
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biodegradable inkjet-printed electrochromic display for sustainable short-lifecycle electronics†
Manuel
Pietsch
ab,
Stefan
Schlisske
 ab,
Martin
Held
ab,
Noah
Strobel
ab,
Alexander
Wieczorek
ab and
Gerardo
Hernandez-Sosa
*ab
ab,
Martin
Held
ab,
Noah
Strobel
ab,
Alexander
Wieczorek
ab and
Gerardo
Hernandez-Sosa
*ab
aLight Technology Institute, Karlsruhe Institute of Technology, Engesserstr. 13, 76131 Karlsruhe, Germany. E-mail: gerardo.sosa@kit.edu
bInnovationLab, Speyererstr. 4, 69115 Heidelberg, Germany
First published on 4th November 2020
Abstract
The fabrication of electronics on the basis of biofriendly materials aims to counterbalance the negative trends conveyed by the short life-cycle of electronics. Furthermore, these materials open the possibility to develop optoelectronic technologies which will be in contact with the human body. In this work, we present an electrochromic display fabricated by resource- and energy-efficient digital printing techniques. The biodegradation of the device is certified under the ISO 14855 standard. The display comprises of a poly(3,4-ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS) electrochromic layer, a gelatin-based electrolyte and Au electrodes deposited on a cellulose di-acetate substrate. We investigate the impact of various naturally sourced ionic species on the ionic conductivity of the electrolyte and the figures of merit of the display. The printed devices show an electrochromic contrast of 32 ± 4% and switching times of 3.0 ± 1.4 s, comparable to the spincoated reference devices. The utilization of inkjet printing enables the fabrication of different device designs with individually addressable pixels. The display can be worn innocuously on the skin without loss of performance thanks to the self-adhesion properties of the gelatin hydrogel. The present work highlights the use of industrial relevant technology for the fabrication of truly ecofriendly optoelectronic systems.
An ecofriendly production and appropriate end-of-life of electronics are vital for a world which is currently being shaped by anthropogenic waste and its influence on climate and quality of life.1,2 In the coming years, the increasing use of electronic devices in commodity items as well as in upcoming internet of things (IoT) related technologies threaten to increase the already unsustainable production of electronic waste (e-waste).3 Among the new technologies currently being developed, wearable electronics are a fast-growing branch of research that promises to open new fields of applications by combining light-weight, conformability and mechanical robustness with sensing or optoelectronic functionalities.4,5 Some of these potential uses can be found in the field of electronic skin,6,7 electrocardio- and electromyography,8,9 on-skin diagnostics,10 energy harvesting11–13 and displays.14 These electronic devices will benefit from high throughput fabrication methods like printing techniques, which will enable its production in very large numbers. However, it is foreseeable that such electronic devices will be considered disposable as they might entail short-lifecycles due to the extenuating mechanical requirements or their inherent one-way use in biosensing or medical-related applications. In response to this problematic prognosis several approaches have recently been utilized for designing biodegradable and ecofriendly electronics.15 Materials such as silk, polycaprolactone, paper, and cellulose di-acetate have been used as bio-derived substrate alternatives.16–18 Naturally or synthetically acquired biodegradable materials have been used as passive and active components in various thin-film devices.19–22 Herewith enabling transistors,23 capacitors,24 sensors,25 batteries,26,27 or light-emitting devices28–30 with potential biodegradable properties. These examples demonstrate that there is a path and a will for the development of electronics with reduced environmental footprints. However, it is still necessary to develop economically viable fabrication schemes and disposal processes to ensure, under international standards, the sustainability of the devices as a whole.
Herein, we present a fully printed electrochromic (EC) display whose biodegradability was validated according to the International Standard ISO 14855. The electrochromic devices were fabricated on the basis of the biocompatible poly(3,4-ethylenedioxythiophene):polystyrene sulfonate (PEDOT:PSS) active layer and gold electrodes and a biodegradable gelatin-based electrolyte on a cellulose di-acetate (CA) substrate. The choice of inkjet printing provides an industrially established low-material-waste fabrication process with low-energy consumption which delivers the digital freedom of design and personalization necessary in wearable electronic applications.31,32 The composition of the biodegradable electrolyte was investigated and correlated to the figures of merit of the EC device. The finalized EC displays observe a reversible change between a transparent and a deep blue state with high contrast and fast switching time. Furthermore, its mechanical properties and self-adhesion provided by the combination of the CA substrate and the gelatin electrolyte enable its use as a conformable wearable display with potential use as a readout in smart electronics, health-monitoring and sensing platforms.
Displays are an integral element to many electronic devices as they provide a way to communicate the necessary information to the user. The utilization of electrochromic (EC) devices has demonstrated to be a feasible approach to fabricate low-cost displays33,34 exhibiting low operating voltages.35,36 Furthermore, their simple device architecture37,38 have enabled easy access to their fabrication by printing techniques.39–41 The low number of layers and materials necessary to fabricate EC devices also favor the replacement of hazardous components by biodegradable alternatives.42–44 In our case, we focused on the use of a gelatin-based electrolyte which has recently attracted attention in various research fields for its use in hydrogel configurations45,46 and as an ionic transport material.47–49 As an electrochromic layer, we utilized PEDOT:PSS, a material known for its electrochromic properties50–52 and outstanding biocompatibility.53,54 Complementary, the CA substrate and Au electrodes are established biodegradable and inert materials respectively.
