
How does excited-state antiaromaticity affect the acidity strengths of photoacids?†
Zhili
Wen‡
 ,
Lucas José
Karas‡
,
Chia-Hua
Wu
and
Judy I-Chia
Wu
*
,
Lucas José
Karas‡
,
Chia-Hua
Wu
and
Judy I-Chia
Wu
*
Department of Chemistry, University of Houston, Houston, TX 77204, USA. E-mail: jiwu@central.uh.edu
First published on 27th May 2020
Abstract
Photoacids like substituted naphthalenes (X = OH, NH3+, COOH) are aromatic in the S0 state and antiaromatic in the S1 state. Nucleus independent chemical shifts analyses reveal that deprotonation relieves antiaromaticity in the excited conjugate base, and that the degree of “antiaromaticity relief” explains why some photoacids are stronger than others.
Baird first proposed a set of rules suggesting that the electron-counting rules of aromaticity and antiaromaticity reverse in the lowest triplet states of π-conjugated cycles.1 Based on this set of rules, [4n + 2] π-rings are antiaromatic and [4n] π-rings are aromatic in the first ππ* state. These predictions were later extended to the first singlet ππ* states (S1), and explained the reactivities of many Baird-type antiaromatic [4n + 2] π-systems.2–7 Benzene, in the S1 state, is reactive and readily isomerizes to fulvene.7,8 Among other [4n + 2] π-ring systems, salicylic acid, in the S1 state, undergoes intramolecular proton transfer.9,10 Here we show that, in the S1 state, differences in the acidity strengths of photoacids might be rationalized by the effects of “antiaromaticity relief” upon deprotonation, followed by a redistribution of electrons in the excited conjugate base.
Following the early independent works of Förster11,12 and Weller,13–15 it was recognized that some aromatic acids (e.g., with hydroxyl or ammonium groups) can turn into stronger Brønsted acids in their first excited (S1) states.11–16 2-Naphthol, a prototypical organic “photoacid,” is a weak acid in the ground state, but shows enhanced acidity in the S1 state (pKa = 9.5, pKa* = 2.8, ΔpKa = −6.7), and can deprotonate to the solvent producing an electronically excited conjugate base.15 A Stokes’ shift in the fluorescence spectrum of 2-naphthol in water was interpreted by Förster as radiative decay emitting from the excited conjugate base (see depiction of “Förster cycle” in Scheme 1).11 Yet, despite a large body of theoretical and experimental efforts towards understanding excited-state proton transfer reactions in aromatic acids,17–27 reasons underlying the occurrence of photoacidity remain unclear. The disparate effects of substituents on photoacidity are even more puzzling. 2-Naphthylammonium displays increased acidity in the S1 state (pKa = 4.1, pKa* = −0.8, ΔpKa = −4.9), deprotonating from an NH3+ group, but the change in acidity, ΔpKa, is two-folds less than that of 2-naphthol.28 Aromatic acids, like the 2-naphthoic acid, show the opposite effect and exhibit decreased acidity in the S1 state (pKa = 4.2, pKa* = 11.5, ΔpKa = +7.3) (Table 1).29
