxid. Med. Cell. Longevity, 2019, 2019, 2514312 Search PubMed.
- S. A. Bennett, R. Tanaz, S. N. Cobos and M. P. Torrente, Transl. Res., 2019, 204, 19–30 CrossRef CAS.
- H. He, Z. P. Hu, H. Xiao, F. F. Zhou and B. B. Yang, Hum. Genomics, 2018, 12, 31 CrossRef.
- O. Kovalchuk, V. P. Tryndyak, B. Montgomery, A. Boyko, K. Kutanzi, F. Zemp, A. R. Warbritton, J. R. Latendresse, I. Kovalchuk, F. A. Beland and I. P. Pogribny, Cell Cycle, 2007, 6, 2010–2018 CrossRef CAS.
- T. G. Bredfeldt, K. L. Greathouse, S. H. Safe, M. C. Hung, M. T. Bedford and C. L. Walker, Mol. Endocrinol., 2010, 24, 993–1006 CrossRef CAS.
- N. A. Englert, R. J. Turesky, W. Han, E. E. Bessette, S. D. Spivack, M. Caggana, D. C. Spink and B. C. Spink, Biochem. Pharmacol., 2012, 84, 722–735 CrossRef CAS.
- L. F. Chen, Y. T. Lin, D. A. Gallegos, M. F. Hazlett, M. Gomez-Schiavon, M. G. Yang, B. Kalmeta, A. S. Zhou, L. Holtzman, C. A. Gersbach, J. Grandl, N. E. Buchler and A. E. West, Cell Rep., 2019, 26, 1174–1188 CrossRef CAS.
- Y. J. Gao, L. J. Chen, Y. L. Han, F. R. Wu, W. S. Yang, Z. Zhang, T. Huo, Y. M. Zhu, C. T. Yu, H. Kim, M. Lee, Z. Tang, K. Phillips, B. He, S. Y. Jung, Y. C. Song, B. K. Zhu, R. M. Xu and Q. Feng, Commun. Biol., 2020, 3, 165 CrossRef CAS.
- Z. G. Yuan, S. P. Chen, C. M. Gao, Q. Z. Dai, C. L. Zhang, Q. S. Sun, J. S. Lin, C. Guo, Y. Z. Chen and Y. Y. Jiang, Bioorg. Chem., 2019, 87, 200–208 CrossRef CAS.
- W. J. Liu, Y. Cui, W. Ren and J. Irudayaraj, Clin. Epigenet., 2019, 11, 16 CrossRef.
- S. Sidoli, M. Lopes, P. J. Lund, N. Goldman, M. Fasolino, M. Coradin, K. Kulej, N. V. Bhanu, G. Vahedi and B. A. Garcia, Sci. Rep., 2019, 9, 13613 CrossRef.
- S. W. Harshman, M. E. Hoover, C. Huang, O. E. Branson, S. B. Chaney, C. M. Cheney, T. J. Rosol, C. L. Shapiro, V. H. Wysocki, K. Huebner and M. A. Freitas, J. Proteome Res., 2014, 13, 2453–2467 CrossRef CAS.
- J. R. Wisniewski, M. Y. Hein, J. Cox and M. Mann, Mol. Cell. Proteomics, 2014, 13, 3497–3506 CrossRef CAS.
- D. Rangasamy, Curr. Med. Chem., 2010, 17, 3155–3161 CrossRef CAS.
- C. Bonisch, K. Schneider, S. Punzeler, S. M. Wiedemann, C. Bielmeier, M. Bocola, H. C. Eberl, W. Kuegel, J. Neumann, E. Kremmer, H. Leonhardt, M. Mann, J. Michaelis, L. Schermelleh and S. B. Hake, Nucleic Acids Res., 2012, 40, 5951–5964 CrossRef.
- L. Millan-Arino, A. B. Islam, A. Izquierdo-Bouldstridge, R. Mayor, J. M. Terme, N. Luque, M. Sancho, N. Lopez-Bigas and A. Jordan, Nucleic Acids Res., 2014, 42, 4474–4493 CrossRef CAS.
- G. P. Vicent, R. H. G. Wright and M. Beato, Biochim. Biophys. Acta, Gene Regul. Mech., 2016, 1859, 520–525 CrossRef CAS.
- C. L. Woodcock, A. I. Skoultchi and Y. Fan, Chromosome Res., 2006, 14, 17–25 CrossRef CAS.