Fig. 1a shows the device architecture used in this work consisting of individually addressable electrochromic PEDOT:PSS pixels, which are connected to gold working electrodes and surrounded by a gold counter electrode. The lateral structure is covered by the gelatin based electrolyte to enable the ion movement. Fig. 1b presents photographs of inkjet-printed electrochromic devices in the form of a battery indicator and a squid-shape, highlighting the freedom of design offered by digital printing. Furthermore, the circular features of the squid-pattern accentuate the advantages of this additive manufacturing technique over lithographic and mask-based processes, which would require multiple processing steps to produce this enclosed structure. As it can be seen in Fig. 1, the gelatin electrolyte (∼4 mm) is utilized to encapsulate the complete CA substrate beyond its edges, rendering the device self-adhesive. Thereby, we could realize an on-skin wearable device that conforms to the body shape and movement. Fig. 1c and Movie S1 of the ESI,† show the device under operation while mounted on a hand performing different movements without negatively affecting its functionality. Furthermore, Fig. 1d highlights the possible range of motions that can be performed while wearing the device without any detachment. The corresponding Movie S2 can be found in the ESI.†
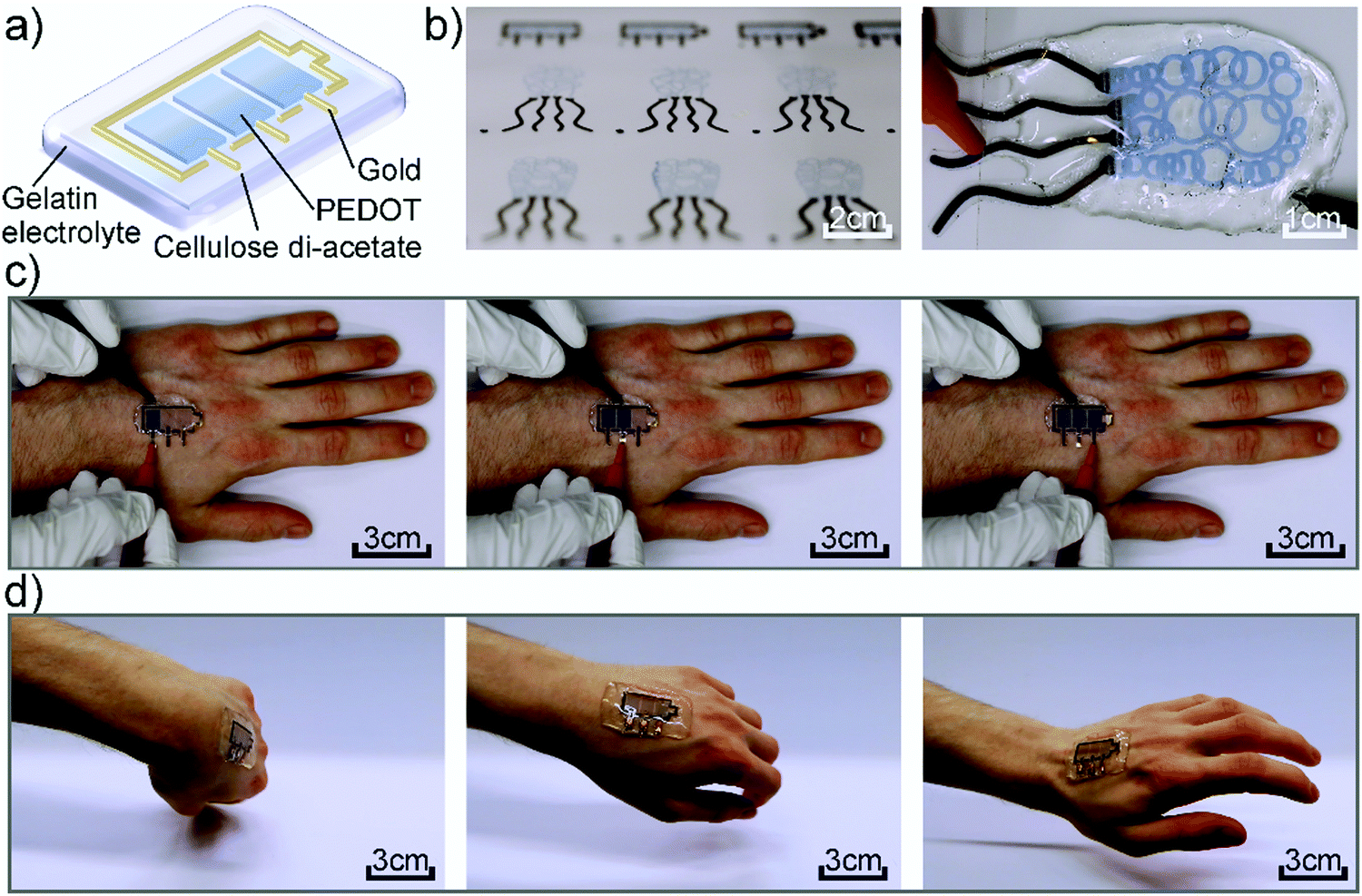 | ||
| Fig. 1 (a) Scheme of the used device architecture. (b) Pictures of multiple other printed devices, as well as the squid-shaped device under operation. (c) Printed battery indicator under operation while attached to a hand. (d) Conformability of the device while moving the hand. | ||
The electrolyte system is a crucial part of the EC device as its properties determine parameters such as switching times and device stability. In our case besides exhibiting high performance it should also be environmentally innocuous. Thus, we tested a variety of natural salts commonly found in food such as NaCl, CaCl2 and K2SO4 or in nutritional supplements such as choline salts. These salts were tested at different ratios in a hydrogel electrolyte based on glycerol, water, and gelatin (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7.5:1). The ionic conductivity at room temperature of the different electrolytes is shown in Fig. 2a. The corresponding Nyquist plots can be found in Fig. S1a in the ESI.† It can be observed that the ionic conductivity rises about two orders of magnitude in comparison to the as-received gelatin system. The high amount of glycerol in the used electrolyte promotes ion transport yielding ionic conductivities on the order of 10−7 S cm−1.
:7.5:1). The ionic conductivity at room temperature of the different electrolytes is shown in Fig. 2a. The corresponding Nyquist plots can be found in Fig. S1a in the ESI.† It can be observed that the ionic conductivity rises about two orders of magnitude in comparison to the as-received gelatin system. The high amount of glycerol in the used electrolyte promotes ion transport yielding ionic conductivities on the order of 10−7 S cm−1.
