
An interlayer composed of a porous carbon sheet embedded with TiO2 nanoparticles for stable and high rate lithium–sulfur batteries†
Yushan
Jiang‡
a,
Yaqian
Deng‡
a,
Bin
Zhang‡
a,
Wuxing
Hua
b,
Xinliang
Wang
a,
Qi
Qi
a,
Qiaowei
Lin
a and
Wei
Lv
 *a
*a
aShenzhen Key Laboratory for Graphene-based Materials, Engineering Laboratory for Functionalized Carbon Materials, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen, 518055, China. E-mail: lv.wei@sz.tsinghua.edu.cn
bNanoyang Group, State Key Laboratory of Chemical Engineering, School of Chemical Engineering and Technology, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin University, Tianjin 300072, China
First published on 12th May 2020
Abstract
The shuttling of lithium polysulfides (LiPSs) in lithium–sulfur (Li–S) batteries results in low sulfur utilization and fast capacity decay, hindering their practical applications. Constructing an interlayer is an efficient way to block the LiPS shuttling, but maintaining a low Li ion diffusion resistance with such an interlayer is hard to achieve. Herein, a thin porous carbon nanosheet embedded with TiO2 nanoparticles (denoted PCNS-TiO2) was used to fabricate an interlayer on the separator, which effectively solves the above problem. The PCNS-TiO2 was prepared by using the Ti3C2Tx MXene as the two-dimensional (2D) template directing the porous carbon sheet formation, and the Ti3C2Tx transformed into TiO2 nanoparticles embedded in the PCNS. The decomposition of the MXene eliminates the ion blocking effect by the 2D nanosheet structure. The thin and hierarchical porous structure allows fast Li ion diffusion across the interlayer, and at the same time, the porous structure and the strong adsorption ability of TiO2 effectively block the polysulfide diffusion. Thus, the Li–S battery with this interlayer shows good rate performance with a high capacity of 627 mA h g−1 at 2 C. Meanwhile, stable cycling performance is also achieved, showing a low capacity decay of 0.063% per cycle after 300 cycles at 0.5 C.
Introduction
Lithium–sulfur (Li–S) batteries are promising next-generation batteries due to the low cost, environmental friendliness, and the high theoretical capacity of sulfur cathodes.1 However, the insulating nature of sulfur and the shuttling of lithium polysulfides (LiPSs) during the charging–discharging process result in low sulfur utilization and fast capacity decay, hindering their practical applications.2,3 Many efforts have been made to solve those problems.4–6 Carbon materials, such as porous carbon,7–9 carbon nanotubes (CNTs) and graphene (GN),10–14 have been widely used as the sulfur host due to their good conductivity and large surface area, which can improve the sulfur utilization and physically restrain the LiPS shuttling. However, even with a complicated carbon structure, it is still hard to efficiently suppress the shuttling of LiPSs due to the weak interaction of polar LiPSs with the nonpolar carbon surface.15,16 Polar noncarbon materials with a large surface area, such as MXenes with a two-dimensional (2D) structure, show strong adsorption ability towards LiPSs and are widely investigated in Li–S batteries.17–21 But the less porous structure lowers the efficiency of suppressing the LiPS shuttling. Recently, it is shown that the cycling performance of Li–S batteries can be significantly improved via an interlayer design that is placed between the cathode and anode or on the separator,22–25 and carbonaceous interlayers have drawn extensive attention as they not only intercept the LiPS migration but also act as a conductive vice-electrode enabling the reuse of trapped active materials.26–29However, some crucial issues remain to be solved for these carbon-based interlayers. First, carbonaceous interlayers usually show weak chemical interaction with LiPSs due to their nonpolar carbon surface. As a result, the polar metal oxides and sulfides are always composited with carbons to enhance the trapping ability toward LiPSs, which leads to a complicated preparation process.30,31 Second, to enhance the blocking efficiency toward LiPS migration, complicated porous and tightly stacked structures are always used. For example, carbons with a 2D or sheet-like structure, such as GN, are very suitable to build a tight interlayer coating on the separator owing to their planar structure, which can effectively block the LiPS diffusion.32,33 However, the ions cannot vertically pass through the planar GN sheet, and this certainly impedes the Li ion diffusion in the charging–discharging process.34 Thus, it is urgent to design a carbon interlayer with strong LiPS trapping ability and low Li ion diffusion resistance for Li–S batteries.
Herein, we prepare a porous carbon nanosheet embedded with TiO2 nanoparticles (NPs) (denoted PCNS-TiO2) and fabricate an interlayer coating on the separator, which well balances the above two points and effectively improves the cycling stability and rate performance of Li–S batteries. The PCNS-TiO2 is synthesized via a hydrothermal treatment of the mixture of Ti3C2Tx MXene nanosheets (NSs) and glucose and following a heat treatment. Ti3C2Tx MXene is used as the 2D template directing the porous carbon sheet formation and decomposes into TiO2 NPs after the heat treatment. The decomposition of the MXene eliminates the ion blocking effect by the 2D NSs mentioned above. At the same time, this process leads to a tight contact between the formed TiO2 and carbon framework, ensuring fast electron transfer for the conversion of LiPSs trapped by TiO2. These 2D carbon NSs are very thin and have a hierarchical porous structure, facilitating the diffusion of Li ions. Besides, they can still effectively confine the polysulfides due to the combination of chemical trapping and physical adsorption abilities by TiO2 and the rich porous structure in the PCNS. Thus, the Li–S battery with this interlayer shows good rate performance and a high capacity of 627 mA h g−1 at 2 C. Meanwhile, high cycling stability is also achieved with a capacity of 718 mA h g−1 after 300 cycles at 0.5 C, showing a low capacity decay of 0.063% per cycle. Even with a higher sulfur loading of 3 mg cm−2, stable cycling performance with a capacity decay of 0.139% per cycle is still achieved for 300 cycles at 0.3 C.
Experimental methods
Preparation process of PCNS-TiO2, TiO2 and PCS
Ti3C2Tx MXene was prepared by etching Ti3AlC2 with a mixture of lithium fluoride (LiF) and hydrochloric acid (HCl).35 The PCNS-TiO2 was prepared through the following process. First, the Ti3C2Tx colloidal solution (1 mg mL−1) was sonicated for 1.5 h. Afterwards, 2 g thiourea and 800 mg glucose were successively dissolved into 20 mL Ti3C2Tx solution under vigorous stirring. Then, the above solution was transferred into a 100 mL Teflon-lined autoclave and then subjected to hydrothermal treatment at 160 °C for 24 h. Then, the obtained product was thoroughly washed with alcohol and water, followed by freeze-drying for 48 h. Finally, PCNS-TiO2 was obtained by annealing the above product at 900 °C for 4 h under an Ar atmosphere. TiO2 was prepared through the same process without the addition of glucose. The porous carbon sphere (denoted as PCS) was prepared through the same process without the addition of Ti3C2Tx.Preparation of the interlayers
30 mg PCNS-TiO2 powder was dispersed into 100 mL N-methyl pyrrolidone (NMP) under sonication for 3 h. Then, the PCNS-TiO2 dispersion was filtered on a commercial polypropylene (PP) separator (Celgard 2400) to form the PCNS-TiO2 coated separator. The modified separator was punched into small discs with a diameter of 19 mm followed by drying at 60 °C for 0.5 h. The TiO2 and PCS coated separators were prepared by the same process. These coatings have the same mass loading of 0.23 mg cm−2.Materials characterization
A field emission scanning electron microscope (FE-SEM, HITACHI SU8010) and a transmission electron microscope (TEM, FEI Tecnai G2 F30) were used to characterize the microstructure of the above products. X-ray diffraction (XRD) patterns were obtained using a Bruker D8 Advance diffractometer with Cu Kα radiation. Thermogravimetric analysis (TG) was conducted on a TG/DTA7300 thermogravimetric/differential thermal analyser in the atmosphere from room temperature to 900 °C with a heating rate of 10 °C min−1. N2 adsorption isotherms were obtained on a BEL mini instrument at 77 K. The specific surface area (SSA) of the product was calculated with the Brunauer–Emmett–Teller (BET) method according to the N2 adsorption isotherm. Pore size distribution (PSD) was calculated with the density functional theory (DFT) method. X-ray photoelectron spectroscopy (XPS, PHI5000VersaProbeII) with a monochromatic Al Kα source was used to obtain the XPS spectrum.Static adsorption test toward LiPSs
The Li2S6 solution (0.5 M) was prepared by mixing Li2S and sulfur with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 in DOL/DME solvent (1:1 by volume) followed by continuous stirring at 70 °C for 24 h. The 0.5 M Li2S6 solution was then diluted into 0.5 mM with a mixture of DOL/DME solvent (1:1 by volume). The static adsorption test was conducted by adding 5 mg PCNS-TiO2, TiO2 and PCS into 2 mL Li2S6 solution (0.5 mM), respectively.
:5 in DOL/DME solvent (1:1 by volume) followed by continuous stirring at 70 °C for 24 h. The 0.5 M Li2S6 solution was then diluted into 0.5 mM with a mixture of DOL/DME solvent (1:1 by volume). The static adsorption test was conducted by adding 5 mg PCNS-TiO2, TiO2 and PCS into 2 mL Li2S6 solution (0.5 mM), respectively.
Symmetric cell assembly and measurements
The PCNS-TiO2 electrodes of the symmetric cell were prepared by dispersing PCNS-TiO2 into ethanol through sonication for 2 h followed by dropping the obtained dispersion on carbon fiber papers (CPs) with a diameter of 12 mm. The CPs loaded with PCNS-TiO2 were then dried at 60 °C. The TiO2 and PCS electrodes were prepared through the same process with the same mass loading of 0.8–1 mg cm−2. The symmetric cells were assembled with the above electrodes and 40 μL Li2S6 solution as the electrolyte (0.5 M). The cyclic voltammetry (CV) measurement of the symmetric cells was conducted at a scan rate of 1 mV s−1 in the voltage range of −0.8 to 0.8 V on a Princeton electrochemical workstation.Asymmetric Li–S battery assembly and measurements
The PCNS-TiO2/S, TiO2/S and PCS/S hybrids (sulfur content: 70 wt%) were prepared by the melt-diffusion method using PCNS-TiO2, TiO2 and PCS as the sulfur host. The mixture of the hybrid, multi-walled carbon nanotubes (MWCNTs) and polyvinylidene fluoride (PVDF) with a mass ratio of 6:2:2 in NMP was stirred overnight and coated on a piece of aluminum foil followed by drying at 60 °C for 12 h to prepare the cathodes (sulfur loading: 0.7–0.8 mg cm−2). An asymmetric CR2032 Li–S battery was assembled with the above cathode, a Li foil anode and 40 μL electrolyte. The Li–S battery electrolyte consists of DOL/DME (1:1 by volume) solvent, LiTFSI (1 M) and LiNO3 (1 wt%). The CV measurement of the above Li–S batteries was conducted at a scan rate of 0.05 mV s−1 in the voltage range of 1.7 to 2.8 V (vs. Li+/Li).
Li2S precipitation experiments
0.2 M Li2S8 solution was prepared by mixing Li2S and sulfur with a molar ratio of 1:7 in tetraglyme followed by continuous stirring at 70 °C for 24 h. The electrode preparation process is the same as that for a symmetric cell electrode. The cell was assembled using the above electrode as the cathode, Li foil as the anode, 15 μL Li2S8 (0.2 M) solution as the electrolyte on the cathode and 15 μL blank electrolyte on the anode side in a CR2032 coin cell. The cell was galvanostatically discharged at 0.134 mA to 2.06 V and then potentiostatically discharged at 2.05 V until the current dropped below 10−5 A. The galvanostatic and potentiostatic discharge tests were performed on a Princeton electrochemical workstation.
LiPS permeation measurement
LiPS permeation measurement was tested with an H-shaped glass cell. First, a PCNS-TiO2 coated separator was placed in the middle of the H-shaped glass cell to separate the two tubes on two sides. Afterward, 12 mL Li2S6 solution (1 mM) was added into the left tube and 12 mL blank solvent (DOL/DME, 1:1 by volume) was added into the right tube.
Li–S battery assembly and electrochemical performance measurements
(1) For the electrochemical impedance spectrum (EIS) measurement, the GN/S hybrid (sulfur content: 70 wt%) prepared by the melt-diffusion method was used as the cathode material, and the battery was assembled with the GN/S cathode (sulfur loading: 1–1.2 mg cm−2), the separator with the interlayer and 50 μL electrolyte. The EIS measurement was conducted in the scan frequency range of 10 mHz to 100 kHz on a Princeton electrochemical workstation. The galvanostatic charge–discharge test was conducted in the voltage range of 1.7 to 2.8 V (vs. Li+/Li). (2) For the cycling performance measurement, the CMK-3/S hybrid (sulfur content: 70 wt%) was prepared as the cathode material. The Li–S batteries were assembled with CMK-3/S cathodes (sulfur loading: 1.6 and 3.0 mg cm−2), the separators with the interlayers and lithium foil anodes in the CR2032 coin cells. The electrolyte/sulfur (E/S) ratio is 15:1. The cycling performance measurement was conducted in the voltage range of 1.7 to 2.8 V (vs. Li+/Li) under the current densities of 0.5 C and 0.3 C, respectively, with a Land 2001A battery testing system.