- M. A. Willcockson, S. E. Healton, C. N. Weiss, B. A. Bartholdy, Y. Botbol, L. N. Mishra, D. S. Sidhwani, T. J. Wilson, H. B. Pinto, M. I. Maron, K. A. Skalina, L. N. Toro, J. Zhao, C. H. Lee, H. Hou, N. Yusufova, C. Meydan, A. Osunsade, Y. David, E. Cesarman, A. M. Melnick, S. Sidoli, B. A. Garcia, W. Edelmann, F. Macian and A. I. Skoultchi, Nature, 2020 DOI:10.1038/s41586-020-3032-z.
- N. Yusufova, A. Kloetgen, M. Teater, A. Osunsade, J. M. Camarillo, C. R. Chin, A. S. Doane, B. J. Venters, S. Portillo-Ledesma, J. Conway, J. M. Phillip, O. Elemento, D. W. Scott, W. Beguelin, J. D. Licht, N. L. Kelleher, L. M. Staudt, A. I. Skoultchi, M. C. Keogh, E. Apostolou, C. E. Mason, M. Imielinski, T. Schlick, Y. David, A. Tsirigos, C. D. Allis, A. A. Soshnev, E. Cesarman and A. M. Melnick, Nature, 2020 DOI:10.1038/s41586-020-3017-y.
- S. Bhan, W. May, S. L. Warren and D. B. Sittman, Gene, 2008, 414, 10–18 CrossRef CAS.
- C. M. Torres, A. Biran, M. J. Burney, H. Patel, T. Henser-Brownhill, A. S. Cohen, Y. Li, R. Ben-Hamo, E. Nye, B. Spencer-Dene, P. Chakravarty, S. Efroni, N. Matthews, T. Misteli, E. Meshorer and P. Scaffidi, Science, 2016, 353, aaf1644 CrossRef CAS.
- Y. F. Zhao, J. He, L. Yang, Q. C. Luo and Z. Liu, J. Breast Cancer, 2018, 21, 112–123 CrossRef.
- R. Ghosh and S. J. Tabrizi, Polyglutamine Disorders, 2018, vol. 1049, pp. 1–28 Search PubMed.
- E. F. Morand, R. Furie, Y. Tanaka, I. N. Bruce, A. D. Askanase, C. Richez, S. C. Bae, P. Z. Brohawn, L. Pineda, A. Berglind, R. Tummala and T.-T. Investigators, N. Engl. J. Med., 2020, 382, 211–221 CrossRef CAS.
- T. Dorner and R. Furie, Lancet, 2019, 393, 2344–2358 CrossRef.
- A. De Simone and A. Milelli, ChemMedChem, 2019, 14, 1067–1073 CrossRef CAS.
- S. S. Yang, R. Zhang, G. Wang and Y. F. Zhang, Transl. Neurodegener., 2017, 6, 19 CrossRef.
- M. Ziemka-Nalecz, J. Jaworska, J. Sypecka and T. Zalewska, J. Neuropathol. Exp. Neurol., 2018, 77, 855–870 CrossRef CAS.
- S. W. Harshman, N. L. Young, M. R. Parthun and M. A. Freitas, Nucleic Acids Res., 2013, 41, 9593–9609 CrossRef CAS.
- Y. Zhao and B. A. Garcia, Cold Spring Harbor Perspect. Biol., 2015, 7, a025064 CrossRef.
- K. Kutanzi and O. Kovalchuk, Cancer Biol. Ther., 2013, 14, 564–573 CrossRef CAS.
- N. Ma, Y. Luo, Y. Wang, C. Liao, W. C. Ye and S. Jiang, Curr. Top. Med. Chem., 2016, 16, 415–426 CrossRef CAS.
- N. S. Banerjee, D. W. Moore, T. R. Broker and L. T. Chow, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E11138–E11147 CrossRef CAS.
- S. Lin and B. A. Garcia, Methods Enzymol., 2012, 512, 3–28 CAS.
- S. Tyanova, T. Temu and J. Cox, Nat. Protoc., 2016, 11, 2301–2319 CrossRef CAS.
- S. Anand, M. Samuel, C. S. Ang, S. Keerthikumar and S. Mathivanan, Methods Mol. Biol., 2017, 1549, 31–43 CrossRef CAS.
- K. T. Rigbolt, J. T. Vanselow and B. Blagoev, Mol. Cell. Proteomics, 2011, 10, O110.007450 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ay02146f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |