
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid sodium periodate cleavage of an unnatural amino acid enables unmasking of a highly reactive α-oxo aldehyde for protein bioconjugation†
Robin L.
Brabham‡
 a,
Tessa
Keenan‡
a,
Annika
Husken
a,
Jacob
Bilsborrow
a,
Ryan
McBerney
b,
Vajinder
Kumar
bc,
W. Bruce
Turnbull
b and
Martin A.
Fascione
*a
a,
Tessa
Keenan‡
a,
Annika
Husken
a,
Jacob
Bilsborrow
a,
Ryan
McBerney
b,
Vajinder
Kumar
bc,
W. Bruce
Turnbull
b and
Martin A.
Fascione
*a
aDepartment of Chemistry, University of York, Heslington, YO10 5DD, York, UK. E-mail: martin.fascione@york.ac.uk
bSchool of Chemistry and Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds LS2 9JT, UK
cAkal University, Talwandi Sabo, Punjab, India
First published on 14th May 2020
Abstract
The α-oxo aldehyde is a highly reactive aldehyde for which many protein bioconjugation strategies exist. Here, we explore the genetic incorporation of a threonine-lysine dipeptide into proteins, harbouring a “masked” α-oxo aldehyde that is rapidly unveiled in four minutes. The reactive aldehyde could undergo site-specific protein modification by SPANC ligation.
Genetic code expansion has revolutionised our ability to site-selectively install unnatural functionality into proteins.1 The use of the pyrrolysine (Pyl) tRNACUA/pyrrolysyl-tRNA synthetase (RS) pair has proven to be a highly successful platform for this purpose, with the introduction of reactive handles such as azides,2 alkenes3–5 and alkynes6–8 into proteins, opening up a wide range of site-specific chemical bioconjugation strategies. The aldehyde is a particularly versatile functional group owing to its unique reactivity, stability and relatively low abundance in nature, however few examples of the site-specific installation of aldehydes into proteins by amber stop codon suppression have been reported, despite the many bioorthogonal methodologies available that utilise these functional handles.9 Notably, through the use of a mutant Pyl tRNA-RS pair from Methanocaldococcus janaschii, an aldehyde-containing phenylalanine analogue was previously shown to be incorporated into a protein, facilitating modification by oxime ligation.10 This finding breathed fresh life into well-established protein carbonyl chemical modification, which had been hampered by the limitations on the positioning of the required reactive aldehydes. Previous methods to install protein aldehydes required an enzymatic tag, such as the use of FGE11 or an exposed N-terminal serine, threonine or glycine residue;9 through the use of amber stop codon suppression, site-specific protein aldehyde modification could now move beyond such sequence limitations.
Aldehydes can differ vastly in their electronic properties, dictated by the method used for their installation.9 The α-oxo aldehyde is a highly reactive aldehyde for which many reliable protein bioconjugation methodologies have been established.9,12 Previously restricted to the N-terminus of a protein and requiring an exposed seryl, threonyl or glycyl residue for its formation, the site-specific incorporation of a thiazolidine-protected α-oxo aldehyde into a protein was recently demonstrated.13 We recently reported a biocompatible method of unmasking a genetically encoded thiazolidine-protected α-oxo aldehyde in a protein, using stoichiometric allylpalladium(II) chloride.14 However, this procedure requires some optimisation for individual proteins, in order to balance the reactivity of the palladium complex to open the thiazolidine with potential side-reactions including protein precipitation. In this work, we explore an alternative route to the site-specific installation of a α-oxo aldehyde, through the genetic incorporation of a threonine-lysine derivative, and its rapid unmasking within four minutes (Fig. 1). The reactivity of the α-oxo aldehyde in site-selective protein modification was subsequently demonstrated by Strain-Promoted Alkyne-Nitrone Cycloaddition (SPANC) ligation.
