
Photoinduced strategies towards strained molecules
Mukund M. D.
Pramanik
,
Hao
Qian
,
Wen-Jing
Xiao
 * and
Jia-Rong
Chen
*
* and
Jia-Rong
Chen
*
CCNU-uOttawa Joint Research Center, Key Laboratory of Pesticides & Chemical Biology, Ministry of Education, College of Chemistry, Central China Normal University, 152 Luoyu Road, Wuhan, Hubei 430079, China. E-mail: wjxiao@mail.ccnu.edu.cn; chenjiarong@mail.ccnu.edu.cn
First published on 22nd July 2020
Abstract
New photoinduced strategies towards radical reactions of strained molecules such as [1.1.1]propellane and bicyclo[1.1.0]butanes by photoredox or metallaphotoredox catalysis have recently been disclosed. Such strategies enable the controllable construction of 1,3-difunctionalized bicyclo[1.1.1]pentanes, cyclobutanes, and drug-like analogues, and offer new opportunities for reaction design of [1.1.1]propellane and other strained molecules.
The selective incorporation of three-dimensional, strained cyclic carbon scaffolds into bioactive molecules can often dramatically alter their pharmacokinetic profiles such as lipophilicity and passive permeability, compared to their parent compounds, while rendering similar or improved levels of potency.1 Not surprisingly, therefore, the rigid, linear, and sp3-rich bicyclo[1.1.1]pentane (BCP) has recently been extensively explored as a robust bioisostere for arene, internal alkyne, and tert-butyl groups in modern medicinal chemistry and drug discovery.2
From the synthetic point of view, [1.1.1]propellane is the most promising precursor for the construction of BCP derivatives because of its unique fragile central carbon–carbon σ-bond.3 As a result, a wide range of radical or ionic ring-opening reactions of [1.1.1]propellane have been developed for the assembly of various 1,3-difunctionalized BCP derivatives using appropriate radicals or metal-based reagents by a single or multi-step procedure (Scheme 1a).4 Despite the high efficiency, however, in most cases, relatively forcing reaction conditions, sensitive organometallic reagents, or irradiation of a high-pressure mercury lamp results in limitation of the substrate scope, poor functional group tolerance, and/or low diversity of BCP products. In particular, the preparation of structurally complex 1,3-disubstituted BCP derivatives remains largely difficult and still requires long multiple chemical steps.5
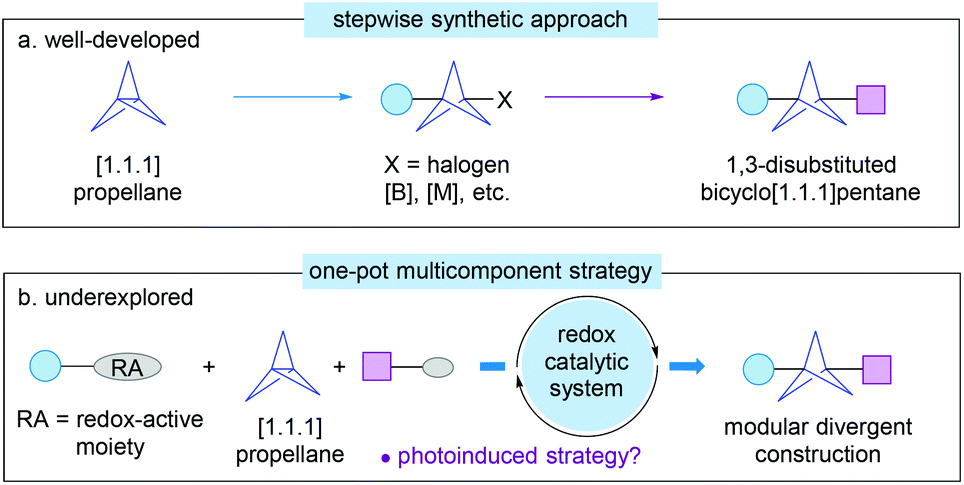 | ||
| Scheme 1 State-of-the-art strategies for the synthesis of 1,3-disubstituted bicyclo[1.1.1]pentanes from strained [1.1.1]propellane. | ||
Recently, Anderson et al. disclosed that triethylborane-initiated atom transfer radical addition (ATRA) reactions of [1.1.1]propellane with organic halides provided an efficient access to a variety of 1-halo-3-substituted bicyclopentanes.6a Combination of this protocol with transition metal-catalyzed coupling allows further introduction of functional groups at the C–halogen bond. Shortly thereafter, Anderson, Duarte and coworkers developed the first example of the photoredox-catalyzed atom-transfer radical addition (ATRA) reaction of organic halides 2 to [1.1.1]propellane 1 under mild conditions (Scheme 2).6b This reaction demonstrated an exceptionally broad substrate scope and functional group tolerance. A wide range of (hetero)aryl, 1°/2° alkyl iodides, and α-EWG halides are all well tolerated, giving the corresponding diversely functionalized 1-iodo-bicyclo[1.1.1]pentanes 3 (iodo-BCPs) with moderate to excellent yields. This protocol could also be applied to late-stage modification of biomolecules and drug-like compounds. The iodo-BCPs are versatile precursors of other derivatives. Building on this methodology, the Anderson group recently developed a generally applicable iron-catalyzed Kumada cross-coupling of iodo-BCPs with aryl and heteroaryl Grignard reagents, enabling the construction of various all-carbon 1,3-disubstituted bicyclo[1.1.1]pentanes.6c Notably, the Zhu group recently disclosed an elegant photoredox-catalyzed ATRA reaction of [1.1.1]propellane using bromoalkyl heteroarylsulfones, providing atom economy and general access to diverse alkylheteroarylsulfone-substituted BCP derivatives.7
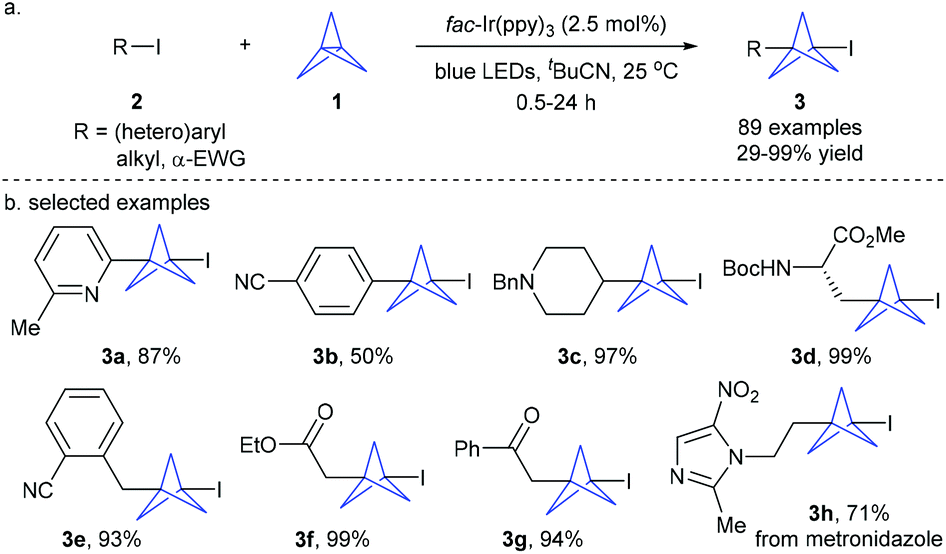 | ||
| Scheme 2 Anderson and Duarte's photoredox-catalyzed ATRA reaction between organic iodides and [1.1.1]propellane. | ||
On the other hand, the one-pot radical multicomponent reaction (MCR) of [1.1.1]propellane is a synthetically more advantageous and versatile strategy for the one-step, modular construction of diversely 1,3-disubstitued BCP derivatives (Scheme 1b).8 A breakthrough in this field has come from the group of Uchiyama in 2017, who first disclosed that a combination of iron(II) phthalocyanine (Fe(Pc)) as a catalyst and tert-butyl hydroperoxide (TBHP) as an oxidant enabled an efficient radical multicomponent carboamination of [1.1.1]propellane using methyl hydrazinecarboxylate and (hetero)arylhydrazines 4 as carbon-radical precursors and di-tert-butyl azodicarboxylate (DBAD) 5 as a radical acceptor (Scheme 3).9a The radical mechanism and overall thermodynamically and kinetically favourable process have also been supported by experimental and computational studies. Though this protocol has not been extended to any other radical traps, this tin-free radical multicomponent strategy opened up a new way to the direct preparation of unsymmetrically 1,3-disubstituted BCP derivatives 6, particularly the valuable 3-substituted BCP-amines (BCPAs). Quite recently, the Kanazawa and Uchiyama group further developed an interesting UV light-driven silaboration of [1.1.1]propellane using MesPhSi-Bpin for the first time.9b This protocol enabled the direct introduction of B and Si functional groups into the [1.1.1]propellane scaffold by an ATRA process. However, the current catalytic system was exclusively limited to Me2PhSi-Bpin and MePh2Si-Bpin.
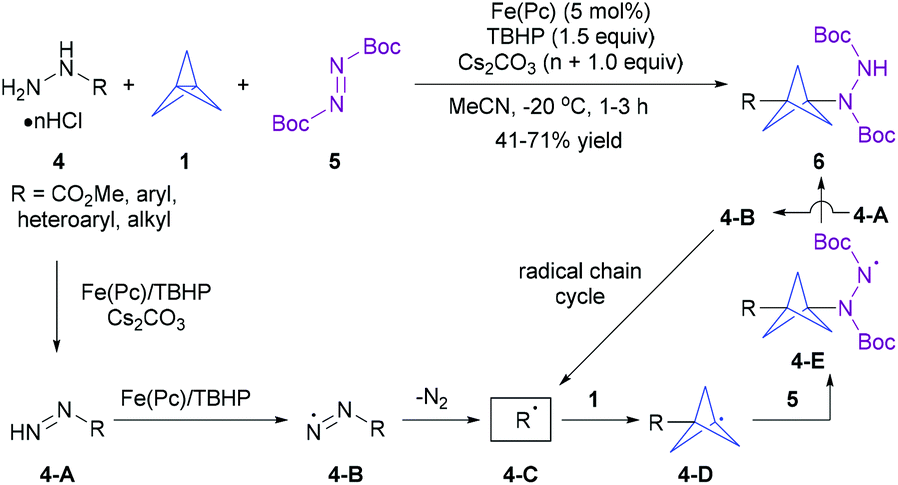 | ||
| Scheme 3 Uchiyama's radical multicomponent carboamination of [1.1.1]propellane. | ||
In order to develop a more general and direct approach for the divergent synthesis of functionalized bicyclo[1.1.1]pentylamines, Leonori, Sheikh, and co-workers recently developed the first example of the photoredox-catalyzed radical multicomponent reaction of [1.1.1]propellane with amide-derived electrophilic nitrogen-radicals and SOMOphiles (Scheme 4a).10,11 The key to the success of this strategy was the smart exploration of the electrophilic property of amidyl radicals and the electron-rich nature of [1.1.1]propellane 1 as well as the strong polar effects existing in their C–N bond-forming/ring-opening transition state. The feasibility of this step was also confirmed by detailed computational studies. Furthermore, kinetically preferred atom/group-transfer from the SOMOphile to the initially formed BCP radicals over the competing BCP radical oligomerization is also critical to this protocol.
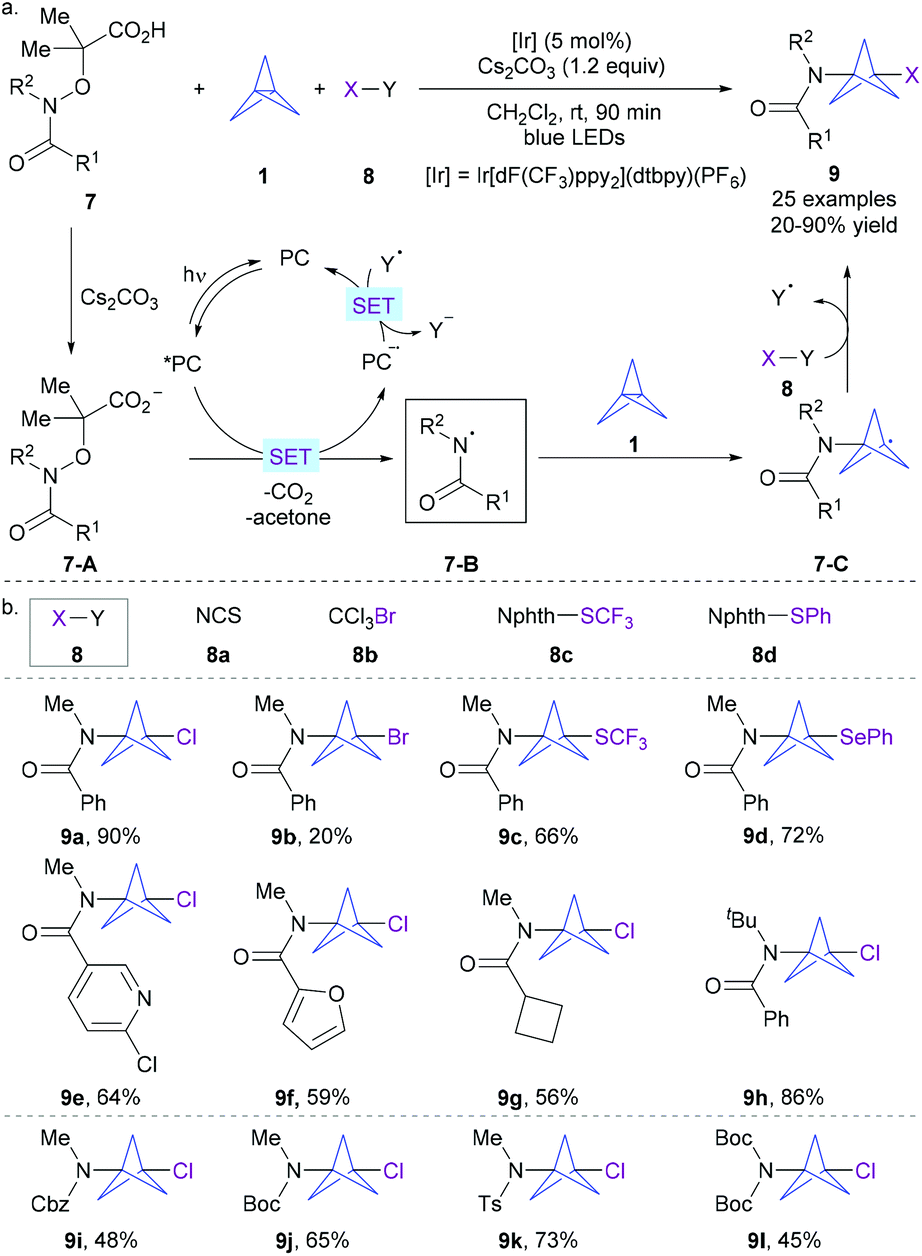 | ||
| Scheme 4 Leonori and Sheikh's visible light-induced photoredox-catalyzed divergent amino-functionalization of [1.1.1]propellane. | ||
Guided by DFT calculation, Leonori, Sheikh, and co-workers first developed a practical procedure for reproducible and large-scale preparation of [1.1.1]propellane 1 as a 0.5 M solution in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 benzene/CH2Cl2 mixture, and identified that a combination of Ir[dF(CF3)ppy2](dtbpy)(PF6) as a photocatalyst and Cs2CO3 as a base in CH2Cl2 under irradiation of blue LEDs enabled an efficient radical multicomponent reaction of α-imino-oxy acetic acid 7, [1.1.1]propellane 1, and SOMOphiles 8 (Scheme 4a). Mechanistically, it was postulated that a reductive quenching photoredox cycle was involved. The in situ-formed carboxylate 7-A undergoes single electron transfer (SET) oxidation by the visible light-excited photocatalyst *PC to form the key amidyl radical 7-B upon extrusion of CO2 and acetone, together with the reduced form of the photocatalyst PC˙−. Then, the electrophilic amidyl radical 7-B undergoes addition across electron-rich 1 to trigger the cleavage of its central Csp3–Csp3 bond, resulting in stabilized bicyclopentyl radical 7-C. Interception of 7-C by phthalimide-based reagents 8 through an atom/group-transfer process gives rise to the final product 9 with the release of the electron-poor radical Y˙. Ultimately, a SET from the reduced form of the photocatalyst to the intermediate Y occurred to regenerate the ground state photocatalyst PC, rendering a redox-neutral process and closing the photocatalytic cycle.
:1 benzene/CH2Cl2 mixture, and identified that a combination of Ir[dF(CF3)ppy2](dtbpy)(PF6) as a photocatalyst and Cs2CO3 as a base in CH2Cl2 under irradiation of blue LEDs enabled an efficient radical multicomponent reaction of α-imino-oxy acetic acid 7, [1.1.1]propellane 1, and SOMOphiles 8 (Scheme 4a). Mechanistically, it was postulated that a reductive quenching photoredox cycle was involved. The in situ-formed carboxylate 7-A undergoes single electron transfer (SET) oxidation by the visible light-excited photocatalyst *PC to form the key amidyl radical 7-B upon extrusion of CO2 and acetone, together with the reduced form of the photocatalyst PC˙−. Then, the electrophilic amidyl radical 7-B undergoes addition across electron-rich 1 to trigger the cleavage of its central Csp3–Csp3 bond, resulting in stabilized bicyclopentyl radical 7-C. Interception of 7-C by phthalimide-based reagents 8 through an atom/group-transfer process gives rise to the final product 9 with the release of the electron-poor radical Y˙. Ultimately, a SET from the reduced form of the photocatalyst to the intermediate Y occurred to regenerate the ground state photocatalyst PC, rendering a redox-neutral process and closing the photocatalytic cycle.
This protocol allowed simultaneous installation of the C–N bond together with the C–Cl, C–Br, C–S and C–Se bonds when using NCS, CCl3Br, and phthalimide-based reagents 8c–d (Scheme 4b). Markedly, significant variation of the substitution pattern of amidyl radical precursors can also be well tolerated. N-Me-benzamides with various functional groups on the phenyl ring, as well as those containing heteroaryl and alkyl groups, are all amenable to the reaction. Other electrophilic N-radicals based on easily removable protecting groups (e.g., Cbz, Boc, Ts) also performed well to give the target products 9l–i with good yields. This strategy significantly shortened the access to BCPAs and can be potentially extended to other electrophilic radicals.
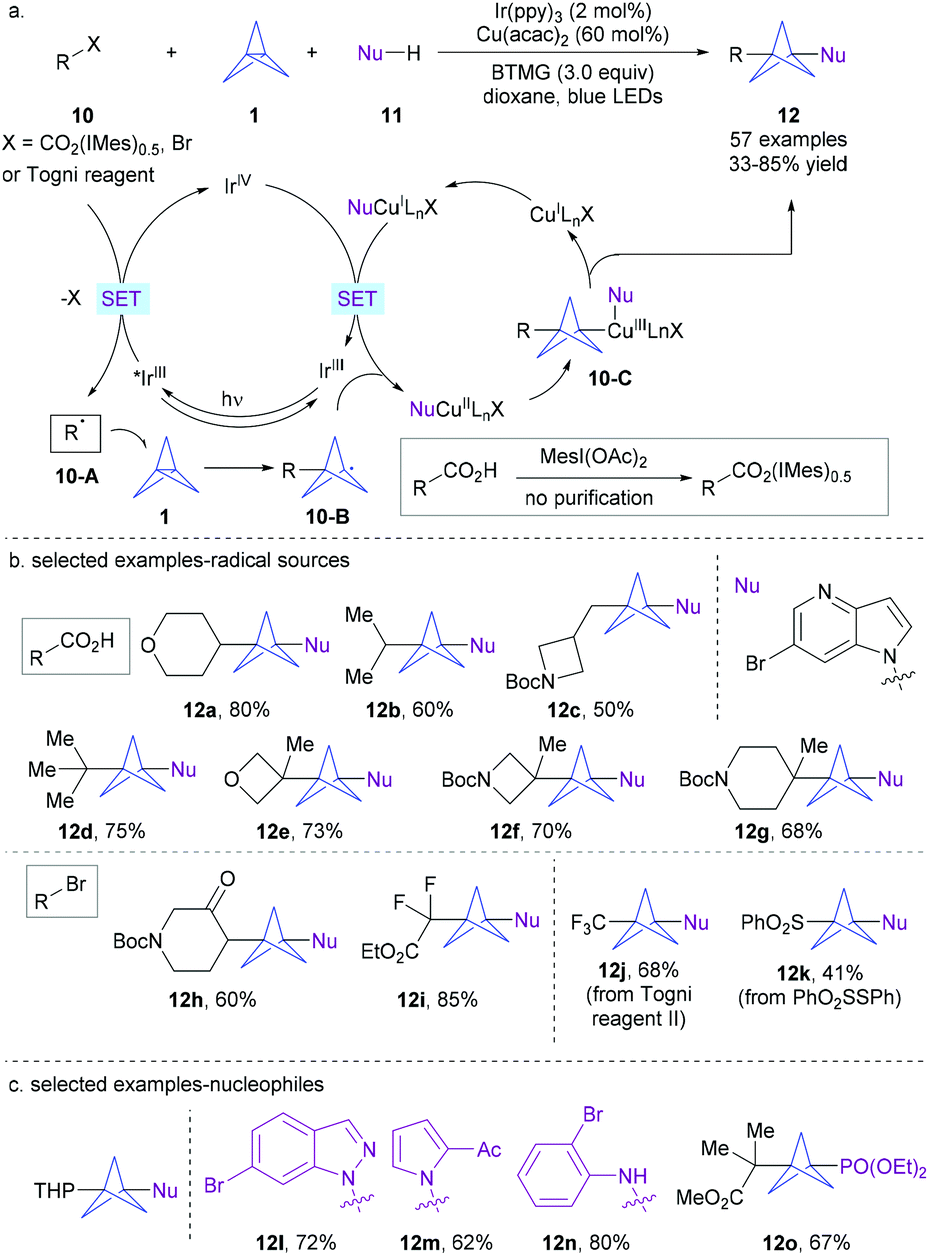
Metallaphotoredox catalysis has recently emerged as a powerful platform for the controllable generation of radicals from diverse readily accessible precursors and their ensuing engagement in transition metal-catalyzed cross-coupling.12 Building on their previous work on dual photoredox and copper-catalyzed carboxylic acid chemistry,13 the group of MacMillan disclosed an elegant metallaphotoredox-catalyzed one-step radical multicomponent reaction of [1.1.1]propellane using various activated carboxylic acids as the radical source and heteroatom nucleophiles (Scheme 5a).14 Despite the potential competing two-component cross-coupling between alkyl radicals and nucleophiles as well as BCP oligomerization, it was established that a combination of Ir(ppy)3 (2 mol%) as a photocatalyst and Cu(acac)2 (60 mol%) as a coupling catalyst in the presence of 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG) as a base enabled the target reaction to proceed smoothly with excellent chemoselectivity under irradiation of blue LEDs.
 | ||
| Scheme 5 MacMillan's visible light-induced dual photoredox and copper-catalyzed radical multicomponent coupling of [1.1.1]propellane. | ||
In contrast to Leonori and Sheikh's approach, herein, the key alkyl radicals were generated through a photocatalytic oxidative quenching cycle. It was proposed that the photoexcited strong reducing photocatalyst *IrIII initially reduced the in situ-formed iodonium dicarboxylate 10 to form alkyl radical 10-A after extrusion of CO2, together with the oxidized form of the catalyst IrIV. Next, a ring-opening radical addition of species 10-A to [1.1.1]propellane 1 affords BCP radical 10-B. Then, 10-B can be intercepted by a nucleophile-ligated copper complex (NuCuIILnX) to form the CuIII complex that further undergoes reductive elimination to furnish the final product 12. This decarboxylative radical multicomponent coupling demonstrates a broad substrate scope and high functional group compatibility (Scheme 5b). Notably, the radical precursors, iodonium dicarboxylates, can be generated directly from a wide range of easily available carboxylic acids without purification. As such, a variety of drug-like bicyclopentanes are obtained with moderate to good yields. The scope of the radical source could be expanded to alkyl bromides, Togni reagent II and thiosulfonate. Moreover, a series of N-nucleophiles including many medicinally relevant N-heterocycles, and anilines, as well as P- and S-nucleophiles are all amenable to this protocol (Scheme 5c). Successful application of this method to natural products and pharmaceuticals also highlights the potential of this MCR strategy.
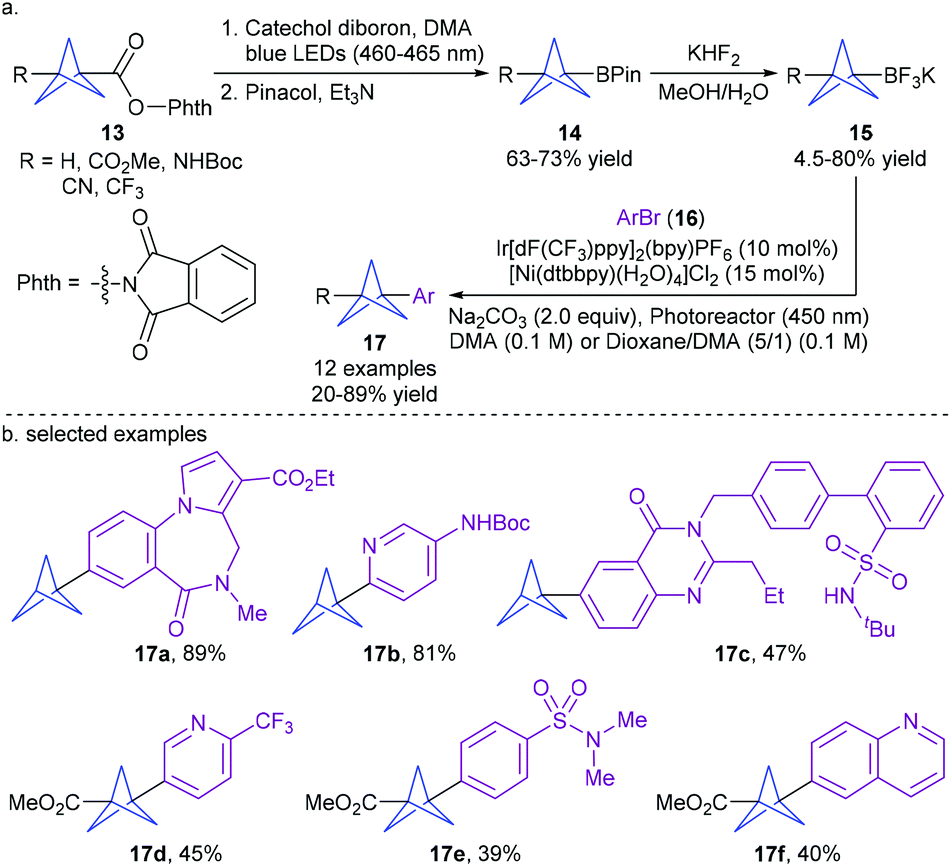
Drawing inspiration from the extensive portfolio of dual photoredox and nickel-catalyzed radical cross-coupling,15 the Merck researchers VanHeyst and Qi et al.16a recently disclosed a practical and scalable continuous flow synthesis of bicyclo[1.1.1]pentane trifluoroborate salts (BCP BF3K 15) by photoinduced decarboxylative borylation of N-hydroxyphthalimide (NHPI) esters 13 under irradiation of 100 W 460–465 nm blue LEDs and treatment with KHF2 (up to 200 g scale) (Scheme 6).16b Thereby, they developed a general visible light-induced cross-coupling of BCP BF3K 15 with a variety of complex aryl halides using Ir[dF(CF3)ppy]2(bpy)PF6 and [Ni(dtbbpy)(H2O)4]Cl2 as catalysts. This protocol enables the efficient installation of the BCP scaffold into a wide range of drug-like molecules, highlighting its potential in drug discovery.
 | ||
| Scheme 6 Merck's photo-induced cross-coupling of bicycle[1.1.1]pentane trifluoroborate salts with (hetero)aryl halides. | ||
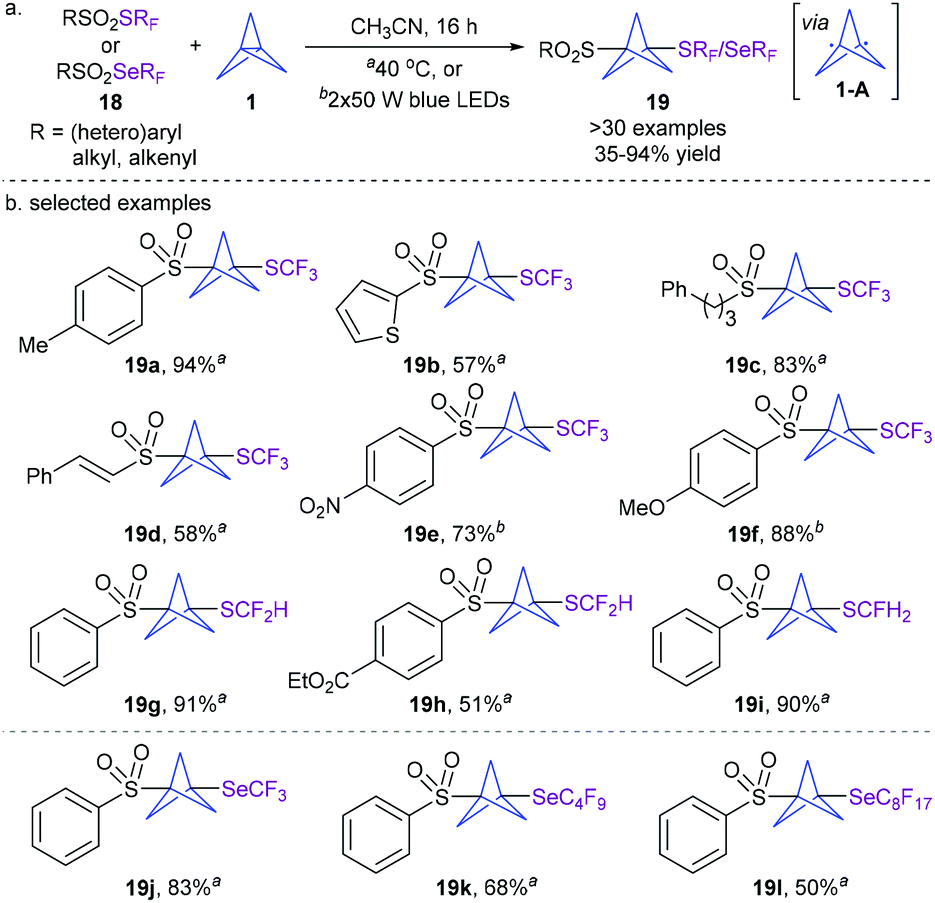
Given the biological significance of the fluoroalkylthio(seleno) moiety, the Zhu group recently disclosed for the first time an efficient approach for the synthesis of fluoroalkylthio(seleno)-functionalized BCPs under simple and practical thermal or photoinduced conditions without any photocatalyst (Scheme 7).17 A range of readily available reagents 18 containing SCF3, SCF2H, SCFH2, SeCF3, SeC4F9, or SeC8F17 reacted well with [1.1.1]propellane 1, furnishing a variety of 1,3-disubstituted BCPs 19 in generally good yields. A series of control experiments suggest that the reaction involves a radical process and proceeds via diradical intermediate 1-A formed by the homolysis of the strained central C–C bond of [1.1.1]propellane 1. This operationally simple and atom-economical method provides a novel access to new BCP scaffolds.
 | ||
| Scheme 7 Zhu's practical approach to fluoroalkylthio(seleno)-functionalized bicyclo[1.1.1]pentanes. | ||
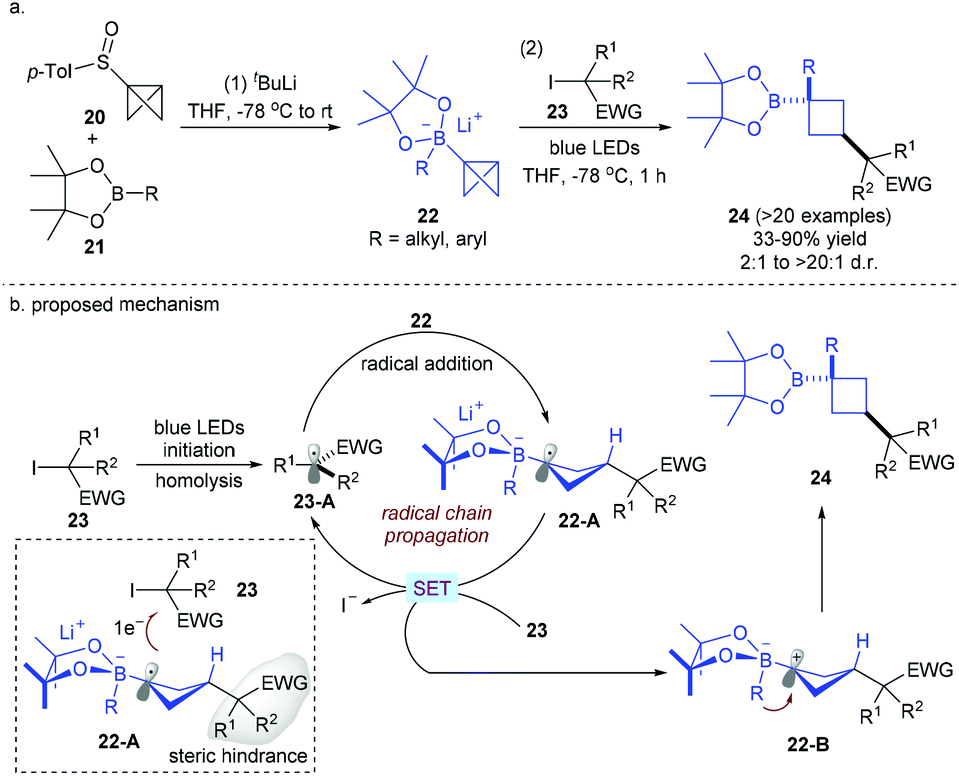
Functionalized cyclobutanes are another class of attractive carbocycles since they can serve as conformationally restricted structural scaffolds and C(sp3)-rich building blocks. Direct functionalization with bicyclo[1.1.0]butane (BCB) derivatives provides a powerful method for the construction of cyclobutane-containing natural products and drug-like compounds. Building on the reactivity mode between vinyl boronates and electrophilic radicals,18a,b in 2019, the Aggarwal group developed for the first time a radical addition/rearrangement reaction between bicyclobutyl (BCB)–boronate complexes 22, easily prepared from BCB-sulfoxides 20 and boronic acid pinacol esters 21, and electron-deficient radicals derived from alkyl iodides under visible light irradiation without an external photocatalyst (Scheme 8).18c This protocol shows a broad substrate scope and functional group tolerance with respect to both starting boronic esters and radical sources, providing the corresponding 1,3-disubstituted cyclobutyl boronic esters in good yields with excellent stereoselectivity. Successful extension to biologically active or complex compound-derived boronic esters and alkyl iodides also highlights the potential of this stereospecific strategy. As for the mechanism, it is postulated that alkyl radical 23-A