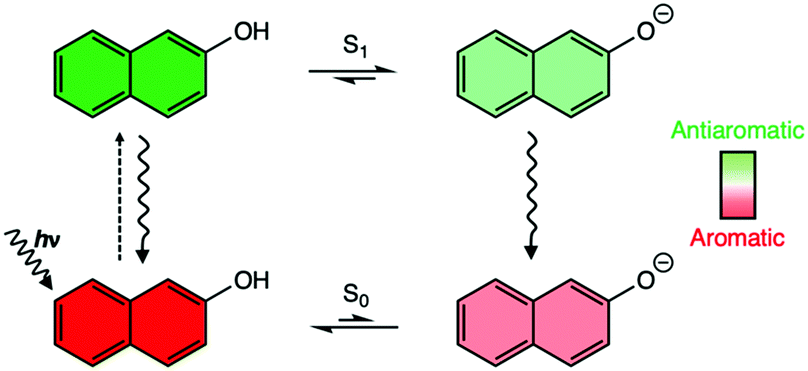 | ||
| Scheme 1 The Förster cycle of 2-naphthol. As indicated by the color scheme above, 2-naphthol is [4n + 2] π-aromatic in the ground state (red) but becomes [4n + 2] π-antiaromatic in the S1 state (green). Deprotonation relieves excited-state antiaromaticity, stabilizing the excited conjugate base (light green). In the S0 state, the conjugate base (light red) is only moderately less aromatic than the acid. | ||
Why do substituents have such disparate effects on the photoacidities of aromatic acids? Here, we relate the effects of photoacidity to a switch in the ground and excited-state (anti)aromatic character of aromatic acids. According to the Hückel rule, cyclic π-conjugated rings with [4n + 2] π-electrons are aromatic, and those with [4n] π-electrons are antiaromatic.30 But this electron-counting rule reverses in the first ππ* state following Baird's rule.1–7 2-Naphthol is [4n + 2] Hückel aromatic (ten π-electrons in naphthalene) in the ground state, but becomes [4n + 2] antiaromatic in the S1 state. Upon deprotonation, an excited conjugate base forms, and negative charge on the O− delocalizes into the ring, giving rise to a resonance structure with breached cyclic [4n + 2] π-electron delocalization—this alleviates antiaromaticity in the S1 state of the acid (see Scheme 1 and resonance structure for the excited conjugate base in Fig. 1a, note delocalization of the negative charge into the ring). In this way, photoacidity might be considered as a consequence of antiaromaticity relief in the S1 states of aromatic acids.6 Based on a more bond equalized S1vs. S0 state of 2-naphtholate, Agmon et al. pointed out similarly that the excited conjugate base of 2-naphthol might be stabilized by increased aromatic character.19,20 The effects of ground and excited-state (anti)aromaticity also have been recognized in other excited-state proton transfer processes.31,32
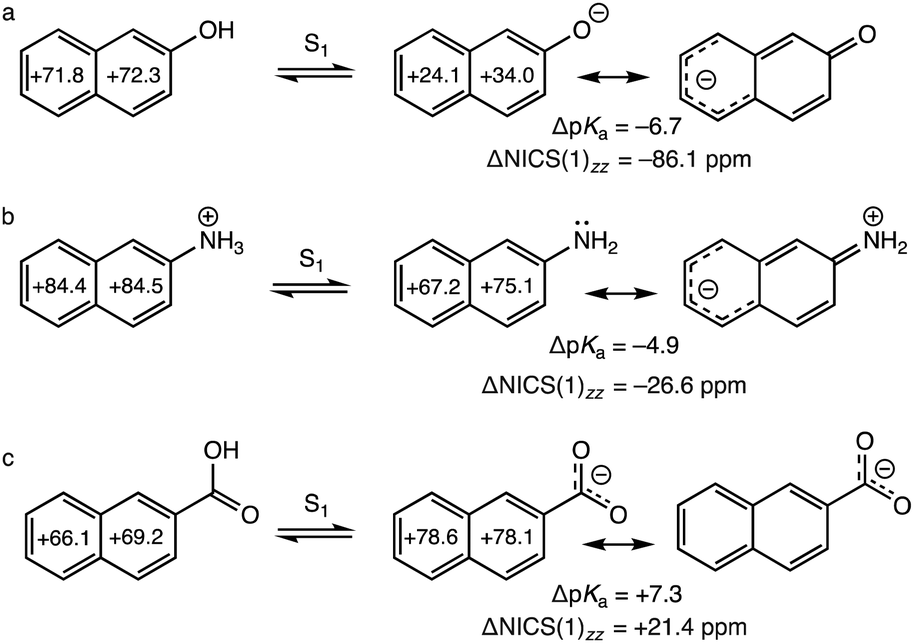 | ||
| Fig. 1 Computed NICS(1)zz and ΔNICS(1)zz (sum of NICS(1)zz values of the conjugate base minus that of the acid) (in ppm) values for the S1 states of the acid and conjugate base at CASSCF(12,12)/6-311+G(d,p), for (a) 2-naphthol, (b) 2-naphthylammonium, and (c) 2-naphthoic acid. Negative ΔNICS(1)zz values indicate antiaromaticity relief, and positive values indicate antiaromaticity gain, upon formation of the excited conjugate base. Experimental ΔpKa values are included for reference. | ||
Even though not realized at the time, the possible effects of excited-state antiaromaticity relief were implied in Weller's original explanation (1950's) of photoacidity—argued based on a redistribution of ring π-electrons in the S1 states of the aromatic acids;13–15 notably, these ideas were published roughly ten years prior to Dewar33 and Zimmerman's34 independent works and Baird's1 proposal (60's–70's) of a reversed Hückel π-electron-counting rule for aromaticity and antiaromaticity in the first ππ* states of transition states33,34 and of π-conjugated rings.1 Weller reasoned that when 2-naphthol is electronically excited to the S1 state, intramolecular charge transfer from the hydroxyl oxygen to the aromatic ring increases acidity of the OH group. Later, it was suggested that even more pronounced charge redistribution happens upon deprotonation (as indicated by shortened C–O bond lengths and changes in dipole moments),21 stabilizing the excited conjugate base. We now relate the effects of “charge redistribution” in the excited conjugate base to “relief of excited-state antiaromaticity.” This rationale also may explain why photoacidity only is observed for aromatic acids (i.e., with [4n + 2] ring π-electrons), but not for other hydroxyl, amine, or ammonium compounds.
Compared to 2-naphthol, the effect of charge redistribution for alleviating antiaromaticity in the excited conjugate base of 2-naphthylammonium is much weaker, since delocalization of a neutral nitrogen lone pair into the naphthalene ring is less effective (see Fig. 1b, note charge separated resonance form). In the S1 state of 2-naphthylammonium, deprotonation produces a neutral amine (NH2); proton transfer alleviates antiaromaticity of the excited naphthalene ring, but to a lesser degree compared to that of 2-naphthol. Notably, compounds with competing deprotonation sites like salicylamide35,36 and 3-ammonium-2-naphthol37 undergo proton transfer from NH3+ in the ground state (i.e., to retain aromaticity of the π-ring), but deprotonate from the OH group in the S1 state (i.e., to alleviate excited-state antiaromaticity of the π-ring) when solvated in water. In the S1 state of 2-naphthoic acid, deprotonation of the carboxylic group gives a carboxylate (COO−). But negative charge is mostly delocalized between the two oxygen atoms, and does not help lessen excited-state antiaromaticity in the naphthalene ring (Fig. 1c).
We performed dissected nucleus independent chemical shifts,38,39 NICS(1)zz, to quantify excited-state antiaromaticity in the S1 states40 of the acids and conjugate bases of 2-naphthol, 2-naphthylammonium, and 2-naphthoic acid (Fig. 1). The computed ring NICS(1)zz values of excited 2-naphthol are large and positive (+71.8, +72.3 ppm, strongly antiaromatic) but become much less so in the excited conjugate base (+24.1, +34.0 ppm, weakly antiaromatic), suggesting decreased antiaromaticity upon deprotonation of the excited acid (ΔNICS(1)zz = −86.1 ppm, Fig. 1a). 2-Naphthylammonium reveals a lesser degree of antiaromaticity relief upon deprotonation (ΔNICS(1)zz = −26.6 ppm, Fig. 1b). Accordingly, the computed exocyclic CO and CN bond distances of 2-naphthol (1.350 Å) and 2-naphthylammonium (1.476 Å), are longer in the S1 state acid, and shorter in the excited conjugate base (1.247 Å and 1.373 Å, respectively), indicative of electron delocalization from the deprotonated site into the excited naphthalene ring (see optimized geometries in the ESI†).
In contrast, computed ring NICS(1)zz values for 2-naphthoic acid in the S1 state are positive for the acid (+66.1, +69.2 ppm, strongly antiaromatic) but become even more so in the excited conjugate base (+78.6, +78.1 ppm, strongly antiaromatic), suggesting increased antiaromaticity upon deprotonation of the excited acid (ΔNICS(1)zz = +21.4 ppm, Fig. 1c). The exocyclic C–C bond of the excited acid is 1.471 Å (cf. 1.40 Å CC length of benzene), indicating modest π-conjugation between the carboxylic group and the naphthalene ring. In the excited conjugate base, the exocyclic C–C bond lengthens to 1.534 Å (cf. 1.53 Å CC length of ethane), suggesting little resonance between the exocyclic carboxylate group and the excited (antiaromatic) naphthalene (see geometries in the ESI†). In the ground state, deprotonation has less effect on the 10 π-electron aromatic character of the naphthalene ring, in 2-naphthol (ΔNICS(1)zz = +8.5 ppm), 2-naphthylammonium (ΔNICS(1)zz = +2.2 ppm), and 2-naphthoic acid (ΔNICS(1)zz = −1.2 ppm); positive/negative values indicate aromaticity loss/gain (see full data in the ESI†).
Naphthols with cyano (CN) substituents at the C5 and C8 positions are very strong photoacids: 8-cyano-2-naphthol (pKa = 8.4, pKa* = −0.8, ΔpKa = −9.2) and 5,8-dicyano-2-naphthol (pKa = 7.8, pKa* = −4.5, ΔpKa =−–12.3) show increased acidities of up to 12 units in the S1 state.17,18,41,42 These strong photoacids can undergo excited-state proton transfer reactions in methanol, methylsulfonyl, and other organic solvents in the absence of water, first expanding the possibility of studying proton transfer kinetics in non-aqueous solvents.41 Computed ring NICS(1)zz values for the S1 state of 8-cyano-2-naphthol and its excited conjugate base (ΔNICS(1)zz = −137.2 ppm, Fig. 2a) show significant excited-state antiaromaticity relief upon deprotonation, and the effect in 5,8-dicyano-2-naphthol (ΔNICS(1)zz = −167.3 ppm, Fig. 2b) is even greater (cf. ΔNICS(1)zz = −86.1 ppm, for 2-naphthol). As suggested by the resonance forms in Fig. 2, the electron-withdrawing CN groups help increase charge redistribution in the excited conjugate base, by inductive effects, but also by resonance stabilization (see resonance contributors with negative charges delocalized to the nitrogen atoms). We note that other known strong photoacids with electron-withdrawing groups, e.g., sulfonyls, also have π-systems on the substituents capable of delocalizing negative charge of the excited conjugate base.