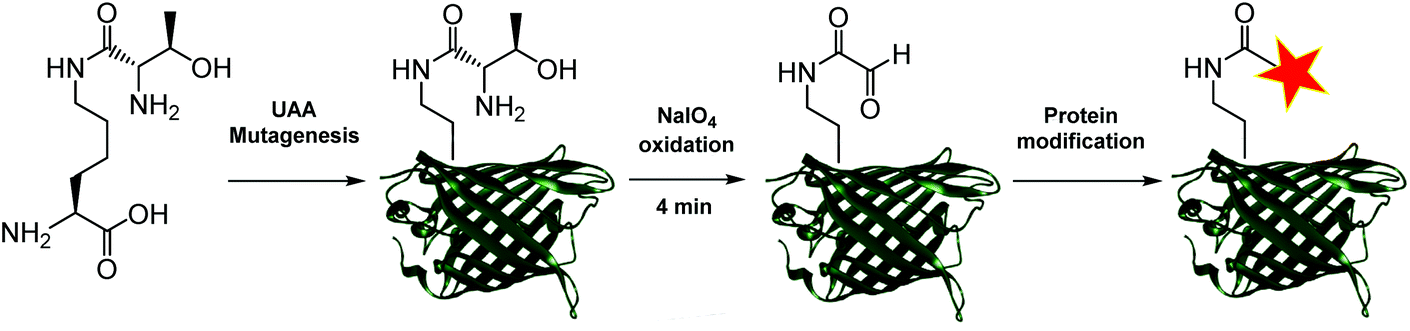 | ||
| Fig. 1 New route for the site-selective installation of a α-oxo aldehyde into proteins. | ||
To generate a protein α-oxo aldehyde, we explored the periodate-mediated cleavage of a genetically encoded ε-lysine dipeptide harbouring a 1,2-amino alcohol motif, arising from a serine or threonine residue. In order to maximise the chances of discovering a suitable substrate for amber stop codon suppression, four dipeptides (1–4) were synthesised, differing in the absence/presence of a β-methyl group (serine/threonine respectively) and the α-configuration (S/R, “natural” vs. “unnatural” respectively) (Fig. 2). The modulation of α-configuration and β-methyl groups was considered to offer insight into the amber stop codon suppression process, with these changes subtly altering amino acid polarity, steric bulk, and positioning within the pylS active site. An activation-coupling-deprotection strategy was used to synthesise all four dipeptides in three steps with cumulative yields of 39% (1), 50% (2), 32% (3) and 56% (4) respectively (Schemes S1 and S2, ESI†). Considering that the promiscuity of the wild type M. mazei Pyl tRNA-RS pair had been shown to extend to several lysine dipeptides,14–16 we chose to investigate this pair for the genetic incorporation of our dipeptides 1–4. To screen the suitability of the dipeptides 1–4 as substrates for the M. mazei Pyl tRNA-RS pair, an expression trial using EGFP(Y39TAG)-His6 as a reporter protein was carried out and the samples were analysed by SDS-PAGE (Fig. 2). EGFP(Y39TAG), containing an amber mutation at surface-exposed position Y39,17 was selected due to its ease of visualisation and highly optimised expression system for use with unnatural amino acid (UAA) mutagenesis. The known Pyl tRNA-RS pair substrate Nε-propargyloxycarbonyl-L-lysine 5 served as a positive control for the expression trial.18 As expected, a thick band corresponding to the overexpression of EGFP was observed in the positive control lane 5, at the expected molecular mass of ∼28 kDa. Interestingly, only the Thr-Lys dipeptide 3 appeared to be genetically incorporated into EGFP as demonstrated by a band of overexpression observed at ∼28 kDa, albeit far less intense than the corresponding band in the positive control 5. To further validate these observations, the cell lysates were observed under fluorescent light (Fig. S1, ESI†). As expected, strong fluorescence was observed in the positive control 5 and mild fluorescence in Thr-Lys dipeptide 3 cell lysates respectively, while no fluorescence was observed in all other cell lysates. Genetic incorporation of dipeptides 1–4 into EGFP was also attempted with the Pyl tRNA-RS pair Y306A Y384F mutant,19 however none of the dipeptides were successfully incorporated (data not shown). A possible explanation for a threonine derivative being a superior PylRS substrate to a serine derivative, is that the extra methyl group can better occupy the hydrophobic space within the substrate binding pocket.20
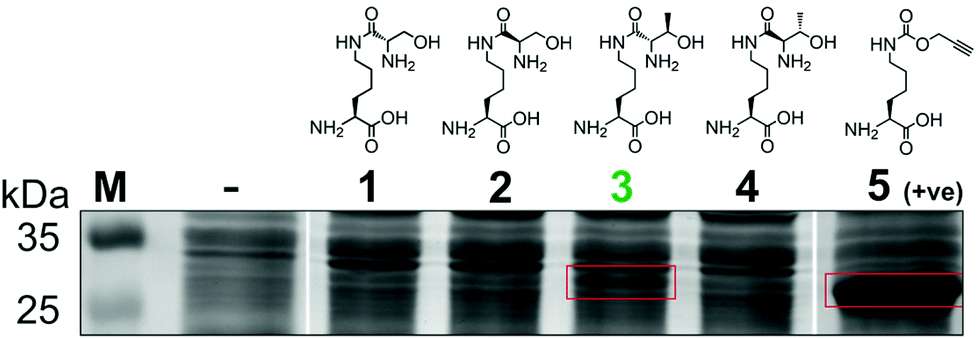 | ||
| Fig. 2 Screening of lysine dipeptides 1–4 for their incorporation into EGFP at position Y39 by the M. mazei Pyl tRNA-RS pair and analysis by SDS-PAGE. The full uncropped SDS-PAGE gel is included in the ESI (Fig. S7†). | ||
Following the identification of a suitable dipeptide substrate for the M. mazei Pyl tRNA-RS pair to facilitate the site-specific installation of a α-oxo aldehyde into a protein, the Thr-Lys dipeptide 3 was also introduced into sfGFP at the surface exposed position N150,21 using a method of amber stop codon suppression adapted for GFP expression.14 The cells were cultivated in Terrific Broth medium supplemented with 0.02% arabinose and 1.5 mM of Thr-Lys dipeptide 3, promoting the expression of the full-length sfGFP(N150ThrK)-His6 protein 6. Following purification by Ni2+ affinity chromatography, the purity and molecular mass of 6 was validated by SDS-PAGE and ESI-FTICR-MS (Fig. S2 and S3, ESI†). Following the successful incorporation of the Thr-Lys dipeptide 3, periodate-mediated oxidation of the 1,2-aminoalcohol motif in the threonine residue was explored (Fig. 3). Whilst periodate-mediated oxidation of 1,2-aminoalcohols is generally fast, periodate will also oxidise other amino acid residues in proteins, including cysteine, methionine, tryptophan, tyrosine, and histidine, albeit more slowly and dependent on the experimental conditions.12,22 However, protein over-oxidation can be avoided by performing the reaction at neutral pH, controlling the reaction stoichiometry and minimising the reaction time. Additionally, excess methionine or ethylene glycol should be added either during or after the reaction, to quench the unreacted periodate.12 Based on conditions previously optimised for oxidising an N-terminal serine residue in an EGFP mutant,23 oxidation was first attempted in phosphate buffered saline (PBS) using 3, 5 and 10 eq. of NaIO4, and 6, 10 and 20 eq. of L-methionine, respectively (Fig. 3a). Oxidation reactions were allowed to proceed for 4–10 min, after which samples were desalted and the amount of oxidised protein determined after analysis by ESI-FTICR-MS. The addition of 3 eq. of NaIO4 and 6 eq. of L-methionine led to ∼59% conversion in 4 min. Attempts to increase this conversion were largely unsuccessful. Longer reaction times and greater eq. of NaIO4 and L-methionine led to no improvement in conversion, and in some cases undesired off-target oxidation of the protein was observed. Reviewing literature protocols led to closer inspection of the buffer composition used. Sodium phosphate buffers are commonplace, as is the use of NaCl, but potassium ions are seldom encountered. One hypothesis is that potassium cations may interact with the periodate anion and precipitate as potassium periodate, poorly soluble in water (ca. 8 mM) at the typical 0 °C of periodate oxidation, as was observed in early applications of periodate oxidation.24 Given that periodate was only used in 5 eq., even minor precipitation of periodate could noticeably affect the extent and rate of oxidation. With this consideration in mind, the oxidation was repeated in a potassium-free system using 20 mM sodium phosphate (PB), 150 mM NaCl buffer pH 7.4 with 5 eq. of NaIO4 and 10 eq. of L-methionine, for 4 min. These conditions resulted in complete oxidation of 6 to 7 (Fig. 3b). In this example 7 exists almost completely as the hydrate. The effect of removing potassium, a seemingly innocuous buffer component, is easily overlooked yet remarkably profound here.
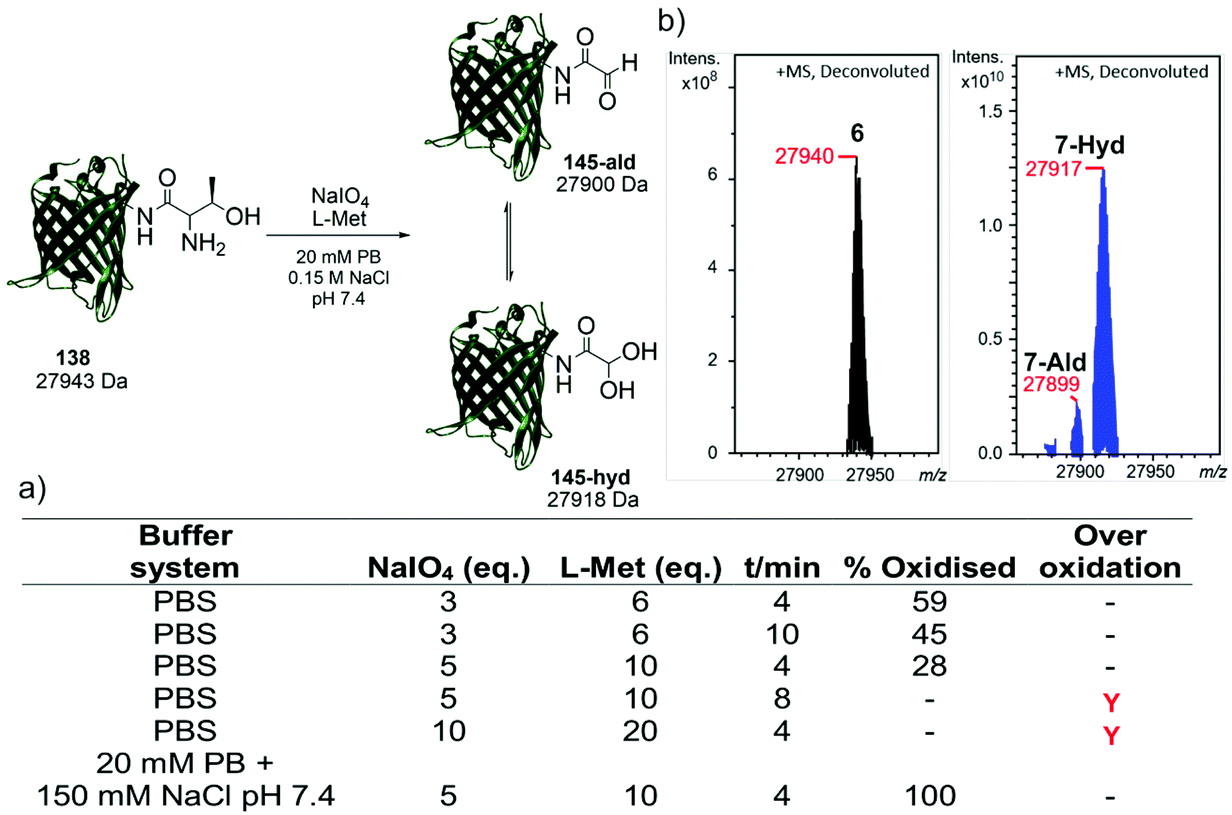 | ||
| Fig. 3 (a) Optimisation of oxidation conditions to unmask the α-oxo aldehyde in sfGFP(N150ThrK)-His66. % oxidised product was determined by ESI-FTICR-MS analysis (b) deconvoluted ESI-FTICR mass spectrum showing the complete oxidation of 6 (left) to form 7 (right) (experimental masses shown). | ||
To demonstrate the reactivity of the newly exposed α-oxo aldehyde, SPANC ligation was explored. SPANC ligation is a one-pot modification strategy in which a nitrone, formed in situ from a protein glyoxyl aldehyde and an N-substituted hydroxylamine, undergoes a [3 + 2] cycloaddition with a strained alkyne, typically a cyclooctyne or a BCN.25,26 SPANC ligations are generally performed at pH 6.8 in NH4OAc buffer, with the addition of p-anisidine which has been shown to enhance the rate of the SPANC ligation.26 SPANC ligation was used to modify 7 with a biotin bicyclononyne (BCN) probe 8, to afford biotinylated protein 9. Complete conversion was observed within 18 h, as determined by ESI-FTICR-MS (Fig. 4a). The successful incorporation of the biotin in probe 9 was further validated by western blotting (Fig. 4c). To further showcase the utility of the unmasked α-oxo aldehyde in 7, the protein was also fluorescently labelled by SPANC ligation using dansyl BCN probe 10, to afford dansylated protein 11. Again, complete conversion was validated by ESI-FTICR-MS (Fig. 4b) and the successful incorporation of the dansyl group into 11 was visualised through denatured protein in-gel fluorescence (Fig. 4c). The unmasked protein α-oxo aldehyde has been successfully shown to undergo bioconjugation by SPANC, which was previously only accessible on N-terminal α-oxo aldehydes.
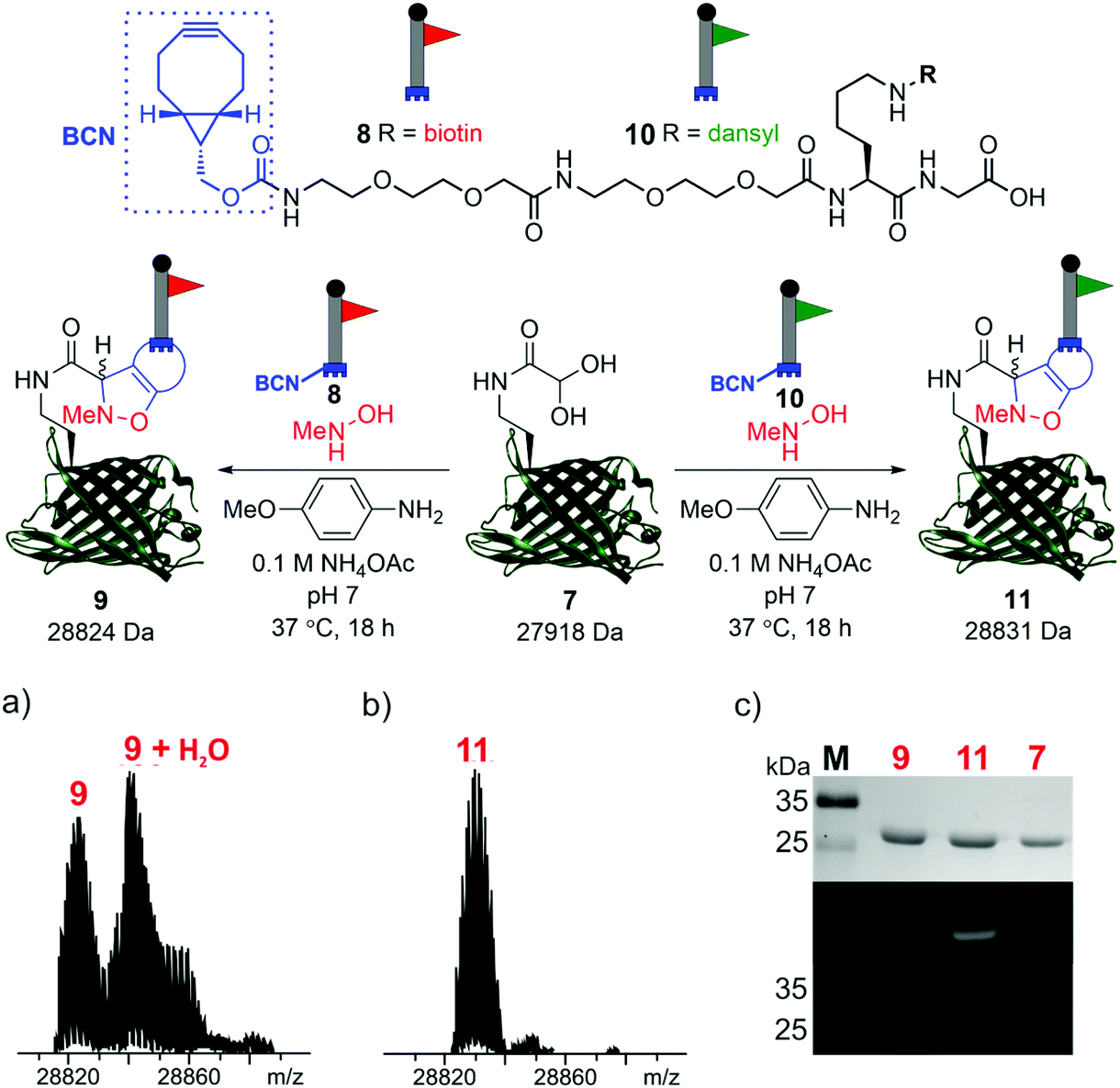 | ||
| Fig. 4 SPANC ligation of protein aldehyde 7 with BCN biotin probe 8 or BCN dansyl probe 10 (top). In both cases, ESI-FTICR-MS data show the complete consumption of 7-ald to afford biotinylated protein 9 (a) (calc. 28822 Da, found 28824 Da) and dansylated protein 11 (b) (calc. 28829 Da, found 28831 Da). (c) Protein modification validated by Coomassie-stained SDS-PAGE (upper) and protein in-gel fluorescence (middle, 11) or western blotting with an anti-biotin probe (lower, 9). The uncropped SDS-PAGE gels and western blot are shown in the ESI (Fig. S9†). | ||
In summary, we have shown the genetic incorporation of a new NCAA 3 into proteins, harbouring a “masked” α-oxo aldehyde, by using the highly promiscuous M. mazei Pyl tRNA-RS pair. The NCAA 3 was produced by using a straightforward three-step synthesis. We have demonstrated the rapid unmasking of the highly reactive α-oxo aldehyde by using NaIO4 oxidation within 4 min and demonstrated its utility in protein bioconjugation by using SPANC ligation to confer avidin affinity or fluorescent functionality, through the use of reactive probes bearing biotin or dansyl groups. This work has shown that SPANC bioconjugations are not limited in scope to the N-terminus, removing a heavy restriction on the use of this chemistry. The “internal α-oxo aldehyde” exhibits comparable reactivity to an N-terminal α-oxo aldehyde and presents new opportunities for site-selective protein modification beyond the protein N-terminus by a range of bioconjugation strategies including hydrazone ligation, thiazolidine ligation, oxime ligation and organocatalyst-mediated protein aldol ligation (OPAL) chemistry.12,23
Conflicts of interest
There are no conflicts of interests.References
- A. Dumas, L. Lercher, C. D. Spicer and B. G. Davis, Chem. Sci., 2015, 6, 50–69 RSC.
- Z. A. Wang, Y. Kurra, X. Wang, Y. Zeng, Y. J. Lee, V. Sharma, H. Lin, S. Y. Dai and W. R. Liu, Angew. Chem., Int. Ed., 2017, 56, 1643–1647 CrossRef CAS PubMed.
- Y. Kurra, K. A. Odoi, Y.-J. Lee, Y. Yang, T. Lu, S. E. Wheeler, J. Torres-Kolbus, A. Deiters and W. R. Liu, Bioconjugate Chem., 2014, 25, 1730–1738 CrossRef CAS PubMed.
- J. Li, S. Jia and P. R. Chen, Nat. Chem. Biol., 2014, 10, 1003 CrossRef CAS PubMed.
- K. Lang, L. Davis, J. Torres-Kolbus, C. Chou, A. Deiters and J. W. Chin, Nat. Chem., 2012, 4, 298 CrossRef CAS PubMed.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed.
- Y.-S. Wang, X. Fang, A. L. Wallace, B. Wu and W. R. Liu, J. Am. Chem. Soc., 2012, 134, 2950–2953 CrossRef CAS PubMed.
- S. Wallace and J. W. Chin, Chem. Sci., 2014, 5, 1742–1744 RSC.
- R. J. Spears and M. A. Fascione, Org. Biomol. Chem., 2016, 14, 7622–7638 RSC.
- A. Tuley, Y.-J. Lee, B. Wu, Z. U. Wang and W. R. Liu, Chem. Commun., 2014, 50, 7424–7426 RSC.
- I. S. Carrico, B. L. Carlson and C. R. Bertozzi, Nat. Chem. Biol., 2007, 3, 321–322 CrossRef CAS PubMed.
- O. a. El-Mahdi and O. Melnyk, Bioconjugate Chem., 2013, 24, 735–765 CrossRef CAS PubMed.
- X. Bi, K. K. Pasunooti, J. Lescar and C.-F. Liu, Bioconjugate Chem., 2017, 28, 325–329 CrossRef CAS PubMed.
- R. L. Brabham, R. J. Spears, J. Walton, S. Tyagi, E. A. Lemke and M. A. Fascione, Chem. Commun., 2018, 54, 1501–1504 RSC.
- X. Li, T. Fekner, J. J. Ottesen and M. K. Chan, Angew. Chem., Int. Ed., 2009, 48, 9184–9187 CrossRef CAS.
- D. P. Nguyen, T. Elliott, M. Holt, T. W. Muir and J. W. Chin, J. Am. Chem. Soc., 2011, 133, 11418–11421 CrossRef CAS PubMed.
- T. Plass, S. Milles, C. Koehler, C. Schultz and E. A. Lemke, Angew. Chem., Int. Ed., 2011, 50, 3878–3881 CrossRef CAS PubMed.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia and X. Zhang, Nat. Chem., 2014, 6, 352 CrossRef CAS.
- T. Yanagisawa, R. Ishii, R. Fukunaga, T. Kobayashi, K. Sakamoto and S. Yokoyama, Chem. Biol., 2008, 15, 1187–1197 CrossRef CAS PubMed.
- J. M. Kavran, S. Gundllapalli, P. O'Donoghue, M. Englert, D. Söll and T. A. Steitz, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11268–11273 CrossRef CAS PubMed.
- S. J. Miyake-Stoner, C. A. Refakis, J. T. Hammill, H. Lusic, J. L. Hazen, A. Deiters and R. A. Mehl, Biochemistry, 2010, 49, 1667–1677 CrossRef CAS PubMed.
- H. Dixon, Biochem. J., 1962, 83, 91 CrossRef CAS PubMed.
- R. J. Spears, R. L. Brabham, D. Budhadev, T. Keenan, S. McKenna, J. Walton, J. A. Brannigan, A. M. Brzozowski, A. J. Wilkinson and M. Plevin, Chem. Sci., 2018, 9, 5585–5593 RSC.
- F. Brown, S. Dunstan, T. Halsall, E. Hirst and J. Jones, Nature, 1945, 156, 785–786 CrossRef CAS.
- R. P. Temming, L. Eggermont, M. B. van Eldijk, J. C. van Hest and F. L. van Delft, Org. Biomol. Chem., 2013, 11, 2772–2779 RSC.
- X. Ning, R. P. Temming, J. Dommerholt, J. Guo, D. B. Ania, M. F. Debets, M. A. Wolfert, G. J. Boons and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 3065–3068 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob00972e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |