Nitrile-substituted 2-(oxazolinyl)-phenols: minimalistic excited-state intramolecular proton transfer (ESIPT)-based fluorophores†
Received
14th February 2020
, Accepted 30th April 2020
First published on 4th May 2020
Abstract
Herein, we present minimalistic single-benzene, excited-state intramolecular proton transfer (ESIPT)-based fluorophores as powerful solid-state emitters. The very simple synthesis gave access to all four regioisomers of nitrile-substituted 2-(oxazolinyl)-phenols (MW = 216.1). In respect of their emission properties they can be divided into aggregation-induced emission enhancement (AIEE) luminophores (1-CN and 2-CN), dual state emission (DSE) emitters (3-CN) and aggregation-caused quenching (ACQ) fluorophores (4-CN). Remarkably, with compound 1-CN we discovered a minimalistic ESIPT-based fluorophore with extremely high quantum yield in the solid state ΦF = 87.3% at λem = 491 nm. Furthermore, quantum yields in solution were determined up to ΦF = 63.0%, combined with Stokes shifts up till 11.310 cm−1. Temperature dependent emission mapping, crystal structure analysis and time-dependent density functional theory (TDDFT) calculations gave deep insight into the origin of the emission properties.
Introduction
Highly emissive organic fluorophores have received great attention due to their intriguing and multifarious properties in the fields of biological imaging,1 fluorescent sensors2 and organic light-emitting diodes (OLEDs).3 Commonly, three general strategies for the design of organic fluorescent frameworks are widely used – extended π-conjugated systems,4 donor–acceptor (D–A) systems5 and aggregation induced emission (AIE).6,7 Extended π-conjugated systems based on a rigid and flat scaffold suffer from decreased solubility in conventional solvents. Furthermore, upon aggregation either in highly concentrated solution or in the solid state, their tendency of intermolecular π–π-stacking leads to radiationless quenching processes lowering the emission efficiency which results in low quantum yields. Contrarily, D–A systems are mostly constructed as small π-conjugated scaffolds, which are extensively used as fluorescent dyes and as luminophores in optoelectronic devices. The design and application of AIE-based luminophores have captured great attention in the recent two decades, since such structures such as siloles,8 stilbenes9 or arylethylenes,10 show no or solely weak emission in solution but strong luminescence in the aggregated-state. With regard to biological application of organic fluorophores various properties are desirable. Small structures avoid impairing metabolite traffic within cells,11 solid-state emission prevent fluorescent quenching12 and a large Stokes’ shifted emission minimizes the tendency of self-reabsorption.
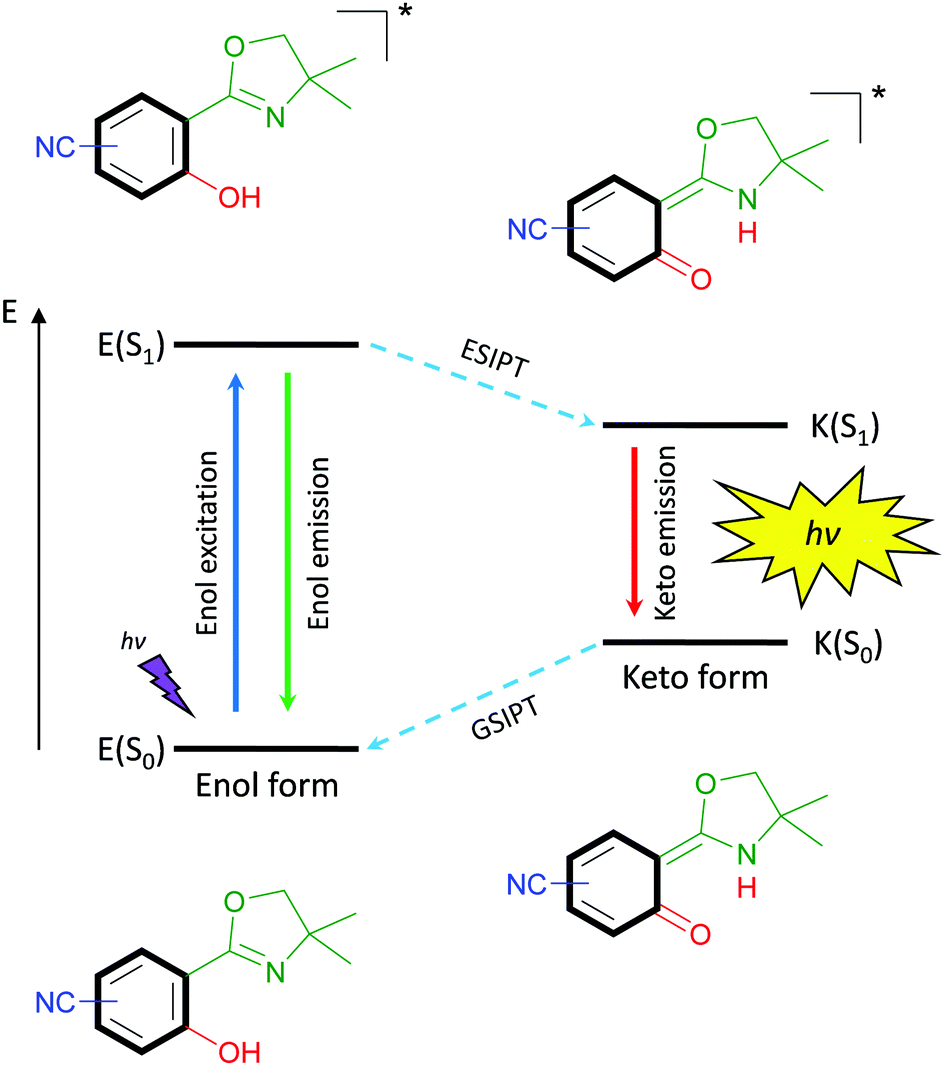
The latter property is remarkably strong for excited-state intramolecular proton transfer (ESIPT)-based luminophores.13 Such fluorophores are underlying to an enol–keto-phototautomerism cycle consisting of four levels, which causes the bathochromic-shifted emission and dual emission behavior (Scheme 1). Within this cycle, upon light absorption in the enol form, a phototautomerization on the subpicosecond timescale,14 emission of the keto form and ground-state intramolecular proton transfer (GSIPT) takes place.
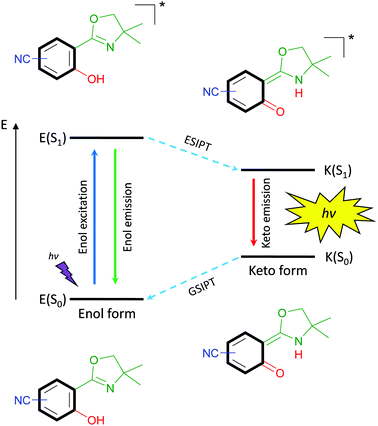 |
| | Scheme 1 Schematic representation of the ESIPT-process. | |
If the proton transfer in the excited state does not occur in a quantitative manner, dual emission15 can be observed, which may result in white light generation.16 Also, ESIPT-based luminophores have been identified as polymorph-dependent emitters,17,18 color-tuning by substitution-effect luminophores19 and amplified spontaneous emission molecules.20,21
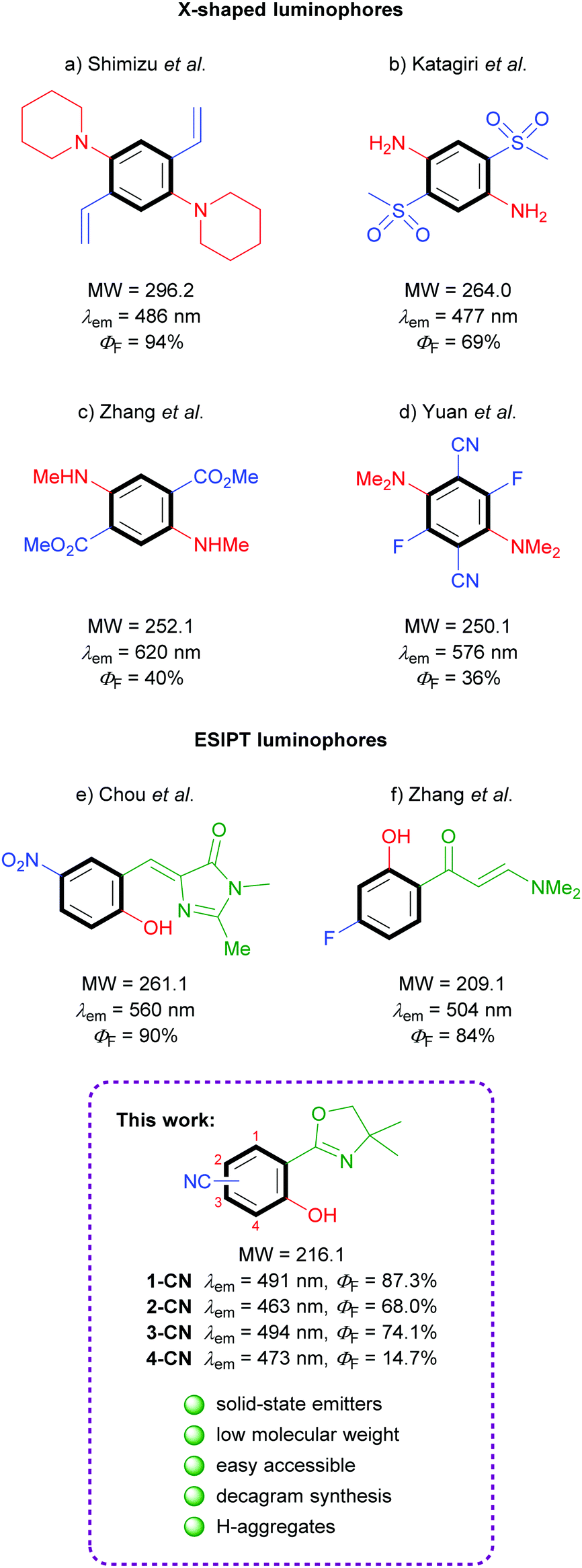
Benzene is the simplest π-unit for the construction of luminescent systems. In general, emissive organic structures contain more than just one benzene ring either for conjugation or as substituent. The design of minimalistic, single-benzene based, organic molecules exhibiting bright solid-state fluorescence is of fundamental interest and extremely challenging. Despite the supposedly straightforward synthesis of such simple structures, literature examples of low molecular weight, highly emissive molecules are extremely rare.
Efficacious known single-benzene luminophores can be divided into two categories – D–A based X-shaped structures and ESIPT-based molecules (Fig. 1). The popular tetrasubstituted X-shaped design of benzene with two electron-donating groups (EDGs) and two electron-withdrawing groups (EWGs) was first reported by Shimizu's group with the development of 1,4-bis(alkenyl)-2,5-dipiperidinobenzenes (Fig. 1a).22 Through the judicious choice of EWGs on the alkenyl side arm, the solid-state emission could be tuned across the full-color spectrum. In the following, Katagiri's group published 2,5-bis(methylsulfonyl)-1,4-phenyldiamine, which displayed green pH-independent and blue solid-state emission properties (Fig. 1b).23 In 2017, Zhang and coworkers developed dimethyl 2,5-bis-(methylamino)terephthalate, exhibiting a strong red emission (Fig. 1c).24 Very recently, full color range tuning was also achieved by Yuan's group through fully substituted 2,5-bis(alkylamino)-3,6-difluoroterephthalonitriles (Fig. 1d).25 X-shaped single-benzene fluorophores suffer from drawbacks like strong intra- and intermolecular hydrogen-bonding and the highly substituted character, which strongly restrict further derivatization and emission tuning. Potent low molecular weight ESIPT-based luminophores are truly rare, since potent structures are commonly constructed using π-extended proton acceptors such as benzazoles.26 Chuo and co-workers reported hydroxylated analogues of the green fluorescent protein chromophore, which display rather weak emission in solution (up to ΦF = 12%) but strong yellow emission in the solid state (Fig. 1e).27 (2-Hydroxyphenyl)propenone derivatives exhibiting no emission in solution but efficient green crystal emission were published by Zhang's group (Fig. 1f).21 While all these investigations were based on the search of efficient functional groups achieving high solid-state quantum yields for the given chromophore, the power of different regioisomers was never examined in detail.
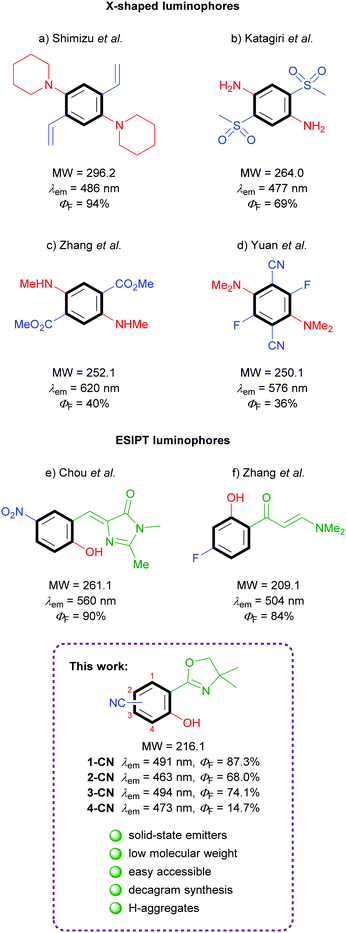 |
| | Fig. 1 Literature examples of single-benzene solid-state emitters. | |
In the course of our investigation toward minimalistic and highly emissive luminophores we observed phenol generation of magnesiated 2-aryloxazolines upon exposure to air or molecular oxygen as sustainable oxygen sources.28,29 The so developed deprotonative hydroxylation protocol delivered full colored ESIPT-based fluorophores as already shown in initial examinations. Herein, we report nitrile-substituted ortho-hydroxy-2-phenyloxazolines as extremely efficient, low molecular weight, single-benzene fluorophores (Fig. 1; bottom). All four possible regioisomers were synthesized and analyzed comprehensively in terms of photophysical properties, crystal structures and theoretical calculations.
Results and discussion
Syntheses
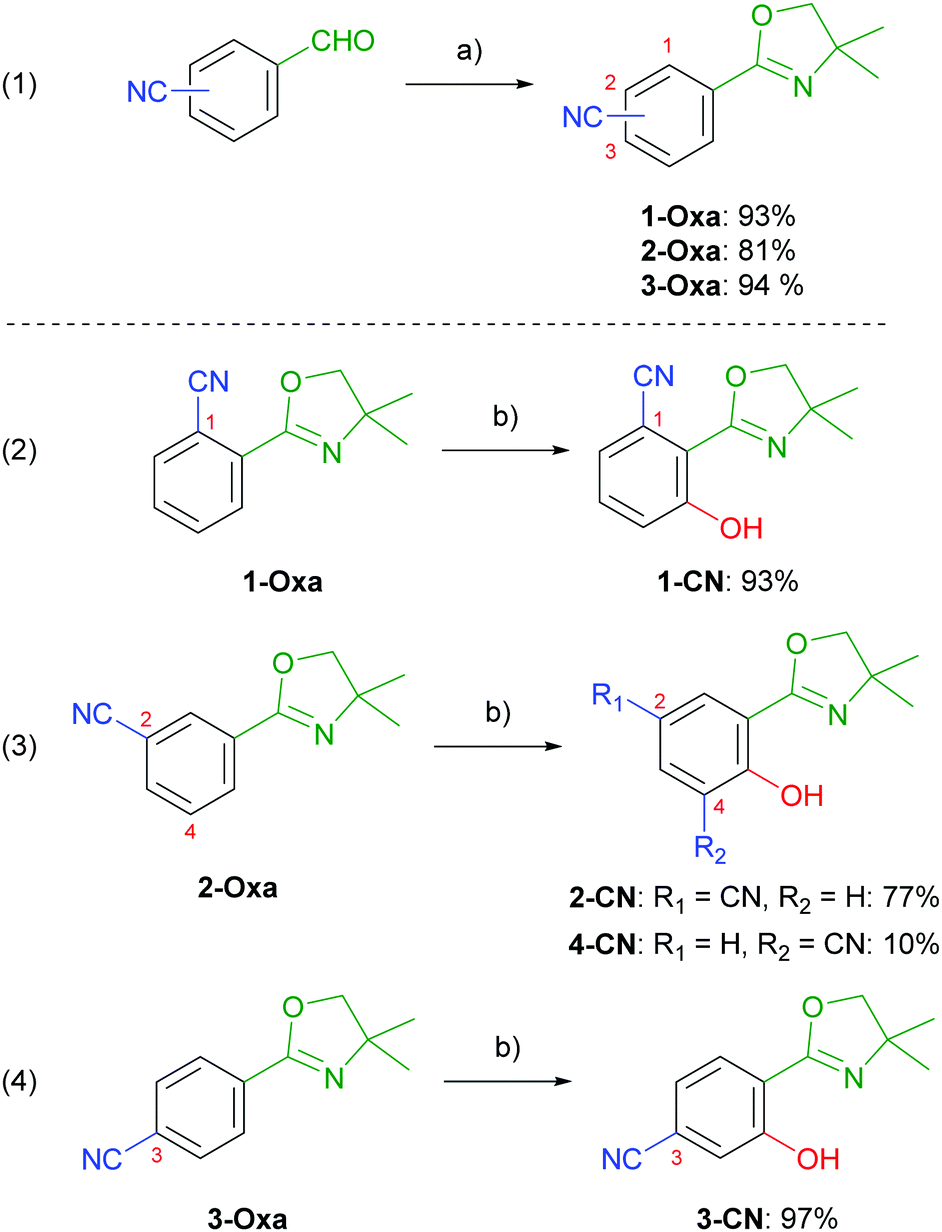
Nitrile-substituted phenols were achieved starting from formylbenzonitriles, which were transformed to the corresponding 2-aryloxazolines (1-Oxa, 2-Oxa and 3-Oxa) in very good yields via a two steps sequence including condensation with 2-amino-2-methylpropan-1-ol and subsequent oxidation using NBS (Scheme 2, entry 1).30 Phenol incorporation was accomplished by ortho-directed metalation using TMPMgCl·LiCl and ensuing smooth hydroxylation with molecular oxygen providing the desired phenols (1-CN, 2-CN and 3-CN) in excellent yields (Scheme 2, entries 2–4).28 To our delight, hydroxylation of 2-Oxa delivered besides the major isomer 2-CN also the minor regioisomer 4-CN, even though in low yields. Summarizing, this synthetic route demonstrates the simplicity of achieving these highly potent luminophores. To further demonstrate the robustness of our synthetic approach we performed the synthesis of 1-CN in a scale-up reaction and achieved this structure on a decagram scale (see the ESI,† Chapter 2.4).
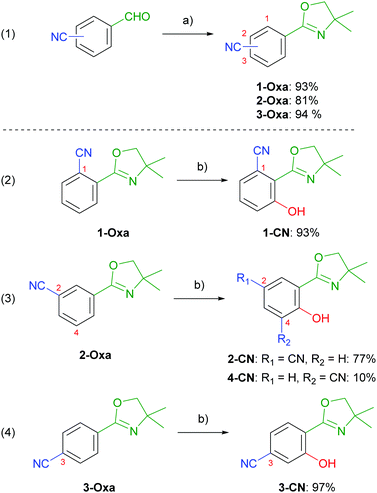 |
| | Scheme 2 (a) (1) 2-amino-2-methylpropan-1-ol, 4 Å MS, CH2Cl2, 25 °C, 20 h; (2) NBS, CH2Cl2, 25 °C, 4 h; (b) (1) TMPMgCl·LiCl, THF, 25 °C, 1 h; (2) O2 (1 atm), 25 °C, 24 h. NBS = N-bromosuccinimide, TMP = 2,2,6,6-tetramethylpiperidinyl. | |
Absorption properties
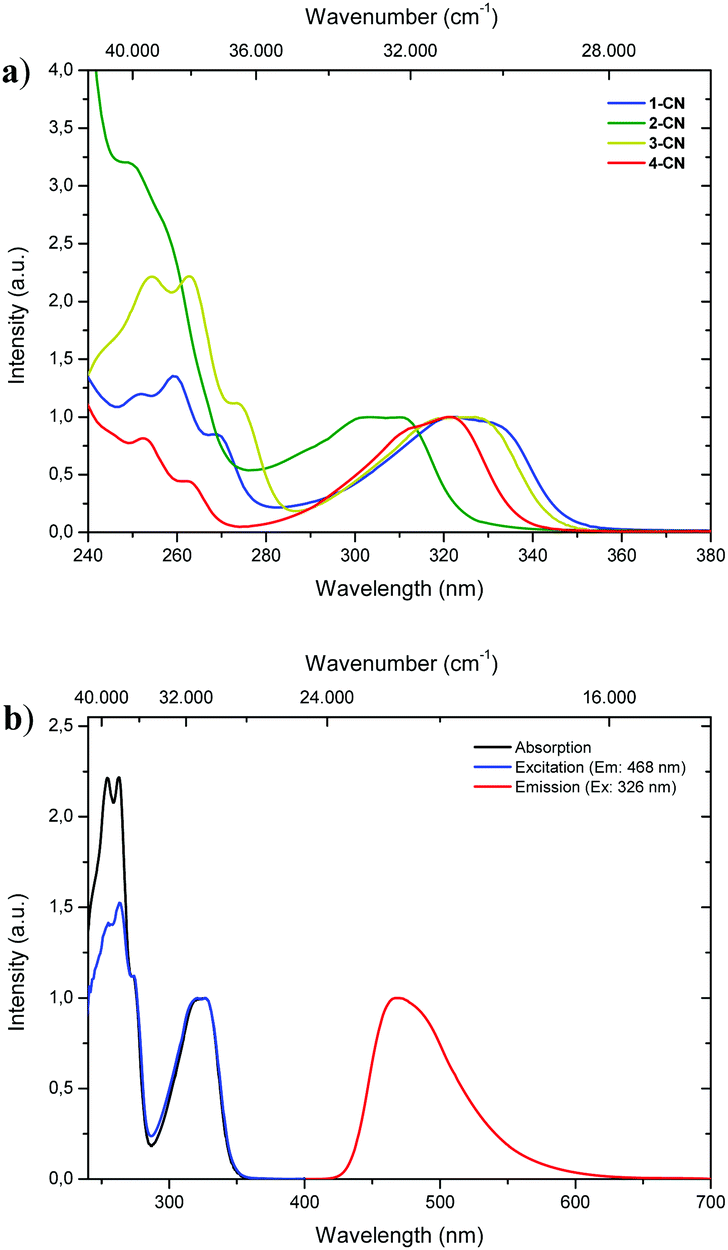
With all regioisomers of nitrile-substituted phenols (1-CN, 2-CN, 3-CN and 4-CN) in hand, we started to investigate their photophysical properties. UV-vis absorption spectra were measured in seven solvents exhibiting differing polarities, i.e., methanol (MeOH), acetonitrile (ACN), ethyl acetate (EtOAc), 2-methyltetrahydrofuran (2-MeTHF), dichloromethane (DCM), toluene (PhMe) and cyclohexane (CH). Generally, the absorption properties of the respective luminophore are widely solvent independent (Table 1; see the ESI,† Fig. S22, S48, S72, S96 and Table S1, ESI†), while slight differences are detectable in the same solvent (Fig. 2a). In DCM solution, intense absorption bands deriving from the single-benzene core are visible below 280 nm, which are indicative of typical π–π* transitions. Furthermore, broad and unstructured absorption bands are detected with maxima λabs between 303 and 326 nm and extinction coefficients ranging from 5300 to 8850 mol L−1 cm−1 (Fig. 2a and Table 1). The excitation spectrum of 3-CN in CH2Cl2 indicates a maximum population of the excited-state upon irradiation in the absorption band with a maximum at 326 nm, while bands below 280 nm show diminished transitions for emission at 468 nm (Fig. 2b).
Table 1 Photophysical data of 1-CN, 2-CN, 3-CN and 4-CN measured in aerated solution at 295 K
| Compd |
Solvent |
λ
abs [nm] (ε [mol L−1 cm−1]) |
λ
em [nm] |
Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) [cm−1] [cm−1] |
Φ
F
[%] |
τ
[ns] (Rel%) |
k
r
[108 s−1] |
k
nr
[108 s−1] |
|
Absolute quantum yields were determined by using an integration sphere.
Relative ratio of the species of a double exponential function are given in parentheses.
k
r (108 s−1) and knr (108 s−1) were calculated using the equations kr = ΦF/τ and knr = (1 − ΦF)/τ.
Half lifetimes were measured after bubbling with argon for 30 minutes.
|
|
1-CN
|
MeOH |
321 |
467 |
9740 |
13.7 |
1.87 (71.6) |
0.64 |
4.00 |
| 2.87 (28.4) |
| ACN |
320 |
471 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 020 020 |
15.2 |
2.08 |
0.73 |
4.07 |
| EtOAc |
320 |
473 |
10110 |
15.6 |
2.34 |
0.67 |
3.61 |
| 2-MeTHF |
321 |
476 |
10140 |
14.0 |
2.07 |
0.68 |
4.16 |
| 2.11d |
| THF |
321 |
477 |
10190 |
17.2 |
2.38 (92.5) |
0.67 |
3.21 |
| 5.10 (7.5) |
| DCM |
322 (5790) |
471 |
9820 |
24.9 |
3.09 |
0.81 |
2.43 |
| PhMe |
322 |
477 |
10090 |
18.1 |
2.31 (21.5) |
0.30 |
1.35 |
| 7.09 (78.5) |
| CH |
322 |
481 |
10270 |
8.2 |
1.13 |
0.73 |
8.16 |
|
|
|
2-CN
|
MeOH |
303 |
445 |
10530 |
20.5 |
2.71 |
0.76 |
2.94 |
| ACN |
301 |
449 |
10950 |
16.0 |
2.35 |
0.68 |
3.57 |
| EtOAc |
303 |
454 |
10980 |
24.7 |
2.86 |
0.86 |
2.64 |
| 2-MeTHF |
304 |
455 |
10920 |
22.1 |
2.88 |
0.77 |
2.70 |
| DCM |
303 (5300) |
452 |
10880 |
25.2 |
3.30 |
0.76 |
2.27 |
| PhMe |
306 |
462 |
11040 |
22.3 |
6.74 |
0.33 |
1.15 |
| CH |
306 |
468 |
11310 |
15.1 |
2.04 |
0.74 |
4.17 |
|
|
|
3-CN
|
MeOH |
323 |
465 |
9450 |
51.0 |
2.53 (5.5) |
0.70 |
0.67 |
| 7.60 (94.5) |
| ACN |
324 |
465 |
9360 |
49.4 |
7.29 |
0.68 |
0.69 |
| EtOAc |
326 |
468 |
9310 |
47.4 |
6.75 |
0.70 |
0.78 |
| 2-MeTHF |
327 |
469 |
9260 |
47.0 |
6.53 |
0.72 |
0.81 |
| 7.67d |
| DCM |
326 (5790) |
468 |
9310 |
55.6 |
8.10 |
0.69 |
0.55 |
| PhMe |
328 |
479 |
9610 |
55.0 |
8.95 |
0.61 |
0.50 |
| CH |
327 |
487 |
10050 |
39.7 |
6.02 |
0.66 |
1.00 |
|
|
|
4-CN
|
MeOH |
320 |
454 |
9220 |
49.8 |
5.27 |
0.95 |
0.95 |
| ACN |
319 |
456 |
9420 |
40.5 |
5.30 |
0.76 |
1.12 |
| EtOAc |
320 |
459 |
9460 |
63.0 |
5.22 |
1.21 |
0.71 |
| 2-MeTHF |
321 |
462 |
9510 |
48.7 |
5.14 |
0.95 |
1.00 |
| DCM |
321 (8850) |
457 |
9270 |
52.9 |
5.52 |
0.96 |
0.85 |
| PhMe |
322 |
469 |
9730 |
52.4 |
7.92 |
0.66 |
0.60 |
| CH |
322 |
479 |
10180 |
44.8 |
5.10 |
0.88 |
1.08 |
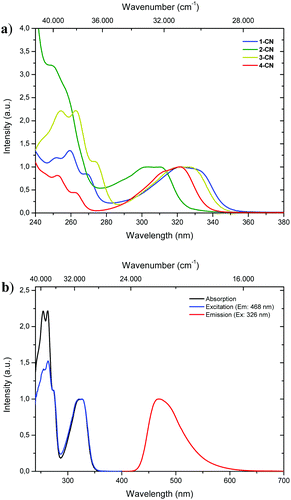 |
| | Fig. 2 (a) Normalized UV-vis absorption spectra of 1-CN (blue line), 2-CN (green line), 3-CN (yellow line) and 4-CN (red line) in CH2Cl2. (b) Normalized absorption (black line), excitation (blue line) and emission spectra (red line) of 3-CN in CH2Cl2. | |
Fluorescence properties in solution
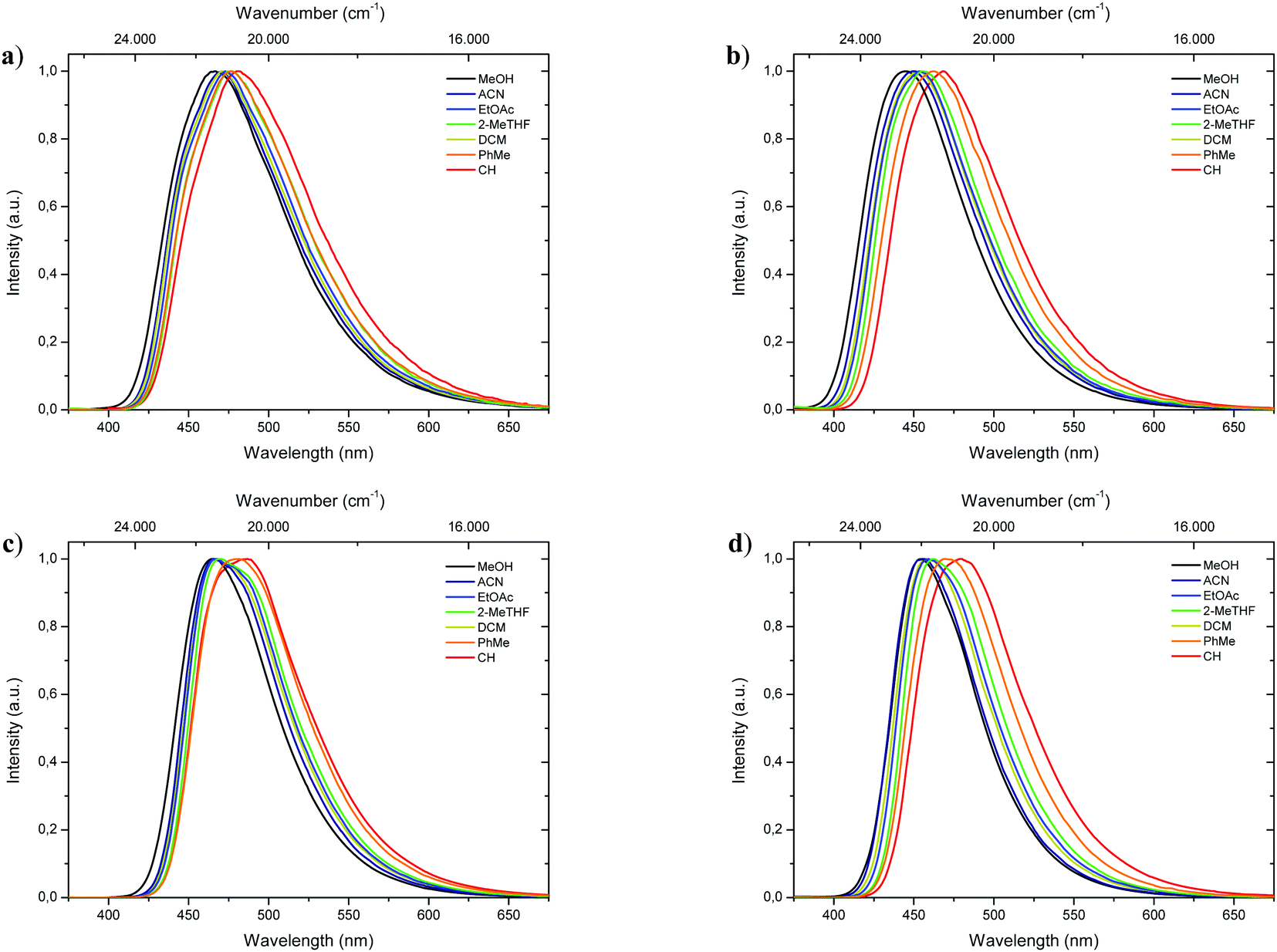
Complementary to absorption measurements, fluorescence properties 1-CN, 2-CN, 3-CN and 4-CN were studied in solvents with distinctive dipole moments – i.e., MeOH, ACN, EtOAc, 2-MeTHF, DCM, PhMe, CH. The results are listed in Table 1. Excitation in the respective absorption maxima resulted in blue to cyanic emission maxima λem (445 nm to 487 nm) depending on the structure and the utilized solvent (Fig. 3). Notably, 1-CN and 3-CN emit in average 10 to 20 nm more red-shifted than 2-CN and 4-CN. Dual emission behavior, which could be caused by the enol–keto-phototautomerization, was never identified. Slight solvatochromism is observed as expected for ESIPT-based structures. Protic, polar solvents such as methanol result in blue-shifted emission due to phenol coordination, while with decreasing solvent polarity a red-shifted emission can be detected as displayed for 1-CN, 2-CN, 3-CN and 4-CN (Fig. 4). Due to their ESIPT-based character, all phenols exhibit large Stokes’ shifted emission of up to 11.310 cm−1, which resulted in baseline separated absorption and emission bands (Fig. 2b). Hence, highly efficient proton transfer in the excited state takes place for all phenols in all solvents, indicating that emission exclusively originate from the keto tautomer. Quantum yields (ΦF) in solution show a significant solvent-dependency as well. Lowest quantum yields are usually observed for CH, while highest values are observed for DCM, ACN and EtOAc. For 1-CNΦF ranges from 8.2% (CH) to 24.9% (DCM), while for 2-CNΦF ranges from 15.1% (CH) to 25.2% (DCM). Significantly higher quantum yields in solution were measured for 3-CN ranging from 39.7% (CH) to 55.6% (DCM) and for 4-CN from 40.5% (ACN) to 63.0% (EtOAc). Fluorescence half lifetimes (τ) of the excited state were determined by time correlated single photon counting (TCSPC). Similar tendencies were observed for all phenols, with lowest half lifetime values in CH, ranging from 1.13 ns (1-CN) to 6.02 ns (3-CN), and highest values in DCM, ranging from 3.09 ns (1-CN) to 8.10 ns (3-CN). Fluorescence half lifetimes are significant higher in PhMe, ranging from 7.09 ns (1-CN, major species of biexponential decay) to 8.95 ns (3-CN). To investigate the role of aerated vs. deaerated media, 2-MeTHF solutions of 1-CN and 3-CN were bubbled with argon for 30 minutes and measured again. For 1-CN no prolonged fluorescence half lifetimes were observed (see Fig. S13, ESI†), while the 3-CN solution shows a significant half lifetime increase by a factor of 1.17 (6.53 ns to 7.67 ns) (see Fig. S63, ESI†). With values of ΦF and τ in hand, the radiative (kr) and nonradiative (knr) decay rate constants were determined according to the equations kr = ΦF/τ and knr = (1 − ΦF)/τ.
 |
| | Fig. 3 Representative images of 1-CN, 2-CN, 3-CN and 4-CN in CH2Cl2 solution at c = 10−4 mol L−1 under 366 nm irradiation. | |
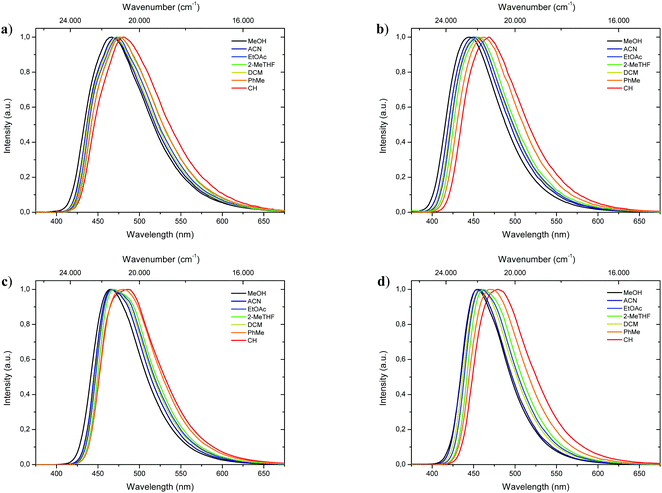 |
| | Fig. 4 Normalized emission spectra of 1-CN (a), 2-CN (b), 3-CN (c) and 4-CN (d) in various solvents. | |
For phenols showing high emission in solution radiative decay rates kr ranging from 0.68 × 108 s−1 (3-CN in ACN) to 1.21 × 108 s−1 (4-CN in EtOAc) and nonradiative decay rates knr ranging from 0.50 × 108 s−1 (3-CN in PhMe) to 1.12 × 108 s−1 (4-CN in ACN) were observed. These values indicate the suppression of nonradiative decay pathways and preferred radiative decays for 3-CN and 4-CN in solution.
Fluorescence at low temperatures
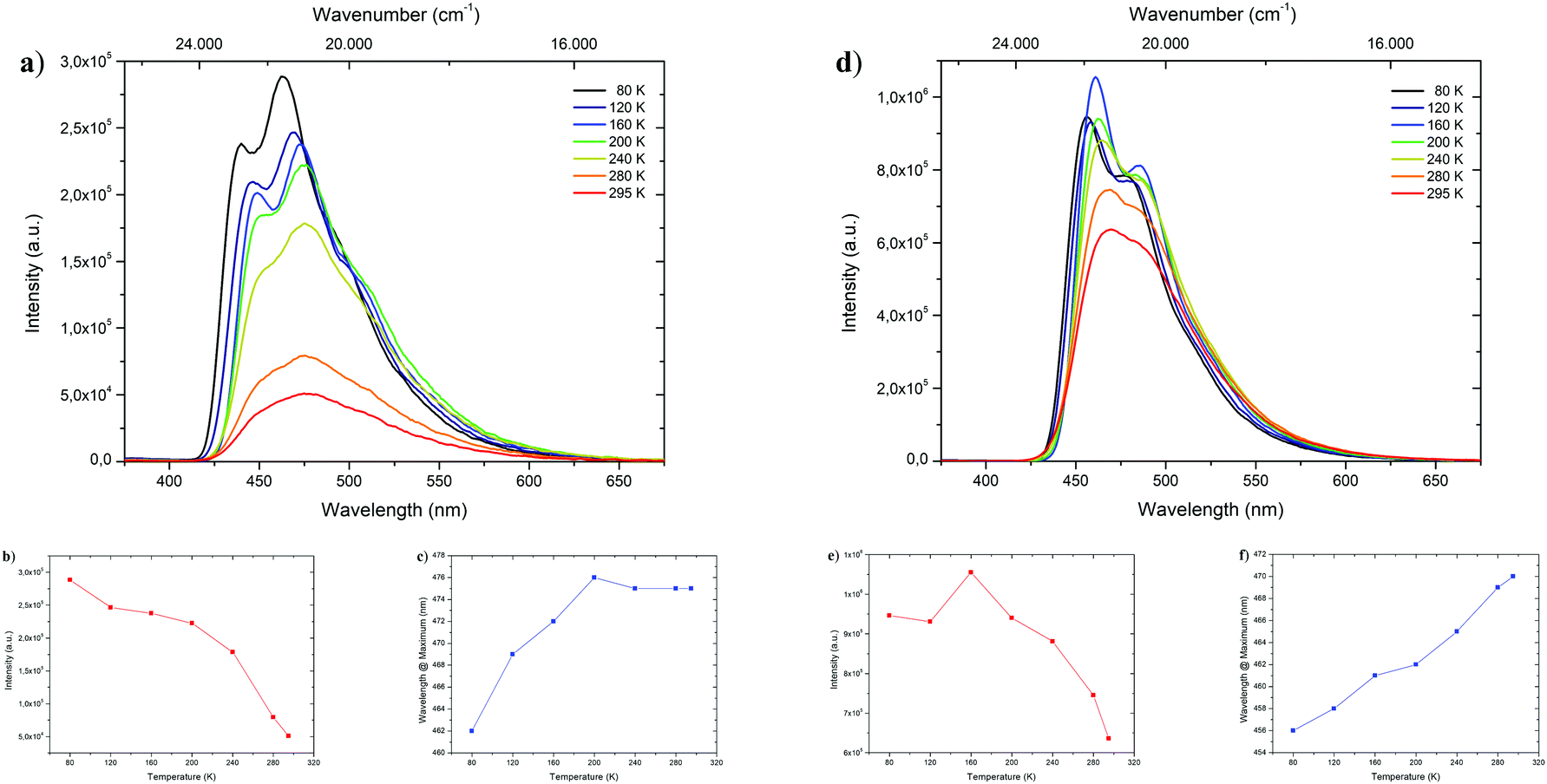
To shed light on the temperature dependent emission behavior, deaerated 2-MeTHF solutions of 1-CN, 2-CN, 3-CN and 4-CN were examined and temperature maps were created by measuring in 40 K intervals down to 80 K (Fig. 5 and Table 2). A steady increase in photoluminescence intensity was observed upon cooling of 1-CN solution with maximum intensity detected at 80 K (Fig. 5a and b). This occurrence is unusual for ESIPT-based luminophores due to the typical suppression of the emission process at deep temperatures. Compared to 295 K the emission intensity is multiplied by the factor of 5.65 at 80 K. Additionally, a significant blue-shifted emission was detected while cooling below 200 K with a wavelength of 462 nm at 80 K emission maximum (Fig. 5c). Analyzing the temperature dependent emission of 3-CN in 2-MeTHF indicates an increasing intensity up to 160 K (factor 1.66 compared to 295 K) followed by a decrease in intensity (Fig. 5d and e). This behavior of a lowered intensity below a certain temperature is in accordance with known ESIPT-based emitters.17
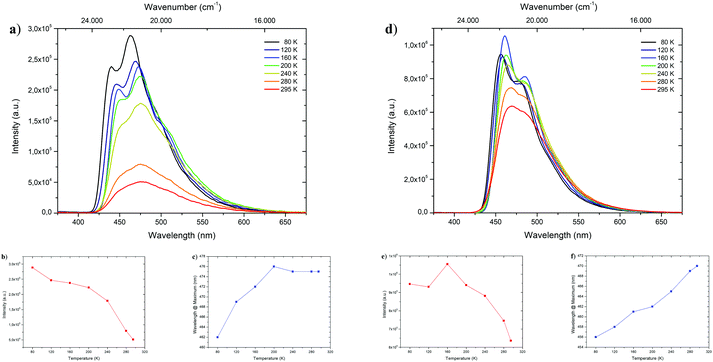 |
| | Fig. 5 Top: Temperature dependent emission spectra of 1-CN (a) and 3-CN (d) in deaerated 2-methyl tetrahydrofuran during heating from 80 K to 295 K with excitation at 321 and 327 nm, respectively. Bottom: Temperature dependent trends of intensity (1-CN (b) and 3-CN (e)) and wavelength (1-CN (c) and 3-CN (f)). | |
Table 2 Photophysical data of 1-CN, 2-CN, 3-CN and 4-CN measured in deaerated 2-MeTHF at 80 K
| Compd |
λ
em [nm] |
τ [ns] (Rel%) |
|
1-CN
|
440, 462 |
2.22 (36.4), 8.17 (51.7), 44.0 (11.9) |
|
2-CN
|
444, (432) |
1.22 (24.2), 5.58 (61.6), 23.0 (14.2) |
|
3-CN
|
456, 476 |
1.95 (20.2), 7.29 (63.8), 24.1 (16.0) |
|
4-CN
|
444, (462) |
5.43 (100) |
Again, linear blue-shifted emission was detected with a minimal emission wavelength of 456 nm at 80 K (Fig. 5f). Importantly, both temperature maps (Fig. 5a and d) display the disappearance of the broad emission band at 295 K and splitting into at least two definite bands. Owing to suppressions of intramolecular degrees of freedom line broadening is significantly reduced, resulting in the given pattern. Temperature maps of 2-CN and 4-CN show similar behavior as 3-CN (see the ESI,† Chapters 3.2.2 and 3.4.2).
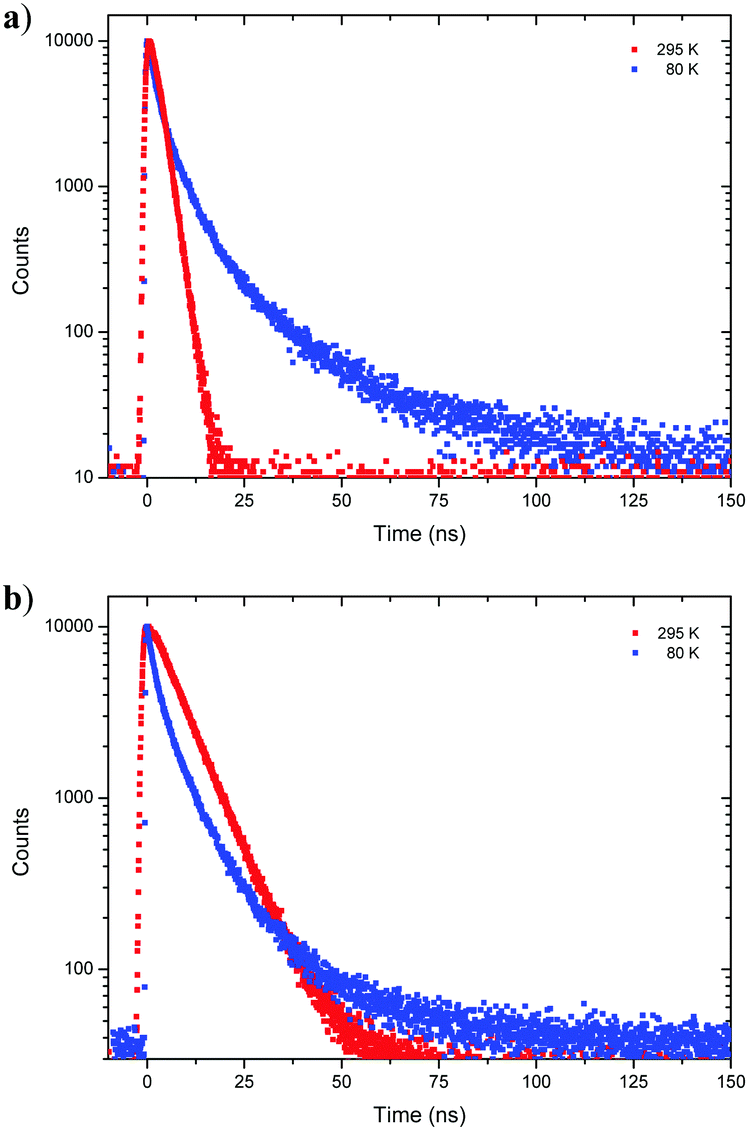
Next, fluorescence half lifetimes (τ) of the excited states at 80 K were determined for all phenols in deaerated 2-MeTHF by TCSPC. Remarkably, half lifetimes were prolonged in average and triple exponential decays were observed for 1-CN, 2-CN and 3-CN (Fig. 6 and Table 2). Long living species with half lifetimes ranging from 23.0 ns (2-CN) to 44.0 ns (1-CN) were detected in lower population. Only, 4-CN showed a contrary behavior with only one single emitting species, which exhibits a slightly increased half lifetime compared to measurements at 295 K. It is worth mentioning that long living triplet emission (phosphorescence) or thermally activated delayed fluorescence (TADF) was never observed during our investigations, neither at rt nor at 80 K.
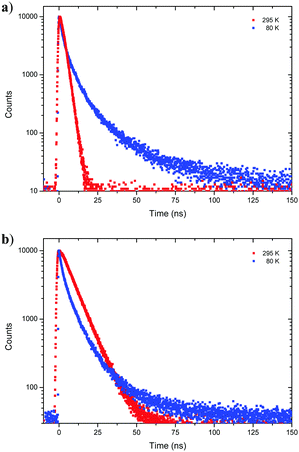 |
| | Fig. 6 Time resolved emission decay curves of 1-CN (a) and 3-CN (b) in deaerated 2-MeTHF at 80 K and 295 K. | |
Aggregation induced emission enhancement
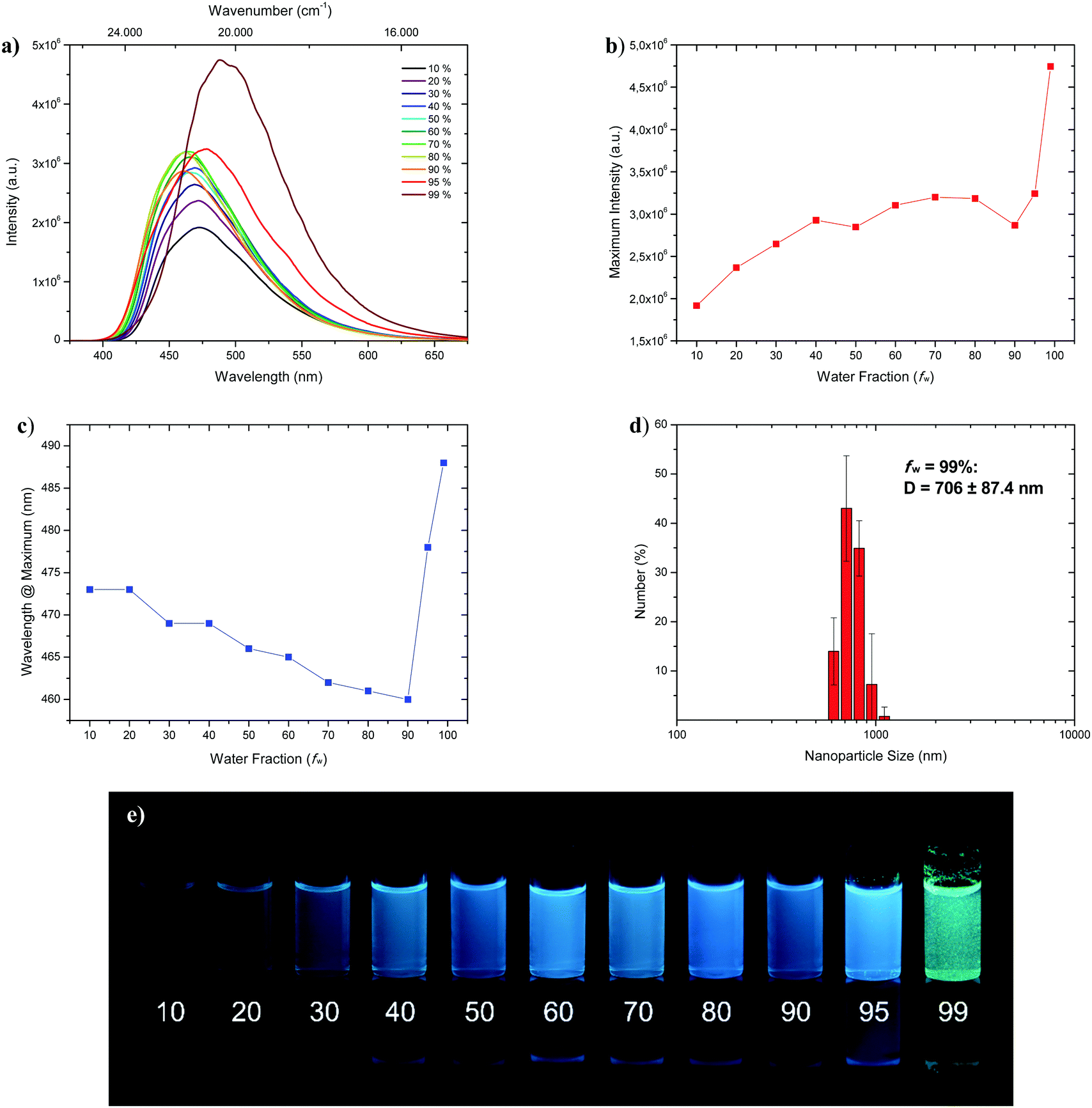
Fluorophores which show no emissive or low emissive properties in solution can become highly emissive in the solid state. Several mechanisms for the emission enhancement like the restriction of intramolecular vibrational and rotational motions (RIM), intramolecular charge transfer (ICT)31 or twisted intramolecular charge transfer (TICT) are well known and result in the phenomenon called aggregation-induced emission (AIE) and aggregation-induced emission enhancement (AIEE).6,7 We chose 1-CN for further investigations towards aggregation-induced emission and utilized tetrahydrofuran (THF) as a non-aggregating solvent and water as the aggregating solvent.32 In the course of the investigation, the concentration was kept at 4 × 10−5 mol L−1. Initially, as depicted in Fig. 7a, fluorescence is already present at low water fractions (fw = 10%), as confirmed by emission properties of pure THF solution (λem = 477 nm, ΦF = 17.2% (Table 1)).
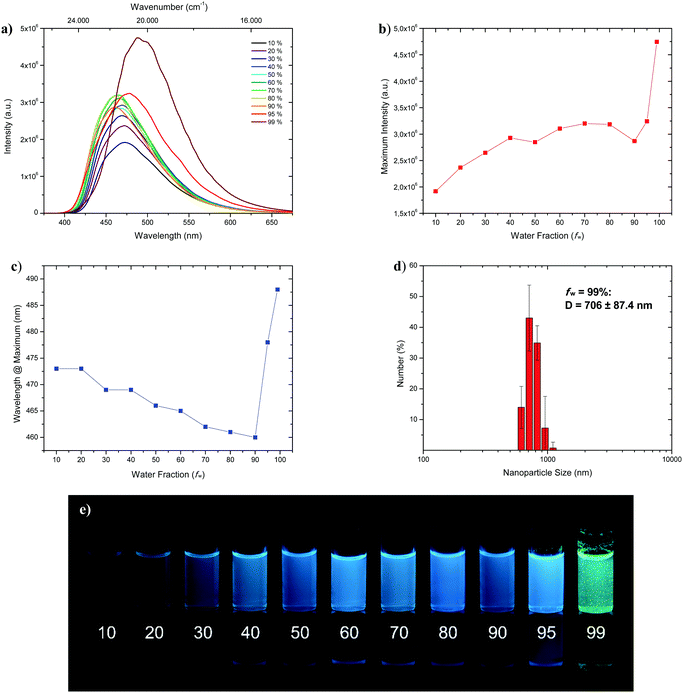 |
| | Fig. 7 (a) Emission spectra of 1-CN in THF/water mixtures with differing water fraction (fw in %) at a concentration of 4 × 10−5 mol L−1 under excitation with 321 nm. (b) Plot of maximum intensity versus water fraction (fw). (c) Plot of wavelength at maximum versus water fraction (fw). (d) DLS analysis of nanoparticle size distribution of fraction fw = 99%. (e) Representative AIEE image of 1-CN in THF/water mixtures with given water content (in %) under 366 nm irradiation. | |
With increasing water fractions of up to fw = 90%, a moderate emission enhancement is detected with a slight drop of emission intensity (Fig. 7b) at fw = 50% (λem = 466 nm) and fw = 90% (λem = 460 nm) including a blue-shifted emission (Fig. 7c). This hypsochromic shift, starting from fw = 10% to fw = 90% can be explained by the increasing polarity of the surrounding medium, which is in accordance with emission wavelength tendencies for polar solvents like methanol, reasoned by phenol coordination. Interestingly, further water addition to fw = 95% resulted in a red-shifted emission (λem = 478 nm) and an increase of intensity. Maximum emission boosting was obtained at fw = 99% with another significant red-shift (λem = 488 nm) and almost doubling of fluorescence intensity, which resulted in a quantum yield of ΦF = 41.3%. Within the last two analyzed fractions visible nano-aggregates have been formed, while massive nanoparticle formation has only taken place at fw = 99% (Fig. 7; bottom). The nano-aggregated particles in fw = 99% were analyzed in respect of their size using dynamic light scattering (DLS), revealing an average hydrodynamic radius of 706 ± 87.4 nm (Fig. 7b). Additionally, the emission properties of doped PMMA films with different weight concentration of 1-CN (1 wt%, 10 wt% and 50 wt%) were examined (see the ESI,† Chapter 3.1.3). While films with 1 wt% and 10 wt% of 1-CN exhibit similar blue emission, PMMA film with 50 wt% of 1-CN showed bright green emission.
Solid-state emission
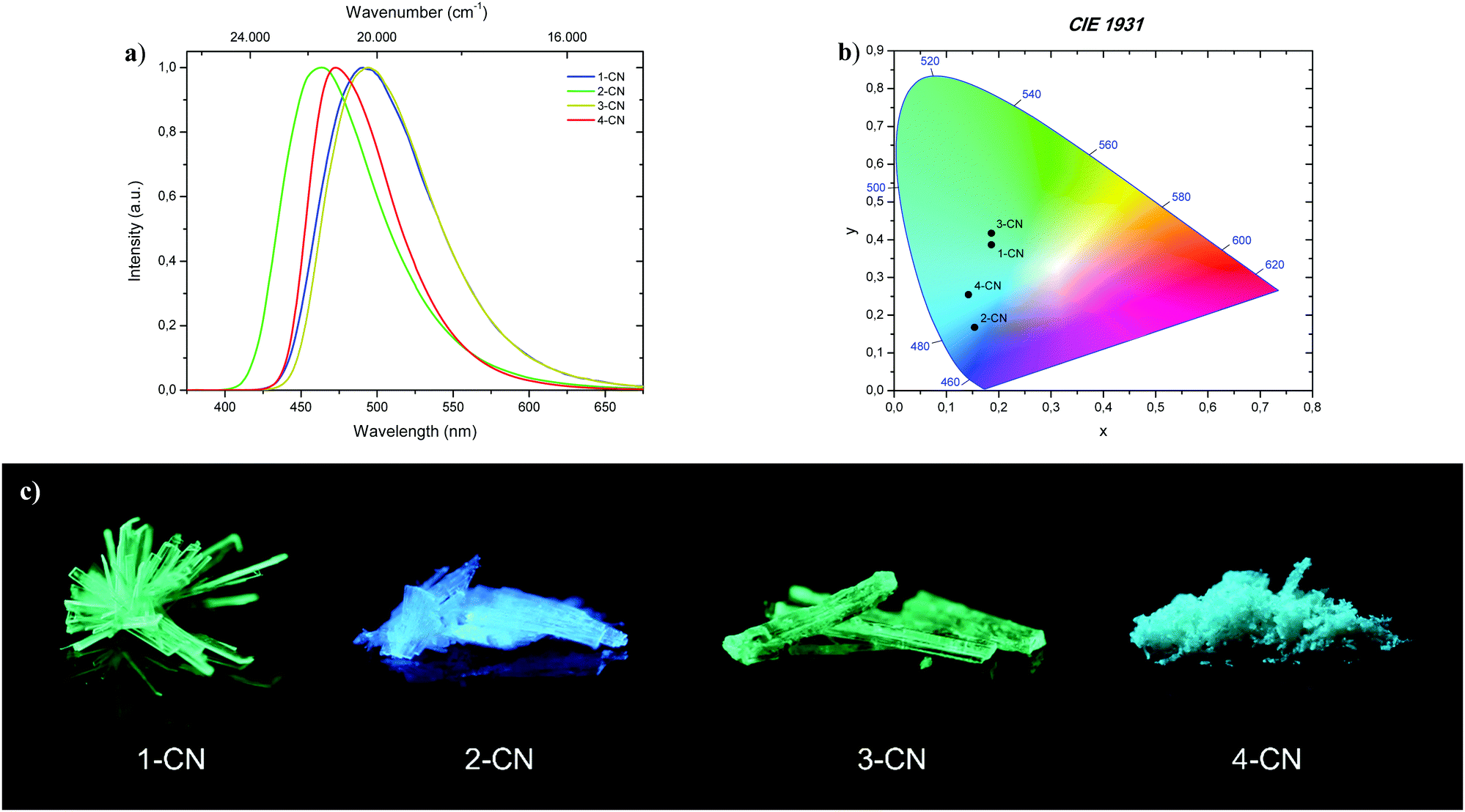
All phenols (1-CN, 2-CN, 3-CN and 4-CN) exhibit bright luminescence in the solid-state to the naked eye by simple irradiation with a 366 nm UV-handlamp (Fig. 8; bottom). Therefore, investigations of the photophysical properties were conducted in the crystalline state for 1-CN, 2-CN, 3-CN and in the microcrystalline state for 4-CN (Fig. 8a). Optical properties of all phenols in the solid-state are listed in Table 3.
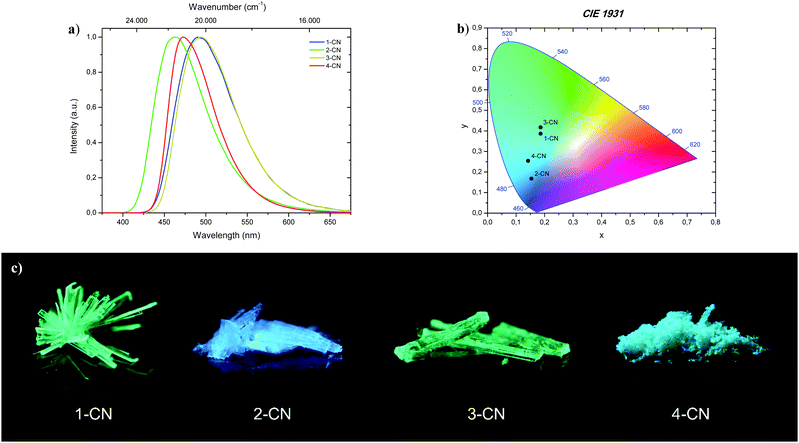 |
| | Fig. 8 (a) Normalized emission spectra of 1-CN, 2-CN, 3-CN and 4-CN in the solid-state. (b) CIE 1931 chromaticity plot with emission color coordinates of 1-CN, 2-CN, 3-CN and 4-CN. (c) Representative images of 1-CN, 2-CN, 3-CN and 4-CN in the solid-state under 366 nm irradiation. | |
Table 3 Photophysical data of 1-CN, 2-CN, 3-CN and 4-CN measured in the solid-state at 295 K
| Compd |
λ
exc [nm] |
λ
em [nm] |
Δ [cm−1] |
Φ
F
[%] |
τ [ns] |
k
r
[108 s−1] |
k
nr
[108 s−1] |
x; y (CIE 1931) |
|
Absolute quantum yields were determined by using an integration sphere.
k
r (108 s−1) and knr (108 s−1) were calculated using the equations kr = ΦF/τ and knr = (1 − ΦF)/τ.
|
|
1-CN
|
347 |
491 |
8.450 |
87.3 |
13.3 |
0.66 |
0.10 |
0.186; 0.387 |
|
2-CN
|
326 |
463 |
9.080 |
68.0 |
7.35 |
0.92 |
0.44 |
0.153; 0.168 |
|
3-CN
|
348 |
494 |
8.490 |
74.1 |
10.8 |
0.69 |
0.24 |
0.186; 0.417 |
|
4-CN
|
346 |
473 |
7.760 |
14.7 |
7.76 |
0.19 |
1.10 |
0.142; 0.254 |
The brightest luminophore was 1-CN, which showed a strong green-cyan emission with λem = 491 nm and ΦF = 87.3% in the crystalline state. The fluorescence half lifetime was determined to τ = 13.3 ns. Dynamics studies (kr = 0.66 × 108 s−1 and knr = 0.10 × 108 s−1) revealed that the negligible nonradiative decay pathways yielded the high ΦF. Noteworthily, semicrystalline powder of 1-CN showed the same emission properties, indicating that no crystallization-induced emission enhancement (CIEE) has taken place (see the ESI,† Fig. S21). To the best of our knowledge 1-CN represents the lowest molecular weight ESIPT-based fluorophore with a quantum yield above 85% in the solid-state. Also, 2-CN displays strong blue emission with λem = 463 nm and ΦF = 68.0% in the crystalline state and a high Stokes shift of 9.080 cm−1. Based on these powerful solid-state emission properties and the weak emission in solution, both, 1-CN and 2-CN show a typical AIEE. Powerful green-cyan emission is observed for 3-CN with λem = 494 nm and ΦF = 74.1% in the crystalline state. A high fluorescence half lifetime of τ = 10.8 ns and low radiationless decay pathways (kr = 0.69 × 108 s−1 and knr = 0.24 × 108 s−1) verified the highly potent solid-state emission nature of 3-CN. Together with its strong emission properties in solution, 3-CN represents a dual state emission (DSE) luminophore. In comparison, 4-CN showed relatively weak blue-cyan fluorescence in the microcrystalline state with λem = 473 nm and ΦF = 14.7%. In consideration of the remarkable emission properties in solution, 4-CN demonstrates aggregation-caused quenching (ACQ) character. As in solution, the large Stokes shifted emission is caused by an efficient proton transfer in the excited state, resulting in exclusive emission from the keto tautomer. CIE 1931 chromaticity coordinates (Fig. 8b) were calculated from the corresponding photoluminescence spectra, giving the following (x; y) values for 1-CN (0.186; 0.387), 2-CN (0.153; 0.168), 3-CN (0.186; 0.417) and 4-CN (0.142; 0.254). In the course of photophysical investigations, none of these analyzed emitters showed the tendency for photobleaching, neither in solution nor in the solid-state.
Single crystal packing
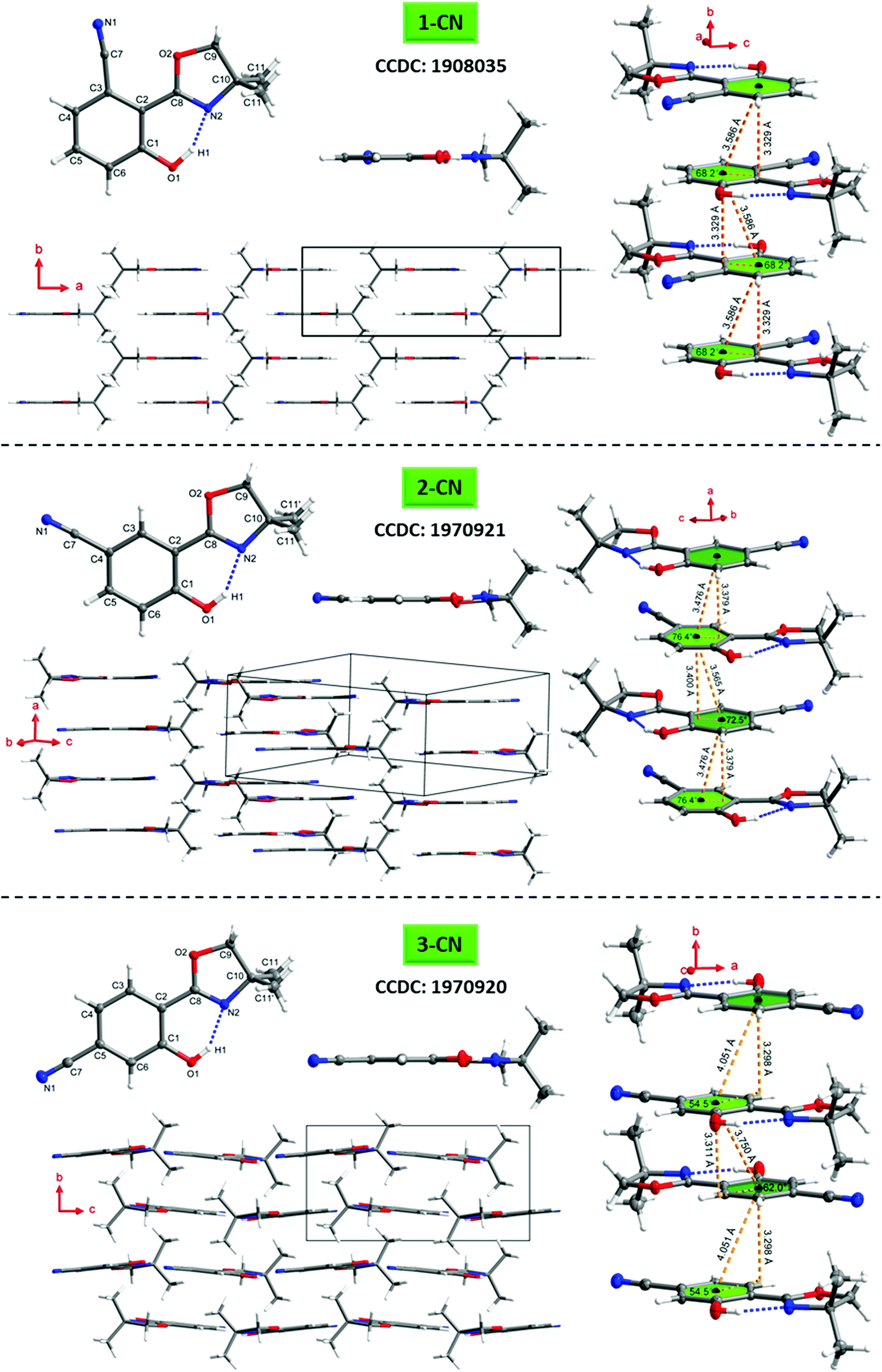
To better understand the emission characteristics of these highly efficient luminophores, suitable single crystals for X-ray diffraction were obtained for 1-CN, 2-CN and 3-CN by evaporation of a concentrated dichloromethane solution. Unfortunately, 4-CN showed only microcrystallite formation, which were not suitable for single crystal analysis. Crystallographic data are summarized in Table 4 and Tables S2–S19 (ESI†) and depicted in Fig. 9 and Fig. S108–S147 (ESI†).
Table 4 Hydrogen bond (⋯) and crystal parameters of 1-CN, 2-CN and 3-CN
| Compd |
O1–H1⋯N2 |
Dihedral angle C1–C2–C8–N2 [°] |
Slip angles [°] |
Plane distance [Å] with corresponding centroid–centroid distance ([Å]) |
| Length [Å] |
Angle [°] |
|
1-CN
|
1.6704(3) |
153.294(3) |
0 |
68.2 |
3.329 (3.586) |
|
2-CN
|
1.8592(19) |
147.790(12) |
−5.6(3); −3.8(3) |
72.5; 76.4 |
3.379 (3.476); 3.400 (3.565) |
|
3-CN
|
1.8289(11) |
148.005(8) |
−0.05(17) |
54.5; 62.0 |
3.298 (4.051); 3.311 (3.750) |
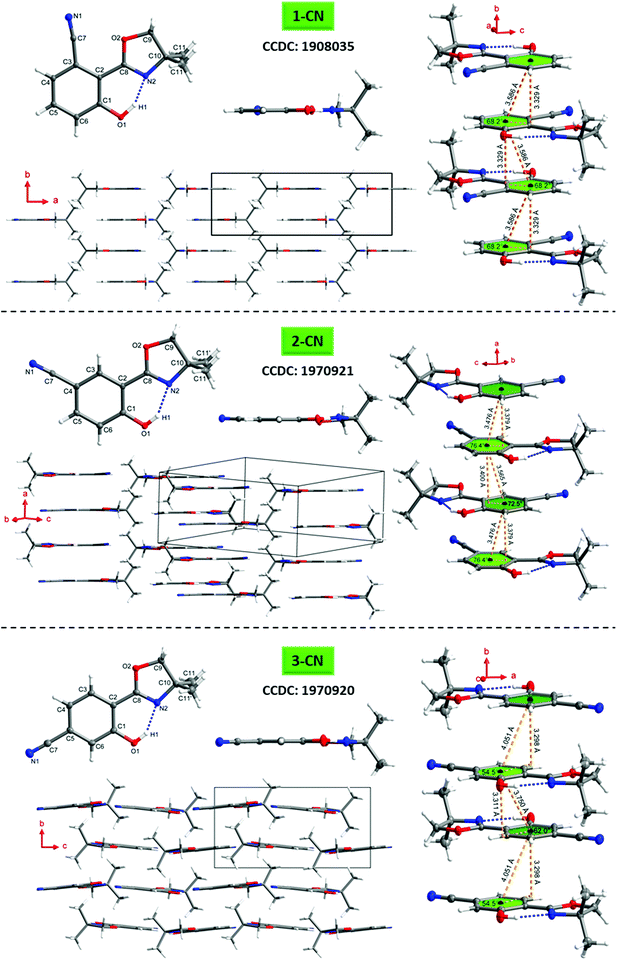 |
| | Fig. 9 Molecular structures of 1-CN (top), 2-CN (middle) and 3-CN (bottom) showing 50% probability ellipsoids, including top view and sideview of the single structure, molecular packing as well as the π–π-interactions of adjacent molecules. | |
The O1–H1⋯N2 hydrogen bond distances in 1-CN, 2-CN and 3-CN were determined to be 1.670, 1.859 and 1.829 Å, respectively, while the corresponding angles are assigned to be 153.29°, 147.79°, and 148.01°. Dihedral angles between the phenol and oxazoline rings (i.e., the C1–C2–C8–N2 angle) were found to be 0° for 1-CN, −5.6° and −3.8° (C21–C22–C28–N22) for 2-CN and −0.05° for 3-CN, respectively. These crystal data revealed that the O–H⋯N hydrogen bond in 1-CN is the strongest, which in turn facilitates intramolecular proton transfer to the adjacent nitrogen atom and rationalizes the extremely high ΦF. To gain a deeper understanding of the increased ΦF values in the solid-state, the crystal stacking modes were examined. 1-CN (orthorhombic, Pnma), 2-CN (triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) and 3-CN (monoclinic, P21/c) exhibit different packing modes in their respective crystal lattice, including various types of intermolecular interactions. This variety of contacts leads to a restriction of intramolecular rotation and explains the increased ΦF values in the solid-state. For 1-CN, several intermolecular interactions,33 like C–H⋯O (2.679 Å) or C–H⋯N (2.658 Å) are identified (see the ESI,† Fig. S120), while antiparallel molecule cross-stacking takes place along the b-axis in a sheet structure. Interplanar spacing is determined to 3.329 Å, whereas the adjacent π-skeleton distance is 3.586 Å with a slip angle of 68.2° (Fig. 9). Thus, relatively strong π–π-interactions lead to intense H-aggregation. Similarly, numerous intermolecular interactions, like C–H⋯O (2.584 Å) or C–H⋯N (2.627 Å) were detected for 2-CN (see the ESI,† Fig. S131). Molecules are cross-stacked in an antiparallel sheet structure along the a-axis with interplanar spacing of 3.379 and 3.400 Å to the adjacent molecules, respectively. π-Skeleton distances of 3.476 Å and 3.565 Å with slip angles of 76.4° and 72.5° were determined, indicating strong H-aggregation.
) and 3-CN (monoclinic, P21/c) exhibit different packing modes in their respective crystal lattice, including various types of intermolecular interactions. This variety of contacts leads to a restriction of intramolecular rotation and explains the increased ΦF values in the solid-state. For 1-CN, several intermolecular interactions,33 like C–H⋯O (2.679 Å) or C–H⋯N (2.658 Å) are identified (see the ESI,† Fig. S120), while antiparallel molecule cross-stacking takes place along the b-axis in a sheet structure. Interplanar spacing is determined to 3.329 Å, whereas the adjacent π-skeleton distance is 3.586 Å with a slip angle of 68.2° (Fig. 9). Thus, relatively strong π–π-interactions lead to intense H-aggregation. Similarly, numerous intermolecular interactions, like C–H⋯O (2.584 Å) or C–H⋯N (2.627 Å) were detected for 2-CN (see the ESI,† Fig. S131). Molecules are cross-stacked in an antiparallel sheet structure along the a-axis with interplanar spacing of 3.379 and 3.400 Å to the adjacent molecules, respectively. π-Skeleton distances of 3.476 Å and 3.565 Å with slip angles of 76.4° and 72.5° were determined, indicating strong H-aggregation.
In 3-CN crystals, molecules are cross-stacked along the b-axis with an interplanar distance of 3.298 and 3.311 Å. Slip angles of 54.5° and 62.0° result in π-skeleton distances of 4.051 Å and 3.750 Å. In contrast to the previously discussed solid-state structures, sheet structure distortion of 12.4° is detected (see the ESI,† Fig. S145). Again, intermolecular interactions such as C–H⋯O (2.413 Å) or C–H⋯N (2.729 Å) lead to a restriction of intramolecular rotation, pointing to weak H-aggregation (see the ESI,† Fig. S146).
Interestingly, 1-CN exhibits a bent nitrile bond (N1–C7–Cipso) with an angle of 173.01°, whereas the respective bond in 2-CN and 3-CN is almost linear (see the ESI,† Tables S11 and S17). This observation can be explained through adjacent orbital interactions of the nonbonding orbital of the oxazoline-oxygen (O2) and a π*-orbital of the nitrile bond. The interaction energy was calculated by means of natural bond orbital (NBO) analysis and determined to 1.19 kcal mol−1 (see the ESI,† Chapter 6).
Commonly, H-aggregation results in diminished quantum yields, whereas J-aggregation results in improved emission.341-CN, 2-CN and 3-CN exhibit high to extremely high quantum yields (ΦF) in the solid-state, as already reported for other H-aggregated structures.35 However, those literature known luminophores exhibit larger π-conjugated systems and are not constructed on a minimalistic single-benzene scaffold like the presented emitters. Usually, highly potent solid-state emitters based on a single-benzene core are commonly showing J-aggregation.21–241-CN demonstrates its powerful emission (ΦF = 87.3%) with a well-arranged crystal lattice, including a crystal system of relatively high symmetry (orthorhombic), several short intermolecular contacts (C–H⋯O/N) and strong π–π-interactions (centroid–centroid-distance = 3.586 Å). A short hydrogen bond distance (1.670 Å), a high hydrogen bond angle (153.29°) as well as a zero dihedral angle confirms the great solid-state emission potential of 1-CN. The origin of the high photoluminescence quantum yield of 1-CN can be multifarious. Intermolecular interactions in the crystal lattice such as C–H⋯N interactions or antiparallel coupling of local dipoles as described by Park and Adachi for styrylbenzene derivatives can cause such unusual optical behavior.35b,d
Finally, TGA/DSC measurements were conducted to investigate the thermal stability of the discussed luminophores. All of these novel low molecular weight phenols (MW = 216.1), exhibit melting points TM between 121 and 170 °C and decomposition temperatures Tdecomp above 170 °C as determined by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (Table 5). The corresponding spectra are shown in the ESI† (Fig. S149–S152).
Table 5 Thermal stability properties
| Compd |
Onset/endset T [°C] |
T
M [°C] |
T
decomp [°C] |
|
1-CN
|
199/233 |
172.4 |
229.5 |
|
2-CN
|
188/220 |
134.5 |
216.5 |
|
3-CN
|
190/224 |
122.2 |
219.9 |
|
4-CN
|
198/229 |
122.8 |
224.8 |
Calculated emission spectra
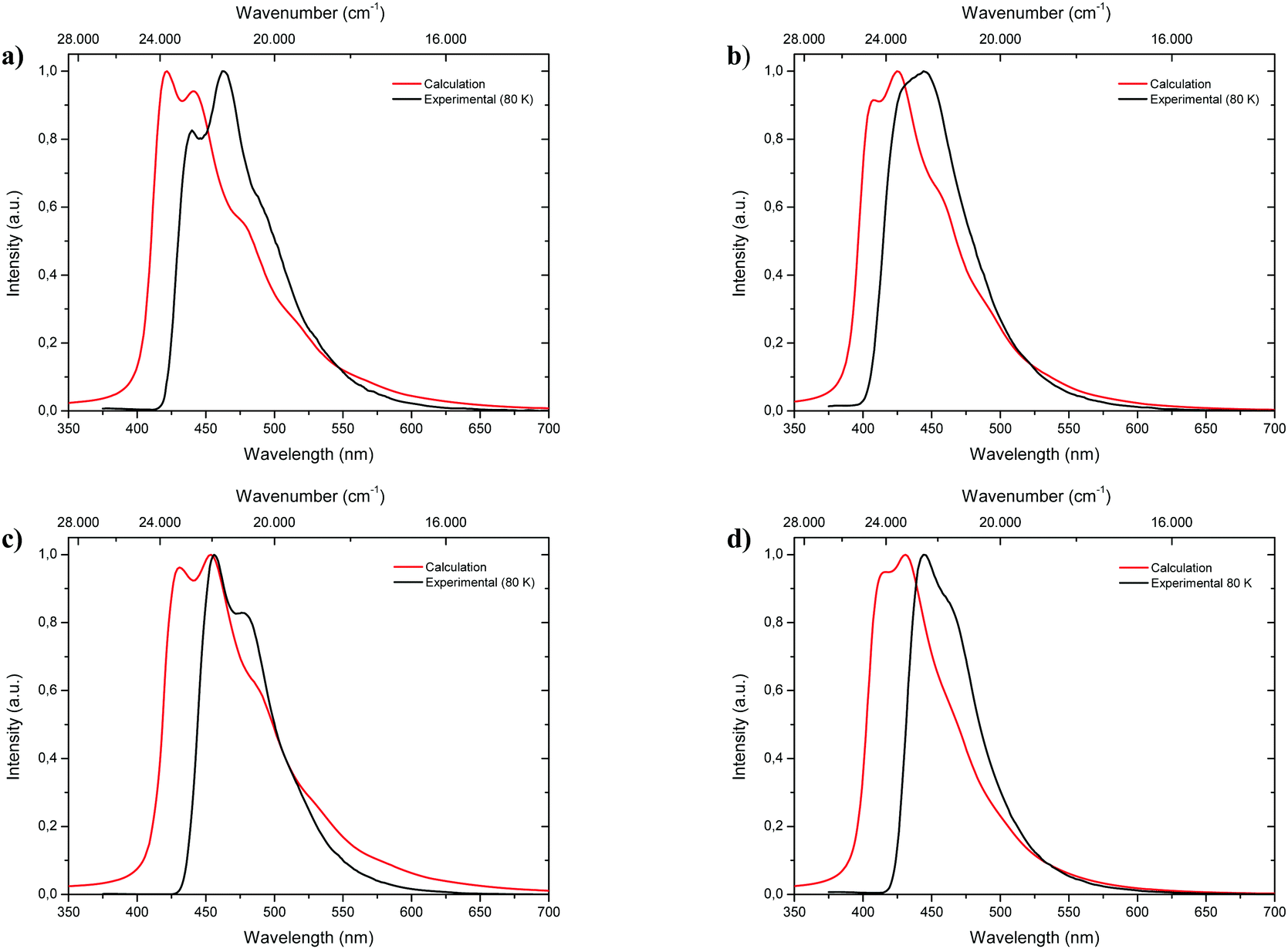
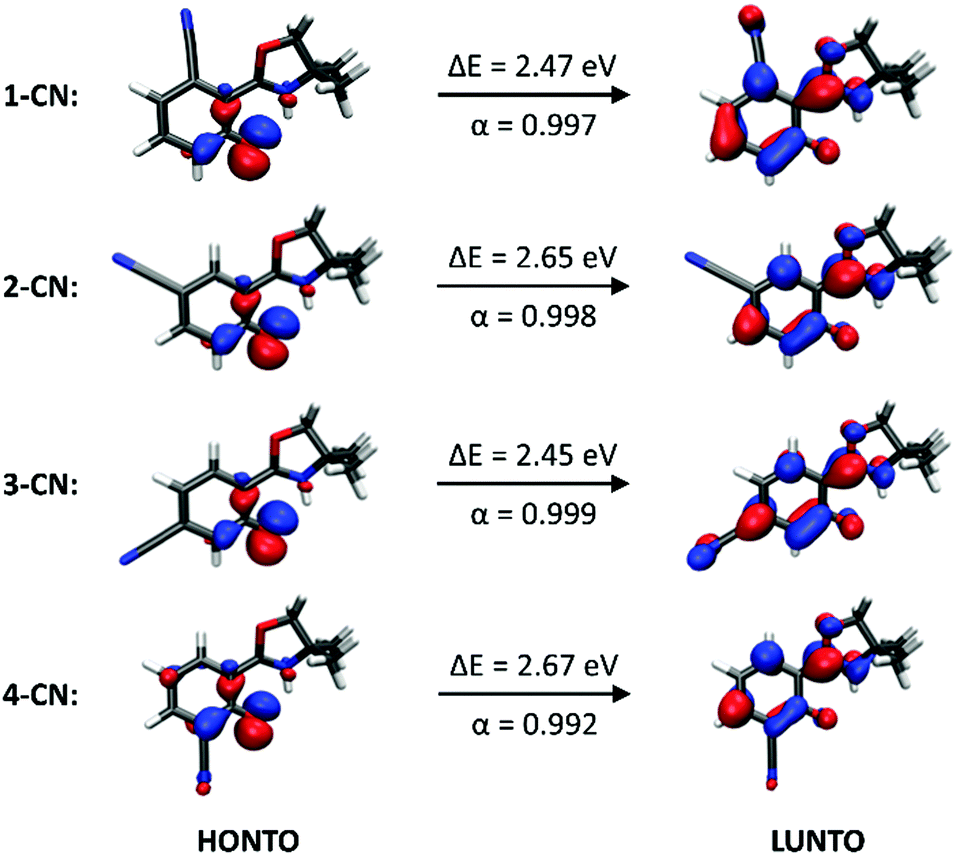
Fluorescence spectra of the keto tautomers of 1-CN, 2-CN, 3-CN and 4-CN were calculated with Time-Dependent Density Functional Theory (TDDFT)36 at the TD-B9737/cc-pVDZ38 level of theory to rationalize the experimental emission spectra. This level of theory was chosen because of the accurate reproduction of the experimental emission data (Fig. 10). Natural transition orbitals (NTOs)39 were applied to characterize the first electronically excited singlet (S1) state at the S1 equilibrium geometry of the keto tautomer, out of which fluorescence takes place. NTOs typically allow the characterization of an electronically excited state via a single excitation from the Highest Occupied Natural Transition Orbital (HONTO) into the Lowest Unoccupied Natural Transition Orbital (LUNTO) with a remarkably high eigenvalue. In all cases considered here, the S1 state can be characterized as an n–π*-state with eigenvalues>0.99 (Fig. 11). The excitations are quite local, signifying only insignificant charge-transfer character. Due to the accurate reproduction of the experimental emission spectra by TDDFT, the calculated spectra provide further evidence that emission takes place from the S1 state of the keto tautomer of each molecule presented here.
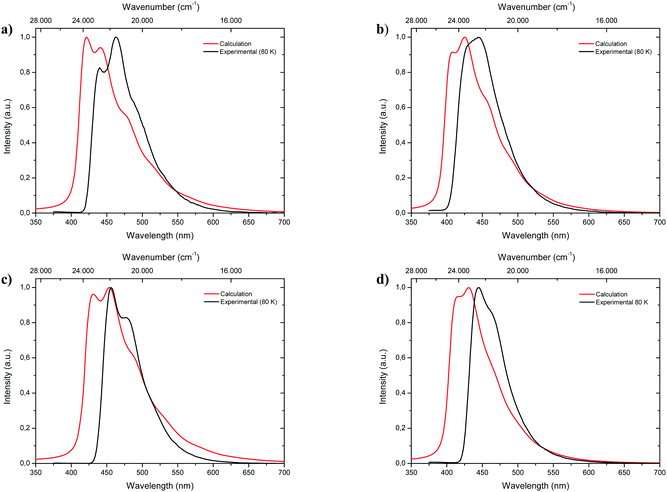 |
| | Fig. 10 Comparison of calculated fluorescence spectra (TD-B97/cc-pVDZ level of theory; red lines) with experimental emission spectra (black lines) measured in 2-MeTHF at 80 K of 1-CN (a), 2-CN (b), 3-CN (c) and 4-CN (d). | |
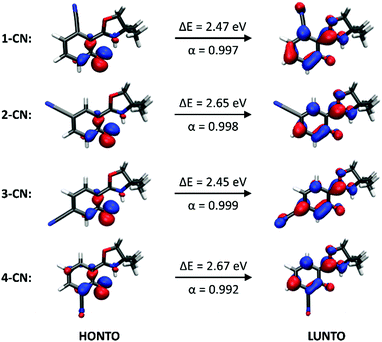 |
| | Fig. 11 Excitations from the Highest Occupied Natural Transition Orbital (HONTO) into the Lowest Unoccupied Natural Transition Orbital (LUNTO) that characterize the first electronically excited singlet (S1) state at the S1 equilibrium geometry of the keto tautomer, out of which the fluorescence of each of the four isomers takes place. ΔE is the electronic excitation energy of the S1 state in the keto form and α is the eigenvalue of the HONTO → LUNTO transition. | |
Conclusions
In summary, minimalistic, nitrile-substituted single-benzene ESIPT-based luminophores exhibiting powerful emission in the solid-state have been successfully synthesized and investigated towards their emission properties. Depending on the nitrile position, AIEE (1-CN and 2-CN), DSE (3-CN) or ACQ characteristics (4-CN) with blue to green-cyan emission colors and high Stokes shifts were identified for all investigated fluorophores. Quantum yields are remarkably high and with 1-CN the lowest molecular weight ESIPT-based luminophore with a quantum yield above 85% in the solid-state was discovered (MW = 216.1; ΦF = 87.3%). Through careful single crystal analysis, we identified the well-shaped molecular structure and the eminent organized crystal lattice as the origin of these outstanding photoluminescence properties. TDDFT calculated emission spectra gave evidence for emission from the S1 state of the keto tautomer. This exceptionally straightforward concept for the creation of minimalistic, highly emissive fluorophores opens up the opportunity for future design studies yielding luminophores with superior properties. Applications of the discussed fluorophores in optoelectronic materials are underway in our laboratories.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank Prof. Dr Nadja-C. Bigall, Dr Dirk Dorfs, and Pascal Rusch (all from Leibniz University Hannover) for supporting the photophysical measurements.
References
-
(a) R. Long, C. Tang, J. Xu, T. Li, C. Tong, Y. Guo, S. Shi and D. Wang, Chem. Commun., 2019, 55, 10912 RSC;
(b) R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228 RSC;
(c) H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620 CrossRef CAS PubMed;
(d) L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142 CrossRef CAS PubMed.
-
(a) X. Li, X. Gao, W. Shi and H. Ma, Chem. Rev., 2014, 114, 590 CrossRef CAS PubMed;
(b) S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS PubMed;
(c) L. Basabe-Desmonts, D. N. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 993 RSC;
(d) R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419 CrossRef PubMed;
(e) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef CAS PubMed.
-
(a)
H. Yersin, Highly Efficient OLEDs with Phosphorescent Materials, Wiley-VCH, Hoboken, 2008 Search PubMed;
(b)
K. Müllen and U. Scherf, Organic Light-Emitting Devices. Synthesis, Properties, and Applications, Wiley-VCH, Weinheim, 2006 Search PubMed;
(c) R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. D. Santos, J. L. Brédas, M. Lögdlund and W. R. Salaneck, Nature, 1999, 397, 121 CrossRef CAS.
-
(a) M. D. Watson, A. Fechtenkötter and K. Müllen, Chem. Rev., 2001, 101, 1267 CrossRef CAS PubMed;
(b) R. E. Martin and F. Diederich, Angew. Chem., Int. Ed., 1999, 38, 1350 CrossRef.
-
(a) O. Ostroverkhova, Chem. Rev., 2016, 116, 13279 CrossRef CAS PubMed;
(b) H. Meier, Angew. Chem., Int. Ed., 2005, 44, 2482 CrossRef CAS PubMed;
(c) R. Gompper and H.-U. Wagner, Angew. Chem., Int. Ed. Engl., 1988, 27, 1437 CrossRef.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
-
B. Z. Tang and A. Qin, Aggregation-Induced Emission. Fundamentals, Wiley, John Wiley & Sons Inc., Chichester, 2013 Search PubMed.
-
(a) B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544 CrossRef CAS PubMed;
(b) D. Oelkrug, A. Tompert, J. Gierschner, H.-J. Egelhaaf, M. Hanack, M. Hohloch and E. Steinhuber, J. Phys. Chem. B, 1998, 102, 1902 CrossRef CAS.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726 RSC.
- S. Benson, A. Fernandez, N. D. Barth, F. de Moliner, M. H. Horrocks, C. S. Herrington, J. L. Abad, A. Delgado, L. Kelly, Z. Chang, Y. Feng, M. Nishiura, Y. Hori, K. Kikuchi and M. Vendrell, Angew. Chem., Int. Ed., 2019, 58, 6911 CrossRef CAS PubMed.
- M. Shimizu and T. Hiyama, Chem. – Asian J., 2010, 5, 1516 CrossRef CAS PubMed.
-
(a) A. C. Sedgwick, L. Wu, H.-H. Han, S. D. Bull, X.-P. He, T. D. James, J. L. Sessler, B. Z. Tang, H. Tian and J. Yoon, Chem. Soc. Rev., 2018, 47, 8842 RSC;
(b) V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169 RSC;
(c) A. P. Demchenko, K.-C. Tang and P.-T. Chou, Chem. Soc. Rev., 2013, 42, 1379 RSC;
(d) J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615 CrossRef CAS PubMed.
- P. F. Barbara, P. K. Walsh and L. E. Brus, J. Phys. Chem., 1989, 93, 29 CrossRef CAS.
- H.-Q. Yin, F. Yin and X.-B. Yin, Chem. Sci., 2019, 10, 11103 RSC.
-
(a) K. Benelhadj, W. Muzuzu, J. Massue, P. Retailleau, A. Charaf-Eddin, A. D. Laurent, D. Jacquemin, G. Ulrich and R. Ziessel, Chem. – Eur. J., 2014, 20, 12843 CrossRef CAS PubMed;
(b) H. Shono, T. Ohkawa, H. Tomoda, T. Mutai and K. Araki, ACS Appl. Mater. Interfaces, 2011, 3, 654 CrossRef CAS PubMed;
(c) K.-C. Tang, M.-J. Chang, T.-Y. Lin, H.-A. Pan, T.-C. Fang, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Am. Chem. Soc., 2011, 133, 17738 CrossRef CAS PubMed;
(d) S. Park, J. E. Kwon, S. H. Kim, J. Seo, K. Chung, S.-Y. Park, D.-J. Jang, B. Milián Medina, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 14043 CrossRef CAS PubMed.
- Y. Zhang, H. Yang, H. Ma, G. Bian, Q. Zang, J. Sun, C. Zhang, Z. An and W.-Y. Wong, Angew. Chem., Int. Ed., 2019, 58, 8773 CrossRef CAS PubMed.
-
(a) T. Mutai, H. Shono, Y. Shigemitsu and K. Araki, CrystEngComm, 2014, 16, 3890 RSC;
(b) T. Mutai, H. Tomoda, T. Ohkawa, Y. Yabe and K. Araki, Angew. Chem., Int. Ed., 2008, 47, 9522 CrossRef CAS PubMed.
-
(a) T. Mutai, T. Ohkawa, H. Shono and K. Araki, J. Mater. Chem. C, 2016, 4, 3599 RSC;
(b) T. Mutai, H. Sawatani, T. Shida, H. Shono and K. Araki, J. Org. Chem., 2013, 78, 2482 CrossRef CAS PubMed.
- S. Park, J. E. Kwon, S.-Y. Park, O.-H. Kwon, J. K. Kim, S.-J. Yoon, J. W. Chung, D. R. Whang, S. K. Park, D. K. Lee, D.-J. Jang, J. Gierschner and S. Y. Park, Adv. Opt. Mater., 2017, 5, 1700353 CrossRef.
- B. Tang, H. Liu, F. Li, Y. Wang and H. Zhang, Chem. Commun., 2016, 52, 6577 RSC.
- M. Shimizu, Y. Takeda, M. Higashi and T. Hiyama, Angew. Chem., Int. Ed., 2009, 121, 3707 CrossRef.
- T. Beppu, K. Tomiguchi, A. Masuhara, Y.-J. Pu and H. Katagiri, Angew. Chem., Int. Ed., 2015, 54, 7332 CrossRef CAS PubMed.
- B. Tang, C. Wang, Y. Wang and H. Zhang, Angew. Chem., Int. Ed., 2017, 56, 12543 CrossRef CAS PubMed.
- Z. Xiang, Z.-Y. Wang, T.-B. Ren, W. Xu, Y.-P. Liu, X.-X. Zhang, P. Wu, L. Yuan and X.-B. Zhang, Chem. Commun., 2019, 55, 11462 RSC.
- D. Yao, S. Zhao, J. Guo, Z. Zhang, H. Zhang, Y. Liu and Y. Wang, J. Mater. Chem., 2011, 21, 3568 RSC.
- W.-T. Chuang, C.-C. Hsieh, C.-H. Lai, C.-H. Lai, C.-W. Shih, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Org. Chem., 2011, 76, 8189 CrossRef CAS PubMed.
- D. Göbel, N. Clamor, E. Lork and B. J. Nachtsheim, Org. Lett., 2019, 21, 5373 CrossRef PubMed.
- D. Göbel, N. Clamor and B. J. Nachtsheim, Org. Biomol. Chem., 2018, 16, 4071 RSC.
- K. Schwekendiek and F. Glorius, Synthesis, 2006, 2996 CAS.
-
R. Misra and S. P. Bhattacharyya, Intramolecular Charge Transfer, Wiley-VCH, Weinheim, 2018 Search PubMed.
-
(a) Q. Yu, X. Zhang, S.-T. Wu, H. Chen, Q.-L. Zhang, H. Xu, Y.-L. Huang, B.-X. Zhu and X.-L. Ni, Chem. Commun., 2020, 56, 2304 RSC;
(b) T. Zhang, L. Wen, G. Liu, J. Yan, X. Liu, K. Zheng and N. Zhang, Chem. Commun., 2019, 55, 13713 RSC.
- H. Lin, X. Chang, D. Yan, W.-H. Fang and G. Cui, Chem. Sci., 2017, 8, 2086 RSC.
-
(a) N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069 CrossRef CAS PubMed;
(b) D. Möbius, Adv. Mater., 1995, 7, 437 CrossRef.
-
(a) Y. Niu, R. Wang, P. Shao, Y. Wang and Y. Zhang, Chem. – Eur. J., 2018, 24, 16670 CrossRef CAS PubMed;
(b) S.-J. Yoon and S. Park, J. Mater. Chem., 2011, 21, 8338 RSC;
(c) S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675 CrossRef CAS PubMed;
(d) R. Kabe, H. Nakanotani, T. Sakanoue, M. Yahiro and C. Adachi, Adv. Mater., 2009, 21, 4034 CrossRef CAS;
(e) U. Rösch, S. Yao, R. Wortmann and F. Würthner, Angew. Chem., Int. Ed., 2006, 45, 7026 CrossRef PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data, photophysical data, X-ray crystallographic data, and copies of 1H-, 13C-NMR spectra. CCDC 1908035, 1970920 and 1970921. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0tc00776e |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *cde and
Boris J.
Nachtsheim
*cde and
Boris J.
Nachtsheim