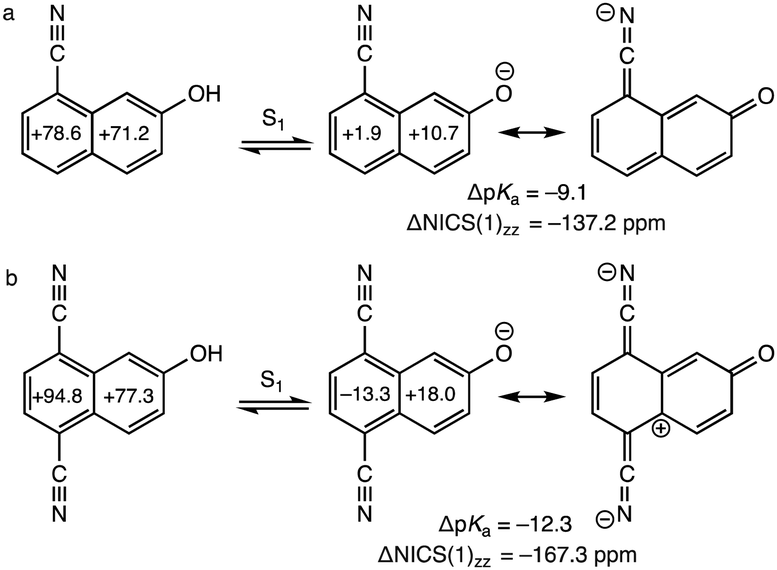 | ||
| Fig. 2 Computed NICS(1)zz and ΔNICS(1)zz (sum of NICS(1)zz values of the conjugate base minus that of the acid) (in ppm) values for the S1 states of the acid and conjugate base at CASSCF(12,12)/6-311+G(d,p), for (a) 8-cyano-2-naphthol and (b) 5,8-dicyano-2-naphthol. Negative ΔNICS(1)zz values indicate antiaromaticity relief upon formation of the excited conjugate base. Experimental ΔpKa values are included for reference. Note resonance form showing delocalization of the negative charge into the CN groups. | ||
Computed deprotonation reaction energies (ΔE) based on the equation: ArOH + H2O → ArO− + H3O+, document the energetic effects of (anti)aromaticity loss in the S0 and S1 states upon deprotonation, and agree with the conclusions based on NICS analyses. Compared to computed ΔE values in the S0 state, deprotonation is less endothermic for the S1 states of 2-naphthol (ΔΔE = −17.99 kcal mol−1, ΔΔE = ΔE(S1) − ΔE(S0)), 2-naphthylammonium (−2.77 kcal mol−1), 8-cyano-2-naphthol (−28.85 kcal mol−1), and 5,8-dicyano-2-naphthol (−29.28 kcal mol−1), but more endothermic for the S1 states of 2-naphthoic acid (+1.39 kcal mol−1) (see full data in the ESI†).
Aromatic acids with competing deprotonation sites like salicylamide undergo proton transfer from NH3+ in the S0 state, but deprotonate from the OH group in the S1 state.35,36 In the S0 state, computed NICS(1)zz for the acid (−24.4 ppm) and conjugate base (−24.5 ppm, deprotonated at NH3+) give nearly the same values (ΔNICS(1)zz = +0.1 ppm) (Fig. 3, bottom). But in the S1 state, the ring NICS(1)zz value for protonated salicylamide is large and positive (+40.2 ppm) while that for the excited conjugate base is modestly negative (−4.2 ppm, deprotonated at OH), documenting the effects of excited-state antiaromaticity relief (ΔNICS(1)zz = −44.4 ppm) (Fig. 3, top). Notably, when a proton is removed from the NH3+ site of the electronically excited acid, computed NICS(1)zz for the excited conjugate base (+65.8 ppm) show increased excited-state antiaromaticity in the benzene ring.
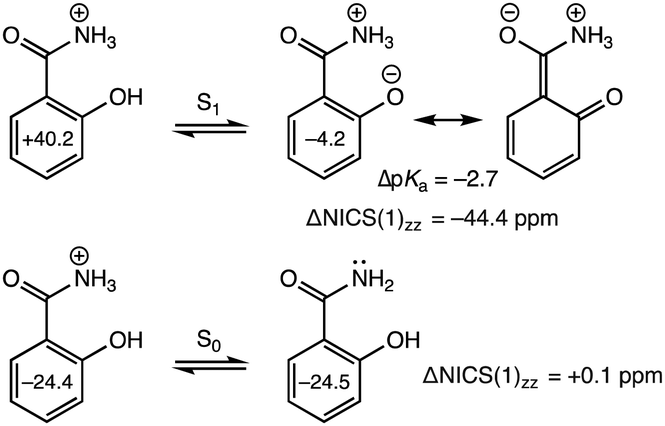 | ||
| Fig. 3 Computed NICS(1)zz and ΔNICS(1)zz (sum of NICS(1)zz values of the conjugate base minus that of the acid) (in ppm) values for the S0 and S1 states of protonated salicylamide an its conjugate base at CASSCF(12,12)/6-311+G(d,p). Negative ΔNICS(1)zz values indicate antiaromaticity relief upon formation of the excited conjugate base. Positive ΔNICS(1)zz values in the ground state indicate aromaticity loss upon formation of the conjugate base. An experimental ΔpKa value is included for reference. | ||
A long-standing and much debated anomaly is the stronger photoacidity of 1-naphthol (pKa = 9.2, pKa* = −0.5, ΔpKa = −9.7) compared to its structurally similar 2-naphthol isomer (pKa = 9.5, pKa* = 2.8, ΔpKa = −6.7); note 3-fold ΔpKa difference!43–45 Based on time-resolved emission spectroscopy and steady state spectrofluorometry experiments, 1-naphthol displayed a rate of deprotonation (k1*) greater than 2-naphthol by 280 times.44 It was proposed that the stronger photoacidity of 1-naphthol was a consequence of populating and emitting from the 1La state, while 2-napththol shows near degenerate 1La and 1Lb states, emitting from the 1Lb state.17,46,47 We speculate that naphthols promoted to the 1La state deprotonate more readily because of pronounced antiaromatic character in the 1La state of naphthalene. Computed geometric indices of aromaticity for excited naphthalene show a more bond alternated 1La state and a more bond equalized 1Lb state (see data in the ESI†). See also resonance structures of naphthalene in the 1La (B2u) state (“diradical form,” note “antiaromatic” Clar sextet structure in the 1La) and 1Lb (B3u) state (“allyl radical form”) (Scheme 2). We note that Baird's original paper on the effects of triplet (anti)aromaticity also recognized a more antiaromatic “diradical form” for triplet benzene.1
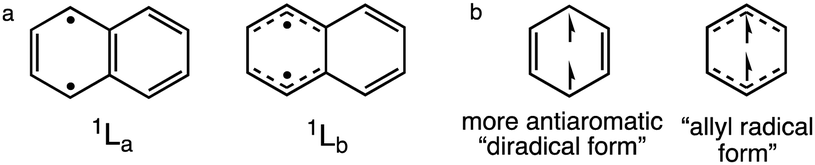 | ||
| Scheme 2 (a) Resonance structures for the 1La and 1Lb states of excited state naphthalene, and (b) the “diradical” (B1u) and “allyl radical” (B2u) forms of triplet benzene. | ||
Although substituents are typically considered to have negligible effects on aromaticity (unless charged or in the presence of other push/pull substituents),48,49 they can easily perturb the excited-state antiaromaticity of organic compounds,50 having tremendous effects on reactions such as the excited-state proton transfer of photoacids. These findings are another manifestation of the increasingly recognized effects of excited-state (anti)aromaticity on the photochemical reactivity of organic compounds.
Computational methods: Geometry optimizations for the ground (S0) and excited (S1, 1ππ*) states of all acid and conjugate base structures were performed at CASSCF(12,12)/6-311+G(d,p) with Cs symmetry, employing Molpro2012.1.51 The S0 and S1 geometries of 5,8-dicyano-2-naphthol and its conjugate base were computed at CASSCF(10,10)/6-311+G(d,p) with Cs symmetry. Computed dissected nucleus-independent chemical shifts, NICS(1)zz,38,39 were performed at CASSCF(12,12)/6-31G(d,p), employing the Dalton2016 program,52 to quantify the magnetic effects of aromaticity and antiaromaticity in the S0 and S1 states40 of the acids and conjugate bases. NICS(1)zz values were computed at 1 Å above each of the six membered ring centers and include only contributions from the “out-of-plane” (zz) tensor component perpendicular to the ring plane. ΔNICS(1)zz values were calculated based on the sum of ring NICS(1)zz values of the conjugate base minus that of the acid.
J. I. W. thanks the National Science Foundation (NSF) (CHE-1751370) and the National Institute of General Medical Sciences (NIGMS) of the National Institute of Health (R35GM133548) for grant support. The authors acknowledge the use of the Sabine cluster and support from the Research Computing Data Core at the University of Houston.
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. C. Baird, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef CAS.
- J.-I. Aihara, Bull. Chem. Soc. Jpn., 1978, 51, 1788–1792 CrossRef CAS.
- P. B. Karadakov, J. Phys. Chem. A, 2008, 112, 7303–7309 CrossRef CAS PubMed.
- P. B. Karadakov, J. Phys. Chem. A, 2008, 112, 12707–12713 CrossRef CAS PubMed.
- F. Feixas, J. Vandenbussche, P. Bultinck, E. Matito and M. Solà, Phys. Chem. Chem. Phys., 2011, 13, 20690–20703 RSC.
- M. Rosenberg, C. Dahlstrand, K. Kilså and H. Ottosson, Chem. Rev., 2014, 114, 5379–5425 CrossRef CAS PubMed.
- R. Papadakis and H. Ottosson, Chem. Soc. Rev., 2015, 44, 6472–6493 RSC.
- L. Kaplan and K. E. Wilzbach, J. Am. Chem. Soc., 1968, 90, 3291–3292 CrossRef CAS.
- S. Nagaoka, U. Nagashima, N. Ohta, M. Fujita and T. Takemura, J. Phys. Chem., 1988, 92, 166–171 CrossRef CAS.
- S. Nagaoka and U. Nagashima, Chem. Phys., 1989, 136, 153–163 CrossRef CAS.
- T. Förster, Naturwissenschaften, 1949, 36, 186–187 CrossRef.
- T. Förster, Z. Elektrochem., 1950, 54, 531–535 Search PubMed.
- A. Weller, Z. Elektrochem., 1952, 56, 662–668 CAS.
- A. Weller, Prog. React. Kinet., 1961, 1, 187–214 CAS.
- A. Weller, Z. Phys. Chem., 1958, 15, 438–453 CrossRef CAS.
- J. F. Ireland and P. A. H. Wyatt, Adv. Phys. Org. Chem., 1976, 12, 131–221 CrossRef CAS.
- L. M. Tolbert and J. E. Haubrich, J. Am. Chem. Soc., 1994, 116, 10593–10600 CrossRef CAS.
- L. M. Tolbert and K. M. Solntsev, Acc. Chem. Res., 2002, 35, 19–27 CrossRef CAS PubMed.
- N. Agmon, W. Rettig and C. Groth, J. Am. Chem. Soc., 2002, 124, 1089–1096 CrossRef CAS PubMed.
- N. Agmon, J. Phys. Chem. A, 2005, 109, 13–35 CrossRef CAS PubMed.
- G. Granucci, J. T. Hynes, P. Millié and T.-H. Tran-Thi, J. Am. Chem. Soc., 2000, 122, 12243–12253 CrossRef CAS.
- L. N. Silverman, D. B. Spry, S. G. Boxer and M. D. Fayer, J. Phys. Chem. A, 2008, 112, 10244–10249 CrossRef CAS PubMed.
- H. Shizuka, Acc. Chem. Res., 1985, 18, 141–147 CrossRef CAS.
- P. Wan and D. Shukla, Chem. Rev., 1993, 93, 571–584 CrossRef CAS.
- M. Rini, B.-Z. Magnes, E. Pines and E. T. J. Nibbering, Science, 2003, 301, 349–352 CrossRef CAS PubMed.
- O. F. Mohammed, D. Pines, J. Dreyer, E. Pines and E. T. J. Nibbering, Science, 2005, 310, 83–86 CrossRef CAS PubMed.
- D. Pines, E. T. J. Nibbering and E. Pines, Isr. J. Chem., 2015, 55, 1240–1251 CrossRef CAS.
- K. Tsutsumi and H. Shizuka, Chem. Phys. Lett., 1977, 52, 485–488 CrossRef CAS.
- E. L. Wehry and L. B. Rogers, J. Am. Chem. Soc., 1966, 88, 351–354 CrossRef CAS.
- E. Hückel, Z. Phys., 1931, 70, 204–286 CrossRef.
- C. H. Wu, L. J. Karas, H. Ottosson and J. I. Wu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 20303–20308 CrossRef CAS PubMed.
- B. J. Lampkin, Y. H. Nguyen, P. B. Karadakov and B. VanVeller, Phys. Chem. Chem. Phys., 2019, 21, 11608–11614 RSC.
- M. J. S. Dewar, Angew. Chem., Int. Ed. Engl., 1971, 10, 761–776 CrossRef CAS.
- H. E. Zimmerman, Acc. Chem. Res., 1971, 4, 272–280 CrossRef CAS.
- S. G. Schulman, P. J. Kovi and J. F. Young, J. Pharm. Sci., 1973, 62, 1197–1199 CrossRef CAS PubMed.
- G. J. Woolfe and P. J. Thistlethwaite, J. Am. Chem. Soc., 1980, 102, 6917–6923 CrossRef CAS.
- K. J. Nelson, P. J. Brown, H. E. Rudel and K. Takematsu, Phys. Chem. Chem. Phys., 2019, 21, 24383–24392 RSC.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- C. Corminboeuf, T. Heine, G. Seifert, P. v. R. Schleyer and J. Weber, Phys. Chem. Chem. Phys., 2004, 6, 273–276 RSC.
- V. Gogonea, P. v. R. Schleyer and P. R. Schreiner, Angew. Chem., Int. Ed., 1998, 37, 1945–1948 CrossRef CAS.
- L. M. Tolbert and J. E. Haubrich, J. Am. Chem. Soc., 1990, 112, 8163–8165 CrossRef CAS.
- D. Huppert, L. M. Tolbert and S. Linares-Samaniego, J. Phys. Chem. A, 1997, 101, 4602 CrossRef CAS.
- C. M. Harris and B. K. Selinger, J. Phys. Chem., 1980, 84, 1366–1371 CrossRef CAS.
- S. P. Webb, S. W. Yeh, L. A. Philips, L. M. Tolbert and J. H. Clark, J. Am. Chem. Soc., 1984, 106, 7286–7288 CrossRef CAS.
- S. P. Webb, L. A. Philips, S. W. Yeh, L. M. Tolbert and J. H. Clark, J. Phys. Chem., 1986, 90, 5154–5164 CrossRef CAS.
- R. Knochenmuss, I. Fischer, D. Lührs and Q. Lin, Isr. J. Chem., 1999, 39, 221–230 CrossRef CAS.
- F. Messina, M. Prémont-Schwarz, O. Braem, D. Xiao, V. S. Batista, E. T. J. Nibbering and M. Chergui, Angew. Chem., Int. Ed., 2013, 52, 6871–6875 CrossRef CAS PubMed.
- T. M. Krygowski, K. Ejsmont, B. T. Stępień, M. K. Cyrański, J. Poater and M. Solà, J. Org. Chem., 2004, 69, 6634–6640 CrossRef CAS PubMed.
- T. M. Krygowski and B. T. Stępień, Chem. Rev., 2005, 105, 3482–3512 CrossRef CAS PubMed.
- M. Baranac-Stojanović, J. Org. Chem., 2020, 85, 4289–4297 CrossRef PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, WIREs Comput. Mol. Sci., 2011, 2, 242–253 CrossRef.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansik, H. J. Aa. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. Rybkin, P. Salek, C. C. M. Samson, A. Sánchez de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, WIREs Comput. Mol. Sci., 2014, 4, 269–284 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Cartesian coordinates for all structures, computed ground state NICS data, HOMA analyses, and bond alternation analysis for the 1La and 1Lb states of naphthalene. See DOI: 10.1039/d0cc02952a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |