
From terpenes to sustainable and functional polymers
Francesco
Della Monica
 *a and
Arjan W.
Kleij
*ab
*a and
Arjan W.
Kleij
*ab
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute for Science & Technology (BIST), Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: fdellamonica@iciq.es; akleij@iciq.es
bCatalan Institute for Research and Advanced Studies (ICREA), Pg. Lluis Companys 23, 08010 Barcelona, Spain
First published on 27th July 2020
Abstract
Our dependence on fossil-fuel based raw materials and alarming plastics accumulation in the environment has inspired many scientists to design and implement more sustainable approaches for polymer synthesis. The identification and assessment of new and renewable biobased monomers has become a major topic of interest en route to a circular economy. In this respect, terpenes and terpenoid scaffolds have attracted much attention due to their ubiquitous nature and high functionality. This minireview uncovers recent advancements in the synthesis of terpene-based polymers, and specifically discusses the formation of terpene-derived polycarbonates, polyesters, polyurethanes and polyamides. The potential of these polymers in post-synthetic curing and modifications is also highlighted to illustrate the opportunities that exist to develop new polymer formulations beyond bench scale.
 Francesco Della Monica | Dr Francesco Della Monica obtained his Master degree in Chemistry (2013) and PhD in Chemistry (2018) at the University of Salerno (Italy) in the group of Prof. Carmine Capacchione. His work was mainly devoted to the development of new iron-based catalysts for the reaction of carbon dioxide with epoxides. He spent one year as postdoctoral researcher at the same institute, working on the green, catalytic production of value-added products from renewable resources. In July 2019, he joined the research group of Prof. Arjan W. Kleij as a MSCA-IF fellow to develop new terpenoid monomers for the synthesis of sustainable polymers. |
 Arjan W. Kleij | Prof. Arjan W. Kleij received his PhD degree from the University of Utrecht, the Netherlands in 2000 under the supervision of Prof. Gerard van Koten. After two PDRA positions in Madrid (Spain) and the University of Amsterdam, and two appointments in industry (Avantium, Hexion), he started his independent career at ICIQ (Tarragona) and ICREA (Barcelona) in 2006. His main research interests are the catalytic valorization of carbon dioxide, stereo- and enantioselective synthesis using functionalized heterocycles, and the development of biobased polymers derived from carbon dioxide and/or terpene monomers. |
1. Introduction
Modern society is facing severe environmental problems due to uncontrolled disposal of large quantities of single-use plastic waste.1,2 A clear testament of the global nature of this issue is and continues to be plastic accumulation in the oceans also referred to as “plastic soup”.3 This consequently impacts marine life negatively,4–7 while potentially causing health hazards due to the formation of micro-plastic particles. Nowadays the production of polymeric materials is still largely based on oil-dependent chemistry. In our era, with climate change gaining importance on the political and social agendas of many countries, there is a growing awareness that we should lower the dependence of both our energy consumption and chemical production on natural fossil resources.7–9 Hence, it is clear that a transition toward the use of renewable resources may help to meet the growing environmental concerns, and several regulations and restrictions with respect to the use of certain chemicals and polymeric materials have already been adopted.10–12Since natural polymers such as cellulose,13 and poly(hydroxyalkanoates)14–20 are well-known and have been extensively used,21 nature may further serve as a source of inspiration for the identification of other types of bio-based raw materials for the production of sustainable polymers.22–26 Various research communities have directed their attention to the production and utilisation of new types of starting materials derived from renewable feedstock.27–29 An illustrative example is presented by the production of L-lactide from starch, and its use in the industrial production of poly(lactic acid), PLA.30 This role model example has spurred numerous studies focussing on the creation of renewable monomers starting from starch,31 sugar,32 vegetable oils,33 cellulose34 and lignin.35 In parallel, extensive efforts have been made in the valorisation of other molecules (often considered as waste) such as carbon dioxide in sustainable polymer development.36–38
Within this framework, bio-derived terpene-based monomers are attracting growing attention.39–44 Polyisoprene (a major constituent of natural rubber) has been known for more than a century,45–47 and other terpenes (such as pinene,48–50 limonene,51–54 pirocarvone,55 myrcene,56–64 alloocimene,65 ocimene66,67 and farnesene68,69) have also been used in the preparation of polyolefins (for an example see Fig. 1, top).
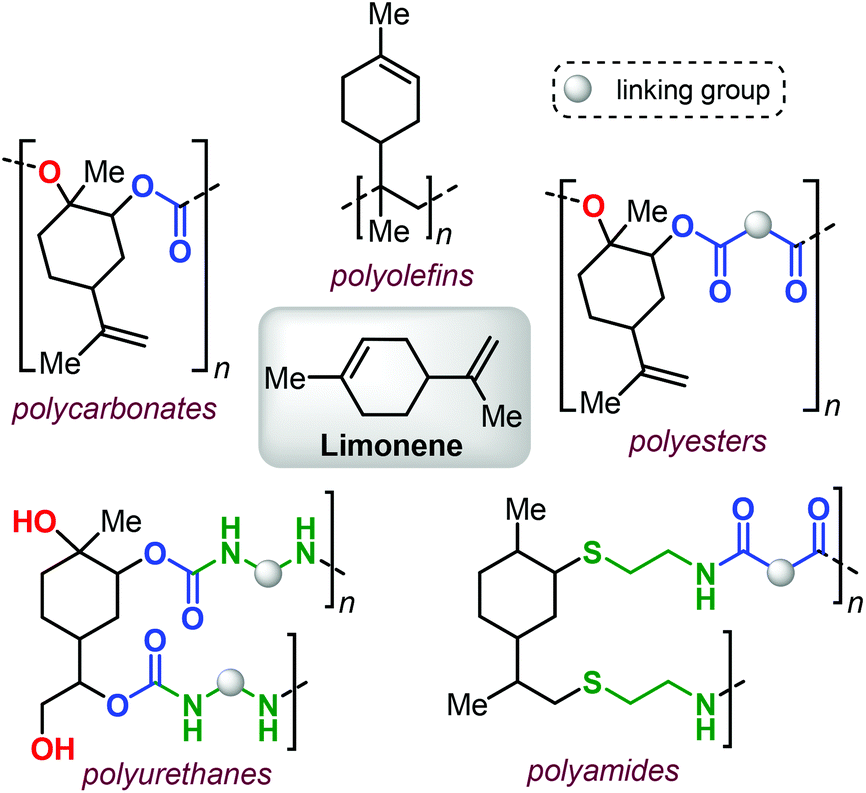 | ||
| Fig. 1 Polymer diversity created from limonene. | ||
Additional advances in the synthesis of different terpene-based polymers have been achieved in relatively recent times. Their structural diversity (i.e., being linear, cyclic or polycyclic) and the presence of double bonds with different reactivity render terpenes as highly versatile and easy to functionalize building blocks.70–72
Limonene, a commercial monoterpene that can be isolated from citrus fruits (Fig. 1),73 represents a terpene structure that comprises of two distinct double bonds that can be easily modified, for instance by epoxidation or thiol-ene click reactions. These features, together with the availability of this terpene feedstock, has allowed forging structurally diverse polymeric materials thus showcasing limonene as a potential platform molecule (Fig. 1).
In this minireview, we describe recent developments in the use of terpene-based monomers in the construction of more sustainable and functional polymers. Specifically, the most important types of polymers that have been targeted are addressed and their prospective post-modification is also highlighted to demonstrate the opportunities that exist to design polymer properties that can be advantageous in the realisation of future materials.
2. Polycarbonates
Polycarbonates (PCs) can be divided into aromatic and aliphatic PCs on the bases of the chemical structure of their repeating units. Commercial PC is based on bis-phenol-A (BPA), and displays high-performance optical, thermal and above all mechanical properties.74 BPA-based PCs find application as engineering plastics in construction, in the automotive industry and in electronics. The preparation and use of BPA derived PCs has two significant problems: (1) the industrial production involves the use of highly toxic phosgene;75 and (2) polymer hydrolysis/degradation may release BPA metabolites accumulating in the environment, with some of these metabolites believed to act as endocrine disruptors.76,77 On the contrary, aliphatic PCs are generally bio-compatible and -degradable, and they have emerged as possible substitutes of BPA-based PC.78,79Unfortunately, applications of aliphatic PCs, such as poly(propylene carbonate) (PPC) are currently limited as they display a narrow window of mechanical and thermal properties,80 and hence are mainly employed as oligomeric polyols (cf., chain extension) in polyurethane production.81,82 Thus, the identification of new, structurally rigid monomers derived from renewable and non-toxic resources can help to increase the application potential of aliphatic PCs by expanding their properties as to find suitable alternatives for commercial PC.
In this context, preparation of PCs by ring-opening copolymerization (ROCOP) reaction of CO2 with epoxides has demonstrated high versatility in the last decades.83–87 Typically, cyclic epoxides such as cyclohexene oxide (CHO) are more prone to produce PCs by reaction with CO2.88 Due to their structural resemblance with CHO, α-pinene oxide (APO) and limonene oxide (LO) are attractive, sustainable alternatives to CHO when focusing on new types of aliphatic PCs (Fig. 2). In fact, pinenes are monoterpene bicyclic compounds present in turpentine oil, with the main being α-pinene (45–97%), followed by β-pinene (0.5–28%) and traces of other terpenes such as camphene, Δ3-carene and limonene.89,90
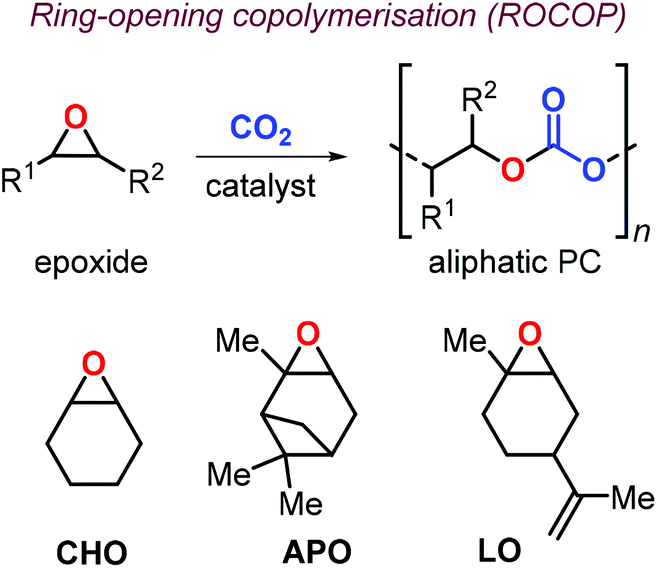 | ||
| Fig. 2 Ring-opening copolymerisation of epoxides and CO2 toward aliphatic polycarbonates, and structures of cyclohexene oxide (CHO), α-pinene oxide (APO) and limonene oxide (LO). | ||
Turpentine is the volatile fraction of oleoresin obtained from conifers and, depending on the production method, can be divided into sulfate turpentine, gum turpentine, and wood turpentine.91 Turpentine global production is estimated to be 330 kton per year mainly obtained (70%) as by-product in the pulp industry during the Kraft process.92,93 The main uses of turpentine oil are in the production of pine oil, flavours, fragrances and resins,94 and is also attracting interest in the production of biofuels.95
Epoxidation of α-pinene, leads to the formation of APO (Fig. 3). To date, the only example of a synthesis of poly(α-pinene carbonate) is based on the ROCOP of APO and CO2 promoted by the binary catalyst CrIII(salen) complex 1/bis-(triphenylphosphine)iminium chloride ([PPN]Cl).96 In the last few years, much more efforts have been dedicated to the use of LO (Fig. 2) as a renewable terpene feedstock in polymer synthesis. The two enantiomers of limonene can be isolated from different natural sources.97 For example, (S)-limonene is present in fir cone oil while (R)-limonene is a major constituent in orange peel. Epoxidation of either enantiomer gives LO as a cis/trans mixture (Fig. 4). Since (R)-limonene extraction is economically more attractive, (R)-LO is relatively inexpensive having a cis![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :trans ratio of around 46:54.
:trans ratio of around 46:54.
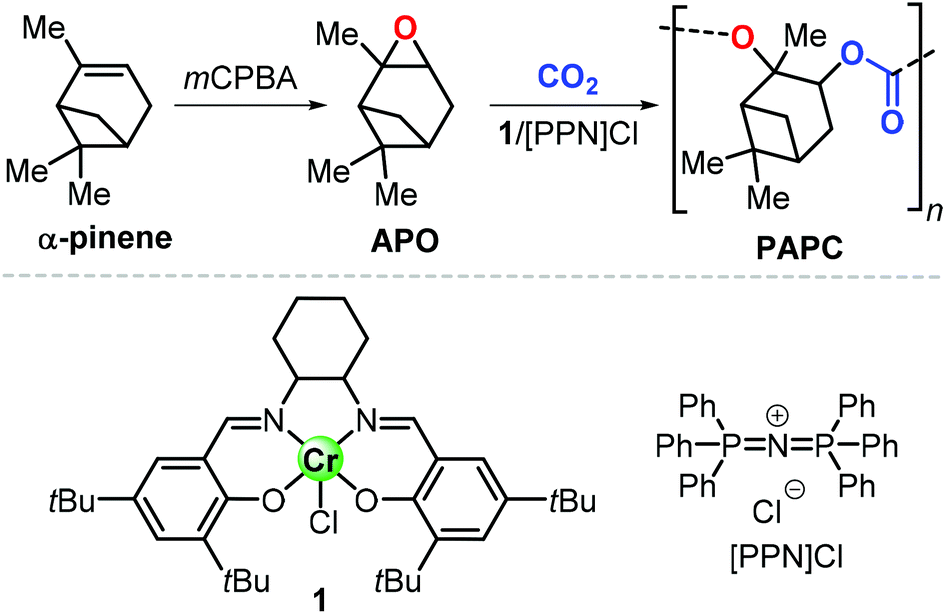 | ||
| Fig. 3 Synthesis of poly(α-pinene carbonate), PAPC, from α-pinene, and structures of CrIII(salen) complex 1 and [PPN]Cl. mCPBA stands for meta-chloro-perbenzoic acid. | ||
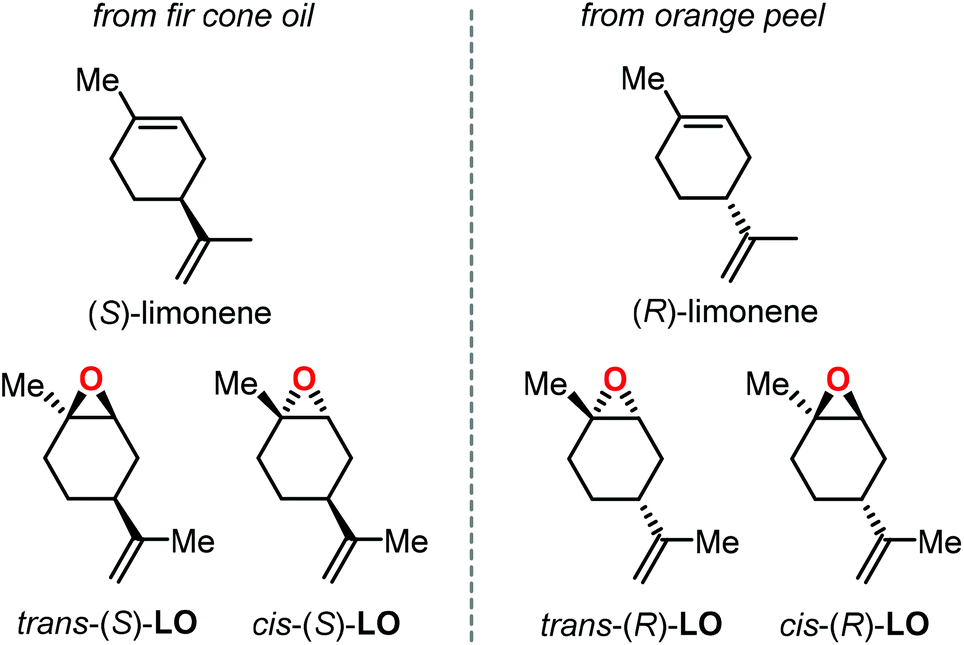 | ||
| Fig. 4 Structure of limonene enantiomers and limonene oxide stereoisomers. | ||
Consequently, the major part of recent polymerisation studies are based on the use of (R)-LO rather than (S)-LO. To date, only two catalytic systems are known to promote the formation of poly(limonene carbonate), PLC, and both exhibit different features. Copolymerisation of LO with CO2 was described for the first time by Coates et al. using the β-diiminate zinc complex 2a (Fig. 5).98 Starting from (R)-LO, 2a produces PLC in a stereoselective fashion (with 98.3% trans-configured repeat units) under mild conditions (25 °C, 6.9 bar of CO2). Deviation from these optimised conditions results into lower degrees of stereocontrol and a strong decrease of catalytic activity. Control experiments revealed that copolymerisation of pure trans-(R)-LO proceeds smoothly, while reaction with pure cis-(R)-LO was unsuccessful. The analysis of the depolymerisation products was performed to examine the origin of the stereoselective conversion of trans-(R)-LO. Starting from PLC samples with different content of trans units, (1S,2S,4R)-1-methyl-4-(1-methylethenyl)-1,2-cyclohexanediol was isolated as the only product after a hydroxide-induced hydrolysis. This finding supports the hypothesis that the ring-opening step proceeds by an axial attack which, in the trans isomer, occurs on the less hindered β-position (i.e., at a secondary carbon centre), which is kinetically preferred.