Results and discussion
The preparation process of PCNS-TiO2 is schematically shown in Fig. 1. First, thiourea and glucose were added into the Ti3C2Tx dispersion to form a uniform mixture, and then, the mixture underwent a hydrothermal treatment at 160 °C. During this process, the hydrothermal carbonization of glucose occurred on the Ti3C2Tx NS surface, forming a sandwich structure with the Ti3C2Tx NS in the middle, and the product was denoted as CNS-Ti3C2Tx. The formation of the sandwich structure should be ascribed to the strong interaction of glucose with the functional groups on the Ti3C2Tx NS surface. The addition of thiourea prevents the oxidation of Ti3C2Tx and helps maintain the NS structure during this process. Then, a heat treatment at 900 °C for 4 h was conducted under an Ar atmosphere, and in this process, the hierarchical porous carbon NSs were formed, and Ti3C2Tx was oxidized to TiO2 NPs embedded in the middle. In contrast, the PCS was obtained without the Ti3C2Tx template under the same conditions, and only TiO2 NPs can be obtained only with Ti3C2Tx.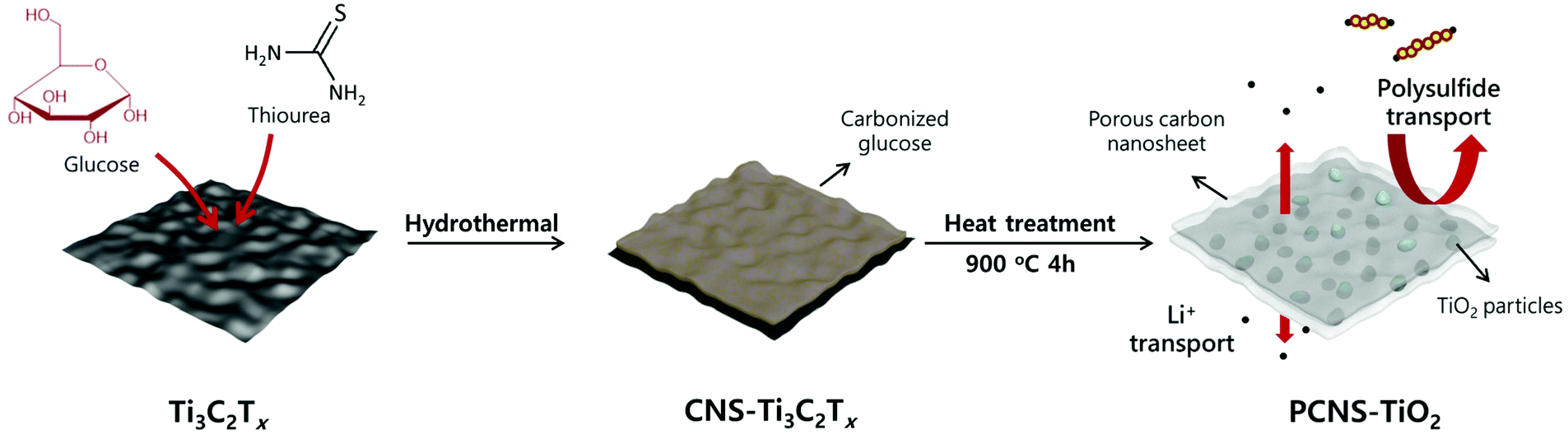 | ||
| Fig. 1 Scheme of the preparation process of PCNS-TiO2. | ||
The SEM images in Fig. S1† illustrate the different morphologies of products in the preparation process. The carbon spheres with a diameter of about 4 μm were obtained after the hydrothermal treatment of glucose and thiourea (Fig. S1a†), and the sphere structure was well retained after the treatment at 900 °C (Fig. S1b†), which is consistent with the previous reports for the preparation of PCSs.36,37 After the introduction of the Ti3C2Tx NS template, the obtained CNS-Ti3C2Tx after hydrothermal treatment shows a sheet-like structure (Fig. S1c†), which is different from the sphere structure obtained without Ti3C2Tx, suggesting that the Ti3C2Tx as the 2D template directs the hydrothermal carbonization of glucose on its surface. The strong interaction between glucose and the oxygen functional groups (-OH, -O-) on MXene NSs prevents the self-aggregation of glucose during hydrothermal carbonization to form the sphere structure, which is similar to the phenomenon of preparing carbon NSs using graphene oxide as the template.38 After annealing at 900 °C, TiO2 NPs in the carbon sheets can be observed and the sheet-like structure was well preserved (Fig. 2a). The TEM images of PCNS-TiO2 further demonstrate that TiO2 NPs with a size ranging from 10 to 50 nm are embedded in a very thin carbon sheet (Fig. 2b). Fig. 2c shows a lattice spacing of 0.25 nm of the NPs, which corresponds to the (101) plane of rutile TiO2. Besides, the hydrothermal treatment of Ti3C2Tx without glucose results in the formation of TiO2 NPs with a size of about 50 nm, which grow into larger NPs with a size of around 200 nm and change to pure rutile TiO2 after annealing at 900 °C (Fig. S1d–S1f and S2†). In contrast, the TiO2 NPs in PCNS-TiO2 have a much smaller size.