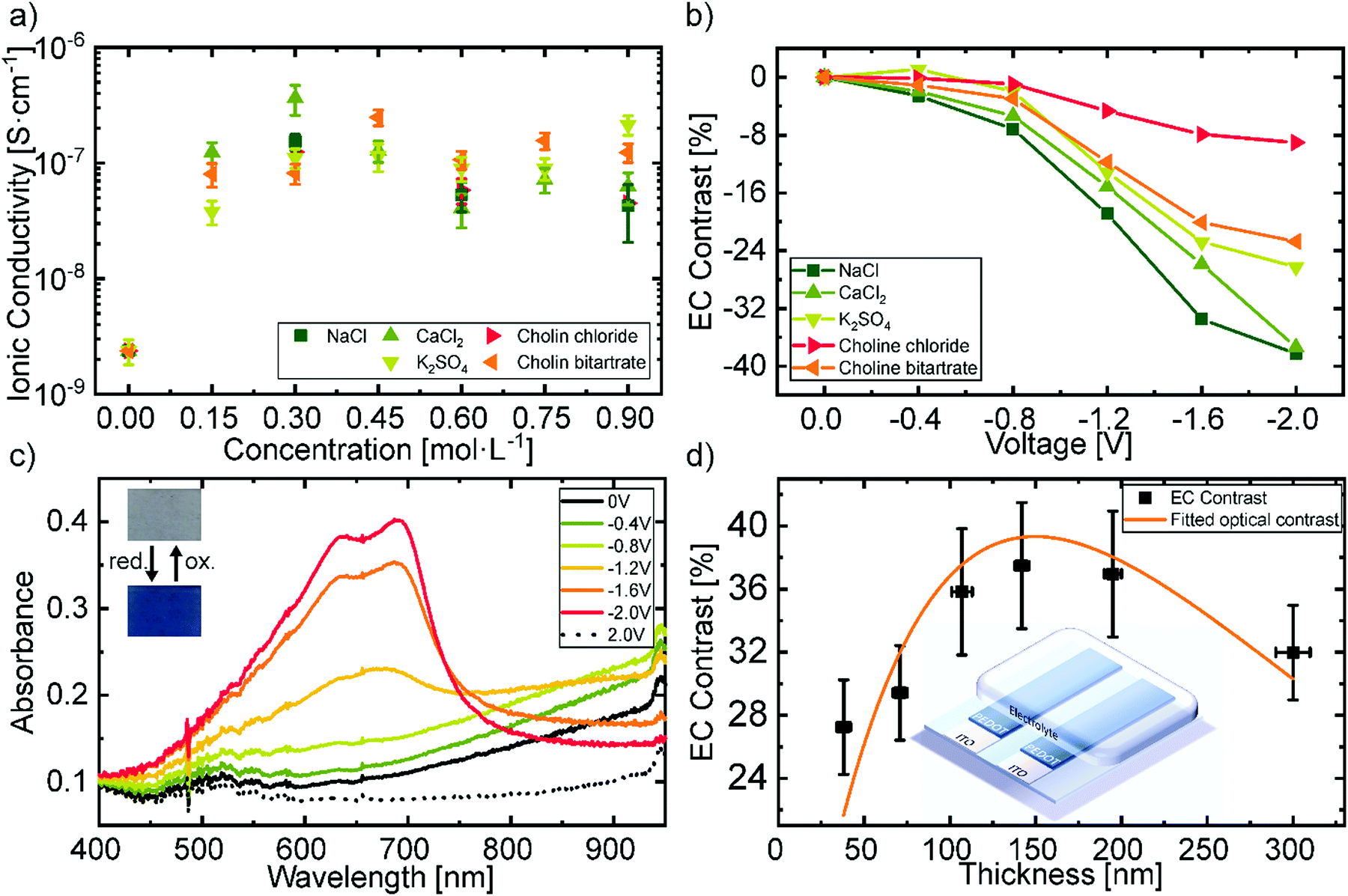 | ||
| Fig. 2 (a) Ionic conductivity of different salts in the gelatin electrolyte. (b) Electrochromic contrast at a wavelength of 685 nm by applied voltage with different electrolytes. (c) Change in absorbance of the PEDOT:PSS film under varying electrical potentials with NaCl in the electrolyte. (d) Electrochromic contrast (685 nm) at different thicknesses of the PEDOT:PSS layer with NaCl in the electrolyte. The inset shows the used device architecture. | ||
A reduction of up to two orders of magnitude in the ionic conductivity as well as a trend depending on the ion size was observed when utilizing only a tenth of the amount of glycerol (ESI,† Fig. S1b). Furthermore, the high glycerol content improves the ultimate tensile strength of the electrolyte against strain from 100% to 150%, as it can be seen in the destructive tensile test shown in Fig. S1c (ESI†). The results correspond well to previous reported studies, which identified the glycerol content as a crucial parameter for improving the ionic conductivity of gelatin electrolytes via better ion solvation,55 as well as for enhancing the mechanical properties of the gel by acting as a plasticizer.56
The concentration of the different biodegradable salt species was fixed at 0.15 mol L−1, to further investigate their influence on the change in transmittance. Fig. 2b shows the electrochromic contrast of the devices as a function of salt type and voltage. The EC is defined as the change in transmittance and was derived from the measurements shown in Fig. S2 in the ESI† at a wavelength of 685 nm. It can be observed that, larger ions like the choline salts need higher voltages to reach the same contrast as smaller sized salts, such as NaCl. This can be attributed to the ionic transport properties of the electrochromic layer depending on the ionic species.57 We chose NaCl as the salt in the hydrogel electrolyte for the printed devices, since it revealed the highest change in contrast at low voltages and did not greatly differ from the other salts in terms of ionic conductivity. Additionally, NaCl is one of the most common salts used in our daily life and is also available in large quantities which would be advantageous for a future upscaling. The cyclic voltammetry measurement of this electrolyte is shown in Fig. S1d in the ESI.† The redox potentials vs. an Ag/AgCl reference electrode are −1.9 V and 1.9 V respectively. Therefore, we assume sufficient stability of the electrolyte under operation, even though the potentials are not directly transferrable to the two-electrode system in the presented devices.
Fig. 2c displays the electrochromic behavior with the best contrast of the biocompatible PEDOT:PSS film utilizing the gelatin based electrolyte with 0.15 mol L−1 NaCl. In its initial state, the doping of PEDOT by the PSS typically results in the observed optical transmittance of ∼80% between 400 nm and 700 nm and an absorption band in the near-infrared region. Increasing the negative voltage at the attached electrode leads to a drift of cations into the electrochromic layer and the consequent reduction of PEDOT to its neutral state. At −2 V the absorbance peak in the near-infrared is minimized and two new peaks in the visible region rise, resulting in a blue coloration of the film. The absorbance peaks of the neutral PEDOT have their maximum at 695 ± 4 nm with a full-width half maximum (FWHM) of 32 ± 9 nm, as well as at 640 ± 2 nm with a FWHM of 140 ± 8 nm. The change in absorbance is reversible by changing the polarity of the applied voltage.
An important figure of merit for electrochromic display applications is the contrast between the colorized and the bleached state which exhibits a maximum at a specific layer thickness.58Fig. 2d shows the obtained EC contrast when varying layer thicknesses through different spincoating rotation speed. The difference of two exponential decreases, which corresponds to the absorption of the polymer in its bleached and colored state, was fitted to the values to obtain the layer thickness at which the contrast is maximal. The maximum electrochromic contrast was obtained for a layer thickness of 150 ± 6 nm. The initial values of the fitting parameters were taken from the exponential decrease of the transmittance with increasing thickness as shown in Fig. S3 in the ESI.†
In order to define the most suitable device architecture for the printed devices, we characterized the contact angles of the PEDOT:PSS dispersion and the gold nanoparticle ink on top of the as-received CA substrate and on top of each other (Fig. 3a and c). Both PEDOT:PSS and the Au ink are based on water and therefore exhibit low contact angles ∼7° on top of CA. However, when depositing the Au ink on top of PEDOT:PSS the contact angle increases to 35.4°. This difference of the contact angles corresponds to a gradient in surface free energies which causes a material-flow towards the higher SFE surface and therefore a dewetting of the Au ink on the edge of the PEDOT:PSS patterns. This results in a discontinuous Au film on the edge of PEDOT and CA indicated in the red circles in Fig. 3b. In contrast, the contact angles of the PEDOT:PSS dispersion on top of the printed and sintered Au layer yield 15.0° ± 0.5° (Fig. 3c), which do not produce an appreciable dewetting effect resulting in homogenous and smooth pattern formation (Fig. 3d). We, therefore, chose to first print the Au electrode structures and subsequently deposit the PEDOT:PSS for all printed devices to ensure a reliable process route and high yield. The layer thickness of the printed PEDOT:PSS is 150 ± 20 nm and 90 ± 10 nm for the printed gold.
 | ||
| Fig. 3 (a) Wetting behavior of the gold ink on CA (left) and on PEDOT:PSS film (right). (b) Photograph of the dewetting of the gold ink on the edge of the PEDOT:PSS layer indicated by the red circles. (c) Wetting behavior of PEDOT:PSS dispersion on CA (left) and on printed gold (right). (d) Homogenous layer formation on the edge of the PEDOT layer indicated by the circles when the gold electrode was printed first. (e) Coloration efficiency and sheet resistance of the electrodes of differently produced devices. (f) Microscope image of the printed gold electrode. | ||
The figures of merit of the printed devices and spincoated references are shown in Table 1. The EC contrast of 32 ± 4% at 685 nm, as well as the contrast ratio of 1.7 ± 0.3 of the printed devices, are comparable to the reference (Fig. 2d), and literature values.58 However, the coloration efficiency of 99 ± 6 cm2 C−1 is about 2.3 times lower than the coloration efficiency of the reference device. We assume, that this can be attributed to the different geometry and conductivity of the electrodes. Note, that the ITO electrode of the reference devices extends underneath the entire area of the EC pixel, whereas the printed gold electrode only connects to the pixels in a small area.
| EC contrast [%] | Contrast ratio | Coloration efficiency [cm2 C−1] | Switching timecolored [s] | Switching timebleached [s] | |
|---|---|---|---|---|---|
| Reference | 38 ± 2 | 1.6 ± 0.2 | 230 ± 13 | 2.5 ± 0.2 | 0.9 ± 0.1 |
| Printed | 32 ± 4 | 1.7 ± 0.3 | 99 ± 6 | 3.0 ± 1.4 | 1.6 ± 0.2 |
The coloration efficiencies, as well as the electrodes sheet resistance of a series of reference devices on glass and CA, are shown in Fig. 3e. In the case of evaporated gold electrodes on glass, the coloration efficiency of the devices with spincoated and printed PEDOT:PSS does not differ from the reference device on ITO. The same holds for the reference device with evaporated gold electrodes on CA. Only for the fully printed device with printed gold electrodes the coloration efficiency drops. At the same time, the sheet resistance of the electrode rises from 1.9 ± 0.2 Ω □−1 about two orders of magnitude to 112 ± 5 Ω □−1. Therefore, we conclude that the conductivity of the electrode has a greater influence on the coloration efficiency than the different geometries. As the microscope pictures of the printed gold layer in Fig. 3f indicate a fully closed layer, we relate the increase of sheet resistance to the low drying temperature of 60°, which is necessary to not melt the CA substrate. The higher sheet resistance of the printed Au electrode did affect the coloring and bleaching switching times as both were increased by more than 0.5 s for the printed device. The time at which the absorbance reached 90%, respectively 10% of its maximum was taken into account for this calculation.59
Fig. 4 presents a wearable display prototype incorporating the battery indicator beside a weather application, a watch, and a Wi-Fi indicator, to demonstrate the scalability of the devices and the control of the fabrication process. The encapsulation by the gelatin electrolyte beyond the CA substrate enables its wearability. Even though it is not stretchable, the display can be worn on an arm in an everyday-life situation due to the flexibility of the substrate in combination with the conformability and adhesion of the gelatin hydrogel, as depicted in Fig. 4a. The long term effects of the display on the skin, including breathability, water vapor transmission and irritations are still to be tested in collaboration with dermatological specialists.
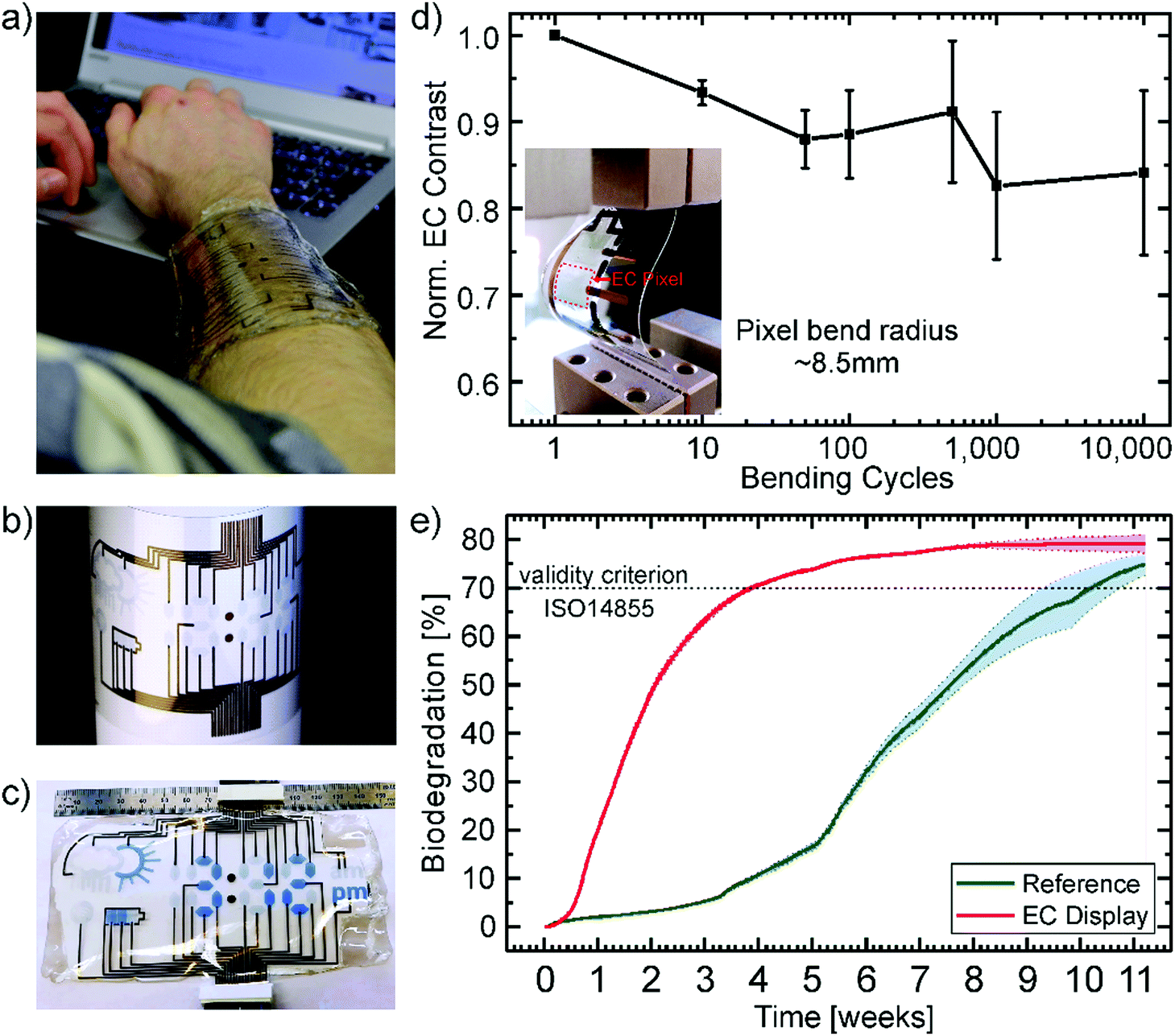 | ||
| Fig. 4 (a) Photograph of the display attached directly onto the skin in an everyday-life situation demonstrating its wearability and transparency. (b) Photograph of the inkjet-printed displays highlighting the scalability and control of the process to produce a multisegment display. (c) The final display under operation showing the time, the battery status and sunny weather. (d) Electrochromic contrast (average of 3 devices) normalized to the starting value after bending the device several times by a bend radius of 8.5 mm. The inset shows a photograph of the bended device. (e) Biodegradation of the EC display and microcellulose (particle size 20 μm) as a reference. | ||
Printed gold electrodes address each of the 33 individual segments of the flexible display for separate on/off switching (Fig. 4b). To demonstrate the control of the production processes and the potential for up-scaling, we printed up to four displays on one single A4-size substrate (ESI,† Fig. S4). Under operation, the bleached (OFF) PEDOT:PSS pixels, as well as their gold electrodes act as counter electrodes for the colored (ON) segments. As there will always be a combination of ON and OFF pixels, there is no necessity for individual counter electrodes for each pixel. An additional gold electrode in the form of the battery shape acts as the counter electrode for all other electrochromic segments to turn all pixels OFF. In Fig. 4c the display is depicted under operation. The segments which are OFF, appear transparent with a slight blueish hue since the printed PEDOT layers show transmission of around 80% in the visible region and the segments which are ON show a bright blue color.