could be first generated from alkyl iodides by photolytic initiation. Then, electrophilic radical 23-A undergoes addition to the strained central σ-C–C bond of 22 to give electron-rich radical anion 22-A. A SET oxidation of 22-A by another molecule of alkyl iodide 23 occurs to regenerate radical 23-A and zwitterionic species 22-B. Note that it is speculated that this step is also a stereoselectivity-determining process due to the steric hindrance between the alkyl iodide-derived substituent and alkyl iodide 23. Finally, 22-B undergoes facile 1,2-migration of the R group on the opposite lobe of the p orbital before the bond formation of the C+–B bond, leading to the desired product 24 with the cis-configuration.
 | ||
| Scheme 8 Aggarwal's stereocontrolled synthesis of cyclobutyl boronic esters by photoinduced radical addition to bicyclobutyl (BCB)–boronate complexes. | ||
On the basis of their previous photocatalytic addition of nonactivated amines to peptides,19a the Jui group recently developed an interesting photoredox-catalyzed addition of dimethylaniline derivatives 25 to bench-stable BCB reagent 26, giving the corresponding α-cyclobutyl N-alkylaniline products 27 in moderate to good yields with moderate diastereoselectivity (Scheme 9).19b In this redox- and proton-neutral process, α-amino radicals 26-B, formed by photo-induced SET oxidation of amines substrates 25, are involved as the key intermediates and undergo a formal Giese-type addition to the highly strained phenyl sulfonyl BCB 26via carbon radical intermediate 25-C.
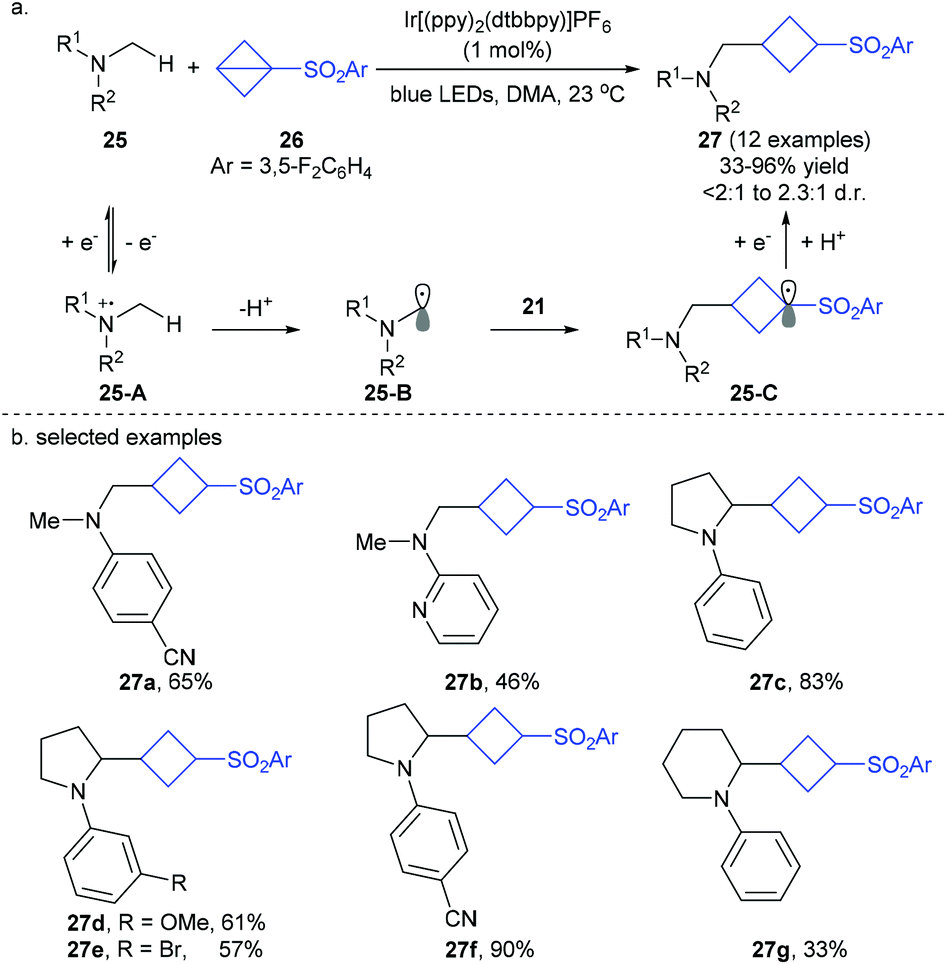 | ||
| Scheme 9 Jui's photoredox-catalyzed Giese-type addition of α-amino carbon radicals to bicyclo[1.1.0]butane (BCB). | ||
At almost the same time, Cintrat, Ernouf, and co-workers disclosed a robust photoredox-catalyzed decarboxylative radical addition of α-amino and α-oxy carboxylic acids 28 to sulfonyl BCB derivatives 29 (Scheme 10).20 It is also proposed that the reaction involves α-amino and α-oxy C(sp3)-centered radicals 28-A as the key intermediates, and proceeds through a formal Giese-type addition of such radicals to easily available and bench stable BCB derivatives 29. This mild and redox-neutral protocol exhibited a broad substrate scope and excellent functional group tolerance with respect to each component including complex radical precursors such as peptides, thus providing a concise access to diverse highly valuable 1,3-disubstituted cyclobutanes 30.
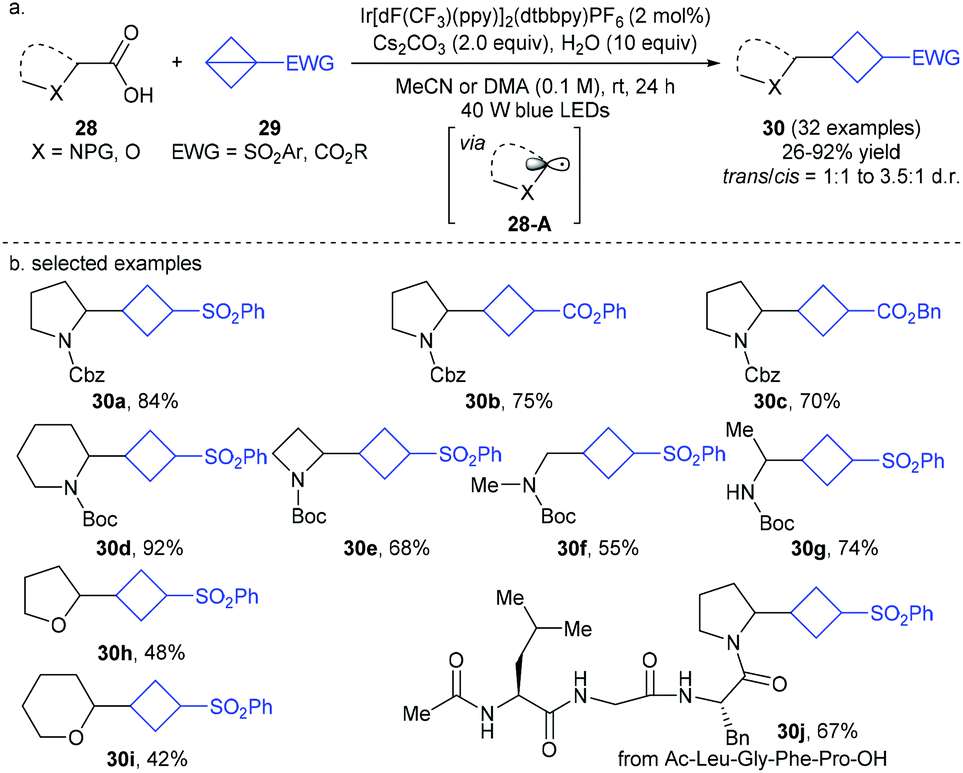 | ||
| Scheme 10 Cintrat and Ernouf's photoredox-catalyzed decarboxylative radical addition of α-amino and α-oxy carboxylic acids to bicyclo[1.1.0]butanes (BCBs). | ||
In summary, we have highlighted some recent inspiring advances in the use of photoinduced strategies toward the development of new reactions of strained [1.1.1]propellane and bicyclo[1.1.0]butane (BCB) derivatives. These reactions enabled the efficient and modular construction of diverse highly valuable polyfunctionalized bicyclo[1.1.1]pentanes and cyclobutanes. These studies offer new opportunities for reaction design of strained [1.1.1]propellane, bicyclo[1.1.0]butanes (BCBs), and other strained molecules such as azetidines and bicyclo[2.1.0]pentanes. However, some challenges still remain in this emerging field. For instance, the scope of radical precursors is relatively narrow and awaits further expansion. Moreover, under the above-mentioned photoinduced conditions, all of the bicyclo[1.1.1]pentanes, bicyclo[1.1.0]butanes, and their analogues are prepared only with 1,3-“para”-substituents. Therefore, further investigations toward the incorporation of valuable functionalities into their “ortho/meta-”position are desirable and can be expected.21 Another major issue involves the development of stereoselective variants due to the inherent radical properties of these processes. We hope that this highlight will attract more research interest to this field.22
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of our own work in photochemistry from the NSFC (21971081, 91856119, 21820102003, and 91956201) is gratefully acknowledged.Notes and references
- (a) E. M. Carreira and T. C. Fessard, Four-Membered Ring-Containing Spirocycles: Synthetic Strategies and Opportunities, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS PubMed; (b) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Visible Light-Driven Radical-Mediated C–C Bond Cleavage/Functionalization in Organic Synthesis, Chem. Rev., 2020 DOI:10.1021/acs.chemrev.0c00030.
- (a) G. M. Locke, S. S. R. Bernhard and M. O. Senge, Nonconjugated Hydrocarbons as Rigid-Linear Motifs: Isosteres for Material Sciences and Bioorganic and Medicinal Chemistry, Chem. – Eur. J., 2019, 25, 4590–4647 CrossRef CAS PubMed; (b) P. K. Mykhailiuk, Saturated Bioisosteres of Benzene: Where to Go Next?, Org. Biomol. Chem., 2019, 17, 2839–2849 RSC.
- (a) A. M. Dilmac, E. Spuling, A. de Meijere and S. Brase, Propellanes-From a Chemical Curiosity to “Explosive” Materials and Natural Products, Angew. Chem., Int. Ed., 2017, 56, 5684–5718 CrossRef CAS PubMed; (b) M. D. Levin, P. Kaszynski and J. Michl, Bicyclo[1.1.1]pentanes, [n]Staffanes, [1.1.1]Propellanes, and Tricyclo[2.1.0.0(2,5)]pentanes, Chem. Rev., 2000, 100, 169–234 CrossRef CAS PubMed.
- (a) R. Gianatassio, J. M. Lopchuk, J. Wang, C. M. Pan, L. R. Malins, L. Prieto, T. A. Brandt, M. R. Collins, G. M. Gallego, N. W. Sach, J. E. Spangler, H. Zhu, J. Zhu and P. S. Baran, Strain-Release Amination, Science, 2016, 351, 241–246 CrossRef CAS PubMed; (b) I. S. Makarov, C. E. Brocklehurst, K. Karaghiosoff, G. Koch and P. Knochel, Synthesis of Bicyclo[1.1.1]pentane Bioisosteres of Internal Alkynes and para-Disubstituted Benzenes from [1.1.1]Propellane, Angew. Chem., Int. Ed., 2017, 56, 12774–12777 CrossRef CAS PubMed.
- R. Pellicciari, M. Raimondo, M. Marinozzi, B. Natalini, G. Costantino and C. Thomsen, (S)-(+)-2-(3′-Carboxybicyclo[1.1.1]pentyl)-glycine, a Structurally New Group I Metabotropic Glutamate Receptor Antagonist, J. Med. Chem., 1996, 39, 2874–2876 CrossRef CAS PubMed.
- (a) D. F. J. Caputo, C. Arroniz, A. B. Durr, J. J. Mousseau, A. F. Stepan, S. J. Mansfield and E. A. Anderson, Synthesis and Applications of Highly Functionalized 1-Halo-3-substituted Bicyclo[1.1.1]pentanes, Chem. Sci., 2018, 9, 5295–5300 RSC; (b) J. Nugent, C. Arroniz, B. R. Shire, A. J. Sterling, H. D. Pickford, M. L. J. Wong, S. J. Mansfield, D. F. J. Caputo, B. Owen, J. J. Mousseau, F. Duarte and E. A. Anderson, A General Route to Bicyclo[1.1.1]pentanes through Photoredox Catalysis, ACS Catal., 2019, 9, 9568–9574 CrossRef CAS; (c) J. Nugent, B. R. Shire, D. F. J. Caputo, H. D. Pickford, F. Nightingale, I. T. T. Houlsby, J. J. Mousseau and E. A. Anderson, Synthesis of All-Carbon Disubstituted Bicyclo[1.1.1]pentanes by Iron-Catalyzed Kumada Cross-Coupling, Angew. Chem., Int. Ed., 2020, 59, 11866–11870 CrossRef CAS PubMed.
- H. Zhang, M. Wang, X. Wu and C. Zhu, Radical-mediated Bromoalkylation of [1.1.1]Propellane: Synthesis of Bromo-substituted Bicyclo[1.1.1]pentane (BCP) Derivatives, Chin. J. Org. Chem., 2020 DOI:10.6023/cjoc202005001.
- A. Domling, W. Wang and K. Wang, Chemistry and Biology of Multicomponent Reactions, Chem. Rev., 2012, 112, 3083–3135 CrossRef CAS.
- (a) J. Kanazawa, K. Maeda and M. Uchiyama, Radical Multicomponent Carboamination of [1.1.1]Propellane, J. Am. Chem. Soc., 2017, 139, 17791–17794 CrossRef CAS PubMed; (b) M. Kondo, J. Kanazawa, T. Ichikawa, T. Shimokawa, Y. Nagashima, K. Miyamoto and M. Uchiyama, Silaboration of [1.1.1]Propellane: A Storable Feedstock for Bicyclo[1.1.1]pentane Derivatives, Angew. Chem., Int. Ed., 2020, 59, 1970–1974 CrossRef CAS PubMed.
- J. H. Kim, A. Ruffoni, Y. S. S. Al-Faiyz, N. S. Sheikh and D. Leonori, Divergent Strain-Release Amino-Functionalization of [1.1.1]Propellane with Electrophilic Nitrogen-Radicals, Angew. Chem., Int. Ed., 2020, 59, 8225–8231 CrossRef CAS PubMed.
- S. Garbarino, D. Ravelli, S. Protti and A. Basso, Photoinduced Multicomponent Reactions, Angew. Chem., Int. Ed., 2016, 55, 15476–15484 CrossRef PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The Merger of Transition Metal and Photocatalysis, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS.
- Y. Liang, X. Zhang and D. W. C. MacMillan, Decarboxylative sp3 C–N Coupling via Dual Copper and Photoredox Catalysis, Nature, 2018, 559, 83–88 CrossRef CAS PubMed.
- X. Zhang, R. T. Smith, C. Le, S. J. McCarver, B. T. Shireman, N. I. Carruthers and D. W. C. MacMillan, Copper-Mediated Synthesis of Drug-like Bicyclopentanes, Nature, 2020, 580, 220–226 CrossRef CAS PubMed.
- (a) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Alkyl Carbon-Carbon Bond Formation by Nickel/Photoredox Cross-Coupling, Angew. Chem., Int. Ed., 2019, 58, 6152–6163 CrossRef CAS PubMed; (b) D. N. Primer and G. A. Molander, Enabling the Cross-Coupling of Tertiary Organoboron Nucleophiles through Radical-Mediated Alkyl Transfer, J. Am. Chem. Soc., 2017, 139, 9847–9850 CrossRef CAS PubMed.
- (a) M. D. VanHeyst, J. Qi, A. J. Roecker, J. M. E. Hughes, L. Cheng, Z. Zhao and J. Yin, Continuous Flow-Enabled Synthesis of Bench-Stable Bicyclo[1.1.1]pentane Trifluoroborate Salts and Their Utilization in Metallaphotoredox Cross-Couplings, Org. Lett., 2020, 22, 1648–1654 CrossRef CAS PubMed; (b) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Photoinduced Decarboxylative Borylation of Carboxylic Acids, Science, 2017, 357, 283–286 CrossRef CAS PubMed.
- Z. Wu, Y. Xu, J. Liu, X. Wu and C. Zhu, A practical Access to Fluoroalkylthio(seleno)-functionalized Bicyclo[1.1.1]pentanes, Sci. China: Chem., 2020, 63, 1025–1029 CrossRef CAS.
- (a) M. Kischkewitz, K. Okamoto, C. Muck-Lichtenfeld and A. Studer, Radical-Polar Crossover Reactions of Vinylboronate Complexes, Science, 2017, 355, 936–938 CrossRef CAS PubMed; (b) M. Silvi, C. Sandford and V. K. Aggarwal, Merging Photoredox with 1,2-Metallate Rearrangements: The Photochemical Alkylation of Vinyl Boronate Complexes, J. Am. Chem. Soc., 2017, 139, 5736–5739 CrossRef CAS PubMed; (c) M. Silvi and V. K. Aggarwal, Radical Addition to Strained σ-Bonds Enables the Stereocontrolled Synthesis of Cyclobutyl Boronic Esters, J. Am. Chem. Soc., 2019, 141, 9511–9515 CrossRef CAS PubMed.
- (a) R. A. Aycock, C. J. Pratt and N. T. Jui, Aminoalkyl Radicals as Powerful Intermediates for the Synthesis of Unnatural Amino Acids and Peptides, ACS Catal., 2018, 8, 9115–9119 CrossRef CAS; (b) C. J. Pratt, R. A. Aycock, M. D. King and N. T. Jui, Radical alpha-C-H Cyclobutylation of Aniline Derivatives, Synlett, 2020, 51–54 CAS.
- G. Ernouf, E. Chirkin, L. Rhyman, P. Ramasami and J.-C. Cintrat, Photochemical Strain-Release-Driven Cyclobutylation of C(sp3)-Centered Radicals, Angew. Chem., Int. Ed., 2020, 59, 2618–2622 CrossRef CAS PubMed.
- (a) X. Ma, D. L. Sloman, Y. Han and D. J. Bennett, A Selective Synthesis of 2,2-Difluorobicyclo[1.1.1]pentane Analogues: “BCP-F2”, Org. Lett., 2019, 21, 7199–7203 CrossRef CAS PubMed; (b) R. M. Bychek, V. Hutskalova, Y. P. Bas, O. A. Zaporozhets, S. Zozulya, V. V. Levterov and P. K. Mykhailiuk, Difluoro-Substituted Bicyclo[1.1.1]pentanes for Medicinal Chemistry: Design, Synthesis, and Characterization, J. Org. Chem., 2019, 84, 15106–15117 CrossRef CAS PubMed.
- M. O. Senge and N. Grover, Synthetic Advances in the C–H Activation of Rigid Scaffold Molecules, Synthesis, 2020 DOI:10.1055/s-0040-1707884.
| This journal is © the Partner Organisations 2020 |