
Mechanistic insights of the reduction of gold salts in the Turkevich protocol†
Yunhu
Gao
 and
Laura
Torrente-Murciano
*
and
Laura
Torrente-Murciano
*
Department of Chemical Engineering and Biotechnology, University of Cambridge, Cambridge, CB3 0AS, UK. E-mail: lt416@cam.ac.uk
First published on 28th December 2019
Abstract
This paper presents fundamental understanding of the mechanism of the Turkevich protocol, the method recommended by the National Institute of Standards and Technology for the synthesis of gold nanoparticles using sodium citrate as reducing agent. Herein, we reveal that the Turkevich mechanism consists of two consecutive reduction steps (Au3+ → Au+ → Au0) rather than a reduction followed by the disproportionation reaction as conventionally believed. This new understanding has profound implications: i. the second reduction step (Au+ → Au0), rather than the previously postulated first reduction step, is the rate-limiting reduction step and ii. the formation of acetone dicarboxylate (DC2−) as an intermediate product through the oxidation of citrate has a key role as stabilizer and as a reducing agent (stronger than sodium citrate). This knowledge enables the synthesis of monodispersed gold nanoparticles with sizes ranging from 5.2 ± 1.7 nm to 21.4 ± 3.4 nm, with the lower end considerably smaller than previously reported through the Turkevich route. This work provides fundamental guidance for the controllable synthesis of nanoparticles using DC2− as a reducing agent directly applicable to other precious metals.
1. Introduction
Gold nanoparticles (Au NPs) have potential applications in medicine, catalysis, electronics and photonics due to their unique chemical and physical properties compared with their bulk counterpart.1–6 Au NPs are usually synthesized by the reduction of chloroauric acid (HAuCl4) with a reducing agent such as sodium borohydride,7 sodium citrate,8 and ascorbic acid.9 The Turkevich protocol is the most reliable and popular method for the synthesis of Au NPs,10 and consequently the recommended method by the National Institute of Standards and Technology for reference material preparation.11 It consists of the use of sodium citrate to reduce chloroauric acid at elevated temperatures in an aqueous medium under various conditions. The citrate ion also acts as a stabilizer, which can be easily replaced post-synthesis by other ligands, in favor of the functionalization of Au NPs.12The accepted mechanism of the Turkevich method for the synthesis of gold nanoparticles consists of the initial redox reaction (R1) (Scheme 1),10,13,14 where trivalent gold gets reduced to monovalent gold by citrate which in turn gets oxidised to acetone dicarboxylate (DC2−), the conjugated base of dicarboxyacetone (DCA). This first redox step is deemed as the rate-determining step.15 Consecutively, the disproportionation reaction (R2) (Scheme 1) takes place where metallic gold and trivalent gold are produced. In the original paper by Gammons et al. investigating the disproportionation reaction,16 the reaction was conducted in the absence of any reducing agent and stabilizer. It is thus expected that in the presence of reducing agents, such as sodium citrate and DC2− present in the conventional Turkevich protocol, a competitive parallel reduction of Au+ to metallic gold might take place, however, to the best of our knowledge, this parallel reduction reaction has not been investigated.
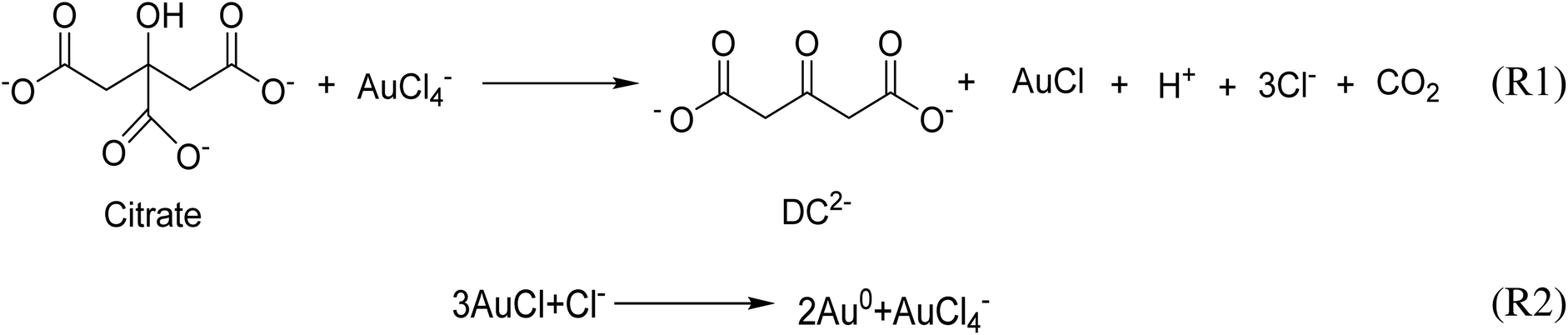 | ||
| Scheme 1 The accepted mechanism for Turkevich protocol, consisting of the initial redox reaction (R1) and the disproportionation reaction (R2). | ||
Much research has been done to unravel the mechanism of Turkevich protocol and optimize its synthesis conditions in order to obtain monodispersed Au NPs with controllable sizes.8,10,13–15,17–19 The sodium citrate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HAuCl4 molar ratio has shown to have a great effect on the particle size,20 while the temperature does not play a considerable role from 50 °C to 100 °C.21 Increasing the sodium citrate:HAuCl4 molar ratio leads to smaller Au NPs due to higher nucleation rate and consequently higher concentration of generated nuclei.22–24 The pH value of the reaction medium has also been shown to have an effect on both the particle size and kinetics.10,14,17,18,25,26 This pH effect is related to the hydrolysis of the citrate and chloroauric species,14 as HCit2− was found to be the strongest reducing species whose concentration is maximised at pH 5.6.10,17,18,26 Similarly, AuCl4− is the most reactive gold precursor compared with its hydrolysed species at high pH values, AuCl3(OH)−, AuCl2(OH)2−, AuCl(OH)3− and Au(OH)4−. As a result, the reaction, especially the nucleation stage, is much faster at pH values between ∼3.7–6.5, compared with pH of ∼6.5–7.7.17 The Au NPs size consequently decreases from 22 nm to 11 nm when pH increases from 3.0 to 5.4, then increases to 28 nm when pH further increases from 5.4 to 7.5. The pH has also a dominating effect on the polydispersity of the Au NPs, being around 10% at pH values between 5.4 and 6.8, increasing to ∼20% when pH is below 4.5 or higher than 7.5.18 The optimization of the reaction (1) by adjusting the pH range of chloroauric acid solution to 3–3.6, and the pH of the mixed solution between 5.4–6.0, at boiling temperature, enabled the synthesis of the smallest Au particles using the conventional Turkevich method to date (11.0 ± 0.4 nm).18,27
:HAuCl4 molar ratio has shown to have a great effect on the particle size,20 while the temperature does not play a considerable role from 50 °C to 100 °C.21 Increasing the sodium citrate:HAuCl4 molar ratio leads to smaller Au NPs due to higher nucleation rate and consequently higher concentration of generated nuclei.22–24 The pH value of the reaction medium has also been shown to have an effect on both the particle size and kinetics.10,14,17,18,25,26 This pH effect is related to the hydrolysis of the citrate and chloroauric species,14 as HCit2− was found to be the strongest reducing species whose concentration is maximised at pH 5.6.10,17,18,26 Similarly, AuCl4− is the most reactive gold precursor compared with its hydrolysed species at high pH values, AuCl3(OH)−, AuCl2(OH)2−, AuCl(OH)3− and Au(OH)4−. As a result, the reaction, especially the nucleation stage, is much faster at pH values between ∼3.7–6.5, compared with pH of ∼6.5–7.7.17 The Au NPs size consequently decreases from 22 nm to 11 nm when pH increases from 3.0 to 5.4, then increases to 28 nm when pH further increases from 5.4 to 7.5. The pH has also a dominating effect on the polydispersity of the Au NPs, being around 10% at pH values between 5.4 and 6.8, increasing to ∼20% when pH is below 4.5 or higher than 7.5.18 The optimization of the reaction (1) by adjusting the pH range of chloroauric acid solution to 3–3.6, and the pH of the mixed solution between 5.4–6.0, at boiling temperature, enabled the synthesis of the smallest Au particles using the conventional Turkevich method to date (11.0 ± 0.4 nm).18,27
The order of addition of chloroauric acid and sodium citrate during the synthesis has a great effect on the particle size distribution.11,28 In the standard Turkevich method, a concentrated sodium citrate solution is added to a diluted HAuCl4 solution. However, in the so-called reversed Turkevich method, where the concentrated acidic HAuCl4 solution is injected to a diluted sodium citrate solution, localised low pH values promote AuCl4− species which contributes to the generation of more seeds and leads to smaller average particle sizes.11
Despite the numerous mechanistic studies of the Turkevich method, most of the researchers have focused on the optimisation of the first Au3+ to Au+ reduction step (R1).29,30 However, some authors, including Turkevich, identified the potential role of the intermediate product DC2− as a reducing agent.8 Doyen et al. confirmed the formation of DCA (at 75 °C) and its derived products acetoacetic acid, acetic acid and acetone by NMR.31 The substitution of 10% sodium citrate by DCA was found to accelerate the reaction and decrease the size and Au NPs in the standard Turkevich protocol. It was postulated that DCA forms a multidentate complex with Au+ which favors the disproportionation reaction, associated to the smaller Au NPs with narrower size distribution obtained in the reversed Turkevich method.28
The morphology of the gold nanoparticles can be controlled by the external addition of chemicals, for example, the presence of L-tyrosine leads to the formation of gold nanoflowers.32 Gold nanowires, which self-assemble to gold foams, can be obtained by halting the Turkevich reaction or controlling low sodium citrate concentration.33–35 Similarly, the addition of trace amounts of tannic acid in the presence of poly-vinyl-pyrrolidone (PVP) can lead to sub-10 nm particles.36,37
This paper presents a systematic study to shed light upon the mechanism of Turkevich for the synthesis of Au NPs by revealing the actual role of DC2− intermediate as a reducing agent and confirming the reduction of Au+ over the previously believed disproportionation reaction. This new knowledge enables the design of the synthetic Turkevich conditions to tune the gold particle size while obtaining narrow size dispersions.
2. Experimental procedures
Materials
Gold(I) chloride AuCl (99.9% trace metal basis), DCA (technical grade), sodium citrate dehydrate (>99%), chloroauric acid (99.99% trace metal basis, 30 wt%), citric acid monohydrate (>99%) and bovine serum albumin (BSA) are purchased from Sigma Aldrich. Sodium hydroxide (0.1 M) is purchased from Fisher Chemical. All chemicals are used without further purifications. 18.2 MΩ cm pure water is from the MilliQ ultra-purification system.Synthesis of Au NPs
The synthesis of Au NPs is carried out following the reversed Turkevich protocol in a 100 mL glass round-bottom flask reactor equipped with a glass condenser. The water bath temperature is controlled by an ETS-D5, IKA hotplate. In a typical Turkevich synthesis, 48.0 mL of water are pre-heated to 80 °C in a stirred round-bottom flask where a thermometer is introduced. Once the temperature is stable, 672 μL 0.125 M sodium citrate solution and 328 μL 0.125 M citric acid solution are added to the reaction vessel. The reaction starts when 1.0 mL 0.0125 M HAuCl4 solution is added to reaction medium. In the reduction of HAuCl4 by DC2− at 80 °C, 46.0 mL of water are pre-heated in a stirred round bottom flask, once the temperature is stable, 2.5 mL 0.1 M NaOH are added to adjust the pH at 4.8 (for comparison purposes with other experiments). 1 mL 0.0125 M HAuCl4 is added to start the reaction about 3 seconds after the addition of 0.5 mL 0.25 M DCA. 1.5 mL samples are collected periodically (every one minute), cooled down in an ice water bath to quench the reaction and analyzed by UV-vis spectroscopy. The reaction is considered completed when the UV-vis spectrum does not change with time. High reproducible results are obtained in all cases, confirmed by doing the experiments in duplicates and comparing their respective final UV-vis spectra.Characterization
The UV-vis spectrum of the Au NP colloidal suspensions is measured using an Agilent Cary-60 UV-Vis spectrophotometer. pH is measured using a HI98100 Checker Hanna pH indicator, with an accuracy of 0.2. Transmission electron microscopy (TEM) images are obtained using a FEI Tecnai 20 equipment. ImageJ software is used to analyze the area-equivalent diameter of more than 200 particles. TEM samples are prepared according to Michen et al.'s protocol,38 which adopts BSA to prevent post-synthesis agglomeration. 1 mL of colloidal solution is mixed with 1.0 mL 0.3 mg mL−1 BSA solution before kept at 5 °C for 2 h. Then, 5 μL mixed sample is drop on a 400 mesh carbon coated copper TEM grid and left to dry at room temperature. Scanning electron microscopy is carried out using a FEI Philips XL30 sFEG equipped with energy dispersive X-ray microscopy (EDS). X-ray photoelectron spectroscopy (XPS) is used to analyse the oxidation state of gold precursor using a Thermo Fischer Escalab 250 Xi with Al K alpha as source with 1486.68 eV energy and 50 eV pass energy. Dwell time is set as 50 ms and step size is 0.1 eV. X-ray spot size is 200 micrometres. The binding energy scale is calibrated by setting the Au4f7/2 binding energy to 84.0 eV.39,40 XPS samples are prepared dropping 5 μL of the Au NP colloidal solution into a silicon sample wafer, dried in a vacuum tank and then stored under argon atmosphere before characterization. Electrochemical experiments are performed to measure the oxidation potentials of sodium citate and DC2− utilising a standard three-electrode setup connected to a potentiostat (VSP, Bio-Logic). A glassy carbon rod (Alfa Aesar, 2 mm diameter) is utilised as working electrode, with an exposed geometric surface area of 0.0314 cm2. Pt wire (Alfa Aesar, >99.997% purity, 0.5 mm diameter) is utilised as counter electrode, Hg/HgSO4 (SI Analytics) is utilised as reference electrode. Prior to each experiment, glassy carbon and Pt electrodes are thoroughly rinsed in sequence with isopropanol and ultrapure water. All cyclic voltammetry experiments are performed at a scan speed of 10 mV s−1.3. Results and discussion
To understand the mechanism of the Turkevich method for the synthesis of Au NPs, a first series of reactions at 25 and 80 °C is carried out following the reversed Turkevich method to investigate the actual role of citrate and its in situ formed oxidation form, dycarboxyacetone (DCA). Choloroauric acid (HAuCl4) is used as gold precursor. The Au:reducing agent molar ratio is kept constant at 1:10 at an initial pH 4.8–4.9 adjusted by addition of sodium citrate and citric acid mixtures or by addition of NaOH (Table 1).
| No | [HAuCl4]/mM | [AuCl]/mM | [DCA]/mM | [Sodium citrate]/mM | [Citric acid]/mM | [NaOH]/mM | PVP/mM | T/°C | Initial pHa | Final pH | Diameterb/nm |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a The initial pH was always measured at 25 °C for comparison purposes. b Measured by TEM. | |||||||||||
| A | 0.25 | — | — | 1.68 | 0.82 | — | — | 25 | 4.9 | 5.0 | 26.0 ± 6.3 |
| B | 0.25 | — | — | 1.68 | 0.82 | — | — | 80 | 4.9 | 4.9 | 15.2 ± 4.2 |
| C | 0.25 | — | 2.5 | — | — | 5 | — | 25 | 4.9 | 7.8 | 21.1 ± 6.9 |
| D | 0.25 | — | 2.5 | — | — | 5 | — | 80 | 4.9 | 7.5 | 10.7 ± 1.4 |
| E | 0.25 | — | 2.5 | 2.5 | — | 1.62 | — | 25 | 4.8 | 6.0 | 23.0 ± 5.6 |
| F | 0.25 | — | 2.5 | 2.5 | — | 1.62 | — | 80 | 4.8 | 6.7 | 13.9 ± 3.0 |
| G | — | 0.25 | — | — | — | 0.01 | 5 | 80 | 4.8 | 3.9 | 11.9 ± 4.2 |
| H | — | 0.25 | — | 1.35 | 1.15 | — | — | 80 | 4.8 | 4.9 | 14.8 ± 4.3 |
| I | — | 0.25 | 2.5 | — | — | 4 | — | 25 | 4.8 | 7.0 | 9.8 ± 3.2 |
| J | — | 0.25 | 2.5 | — | — | 4 | — | 80 | 4.8 | 7.8 | 6.8 ± 1.5 |
Although the Au NPs synthesis is very slow at room temperature using citrate as reducing agent (A), with negligible formation of Au NPs after 20–25 min, changes in absorbance in the UV-region are observed (Fig. S1†), which are believed to be related to the fast substitution of one of the chloride ions by citrate in the Au environment as previously suggested by Ojea-Jiménez et al.14 Increasing the reaction time to 24 h leads to the slow formation of Au NPs (26.0 ± 6.3 nm). On the other hand, the faster nucleation at 80 °C (B) leads to smaller Au NPs (15.2 ± 4.2 nm). It is important to note that the UV-vis spectra of the particles synthesized at room temperature show a significant absorbance at ∼700 nm (Fig. S1†), previously associated to near-surface nucleation events during particle growth41 a phenomena probably responsible of the irregular Au NPs shape in Fig. 3a (see below) and not visible during the synthesis at 80 °C. It is likely that autocatalysed secondary nucleation on the surface of existing gold surface dominates the process under slow reduction conditions at room temperature.41 This effect of temperature on size is associated to the kinetics of reduction of the gold precursor from Au3+ → Au+ → Au0 and consequently the generation of nuclei and/or seeds. A number of synthesis mechanisms have been reported to date from the original LaMer's nucleation theory42 to the more comprehensive seed-mediated growth mechanism.18,25,43 In both cases, when colloidal stability is guaranteed, the resulting size of the particles is directly related to kinetics of the reduction stage which determines not only the concentration of nuclei/seeds where growth take place, but also the amount of remaining precursor. Thus, faster reduction of the gold precursor at higher temperatures leads to a higher rate of nuclei/seeds formation and a smaller particle size in agreement with our observations. In addition, the resulting dispersity of the particles will depend on the degree of separation between the nucleation/seed formation and growth stages.44
Reduction of gold precursor by citrate leads to its oxidation and thus the formation of DC2−. Although the concentration of the latter is normally comparatively small to that of citrate ions during the conventional Turkevich method, for comparison purposes, gold nanoparticles are also synthesized by using directly DC2− as reducing agent rather than sodium citrate (C–D, note that initial pH has been kept constant by adding NaOH into the reaction medium). In this case, the formation of Au NPs can take place even at room temperature (e.g. 25 °C) while increasing the temperature to 80 °C leads to faster kinetics (as expected). Due to the faster nucleation at 80 °C considerably smaller particles (10.7 ± 1.4 nm) are obtained compared with 25 °C (21.1 ± 6.9 nm).18,25
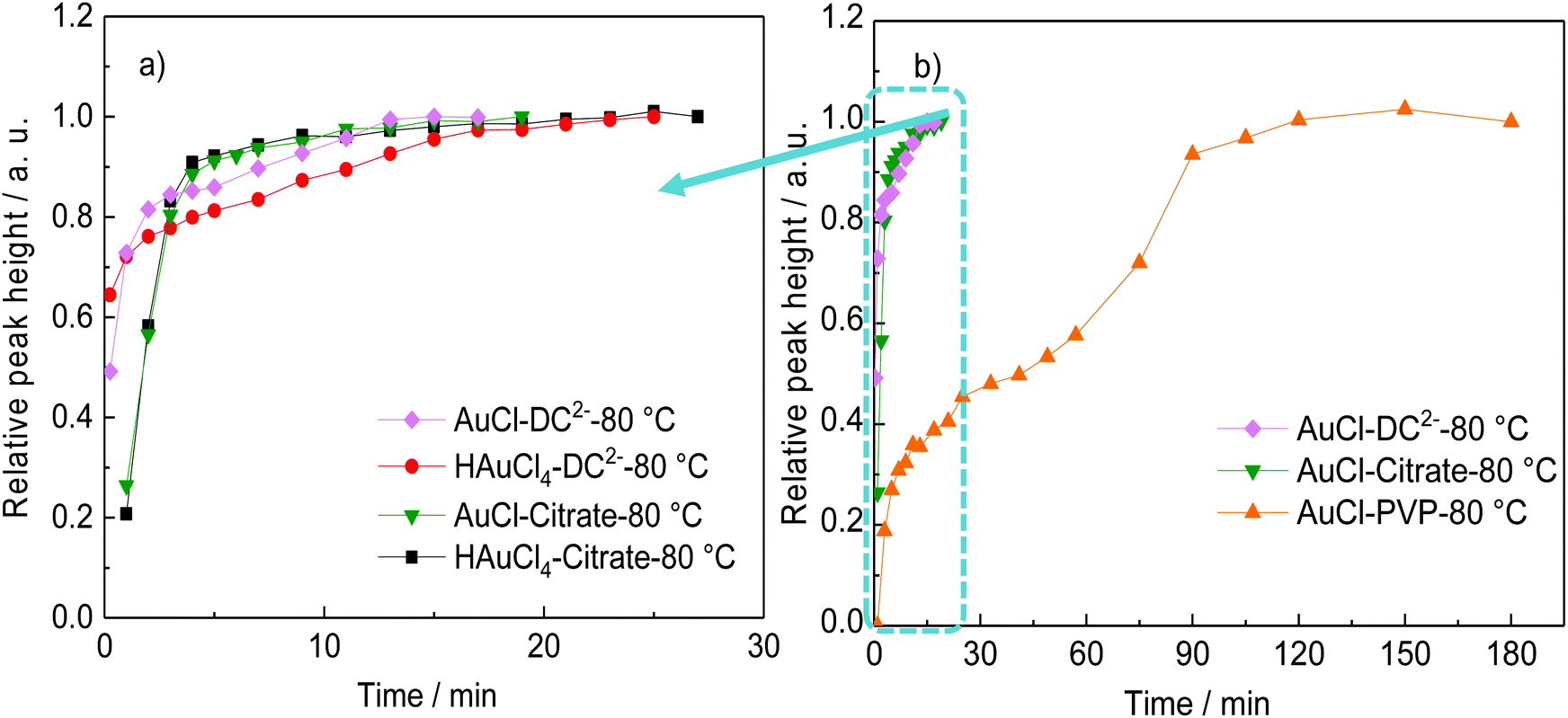
Fig. 1 shows the time-evolution of the formation of Au NPs using the absorption data. At 25 °C, while the reduction of gold is considerably quicker during the first ∼30 min of reaction when DC2− is used as reducing agent compared with citrate, the kinetics of the reduction greatly slows down after this time. Indeed, it requires ∼20 days to achieve full conversion. On the other hand, full reduction of the gold precursor is achieved in ∼24 h in the presence of citrate despite the slower initial kinetics. The decrease of reaction kinetics using DC2− as the reducing agent is likely to its adsorption on the surface of the formed Au NPs, resulting in a decrease of its concentration in the solution.12 In addition, changes in pH (∼0.1) in the first ∼30 min could also affect the hydrolysis of the gold precursor from AuCl4− at pH of 4.9 to AuCl3(OH)−, which is less reactive.26 Indeed, after 20 days, pH increases from the initial value of 4.9 to 7.8 when DC2− is used as the reducing agent, a rise caused by the hydroxide ions resulting from the decomposition of DC2−, as shown in reaction (3), Scheme 2.13 In contrast, sodium citrate acts as a buffer keeping the pH constant during the reaction. In any case, since the initial seed particle formation is faster when DC2− is used as the reducing agent, the resulting Au NPs are smaller, 21.1 ± 6.9 nm, compared with 26.0 ± 6.3 nm when sodium citrate is the reducing agent.