Additionally, the display should withstand stress caused by body movement to be useful as a wearable device. The stress was simulated by bending the device with a bend radius of ∼8.5 mm. The devices were bent up to 10000 times while measuring the electrochromic contrast after certain steps. Fig. 4d shows the average value over three devices normalized to the initial EC contrast (43%). The contrast only decreased about 15.9% from its initial value after 10000 cycles of continuous bending. The inset shows a photograph of the bent device. Since there's no distinct linear, elastic regime the destructive tensile test data of the gelatin electrolyte in Fig. S1c (ESI†), we assume mainly plastic deformation of the electrolyte. This could be a possible explanation for the decrease in contrast after 10000 cycles of bending. Besides mechanical stretching, mechanical compression could occur for some wearable applications (e.g. around the wrist). The combination of both effects on the performance of the presented material system would need to be investigated in future work. Additionally, a degradation of the conductivity of PEDOT:PSS, which is about a few percent at these bending rations,60 could be partially responsible for the decrease in contrast. Alongside its mechanical stability, the lifetime of the devices was tested by applying a square wave between −2.4 V and 0.2 V with a switching period of 20 s. The electrochromic contrast over time is shown in Fig. S5 in the ESI.† It can be observed that after 30 min of stable operation, the contrast starts decreasing to around 12% at 60 min. It levels around 10% for the next 3 hours until it rises again. After 6 hours of continuous switching, the device contrast stabilizes to ∼16% from its initial value (34%). These behaviour is probably caused by the combination of several mechanisms under operation. This includes the degradation of the electrochromic material, the trapping of ions and the influence of the water inside the electrolyte.61–63 Furthermore, the drying-out of the electrolyte gel (i.e. loss of water content) under normal environmental conditions could affect the performance and adhesiveness of the device. Determining if the timescale of this effect is relevant to the device life-cycle would need to be investigated for the specific application.
The mechanical robustness of the displays and their performance under continuous operation demonstrate their suitability for short-lifecycle electronics applications in the fields of wearables. However, an ecofriendly end-of-life scenario of such devices is highly desirable to counteract the negative influence of their disposal on the environment. In particular, we consider this is a necessary feature for electronic technologies which could be fabricated in large numbers through up-scalable industrial methods such as printing technology. Therefore, we subjected the display to an independently-conducted biodegradability test according to the international standard ISO 14855 performed in a compost milieu at 25 °C (see report in the ESI†). This international standard is the closest to industrial relevant biodegradation methods for solid waste. Herein, the display and a microcellulose reference are inserted into bioreactors containing the equal amount of compost inoculum. The reference serves the purpose of validating the test. The carbon dioxide (CO2) produced by microorganisms metabolizing the organic material is measured over time. Additionally, bioreactors with sole inoculum serve to determine the CO2 production baseline originating from the compost itself. The absolute biodegradation is given by the ratio between the theoretically expected and the cumulated net CO2 production. The theoretical amount is calculated from the total amount of organic carbon of the sample. Fig. 4e shows the percentage of biodegradation as a function of time for the electrochromic display and the microcellulose (20 μm particle size) reference. The results clearly prove that the EC display exhibits a higher degradation rate than the reference finally reaching a plateau at 79.1% after about 9 weeks. The reference overcomes the threshold of 70% for a valid verification under the ISO 14855 standard after two additional weeks. Consequently, the biodegradability of the presented electrochromic display is confirmed under this international standard by showing higher biodegradation than the reference. This is to the best of our knowledge the first demonstration of an electronic display that has been certified as biodegradable by an independently made standardized test. Additional information can be found in the Experimental and the full official report is presented in the ESI.†
The remaining 20% of the display which has not degraded can be attributed to various materials. Even though PEDOT:PSS is presented as biocompatible in many literature reports, it failed a biodegradability test according to the ISO 14852. This test needs smaller amounts of material and is done in an aqueous environment, further details can be found in Fig. S6 in the ESI.† Nevertheless, the PEDOT:PSS only accounts for about 10−4% of the carbon in the entire device. Another material, whose degradation process is quite complex, is cellulose di-acetate. It is commonly stated to be a biodegradable material, however the acetyl side groups hinder the direct degradation by cellulase. Therefore, the biodegradation of CA often requires an additional deacetylation in advance.64 It is reasonable, that limited deacetylation happened in the compost environment at the set temperature of 25 °C and some part of the CA substrate did not degrade. The substrate itself contained around 10% of the total carbon in the display. The remaining 10% may be attributed to not yet degraded amounts of glycerol and gelatin by the time the experiment was stopped.
Conclusions
In summary, we present the successful fabrication of a digitally printed electrochromic display with an ISO-certified biodegradability demonstrating the usage of ecofriendly materials in electronics for short-lifecycle electronics. Furthermore, its flexibility, conformability and mechanical robustness render it ideal for its use in wearable applications. The electrochromic characteristics are consistent with literature values for PEDOT:PSS devices, in our case a contrast of 32 ± 4% at 685 nm and switching times of 3.0 ± 1.4 s by changing from a transparent state to a deep blue colored state. However, the device performance could still be enhanced in future work by optimizing the electrodes and increasing the color gamut by adding other biodegradable EC materials. Most importantly, the utilization of an industrial relevant fabrication such as inkjet printing underlines the potential and opportunities of biomaterials in disposable electronics and the potential of reducing the amount of electronic waste. Besides the obvious advantages that this digital printing technique offers for the personalization of wearable electronics, it also conveys a limited waste of resources as it only deposits materials locally on demand. This resource-efficiency adds up to the overall sustainability of the presented biodegradable display.To the best of our knowledge, this is the first demonstration of a biodegradable display which undergoes and successfully passed a standardized, independently performed degradation test. We believe that the exhibited innocuous degradability of this device could be extended to other electronic elements, which in combination with recyclability and reusability schemes, could help to reverse or to prevent some of the environmental impacts of e-waste paving the way towards greener electronics.
Experimental section
Materials
Choline chloride (99%), choline bitartrate (99%), potassium sulfate (K2SO4, 98%), calcium chloride (CaCl2, anhydrous 99%), sodium chloride (NaCl, 99%), gelatin (from porcine skin) and glycerol (99%) were purchased from Sigma Aldrich. Poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS, FHC Solar) was purchased from Heraeus. The gold ink (DryCure Au-J, 1010B) was obtained from C-Ink. The materials were used as received. The ITO coated glass substrates were purchased from Kintec Inc., the cellulose di-acetate foil (CA, 150 μm, clear) from Rachow-Kunststofffolien GmbH.Fabrication of the reference devices
The pre-patterned ITO coated glass substrates were cleaned in acetone and isopropanol in an ultrasonication bath for 10 minutes each. Subsequently, the substrates were treated for 5 minutes with argon plasma (Tetra 30, Diener electronics). PEDOT:PSS was deposited on the ITO coated substrates by spincoating at different rotation speeds (500–4000 rpm) for 60 s with an initial acceleration of 1000 rpm s−1 to obtain electrochromic layers of different thickness. The gelatin electrolyte was deposited while hot on the spincoated layer by drop-casting and cooled to room temperatures afterward. The device architecture is shown schematically in Fig. S7a in the ESI.† Devices with the same geometry as the printed ones were prepared by evaporating 80 nm of gold on previously cleaned glass substrates. PEDOT:PSS was spincoated with 2200 rpm and structured afterward or deposited by inkjet-printing. The device architecture is shown schematically in Fig. S7b (ESI†). The EC devices were characterized by UV-vis spectroscopy alongside with its corresponding I–V characteristics.Fabrication of the gelatin electrolyte
Gelatin was dissolved in deionized water at a concentration of 133 g L−1 together with the corresponding amount of salt at 50 °C on a hotplate. When the gelatin was fully dissolved, glycerol was added to achieve a 6:1 weight ratio of glycerol to gelatin (798 g L−1 glycerol). The solution was stirred for 30 min on a hotplate at 50 °C to achieve a homogeneous mixture. For the characterization of the ionic conductivity, the electrolyte is sandwiched between ITO covered glass substrates (ESI,† Fig. S7c).
Inkjet-printed devices
The CA was cleaned with isopropanol and treated with argon plasma for 5 minutes. The inkjet printing was performed at ambient cleanroom conditions with a Pixdro LP50 (Meyer-Burger). PEDOT:PSS was used for the electrochromic layer as received and was printed with a Fujifilm Dimatix 10pl cartridge with a printhead temperature of 27 °C and a custom-designed waveform. The substrate temperature was kept at room temperature. The gold ink was printed as received on the same printer with a Fujifilm Dimatix 10pl cartridge at a printhead temperature of 27 °C. The substrate was heated to 45 °C while printing the gold ink. The printed gold layer was sintered at 60 °C for 24 h in vacuum. The printed layers were additionally characterized by microscopy.Characterization methods
A PGSTAT128N (AutoLab) was used for the measurements of ionic conductivity by impedance spectroscopy and for the I–V characteristics of the EC devices in a two-electrode setup. The impedance measurement was conducted at a voltage of 500 mV in a frequency range from 100 kHz to 4 mHz. The used equivalent circuit is shown in the inset of Fig. S1a (ESI†). Furthermore, this equipment was used for the cyclic voltammetry measurement in a three-electrode setup with an Ag/AgCl reference electrode. For the UV-vis measurements an AvaSpec-ULS3648 (Avantes), a LR-2 spectrometer (LaserTack) and a light source ranging from 300 nm to 1100 nm were used in transmissive mode. The measurements of the contact angles were conducted with a DSA-100 measurement setup (Krüss). The layers were investigated by microscopy with a Nikon Eclipse 80i. The layer thickness was measured with a profilometer (Veeco Dektak 150). Tensile and bending tests were conducted with a force-displacement tester (FMT-317) from Alluris. The biodegradability test was done according to the international standard ISO 14855 at 25 °C by ISEGA Forschungs- und Untersuchungsgesellschaft mbh. In the official report the display is referred as “sample 1: Polymer- und Gold-beschichtete Cellulose-Di-Acetat Folie (eingegossen in Gelatine)”. The biodegradability test of PEDOT:PSS according to the international standard ISO 14851 was conducted by Frauenhofer UMSICHT.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of the German Federal Ministry of Education and Research (BMBF) through Grant FKZ: 03X5526 and thank M. Ruscello for fruitful discussion.Notes and references
- C. P. Balde, V. Forti, V. Gray, R. Kuehr and P. Stegmann, The global e-waste monitor 2017, 2017 Search PubMed.
- Z. Wang, B. Zhang and D. Guan, Nature, 2016, 536, 23–25 CrossRef CAS.
- B. Tansel, Environ. Int., 2017, 98, 35–45 CrossRef.
- T. Someya, Z. Bao and G. G. Malliaras, Nature, 2016, 540, 379–385 CrossRef CAS.
- J. C. Yang, J. Mun, S. Y. Kwon, S. S. Park, Z. Bao and S. S. Park, Adv. Mater., 2019, 31, 1904765 CrossRef CAS.
- T. Someya, T. Sekitani, S. Iba, Y. Kato, H. Kawaguchi and T. Sakurai, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9966–9970 CrossRef CAS.
- M. Kaltenbrunner, T. Sekitani, J. Reeder, T. Yokota, K. Kuribara, T. Tokuhara, M. Drack, R. Schwödiauer, I. Graz, S. Bauer-Gogonea, S. Bauer and T. Someya, Nature, 2013, 499, 458–463 CrossRef CAS.
- E. Bihar, T. Roberts, E. Ismailova, M. Saadaoui, M. Isik, A. Sanchez-Sanchez, D. Mecerreyes, T. Hervé, J. B. De Graaf and G. G. Malliaras, Adv. Mater. Technol., 2017, 2, 1600251 CrossRef.
- L. Inzelberg, M. D. Pur, S. Schlisske, T. Rödlmeier, O. Granoviter, D. Rand, S. Steinberg, G. Hernandez-Sosa and Y. Hanein, Flexible Printed Electron., 2018, 3, 045001 CrossRef CAS.
- J. Kim, A. S. Campbell and J. Wang, Talanta, 2018, 177, 163–170 CrossRef CAS.
- P. Cataldi, M. Cassinelli, J. A. Heredia-Guerrero, S. Guzman-Puyol, S. Naderizadeh, A. Athanassiou and M. Caironi, Adv. Funct. Mater., 2020, 30, 1907301 CrossRef CAS.
- Z. Wen, M. H. Yeh, H. Guo, J. Wang, Y. Zi, W. Xu, J. Deng, L. Zhu, X. Wang, C. Hu, L. Zhu, X. Sun and Z. L. Wang, Sci. Adv., 2016, 2(10), e1600097 CrossRef.
- T. F. O’Connor, A. V. Zaretski, S. Savagatrup, A. D. Printz, C. D. Wilkes, M. I. Diaz, E. J. Sawyer and D. J. Lipomi, Sol. Energy Mater. Sol. Cells, 2016, 144, 438–444 CrossRef.
- T. Yokota, P. Zalar, M. Kaltenbrunner, H. Jinno, N. Matsuhisa, H. Kitanosako, Y. Tachibana, W. Yukita, M. Koizumi and T. Someya, Sci. Adv., 2016, 2(4), e1501856 CrossRef.
- M. Irimia-Vladu, E. D. Głowacki, G. Voss, S. Bauer and N. S. Sariciftci, Mater. Today, 2012, 15, 340–346 CrossRef CAS.
- D.-H. Kim, J. Viventi, J. J. Amsden, J. Xiao, L. Vigeland, Y.-S. Kim, J. A. Blanco, B. Panilaitis, E. S. Frechette, D. Contreras, D. L. Kaplan, F. G. Omenetto, Y. Huang, K.-C. Hwang, M. R. Zakin, B. Litt and J. A. Rogers, Nat. Mater., 2010, 9, 511–517 CrossRef CAS.
- A. J. Morfa, T. Rödlmeier, N. Jürgensen, S. Stolz and G. Hernandez-Sosa, Cellulose, 2016, 23, 3809–3817 CrossRef CAS.
- S. W. Hwang, J. K. Song, X. Huang, H. Cheng, S. K. Kang, B. H. Kim, J. H. Kim, S. Yu, Y. Huang and J. A. Rogers, Adv. Mater., 2014, 26, 3905–3911 CrossRef CAS.
- M. Irimia-Vladu, E. D. Głowacki, P. A. Troshin, G. Schwabegger, L. Leonat, D. K. Susarova, O. Krystal, M. Ullah, Y. Kanbur, M. A. Bodea, V. F. Razumov, H. Sitter, S. Bauer and N. S. Sariciftci, Adv. Mater., 2012, 24, 375–380 CrossRef CAS.
- V. R. Feig, H. Tran and Z. Bao, ACS Cent. Sci., 2018, 4(3), 337–348 CrossRef CAS.
- H. Tran, V. Rachel Feig, K. Liu, H.-C. Wu, R. Chen, J. Xu, K. Deisseroth and Z. Bao, ACS Cent. Sci., 2019, 5, 1884–1891 CrossRef CAS.
- T. Lei, M. Guan, J. Liu, H.-C. Lin, R. Pfattner, L. Shaw, A. F. McGuire, T.-C. Huang, L. Shao, K.-T. Cheng, J. B.-H. Tok and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5107–5112 CrossRef CAS.
- M. Irimia-Vladu, P. A. Troshin, M. Reisinger, L. Shmygleva, Y. Kanbur, G. Schwabegger, M. Bodea, R. Schwödiauer, A. Mumyatov, J. W. Fergus, V. F. Razumov, H. Sitter, N. S. Sariciftci and S. Bauer, Adv. Funct. Mater., 2010, 20, 4069–4076 CrossRef CAS.
- G. Lee, S.-K. Kang, S. M. Won, P. Gutruf, Y. R. Jeong, J. Koo, S.-S. Lee, J. A. Rogers and J. S. Ha, Adv. Energy Mater., 2017, 7, 1700157 CrossRef.
- C. M. Boutry, A. Nguyen, Q. O. Lawal, A. Chortos, S. Rondeau-Gagné and Z. Bao, Adv. Mater., 2015, 27, 6954–6961 CrossRef CAS.
- X. Huang, D. Wang, Z. Yuan, W. Xie, Y. Wu, R. Li, Y. Zhao, D. Luo, L. Cen, B. Chen, H. Wu, H. Xu, X. Sheng, M. Zhang, L. Zhao and L. Yin, Small, 2018, 14, 1800994 CrossRef.
- X. Jia, C. Wang, V. Ranganathan, B. Napier, C. Yu, Y. Chao, M. Forsyth, F. G. Omenetto, D. R. Macfarlane and G. G. Wallace, ACS Energy Lett., 2017, 2, 831–836 CrossRef CAS.
- N. Jürgensen, M. Ackermann, T. Marszalek, J. Zimmermann, A. J. Morfa, W. Pisula, U. H. F. Bunz, F. Hinkel and G. Hernandez-Sosa, ACS Sustainable Chem. Eng., 2017, 5, 5368–5372 CrossRef.
- N. Jürgensen, J. Zimmermann, A. J. Morfa and G. Hernandez-Sosa, Sci. Rep., 2016, 6, 36643 CrossRef.
- J. Zimmermann, L. Porcarelli, T. Rödlmeier, A. Sanchez-Sanchez, D. Mecerreyes and G. Hernandez-Sosa, Adv. Funct. Mater., 2018, 28, 1705795 CrossRef.
- M. Gao, L. Li and Y. Song, J. Mater. Chem. C, 2017, 5, 2971–2993 RSC.
- S. Khan, L. Lorenzelli and R. S. Dahiya, IEEE Sens. J., 2015, 15, 3164–3185 Search PubMed.
- G. Cai, X. Cheng, M. Layani, A. W. M. Tan, S. Li, A. L. S. Eh, D. Gao, S. Magdassi and P. S. Lee, Nano Energy, 2018, 49, 147–154 CrossRef CAS.
- M. Pietsch, T. Rödlmeier, S. Schlisske, J. Zimmermann, C. Romero-Nieto and G. Hernandez-Sosa, J. Mater. Chem. C, 2019, 7, 7121–7127 RSC.
- K. Krishnamoorthy, A. V. Ambade, M. Kanungo, A. Q. Contractor and A. Kumar, J. Mater. Chem., 2001, 11, 2909–2911 RSC.
- R. H. Bulloch, J. A. Kerszulis, A. L. Dyer and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2015, 7, 1406–1412 CrossRef CAS.
- M. Pan, Y. Ke, L. Ma, S. Zhao, N. Wu and D. Xiao, Electrochim. Acta, 2018, 266, 395–403 CrossRef CAS.
- H. Kai, W. Suda, Y. Ogawa, K. Nagamine and M. Nishizawa, ACS Appl. Mater. Interfaces, 2017, 9, 19513–19518 CrossRef CAS.
- X. Cao, C. Lau, Y. Liu, F. Wu, H. Gui, Q. Liu, Y. Ma, H. Wan, M. R. Amer and C. Zhou, ACS Nano, 2016, 10, 9816–9822 CrossRef CAS.
- M. Layani, P. Darmawan, W. L. Foo, L. Liu, A. Kamyshny, D. Mandler, S. Magdassi and P. S. Lee, Nanoscale, 2014, 6, 4572–4576 RSC.
- G. Cai, P. Darmawan, M. Cui, J. Chen, X. Wang, A. L. S. Eh, S. Magdassi and P. S. Lee, Nanoscale, 2016, 8, 348–357 RSC.
- M. Pan, S. Zhao, L. Ma, N. Wu and D. Xiao, Sol. Energy Mater. Sol. Cells, 2019, 189, 27–32 CrossRef CAS.
- M. Fernandes, R. Leones, A. M. S. Costa, M. M. Silva, S. Pereira, J. F. Mano, E. Fortunato, R. Rego and V. De Zea Bermudez, Electrochim. Acta, 2015, 161, 226–235 CrossRef CAS.
- M. M. Silva, P. C. Barbosa, L. C. Rodrigues, A. Gonçalves, C. Costa and E. Fortunato, Opt. Mater., 2010, 32, 719–722 CrossRef CAS.
- A. Tijore, S. A. Irvine, U. Sarig, P. Mhaisalkar, V. Baisane and S. Venkatraman, Biofabrication, 2018, 10, 025003 CrossRef.
- X. Zhao, Q. Lang, L. Yildirimer, Z. Y. Lin, W. Cui, N. Annabi, K. W. Ng, M. R. Dokmeci, A. M. Ghaemmaghami and A. Khademhosseini, Adv. Healthcare Mater., 2016, 5, 108–118 CrossRef CAS.
- R. Ramadan, H. Kamal, H. M. Hashem and K. Abdel-Hady, Sol. Energy Mater. Sol. Cells, 2014, 127, 147–156 CrossRef CAS.
- H. Li, C. Han, Y. Huang, Y. Huang, M. Zhu, Z. Pei, Q. Xue, Z. Wang, Z. Liu, Z. Tang, Y. Wang, F. Kang, B. Li and C. Zhi, Energy Environ. Sci., 2018, 11, 941–951 RSC.
- H. Qin, R. E. Owyeung, S. R. Sonkusale and M. J. Panzer, J. Mater. Chem. C, 2019, 7, 601–608 RSC.
- R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2–18 CrossRef CAS.
- T. H. Lin and K. C. Ho, Sol. Energy Mater. Sol. Cells, 2006, 90, 506–520 CrossRef CAS.
- P. Andersson, R. Forchheimer, P. Tehrani and M. Berggren, Adv. Funct. Mater., 2007, 17, 3074–3082 CrossRef CAS.
- S. Stříteský, A. Marková, J. Víteček, E. Šafaříková, M. Hrabal, L. Kubáč, L. Kubala, M. Weiter and M. Vala, J. Biomed. Mater. Res., Part A, 2018, 106, 1121–1128 CrossRef.
- L. D. Garma, L. M. Ferrari, P. Scognamiglio, F. Greco and F. Santoro, Lab Chip, 2019, 19, 3776–3786 RSC.
- D. F. Vieira and A. Pawlicka, Electrochim. Acta, 2010, 55, 1489–1494 CrossRef CAS.
- M. Thomazine, R. A. Carvalho and P. J. A. Sobral, J. Food Sci., 2005, 70, 172–176 CrossRef.
- D. G. Frolov, M. M. Petrov, E. E. Makhaeva, M. L. Keshtov and A. R. Khokhlov, Synth. Met., 2018, 239, 29–35 CrossRef CAS.
- J. Padilla, V. Seshadri, G. A. Sotzing and T. F. Otero, Electrochem. Commun., 2007, 9, 1931–1935 CrossRef CAS.
- J. Jensen, M. Hösel, A. L. Dyer and F. C. Krebs, Adv. Funct. Mater., 2015, 25, 2073–2090 CrossRef CAS.
- C.-K. Cho, W.-J. Hwang, K. Eun, S.-H. Choa, S.-I. Na and H.-K. Kim, Sol. Energy Mater. Sol. Cells, 2011, 95, 3269–3275 CrossRef CAS.
- Y. Ren, W. K. Chim, L. Guo, H. Tanoto, J. Pan and S. Y. Chiam, Sol. Energy Mater. Sol. Cells, 2013, 116, 83–88 CrossRef CAS.
- M. Moser, T. C. Hidalgo, J. Surgailis, J. Gladisch, S. Ghosh, R. Sheelamanthula, Q. Thiburce, A. Giovannitti, A. Salleo, N. Gasparini, A. Wadsworth, I. Zozoulenko, M. Berggren, E. Stavrinidou, S. Inal and I. McCulloch, Adv. Mater., 2020, 32, 2002748 CrossRef CAS.
- X. Wang and E. Smela, J. Phys. Chem. C, 2009, 113, 369–381 CrossRef CAS.
- J. Puls, S. A. Wilson and D. Hölter, J. Polym. Environ., 2011, 19, 152–165 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0tc04627b |
| This journal is © The Royal Society of Chemistry 2020 |