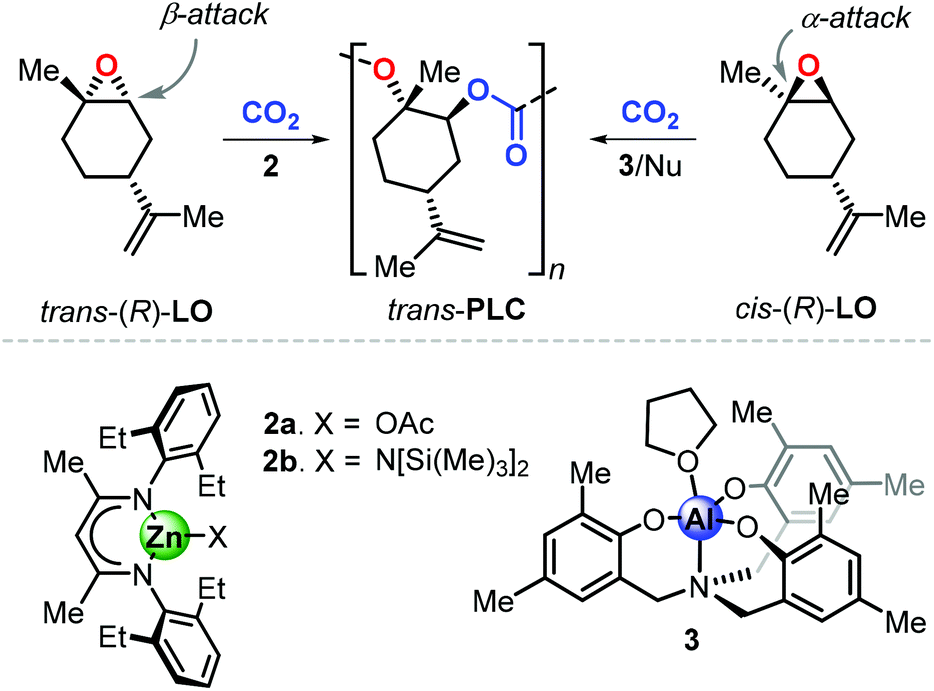 | ||
| Fig. 5 Preparation of trans-PLC from trans-(R)-LO and cis-(R)-LO using catalysts 2a–b or 3. | ||
More recently, Kleij et al. reported on the synthesis of PLC promoted by complex 3 in combination with a nucleophilic (Nu) cocatalyst.99 Copolymerisation of commercial (R)-LO with CO2 under relatively mild conditions (42 °C, 5–10 bar of CO2) resulted in the formation of PLC with significant content of cis-units (up to 33%), suggesting that the catalyst is able to convert both LO stereoisomers. Control experiments revealed that copolymerisation of pure cis-(R)-LO proceeds faster than pure trans-(R)-LO, with a higher degree of stereoselectivity leading to up to 98% trans units in the PLC. This result, in contrast to the observations done with 2, was explained by suggesting that the halide-mediated ring-opening of cis-LO occurs by attack on the more hindered position of the oxirane unit. This hypothesis was supported by detailed computational investigations, which indeed showed that nucleophilic attack on the α-position (at the tertiary carbon centre) is energetically favoured for both LO stereoisomers on the basis of an electronic bias.
Complex 3 was later on also used for the preparation of LO/CHO/CO2 terpolymers.100 Depending on the monomer feed composition, gradient terpolymers with an incorporation level of LO between 10 and 42% were obtained, and with number average molecular weights (Mn) between 3.6 and 8.2 kDa.
In recent years, several groups studied the material properties of PLC. Interestingly, Auriemma, Coates and co-workers described the properties of a PLC stereocomplex and reported on its crystal structure.101,102 Complex 2b was used to prepare stereoregular poly(R-limonene carbonate) and poly(S-limonene carbonate) from (R)-LO and (S)-LO, respectively. The individual polymer samples were found to be amorphous, but after mixing solutions of the two, a precipitate was observed. The thus formed PLC stereocomplex displays the same glass transition temperature (Tg = 120 °C) as the amorphous samples but has a higher decomposition temperature (Td). A melting transition (typical for semi-crystalline polymers) was not observed probably because the melting temperature (Tm) of the PLC stereocomplex is higher than its Td.
Other types of limonene-containing polycarbonates were reported by Greiner, Schmalz and co-workers. They used a living, sequential ROCOP of LO and CHO with CO2 using catalyst 2a which afforded the diblock copolymers poly(limonene carbonate)-block-poly(cyclohexene carbonate)s abbreviated as (PLC-b-PCHC)s.103 Despite the chemical similarity of the two blocks, transmission electron microscopy (TEM) analyses revealed that these copolymers are able to self-assembly into well-defined cylindrical, lamellar, and hexagonally organised morphologies depending on the composition and molecular weight of the PLC-b-PCHC copolymer.
Further to this, Greiner, Rieger and co-workers reported a significant improvement of the preparation of PLC providing high molecular weights (Mn > 100 kDa) under excellent chemoselectivity control (i.e., carbonate linkages >99%) using catalyst 2a.104 Since this catalyst is known to be unreactive towards the incorporation of cis-LO, they devised a stereoselective synthesis for trans-(R)-LO (Fig. 6).105
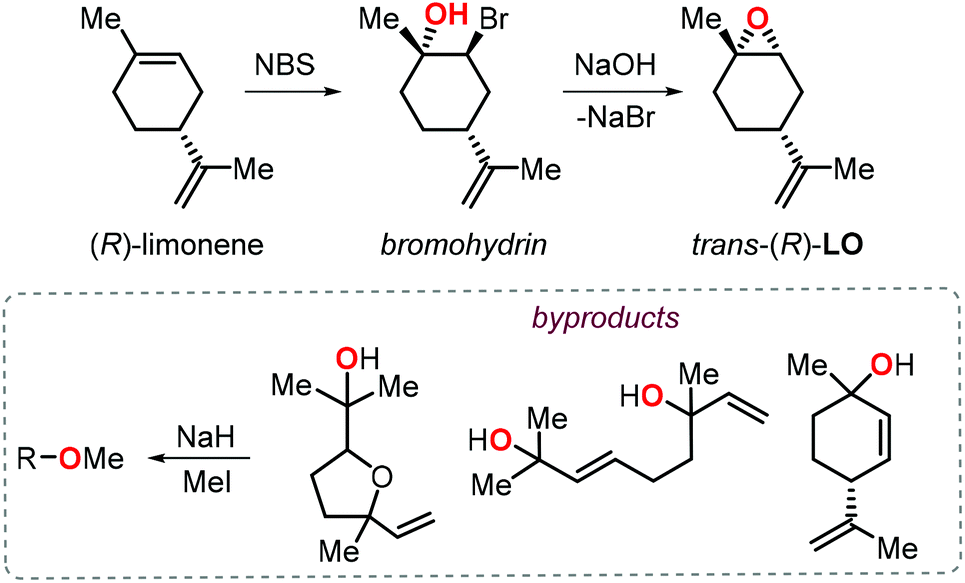 | ||
| Fig. 6 Stereoselective synthesis of trans-(R)-LO and protection of the alcohol groups in the by-products. | ||
Notably, the procedure was scaled up to kilogram quantitaties while obtaining (R)-LO containing 85% of trans isomer. Since protic impurities are well-known to act as chain-transfer agents in the ROCOP process,106 thereby lowering the molecular weight of the polymers, the monomer was treated with NaH and MeI prior to the copolymerisation with CO2 to minimise any free alcohol groups present in the by-products. As a result of this protection procedure, the ROCOP proceeded with excellent control over the molecular weight and by tuning the monomer/catalyst ratio, copolymers with Mn values between 25.4 and 108.6 kDa and low dispersity indices (Đ < 1.2) were obtained. The thermal, mechanical and optical properties of these latter PLCs were determined and revealed that this type of amorphous polymer shows higher transparency and pencil hardness than technical BPA based polycarbonate, rendering it thus appealing for coating applications. Later on Abetz, Greiner et al. also investigated the gas permeability of PLC.107 Interestingly, PLC films have a high permeability to gases (68 and 12 barrer for CO2 and O2, respectively), with a high selectivity for CO2 over N2, and PLC was thus proposed as a good candidate for the production of “breathing glass”.
Apart from the attracting properties of pristine PLC, the presence of free vinyl groups triggered the investigation of post-modification reactions to expand the number of possible applications. Two conceptual approaches have been adopted for the functionalisation of PLC with one based on thiol-ene click chemistry, and the other one based on the introduction of epoxide groups providing ample opportunities for well-developed “epoxy” chemistry (Fig. 7). The thiol-ene approach used by Greiner et al. allowed for the preparation of PLC-based polymers with a wide range of properties.108 For instance, the introduction of polar ammonium groups gave access to polymers with antibacterial activity, whereas the introduction of alcohol groups increased the hydrophilicity (i.e., a smaller contact angle to water). The installation of alkyl ester groups onto the PLC backbone decreased both the Tg and Young modulus (E), resulting in rubbery behaviour.
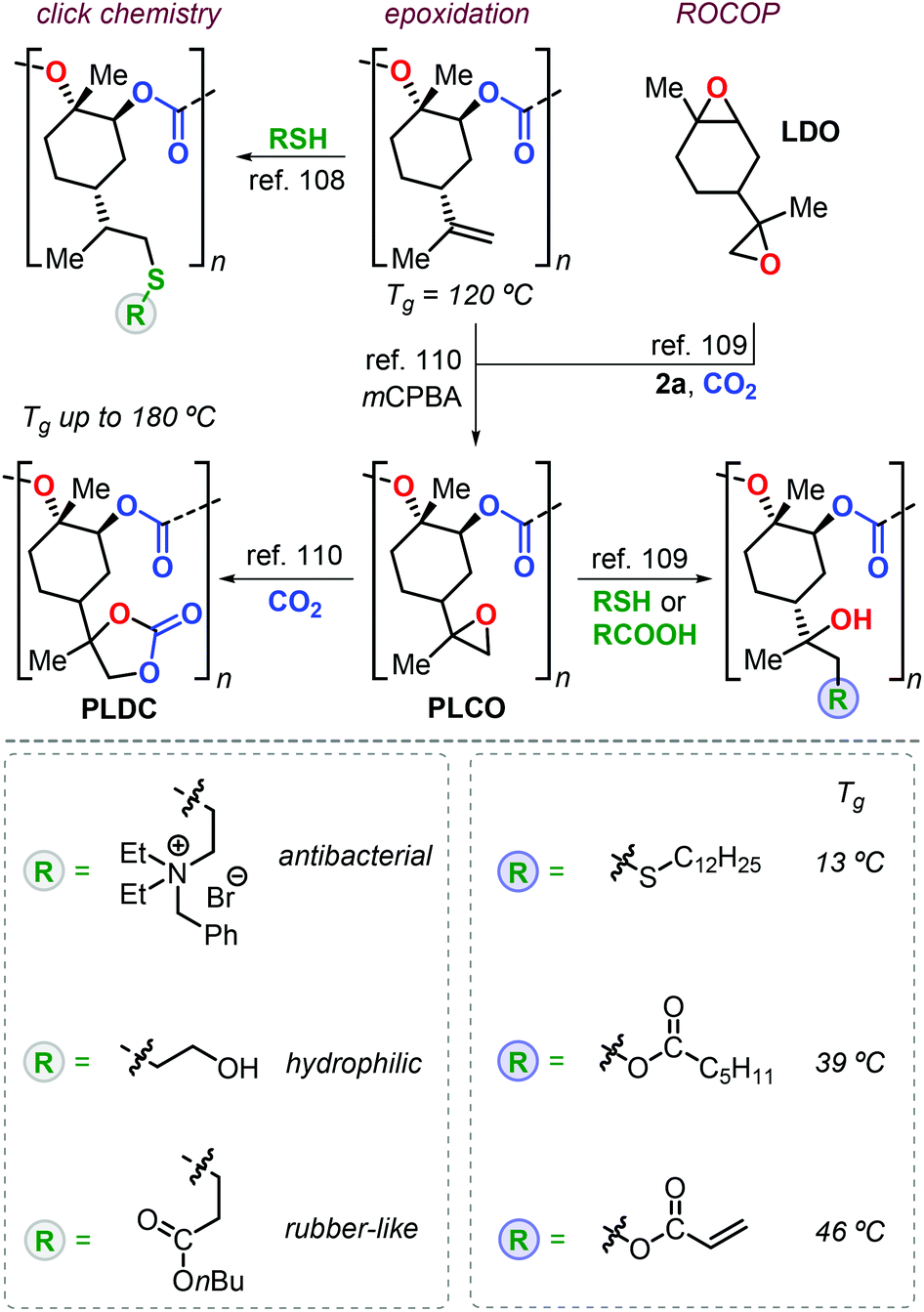 | ||
| Fig. 7 Functional polymers derived from PLC after post-modification using thiol-ene click reactions or epoxy-based chemistry. | ||
The group of Sablong prepared poly(limonene carbonate)oxide, PLCO, by regio-selective copolymerisation of limonene dioxide, LDO, with CO2 promoted by 2a.109
Soon after, Kleij et al. reported a different synthesis of PLCO by simple epoxidation of the double bonds of PLC (Fig. 7).110 Epoxy groups are useful functional groups in ring-opening and cycloaddition reactions giving potential to fine-tune thermal properties. Introduction of long alkyl chains reduces the Tg to 13 °C, while conversion of PLCO to poly(limonene dicarbonate), PLDC, resulted in record-high Tg values of up to 180 °C.
In the last few years, Sablong, Koning and co-workers investigated the use of low molecular weight PLC for coating applications.111–113 In particular, they reported that PLC can be cross-linked through photo-initiated thiol-ene reactions, showing then improved pencil hardness (2H) and solvent resistance when compared against non-cross-linked PLCs.114
End-of-life disposal and polymer reuse are key to sustainable polymer development.115,116 In this context, PLC was proven to be highly stable in a composting environment.108 While more research is warranted to estimate the potential of PLC and its derivatives towards biodegradation, Sablong et al. reported that depolymerisation of hydroxyl-terminated PLC can be achieved using a relatively simple catalyst (TBD, 1,5,7-triazabicyclo[4.4.0]dec-5-ene) which at elevated temperature allowed the selective re-formation of the initial LO monomer (Fig. 8) through a back-biting mechanism.117 Such controlled depolymerisation can set the basis for efficient recycling of PLC and developing sustainable material solutions based on a renewable building block.118
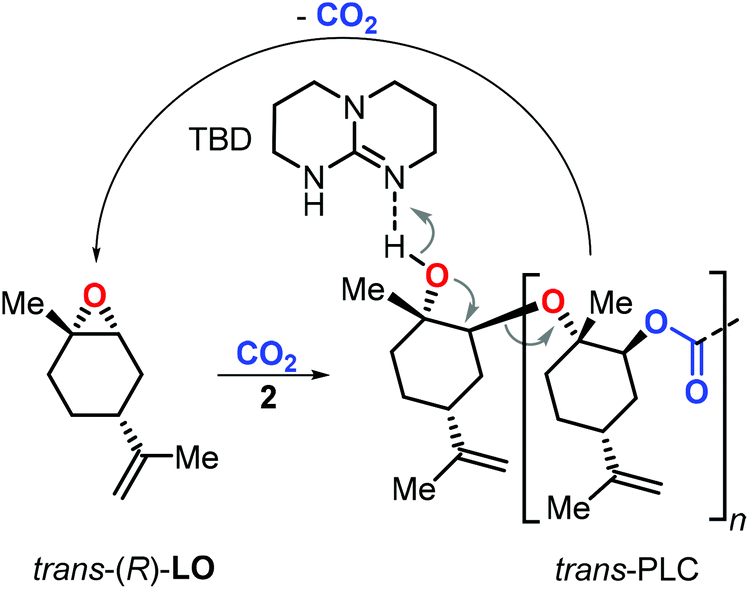 | ||
| Fig. 8 Depolymerisation mode of PLC promoted by TBD. | ||
3. Polyesters
Aliphatic polyesters (APEs) represent one of the most promising classes of sustainable polymers because of their general biocompatibility and facile hydrolytic degradation.119,120 Synthesis of APEs can be achieved by polycondensation of diols with dicarboxylic acids or esters, ring-opening polymerisation (ROP) of cyclic esters and ring-opening copolymerisation (ROCOP) of epoxides with cyclic anhydrides (Fig. 9).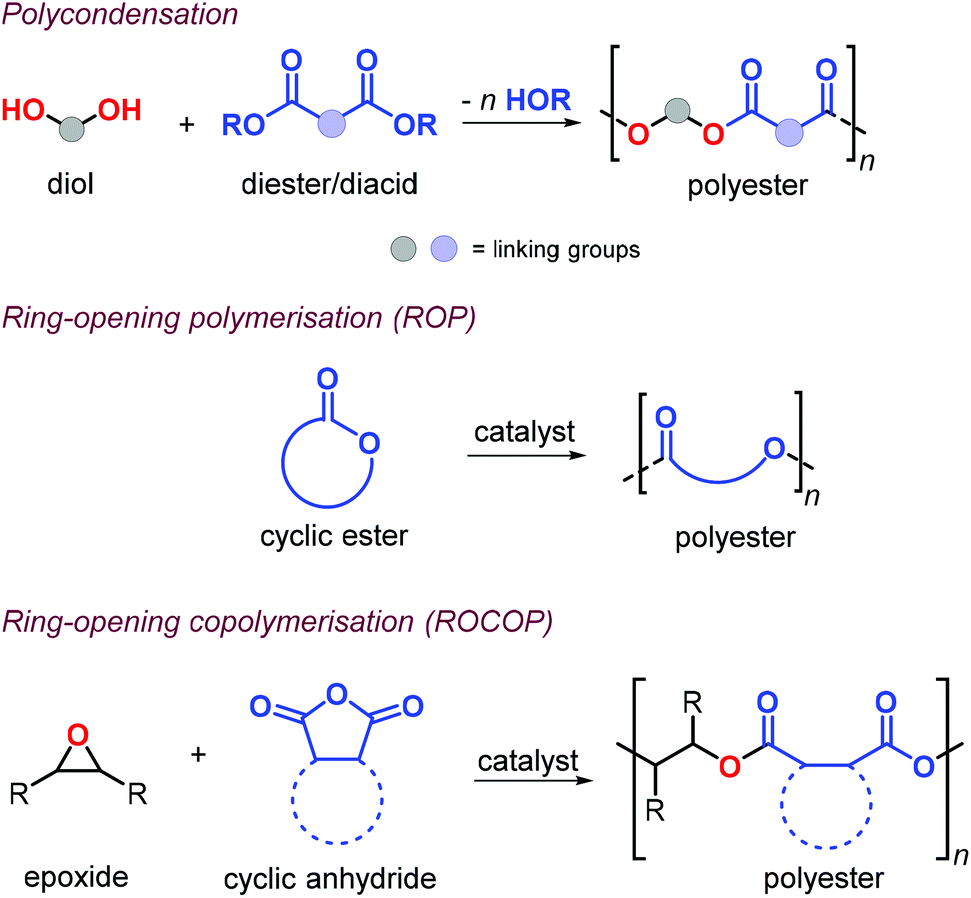 | ||
| Fig. 9 Aliphatic polyester synthesis by polycondensation and ring-opening (co)polymerisation. | ||
During polycondensation, the by-products (water or alcohol) must be removed and very high conversions are required to obtain high molecular weights usually with Đ values around 2.121 The ROP of cyclic esters, usually promoted by metal complexes, is one of the most effective ways toward the formation of APEs.122
This approach allows for excellent control over the molecular weights and typically with narrow polymer size distributions. Unfortunately, the reaction is often limited to six- and seven-membered cyclic monomers and as such may limit the range of mechanical, thermal and post-synthetic properties.123 Alternatively, the alternating ROCOP of epoxides and cyclic anhydrides is particularly attractive.124,125 This method allows for regulating the material properties combining a much larger number of suitable epoxides and anhydride combinations, facilitating thus the incorporation of functional groups useful in post-synthetic modifications.