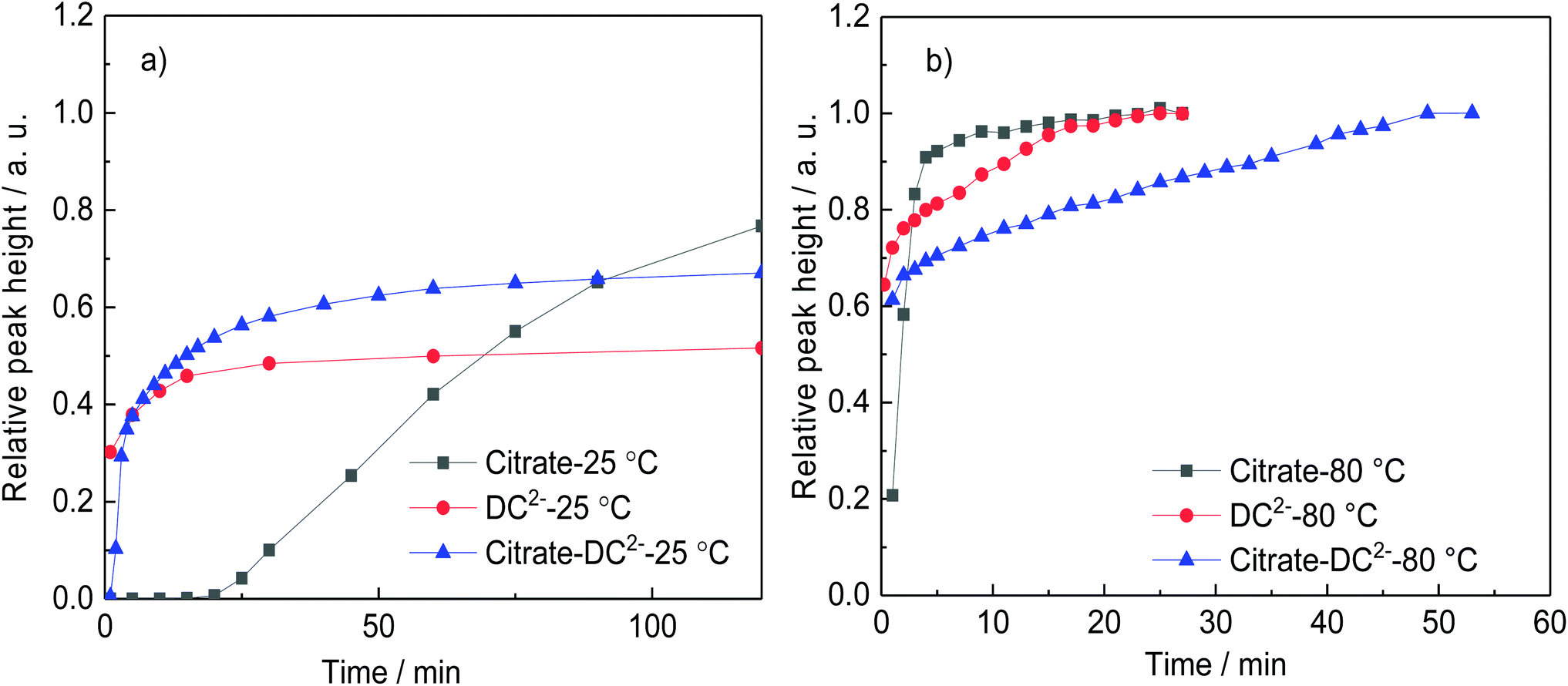 | ||
| Fig. 1 Comparison of the rate of formation of gold nanoparticles (represented by the relative height of the absorption peak) using sodium citrate or DC2− as a reducing agent at a. 25 °C and b. 80 °C. Conditions: [HAuCl4]: 0.25 mM, pH: 4.8–4.9, reducing agent: [sodium citrate]: 2.5 mM, [DC2−]: 2.5 mM; or [sodium citrate] 2.5 mM + [DC2−] 2.5 mM, as in experiments A–F (Table 1). | ||
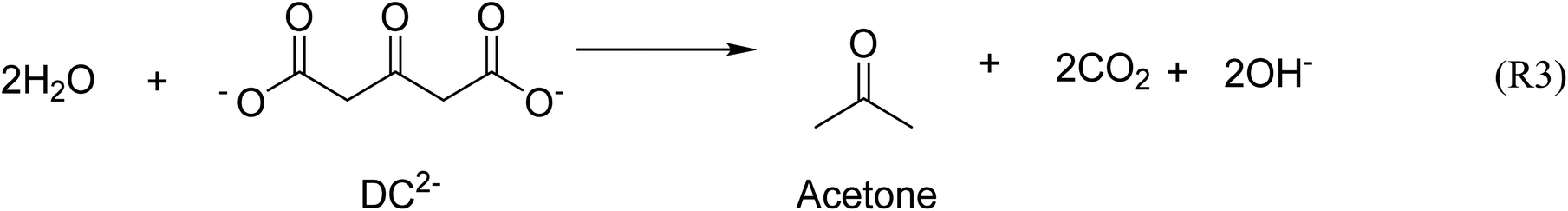 | ||
| Scheme 2 Decomposition of DC2−.13 | ||
The differences in the kinetics of the reduction are also observed at 80 °C, however, in this case, full conversion of the Au NPs is observed in both cases within ∼15 min (Fig. 1b). Similarly than before, when DC2− is used as reducing agent, smaller and more monodispersed Au NPs (10.7 ± 1.4 nm, ±13%, Fig. 3d) are obtained compared with sodium citrate (15.2 ± 4.2 nm, ±28%, Fig. 3b). Both colloidal stability and chemistry of reduction should be taken into consideration to explain the difference.25 As mentioned above, DC2− promotes a faster reduction kinetics of the gold precursor leading to a higher concentration of initial seeds. In addition, DC2− has a stronger stabilization effect compared with citrate according to Grasseschi et al.,12 which may stabilize smaller stable seeds compared with citrate, leading to more seeds even when the same amount of gold precursor is initially reduced.18,25 Both factors simultaneously lead to a higher concentration of seeds and thus, they lead to smaller Au NPs for the same precursor concentration.
These results agree with the measured oxidation potential of the two reducing agents as shown in Fig. 2, where it is shown that DC2− is a stronger reducing agent with an oxidation potential of 0.50 V, compared to sodium citrate at 0.97 V at the studied conditions.
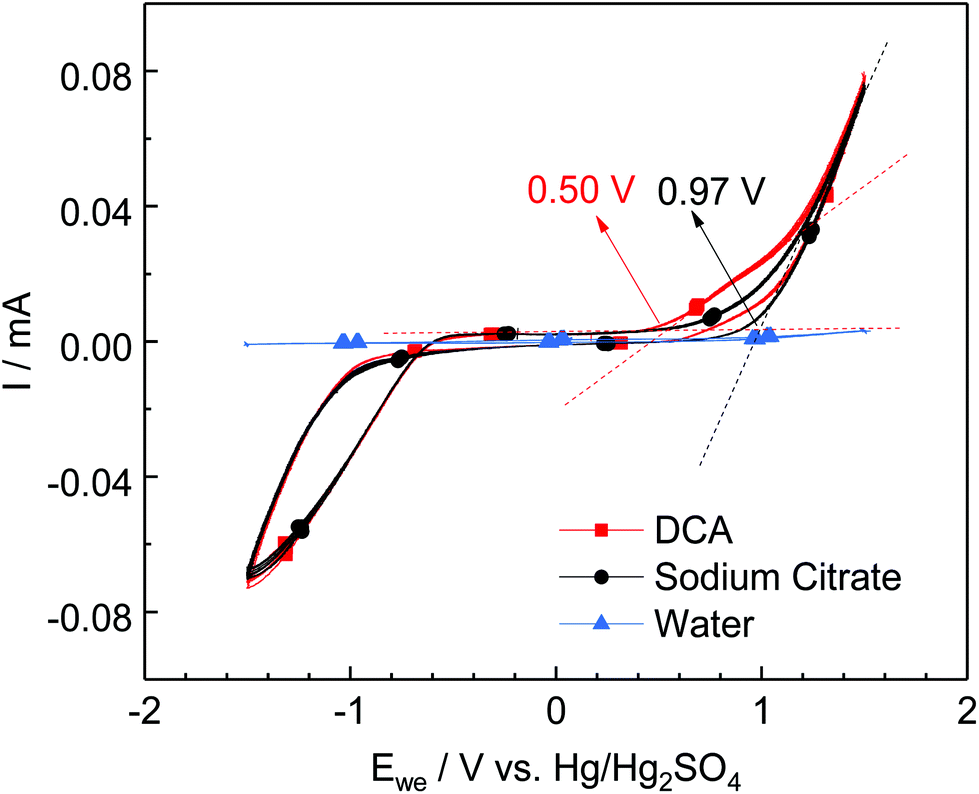 | ||
| Fig. 2 Current vs. voltage response for sodium citrate ([sodium citrate]: 1.35 mM, [citric acid]: 1.15 mM), DC2− ([DCA]: 2.5 mM, [NaOH]: 4 mM), and diluted HCl solution. Conditions: 25 °C, pH 4.9. | ||
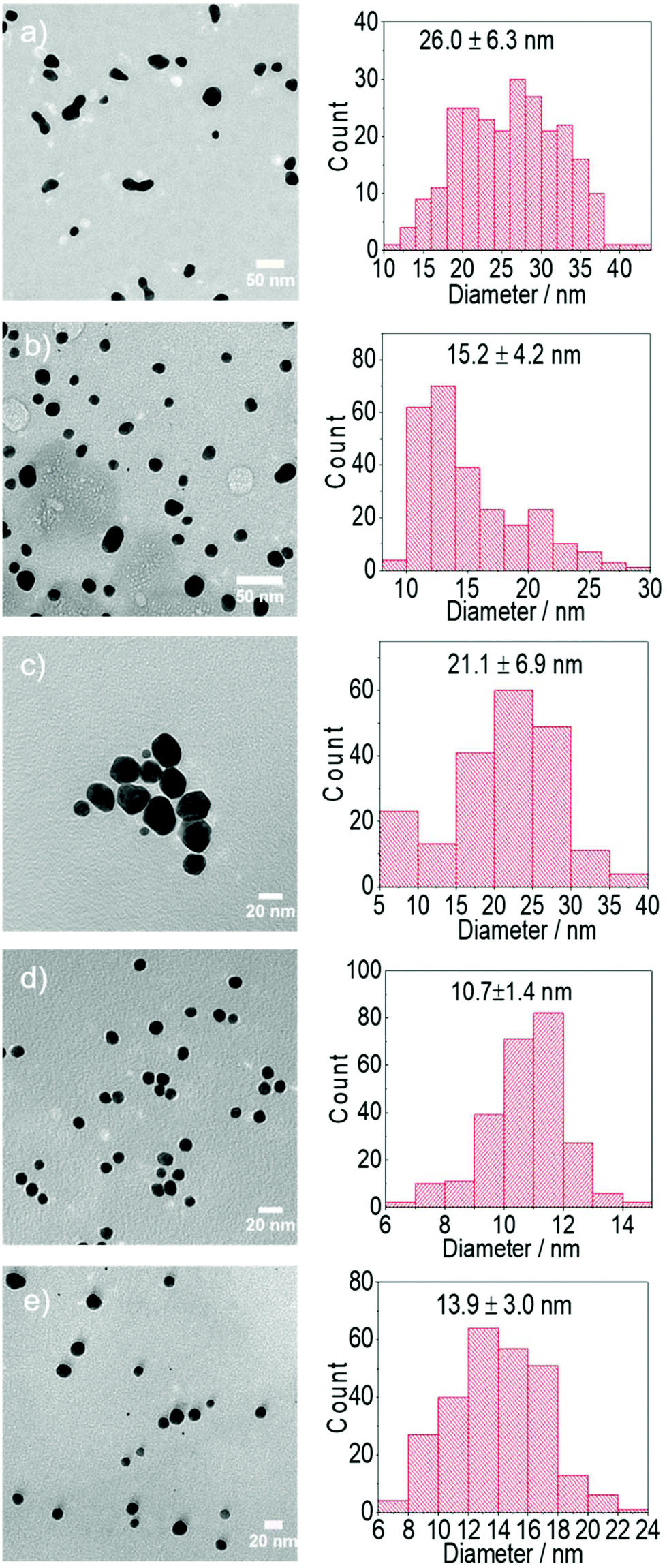 | ||
| Fig. 3 Representative TEM images and particle size distribution of Au NPs synthesised using chloroauric acid as the precursor a. [sodium citrate] 2.5 mM at 25 °C, b. [sodium citrate] 2.5 mM at 80 °C, c. [DC2−] 2.5 mM at 25 °C, d. [DC2−] 2.5 mM at 80 °C, e. [sodium citrate] 2.5 mM and [DC2−] 2.5 mM at 80 °C. Conditions: [HAuCl4]: 0.25 mM, pH: 4.8–4.9, samples are taken after reaction completion. | ||
The reason of the higher monodispersity of the Au NPs formed using DC2− as reducing agent at 80 °C is threefold – firstly, DC2− serves as the stabilizer for Au NPs. Although DC2− is not stable in the aqueous solution, its enol form coordinates with Au NPs forming a complex and consequently stabilising DCA versus its decomposition.12 On the other hand, the enol-Au NPs complex is negatively charged, stabilising the particles electrostatically. Secondly, a fast nucleation takes place due to the high reducing potential of DC2− as discussed above, leading to the formation of seeds to which the remaining DC2− coordinates.12 Both aspects consequently lead to a lower concentration of DC2− in the solution, decreasing the reduction kinetics and thus the formation of new nuclei. Thirdly, an increase in pH due to DC2− decomposition (as discussed above) leads to the hydrolysis of the remaining gold precursor to less reactive forms. These new conditions reduce the seed formation rate to a minimum, separating the seed formation and their growth stages.18,44
Both Au NPs stabilized by DC2− and citrate are stable for at least 5 month as confirmed by UV-vis analysis. Grasseschi et al.12 have previously demonstrated that DC2− have a stronger interaction with the Au surface than citrate ions however, both can be easily replaced by molecules with stronger affinity to gold atoms, e.g. 4-mercaptopyridine, which enables the convenient post-synthesis functionalization of Au NPs.
It is important to note that this high monodispersity of Au NPs achieved with DC2− as reducing agent is only feasible at pH values >∼4.8 when the DCA fully deprotonates into DC2− species. Indeed, in contrast to our observations, Wuithschick et al. concluded that the reduction of chloroauric acid with DCA results on the quick formation of polydispersed and non-reproducible Au NPs,25 while this experiment is done three times to confirm the reproducibility as shown in Fig. S4.† The differences are related to the pH conditions. In their case, pH values <4.8, lead to the protonated form of DCA, which is known to have a low reducing and stabilisation power.11,28 Similarly, Ojea-Jiménez et al. reported that no Au NPs formation is observed at pH 3.7 and 100 °C due to the low reactivity of the protonated form of DCA.28 In addition, it is likely that high temperatures (e.g. boiling conditions) of these two studies lead to the fast decomposition of DCA.
In the presence of both reducing agents, citrate and DC2−, at 80 °C, the seed particle formation rate is faster in comparison with citrate-only but slower than with DC2−-only (Fig. 1b). This is reflected on the resulting Au NPs size with sizes of 10.7 ± 1.4 nm, 15.2 ± 4.2 and 13.9 ± 3.0 nm obtained with only-DC2−, only-citrate and a DC2−–citrate mixture (keeping the individual concentration constant) respectively. A possible explanation is the competitive substitution of chloride ligands in the gold precursor by both DC2− and citrate, which follows the substitution of chloride ligands in the gold precursor by hydroxycarboxylates proposed by Ojea-Jimenez et al. which was later extended to a range of α-hydroxycarboxylate ions with carboxyl and hydroxyl groups by Bartosewicz et al.14,27,45 According to Grasseschi et al.,12 DC2− can isomerize to form a carbon–carbon double bond and a hydroxyl group (enol form) which would be expected to have a similar ligand exchange capability although further mechanistic studies would be needed to verify this. Similar results are obtained at 25 °C; the nucleation rate is faster when only DC2− is used as reducing agent, while the combination of citrate and DC2− gives a faster nucleation than citrate only (Fig. 1a).
According to the accepted mechanism of the Turkevich method, the second reduction step (Au+ to Au0) and consequently the formation of NPs takes place through the disproportionation reaction (reaction (2)). To investigate the role of the reducing agents and specially the DC2− formed as an intermediate species in this second reduction step, a similar series of reactions is carried out but in this case, intermediate Au+ species (AuCl) is used as gold precursor rather than Au3+ present in chloroauric acid (HAuCl4).
Both citrate and DC2− are capable of reducing AuCl to form Au NPs at 80 °C. When AuCl is the gold precursor, DC2− shows a faster a nucleation kinetics, (Fig. 4a), and thus, smaller Au NPs (6.8 ± 1.5 nm, Fig. 5c), compared with sodium citrate (14.8 ± 4.3 nm, Fig. 5b), at the same pH.
 | ||
| Fig. 4 Comparison of the rate of formation of gold nanoparticles (shown by the relative height of the absorption peak) (a) using different gold precursors and reducing agents at 80 °C. Conditions: [HAuCl4]: 0.25 mM, [AuCl]: 0.25 mM, [sodium citrate]: 2.5 mM, [DC2−]: 2.5 mM, pH: 4.8–4.9. Original data are shown in Fig. S3.† (b) By the reduction or the disproportionation reaction of AuCl with DC2− or sodium citrate as a reducing agent (same with (a)), or PVP as stabilizer in the absence of sodium citrate and DC2−. Conditions: [AuCl]: 0.25 mM, [PVP monomer]: 5 mM, pH: 4.8, 80 °C. | ||
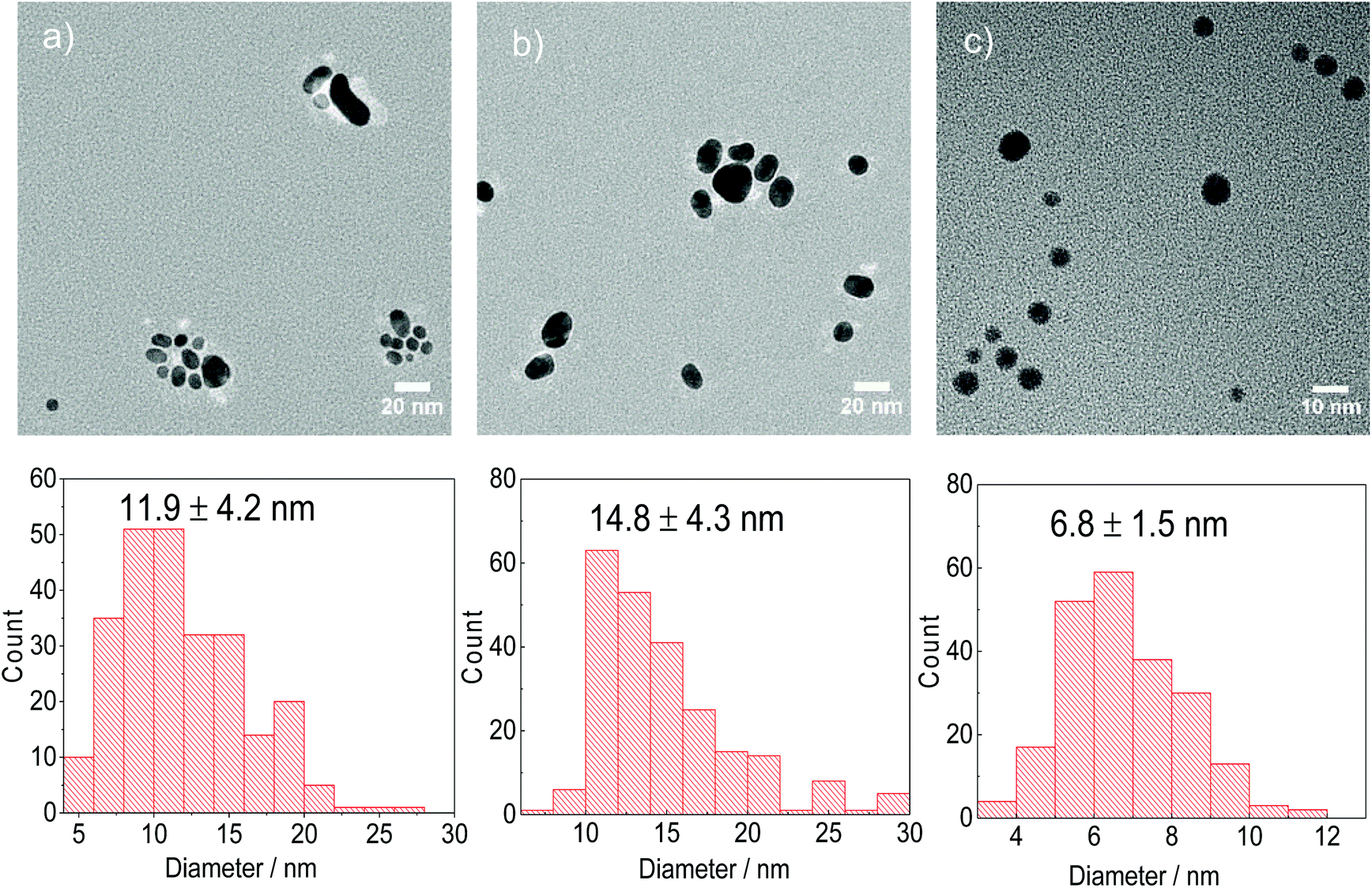 | ||
| Fig. 5 Representative TEM images and particle size distribution of Au NPs synthesised using AuCl as the precursor a. in the absence of reducing agent using PVP as stabilizer, b. sodium citrate and c. DC2−. Conditions: [AuCl]: 0.25 mM, [reducing agent]: 2.5 mM (when applicable), 80 °C pH: 4.8, time: 15–180 min. | ||
Similar results to the ones above are obtained when HAuCl4 is used as gold precursor. Indeed, same kinetics and similar Au NPs size distribution are observed when starting with HAuCl4 (15.2 ± 4.2 nm) or AuCl (14.8 ± 4.3 nm) using sodium citrate as reducing agent (Fig. 4a). Considering that the reduction of gold precursor consists of two reduction steps in series Au3+ → Au+ → Au0 as demonstrated above, the similar rate of reaction shown in Fig. 4a indicate that under these conditions and at 80 °C, the Au+ reduction is considerably slower than the Au3+ reduction to Au+ and the dissolution of AuCl.15
The solubility of AuCl in water is very low, however, DC2− facilitates its dissolution by coordination,13,45 which consequently facilitates its reduction to Au0 and the formation of Au NPs. DC2− drives the reduction of Au+, so indirectly further dissolves the AuCl.
As mentioned above, DC2− is not stable at high temperatures and it decomposes quickly at 80 °C (according to thermal stability studies followed by the characteristic absorption peak of DCA at 240 nm, Fig. S2†). However, as shown in Fig. 4, the reduction of AuCl is quicker than the decomposition of DC2−, successfully leading to the formation of Au NPs. In addition, as discussed before, the coordination of DC2− to Au NPs enhances its stability.12
Despite the low solubility of AuCl, a disproportionation reaction starting with Au+ is carried out (i.e. in the absence of DC2− and citrate) at 80 °C and pH value of 4.8, using PVP as the stabilizer for Au NPs.37 Au NPs with average sizes of 11.9 ± 4.2 nm are synthesized, as shown in Fig. 5a after 150 min. It is important to note that the overall kinetics of the disproportionation reaction and the potential reduction of AuCl by PVP (with a weak reduction potential46) are considerably slower than the reduction of AuCl by both sodium citrate and DC2− (Fig. 4b). These results suggest that in the presence of a reducing agent (sodium citrate or DC2−), the formation of the Au NPs consists of two reduction steps (Au3+ → Au+ → Au0), the second one competing over the much slower disproportionation reaction.
Indeed, our XPS results (Fig. 6) confirm that the extent of the disproportionation reaction in the presence of DC2− is almost negligible. When HAuCl4 is reduced by DC2−, Au3+, Au+ and Au0 are all detected (Fig. 6a). This is consistent with the XPS results of the reduction of HAuCl4 by sodium citrate.40 However, when AuCl is reduced by DC2− (Fig. 6b), only Au+ and Au0 species are present, confirming the direct reduction of Au+ to Au0, with the disproportionation reaction negligible (i.e. absence of Au3+).
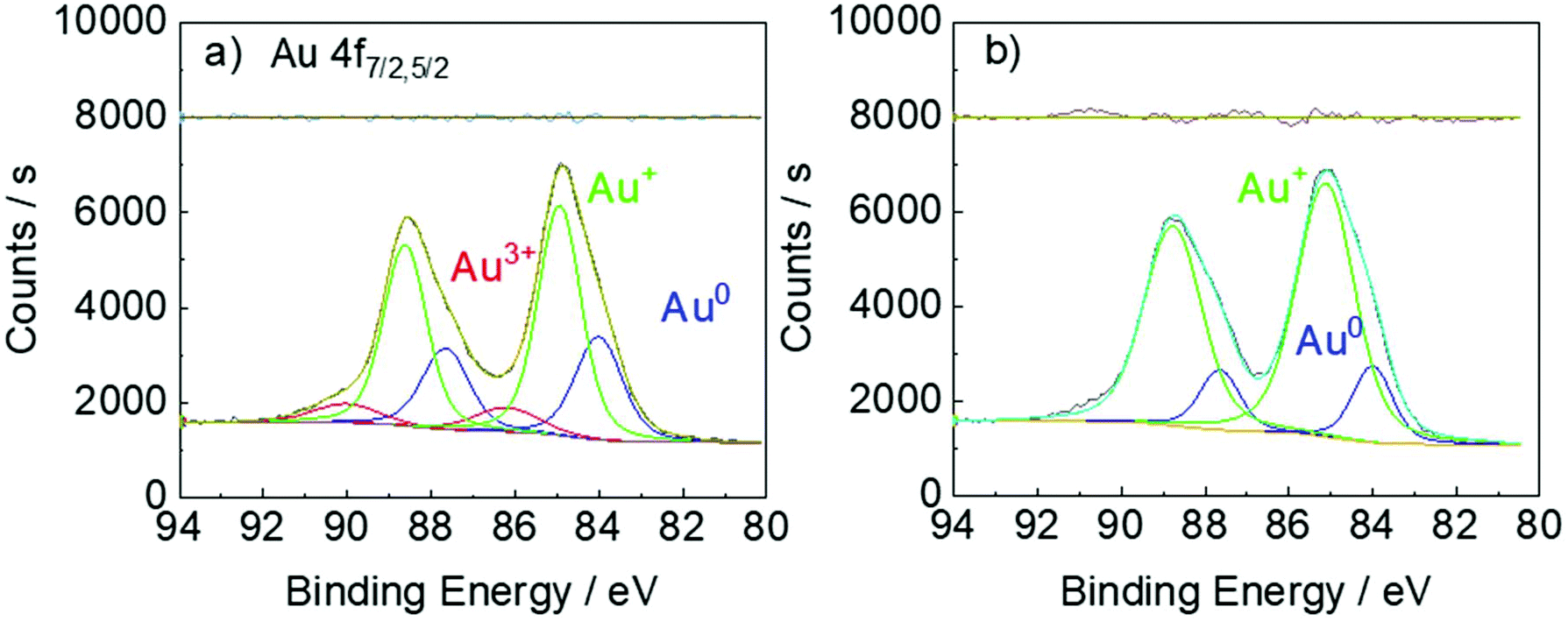 | ||
| Fig. 6 XPS of a. [HAuCl4]: 0.25 mM, [DC2−]: 2.5 mM, sampling time: 5 min; b. [AuCl]: 0.25 mM, [DC2−]: 2.5 mM, sampling time: 15 min. Conditions: pH: 4.8, 25 °C. | ||
Once demonstrated that the second reduction step in the Turkevich method is dominated by the reduction of the Au+ by the reducing agent, further understanding of this step is needed to find out the relationship between reaction conditions and particle size and distribution.
During the conventional Turkevich method, the second reduction step takes place in the presence of both, citrate (present as reactant) and DC2− (present as intermediate product in the first reduction step, reaction (1)). Thus, a new series of reactions is designed (Table 2) to investigate the simultaneous role of both reducing agents.
| No | [AuCl]/mM | [DCA]/mM | [Sodium citrate]/mM | [Citric acid]/mM | [NaOH]/mM | T/°C | Initial pH | Final pH | Diameter/nm |
|---|---|---|---|---|---|---|---|---|---|
| K | 0.25 | 5 | 7.62 | 80 | 4.4 | 9.2 | 6.6 ± 2.1 | ||
| L | 0.25 | 5 | 5 | 80 | 4.4 | 6.5 | 6.9 ± 1.9 | ||
| M | 0.25 | 2.5 | 3.68 | 1.32 | 80 | 4.4 | 5.2 | 7.2 ± 1.6 | |
| N | 0.25 | 0.25 | 2.53 | 2.47 | 80 | 4.4 | 4.4 | 10.8 ± 2.5 | |
| O | 0.25 | 5 | 5 | 80 | 2.5 | 2.8 | 21.4 ± 3.4 | ||
| P | 0.25 | 5 | 3.21 | 1.79 | 80 | 3.6 | 5.1 | 12.6 ± 3.4 | |
| Q | 0.25 | 5 | 5 | 7.72 | 6.2 | 7.6 | 5.2 ± 1.7 | ||
| R | 0.25 | 5 | 5 | 10 | 80 | 9.0 | 8.6 | 6.8 ± 2.5 | |
| S | 0.25 | 5 | 5 | 10.1 | 80 | 9.9 | 9.0 | 7.8 ± 1.8 |
Before that, it is important to compare the results obtained with only DC2− (5 mM) as reducing agent and a mixture of DC2− and citrate (5 mM each) (experiments K and L in Table 2). In both cases, similar Au NPs size and distribution are achieved (6.6 ± 2.1 nm and 6.9 ± 1.9 nm, respectively) which indicates that the presence of DC2− controls the seed particle formation reaction and the stabilisation of the nanoparticles. On the other hand, the presence of sodium citrate has a buffer effect which reduces pH variation during the duration of the reaction.
By keeping constant the concentration of citrate (5 mM) and the pH (4.4), the effect of the [DC2−]:[AuCl] molar ratio on particle size and distribution is investigated. Fig. 7 shows that the rate of gold nanoparticle formation increases rapidly as the [DC2−]:[AuCl] molar ratio increases from 1:1 to 20:1 which simultaneously leads to a smaller size (from 10.8 to 6.9 nm, Fig. 8). These observations indicate that DC2− plays a key role in the nucleation stage as it is known that the quicker the nucleation rate, the higher concentration of nuclei is formed, leading to smaller particle sizes.18,25
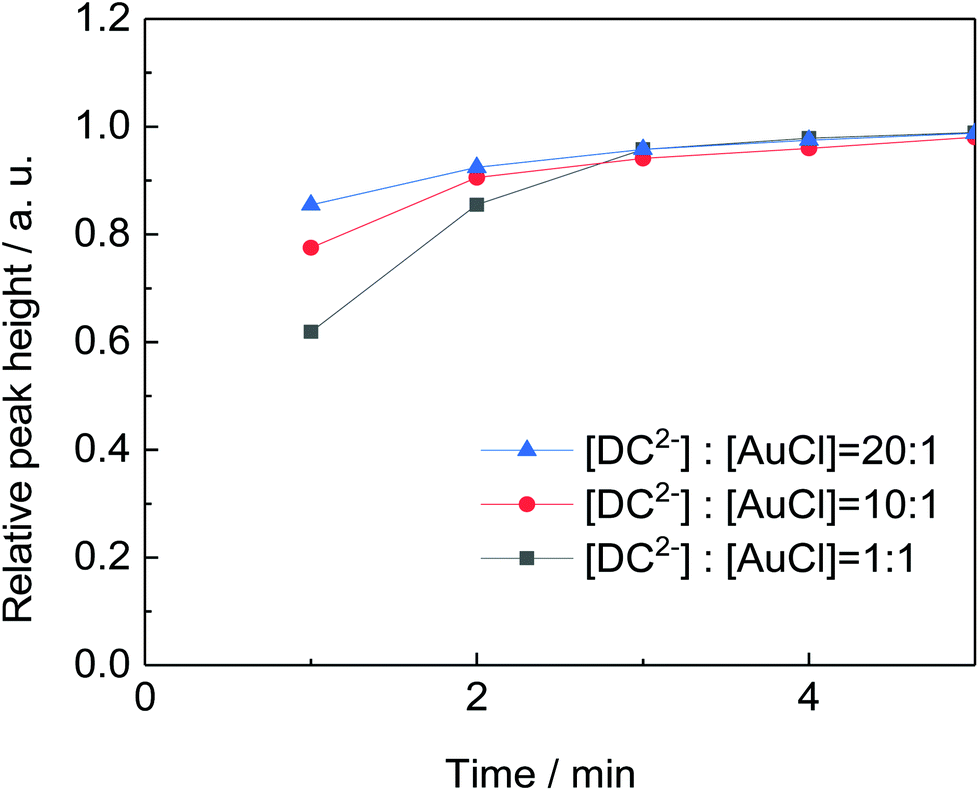 | ||
| Fig. 7 Comparison of the rate of formation of gold nanoparticles (shown by the height of the absorption peak) by varying the [DC2−]:[AuCl] molar ratio at 80 °C. Conditions: [AuCl]: 0.25 mM, [citrate]: 5 mM, pH: 4.4, 80 °C. | ||
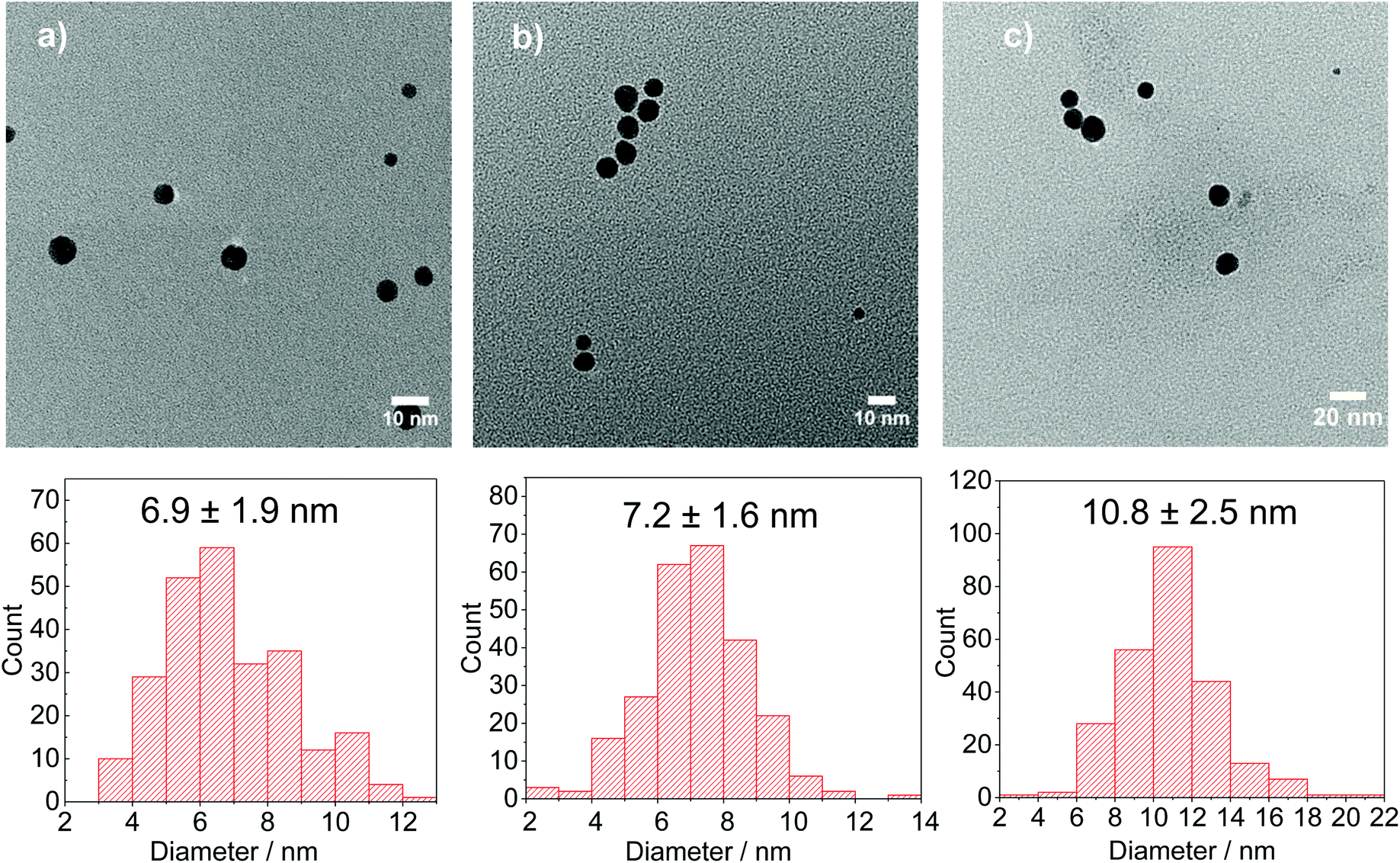 | ||
| Fig. 8 Representative TEM images and particle size distribution of Au NPs synthesised using AuCl as precursor with a DC2−:Au molar ratio of a. 20:1, b. 10:1 and c. 1:1. Conditions: [AuCl]: 0.25 mM, [citrate]: 5 mM, pH: 4.4, 80 °C, time: 7 min–11 min. | ||
The effect of the pH during the reduction of AuCl on the size of the Au NPs is also investigated by keeping constant the concentration of DC2− (5 mM) and varying the ratio of sodium citrate and citric acid, keeping the overall concentration of citrate as 5 mM. NaOH is added to achieve high pH values. Increasing the pH leads to a higher rate of reaction measured by an increase of the absorbance peak associated to the Au NPs as shown in Fig. 9b and a decrease of particle size at values of pH below 4.4. At higher pH values, both the rate of formation of Au NPs and the size (∼6.8 nm) are very similar (Fig. 10). It is important to note that the concentration of gold in the solution is very similar within the whole pH range as confirmed by the absorption of the initial solutions at 400 nm.25,47
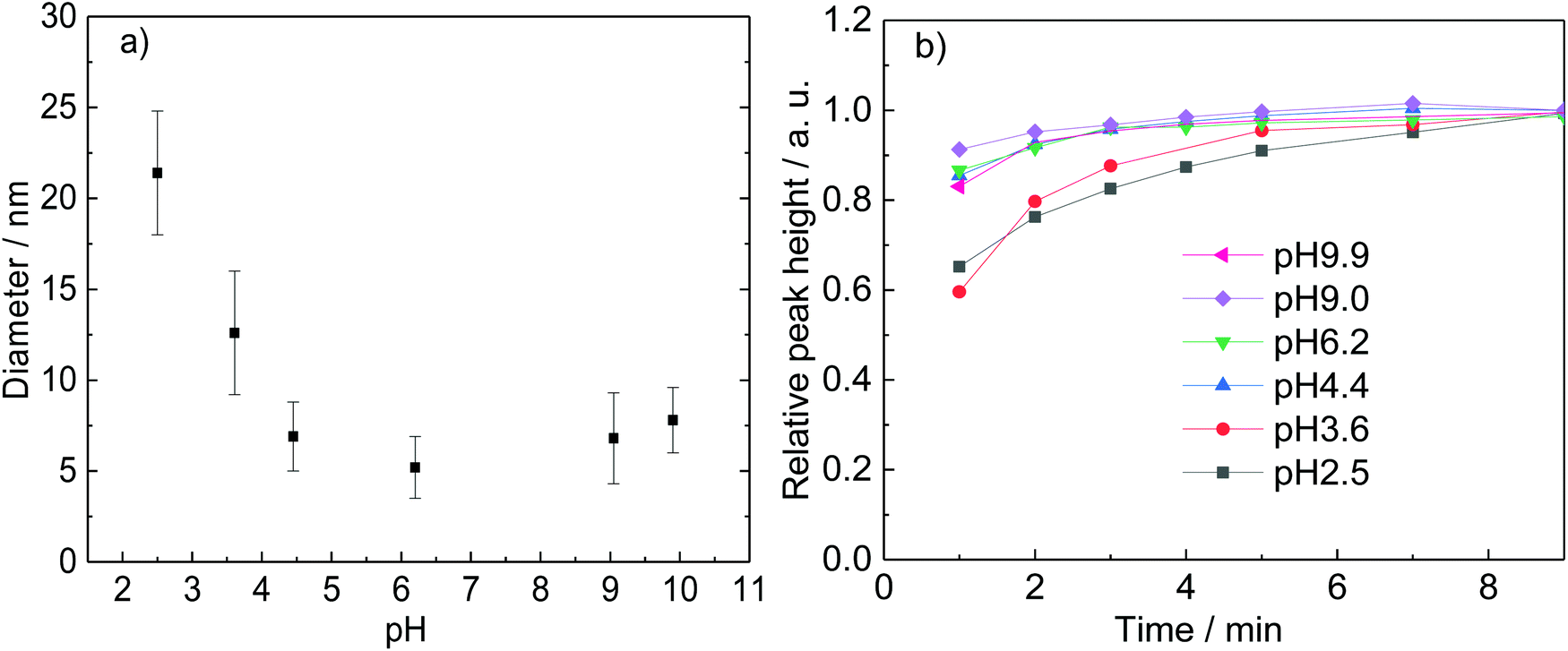 | ||
| Fig. 9 Effect of pH on a. Au NP sizes (the error bars represent the standard deviation) and b. rate of formation. Conditions: [AuCl]: 0.25 mM, [DC2−]: 5 mM, [citrate]: 5 mM, 80 °C, time: 7 min–13 min. | ||
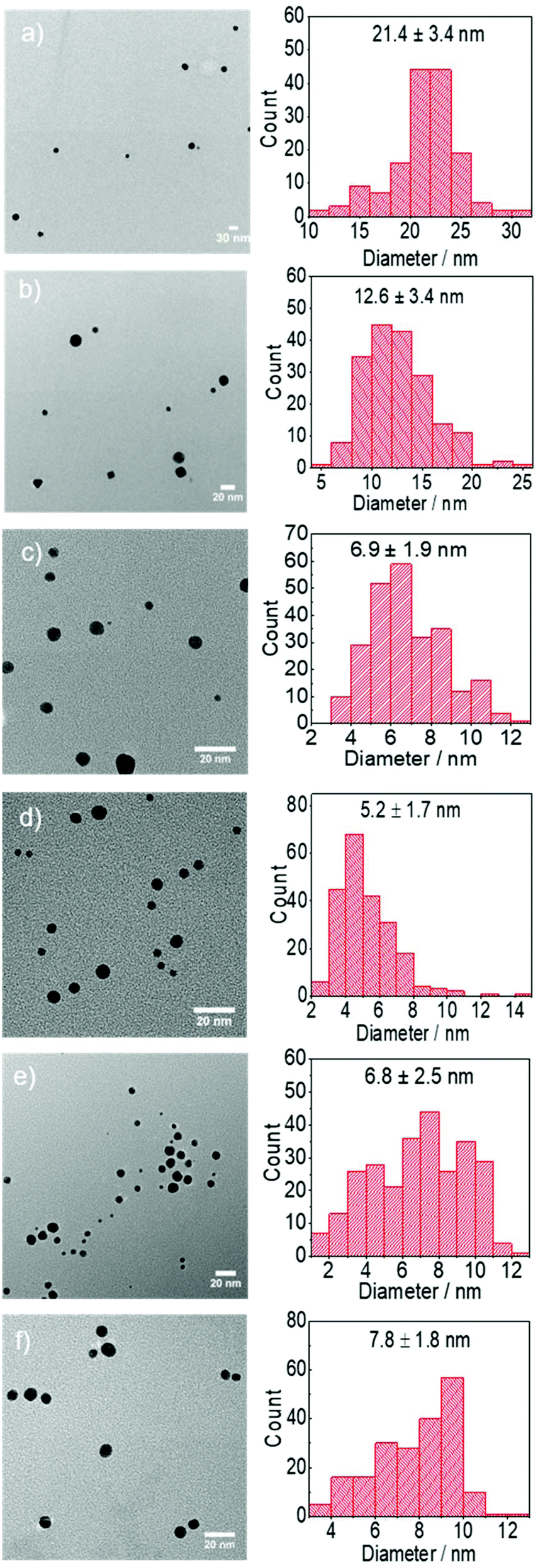 | ||
| Fig. 10 Representative TEM images and particle size distribution of Au NPs synthesised using AuCl as precursor at pH of a. 2.5, b. 3.6, c. 4.4, d. 6.2, e. 9.0 and f. 9.9. Conditions: [AuCl]: 0.25 mM, [DC2−]: 5 mM, [citrate]: 5 mM, 80 °C, time: 7 min–13 min. | ||
These observations demonstrate again the dominating role of DCA over citrate on the second reduction step of Au+ during the Turkevich method. Indeed, both citrate and DCA species are known to protonate/deprotonate in aqueous solutions as shown in reactions (4) and (5) (Scheme 3) as a function of pH as shown in Fig. 11. Increasing the pH from 2.5 to 5.5 leads to the complete deprotonation of DCA into DC2− species, which is believed to be the strongest reducing agent. In accordance, further increase in pH does not lead to any further reduction of particle size nor enhancement of the rate of Au NPs formation (Fig. 9b).
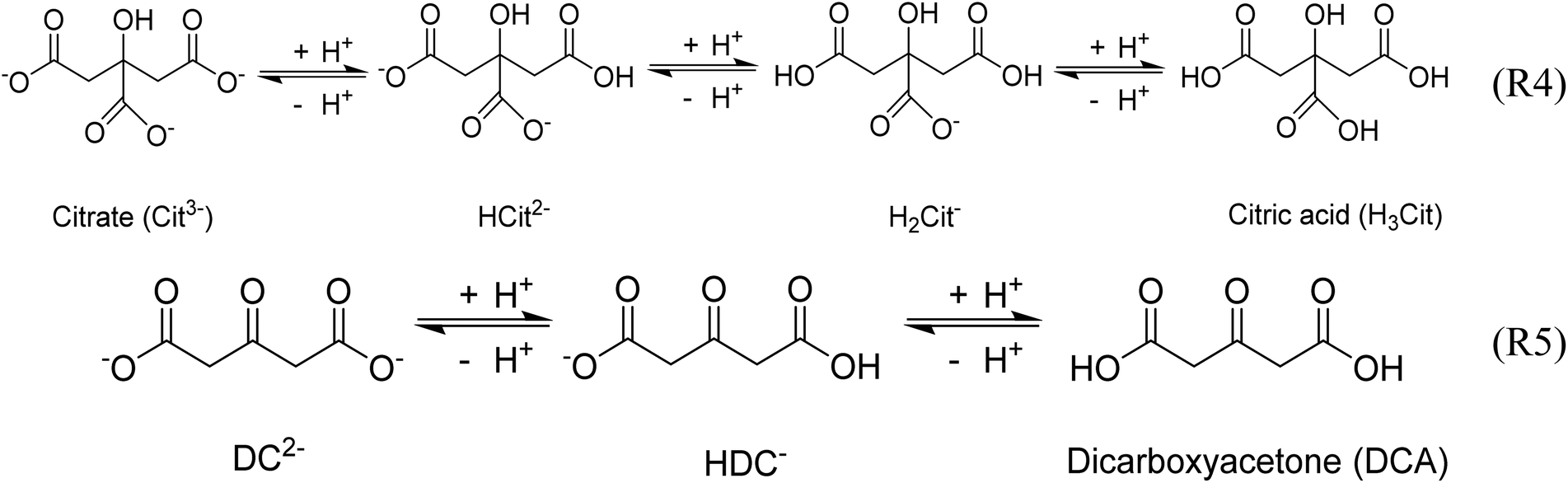 | ||
| Scheme 3 The dissociation of citrate and DCA. | ||
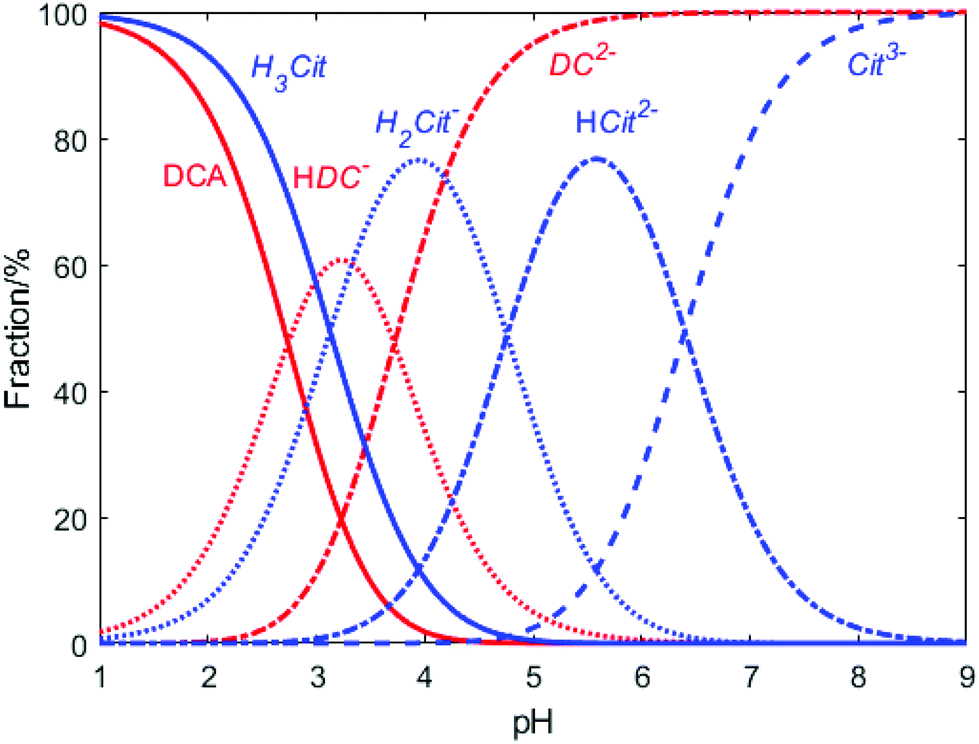 | ||
| Fig. 11 Relative concentration of the different dissociation species of sodium citrate and DCA as a function of pH. H3Cit, H2Cit−, HCit2− and Cit3− represent different forms of citrate. DCA, HDC− and DC2− represent different forms of dicarboxyacetone (according to reactions (3) and (4). The pKa values for citrate hydrolysis are 3.1, 4.76, 6.4, respectively; the pKa values for dicarboxyacetone are 2.74 and 3.72 respectively. | ||
These results are in agreement with the literature where optimum pH values between 5.4 and 6.0 (ref. 18) are demonstrated to maximize the molar fraction of HCit2− in the solution. However, this work also demonstrates that this pH range maximizes both, the HCit2− and the DC2−, both having a critical role in controlling the nucleation step, but the latter having a higher reduction potential as previous shown.
Finally, when AuCl is used as a gold precursor, a black precipitation is always observed, formed from the beginning of the reaction. SEM-EDX indicates it consists of mainly gold with a minimum (∼0.5%) of Cl. Such precipitates are not observed when HAuCl4 is used as precursor suggesting that due to the low solubility of AuCl, its solid form serves as nucleation points during the reduction process, leading to such precipitation.
4. Conclusions
This study challenges the traditional Turkevich mechanism for the synthesis of gold nanoparticles using citrate as reducing agent demonstrating that it consists of two consecutive reduction steps rather than a reduction followed by the disproportionation reaction as commonly believed in the literature. DC2−, formed as intermediate product through the oxidation of citrate, plays a key role in the synthesis as it has a considerably higher reduction potential than citrate and enables the electrostatic stabilization of the Au nanoparticles through coordination of its enol form. As a consequence, high nucleation rates can be achieved in the presence of DC2− leading to monodispersed particles. Directly related, while previous research considered the first reduction (Au3+ → Au+) as the rate-determining step to control the size of Au nanoparticles, in this paper, the second reduction reaction (Au+ → Au0) is inferred as the rate-limiting step. In this way, the size of Au NPs can be tuned from 5.2 ± 1.7 nm to 21.4 ± 3.4 nm by tuning the conditions of the second reduction step.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly acknowledge the financial support from UK Engineering and Physical Science and Research Council (grant number EP/L020443/2) and YH thanks the Chinese Scholarship Council and The Cambridge Trust for his studentship. The authors thank Dr Mauro Malizia for his contribution on electrochemical experiments and Dr Florian Mulks for his discussion.References
- S. E. Lohse and C. J. Murphy, J. Am. Chem. Soc., 2012, 134, 15607 CrossRef CAS PubMed.
- T. García, S. Agouram, A. Dejoz, J. F. Sánchez-Royo, L. Torrente-Murciano and B. Solsona, Catal. Today, 2015, 248, 48 CrossRef.
- L. Torrente-Murciano, Q. He, G. J. Hutchings, C. J. Kiely and D. Chadwick, ChemCatChem, 2014, 6, 2531 CrossRef CAS.
- L. Torrente-Murciano, T. Villager and D. Chadwick, ChemCatChem, 2015, 7, 925 CrossRef CAS.
- A. B. Chinen, C. M. Guan, J. R. Ferrer, S. N. Barnaby, T. J. Merkel and C. A. Mirkin, Chem. Rev., 2015, 115, 10530 CrossRef CAS PubMed.
- L. Torrente-Murciano, B. Solsona, S. Agouram, R. Sanchis, J. M. López, T. García and R. Zanella, Catal. Sci. Technol., 2017, 7, 2886 RSC.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 7, 801 RSC.
- J. Turkevich and P. C. Stevenson, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- D. V. Goia and E. Matijević, Colloids Surf., A, 1999, 146, 139 CrossRef CAS.
- H. Xia, Y. Xiahou, P. Zhang, W. Ding and D. Wang, Langmuir, 2016, 32, 5870 CrossRef CAS PubMed.
- S. K. Sivaraman, S. Kumar and V. Santhanam, J. Colloid Interface Sci., 2011, 361, 543 CrossRef CAS PubMed.
- D. Grasseschi, R. A. Ando, H. E. Toma and V. M. Zamarion, RSC Adv., 2015, 5, 5716 RSC.
- S. Kumar, K. S. Gandhi and R. Kumar, Ind. Eng. Chem. Res., 2007, 46, 3128 CrossRef CAS.
- I. Ojea-Jiménez and J. M. Campanera, J. Phys. Chem. C, 2012, 116, 23682 CrossRef.
- B. Rodríguez-González, P. Mulvaney and L. M. Liz-Marzán, Z. Phys. Chem., 2007, 221, 415 CrossRef.
- C. H. Gammons and Y. Yu, Geochim. Cosmochim. Acta, 1997, 61, 1971 CrossRef CAS.
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, J. Am. Chem. Soc., 2007, 129, 13939 CrossRef CAS PubMed.
- F. Kettemann, A. Birnbaum, S. Witte, M. Wuithschick, N. Pinna, R. Kraehnert, K. Rademann and J. Polte, Chem. Mater., 2016, 28, 4072 CrossRef CAS.
- J. Turkevich, Gold Bull., 1985, 18, 125 CrossRef CAS.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20 CrossRef CAS.
- M. Tran, R. DePenning, M. Turner and S. Padalkar, Mater. Res. Express, 2016, 3, 105027 CrossRef.
- L. Shi, E. Buhler, F. Boué and F. Carn, J. Colloid Interface Sci., 2017, 492, 191 CrossRef CAS PubMed.
- K.-J. Wu, G. M. De Varine Bohan and L. Torrente-Murciano, React. Chem. Eng., 2017, 2, 116 RSC.
- B. Pinho and L. Torrente-Murciano, React. Chem. Eng., 2020, 10.1039/C9RE00452A.
- M. Wuithschick, A. Birnbaum, S. Witte, M. Sztucki, U. Vainio, N. Pinna, K. Rademann, F. Emmerling, R. Kraehnert and J. Polte, ACS Nano, 2015, 9, 7052 CrossRef CAS PubMed.
- W. Patungwasa and J. H. Hodak, Mater. Chem. Phys., 2008, 108, 45 CrossRef CAS.
- B. Bartosewicz, K. Bujno, M. Liszewska and P. Bazarnik, Colloids Surf., A, 2018, 549, 25 CrossRef CAS.
- I. Ojea-Jiménez, N. G. Bastús and V. Puntes, J. Phys. Chem. C, 2011, 115, 15752 CrossRef.
- E. Agunloye, A. Gavriilidis and L. Mazzei, Chem. Eng. Sci., 2017, 173, 275 CrossRef CAS.
- E. Agunloye, L. Panariello, A. Gavriilidis and L. Mazzei, Chem. Eng. Sci., 2018, 191, 318 CrossRef CAS.
- M. Doyen, K. Bartik and G. Bruylants, J. Colloid Interface Sci., 2013, 399, 1 CrossRef CAS PubMed.
- A. Jakhmola and R. Vecchione, Mater. Today Chem., 2019, 1, 100203 CrossRef.
- B. K. Pong, H. I. Elim, J. X. Chong, W. Ji, B. L. Trout and J. Y. Lee, J. Phys. Chem. C, 2007, 111, 6281 CrossRef CAS.
- A. Jakhmola, M. Celentano, R. Vecchione, A. Manikas, E. Battista, V. Calcagno and P. A. Netti, Inorg. Chem. Front., 2017, 4, 1033 RSC.
- L. Pei, K. Mori and M. Adachi, Langmuir, 2004, 20, 7837 CrossRef CAS PubMed.
- J. Piella, N. G. Bastús and V. Puntes, Chem. Mater., 2016, 28, 1066 CrossRef CAS.
- M. Celentano, A. Jakhmola, M. Profeta, E. Battista, D. Guarnieri, F. Gentile, P. A. Netti and R. Vecchione, Colloids Surf., A, 2018, 558, 548 CrossRef CAS.
- B. Michen, C. Geers, D. Vanhecke, C. Endes, B. Rothen-Rutishauser, S. Balog and A. Petri-Fink, Sci. Rep., 2015, 5, 9793 CrossRef CAS PubMed.
- D. G. Castner, K. Hinds and D. W. Grainger, Langmuir, 1996, 12, 5083 CrossRef CAS.
- Y. Mikhlin, A. Karacharov, M. Likhatski, T. Podlipskaya, Y. Zubavichus, A. Veligzhanin and V. Zaikovski, J. Colloid Interface Sci., 2011, 362, 330 CrossRef CAS PubMed.
- Y. Cheng, J. Tao, G. Zhu, J. A. Soltis, B. A. Legg, E. Nakouzi, J. J. De Yoreo, M. L. Sushko and J. Liu, Nanoscale, 2018, 10, 11907 RSC.
- V. K. Lamer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847 CrossRef CAS.
- N. G. Bastús, J. Comenge and V. Puntes, Langmuir, 2011, 27, 11098 CrossRef PubMed.
- K. J. Wu and L. Torrente-Murciano, React. Chem. Eng., 2018, 3, 267 RSC.
- I. Ojea-Jimenez, F. M. Romero, N. G. Bastus and V. Puntes, J. Phys. Chem. C, 2010, 114, 1800 CrossRef CAS.
- C. E. Hoppe, M. Lazzari, I. Pardiñas-Blanco and M. A. López-Quintela, Langmuir, 2006, 22, 7027 CrossRef CAS PubMed.
- T. Hendel, M. Wuithschick, F. Kettemann, A. Birnbaum, K. Rademann and J. Polte, Anal. Chem., 2014, 86, 11115 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr08877f |
| This journal is © The Royal Society of Chemistry 2020 |