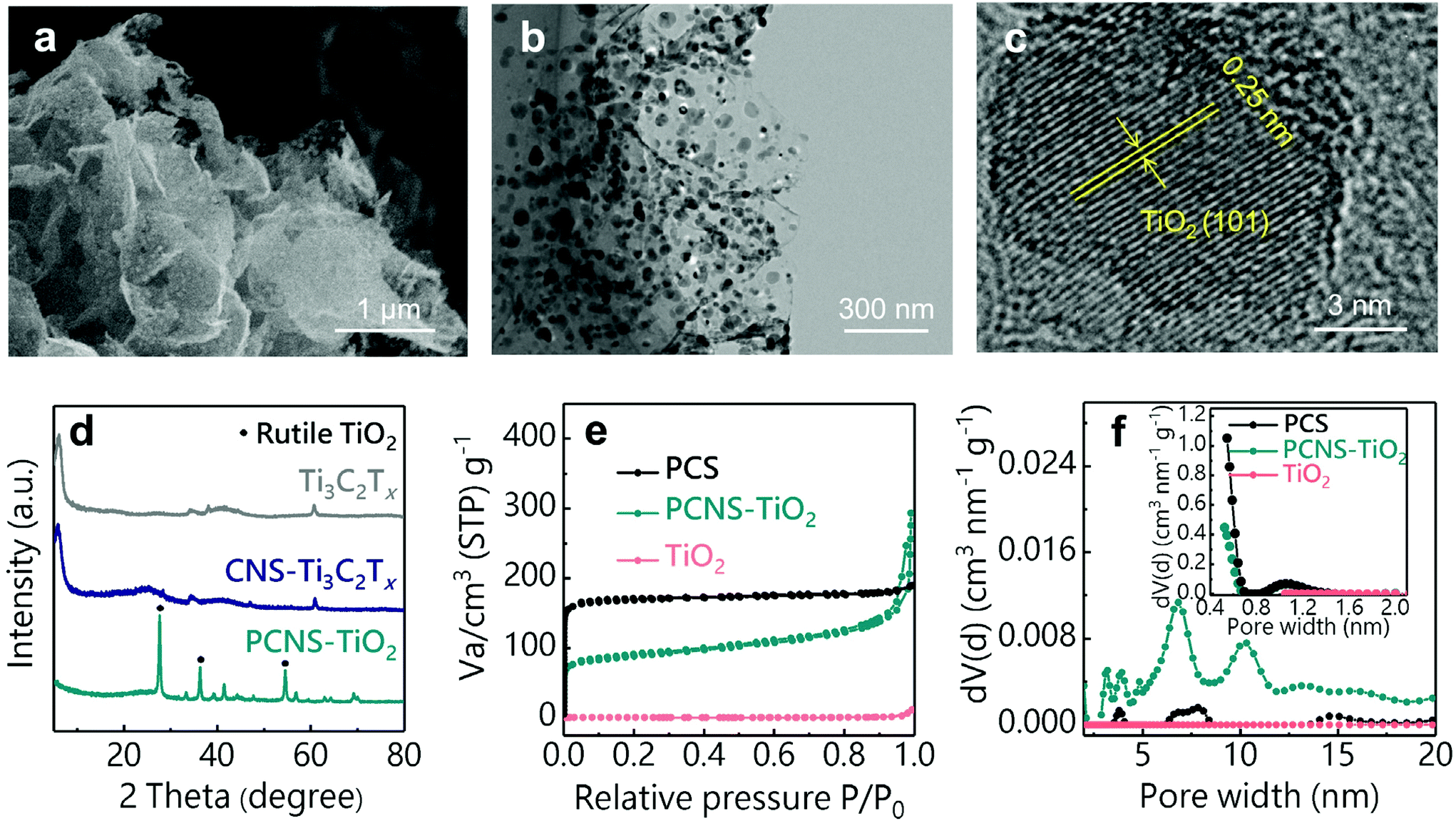 | ||
| Fig. 2 (a) SEM, (b) TEM and (c) HRTEM images of PCNS-TiO2. (d) XRD patterns of Ti3C2Tx, CNS-Ti3C2Tx and PCNS-TiO2. (e) N2 adsorption–desorption isotherms and (f) pore size distributions of the PCS, PCNS-TiO2 and TiO2. | ||
XRD patterns in Fig. 2d show the structural changes of Ti3C2Tx during the preparation process. The pristine Ti3C2Tx shows an obvious (0002) diffraction peak at around 6°, which still exists after hydrothermal treatment, suggesting that the glucose coated on the Ti3C2Tx NS surface can prevent their oxidation. After the heat treatment, the (0002) peak of Ti3C2Tx disappears and new diffraction peaks at 27.4°, 36.1° and 54.3°, which correspond to the (110), (101) and (211) planes of TiO2, appear, indicating that Ti3C2Tx NSs are oxidized into TiO2 NPs. The TiO2 content in the PCNS-TiO2 is about 32.4 wt%, according to the TG profile in Fig. S3.† The nitrogen adsorption–desorption isotherms and the pore size distributions obtained by the DFT method for the PCS, PCNS-TiO2 and TiO2 are shown in Fig. 2e and f. PCNS-TiO2 shows an isotherm with the characters of type I and II, suggesting the existence of micropores and mesopores with a size of about 1 nm and 5–10 nm, respectively. In contrast, the PCS mainly contains micropores with a size of around 1–2 nm, suggested by the type I adsorption isotherm and the pore size distribution, and TiO2 has no pores. The different pore structures between PCNS-TiO2 and PCS should be ascribed to the MXenes as the templates restricting the aggregation of carbonized products, forming the meso- and macropores. The SSAs calculated by the BET method for PCNS-TiO2, PCS and TiO2 are 325, 677 and 3 m2 g−1, respectively. Although the PCS shows a higher SSA than PCNS-TiO2, the small pore size of the microporous structure makes the inner surface hard to be fully used.39
The Li2S6 adsorption test was used to show the adsorption ability of the above samples with the same weight toward LiPSs (Fig. 3a). The Li2S6 solution (0.5 mM) containing PCNS-TiO2 becomes colorless after 8 h, indicating the excellent trapping ability of PCNS-TiO2 toward LiPSs. However, the solutions containing the PCS and TiO2 still show the color of Li2S6, which may be ascribed to the low surface utilization of the PCS and the ultralow SSA of TiO2. The above results show that the hierarchical porous structure can effectively improve the carbon surface utilization, and small TiO2 NPs uniformly distributed in the carbon sheets help further trap LiPSs, resulting in good adsorption ability toward LiPSs. The surface chemistry of PCNS-TiO2 before and after LiPS adsorption measurement was characterized by XPS. The Ti 2p spectrum of pristine PCNS-TiO2 in Fig. S4† shows two typical peaks of Ti4+ in TiO2, which are located at 465.2 eV (Ti 2p1/2) and 459.4 eV (Ti 2p3/2).40 The XPS spectrum of PCNS-TiO2 after the adsorption test is shown in Fig. 3b, the Ti 2p1/2 peak at 465.2 eV shifts to a lower binding energy, and two new peaks at 464.4 and 458.6 eV appear due to the formation of a Ti–S bond, demonstrating the strong chemical adsorption ability of TiO2 NPs toward LiPSs.41
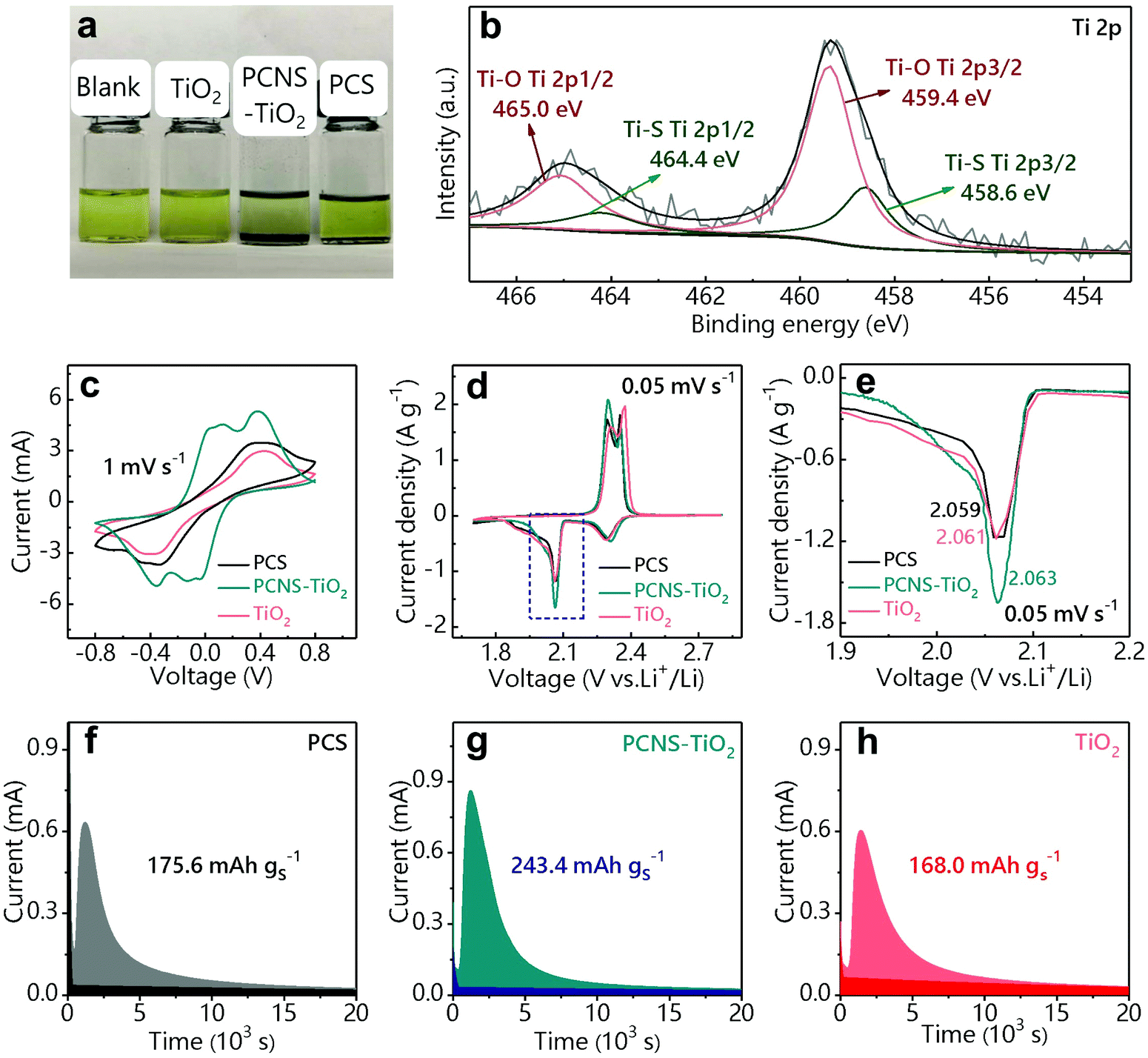 | ||
| Fig. 3 (a) Li2S6 adsorption test of different samples with the same weight. (b) Ti 2p spectrum of PCNS-TiO2 after the Li2S6 adsorption test. (c) CV profiles of symmetrical cells with the PCS, PCNS-TiO2 and TiO2 at a scanning rate of 1 mV s−1. CV profiles of Li–S batteries with PCS/S, PCNS-TiO2/S and TiO2/S as cathodes at a scan rate of 0.05 mV s−1 over a voltage range of 1.7 to 2.8 V (d) and the magnification of the cathodic peak ranging from 1.9 to 2.2 V (e). (f–h) Potentiostatic discharge curves of the Li2S deposition measurement. The light-colored area suggests the precipitation of Li2S, while the dark-colored area indicates the reduction of Li2S8/Li2S6. | ||
Symmetric cells were assembled by loading the PCS, PCNS-TiO2 and TiO2 on CPs as electrodes and Li2S6 solution as the electrolyte to reveal the LiPS conversion ability. Fig. 3c shows the CV profiles of the symmetric cells at a scan rate of 1 mV s−1. It can be observed that the cell with PCNS-TiO2 shows two pairs of well-defined redox peaks. Two anodic peaks located at 0.13 V and 0.37 V correspond to the oxidation of Li2S/Li2S2 to high-order LiPSs and further oxidation from high-order LiPSs to S8, respectively. Two reverse cathodic peaks located at −0.13 V and −0.37 V correspond to a reduction of S8 to high-order LiPSs followed by the conversion to Li2S/Li2S2. Compared with the cell with PCNS-TiO2, the cells with TiO2 and PCS only show one pair of ambiguous peaks, suggesting that PCNS-TiO2 improves the reaction kinetics of LiPS conversion. Besides, the cell with PCNS-TiO2 shows the smallest peak separation, indicating the higher electrochemical reversibility and improved LiPS conversion ability.42,43 Moreover, the higher current for the cell with PCNS-TiO2 indicates the improved utilization of LiPSs.42 The fast reaction kinetics of LiPSs is due to the combined characters of good adsorption ability of TiO2 NPs toward LiPSs and large carbon surface area for Li2S deposition. In addition, the TiO2 NPs are embedded in the PCNS, which ensures fast electron transfer for the conversion of LiPSs trapped by the TiO2 NPs.
To further verify the LiPS conversion ability with the help of PCNS-TiO2, batteries with PCS/S, PCNS-TiO2/S and TiO2/S as cathodes respectively and lithium foil as the anode were assembled. Fig. 3d shows that the CV profiles have two cathodic peaks, which correspond to the reduction of S8 to high-order LiPSs (Li2S4 and Li2S6) and further to low-order Li2S2/Li2S, and the two anodic peaks correspond to the inverse reaction. Both the cathodic peaks and anodic peaks exhibit a higher peak current density for the battery with the PCNS-TiO2 host, indicating higher sulfur utilization. Note that the cathodic peak at ∼2.05 V for the battery with PCNS-TiO2 shows the highest current density and slightly shifts to a higher potential (Fig. 3e), suggesting that the conversion from LiPSs to Li2S2/Li2S is enhanced.41
Li2S precipitation experiments were further conducted to show the LiPS conversion ability on the PCS, PCNS-TiO2 and TiO2 surface. The samples were loaded on CPs as the cathode,44 Li2S8 solution was used as the electrolyte and lithium foil was used as the anode. The precipitation capacity of Li2S is calculated based on the potentiostatic discharge curves in Fig. 3f–h. The cell with PCNS-TiO2 exhibits the largest precipitation capacity (243.4 mA h g−1) compared with the cells with TiO2 (168 mA h g−1) and PCS (175.6 mA h g−1), which is attributed to the good electrical conductivity and excellent LiPS trapping ability of PCNS-TiO2.45 The low conductivity of TiO2 and the weak interaction of the PCS with LiPSs should be the main reasons restricting the conversion of LiPSs for the other two cells.
The interlayers on separators were fabricated respectively by filtering PCS, PCNS-TiO2 and TiO2 dispersions on PP separators. The thicknesses of these interlayers are about 7, 5 and 2.4 μm, respectively, with the same mass loading of 0.23 mg cm−2 (Fig. 4a–c). Fig. 4a and d show that the interlayer composed of PCSs has a loosely aggregated structure and contains numerous microsized voids, which should have a weak ability to block LiPSs. In contrast, the interlayer composed of PCNS-TiO2 shows a compact structure which can prevent the diffusion of polysulfides. In addition, it also contains many large pores to form a hierarchical structure that is beneficial for fast Li ion diffusion (Fig. 4e). However, the interlayer composed of TiO2 is thin and compact, which is not beneficial to the electrolyte infiltration and Li ion diffusion (Fig. 4f). LiPS permeation measurement was also conducted to show the LiPS blocking ability of the PCNS-TiO2 interlayer. The separator with the PCNS-TiO2 interlayer was placed in the middle of an H-shaped glass tube. It can be seen that it is hard for the Li2S6 solution to permeate the PCNS-TiO2 interlayer for at least 8 h (Fig. 4g).
 | ||
| Fig. 4 Cross-sectional and top-view SEM images of (a, d) PCS, (b, e) PCNS-TiO2 and (c, f) TiO2 interlayers on separators; the insets are the photos of the interlayers. (g) LiPS permeation measurement with an H-shaped glass cell for the PCNS-TiO2 coated separator. | ||
Li–S batteries containing the PCS, PCNS-TiO2 and TiO2 interlayers were assembled using lithium foil as the anode and the GN/S hybrid as the cathode (sulfur loading: 1–1.2 mg cm−2). The PCNS-TiO2 interlayer shows the highest electrical conductivity (0.5 S cm−1) compared with TiO2 (lower than 10−10 S cm−1) and PCS (2.2 × 10−3 S cm−1) interlayers measured by the four-point probe method (Fig. S5†), which helps to reuse the LiPSs captured by the interlayer and improve the sulfur utilization. The EIS plots of these batteries are shown in Fig. 5a. Compared with the batteries with TiO2 and PCS interlayers, the battery with the PCNS-TiO2 interlayer shows the smallest semicircle, indicating the lowest charge transfer impedance (Rct) due to the good electrical conductivity and low Li ion diffusion resistance, which facilitates the LiPS conversion. In addition, the calculated Warburg coefficient of the battery with the PCNS-TiO2 interlayer (35.7) is similar to that with a pristine separator (34.6) (Fig. S6a and b†), suggesting a fast ion diffusion even with the interlayer. The rate performance of the batteries with different interlayers is shown in Fig. 5b. It is shown that the battery with the PCNS-TiO2 interlayer delivers much higher capacities under different rates compared with the batteries with TiO2 and PCS interlayers. The galvanostatic charge/discharge voltage profiles of the battery with the PCNS-TiO2 interlayer are shown in Fig. 5c, and two well-defined discharge plateaus are observed even at 2 C. This excellent rate performance is ascribed to the good conductivity of the PCNS-TiO2 interlayer as the vice-electrode and the low Li ion diffusion resistance. Thus, the battery with the PCNS-TiO2 interlayer also shows the lowest polarization indicated by the smallest over-potential (Fig. 5d).
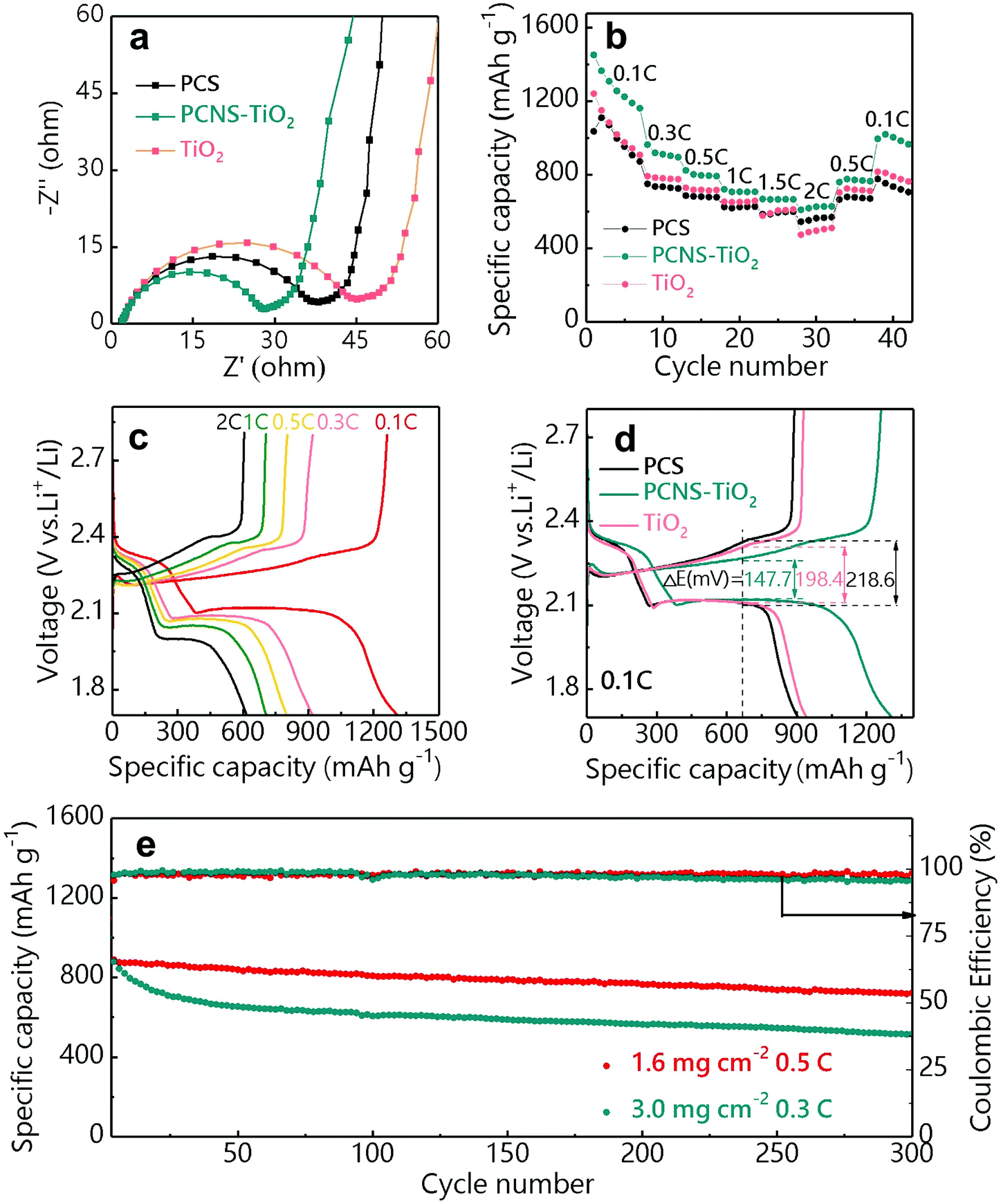 | ||
| Fig. 5 (a) EIS plots and (b) rate performance of Li–S batteries with PCS, PCNS-TiO2 and TiO2 interlayers. (c) Charge/discharge profiles of the Li–S battery with the PCNS-TiO2 interlayer at different rates. (d) Galvanostatic charge/discharge profiles of the batteries with PCS, PCNS-TiO2 and TiO2 interlayers at 0.1 C. (e) Cycling performance of Li–S batteries with the PCNS-TiO2 interlayer under sulfur loadings of 1.6 and 3.0 mg cm−2 at 0.5 and 0.3 C, respectively. | ||
The cycling stability of the battery with the PCNS-TiO2 interlayer was measured under different sulfur loadings of 1.6 and 3 mg cm−2 (Fig. 5e). Under a sulfur loading of 1.6 mg cm−2, a discharge capacity of 723 mA h g−1 was delivered after 300 cycles at 0.5 C, corresponding to a high capacity retention of 81.3% and a low average capacity fading rate of 0.063% per cycle. The good cycling stability is further revealed by the negligible change of over-potential in the charge/discharge profiles at different cycles in Fig. S7.† When the sulfur loading is increased to 3 mg cm−2, stable cycling performance is still achieved with a capacity retention of 58.6% and an average capacity fading rate of 0.139% after cycling for 300 cycles at 0.3 C. The excellent cycling performance of the battery with the PCNS-TiO2 interlayer should be ascribed to the good adsorption ability of TiO2 NPs and high blocking ability of the compact structure toward LiPSs, resulting in improved sulfur utilization.
Conclusions
In summary, a PCNS-TiO2 interlayer with high LiPS blocking ability and low Li ion diffusion resistance is fabricated on the separator, which helps improve the rate performance and cycling stability of Li–S batteries. The 2D structure of the carbon NSs physically suppresses the LiPS diffusion and the embedded TiO2 NPs have strong chemical trapping ability toward LiPSs, effectively suppressing the shuttling effect. At the same time, the thin sheet-like structure containing hierarchical pores eliminates the ion blocking effect of the 2D structure and allows fast Li ion diffusion. In addition, the tight contact between the TiO2 and carbon framework further ensures fast electron transfer for LiPSs and the reuse of captured LiPSs, improving the sulfur utilization. Thus, the Li–S battery with the PCNS-TiO2 interlayer shows good rate performance and cycling stability. A capacity of 718 mA h g−1 is maintained after 300 cycles at 0.5 C with a low capacity decay of 0.063% per cycle. Even with a higher sulfur loading of 3 mg cm−2, stable cycling performance is still achieved for 300 cycles at 0.3 C. Overall, this study provides an interlayer design with low ion resistance for Li–S batteries with stable cycling performance.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2018YFE0124500), the National Natural Science Foundation of China (U1601206 and 51772164), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01N111), Guangdong Natural Science Funds for Distinguished Young Scholars (2017B030306006), Shenzhen Basic Research Project (No. JCYJ20180508152037520) and the Shenzhen Graphene Manufacturing Innovation Center (201901161513 and 201901171523).Notes and references
- A. Manthiram, Y. Fu, S. H. Chung, C. Zu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- S. Xin, Z. Chang, X. Zhang and Y.-G. Guo, Natl. Sci. Rev., 2017, 4, 54–70 CrossRef CAS.
- L. Liu, Y. Wei, C. Zhang, C. Zhang, X. Li, J. Wang, L. Ling, W. Qiao and D. Long, Electrochim. Acta, 2015, 153, 140–148 CrossRef CAS.
- Q. Qi, X. Lv, W. Lv and Q.-H. Yang, J. Energy Chem., 2019, 39, 88–100 CrossRef.
- S. Imtiaz, J. Zhang, Z. A. Zafar, S. Ji, T. Huang, J. A. Anderson, Z. Zhang and Y. Huang, Sci. China Mater., 2016, 59, 389–407 CrossRef CAS.
- H. Ye, D. Lei, L. Shen, B. Ni, B. Li, F. Kang and Y.-B. He, Chin. Chem. Lett., 2020, 31, 570–574 CrossRef CAS.
- Z. Li, L. Yuan, Z. Yi, Y. Sun, Y. Liu, Y. Jiang, Y. Shen, Y. Xin, Z. Zhang and Y. Huang, Adv. Energy Mater., 2014, 4, 1301473 CrossRef.
- J. Zhang, Y. Cai, Q. Zhong, D. Lai and J. Yao, Nanoscale, 2015, 7, 17791–17797 RSC.
- Y. Wei, Y. Tao, C. Zhang, J. Wang, W. Qiao, L. Ling and D. Long, Electrochim. Acta, 2016, 188, 385–392 CrossRef CAS.
- S. Luo, W. Sun, J. Ke, Y. Wang, S. Liu, X. Hong, Y. Li, Y. Chen, W. Xie and C. Zheng, Nanoscale, 2018, 10, 22601–22611 RSC.
- X. Jia, C. Zhang, J. Liu, W. Lv, D. W. Wang, Y. Tao, Z. Li, X. Zheng, J. S. Yu and Q. H. Yang, Nanoscale, 2016, 8, 4447–4451 RSC.
- M. Guo, J. Huang, X. Kong, H. Peng, H. Shui, F. Qian, L. Zhu, W. Zhu and Q. Zhang, New Carbon Mater., 2016, 31, 352–362 CrossRef CAS.
- F. Zhu, Z. Yang, J. Zhao and X. Zhao, New Carbon Mater., 2016, 31, 199–204 CrossRef CAS.
- J. Han, W. Wei, C. Zhang, Y. Tao, W. Lv, G. Ling, F. Kang and Q.-H. Yang, Electrochem. Energy Rev., 2018, 1, 139–168 CrossRef CAS.
- Y. Chen, S. Niu, W. Lv, C. Zhang and Q. Yang, Chin. Chem. Lett., 2019, 30, 521–524 CrossRef CAS.
- W. Ai, W. Zhou, Z. Du, Y. Chen, Z. Sun, C. Wu, C. Zou, C. Li, W. Huang and T. Yu, Energy Storage Mater., 2017, 6, 112–118 CrossRef.
- H. Tang, W. Li, L. Pan, C. P. Cullen, Y. Liu, A. Pakdel, D. Long, J. Yang, N. McEvoy, G. S. Duesberg, V. Nicolosi and C. J. Zhang, Adv. Sci., 2018, 5, 1800502 CrossRef PubMed.
- H. Tang, W. Li, L. Pan, K. Tu, F. Du, T. Qiu, J. Yang, C. P. Cullen, N. McEvoy and C. Zhang, Adv. Funct. Mater., 2019, 29, 1901907 CrossRef.
- Y. Dong, S. Zheng, J. Qin, X. Zhao, H. Shi, X. Wang, J. Chen and Z.-S. Wu, ACS Nano, 2018, 12, 2381–2388 CrossRef CAS PubMed.
- Z. Xiao, Z. Li, P. Li, X. Meng and R. Wang, ACS Nano, 2019, 13, 3608–3617 CrossRef CAS PubMed.
- L. Jiao, C. Zhang, C. Geng, S. Wu, H. Li, W. Lv, Y. Tao, Z. Chen, G. Zhou, J. Li, G. Ling, Y. Wan and Q. H. Yang, Adv. Energy Mater., 2019, 9, 1900219 CrossRef.
- Z. Li, Z. Ma, Y. Wang, R. Chen, Z. Wu and S. Wang, Sci. Bull., 2018, 63, 169–175 CrossRef CAS.
- D. An, L. Shen, D. Lei, L. Wang, H. Ye, B. Li, F. Kang and Y.-B. He, J. Energy Chem., 2019, 31, 19–26 CrossRef.
- J. Yang, L. Xu, S. Li and C. Peng, Nanoscale, 2020, 12, 4645–4654 RSC.
- M. Tian, F. Pei, M. Yao, Z. Fu, L. Lin, G. Wu, G. Xu, H. Kitagawa and X. Fang, Energy Storage Mater., 2019, 21, 14–21 CrossRef.
- Y. S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed.
- W. Kong, L. Yan, Y. Luo, D. Wang, K. Jiang, Q. Li, S. Fan and J. Wang, Adv. Funct. Mater., 2017, 27, 1606663 CrossRef.
- C. L. Lee and I. D. Kim, Nanoscale, 2015, 7, 10362–10367 RSC.
- Y. Cui, X. Wu, J. Wu, J. Zeng, A. P. Baker, F. Lu, X. Liang, J. Ouyang, J. Huang, X. Liu, Z. Li and X. Zhang, Energy Storage Mater., 2017, 9, 1–10 CrossRef.
- J. Xu, Q. Zhang, X. Liang, J. Yan, J. Liu and Y. Wu, Nanoscale, 2020, 12, 6832–6843 RSC.
- N. Zheng, G. Jiang, X. Chen, J. Mao, N. Jiang and Y. Li, Nano-Micro Lett., 2019, 11, 43 CrossRef.
- X. Wang, Z. Wang and L. Chen, J. Power Sources, 2013, 242, 65–69 CrossRef CAS.
- X. Zhou, Q. Liao, T. Bai and J. Yang, J. Mater. Sci., 2017, 52, 7719–7732 CrossRef CAS.
- Y. Yang, C. Chen, J. Hu, Y. Deng, Y. Zhang and D. Yang, Chin. Chem. Lett., 2018, 29, 1777–1780 CrossRef CAS.
- M. Ghidiu, M. R. Lukatskaya, M. Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS PubMed.
- X. Y. Zheng, W. Lv, Y. Tao, J. J. Shao, C. Zhang, D. H. Liu, J. Y. Luo, D. W. Wang and Q. H. Yang, Chem. Mater., 2014, 26, 6896–6903 CrossRef CAS.
- S. Niu, W. Lv, C. Zhang, F. Li, L. Tang, Y. He, B. Li, Q.-H. Yang and F. Kang, J. Mater. Chem. A, 2015, 3, 20218–20224 RSC.
- T. T. Xie, W. Lv, W. Wei, Z. J. Li, B. H. Li, F. Y. Kang and Q. H. Yang, Chem. Commun., 2013, 49, 10427–10429 RSC.
- S. Z. Niu, G. M. Zhou, W. Lv, H. F. Shi, C. Luo, Y. B. He, B. H. Li, Q. H. Yang and F. Y. Kang, Carbon, 2016, 109, 1–6 CrossRef CAS.
- J. Yan, G. Wu, N. Guan, L. Li, Z. Li and X. Cao, Phys. Chem. Chem. Phys., 2013, 15, 10978–10988 RSC.
- T. Zhou, W. Lv, J. Li, G. Zhou, Y. Zhao, S. Fan, B. Liu, B. Li, F. Kang and Q.-H. Yang, Energy Environ. Sci., 2017, 10, 1694–1703 RSC.
- Y. Zhong, L. Yin, P. He, W. Liu, Z. Wu and H. Wang, J. Am. Chem. Soc., 2018, 140, 1455–1459 CrossRef CAS PubMed.
- H. Lin, L. Yang, X. Jiang, G. Li, T. Zhang, Q. Yao, G. W. Zheng and J. Y. Lee, Energy Environ. Sci., 2017, 10, 1476–1486 RSC.
- H. J. Peng, G. Zhang, X. Chen, Z. W. Zhang, W. T. Xu, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2016, 128, 13184–13189 CrossRef.
- G. Zhang, H.-J. Peng, C.-Z. Zhao, X. Chen, L.-D. Zhao, P. Li, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 16732–16736 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr02607g |
| ‡ These authors are equal main contributors. |
| This journal is © The Royal Society of Chemistry 2020 |