In 2017, Sieber et al. reported the preparation of borneol-based polyesters (Fig. 10).126 The (−)-borneol, a natural occurring terpenoid, can be prepared from turpentine oil.127,128 The authors developed a process converting (−)-borneol in 5-exo-hydroxyborneol using Pseudomonas putida KT2440 in a whole-cell biocatalytic approach. The terpene-based diol obtained from this process was successfully copolymerised with succinic acid dimethyl ester obtaining an aliphatic polyester with Mn values in the range 2–4 kDa, and a modest Tg of 70 °C. The use of a Sn-based catalyst was proposed, but its use calls for more sustainable alternatives.
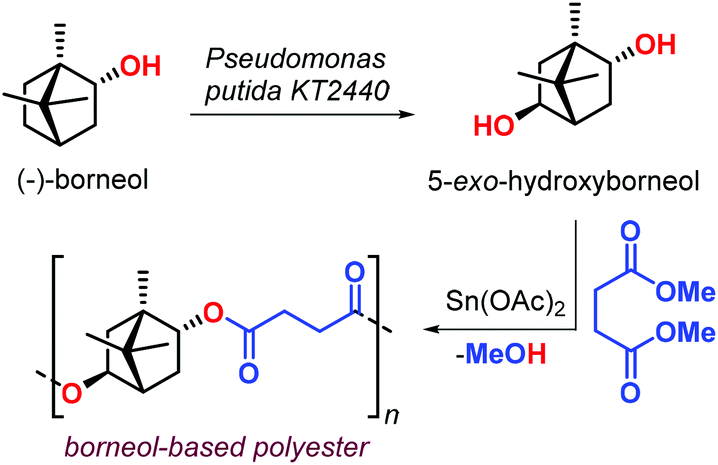 | ||
| Fig. 10 Biotransformation of (−)-borneol in 5-exo-hydroxuborneol and its poly-condensation with succinic acid dimethyl ester. | ||
In 2019, Stockman et al. reported the synthesis of limonene-based diols and hydroxy-acids (Fig. 11).129 Limonene was easily converted into diols a1–2 by a hydroboration-oxidation sequence. Three methods were utilised for the conversion of a1–2 into hydroxy-acids b1–2, and they were classified by a green chemistry metrics evaluation. The most convenient method was the two-step, chemo-selective oxidation of the primary alcohol to carboxylic acid. Diols a1–2 were subjected to copolymerisation with succinic acid in the presence of various Lewis acidic catalysts to give polymer d. Notably, Mn values of up to 30 kDa were obtained using titanium tetra-n-butoxide [Ti(OnBu)4], and Tg values between −7 and 23 °C were observed. Depolymerisation of polymer d regenerating the initial diols was also demonstrated under basic conditions.
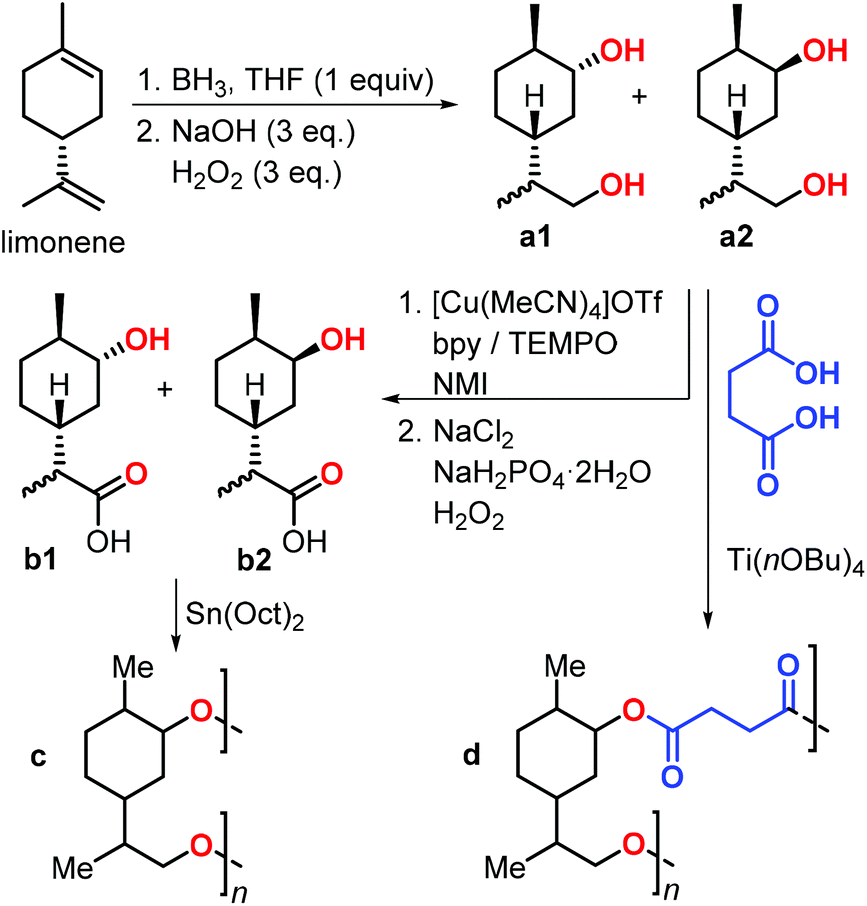 | ||
| Fig. 11 Synthesis of limonene-based diols a1–2 and hydroxy-acids b1–2, and their polycondensation reactions. | ||
Subsequently, polycondensation reactions involving b1 and b2 were investigated. Polymerisation of b1–2 catalysed by tin-octanoate [Sn(Oct)2] led to the formation of oligomers c (Mn < 2.6 kDa) even after very long reaction times (1 month).
In 2019, Nsengiyumva and Miller described the synthesis and thermal behaviour of new camphor-based polyesters (Fig. 12).130
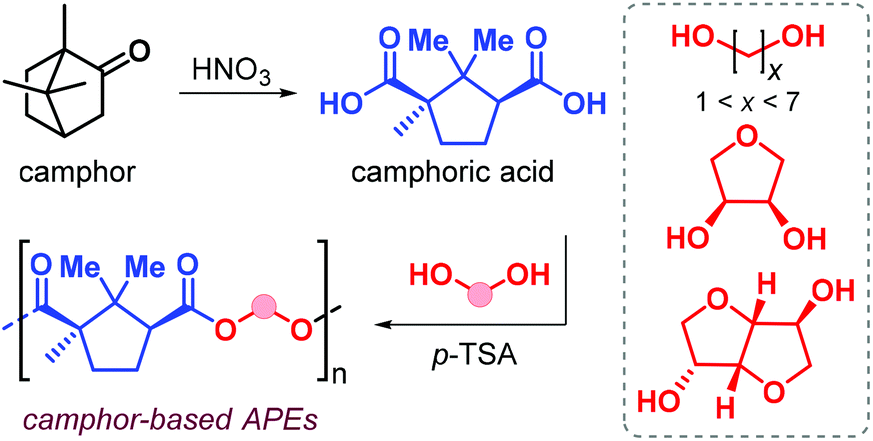 | ||
| Fig. 12 Camphor oxidation to camphoric acid and its polycondensation with diols. | ||
Camphor is a bicyclic terpene that can be extracted from the camphor tree131 or produced in ton-scale from α-pinene,132 and is used as an aroma in perfumes, as a plasticizer and repellent. Oxidation of camphor produces camphoric acid that has been used as co-monomer in polycondensation reaction with various diols catalysed by p-toluenesulfonic acid (p-TFA, Fig. 12). High conversions were obtained after 18 h at 180 °C, and Mn values in the range 7–20 kDa were obtained albeit with broad dispersities (2.7 ≤ Đ ≤ 6.1). When using linear α,ω-diols, the resultant Tg value of the polyester depends on the alkyl chain length; these values range from 51 °C when using ethylene glycol down to −16 °C in the case of 1,6-hexanediol. Upon introduction of conformationally rigid cyclic diols such as erythritol and isosorbide, the Tg values increase to 100 and 125 °C, respectively.
Due to the structural resemblance of poly(ethylene camphorate) (PEC) and poly(ethylene terephthalate) (PET), copolymerisation of bis(hydroxyethyl) camphorate (BEHC) and bis(hydroxyethyl) terephthalate (BHET) was studied (Fig. 13). The PEC-co-PET copolymers were obtained with a wide range of compositions with a PEC content from 7.6 to 82.9%.
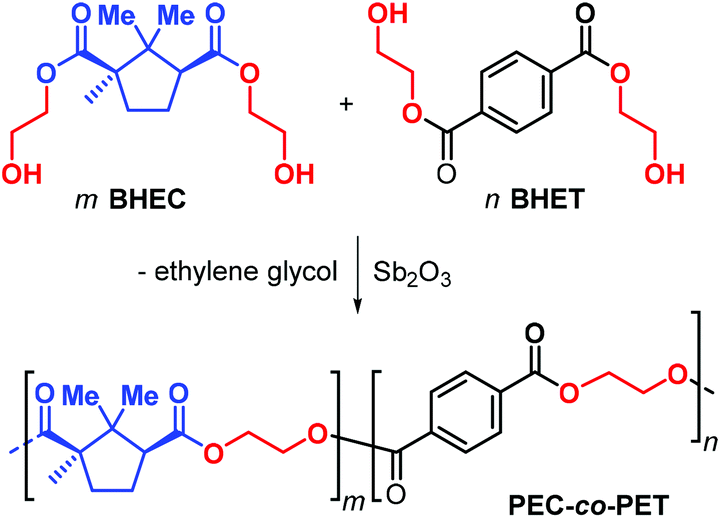 | ||
| Fig. 13 Copolymerisation of bis(hydroxyethyl) camphorate (BEHC) and bis(hydroxyethyl) terephthalate (BHET) toward PEC-co-PET. | ||
Interestingly, copolymers with a PEC incorporation of up to 20% have Tg's in the range 59–66 °C and Tm values between 180 and 209 °C, which are comparable to those of PET. These data clearly emphasize that (partial) exchange of terephthalate for camphorate units does not negative influence the thermal performance parameters.
Some efforts have been devoted to the synthesis of seven-membered lactones from terpenes. Such monomers represent concrete alternatives to petrochemical ε-caprolactone (CL). For example, the PLA properties can be modulated to some extent by copolymerisation of LA with CL.133,134 Biobased lactones are particularly attractive for the synthesis of fully renewable polyesters with improved thermal properties.135
In 2005, Hillmyer, Tolman et al. described the synthesis of a new polyester by ROP of the lactone (−)-menthide, which was derived from menthol (Fig. 14).136 Menthol is a cyclic terpene largely used in the fragrance and flavour industry with several thousands of tons produced every year.137 Baeyer–Villiger oxidation of (−)-menthone, derived from menthol, produces (−)-menthide. The controlled ROP of menthide, promoted by the phenoxy-amine-Zn complex 4 (see Fig. 14), was achieved and poly(menthide)s (PMs) with Mn values in the range 3–91 kDa were obtained by simple variation of the monomer-to-4 ratio. The thermodynamic parameters for this polymerisation process were also determined, and the authors concluded from these studies that the (−)-menthide ring-strain is comparable to that of CL.
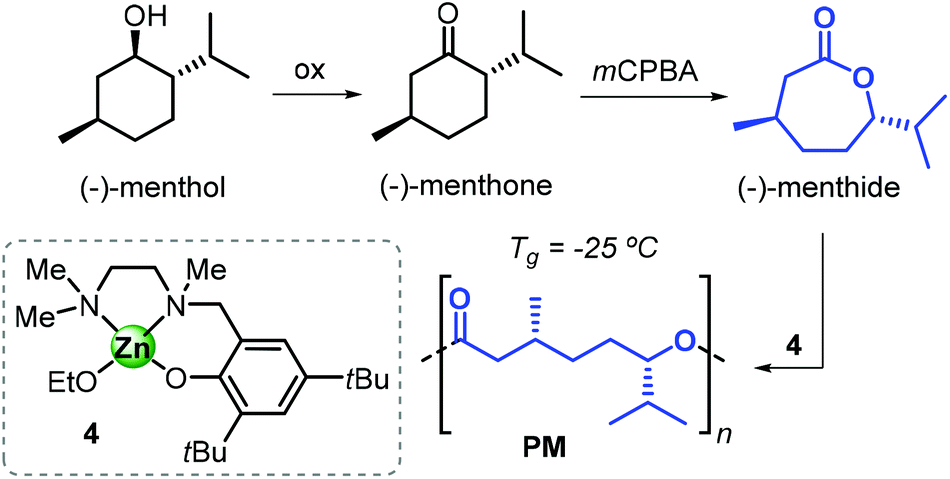 | ||
| Fig. 14 Synthesis of poly(menthide) from (−)-menthol and structure of the phenoxyamino-Zn complex 4. | ||
Triblock copolymers (based on PM) could also be obtained in combination with other biobased monomers such as lactide and tulipalin A.138,139 These block copolymers exhibit very good tensile and elastic properties, and consequently are promising candidates for biobased thermoplastic elastomers.
After these initial reports, the same researchers reported on the use of other terpene-based monomers produced from the oxidation of cyclohexanones. Specifically, dihydrocarvide, dihydrocarvide oxide and carvomenthide can be obtained from biosourced carvone, and their ROP promoted by ZnEt2 and benzyl alcohol provides the corresponding polyesters (Fig. 15).140,141 The presence of functional groups on these monomers has been exploited.142 For example, reaction of the vinyl groups on poly(dihydrocarvide) (PD) with dithiols resulted in the formation of cross-linked gels. Copolymerisation of dihydrocarvide oxide with CL yielded cross-linked networks with good shape-memory properties.
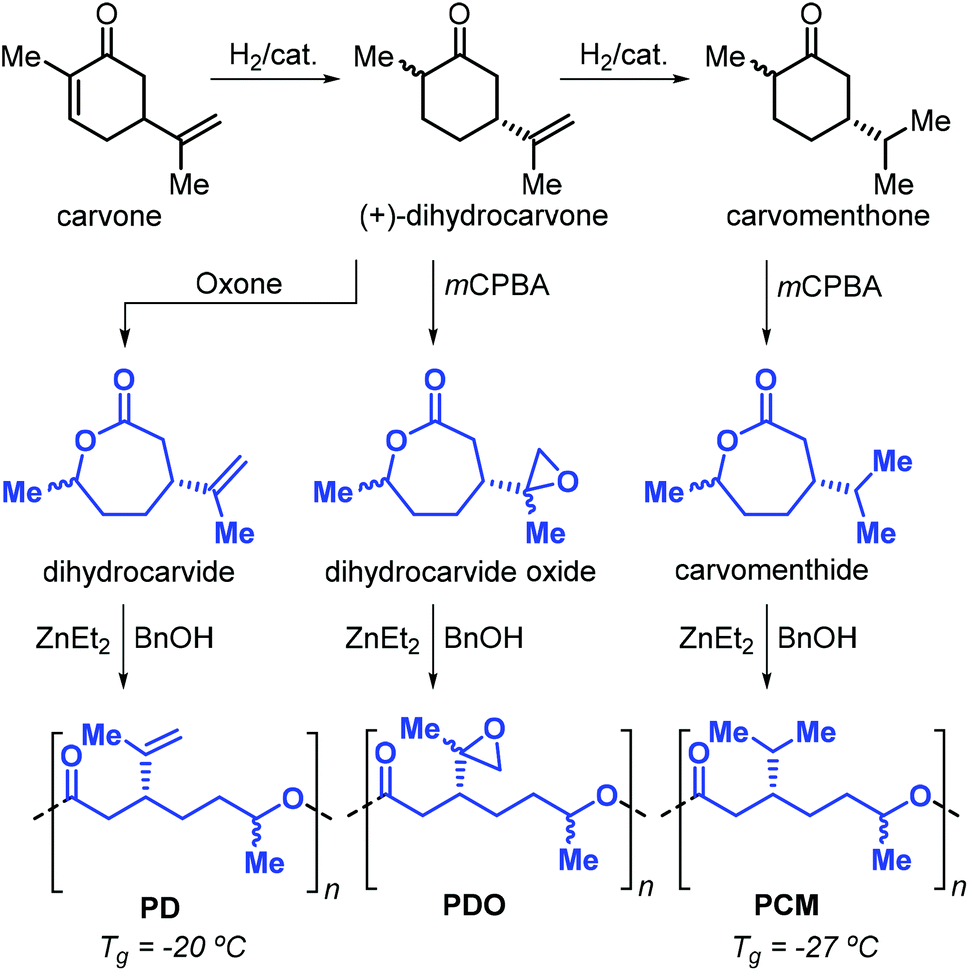 | ||
| Fig. 15 Synthesis of dihydrocarvide, dihydrocarvide oxide and carvomenthide from carvone, and their ROP toward the corresponding polyesters. | ||
More recently, the Jones group reported the synthesis of 4-isopropyl caprolactone from (+)-β-pinene, following a synthetic route similar to that described for carvomenthide (Fig. 16).143 Ring-opening polymerisation of 4-isopropyl caprolactone was efficiently conducted in the presence of amino-phenolate complexes 5 and 6.
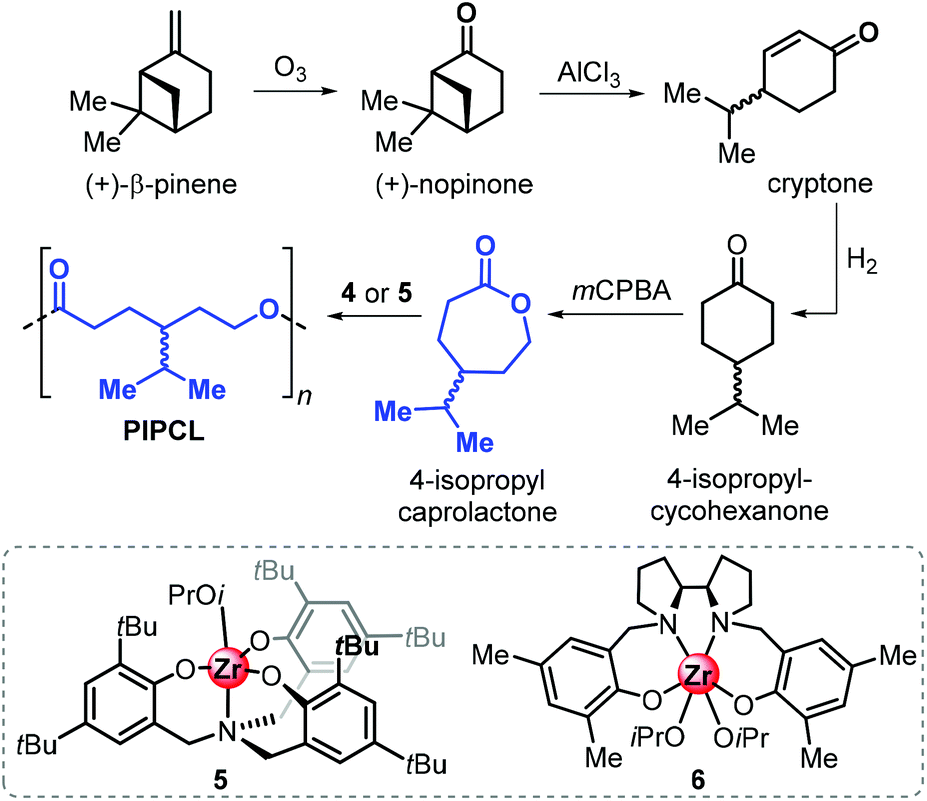 | ||
| Fig. 16 Synthesis of poly(4-isopropyl-caprolactone) from (+)-β-pinene and structures of ROP catalysts aminophenolate Zr complexes 5 and 6. | ||
Importantly, it was found that the conversion of this synthetic, pinene-derived 4-isopropyl caprolactone monomer only produces low molecular weight polymers (Mn up to 4.9 kDa), while the use of the “commercial” 4-isopropyl caprolactone monomer resulted in significantly better polymerisation control allowing to produce polymers with Mn's up to 11.5 kDa. This marked difference was ascribed to the presence of unidentified impurities after the ozonolysis step, which can act as potential chain-transfer agents.
Poly(4-isopropyl caprolactone) (PIPCL) has a low Tg of −50 °C, near the value of polycaprolactone (PCL). One-pot copolymerisation with LA was also feasible and furnished copolymers with a wide range of compositions, but attempts to obtain block-copolymers were unfruitful.
In 2019, Syrén et al. disclosed a chemo-enzymatic synthesis of bicyclic (−)-verbanone lactone (Fig. 17).144 The synthesis starts with the hydrogenation of (−)-verbenone, that can be obtained from (+)-α-pinene through an electrochemical oxidation. Different from the approaches so far described in this section, the traditional Baeyer–Villiger oxidation step was substituted with a biocatalytic oxidation promoted by Baeyer–Villiger monooxygenase (BVMO) and glucose dehydrogenase (GDH). Poly(verbanone lactone) (PVL) was obtained by ROP of (−)-verbanone lactone promoted by methane sulfonic acid (MSA) and benzyl alcohol. With this protocol, low molecular weight polymer was obtained (Mn = 3.3 kDa). Unlike other caprolactone-like polyesters, PVL showed a relatively high Tg of 26 °C likely being the result of the rigid cyclobutane ring incorporated into the polymer chain.
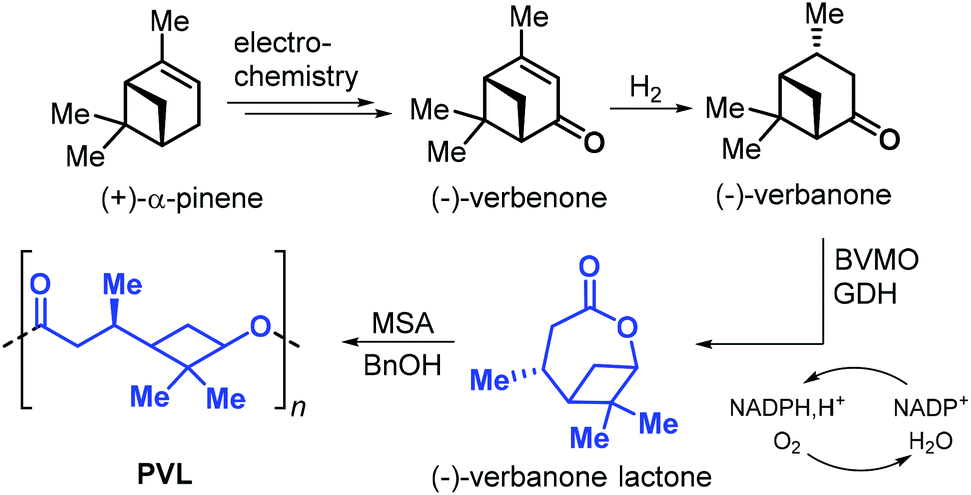 | ||
| Fig. 17 Chemo-enzymatic synthesis of (−)-verbanone lactone from (+)-α-pinene, and ROP toward poly(verbanone lactone). | ||
Due to the success attained in the preparation of polyesters by ROCOP, in the last years more efforts have been focusing on the identification and use of terpene-based epoxide and cyclic anhydride monomers. Since amorphous aliphatic polyesters generally exhibit moderate Tg values (e.g. the Tg of PLA is between 50 and 60 °C), they can hardly compete with well-established commodity polymers such as atactic polystyrene (aPS, Tg of around 100 °C). For this reason, most of the recent research in this subdomain of polyester development has put primary attention towards the identification of structurally rigid monomers that, in principle, could give access to polyesters with higher Tg values.135
In 2011, Thomas et al. described a tandem procedure for the preparation of aliphatic polyesters.145 This procedure relies on the synthesis of cyclic anhydrides from dicarboxylic acids and dimethyldicarbonate (DMDC) catalysed by a series of salen complexes (1, Fig. 3, and 7–9Fig. 18).
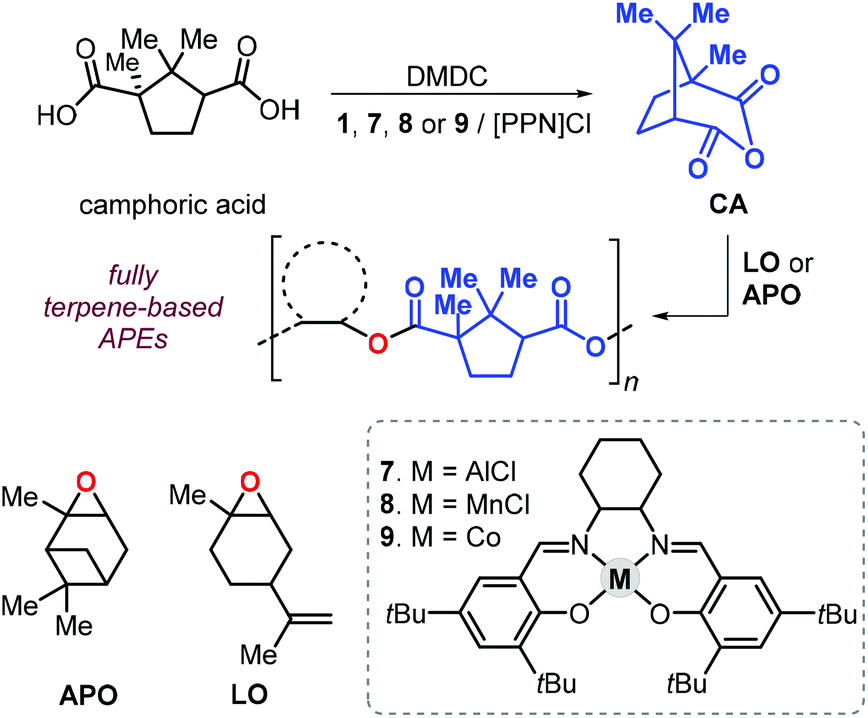 | ||
| Fig. 18 Sequential, one-pot synthesis of fully terpene-based polyesters based on camphoric anhydride and terpene oxides LO or APO, and structure of complexes 7–9. | ||
Thus, addition of an epoxide to the in situ prepared cyclic anhydrides gave the desired polyesters in a one-pot procedure in which both steps are promoted by the same catalyst. With this method, the authors prepared a number of structurally different polyesters with Mn values in the range 4.3–27 kDa and Đ values lower than 1.3. Interestingly, camphoric acid was used for the synthesis of camphoric anhydride (CA) and subsequently for the preparation of fully terpene-based polyesters using APO and LO (Fig. 18). Later, Thomas, Prunet and co-workers employed this tandem methodology for the preparation of APEs with pendant double bonds by reaction of CA and functional epoxides.146 The post-modification of these APEs by ruthenium-catalysed olefin cross-metathesis was explored. The thermal properties of these allyl glycidyl ether-based polyesters, poly(AGE-alt-CA)s, could be tuned by reaction with various olefins, and the resultant Tg's were in a range from 6 to 96 °C (Fig. 19). Unfortunately, functionalisation of fully terpene-based poly(LO-alt-CA), which exhibit a promising Tg of 112 °C, was not successful probably due to steric hindrance.
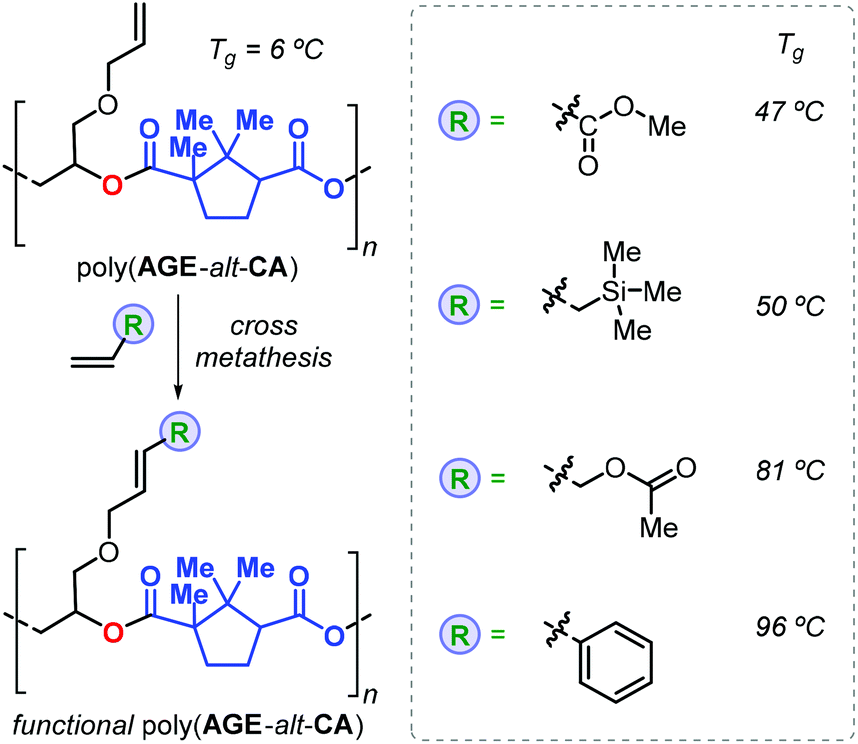 | ||
| Fig. 19 Functional polymers derived from poly(AGE-alt-CA) after post-modification using cross-metathesis reactions. | ||
The efficient preparation of ABA triblock polyesters from decalactone/cyclohexene oxide/cyclic anhydride monomer mixtures using complex 1 as the catalyst was communicated by Williams and Stößer.147 In particular, CA was used for the preparation of the triblock copolymer poly(decalactone)-b-poly(cyclohexene oxide-alt-camphoric anhydride)-b-poly(decalactone) though, in this case, detailed thermal properties were not described.
In 2015, Coates and van Zee described the copolymerisation of propylene oxide (PO) with tricyclic anhydrides TCA1 and TCA2 obtained from the Diels–Alder reaction of α-terpinene or α-phellandrene with maleic anhydride in the presence of Cr, Al and Co salphen complexes 10–12 (Fig. 20).148 The resulting polyesters poly(PO-alt-TCA1) and poly(PO-alt-TCA2) exhibit relatively high Tg values of 109 and 86 °C, respectively. This difference in Tg of more than 20 °C indicates that the spatial orientation of substituents has a strong impact. Remarkably, using a hydrogenated version of TCA1 in the copolymerisation with PO produced a polymer with a Tg of 86 °C, supporting the idea that structural rigidity of the repeating unit strongly influences the thermal behaviour of the polymer.
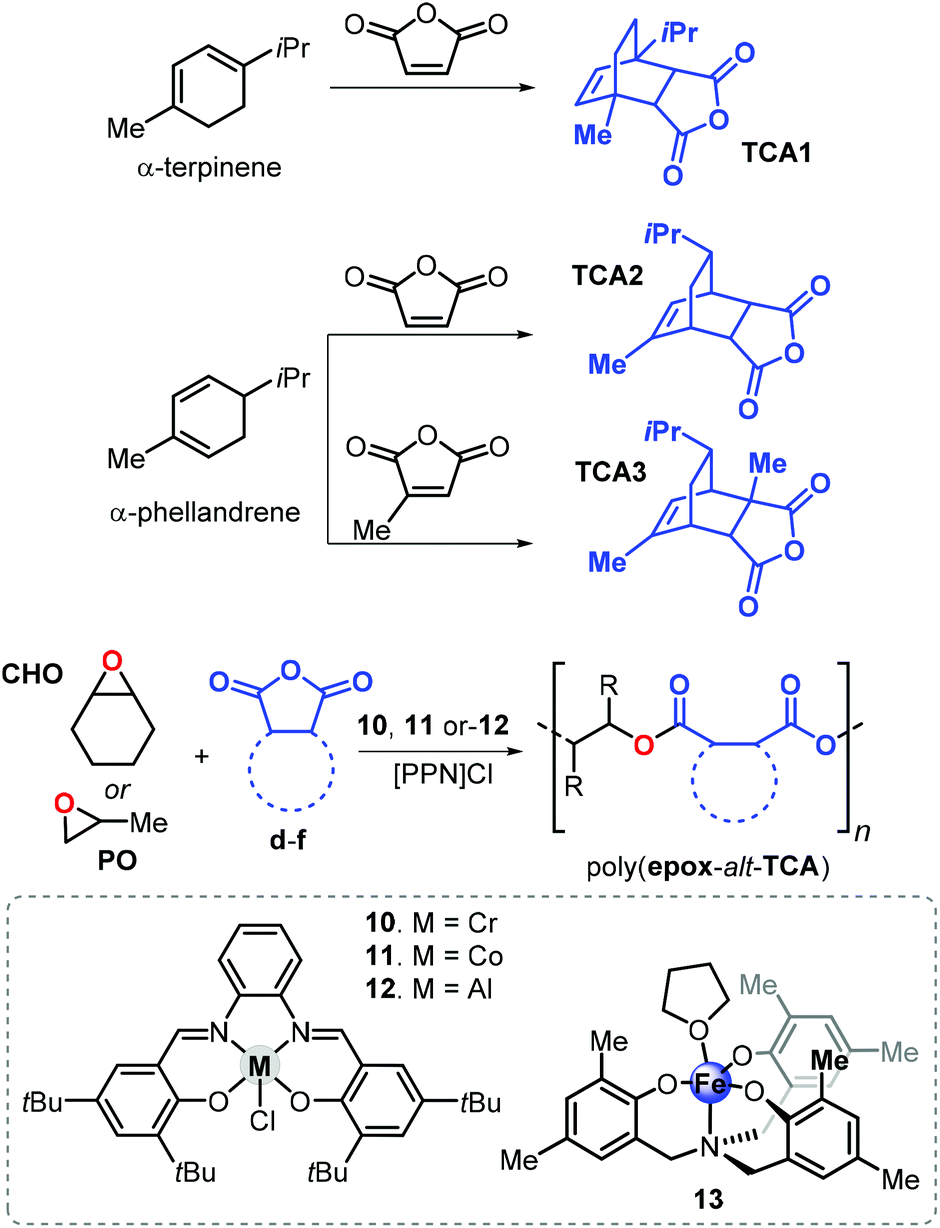 | ||
| Fig. 20 Synthesis of tricyclic anhydrides TCA1–3, their ROCOP affording the corresponding polyesters and structures of complexes 10–13. | ||
The authors also investigated the effect of the catalyst structure on the polymerisation process. It was established that in the presence complexes 10 and 11, transesterification and epimerisation reactions occur at high conversions, resulting in broadening of the molecular weights distributions and lower Tg values. The detrimental effect of epimerisation on the Tg was explained by the inversion of cis-diester into trans-diester units, with the latter believed to be more flexible. On the contrary, the presence of Al-complex 12 led to a well-controlled polymerisation process in which these side-reactions are efficiently suppressed thus maintaining narrow distributions and high Tg values even at high conversions and long reaction time.
A follow-up studies was carried out by Coates and Kleij who envisaged the possibility to obtain polyesters with improved thermal properties by copolymerisation of similar and a wider diversity of biobased tricyclic anhydrides with a more rigid epoxide (CHO).149
The use of TCA1, TCA2, and the new terpene cyclic anhydride TCA3 (Fig. 20), obtained from α-phellandrene and citraconic anhydride, was investigated. Copolymerisation reactions were conducted in the presence of Al-salphen complex 12 and the Fe-aminotriphenolate complex 13. These new polyesters obtained from CHO exhibit higher Tg values when compared with those obtained from PO. For example, poly(CHO-alt-TCA1) has a Tg of 184 °C being 75 °C higher than that of poly(PO-alt-TCA1). However, the CHO-based polymers had typically lower molecular weights (4.1 ≤ Mn ≤ 11.6 kDa) and relatively broader dispersities (1.23 ≤ Đ ≤ 1.58), which may be the result of the larger steric demand and lower rate of the insertion of CHO into the propagating polymer chain. On the basis of end-group analysis, it was concluded that more extensive chain-transfer phenomena occur when CHO was used as epoxide monomer ascribed to the parasitic formation of CHO-based alcohols. By choosing the appropriate epoxide/cyclic anhydride monomer ratio, it was possible to tune the Tg such that a wider temperature range of 66 to 184 °C was enabled for these aliphatic polyesters.
Considering terpene-based epoxides such as APO and LO (Fig. 18) in ROCOP is attractive for the synthesis of renewable polyesters. However, these kinds of monomers are generally sterically congested and therefore relative sluggish to convert, and their application up to now has remained very limited. In 2013, Duchateau et al. reported the copolymerisation of LO with phthalic anhydride (PA) promoted by salphen complex 10.150 This polymerisation process required high temperature (130 °C) and neat conditions, yielding poly(LO-alt-PA) with Mn's up to 7.4 kDa, Đ values of around 1.4 and a maximum Tg of 82 °C.
More recently, Kleij et al. investigated the synthesis of semi-aromatic polyesters by ROCOP of various terpene-based epoxides with both PA and 1,8-naphthalic anhydride (NA) with complex 13 as catalyst (Fig. 21).151 Copolymerisation of LO proceeded under mild condition (65 °C) both under neat and solution conditions and the performance of the catalyst 13 may be explained in terms of the higher geometrical flexibility of the aminotriphenolate complex compared to salen metal complexes. The poly(LO-alt-PA) obtained from cis/trans-LO displayed a Tg of 131 °C (Mn = 10.5 kDa), while that obtained from cis-LO had a higher Tg of 141 °C (Mn = 16.4 kDa). Copolymers obtained from LDO are characterised by low glass transition temperatures. It is likely that this copolymerisation process is not well-controlled due to the presence of two “branching” points in the monomer, and thus a higher potential for cross-linking of growing polymer chains. As a result, the copolymer had a broad molecular weight distribution (Đ ≈ 2) and a relatively low Tg of 59 °C.
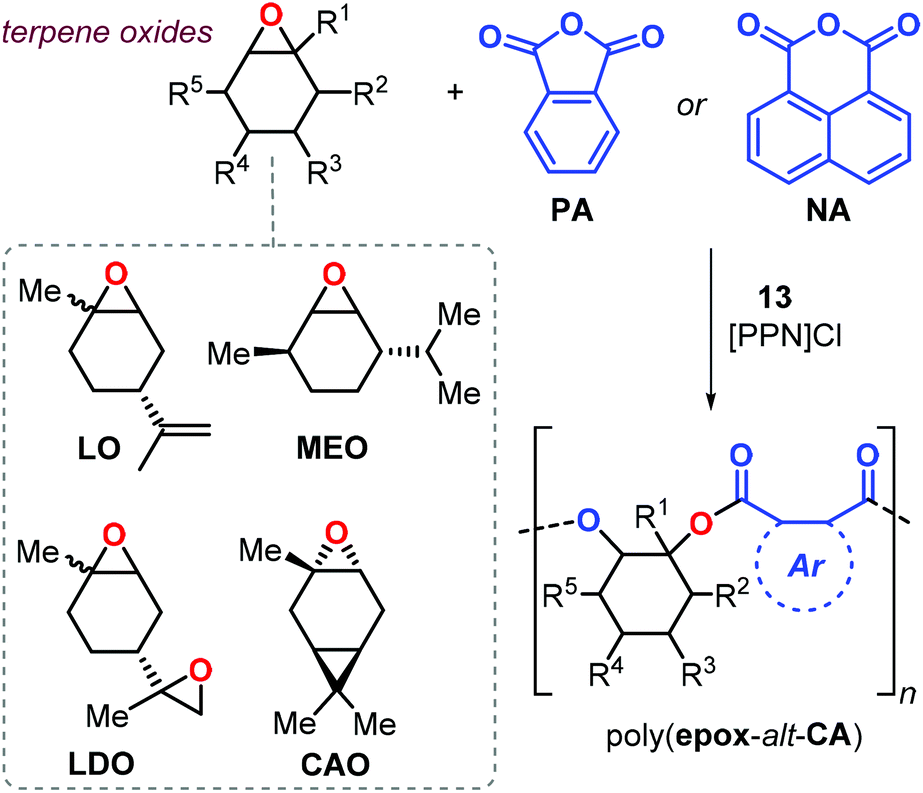 | ||
| Fig. 21 Synthesis of rigid semi-aromatic polyesters from phthalic anhydride and terpene-based epoxides. For 13 see Fig. 20. | ||
Copolymerisation of PA with other structurally different terpene-based epoxides such as menthene oxide (MEO) and carene oxide (CAO) was also performed (Fig. 21). The reaction rate observed for CAO conversion was expectedly lower than noted for LO yielding only oligomers (Mn < 3.7 kDa) with a Tg of 130 °C. Interestingly, poly(MEO-alt-PA) was isolated with an appreciable Mn (12.7 kDa) and a Tg of 165 °C. Remarkably, the structurally more rigid poly(LO-alt-NA) copolymer showed a Tg of 243 °C. However, only low molecular weight oligomers were obtained in this case (Mn < 2.2 kDa) with a decomposition temperature Td10 of 268 °C.
4. Polyurethanes
Polyurethanes (PUs) are employed in a wide range of applications such as foams, insulating materials, paints, liquid coatings, sealants, fibres and elastomers, among others.152 Commercial PU synthesis is based on polyaddition reactions between isocyanates and polyols (Fig. 22a).153 (Aryl)isocyanates are prepared by reaction of amines with highly toxic and hazardous phosgene in an energy-intensive process. Exposure to isocyanates negatively affects human health and as such represent an aspect to be improved in PU production.154 Additionally, the production of commonly employed polyols (such as polyethylene oxide and polypropylene oxide) fully relies on the use of fossil fuels.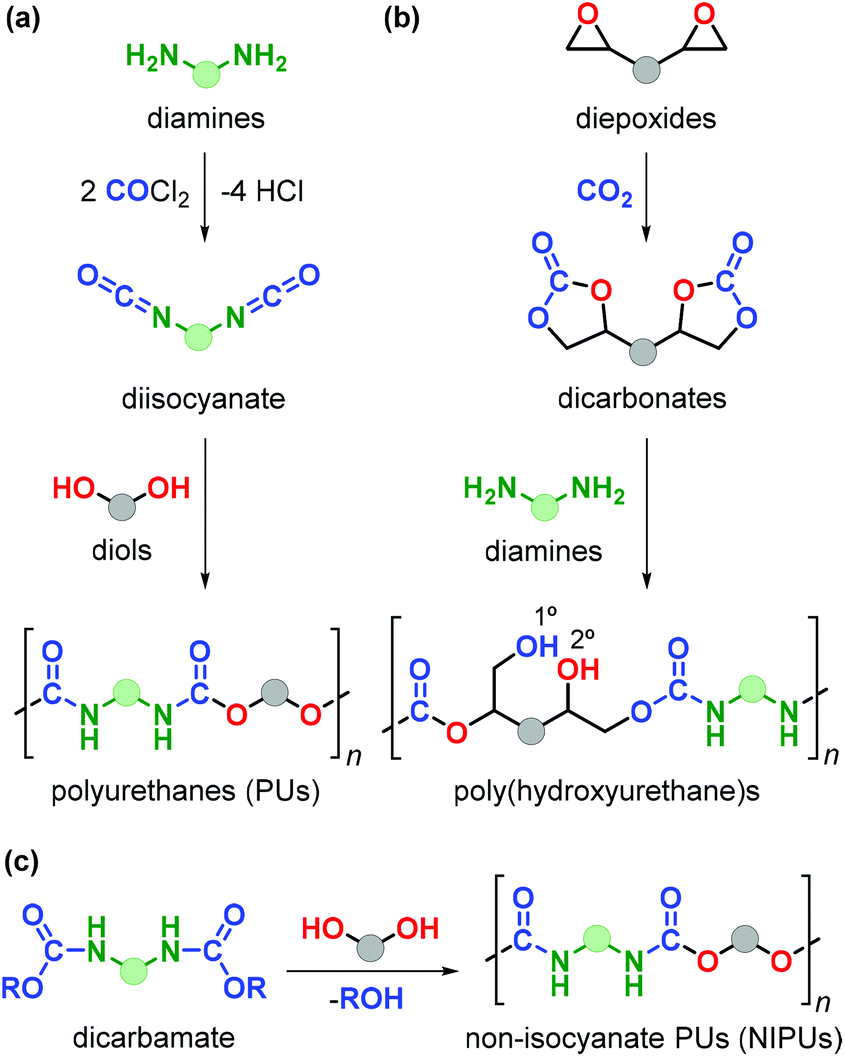 | ||
| Fig. 22 Synthesis of (a) polyurethanes from diisocyanates and diols, (b) poly-hydroxyurethanes from di(cyclic carbonates) and diamines, and (c) non-isocyanate polyurethanes from dicarbamates and diols. The coloured spheres represent connecting units. | ||
The design of non-isocyanate based polyurethanes (NIPUs) has recently emerged as a highly active area in polymer chemistry.155,156 One effective route to NIPUs is the polyaddition reaction between di(cyclic carbonates) and diamines (Fig. 22b). This alternative process has become more appealing and relevant due to the development of more efficient and sustainable methods for the production of cyclic carbonates from CO2 and epoxides.157–161 Furthermore, with many research efforts now being devoted to the production of diamines from renewable sources,162 new opportunities have become available to sustainabilise PU production. NIPUs obtained through this latter route are sometimes also referred to as poly(hydroxyurethane)s because of the presence of primary (1°) and secondary (2°) alcohol groups within the polymer chain with the latter typically prevailing.
The presence of –OH groups results into an additional hydrogen-bonding network increasing the polarity, thermal stability, adhesion properties and water uptake thereby advancing the discovery and development of new materials.153 The synthesis of NIPUs can be achieved by polycondensation reaction of diols with other functional monomers such as dicarbamates (Fig. 22c), however this approach is hampered by the use of phosgene for the production of such monomers.163 In spite of the high attractiveness of this new route towards NIPUs, the number of renewable biosourced terpene-based monomers for NIPU production remains so far highly limited.164
The research group of Mülhaupt investigated the use of limonene dicarbonate (LDC) for the preparation of poly-hydroxyurethanes.165 The LDC was prepared by reaction of LDO with carbon dioxide catalysed by tetrabutyl ammonium chloride (TBAC, Fig. 23). Notably, the reaction was conducted on a kilogram scale, and high temperature and pressure (i.e., 140 °C and 30 bar) were necessary to reach full conversion.
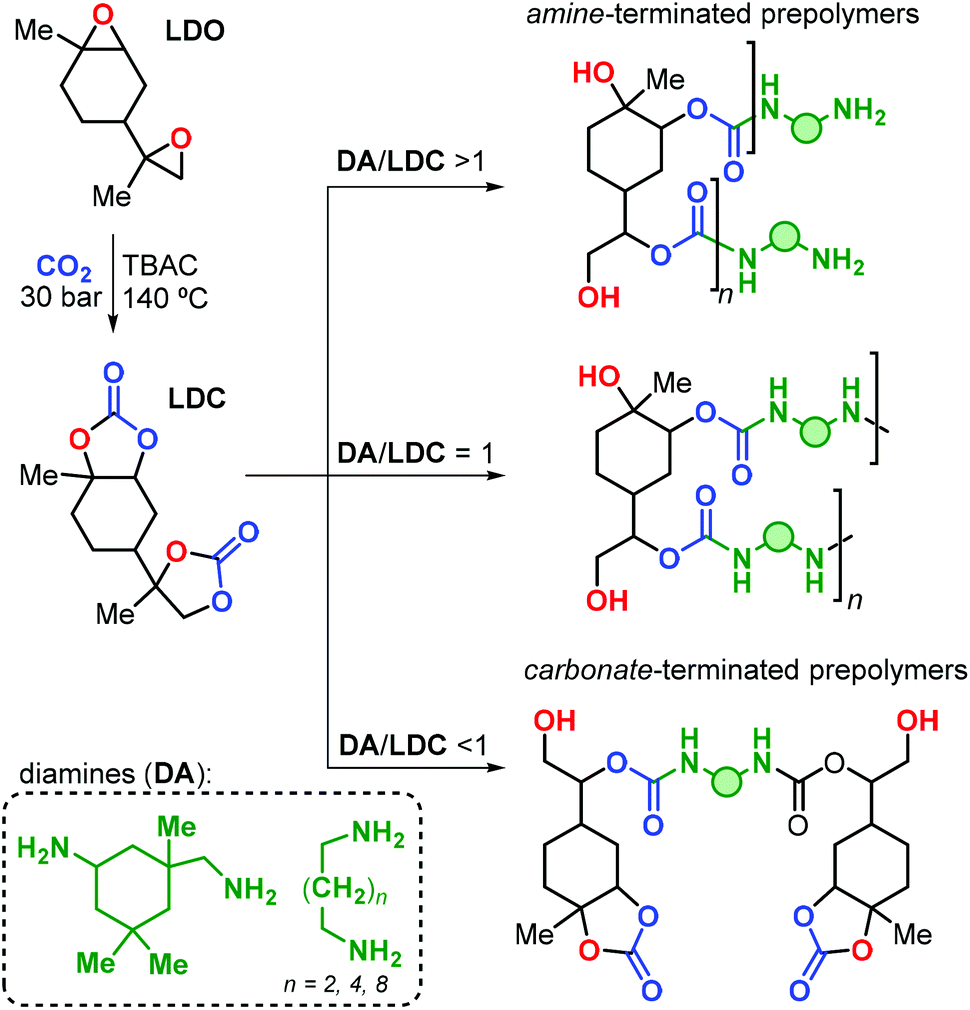 | ||
| Fig. 23 Preparation of limonene dicarbonate (LDC) and its polyaddition reaction with diamines (DA) towards limonene-based NIPU prepolymers. | ||
Linear polyurethanes were afterwards obtained by reaction of LDC with alkyl primary diamine (DA) under neat conditions, with the product features depending on the LDC/DA ratio (Fig. 23). When an equimolar mixture of LDC and DA was used, small oligomers with Mn values in the range 960–1840 Da were obtained. These oligomers exhibit Tg's and Tm's in the range of 33–62 and 80–100 °C, respectively, depending on the nature of the diamine reagent. When DA or LDC was present in excess during the stepwise polyaddition polymerisation, amine- or carbonate-terminated oligomers were formed.
The authors prepared a number of NIPU thermosets by curing LDC with polyfunctional amines including commercial hyperbranched polyethyleneimines (i.e., Lupasol®) with variable content of primary amine groups. Brittle NIPUs were obtained, with high E moduli (2400 ≤ E ≤ 4100 MPa), and low elongation at break (εbreak ≤ 2%). This behaviour was ascribed to the rather rigid nature of the limonene units in these polymer structures.
More recently, the same research group described an improved procedure for the preparation of trans-LDC using consecutive crystallisations,166 and the stereochemical assignment was corroborated by X-ray crystallography. Linear NIPUs were obtained by reaction of LDC-based oligomers (obtained in the presence of excess of diamine, Fig. 23) with carbonated 1,4-diglycidyl ether. Notably, the authors demonstrated the positive impact of the monomer purity on the final properties of the NIPU thermosets prepared from LDC and Lupasol®. Extensively purified LDC led to the formation of colourless and transparent materials, in contrast to the dark samples obtained with crude LDC. Moreover, higher Tg (94 vs. 50 °C), E modulus (4370 vs. 2400 MPa) and tensile strength at break (σB, 53 vs. 7 MPa) were achieved thus pointing at the importance of the impurity profile on the final NIPU performance.
Apart from the application of cyclic carbonate reagents, in 2013 Firdaus and Meier reported the synthesis of limonene-based dicarbamate monomers, and their use in the synthesis of NIPUs by a polycondensation reaction with diols.167 The synthesis starts with a thiol-ene click addition of cysteamine hydrochloride to (R)- and (S)-limonene yielding diamines d1–2 (Fig. 24). Subsequently, carbamate groups were introduced by reaction with dimethyl carbonate (DMC) catalysed by TBD (Fig. 24).
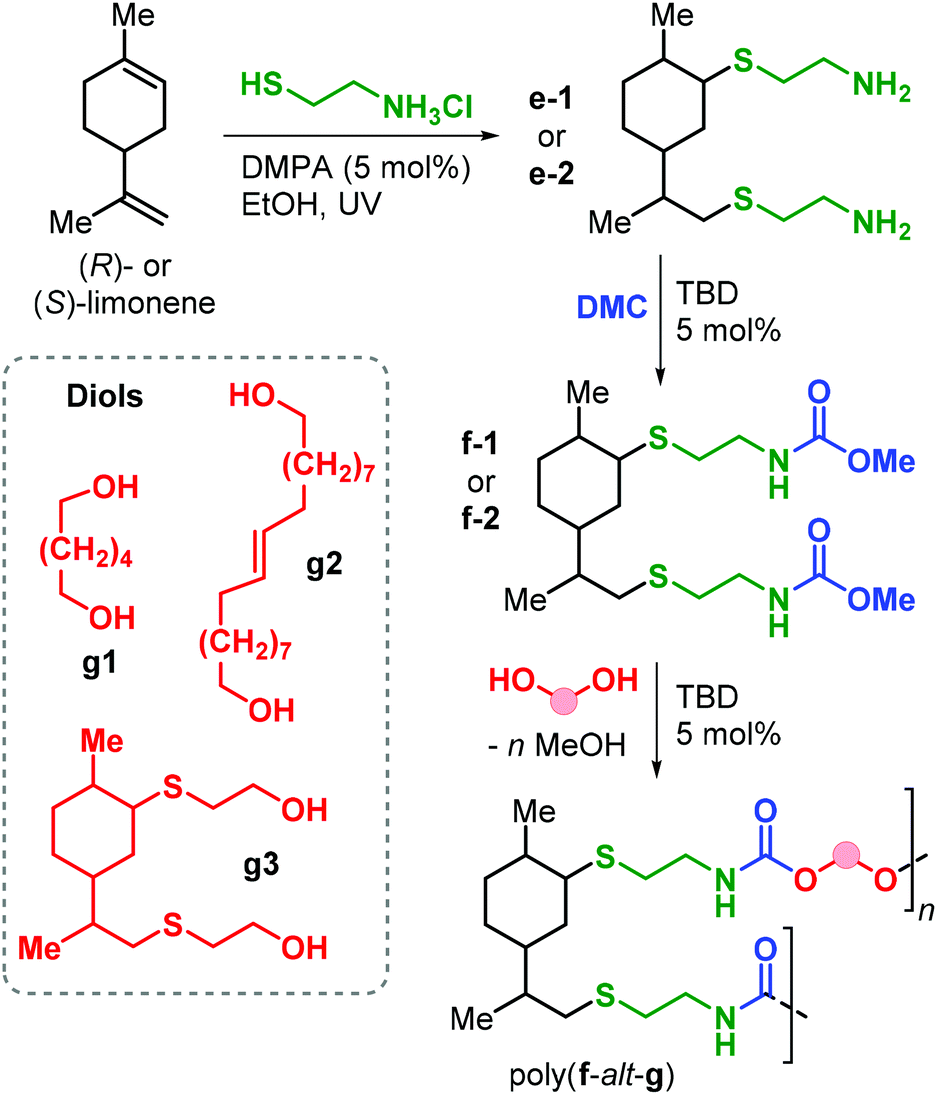 | ||
| Fig. 24 Synthesis of diamine and dicarbamate monomers e1–2 and f1–2, and limonene-based NIPUs poly(f-alt-g). | ||
Polyurethanes of type poly(f-alt-g) were prepared by TDB-catalysed polycondensation reaction of f1–2 with biobased diols g1–3 previously described by the same group.168 Molecular weights as high as 12.6 kDa were obtained with Đ values between 1.8 and 2.2. Interestingly, copolymerisation with g2 (containing flexible alkyl chains) led to semi-crystalline samples with Tm up to 69 °C. On the contrary, NIPUs that are more rigid (obtained from g1 and g2) are amorphous with Tg values in the range 14.6–18.5 °C.
5. Polyamides
Polyamides (PAs) represent a commercially relevant class of polymers. Thanks to excellent mechanical and thermal properties, PAs are mainly used for the production of fibres and engineering plastics employed in transportation, electronics, packaging and customer products.121 Polyamides are generally prepared by polycondensation of diamines with dicarboxylic acids, or by ROP of lactams such as in the production of Nylon-6,6™ and Nylon-6™, respectively (Fig. 25). As for the other polymer types discussed in this review, commercial PA production is currently based on non-renewable raw materials, and efforts are ongoing towards the identification of renewable monomers, especially focusing on novel diamine and diacid reagents.27,162,169 The use of terpene-based monomers in PA polymer formation, however, remains underexplored.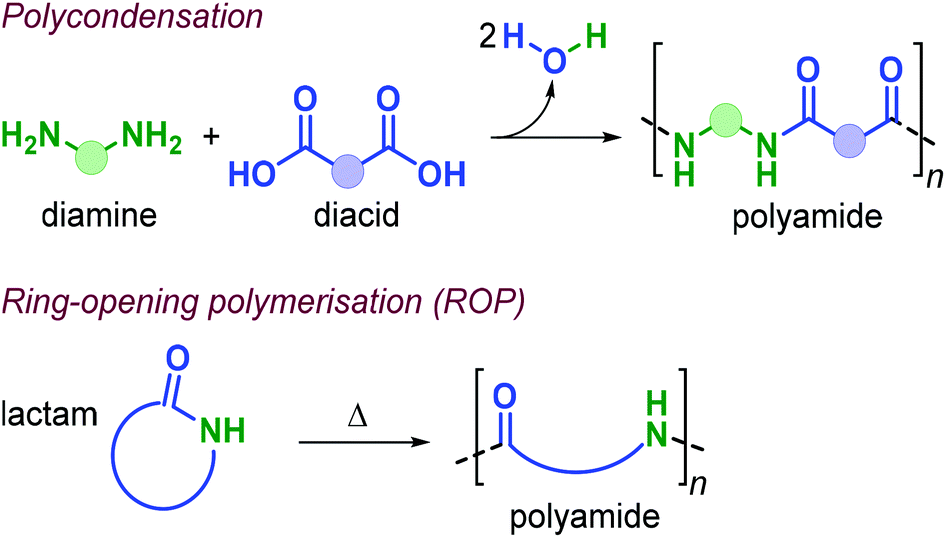 | ||
| Fig. 25 Polyamide (PA) synthesis by polycondensation and ring-opening polymerisation. | ||
Together with renewable PUs, Firdaus and Meier also reported the synthesis of limonene-based PAs using limonene-based diamine monomers e1–2 described in Fig. 24. Specifically, polyamides of type poly(e-alt-h) were prepared by TBD-catalysed polycondensation reaction of e1–2 with biobased diesters such as castor-oil and limonene-derived diesters h1–3 that were previously described by the same group (see Fig. 24).168 In the case of flexible diester h1 (Fig. 26), the resultant PAs had Mn's of up to 10.6 kDa. Copolymerisation of e1–2 with h2 and h3, however, led to significantly lower molecular weights in the range 5.5–7.8 kDa. This difference was ascribed to the formation of crystalline domains when h2 was used as diester as it contains a long saturated alkyl chain, and in the case of h3 it was proposed that the bulkiness of the diester affected propagation.
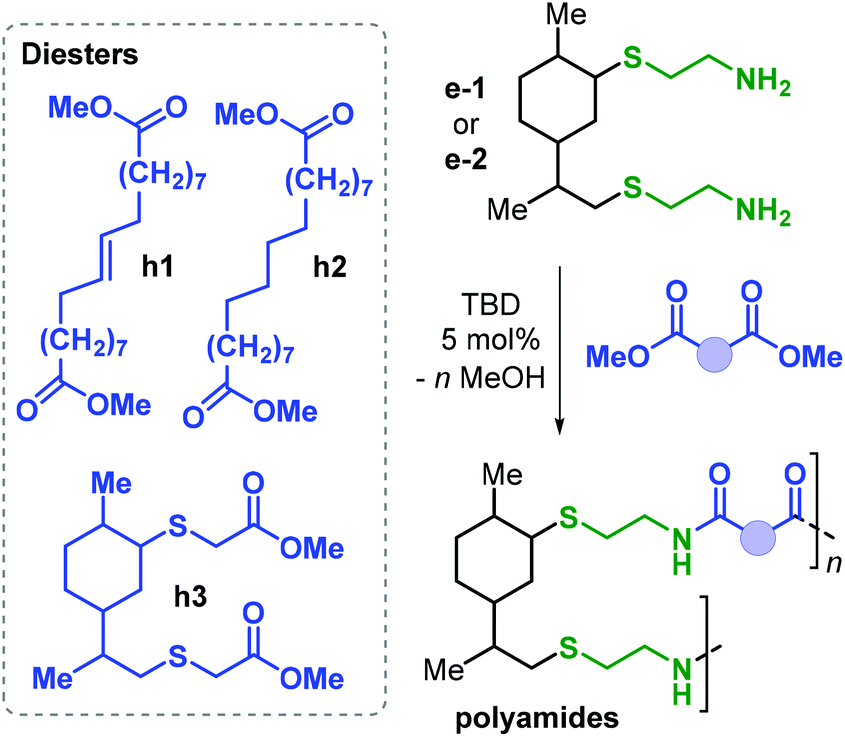 | ||
| Fig. 26 Synthesis of limonene-based polyamides poly(e-alt-h). | ||
Interestingly, the more rigid PA obtained from h3 is amorphous with a Tg of 41.5 °C. Contrary, PAs based on flexible diesters h1 and h2 are semi-crystalline having a Tm as high as 102 °C.
The same authors also explored the effect of limonene-based monomer incorporation in Nylon-6,6™ by copolymerisation of diamines e1–2, adipic acid and hexamethylenediamine. The presence of cycloalkane moieties in the PA polymer affects its crystallisation behaviour. Differential scanning calorimetry (DSC) analyses showed multiple, broad endothermic peaks between 120 and 215 °C depending on the content of e1–2.
The use of terpene-based lactams for the synthesis of polyamides has been investigated by Rieger, Winnacker and co-workers. In 2013, they reported the preparation of stereo-regular polyamide oligomers oligo-i1 and oligo-i2 (Mn < 2.8 kDa), obtained by anionic and acid-catalysed ROP of lactams i1 and i2 (Fig. 27).170 These lactams were prepared from (−)-menthone via Beckmann rearrangement of appropriate oxime precursors.171 The two regio-isomers showed different behaviour with i2 being more reactive. Conceptually, this approach is comparable to the preparation of lactones from cyclic ketones described in section 3 (Fig. 14). Following an early polymerisation attempt reported by Kono and coworkers,172 a regio-selective procedure for the preparation of i1 was developed,173 using hydroxylamine-O-sulfonic acid as catalyst. The thermal properties of oligo-i1, obtained through anionic ROP, were determined.174 Remarkably, semi-crystalline oligo-i1 exhibits a melting temperature of around 300 °C, which is significantly higher than that of Nylon-6™ (220 °C).
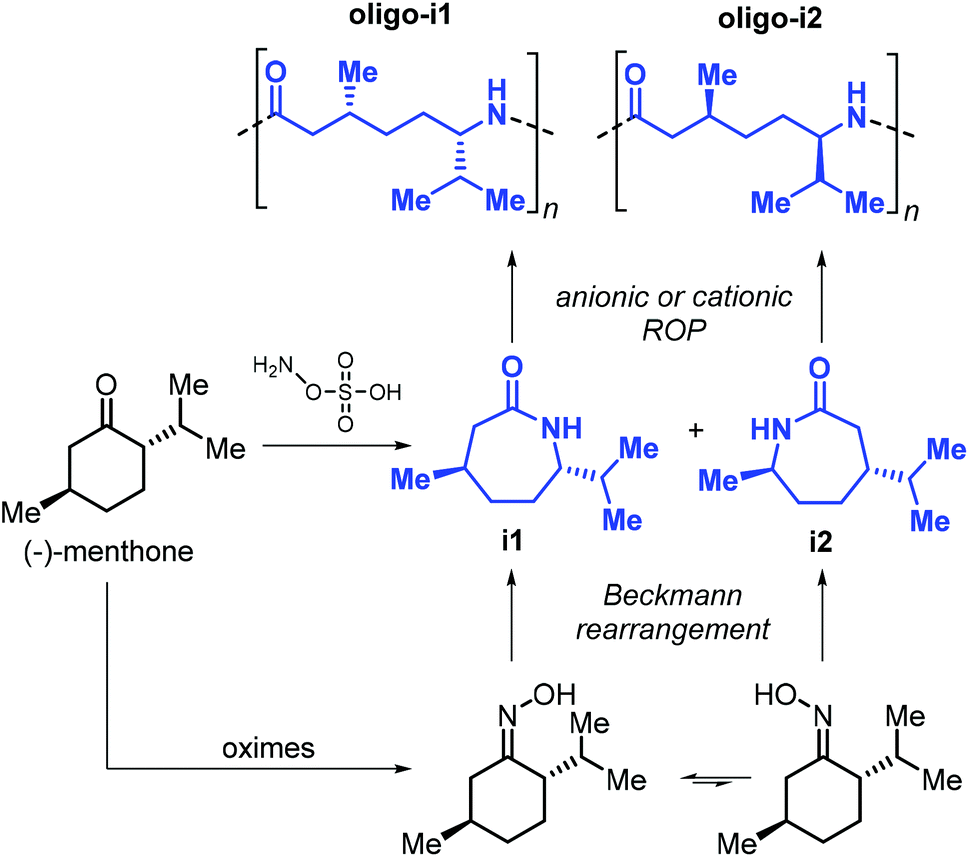 | ||
| Fig. 27 Synthesis of lactams i1 and i2 from (−)-menthone, and structures of polyamide oligomers oligo-i1 and oligo-i2. | ||
Building further on these results, Winnacker et al. described the synthesis of the stereo-regular polyamide poly-j1 starting from nopinone-based monomer j obtained by a procedure similar to that used for lactams i1 and i2 (Fig. 28).175,176
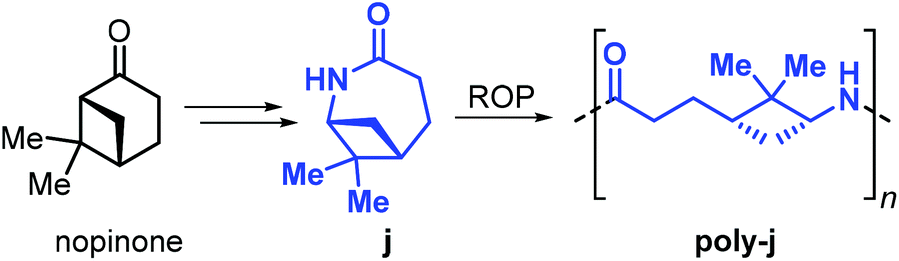 | ||
| Fig. 28 Nopinone derived lactam j and its ROP to form polyamide poly-j. | ||
The rigid and stereo-regular structure of poly-j provides excellent thermal properties with a Tm > 308 °C and a Tg of around 150 °C. Further to that, the interaction of human keratinocytes (HaCat cells) with films of poly-j blended with polyethylene glycol has been examined, demonstrating the potential of biobased polyamides in biomedical applications such as regenerative skin replacement.177,178
In 2019, Sieber and co-workers achieved the ROP of β-lactams k2 and k3, and ε-lactams k1–4 obtained from α-pinene and (+)-carene, respectively (Fig. 29).179 The β-lactams k2 and k3 were synthesised by [2 + 2] cycloaddition of chlorosulfonyl isocyanate to either α-pinene or (+)-carene. The synthesis of ε-lactams k1 and k4 was carried out by a four-step procedure involving the oxidation of the starting materials to their ketones, oxime formation and finally Beckmann rearrangement. The anionic ROP of lactams k1–4 led to the formation of the corresponding PAs poly-k1–5. In the case of poly-k4 a relatively high Mn of 33 kDa was attained.
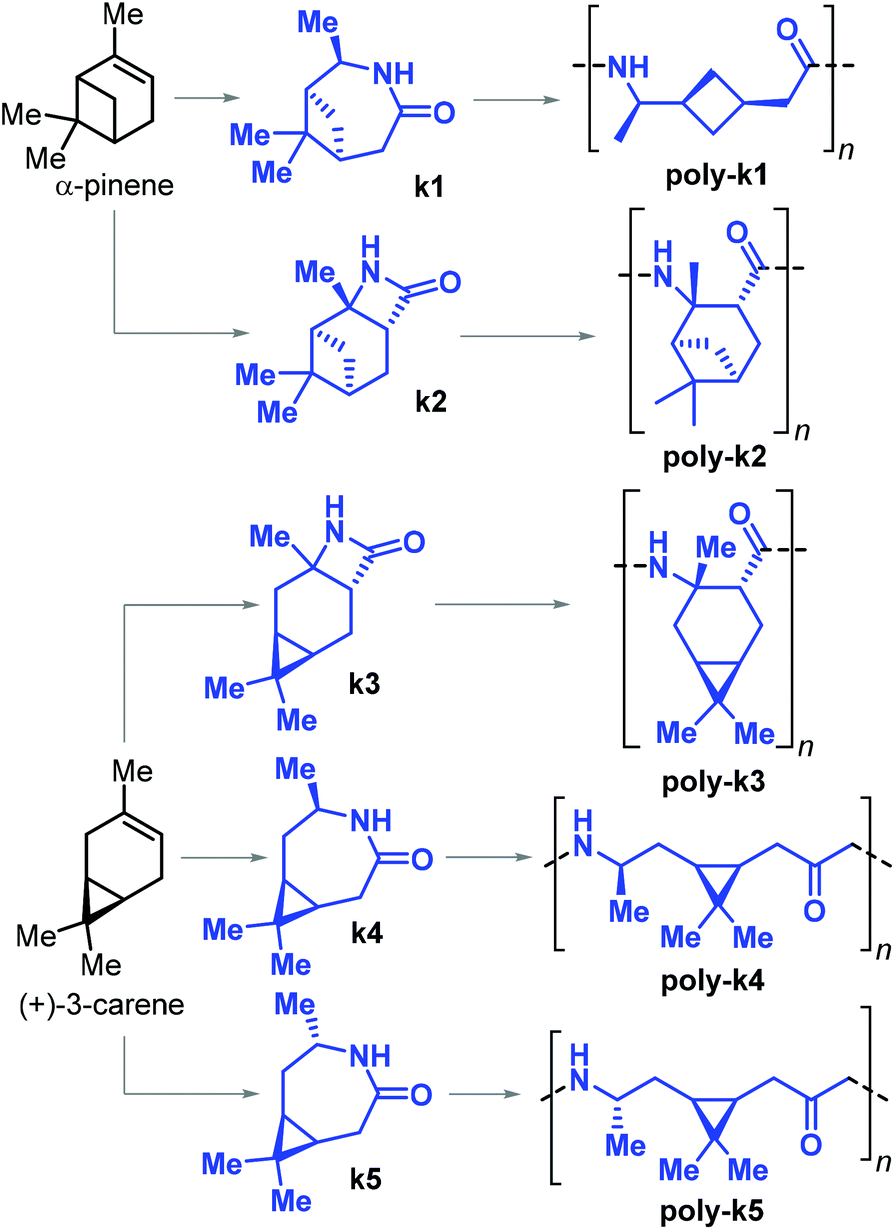 | ||
| Fig. 29 β-Pinene derived lactams k1 and k2, (+)-3-carene derived lactams k3–5 and polyamide polymers poly-k1–5. | ||
Apart from poly-k4 (which showed a Tg of 120 °C), no other thermal transitions could be detected by DSC analysis of all the other pinene- and carene-based polyamides. The authors argued that either the exceptionally bulky structures of these polymers prevent crystallisation, or that these materials have melting temperatures higher than their decomposition temperatures with their Tm's then amounting to or being beyond 400 °C.
In a related contribution from the same group, a modified synthetic procedure for monomer k5 was presented involving the stereoselective chemo-enzymatic oxidation of (+)-carene, producing the monomer with opposite stereochemistry to k4. The syntheses of both monomers were scaled up to molar amounts.180 In the same work, the crystal structure of poly-k5 was reported, and this polyamide was shown to have a Tg and Tm of 105 and 280 °C, respectively.
6. Outlook and conclusion
Despite the enormous and increasing attention for the selection of new, cheap and renewable feedstock for the production of sustainable polymers, the use of terpenes has still been relatively unexplored. With the exception of terpene-based polyolefins which have matured substantially, recent developments have also shown interesting progress in the fabrication of other types of polymers (polycarbonates, polyesters, polyurethanes and polyamides).As a direct consequence of the infancy of this research topic, most research efforts so far have been focussed on the synthesis of terpene-based monomers and their polymers. Much less attention has been devoted to the characterisation of key thermal, mechanical properties and/or surface properties, and demonstration of the full potential of these alternative raw materials and processes is thus an ongoing endeavour. There is obviously still a need for a broad-screen benchmarking of the performance of these terpene-based polymers against petroleum-based materials in future research work surrounding this topic.
At this stage, it is hard to realise a fair comparison between well-known established polymers and the emerging terpene-based alternatives described in this review. However, easy to obtain thermal data (such as Tg and Tm) are commonly used as a first approximation to identify possible applications because of their correlation with the polymer structure. In this respect, the incorporation of terpene monomers (or derivatives thereof) has shown in some cases advantages associated with the rigidity and/or functionality of these compounds. In order to credit these (preliminary) advantages, a collection of thermal data for selected petroleum-based commercial polymers is given in Fig. 30.181–185
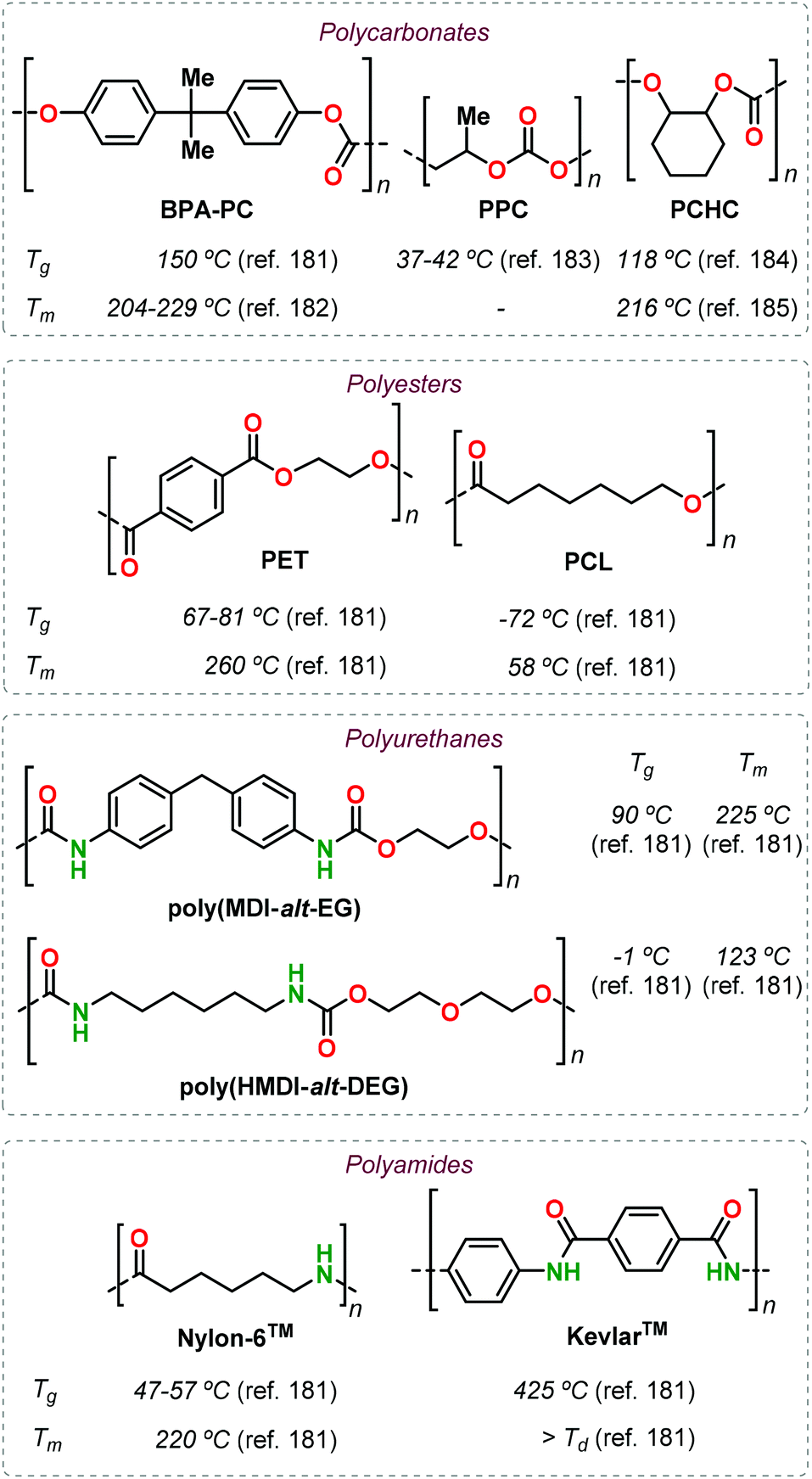 | ||
| Fig. 30 Thermal properties of representative petroleum-based commercial polymers. | ||
The use of LO revealed the possibility to obtain functional polycarbonates with properties similar to that of PCHC in its pristine form, and furthermore can be properly modified post-synthetically to obtain different materials covering a wide range of thermal properties similar to those of PPC and even BPA-PC (Fig. 7). In the case of polyesters, the main driver has so far been the identification of alternatives for PCL. Successful results in this respect have been described starting from different terpenes such as (−)-menthol (Fig. 14), (+)-β-pinene (Fig. 16) and carvone (Fig. 15). Especially in the latter case, the presence of additional functional groups offer the possibility to obtain shape-memory materials that cannot be prepared from PCL. Moreover, owing to the structural rigidity offered by (bi)cyclic terpene scaffolds, it has been possible to obtain polyesters with properties similar to PET (such as in the case of camphor-based polyesters, Fig. 13) or with very high Tg values when combining terpene-based anhydrides and epoxides (Fig. 20 and 21).
The preparation of polyurethanes from terpenes remains largely underexplored, with the only examples known being based on limonene. Nonetheless, the preparation of both poly-hydroxyurethanes (Fig. 23) and polyurethanes (Fig. 24) have been reported featuring properties resembling those of aliphatic PUs such as poly(HMDI-alt-DEG) (Fig. 30) but are still far from those obtained using semi-aromatic PUs (poly(MDI-alt-EG), Fig. 30).
Similar to polyurethanes, few examples of terpene-based polyamides have been reported to date, but polycondensation and ROP approaches have been both considered. Semi-crystalline aliphatic PAs, with thermal properties superior to well-established PAs such as Nylon-6™, have been attained from structurally rigid precursors like nopinone, α-pinene and (+)-3-carene (Fig. 28 and 30). Clearly, the synthesis of bio-based PAs with properties close to those of Kevlar-6™ remains an important goal to achieve.
Concluding, terpenes are typically available on a reasonable scale from inexpensive bio-renewable sources (e.g., pinene and limonene), and do not compete with the food supply chain. The presence of olefin groups gives ample opportunities for chemical modifications such as those realized by addition and epoxidation reactions, providing additional possibilities to forge tailor-made materials with the desired mechanical, thermal, optical, conductive or medicinal features. In addition, exploring further post-functionalisation is facilitated in those cases where more than one type of double bond with different reactivity is embedded in the terpenoid structure. Terpenes additionally offer structural modularity (i.e. they can be linear, cyclic or polycyclic in nature) giving thus potential to use them to fabricate new materials with a wider window of properties than can be realised with most of the conventional monomers currently used in the polymer industry.
This mini-review thus describes the highly attractive nature of terpenes and their versatility in the creation of novel biosourced polymers, and their use in the requisite sustainable materials of the future. The information gathered herein prospectively provides impetus for future research and advances of new terpene-based formulations in the vibrant area of polymer science, and will undoubtedly reveal more, yet unidentified potential of this class of renewable raw materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the CERCA Program/Generalitat de Catalunya, ICREA, the Spanish MINECO (CTQ2017-88920-P) and AGAUR (2017-SGR-232) for financial support. F. D. M. kindly acknowledges the European Community for funding a postdoctoral fellowship through the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 840557.References
- C. M. Rochman, M. A. Browne, B. S. Halpern, B. T. Hentschel, E. Hoh, H. K. Karapanagioti, L. M. Rios-Mendoza, H. Takada, S. Teh and R. C. Thompson, Nature, 2013, 494, 169–171 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2019, 3, e1700782 CrossRef PubMed.
- For more information, please visit: https://www.plasticsoupfoundation.org/en/.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS.
- G. Suaria, C. G. Avio, A. Mineo, G. L. Lattin, M. G. Magaldi, G. Belmonte, C. J. Moore, F. Regoli and S. Aliani, Sci. Rep., 2016, 6, 37551 CrossRef CAS PubMed.
- A. Cózar, F. Echevarría, J. I. González-Gordillo, X. Irigoien, B. Úbeda, S. Hernández-León, Á. T. Palma, S. Navarro, J. García-de-Lomas, A. Ruiz, M. L. Fernández-de-Puelles and C. M. Duarte, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10239–10244 CrossRef PubMed.
- K. L. Law, S. Morét-Ferguson, N. A. Maximenko, G. Proskurowski, E. E. Peacock, J. Hafner and C. M. Reddy, Science, 2010, 329, 1185–1188 CrossRef CAS.
- C.-F. Schleussner, J. Rogelj, M. Schaeffer, T. Lissner, R. Licker, E. M. Fischer, R. Knutti, A. Levermann, K. Frieler and W. Hare, Nat. Clim. Change, 2016, 6, 827–835 CrossRef.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- J. C. Philp, A. Bartsev, R. J. Ritchie, M.-A. Baucher and K. Guy, New Biotechnol., 2013, 30, 635–646 CrossRef CAS PubMed.
- The use of non-biodegradable bags is regulated in the European Community (Directive 2015/720) and banned in Italy (Legge 3 agosto 2017, n. 123).
- The use of bisphenol A in plastic food contact materials is regulated in the European Community by the Commission Regulation (EU) No 10/2011 of 14 January 2011, and Commission Regulation (EU) 2018/213 of 12 February 2018.
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- G.-Q. Chen, Chem. Soc. Rev., 2009, 38, 2434–2446 RSC.
- Z. Li and X. J. Loh, Chem. Soc. Rev., 2015, 44, 2865–2879 RSC.
- C. G. Jaffredo and S. M. Guillaume, Polym. Chem., 2014, 5, 4168–4194 RSC.
- Z. Li, J. Yang and X. J. Loh, NPG Asia Mater., 2016, 8, e265 CrossRef CAS.
- A. Rodriguez-Contreras, Bioengineering, 2019, 6, 82 CrossRef CAS PubMed.
- M. Winnacker, Eur. J. Lipid Sci. Technol., 2019, 121, 1900101 CrossRef CAS.
- M. E. Grigore, R. M. Grigorescu, L. Iancu, R.-M. Ion, C. Zaharia and E. R. Andrei, J. Biomater. Sci., Polym. Ed., 2019, 30, 695–712 CrossRef CAS PubMed.
- Natural Polymers, ed. O. Olatunji, Springer International Publishing, Switzerland, 2016 Search PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- A. Llevot, P.-K. Dannecker, M. von Czapiewski, L. C. Over, Z. Sçyler and M. A. R. Meier, Chem. – Eur. J., 2016, 22, 11510–11521 CrossRef CAS PubMed.
- S. L. Kristufek, K. T. Wacker, Y.-Y. T. Tsao, L. Su and K. L. Wooley, Nat. Prod. Rep., 2017, 34, 433–459 RSC.
- Sustainable Polymers from Biomass, ed. C. Tang and C. Y. Ryu, Wiley-VCH, Weinheim, 2017 Search PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS.
- G. John, S. Nagarajan, P. K. Vemula, J. R. Silverman and C. K. S. Pillai, Prog. Polym. Sci., 2019, 92, 158–209 CrossRef CAS.
- C. Vilela, A. F. Sousa, A. C. Fonseca, A. C. Serra, J. F. J. Coelho, C. S. R. Freire and A. J. D. Silvestre, Polym. Chem., 2014, 5, 3119–3141 RSC.
- Polylactic Acid, ed. L. T. Sin, A. R. Rahmat and W. A. W. A. Rahman, William Andrew Publishing, Oxford, 2012 Search PubMed.
- T. Jiang, Q. Duan, J. Zhu, H. Liu and L. Yu, Adv. Ind. Eng. Polym. Res., 2020, 3, 8–18 Search PubMed.
- J. A. Galbis, M. de Gracia García-Martín, M. V. de Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS PubMed.
- L. Montero de Espinosa and M. A. R. Meier, Eur. Polym. J., 2011, 47, 837–852 CrossRef CAS.
- F. H. Isikgora and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- B. Grignard, S. Gennen, C. Jérôme, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19, 4990–5011 RSC.
- S.-E. Dechent, A. W. Kleij and G. A. Luinstra, Green Chem., 2020, 22, 969–978 RSC.
- P. Sahu, A. K. Bhowmick and G. Kali, Processes, 2020, 8, 553 CrossRef.
- A. W. Kleij, ChemSusChem, 2018, 11, 2842–2844 CrossRef CAS PubMed.
- M. Winnacker, Angew. Chem., Int. Ed., 2018, 57, 14362–14371 CrossRef CAS PubMed.
- K. Satoh, Polym. J., 2015, 47, 527–536 CrossRef CAS.
- M. Winnacker and B. Rieger, ChemSusChem, 2015, 8, 2455–2471 CrossRef CAS PubMed.
- P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8–37 CrossRef CAS PubMed.
- H. Mooibroek and K. Cornish, Appl. Microbiol. Biotechnol., 2000, 53, 355–365 CrossRef CAS PubMed.
- Stereospecific Polymerization of Isoprene, ed. E. Ceausescu, Pergamon Press Ltd, Oxford, 1983 Search PubMed.
- M. J. Shuttleworth and A. A. Watson, Synthetic Polyisoprene Rubbers, in Developments in Rubber Technology-2, ed. A. Whelan and K. S. Lee, Springer, Dordrecht, 1981 Search PubMed.
- T. Higashimura, J. Lu, M. Kamigaito, M. Sawamoto and Y.-X. Deng, Makromol. Chem., 1993, 194, 3455–3465 CrossRef CAS.
- T. Higashimura, J. Lu, M. Kamigaito, M. Sawamoto and Y.-X. Deng, Makromol. Chem., 1993, 194, 3441–3453 CrossRef CAS.
- T. Higashimura, J. Lu, M. Kamigaito, M. Sawamoto and Y.-X. Deng, Makromol. Chem., 1992, 193, 2311–2321 CrossRef CAS.
- M. Ojika, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2017, 56, 1789–1793 CrossRef CAS PubMed.
- B. Sibaja, J. Sargent and M. L. Auad, J. Appl. Polym. Sci., 2014, 131, 41155 CrossRef.
- M. Matsuda, K. Satoh and M. Kamigaito, J. Polym. Sci., Polym. Chem., 2013, 51, 1774–1785 CrossRef CAS.
- A. Singh and M. Kamal, J. Appl. Polym. Sci., 2012, 125, 1456–1459 CrossRef CAS.
- H. Miyaji, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2016, 55, 1372–1376 CrossRef CAS PubMed.
- M. I. Hulnik, I. V. Vasilenko, A. V. Radchenko, F. Peruch, F. Ganachaud and S. V. Kostjuk, Polym. Chem., 2018, 9, 5690–5700 RSC.
- N. Bauer, J. Brunke and G. Kali, ACS Sustainable Chem. Eng., 2017, 5, 10084–10092 CrossRef CAS.
- P. Sarkar and A. K. Bhowmick, ACS Sustainable Chem. Eng., 2016, 4, 5462–5474 CrossRef CAS.
- A. Ávila-Ortega, M. Aguilar-Vega, M. I. Loría Bastarrachea, C. Carrera-Figueiras and M. Campos-Covarrubias, J. Polym. Res., 2015, 22, 226 CrossRef.
- B. Liu, L. Li, G. Sun, D. Liu, S. Li and D. Cui, Chem. Commun., 2015, 51, 1039–1041 RSC.
- J. Hilschmann and G. Kali, Eur. Polym. J., 2015, 73, 363–373 CrossRef CAS.
- S. Georges, A. O. Toure, M. Visseaux and P. Zinck, Macromolecules, 2014, 47, 4538–4547 CrossRef CAS.
- J. M. Bolton, M. A. Hillmyer and T. R. Hoye, ACS Macro Lett., 2014, 3, 717–720 CrossRef CAS.
- J. Raynaud, J. Y. Wu and T. Ritter, Angew. Chem., Int. Ed., 2012, 51, 11805–11808 CrossRef CAS PubMed.
- P. Sahu, P. Sarkar and A. K. Bhowmick, ACS Sustainable Chem. Eng., 2017, 5, 7659–7669 CrossRef CAS.
- M. Naddeo, A. Buonerba, E. Luciano, A. Grassi, A. Proto and C. Capacchione, Polymer, 2017, 131, 151–159 CrossRef CAS.
- D. Peng, G. Du, P. Zhang, B. Yao, X. Li and S. Zhang, Macromol. Rapid Commun., 2016, 37, 987–992 CrossRef CAS PubMed.
- D. H. Lamparelli, V. Paradiso, F. Della Monica, A. Proto, S. Guerra, L. Giannini and C. Capacchione, Macromolecules, 2020, 53, 1665–1673 CrossRef.
- S. B. Luk and M. Marić, Macromol. React. Eng., 2019, 13, 1800080 CrossRef.
- Terpenes: Flavors, Fragrances, Pharmaca, Pheromones, ed. E. Breitmaier, Wiley-VCH, Weinheim, 2006 Search PubMed.
- M. Firdaus, Asian J. Org. Chem., 2017, 6, 1702–1714 CrossRef CAS.
- M. Touaibia, C. Boutekedjiret, S. Perino and F. Chemat, Natural Terpenes as Building Blocks for Green Chemistry, in Plant Based “Green Chemistry 2.0”. Green Chemistry and Sustainable Technology, ed. Y. Li and F. Chemat, Springer, Singapore, 2019 Search PubMed.
- G. Paggiola, S. Van Stempvoort, J. Bustamante, J. M. Vega Barbero, A. J. Hunt and J. H. Clark, Biofuels, Bioprod. Biorefin., 2016, 10, 686–698 CrossRef CAS.
- G. Abts, T. Eckel and R. Wehrmann, Polycarbonates, in Ullmann's Encyclopedia of Industrial Chemistry, 2014 Search PubMed.
- A. Singh, in Encyclopedia of Polymeric Nanomaterials, ed. S. Kobayashi and K. Müllen, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 1–5 Search PubMed.
- E. J. Hoekstra and C. Simoneau, Crit. Rev. Food Sci. Nutr., 2013, 56, 386–402 CrossRef PubMed.
- L. Li, Q. Wang, Y. Zhang, Y. Niu, X. Yao and H. Liu, PLoS One, 2015, 10, e0120330 CrossRef PubMed.
- J. Xu, E. Feng and J. Song, J. Appl. Polym. Sci., 2014, 131, 39822 CrossRef.
- T. Artham and M. Doble, Macromol. Biosci., 2008, 8, 14–24 CrossRef CAS PubMed.
- G. A. Luinstra and E. Borchardt, Adv. Polym. Sci., 2012, 245, 29–48 CrossRef CAS.
- S. H. Lee, A. Cyriac, J. Y. Jeon and B. Y. Lee, Polym. Chem., 2012, 3, 1215–1220 RSC.
- J. Wang, H. Zhang, Y. Miao, L. Qiao, X. Wang and F. Wang, Green Chem., 2016, 18, 524–530 RSC.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460–1479 CrossRef CAS.
- X.-B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS.
- F. Della Monica and A. W. Kleij, Catal. Sci. Technol., 2020, 10, 3483–3501 RSC.
- M. Laska and P. Teubner, Chem. Senses, 1999, 24, 161–170 CrossRef CAS PubMed.
- J. M. Derfer and S. G. Traynor, in Chemistry of Turpentine, ed. D. F. Zinkel and J. Russel, Pulp Chemical Association, New York, 1989, pp. 225–260 Search PubMed.
- M. Gscheidmeier and H. Fleig, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2000, pp. 537–549 Search PubMed.
- J. J. W. Coppen and G. A. Hone, Gum Naval Stores: Turpentine and Rosin from Pine Resin, Food and Agriculture Organization of the United Nations, Rome, Italy, 1995 Search PubMed.
- Y. Nakagawa, M. Tamura and K. Tomishige, Fuel Process. Technol., 2019, 193, 404–422 CrossRef CAS.
- A. J. D. Silvestre and A. Gandini, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Oxford, 2008, pp. 17–38 Search PubMed.
- D. García, F. Bustamante, A. L. Villa, M. Lapuerta and E. Alarcón, Energy Fuels, 2020, 34, 579–586 CrossRef.
- W. Fan, Q. Jia, J. Mu and S. Shan, Method for preparing polycarbonate by copolymerizing carbon dioxide and alpha-pinene derivatives, CN103333329A, 2013 Search PubMed.
- R. Ciriminna, M. Lomeli-Rodriguez, P. Demma Carà, J. A. Lopez-Sanchez and M. Pagliaro, Chem. Commun., 2014, 50, 15288–15296 RSC.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed.
- L. Peña Carrodeguas, J. González-Fabra, F. Castro-Gómez, C. Bo and A. W. Kleij, Chem. – Eur. J., 2015, 21, 6115–6122 CrossRef PubMed.
- C. Martín and A. W. Kleij, Macromolecules, 2016, 49, 6285–6295 CrossRef.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo, W. C. Ellis and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 1215–1218 CrossRef CAS PubMed.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo and G. W. Coates, Macromolecules, 2015, 48, 2534–2550 CrossRef CAS.
- J. Bailer, S. Feth, F. Bretschneider, S. Rosenfeldt, M. Drechsler, V. Abetz, H. Schmalz and A. Greiner, Green Chem., 2019, 21, 2266–2272 RSC.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2015, 18, 760–770 RSC.
- K. N. Gurudutt, S. Rao and P. Srinivas, Flavour Fragrance J., 1992, 7, 343–345 CrossRef CAS.
- D. Darensbourg, Green Chem., 2019, 21, 2214–2223 RSC.
- O. Hauenstein, Md. M. Rahman, M. Elsayed, R. Krause-Rehberg, S. Agarwal, V. Abetz and A. Greiner, Adv. Mater. Technol., 2017, 2, 1700026 CrossRef.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 11862 CrossRef CAS PubMed.
- C. Li, R. J. Sablong and C. E. Koning, Angew. Chem., Int. Ed., 2016, 55, 11572–11576 CrossRef CAS PubMed.
- N. Kindermann, À. Cristòfol and A. W. Kleij, ACS Catal., 2017, 7, 3860–3863 CrossRef CAS.
- C. Li, R. J. Sablong and C. E. Koning, Eur. Polym. J., 2015, 67, 449–458 CrossRef CAS.
- C. Li, S. van Berkel, R. J. Sablong and C. E. Koning, Eur. Polym. J., 2016, 85, 466–477 CrossRef CAS.
- C. Li, M. Johansson, R. J. Sablong and C. E. Koning, Eur. Polym. J., 2017, 96, 337–349 CrossRef CAS.
- T. Stößer, C. Li, J. Unruangsri, P. K. Saini, R. J. Sablong, M. A. R. Meier, C. K. Williams and C. Koning, Polym. Chem., 2017, 8, 6099–6105 RSC.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172–186 RSC.
- C. Li, R. J. Sablong, R. A. T. M. van Benthem and C. E. Koning, ACS Macro Lett., 2017, 6, 684–688 CrossRef CAS.
- F. Parrino, A. Fidalgo, L. Palmisano, L. M. Ilharco, M. Pagliaro and R. Ciriminna, ACS Omega, 2018, 3, 4884–4890 CrossRef CAS PubMed.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS PubMed.
- M. Vert, S. M. Li, G. Spenlehauer and P. Guerin, J. Mater. Sci.: Mater. Med., 1992, 3, 432–446 CrossRef CAS.
- Principles of Polymerization, ed. G. Odian, John Wiley & Sons, Inc., Hoboken, New Jersey, 4th edn, 2004 Search PubMed.
- P. Lecomte and C. Jérôme, Recent Developments in Ring-Opening Polymerization of Lactones, in Synthetic Biodegradable Polymers. Advances in Polymer Science, ed. B. Rieger, A. Künkel, G. Coates, R. Reichardt, E. Dinjus and T. Zevaco, Springer, Berlin, Heidelberg, 2011, vol. 245 Search PubMed.
- Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH Verlag GmbH & Co., Weinheim, 2009 Search PubMed.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- S. Roth, I. Funk, M. Hofer and V. Sieber, ChemSusChem, 2017, 10, 3574–3580 CrossRef CAS PubMed.
- A. Orav, T. Kailas and M. Liiv, Chromatographia, 1996, 43, 215–219 CrossRef CAS.
- Z.-D. Zhao and L.-W. Bi, Biomass Chem. Eng., 2009, 1–8 Search PubMed.
- M. R. Thomsett, J. C. Moore, A. Buchard, R. A. Stockman and S. M. Howdle, Green Chem., 2019, 21, 149–156 RSC.
- O. Nsengiyumva and S. A. Miller, Green Chem., 2019, 21, 973–978 RSC.
- S. Guo, Z. Geng, W. Zhang, J. Liang, C. Wang, Z. Deng and S. Du, Int. J. Mol. Sci., 2016, 17, 1836 CrossRef PubMed.
- D. Ponomarev and H. Mettee, J. Chem. Educ., 2016, 18, 1–4 Search PubMed.
- E. Stirling, Y. Champouret and M. Visseaux, Polym. Chem., 2018, 9, 2517–2531 RSC.
- F. Della Monica, E. Luciano, A. Buonerba, A. Grassi, S. Milione and C. Capacchione, RSC Adv., 2014, 4, 51262–51267 RSC.
- H. T. H. Nguyen, P. Qi, M. Rostagno, A. Feteha and S. A. Miller, J. Mater. Chem. A, 2018, 6, 9298–9331 RSC.
- D. Zhang, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2005, 6, 2091–2095 CrossRef CAS PubMed.
- J. D. Heck, Food Chem. Toxicol., 2010, 48, S1–S38 CrossRef CAS PubMed.
- C. L. Wanamaker, L. E. O'Leary, N. A. Lynd, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2007, 8, 3634–3640 CrossRef CAS PubMed.
- J. Shin, Y. Lee, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2012, 13, 3833–3840 CrossRef CAS PubMed.
- J. R. Lowe, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2009, 10, 2003–2008 CrossRef CAS PubMed.
- J. R. Lowe, M. T. Martello, W. B. Tolman and M. A. Hillmyer, Polym. Chem., 2011, 2, 702–708 RSC.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed.
- H. C. Quilter, M. Hutchby, M. G. Davidson and M. D. Jones, Polym. Chem., 2017, 8, 833–837 RSC.
- A. Stamm, A. Biundo, B. Schmidt, J. Brücher, S. Lundmark, P. Olsén, L. Fogelström, E. Malmström, U. T. Bornscheuer and P.-O. Syrén, ChemBioChem, 2019, 20, 1664–1671 CrossRef CAS PubMed.
- C. Robert, F. de Montigny and C. M. Thomas, Nat. Commun., 2011, 2, 586 CrossRef PubMed.
- L. Fournier, C. Robert, S. Pourchet, A. Gonzalez, L. Williams, J. Prunet and C. M. Thomas, Polym. Chem., 2016, 7, 3700–3704 RSC.
- T. Stößer and C. K. Williams, Angew. Chem., Int. Ed., 2018, 57, 6337–6341 CrossRef PubMed.
- N. J. Van Zee and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 2665–2668 CrossRef CAS PubMed.
- M. J. Sanford, L. Peña Carrodeguas, N. J. Van Zee, A. W. Kleij and G. W. Coates, Macromolecules, 2016, 49, 6394–6400 CrossRef CAS.
- E. H. Nejad, A. Paoniasari, C. G. W. van Melis, C. E. Koning and R. Duchateau, Macromolecules, 2013, 46, 631–637 CrossRef CAS.
- L. Peña Carrodeguas, C. Martín and A. W. Kleij, Macromolecules, 2017, 50, 5337–5345 CrossRef.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- G. Wegener, M. Brandt, L. Duda, J. Hofmann, B. Klesczewski, D. Koch, R.-J. Kumpf, H. Orzesek, H.-G. Pirkl, C. Six, C. Steinlein and M. Weisbeck, Appl. Catal., A, 2001, 221, 303–335 CrossRef CAS.
- A Summary of Health Hazard Evaluations: Issues Related to Occupational Exposure to Isocyanates, 1989 to 2002, Publication No. 2004-116, ed. J. Weber, U. S. National Institute for Occupational Safety and Health, NIOSH Publications Dissemination, Cincinnati, 2004 Search PubMed.
- J. Guan, Y. Song, Y. Lin, X. Yin, M. Zuo, Y. Zhao, X. Tao and Q. Zheng, Ind. Eng. Chem. Res., 2011, 50, 6517–6527 CrossRef CAS.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- F. Della Monica, A. Buonerba and C. Capacchione, Adv. Synth. Catal., 2019, 361, 265–282 CrossRef CAS.
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353 CrossRef.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- A. Cornille, R. Auvergne, O. Figovsky, B. Boutevin and S. Caillol, Eur. Polym. J., 2017, 87, 535–552 CrossRef CAS.
- M. Ghasemlou, F. Daver, E. P. Ivanova and B. Adhikari, Eur. Polym. J., 2019, 118, 668–684 CrossRef CAS.
- M. Bähr, A. Bitto and R. Mülhaupt, Green Chem., 2012, 14, 1447–1454 RSC.
- V. Schimpf, B. S. Ritter, P. Weis, K. Parison and R. Mülhaupt, Macromolecules, 2017, 50, 944–955 CrossRef CAS.
- M. Firdaus and M. A. R. Meier, Green Chem., 2013, 15, 370–380 RSC.
- M. Firdaus, L. M. de Espinosa and M. A. R. Meier, Macromolecules, 2011, 44, 7253–7262 CrossRef CAS.
- P. Radzik, A. Leszczyńska and K. Pielichowski, Polym. Bull., 2020, 77, 501–528 CrossRef CAS.
- M. Winnacker, S. Vagin, V. Auer and B. Rieger, Macromol. Chem. Phys., 2014, 215, 1654–1660 CrossRef CAS.
- N. Komatsu, S. Simizu and T. Sugita, Synth. Commun., 1992, 22, 277–279 CrossRef CAS.
- M. Imoto, H. Sakurai and T. Kono, J. Polym. Sci., 1961, 50, 467–473 CrossRef CAS.
- M. Winnacker, A. Tischner, M. Neumeier and B. Rieger, RSC Adv., 2015, 5, 77699–77705 RSC.
- M. Winnacker, M. Neumeier, X. Zhang, C. M. Papadakis and B. Rieger, Macromol. Rapid Commun., 2016, 37, 851–857 CrossRef CAS PubMed.
- M. Winnacker, J. Sag, A. Tischner and B. Rieger, Macromol. Rapid Commun., 2017, 38, 1600787 CrossRef PubMed.
- M. Winnacker and J. Sag, Chem. Commun., 2018, 54, 841–844 RSC.
- M. Winnacker, A. J. G. Beringer, T. F. Gronauer, H. H. Güngör, L. Reinschlüssel, B. Rieger and S. A. Sieber, Macromol. Rapid Commun., 2019, 40, 1900091 CrossRef PubMed.
- M. Winnacker, D. H. Lamparelli, C. Capacchione, H. H. Güngör, L. Stieglitz, K. S. Rodewald, M. Schmidt and T. F. Gronauer, Macromol. Chem. Phys., 2020, 221, 2000110 CrossRef CAS.
- P. N. Stockmann, D. L. Pastoetter, M. Woelbing, C. Falcke, M. Winnacker, H. Strittmatter and V. Sieber, Macromol. Chem. Phys., 2019, 40, 1800903 Search PubMed.
- P. N. Stockmann, D. Van Opdenbosch, A. Poethig, D. L. Pastoetter, M. Hoehenberger, S. Lessig, J. Raab, M. Woelbing, C. Falcke, M. Winnacker, C. Zollfrank, H. Strittmatter and V. Sieber, Nat. Commun., 2020, 11, 509 CrossRef CAS PubMed.
- Polymer Data Handbook, ed. J. E. Mark, Oxford University Press, Inc., 1999 Search PubMed.
- S. Sohn, A. Alizadeh and H. Marand, Polymer, 2000, 41, 8879–8886 CrossRef CAS.
- G. A. Luinstra, Polym. Rev., 2008, 48, 192–219 CrossRef CAS.
- F. Della Monica, B. Maity, T. Pehl, A. Buonerba, A. De Nisi, M. Monari, A. Grassi, B. Rieger, L. Cavallo and C. Capacchione, ACS Catal., 2018, 8, 6882–6893 CrossRef CAS.
- G.-P. Wu, S.-D. Jiang, X.-B. Lu, W.-M. Ren and S.-K. Yan, Chin. J. Polym. Sci., 2012, 30, 487–492 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |