
CO2 = CO + [O]: recent advances in carbonylation of C–H bonds with CO2
Lei
Song
a,
Yuan-Xu
Jiang
a,
Zhen
Zhang
*ab,
Yong-Yuan
Gui
ac,
Xiao-Yu
Zhou
a and
Da-Gang
Yu
 *a
*a
aKey Laboratory of Green Chemistry & Technology of Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, P. R. China. E-mail: dgyu@scu.edu.cn
bCollege of Pharmacy and Biological Engineering, Chengdu University, Chengdu 610106, P. R. China. E-mail: zhangzhen1@cdu.edu.cn
cCollege of Chemistry and Materials Science, Sichuan Normal University, Chengdu 610068, P. R. China
First published on 12th March 2020
Abstract
Carbon dioxide (CO2) is an ideal one-carbon source owing to its nontoxicity, abundance, availability, and recyclability. Although the thermodynamic stability and kinetic inertness of CO2 bring its utilization challenges, many groups have achieved significant progress in the utilization of CO2 to synthesize valuable carbonyl-containing compounds, which could be also generated via oxidative carbonylation of C–H bonds with CO. As CO2 has a higher oxidation state than CO, it could be considered as a combination of CO and oxidant (CO2 = CO + [O]), thus providing a safe, redox-neutral and economic strategy. In this Feature Article, we have summarized the recent advances in carbonylation of C–H bonds with CO2 based on this concept. The plausible mechanisms of such reactions and future of this field are also discussed.
 Lei Song | Lei Song was born in Hunan, China, in 1993. He received his MS degree in chemistry from Sichuan University in 2018 and has been a PhD student under the supervision of Prof. Da-Gang Yu. His current research focuses on the carboxylation/carbonylation of C–H bonds with CO2. |
 Yuan-Xu Jiang | Yuan-Xu Jiang was born in Sichuan Province, China, in 1996. He received his BS degree in applied chemistry from Huaqiao University in 2018. He is currently pursuing his MS degree at Sichuan University under the guidance of Prof. Da-Gang Yu. His research projects focus on the activation and utilization of CO2. |
 Zhen Zhang | Prof. Dr Zhen Zhang was born in Shanxi Province, China, in 1985. He received his bachelor's degree in 2007 from Sichuan University. In 2012, he joined Prof. Yong-Gang Zhi's group to pursue master and doctoral studies at Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences. From 2015, as a visiting PhD student, he joined Prof. Da-Gang Yu's laboratory at Sichuan University. He received his PhD in organic chemistry in 2017 and then joined Chengdu University as associate professor. His research projects focus on green chemistry and biochemistry. |
 Yong-Yuan Gui | Dr Yong-Yuan Gui was born in Henan, China, in 1987. He received his PhD degree in chemistry from Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences, in 2015 (with Prof. Li-Xin Wang). He then joined the laboratory of Prof. Da-Gang Yu at Sichuan University as a postdoctoral fellow, focusing on photoredox/nickel dual catalysis and asymmetric catalysis with CO2. In 2019, he joined the faculty of the College of Chemistry and Materials Science, Sichuan Normal University. His research interests include photocatalysis, asymmetric catalysis and other sustainable chemistry. |
 Xiao-Yu Zhou | Xiao-Yu Zhou was born in Chongqing, China, in 1996. She received her BS degree in applied chemistry from Northwest A&F University in 2018. She is currently pursuing her MS degree at Sichuan University under the guidance of Prof. Da-Gang Yu. Her research projects focus on the activation and utilization of CO2. |
 Da-Gang Yu | Prof. Dr Da-Gang Yu was born in Jiangxi province, China, in 1986. He received his PhD with Prof. Dr Zhang-Jie Shi from Peking University in 2012. Then he carried out postdoctoral research with Humboldt fellowship in the group of Prof. Dr Frank Glorius, Muenster University. Since 2015, he has been working independently in Sichuan University with support from “The Thousand Young Talents Plan” and National Natural Science Foundation of China-Outstanding Young Scholars. His research interests mainly focus on novel transformations of CO2, radical chemistry and novel transition metal-catalysis. He received the Thieme Chemistry Journal Award in 2017 and the Chinese Chemical Society Youth Award in 2018. |
1. Introduction
CO2 has been regarded as a promising C1 source for a long time as it is a nontoxic, abundant and recyclable building block providing various benefits in organic synthesis.1 Although the utilization of CO2 is restricted due to its thermodynamic stability and kinetic inertness, significant effort has been exerted toward the conversion of CO2 into value-added chemicals.2 Many kinds of organic transformations of CO2 have been developed in the last few decades to generate a variety of valuable organic molecules, including important carbonyl-containing compounds.As it is well known, carbonylation reaction is an important and useful method to synthesize carbonyl-containing compounds, which are widely found in natural products and pharmaceutical molecules.3 Traditionally, phosgene is the main carbonylation reagent due to its high reactivity.4 With the development of transition metal catalysis, the feedstock gas carbon monoxide (CO) has been extensively used as a replacement of phosgene in organic synthesis. Notably, the transition-metal-mediated and/or catalyzed oxidative carbonylation with CO via direct C–H bond functionalization5 attracts more and more attention owing to the high efficiency and economy (Scheme 1A).6 However, both highly toxic CO and stoichiometric oxidants, such as high-valence metal salts or O2, are indispensable in such carbonylation reactions, which causes environmental impact, safety issues and high cost. Therefore, it is highly desirable to develop a novel carbonylation strategy avoiding the use of oxidants and toxic CO. Because the carbon in CO2 shows a higher oxidation state than that in CO, CO2 might act similarly to the combination of CO and oxidant, that is, CO2 = CO + [O] (Scheme 1B).2t Following this concept, if we use CO2 as the carbonylation source in carbonylation of C–H bonds, we could realize such transformations under redox-neutral conditions, and ideally, with “H2O” (or carbonate in the presence of base) as the only byproduct, thus solving the problems remaining in the oxidative carbonylation reaction of C–H bonds with CO. Although it looks simple and fantastic, there are huge challenges to formally cleave the strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, which is highly energy-demanding. In this Feature Article, we have summarized very recent advances in carbonylation of C–H bonds with CO2, such as lactamization and lactonization, based on this concept. The plausible mechanisms of such reactions and future of this field are discussed. Another important strategy, in situ reduction of CO2 to CO and the following carbonylation with CO,6j,k is different from the concept here and will be only briefly mentioned.
O bond, which is highly energy-demanding. In this Feature Article, we have summarized very recent advances in carbonylation of C–H bonds with CO2, such as lactamization and lactonization, based on this concept. The plausible mechanisms of such reactions and future of this field are discussed. Another important strategy, in situ reduction of CO2 to CO and the following carbonylation with CO,6j,k is different from the concept here and will be only briefly mentioned.
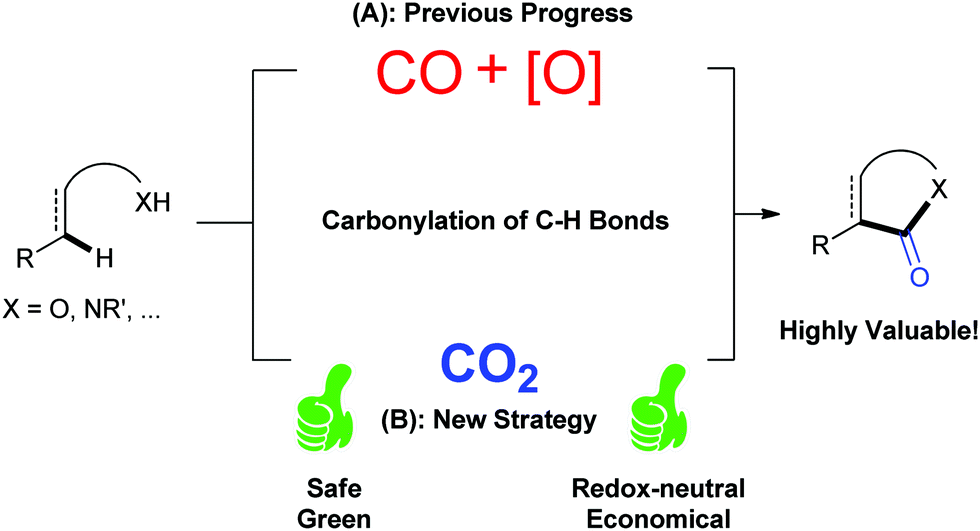 | ||
| Scheme 1 CO2 = CO + [O]: new strategy of carbonylation of C–H bonds with CO2. | ||
2. Lactamization of C–H bonds with CO2
Lactam-containing heterocyclic compounds are common motifs in drugs and bioactive natural products, as well as important intermediates in organic synthesis (Scheme 2).7 Recently, many groups, such as Alper, Zhu, Chuang, and Zhang groups, have developed nice and efficient methodologies to synthesize these compounds through transition-metal-catalyzed oxidative carbonylation of C–H bonds with CO.8 However, the requirement of (sub)stoichiometric metal oxidants (Ag+ or Cu2+ salts) represents a major limitation associated with the methods described above.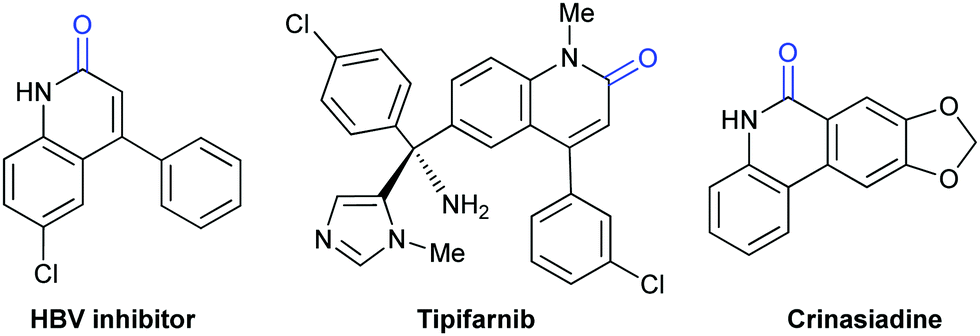 | ||
| Scheme 2 Selected biologically active lactam-containing moieties. | ||
Different from previous strategies, our group reported the direct lactamization of C(sp2)–H bonds in o-alkenyl- or o-heteroaryl-anilines with CO2 in the presence of base (Scheme 3).9 In these reactions, CO2 serves as a carbonylation reagent to avoid the use of external chemical oxidants and toxic CO. These transition metal-free and redox-neutral reactions are simple to operate and user-friendly. By heating o-alkenyl- and o-heteroaryl-anilines with NaOtBu in diglyme under 1 atm of CO2 at 140 °C for 24 hours, a series of important quinolinones and polyheterocyclic compounds were delivered in high efficiency, making it an attractive method for the pharmaceutical industry. We further showed the potential applications of this transformation toward some valuable intermediates and biologically active compounds, including an HBV inhibitor and a key intermediate to Tipifarnib.
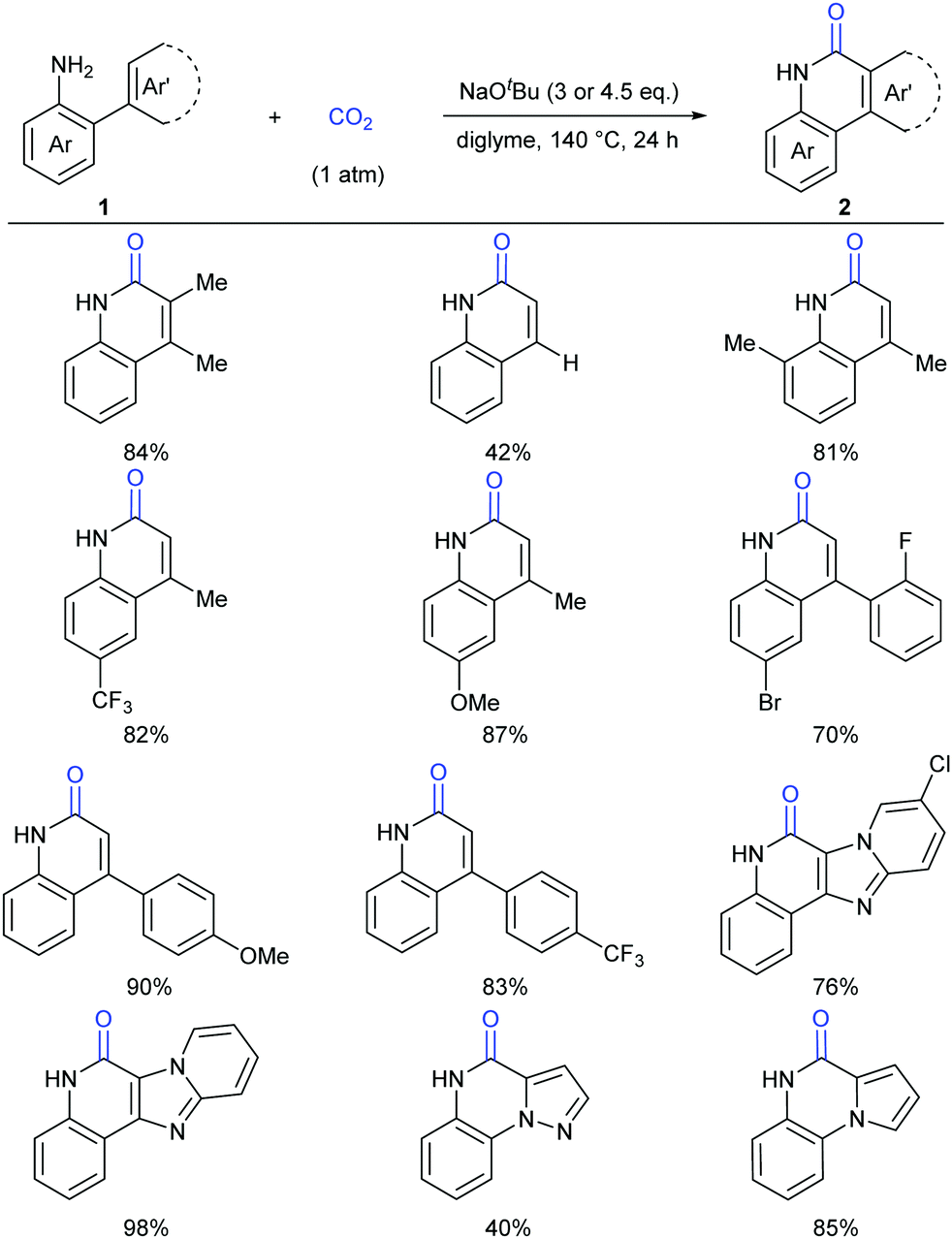 | ||
| Scheme 3 Transition metal-free carbonylation of C(sp2)–H bonds in o-alkenyl- and o-heteroaryl-anilines with CO2 in the presence of NaOtBu. | ||
A series of control experiments were conducted to probe the mechanism of this process. First, NaOtBu-mediated nucleophilic attack of aniline 3 on CO2 under the reaction conditions would generate intermediates 5 and 5′, which could undergo elimination to give the key intermediate isocyanates 6. The following cyclization reaction would efficiently afford the stable products 4 and/or the corresponding deprotonated versions, which might also be a driving force for this transformation (Scheme 4).
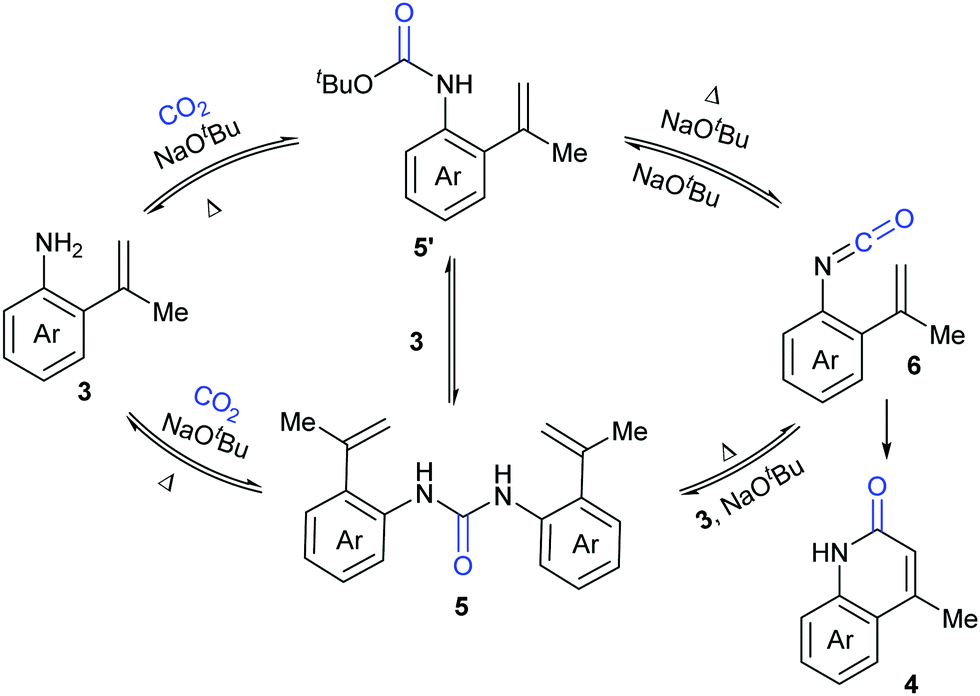 | ||
| Scheme 4 Possible mechanism for the lactamization reaction. | ||
This process was further demonstrated by the Li group.10 Based on the DFT calculation, they speculated that the reaction went through three stages: (i) C–N bond construction to generate the intermediate carbamate species; (ii) deoxygenation of carbamate species to generate isocyanate species 6; (iii) intramolecular cyclization and H migration to yield the final product 4. The deoxygenation of the carbamate species in stage (ii) was the rate-determining step for the entire reaction and the base was very important in overcoming the energetic bottleneck in stages (i) and (iii).
Although our system worked well for those anilines bearing electron-rich alkene or heteroarenes at the ortho position, it was not suitable for the unactivated o-phenylaniline derivatives, which might arise from the low electron density and nucleophilicity of the o-phenyl motif to attack the in situ generated isocyanate intermediate. Soon after the publication of our work,9 Xi's group independently reported the carbonylation of (hetero)aryl C(sp2)–H of o-arylanilines with CO2 by utilizing the combination of methyl triflate (MeOTf) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) (Scheme 5).11 Under such conditions, unactivated o-phenylaniline derivatives worked nicely. Importantly, high temperature and primary amines were also required for the success of this reaction. The reaction went well with electron-rich aromatic substrates to afford the desired products in high yields. In contrast, the electron-poor ones, such as a CF3-substituted o-phenylaniline, only gave a trace amount of the desired products. When a meta-substituted substrate was applied, the regioselectivity of this transformation was difficult to control, providing a mixture of two isomers in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Notably, besides phenanthridinones, seven-membered lactams could also be obtained using this method, representing a significant advance in lactamization.
:1 ratio. Notably, besides phenanthridinones, seven-membered lactams could also be obtained using this method, representing a significant advance in lactamization.
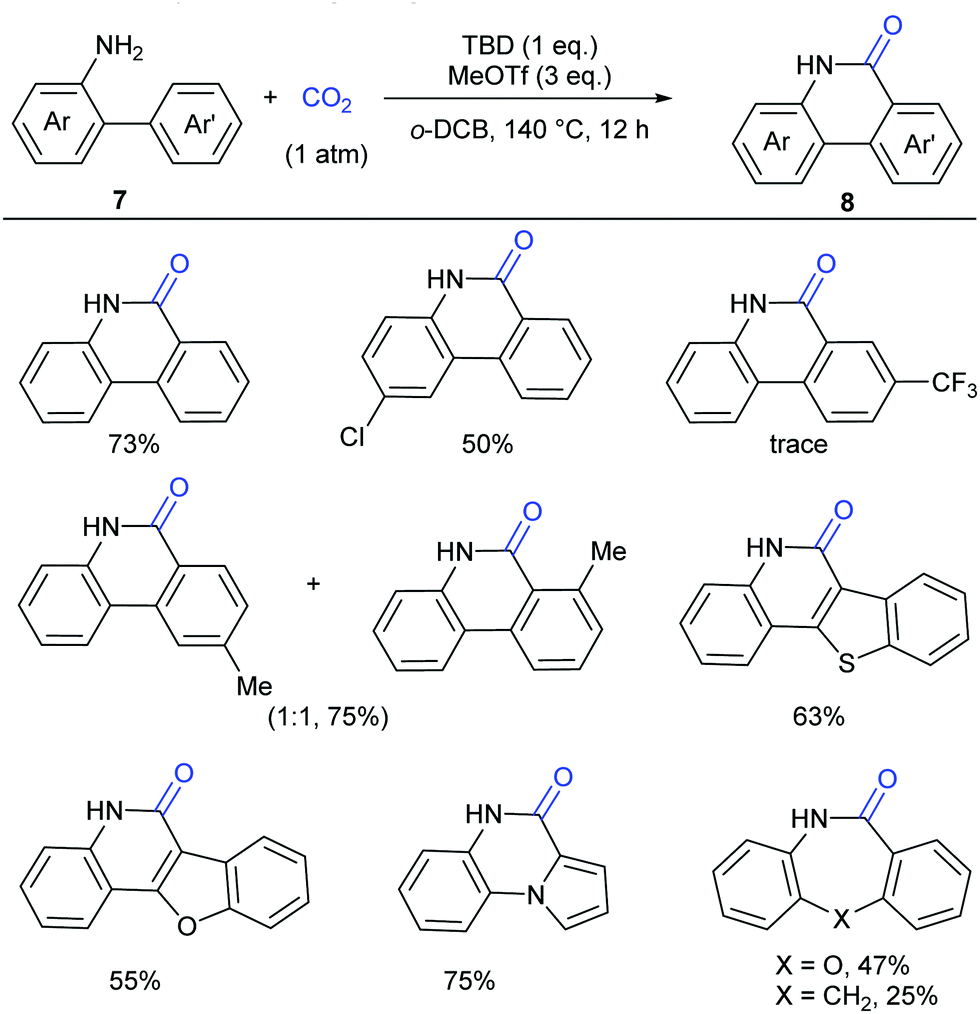 | ||
| Scheme 5 Transition metal-free carbonylation of C(sp2)–H bonds in o-(hetero)arylanilines with CO2 in the presence of TBD and MeOTf. o-DCB = 1,2-dichlorobenzene. | ||
In lactamization by Xi's group, they isolated the methyl carbamate 9 and also observed the formation of isocyanate 10 in the reaction system. As shown in Scheme 6, with the aid of MeOTf, the substrate reacted with the adduct of TBD and CO2 to afford methyl carbamate 9, which underwent subsequent elimination of one molecule of MeOH at high temperature and converted to aryl isocyanate 10. Then, methylation of 10 with MeOTf afforded a highly active species, such as the carbenium ion, which was attacked by the o-phenyl motif to generate 6-methoxyphenanthridine 11. Finally, the desired product 8 was obtained via acidification.
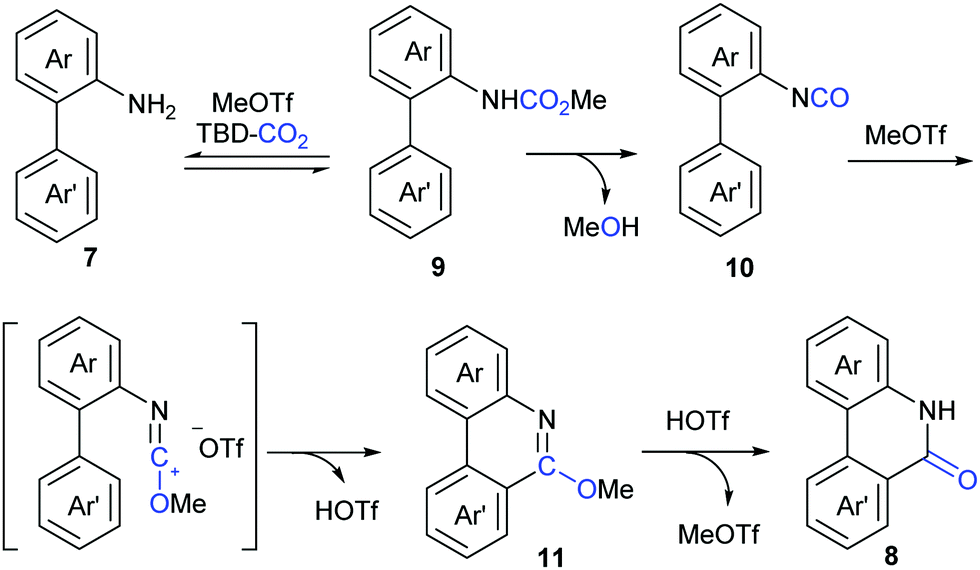 | ||
| Scheme 6 Proposed mechanism by Xi's group. | ||
In the above-mentioned two cases, the lactamization of C–H bonds with CO2 requires the prepared o-alkenyl- or o-(hetero)aryl-anilines as substrates. It has also been interesting whether the lactamization reaction could be realized in cascade reactions through generating such substrates in situ from simple raw materials. Later in 2016, Cheng's group successfully developed a palladium-catalyzed three-component reaction with N-tosylhydrazones, 2-iodoanilines and atmospheric CO2 to generate a series of 4-aryl-2-quinolinones (Scheme 7).12 The procedure allowed the formation of four bonds in one pot synthesis, representing an efficient methodology for incorporation of CO2 into heterocycles.
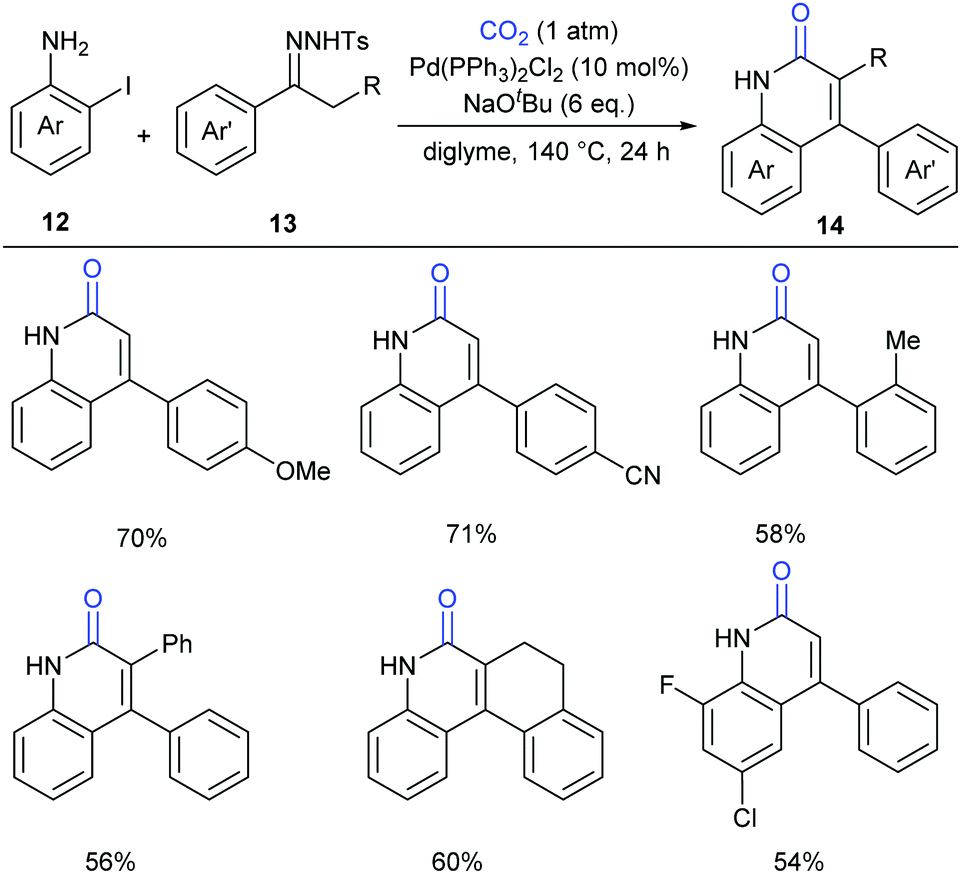 | ||
| Scheme 7 Cascade lactamization by Cheng's group. | ||
A possible reaction mechanism to explain the tandem carbene migratory insertion/lactamization pathway is shown in Scheme 8. The reaction might start with palladium-catalyzed coupling of aryl halides 12 and N-tosylhydrazones 13 to generate a vital intermediate, o-vinyl aniline 15, which further undergoes a similar pathway with CO2 as in our work to deliver the desired product 14.9 Meanwhile, the pathway generating o-iodoisocyanatobenzene prior to o-vinyl aniline 15 formation could not be excluded.
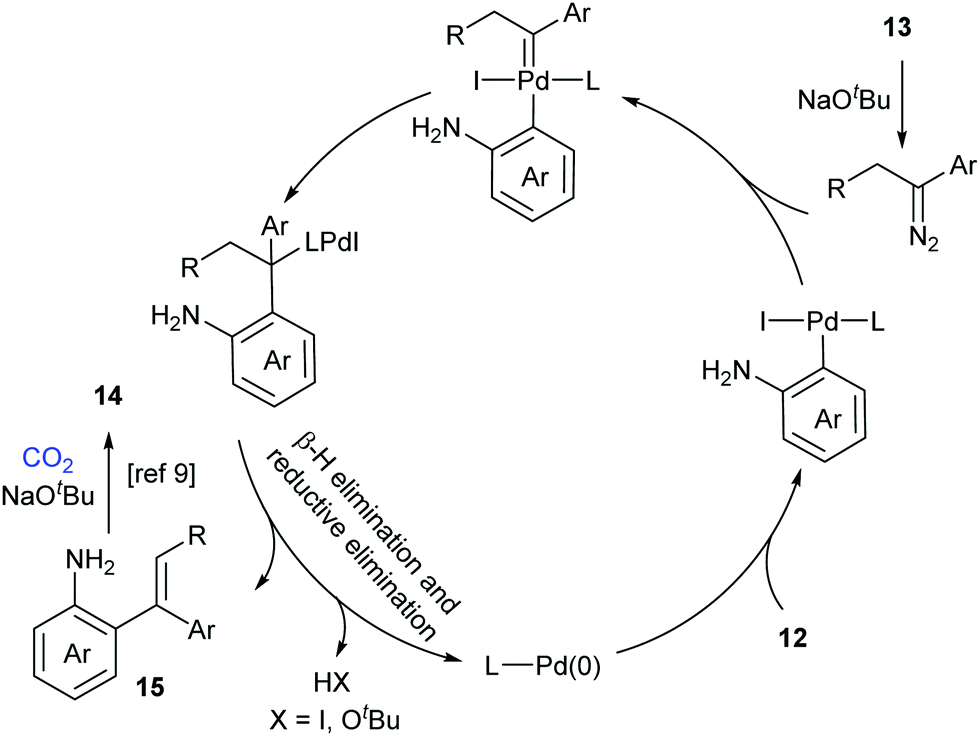 | ||
| Scheme 8 The proposed mechanism by Chen's group. | ||
Although such transition-metal-free lactamization protocols are powerful and efficient, there are still limitations in electron-poor aryl substrates. Recently the Li group have reported a novel Rh(I)-catalyzed, amino-group-directed lactamization of aryl C(sp2)–H bonds in o-(hetero)arylanilines with CO2 in the presence of KOtBu (Scheme 9).13 Based on the previous reports on Rh(I)-catalyzed C–H activation5 and facile insertion of CO2 into the rhodium–carbon bond,14 the authors successfully realized such a Rh(I)-catalyzed carbonylation with generation of various functionalized phenanthridinones under redox-neutral conditions. Both electron-rich and electron-poor substrates showed good reactivity for this transformation, which showed a significant improvement compared to previous works.9,11 Besides o-arylanilines, this reaction could be also extended to o-heteroarylanilines, such as pyridyl-, furanyl- and indolyl-substituted anilines, giving products with potential biological activity.
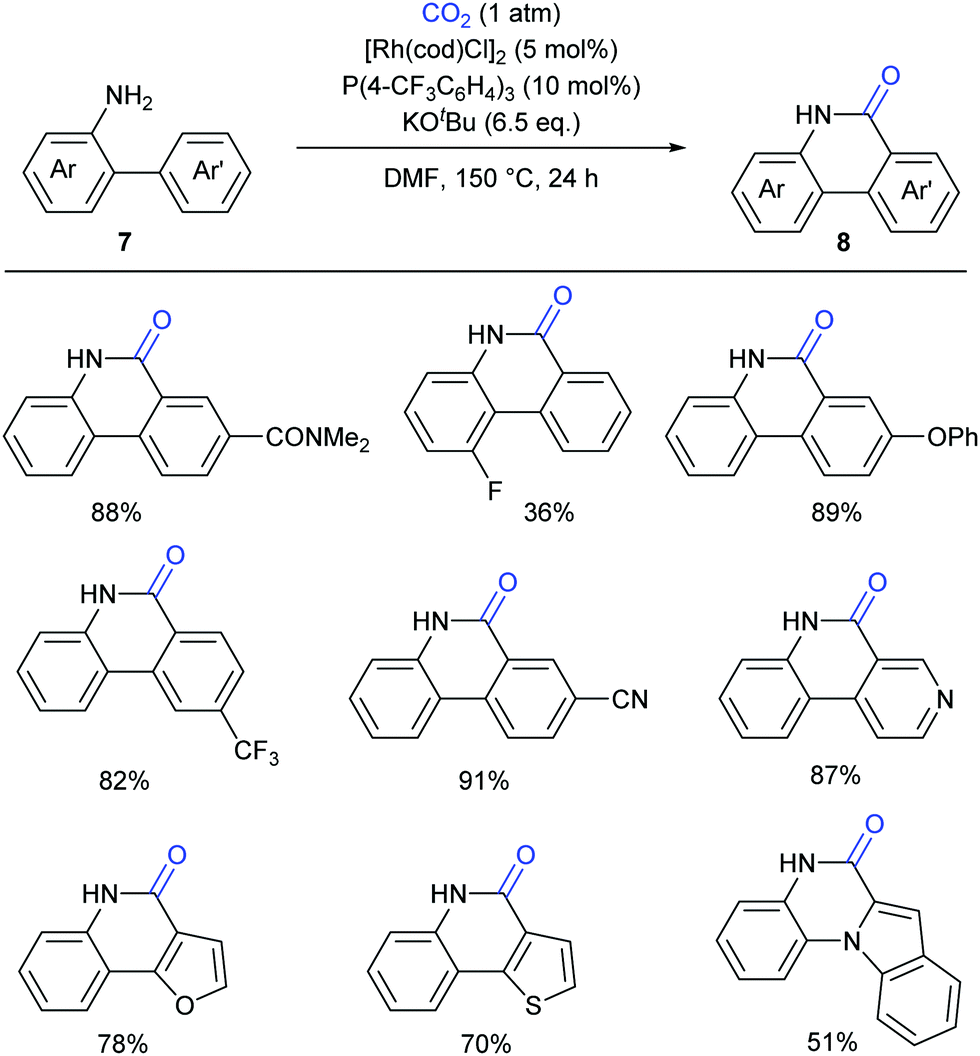 | ||
| Scheme 9 Rh-Catalyzed C–H lactamization of o-arylanilines with CO2. | ||
Based on the control experiments, a possible mechanism involving reversible oxidative addition of the aryl C–H bond to a Rh(I) species 16, reductive elimination of HX, and reversible nucleophilic carboxylation with CO2, followed by irreversible lactamization, was proposed by Li and co-workers (Scheme 10).
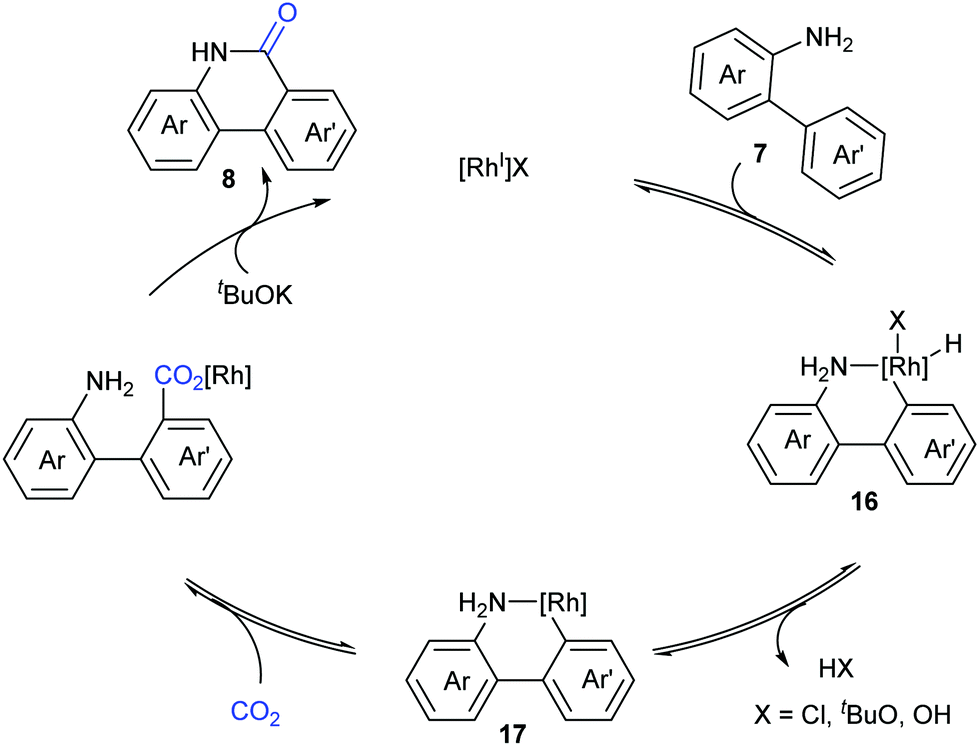 | ||
| Scheme 10 The catalytic cycle of Rh(I)-catalyzed lactamization. | ||
In addition to the construction of six-membered lactam-containing heterocyclic compounds, the core of five-membered phthalimides also attracts much attention.15 Recently, this skeleton has usually been constructed through transition-metal-catalyzed carbonylation of C–H bonds with CO in the presence of oxidants or hydrogen acceptors.16 In 2018, our group reported the first example of palladium-catalyzed carbonylation of unactivated aryl C–H bonds in benzamides with CO2 to afford phthalimides under redox-neutral conditions (Scheme 11a).17a Due to the influence of steric hindrance and directing ability, the substituents on the nitrogen of the benzamides were crucial, only the methyl group featuring good reactivity. Both electron-donating groups and electron-withdrawing groups could be well tolerated in the standard conditions. It is worth mentioning that carbonylation of the C–H bond selectively occurred at the less hindered ortho position for the substrates with meta substituents.
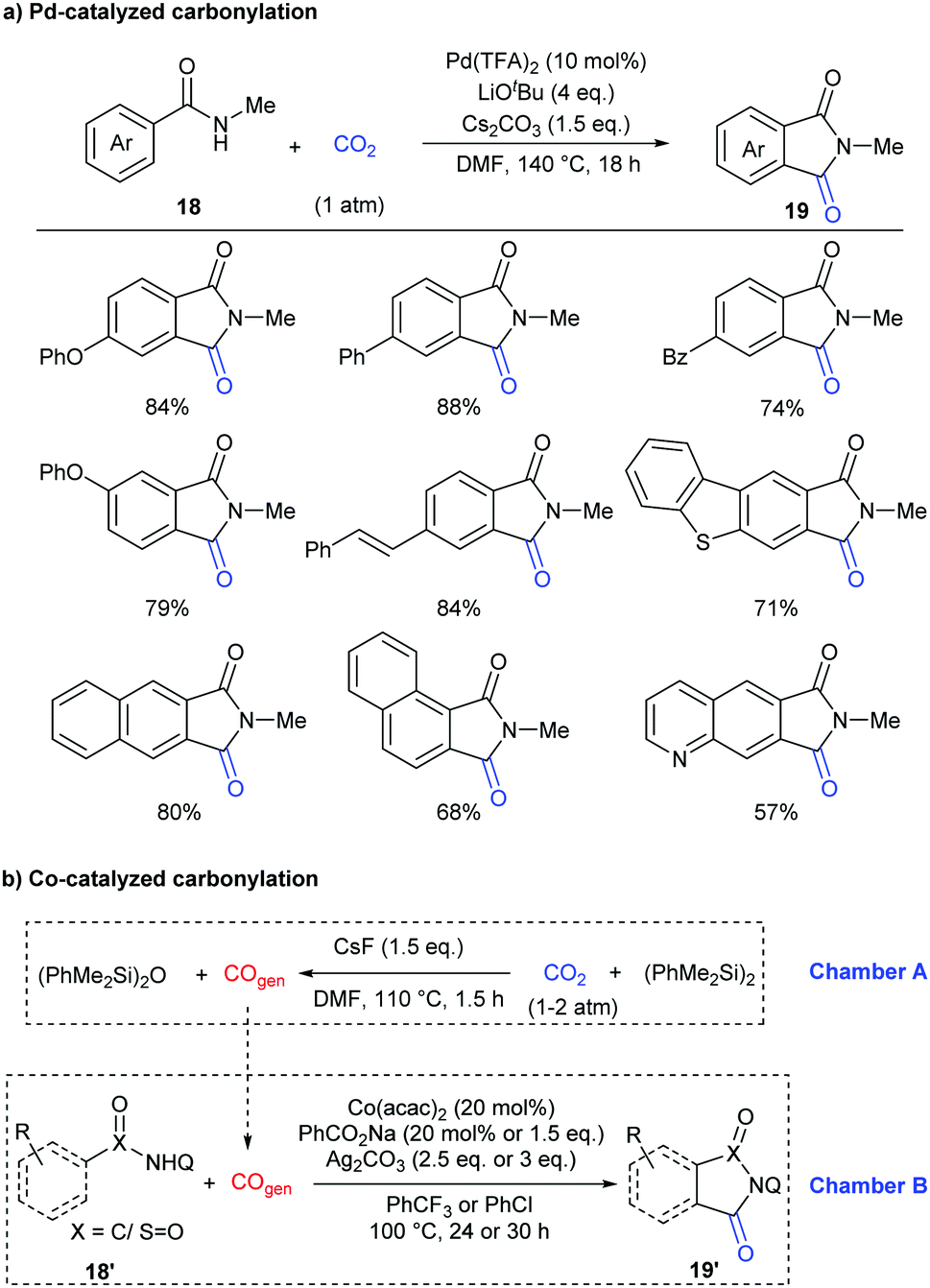 | ||
| Scheme 11 Transition-metal-catalyzed carbonylation of benzamides with CO2. | ||
Some control experiments were further conducted to shed light on the mechanism of the transformation (Scheme 12). We first excluded the possibility of the in situ generation of CO as the active carbonylation reagent and confirmed that the carbonyl group comes from CO2via13C-labeling experiments. The kinetic isotope effect (KIE) value indicated that the C–H activation step might be involved in the rate-determining step. We also postulated that the palladacycle species 20 might act as a vital intermediate in the reaction, as it was detected by ESI-HRMS when nitrogen ligands were employed to increase its stability. Notably, it also showed reactivity in both stoichiometric and catalytic version of such carbonylation.
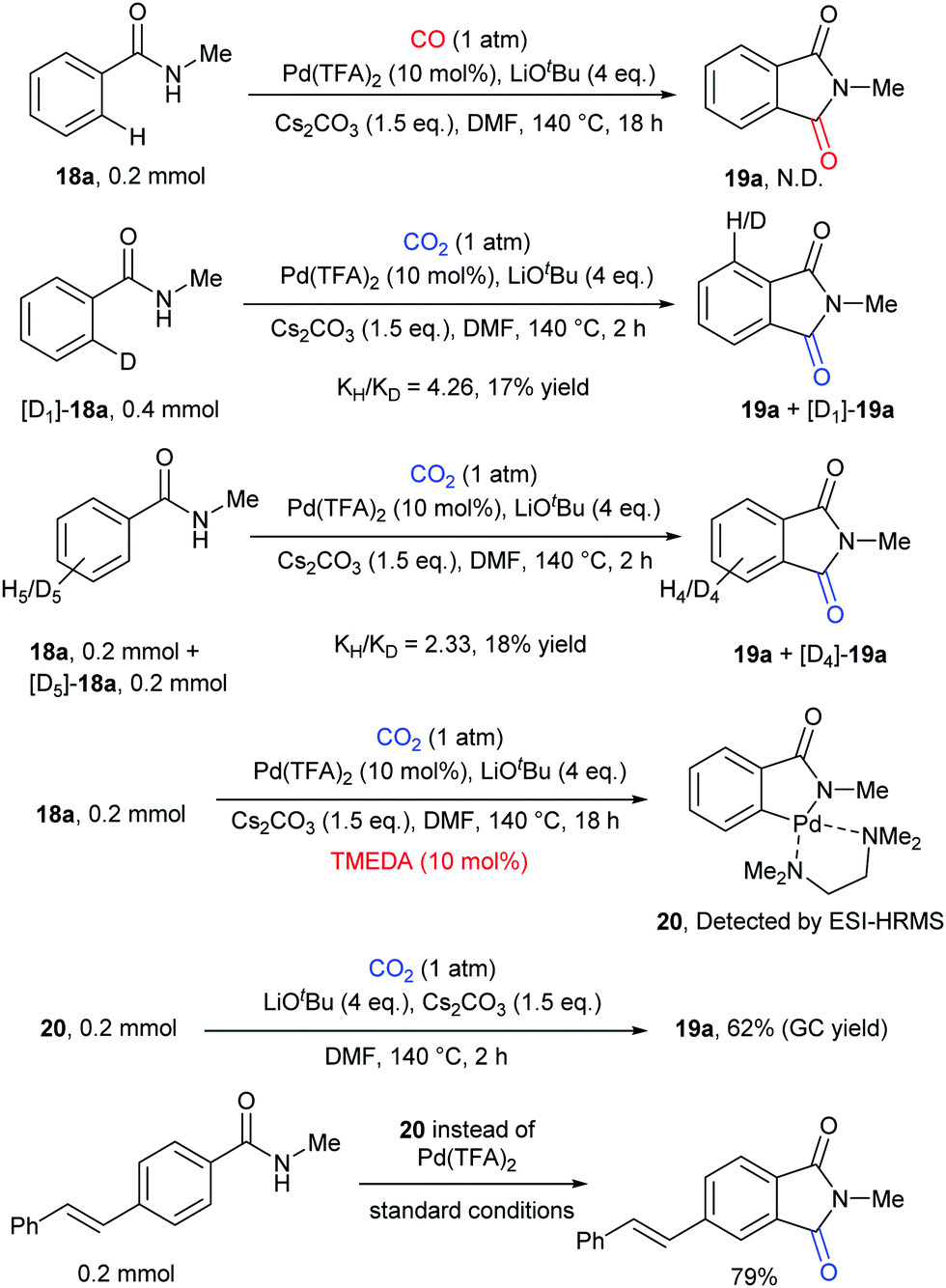 | ||
| Scheme 12 Selected mechanistic studies for Pd-catalyzed carbonylation. | ||
Later, another strategy, in situ reduction of CO2 to CO and the following carbonylation with CO, was also used to construct phthalimide skeletons by Sundararaju's group.17b A tandem reaction was realized in a two-chamber reactor for efficient fluoride-mediated generation of CO from CO2 using disilane as a deoxygenating reagent and utilization of the in situ-produced CO for C–H carbonylation using cobalt catalysis (Scheme 11b). In addition to the C(sp2)–H bond, the C(sp3)–H bond was also compatible in the system.
Compared to most of the research in this field which focused on the lactamization of C(sp2)–H bonds, lactamization of C(sp3)–H bonds with CO2 is more challenging. Very recently, our group reported the lactamization of C(sp3)–H bonds in pyridylamines with one atmosphere of CO2 to synthesize important pyrimidinones (Scheme 13).18a Different from the previous report on Pd-catalyzed lactamization of ketoimines with CO and oxidant,18b these reactions take place under transition-metal-free and redox-neutral conditions with atmospheric CO2 as the carbonyl source. The reactions can be performed on a gram scale, and the product can undergo derivatization easily. Moreover, some bio-active compounds can be quickly prepared by this method.
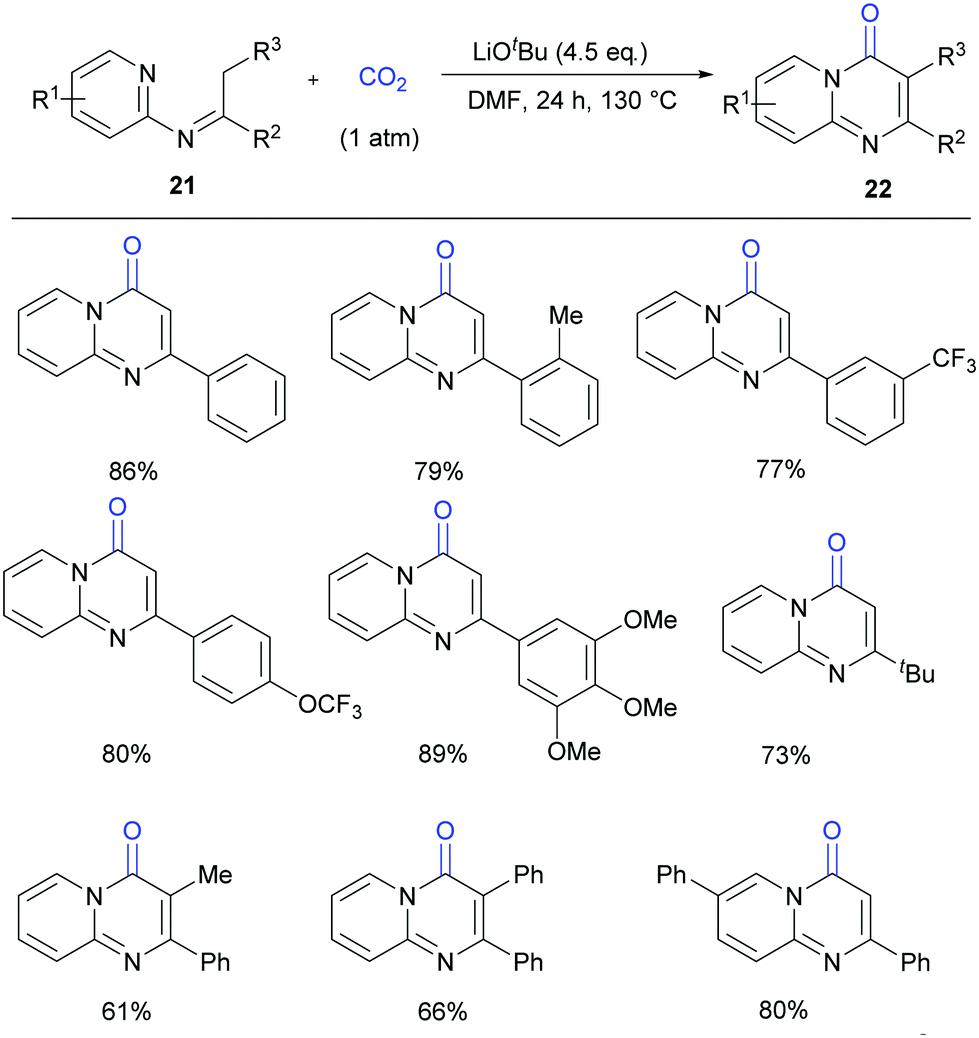 | ||
| Scheme 13 Transition metal-free carbonylation of C(sp3)–H bonds in pyridylamines with CO2 in the presence of LiOtBu. | ||
In the mechanistic studies, we found that 22a was generated in very low yield at 80 °C and the carboxylative product could be detected obviously by ESI-MS. When the reaction was quenched by CH3I, we detected the corresponding methyl ester by ESI-MS and the product 22a in a higher yield, all of which indicated that both the carboxylate intermediate and the ester were important in cyclization reaction to generate the desired product (Scheme 14).
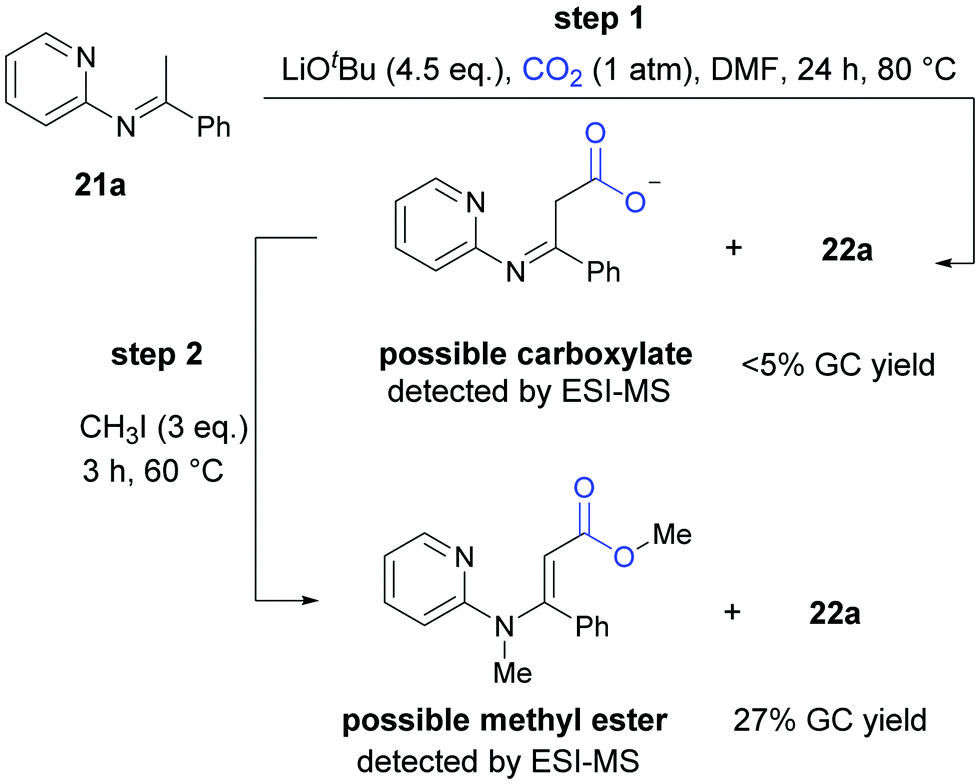 | ||
| Scheme 14 Mechanistic studies for carbonylation of C(sp3)–H bonds in pyridylamines with CO2. | ||
Based on the mechanistic studies and previous reports,19 a possible pathway was proposed (Scheme 15). 21a-1 was formed in the presence of LiOtBu and further reacted with CO2 to generate carboxylate intermediates 21a-2 and 21a-3. Then 21a-3 might react with LiOtBu to generate 21a-4, which transformed to 22a in the presence of base. In addition, another pathway through 21a-5 might be also possible.
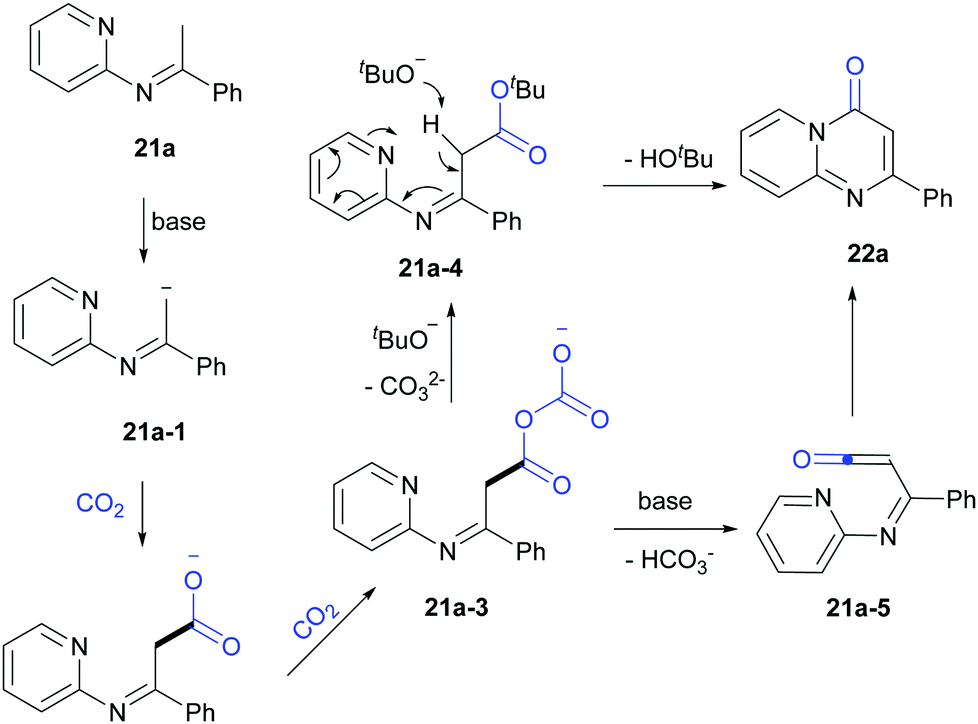 | ||
| Scheme 15 Proposed mechanism for carbonylation of C(sp3)–H bonds in pyridylamines with CO2. | ||
3. Lactonization of C–H bonds with CO2
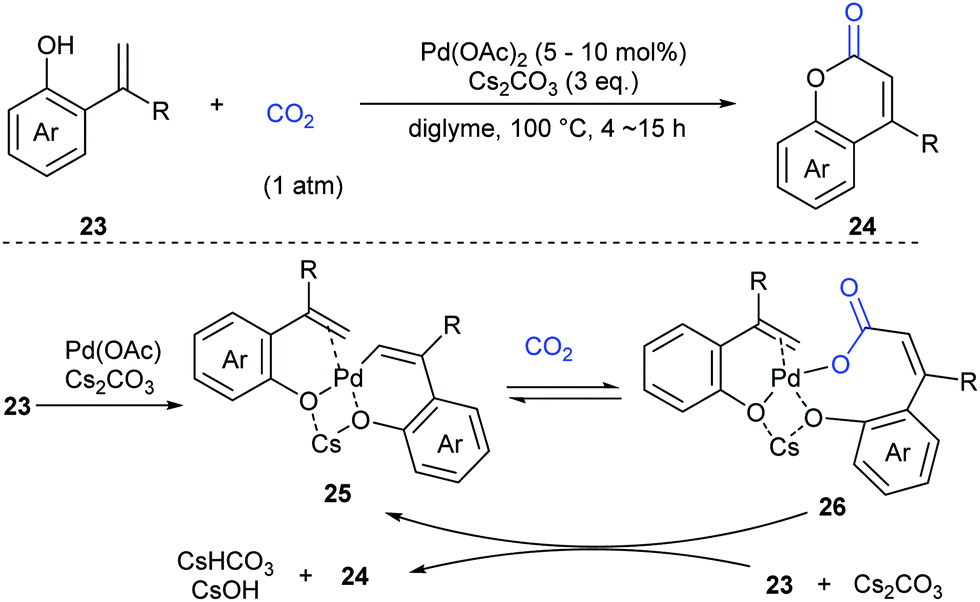
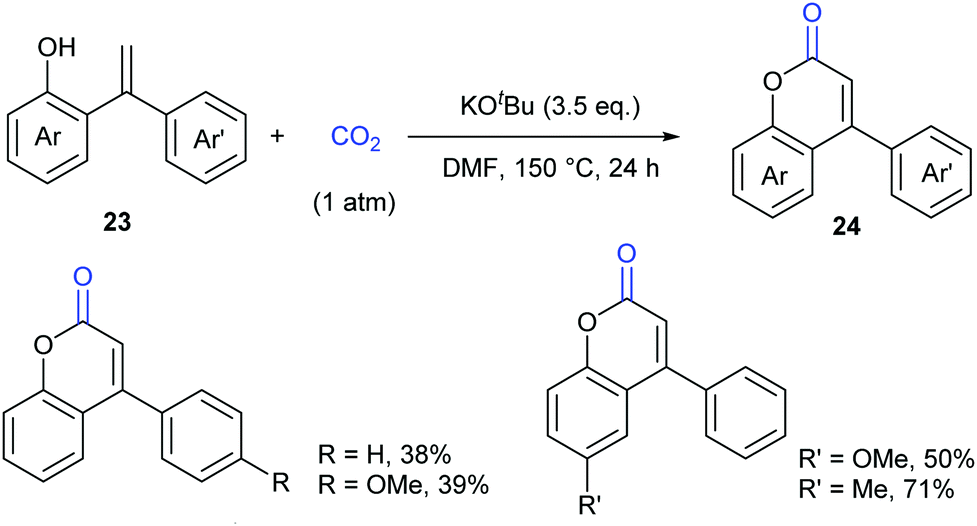
As another important class of carbonyl compounds, lactone-containing compounds are also prevalent in nature and a key structural motif in many bioactive molecules.20 Among the diverse ways to synthesize such structures, it is of great significance to realize carbonylation of C–H bonds. However, the carbonylation reagents are mainly restricted to highly toxic CO.3 Considering the advantages of CO2, it is highly important to develop such reactions with CO2 as the carbonyl source.In 2013, the Iwasawa group pioneered in this field and developed the first palladium-catalyzed lactonization of alkenyl C–H bonds in o-alkenylphenols 23 with CO2 to afford a variety of coumarins 24 with good yields (Scheme 16).21 The use of Pd-catalyst was vital for the reaction and Cs2CO3 was important for the high efficiency. Many functional groups, such as trifluoromethyl, methoxyl and cyano groups, as well as many heterocycles, such as thiophene, pyrrole and pyridine, could be well tolerated under the reaction conditions.
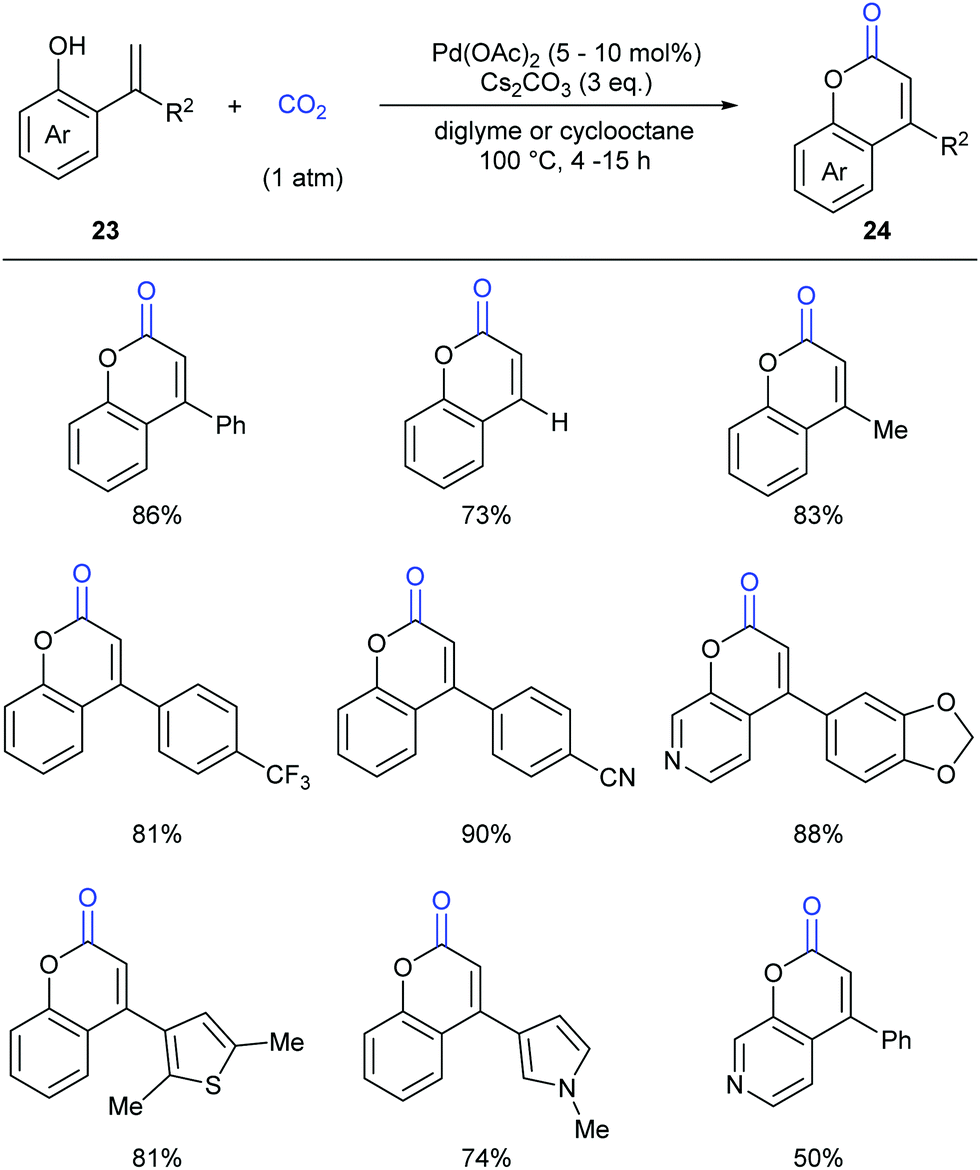 | ||
| Scheme 16 Pd-Catalyzed carbonylation of cyclization of o-alkenylphenols with CO2. | ||
Extensive studies were performed to reveal the mechanism (Scheme 17). A six-membered palladacycle 25, the key intermediate generated via C–H bond cleavage, was isolated and characterized by means of single-crystal X-ray diffraction. Then, the nucleophilic addition of 25 to CO2 gave a palladium carboxylate intermediate, which further reacted with another molecule of o-alkenylphenol 23 and Cs2CO3 to give the desired coumarin product 24 and regenerate the intermediate 25.
 | ||
| Scheme 17 Proposed mechanism for Pd-catalyzed carbonylation reaction. | ||
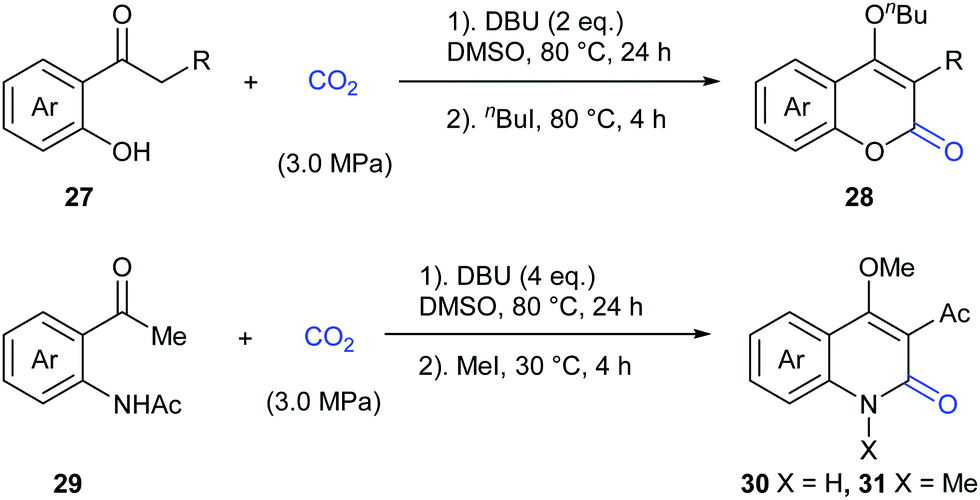
In 2015, the Zhang and Lu group reported a novel transition metal-free carbonylation of C(sp3)–H bonds in o-hydroxyacetophenones and o-aminoacetophenones with CO2 in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as base (Scheme 18).22 The driving force for this transformation mainly relied on the cyclization of the in situ formed β-ketoester with tethered oxygen and nitrogen nucleophiles to form stable heterocycles.
 | ||
| Scheme 18 DBU-mediated carbonylation reaction. | ||
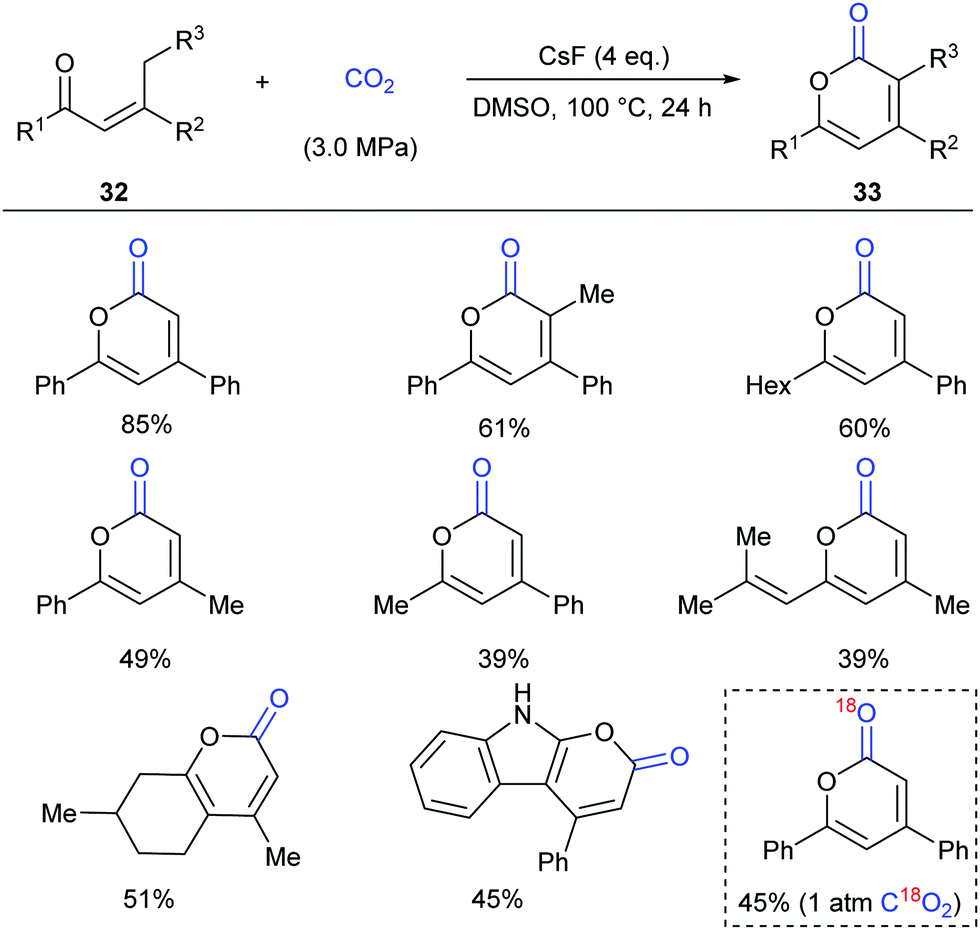
Inspired by the results, in 2016 the same group continuously developed a CsF-promoted carboxylative cyclization of propenyl ketones with CO2 to synthesize α-pyrones (Scheme 19).23 A low concentration of the substrate and a high pressure of CO2 are vital for high conversion and selectivity of the reaction. To exclude the possibility of a trace amount of palladium impurity in base functioning as a catalyst, Pd(OAc)2 was added into the system and decreased reactivity was observed, implying a transition-metal free environment for the reaction conditions. When C18O2 was utilized in this reaction, the product was obtained with only one 18O atom, which clearly demonstrated that the carbonyl was from CO2 in this transformation.
 | ||
| Scheme 19 CsF-mediated carbonylation reaction. | ||
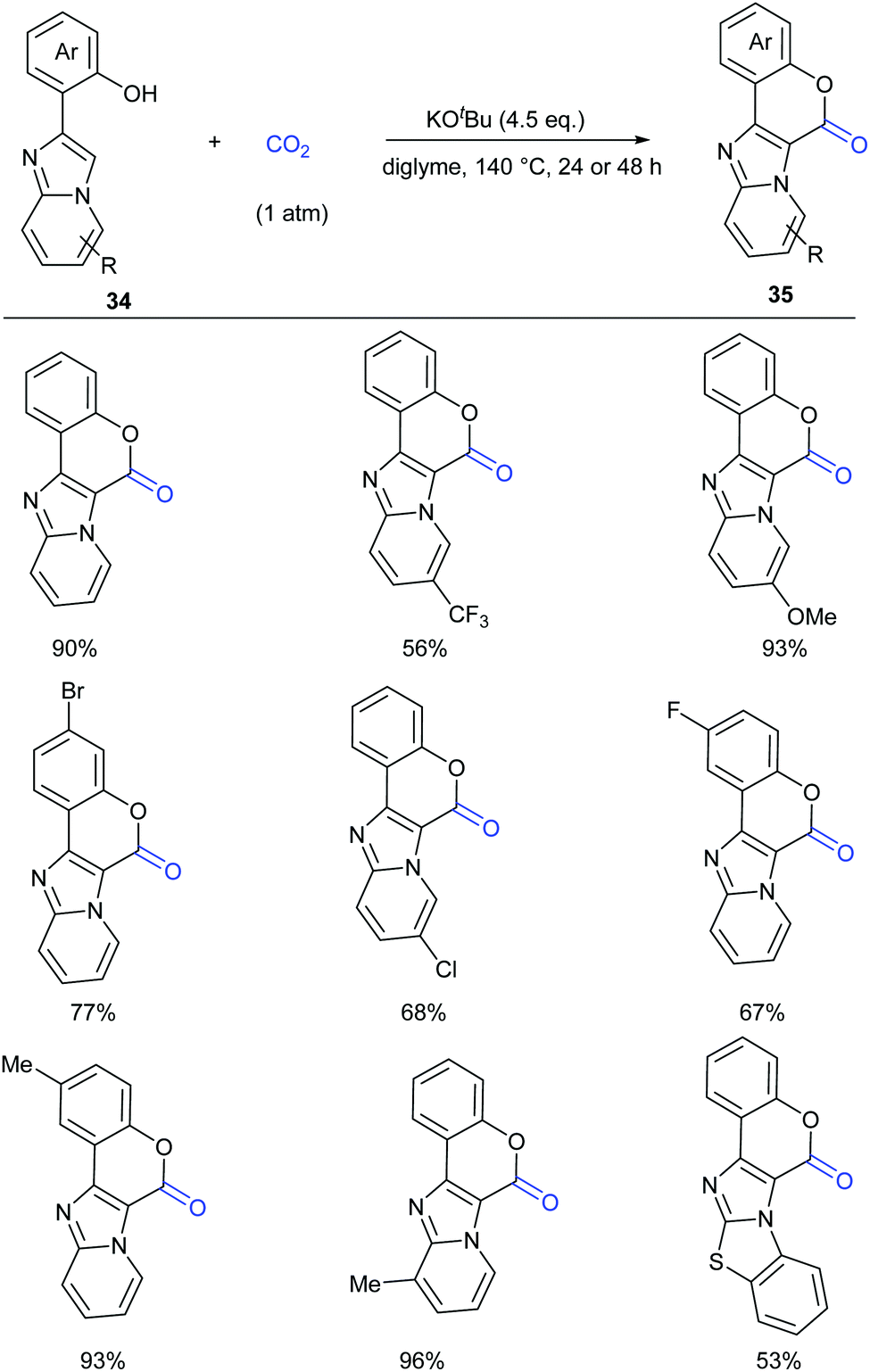
Considering the important biological activity of imidazo [1,2-a]pyridine skeleton-containing compounds, we envisioned whether a series of hybrid structures of coumarin derivatives could be obtained via carbonylation of C–H bonds in imidazo [1,2-a]pyridines with CO2. In 2017, our group successfully developed the KOtBu-promoted lactonization of o-heteroaryl- and o-alkenyl phenols with CO2 to generate coumarin derivatives with good to excellent yields (Scheme 20).24 This transformation featured a broad substrate scope and good functional group tolerance. In addition to o-(imidazole[1,2-α]pyridine-2-yl)phenols, o-(benzo[d]imidazo[2,1-b]thiazol-2-yl)-phenol and o-alkenylphenols could also undergo a similar lactonization reaction with high efficiency (Scheme 21).
 | ||
| Scheme 20 Lactonization of o-(imidazole[1,2-α]pyridine-2-yl)phenols with CO2. | ||
 | ||
| Scheme 21 KOtBu-promoted lactonization of o-alkenylphenols with CO2. | ||
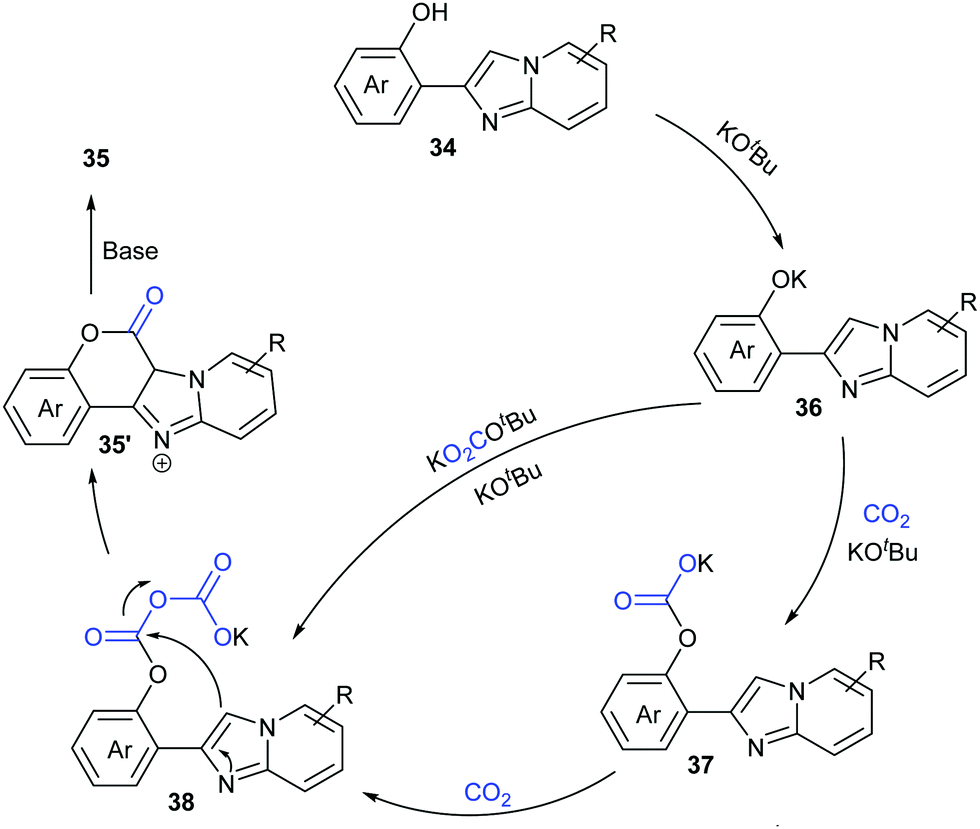
Based on the control experiments, we proposed a possible mechanism (Scheme 22). Deprotonation of compounds 34 with KOtBu formed phenolic potassium 36, which could react with CO2 or with the in situ generated adduct of CO2 and KOtBu (e.g. KOCO2tBu) to afford the carbonate intermediate 37. Then, a similar process took place further to give the intermediate 38. Intramolecular nucleophilic attack with the assistance of base afforded the desired coumarin product 35.
 | ||
| Scheme 22 The plausible mechanism for KOtBu-promoted lactonization of o-(hetero)aryl/alkenyl phenols with CO2. | ||
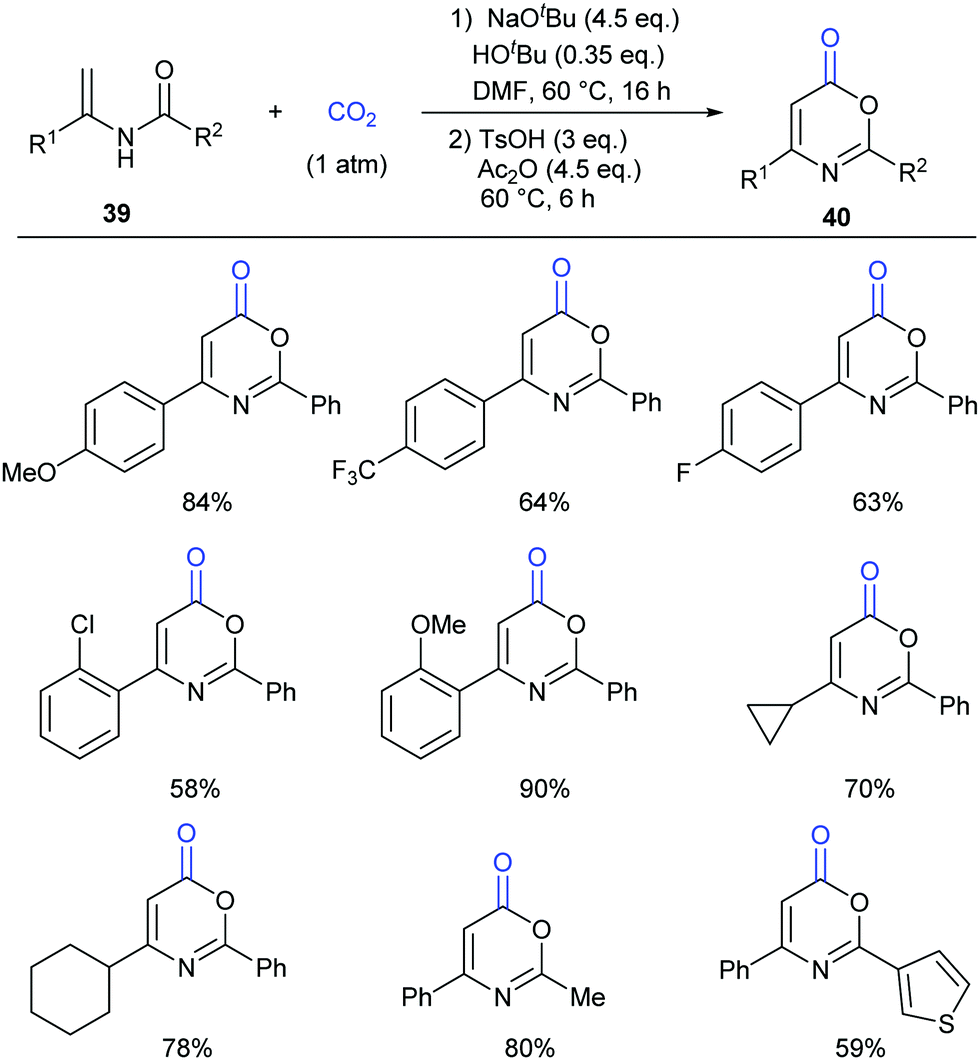
Different from our previous works,9,24 we wondered whether the carbonylation initiated via the nucleophilic attack from the electron-rich carbon sites instead of heteroatom sites (such as nitrogen in aniline and oxygen in phenol) was feasible. Therefore, we turned our attention to electron-rich enamides, which should show higher nucleophilicity in the presence of a strong base. If the carboxylation of C(sp2)–H bonds in enamides with CO2 and the following cyclization happened in sequence, we might be able to construct important oxazinone motifs.25 Indeed, based on this hypothesis, we succeeded in the synthesis of 1,3-oxazin-6-ones in one pot with two steps (Scheme 23).26 Although we did realize the carboxylation of C(sp2)–H bonds in enamides with CO2 in the presence of NaOtBu, the intermediates did not undergo condensation under the basic reaction conditions. Therefore, additional acid and anhydride were vital to promote the condensation to afford the desired products. Notably, such products could be transformed to important pyrimidines and pyridines through decarboxylation [4+2] cycloaddition.
 | ||
| Scheme 23 NaOtBu-promoted carboxylation cyclization of enamides with CO2. | ||
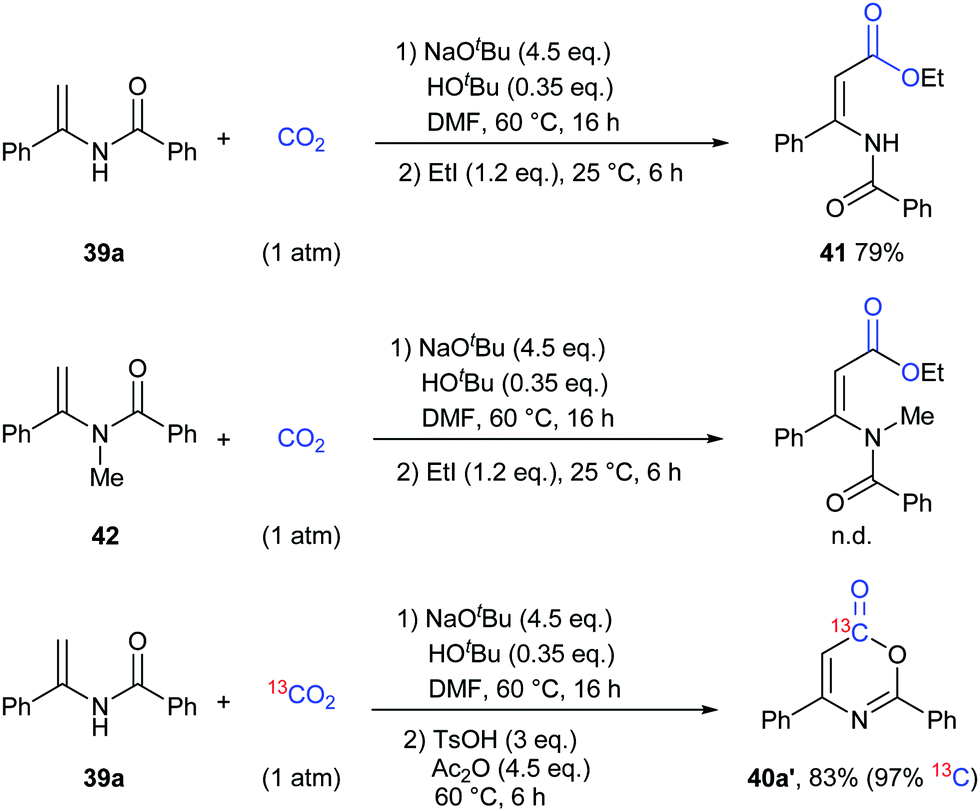
To gain more insight into this transformation, some mechanistic studies were conducted (Scheme 24). First, the corresponding ester 41 could be obtained by adding iodoethane (EtI) into the system, which could confirm the process of nucleophilic attack of electron-rich carbon in enamides on CO2. Then, when the N-methyl substituted enamide 42 was used, this transformation could not occur, which indicated that the interconversion of enamides and imines was important for the reaction under the basic conditions. Moreover, we also ran this reaction with 13CO2 and obtained the 13C-labeled coumarin in high yield, demonstrating that the carbonyl was derived from CO2.
 | ||
| Scheme 24 Mechanistic studies for the carbonylation of enamides with CO2. | ||
On the basis of the results, we proposed a possible pathway as shown in Scheme 25. Deprotonation of enamides by NaOtBu occurred to generate the enamide anions, which could react with CO2 to form carboxylates. Then, the desired product could be achieved by acidification and cyclization in the presence of acid and anhydride.
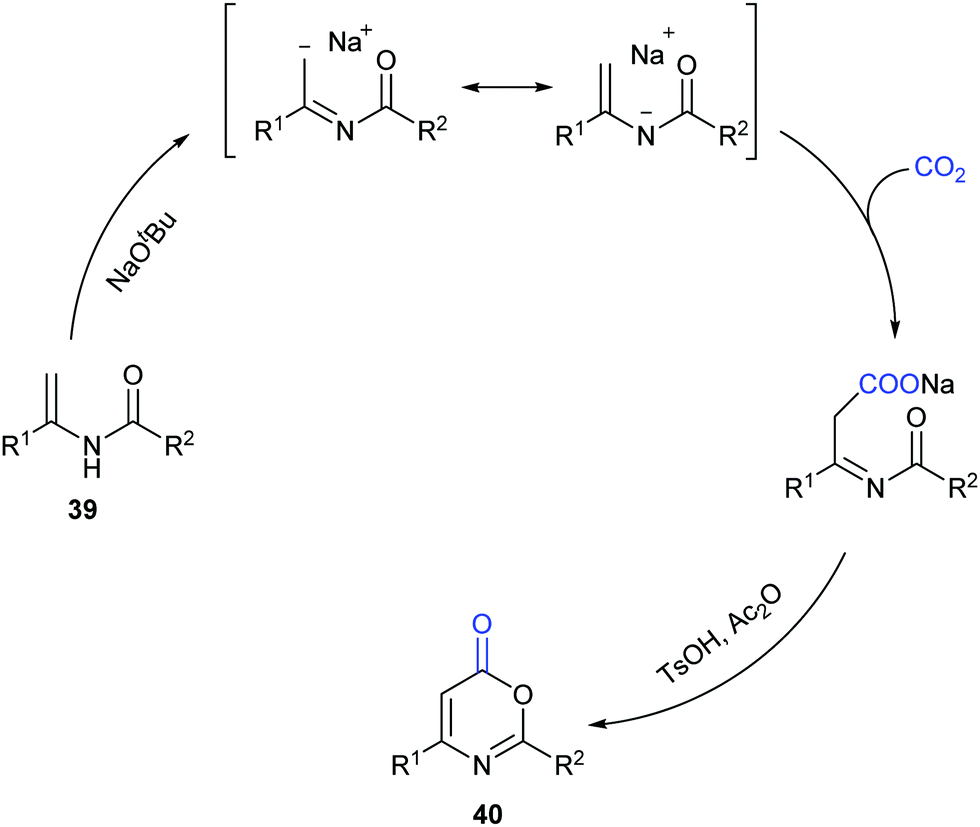 | ||
| Scheme 25 The plausible mechanism of the carbonylation of enamides with CO2. | ||
Compared to the C(sp2)–H bonds in alkenes and electron-rich heteroarenes, the carbonylation of unactivated aryl C–H bonds with CO2 is more difficult due to the lower reactivity and less nucleophilicity. In 2018, our group reported the first palladium-catalyzed carbonylation of the unactivated aryl C–H bond with CO2 to construct phthalides (Scheme 26)27 using tertiary benzylic alcohols as directing groups, which exhibit week coordination to transition metals and might undergo several side reactions, including β-H elimination and C–C bond cleavage. We used a strong base to deprotonate the benzyl alcohol, thus enhancing the coordinative ability of oxygen. However, under such basic conditions, the palladium(II) catalyst was prone to be reduced to inactive palladium black. After an extensive survey of mild oxidants, aryl bromides stood out for maintaining palladium(II) catalyst activity without interfering with the reaction. The selectivity issue arising from the side reactions of tertiary benzylic alcohols was suppressed by lactonization to form the desired products, although the primary and secondary alcohols mainly underwent oxidation via Pd-catalyzed β-H elimination. This reaction was not hampered by ortho substitution and C–H lactonization selectively occurred at the less hindered ortho position for meta substituted substrates. Heterocycles were also tolerated. When 13C-labeled CO2 and an 18O-labeled substrate were used, we could confirm that the carbonyl group was from CO2 and the alcohol oxygen atom was fully incorporated into the product. Based on the mechanistic experiments, we postulated that the five-membered palladacycle species 45 was a plausible intermediate, which could be detected by ESI-HRMS using bipyridine as the ligand.
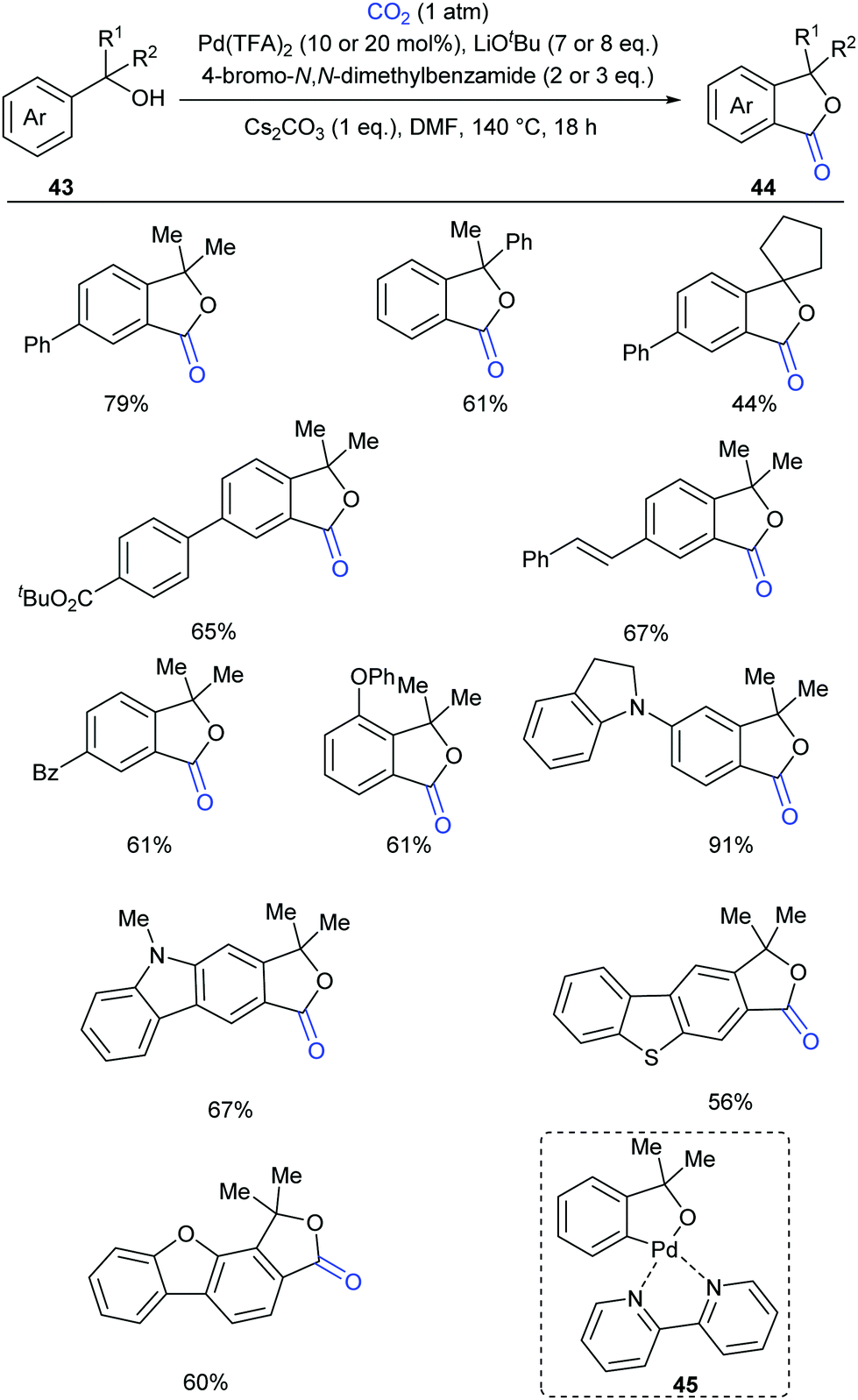 | ||
| Scheme 26 Pd-Catalyzed carbonylation of tertiary benzylic alcohols with CO2. | ||
Based on the density functional theoretical (DFT) calculations and the experimental results, we proposed a possible mechanism (Scheme 27). The active catalytic complex 46 could be generated in situ from Pd(TFA)2 in the presence of LiOtBu and CO2, followed by a counter anion exchange with lithium alcoholate intermediate 47 to afford alcoholate–Pd intermediate 48. Subsequent C–H bond cleavage generated the five-membered palladacycle intermediate 49, followed by two sequential nucleophilic attack on CO2 to afford intermediate 51. Intramolecular attack then leads to complex 52, which further underwent decarbonization and ligand exchange to deliver the desired product 44 and regenerate the catalyst 46. In this catalytic cycle, CO2 has been proven to promote this reaction by decreasing the ring strain in the intramolecular nucleophilic addition and enhancing the leaving ability and provide a novel pathway via decarbonization for C–O bond cleavage.
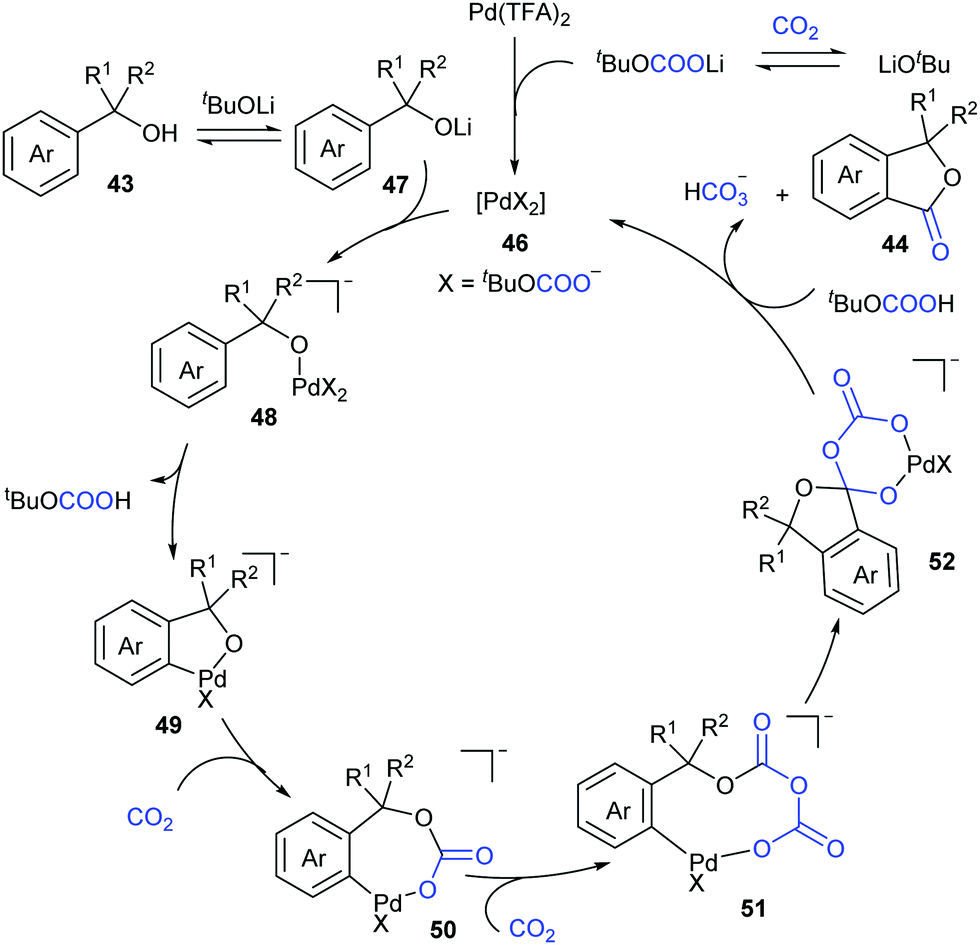 | ||
| Scheme 27 Possible reaction mechanism for carbonylation of tertiary benzylic alcohols with CO2. | ||
At the same time, the Li group independently displayed an elegant Rh-catalyzed lactonization of o-(hetero)arylphenols with CO2 (Scheme 28).14d The utilization of an electron-rich phosphine ligand, 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos), greatly enhanced the reactivity and avoided the undesired Kolbe-Schmitt type reaction. KOtBu could trap CO2 and increase the concentration of CO2 in the solution. Steric hindrance was tolerated in the phenolic part but not in the phenyl part. This transformation occurred at the less hindered ortho position for meta-substituted substrates with unique selectivity.
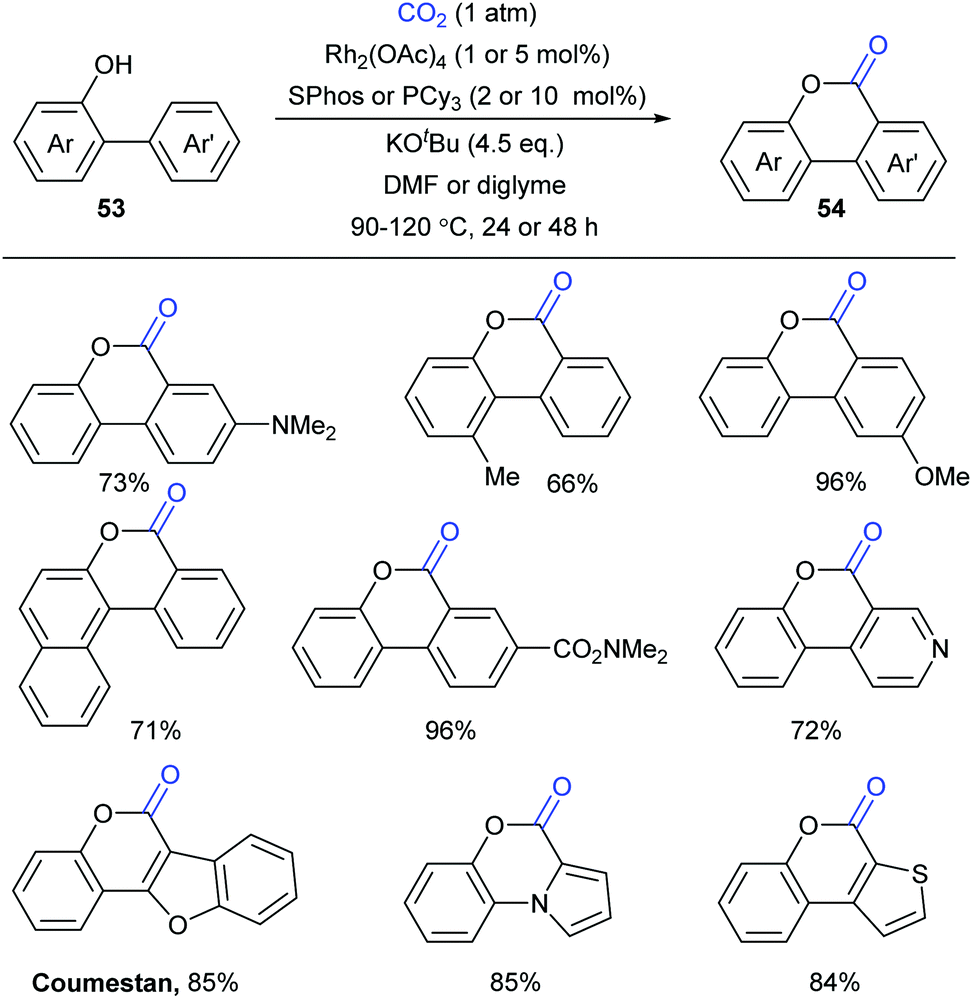 | ||
| Scheme 28 Rh-Catalyzed lactonization of o-(hetero)arylphenols with CO2. | ||
A plausible reaction mechanism was proposed as shown in Scheme 29. The intermediate 55 was formed in situ by coordination of the Rh-catalyst. Then, it coordinated with 56 generated from deprotonation of 53 by KOtBu to afford intermediate 57. With the assistance of an external base, the rhodacycle 58 was generated through the reversible aryl C–H bond activation of 57via a proton abstraction process, followed by nucleophilic attack on CO2 to achieve the eight-membered complex 59. Subsequently, ligand exchange and rapid lactonization delivered the desired product 54 and regenerated the complex 55. It was noteworthy that the shift of the carboxylation–decarboxylation equilibrium to the carboxylation intermediate 59 could be determined by the ligand exchange and lactonization process. This process should be fast and irreversible under the reaction conditions.
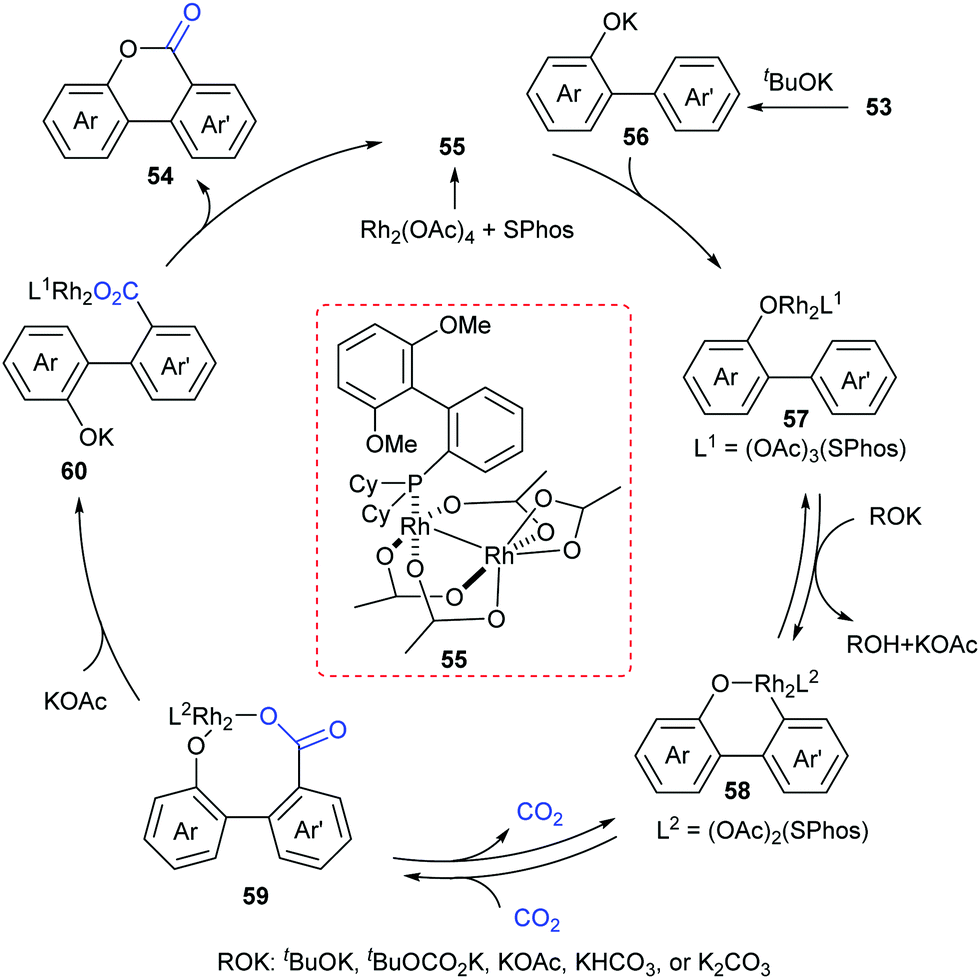 | ||
| Scheme 29 The catalytic cycle of Rh-catalyzed lactonization reaction. | ||
Although the Li group have developed an efficient Rh-catalyzed lactonization method, there is only an isolated example of o-pyridylphenol in this work.14d To extend this strategy, this group further reported Rh-catalyzed C–H carboxylation cyclization of more challenging electron-deficient 2-pyridylphenols with CO2 by tuning ligands (Scheme 30).14e A class of pyrido-coumarin derivatives has been synthesized. Apart from this, the group continuously expanded the catalytic system to o-(imidazo[1,2-a]pyridine-2-yl)phenols through ligand changes, and provided an access to coumarin derivatives based on the structure of imidazo[1,2-a]pyridine (Scheme 31).14f
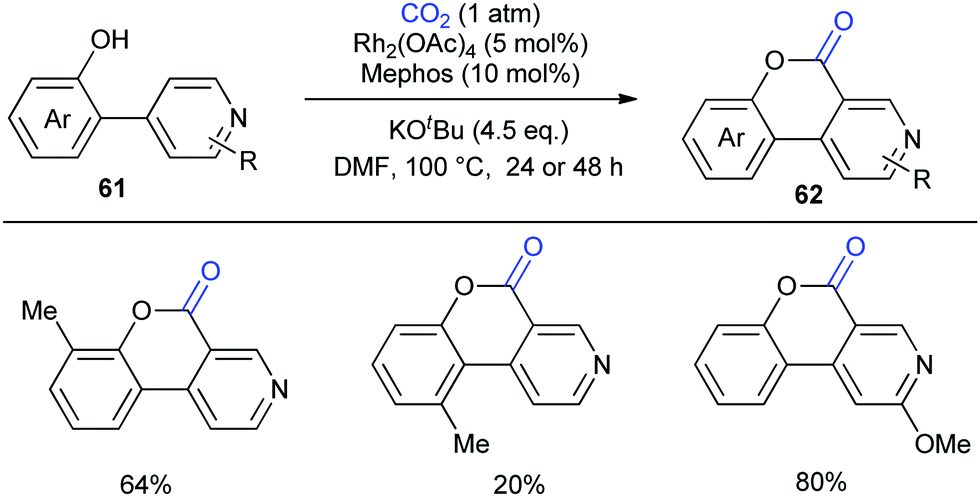 | ||
| Scheme 30 Rh-Catalyzed C–H carbonylation of 2-pyridylphenols with CO2. Mephos = 2-(dicyclohexylphosphino)-2′-methylbiphenyl. | ||
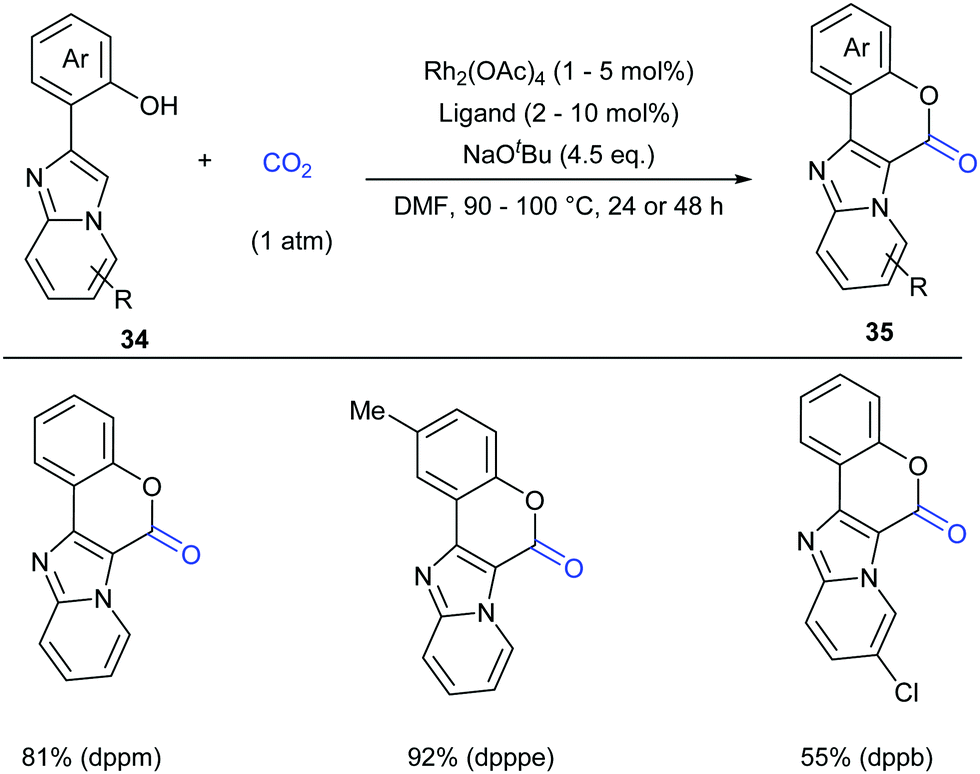 | ||
| Scheme 31 Rh-Catalyzed carbonylation of o-(imidazo[1,2-a]pyridine-2-yl)phenols with CO2. dppm = bis(diphenylphosphino)methane, dpppe = 1,5-bis(diphenylphosphino)pentane, dppb = 1,4-bis(diphenylphosphino)butane. | ||
4. Conclusions
Carbonylation of C–H bonds with CO2 has emerged as an alternative to the traditional oxidative carbonylation in constructing carbonyl-containing heterocycles under redox-neutral conditions. As a carbonylation reagent, CO2 has displayed great superiority over CO used in oxidative carbonylation, which usually poses safety, cost, and environmental issues. The main pathway for the redox-neutral carbonylation with CO2 refers to the successive nucleophilic attack on CO2 with the substrate possessing two nucleophilic sites. In many cases, a carbonate might be generated as a byproduct in the presence of base, facilitating the challenging CO bond cleavage. Although promising, there are still many important challenges in the carbonylation with CO2. For example, high temperature and high loading of a strong base are always required in the process. It would be more favorable to realize CO2 transformation under milder and more environmentally benign conditions. Then, the current catalytic system is mainly focused on noble metal catalysis, and the turnover number/efficiency is still unsatisfactory. It is urgent to develop more efficient and inexpensive transition metal catalytic systems. Moreover, the type of C–H activation is essentially confined to C(sp2)–H bonds and the relatively acidic C(sp3)–H bonds; the more inert C(sp3)–H bonds are less explored. Furthermore, the asymmetric C–H carbonylation should be further developed with suitable chiral ligands for highly enantioselective control. Last but not least, the application of this concept in the generation of ketones via double C–H carbonylation will be also highly attractive in organic synthesis. Taken together, we believe that the concept of CO2 as the combination of CO and oxidant (CO2 = CO + [O]) will continue to show its special advantages and invoke more and more attention in both laboratory and industry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the financial support from the National Natural Science Foundation of China (21822108, 21801025, 21502124), the “973” Project from the MOST of China (2015CB856600) and the Fundamental Research Funds for the Central Universities.Notes and references
- (a) Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Weinheim, Germany, 2010 Search PubMed; (b) Green Carbon Dioxide: Advances in CO2 Utilization, ed. G. Centi and S. Perathoner, Wiley-VCH, Weinheim, Germany, 2014 Search PubMed.
- (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed; (b) L. Ackermann, Angew. Chem., Int. Ed., 2011, 50, 3842 CrossRef CAS PubMed; (c) K. Huang, C.-L. Sun and Z.-J. Shi, Chem. Soc. Rev., 2011, 40, 2435 RSC; (d) R. Martin and A. W. Kleij, ChemSusChem, 2011, 4, 1259 CrossRef CAS PubMed; (e) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956 RSC; (f) W. Zhang and X.-B. Lu, Chin. J. Catal., 2012, 33, 745 CrossRef CAS; (g) M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620 CrossRef CAS PubMed; (h) L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395 RSC; (i) M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709 CrossRef CAS PubMed; (j) C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482 RSC; (k) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (l) D. Yu, S. P. Teong and Y. Zhang, Coord. Chem. Rev., 2015, 293-294, 279 CrossRef CAS; (m) C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353 CrossRef; (n) S. Wang, G. Du and C. Xi, Org. Biomol. Chem., 2016, 14, 3666 RSC; (o) W. Zhang, N. Zhang, C. Guo and X. Lu, Chin. J. Org. Chem., 2017, 37, 1309 CrossRef CAS; (p) Y.-Y. Gui, W.-J. Zhou, J.-H. Ye and D.-G. Yu, ChemSusChem, 2017, 10, 1337 CrossRef CAS PubMed; (q) X.-F. Wu and F. Zheng, Top. Curr. Chem., 2017, 375, 4 CrossRef PubMed; (r) Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707 RSC; (s) J. Luo and I. Larrosa, ChemSusChem, 2017, 10, 3317 CrossRef CAS PubMed; (t) Z. Zhang, T. Ju, J.-H. Ye and D.-G. Yu, Synlett, 2017, 741 Search PubMed; (u) A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948 CrossRef CAS PubMed; (v) S.-S. Yan, Q. Fu, L.-L. Liao, G.-Q. Sun, J.-H. Ye, L. Gong, Y.-Z. Bo-Xue and D.-G. Yu, Coord. Chem. Rev., 2018, 374, 439 CrossRef CAS; (w) Y. Cao, X. He, N. Wang, H.-R. Li and L.-N. He, Chin. J. Chem., 2018, 36, 644 CrossRef CAS; (x) J. Hou, J.-S. Li and J. Wu, Asian J. Org. Chem., 2018, 7, 1439 CrossRef CAS; (y) J. Hu, H. Liu and B. Han, Sci. China: Chem., 2018, 61, 1486 CrossRef CAS; (z) S. Wang and C. Xi, Chem. Soc. Rev., 2019, 48, 382 RSC; (a a) J. Hong, M. Li, J. Zhang, B. Sun and F. Mo, ChemSusChem, 2019, 12, 6 CrossRef CAS PubMed; (a b) L. Cheng and J. Xie, Chin. J. Org. Chem., 2020, 40, 247 Search PubMed.
- (a) Modern Carbonylation Methods, ed. L. Kollár, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef PubMed; (c) L. Wu, X. Fang, Q. Liu, R. Jackstell, M. Beller and X.-F. Wu, ACS Catal., 2014, 4, 2977 CrossRef CAS.
- H. Babad and A. G. Zeiler, Chem. Rev., 1973, 73, 75 CrossRef CAS.
- (a) C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed; (b) S. R. Neufeldt. and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS; (c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (d) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed; (e) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (f) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764 RSC; (g) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053 CrossRef CAS PubMed; (h) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281 CrossRef CAS PubMed; (i) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed; (j) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed; (k) T. Gensch, M. James, T. Dalton and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 2296 CrossRef CAS PubMed; (l) J. Huang, Q. Gu and S. You, Chin. J. Org. Chem., 2018, 38, 51 CrossRef CAS; (m) Q.-L. Yang, P. Fang and T.-S. Mei, Chin. J. Chem., 2018, 36, 338 CrossRef CAS; (n) Y. Liu, H. Yi and A. Lei, Chin. J. Chem., 2018, 36, 692 CrossRef CAS; (o) Z.-T. Jiang, B.-Q. Wang and Z.-J. Shi, Chin. J. Chem., 2018, 36, 950 CrossRef CAS; (p) S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2019, 58, 8304 CrossRef CAS PubMed; (q) Q. Zhang and B.-F. Shi, Chin. J. Chem., 2019, 37, 647 CrossRef CAS.
- (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (b) Q. Liu, H. Zhang and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 10788 CrossRef CAS; (c) X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1 CrossRef CAS PubMed; (d) X.-F. Wu, H. Neumann and M. Beller, ChemSusChem, 2013, 6, 229 CrossRef CAS PubMed; (e) X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041 CrossRef CAS PubMed; (f) B. Liu, F. Hu and B.-F. Shi, ACS Catal., 2015, 5, 1863 CrossRef CAS; (g) Y. Yi, M. Chen and Z. Guan, Chin. Sci. Bull., 2015, 60, 2927 CrossRef; (h) Y. Li, Y. Hu and X. Wu, Chem. Soc. Rev., 2018, 47, 172 RSC; (i) N. Rajesh, N. Barsu and B. Sundararaju, Tetrahedron Lett., 2018, 59, 862 CrossRef CAS; (j) X. He, Y. Cao, X.-D. Lang, N. Wang and L.-N. He, ChemSusChem, 2018, 11, 3382 CrossRef CAS PubMed; (k) L. Wang, W. Sun and C. Liu, Chin. J. Chem., 2018, 36, 353 CrossRef CAS; (l) F. Xu and W. Han, Chin. J. Org. Chem., 2018, 38, 2519 CrossRef CAS.
- (a) P. Cheng, Q. Zhang, Y.-B. Ma, Z.-Y. Jiang, X.-M. Zhang, F.-X. Zhang and J.-J. Chen, Bioorg. Med. Chem. Lett., 2008, 18, 3787 CrossRef CAS PubMed; (b) P. R. Angibaud, M. G. Venet, W. Filliers, R. Broeckx, Y. A. Ligny, P. Muller, V. S. Poncelet and D. W. End, Eur. J. Org. Chem., 2004, 479 CrossRef CAS; (c) J. Miyashiro, K. W. Woods, C. H. Park, X. Liu, Y. Shi, E. F. Johnson, J. J. Bouska, A. M. Olsona, Y. Luo, E. H. Fry, V. L. Giranda and T. D. Penning, Bioorg. Med. Chem. Lett., 2009, 19, 4050 CrossRef CAS PubMed.
- (a) J. Ferguson, F. Zeng, N. Alwis and H. Alper, Org. Lett., 2013, 15, 1998 CrossRef CAS PubMed; (b) D. Liang, Z. Hu, J. Peng, J. Huang and Q. Zhu, Chem. Commun., 2013, 49, 173 RSC; (c) V. Rajeshkumar, T.-H. Lee and S.-C. Chuang, Org. Lett., 2013, 15, 1468 CrossRef CAS PubMed; (d) Z. Liang, J. Zhang, Z. Liu, K. Wang and Y. Zhang, Tetrahedron, 2013, 69, 6519 CrossRef CAS.
- Z. Zhang, L.-L. Liao, S.-S. Yan, L. Wang, Y.-Q. He, J.-H. Ye, J. Li, Y.-G. Zhi and D.-G. Yu, Angew. Chem., Int. Ed., 2016, 55, 7068 CrossRef CAS PubMed.
- W. Li, C. Li and Y. Lyu, Org. Chem. Front., 2018, 5, 2189 RSC.
- S. Wang, P. Shao, G. Du and C. Xi, J. Org. Chem., 2016, 81, 6672 CrossRef CAS PubMed.
- S. Sun, W.-M. Hu, N. Gu and J. Cheng, Chem. – Eur. J., 2016, 22, 18729 CrossRef CAS PubMed.
- Y. Gao, Z. Cai, S. Li and G. Li, Org. Lett., 2019, 21, 3663 CrossRef CAS PubMed.
- (a) H. Mizuno, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2011, 133, 1251 CrossRef CAS PubMed; (b) T. Suga, H. Mizuno, J. Takaya and N. Iwasawa, Chem. Commun., 2014, 50, 14360 RSC; (c) T. Suga, T. Saitou, J. Takaya and N. Iwasawa, Chem. Sci., 2017, 8, 1454 RSC; (d) L. Fu, S. Li, Z. Cai, Y. Ding, X. Guo, L. Zhou, D. Yuan, Q. Sun and G. Li, Nat. Catal., 2018, 1, 469 CrossRef CAS; (e) Z. Cai, S. Li, Y. Gao and G. Li, Adv. Synth. Catal., 2018, 360, 4005 CrossRef CAS; (f) R. Huang, S. Li, L. Fu and G. Li, Asian J. Org. Chem., 2018, 7, 1376 CrossRef CAS.
- (a) A. L. Machado, L. M. Lima, J. X. Araujo, J. C. M. Fraga, V. L. Koatz and E. J. Barreiro, Bioorg. Med. Chem. Lett., 2005, 15, 1169 CrossRef CAS; (b) S. Gobec, I. Plantan, J. Mravljak, U. Svajger, G. S. Wilson, S. L. Besra, R. Soares, D. Appelberg and R. A. Kikelj, Eur. J. Med. Chem., 2007, 42, 54 CrossRef CAS PubMed; (c) V. J. Ragavendran, D. Sriram, K. S. Patel, I. V. Reddy, N. Bharathwajan, J. Stables and P. Yogeeswari, Eur. J. Med. Chem., 2007, 42, 146 CrossRef PubMed.
- (a) S. Inoue, H. Shita, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2009, 131, 6898 CrossRef CAS PubMed; (b) E. J. Yoo, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 17378 CrossRef CAS PubMed; (c) Y. Du, T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 12074 RSC; (d) B. Ma, Y. Wang, J. Peng and Q. Zhu, J. Org. Chem., 2011, 76, 6362 CrossRef CAS PubMed; (e) V. Rajeshkumar, T.-H. Lee and S.-C. Chuang, Org. Lett., 2013, 15, 1468 CrossRef CAS PubMed; (f) L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4688 CrossRef CAS; (g) W. Li, C. Liu, H. Zhang, K. Ye, G. Zhang, W. Zhang, Z. Duan, S. You and A. Lei, Angew. Chem., Int. Ed., 2014, 53, 2443 CrossRef CAS; (h) C. Wang, L. Zhang, C. Chen, J. Han, Y. Yao and Y. Zhao, Chem. Sci., 2015, 6, 4610 RSC; (i) F. Ji, J. Li, X. Li, W. Guo, W. Wu and H. Jiang, J. Org. Chem., 2018, 83, 104 CrossRef CAS.
- (a) L. Song, G. Cao, W. Zhou, J. Ye, Z. Zhang, X. Tian, J. Li and D.-G. Yu, Org. Chem. Front., 2018, 5, 2086 RSC; (b) N. Barsu, D. Kalsi and B. Sundararaju, Catal. Sci. Technol., 2018, 8, 5963 RSC.
- (a) Z. Zhang, X.-Y. Zhou, J.-G. Wu, L. Song and D.-G. Yu, Green Chem., 2020, 22, 28 RSC; (b) Y. Xie, T. Chen, S. Fu, H. Jiang and W. Zeng, Chem. Commun., 2015, 51, 9377 RSC.
- (a) Z. Chen, Y. Wen, H. Ding, G. Luo, M. Ye, L. Liu and J. Xue, Tetrahedron Lett., 2017, 58, 13 CrossRef CAS; (b) X. Zhang, B. Yang, G. Li, X. Shu, D. C. Mungra and J. Zhu, Synlett, 2012, 622 Search PubMed; (c) A. Fiksdahl, C. Plüg and C. Wentrup, Mesoions and Ketene Valence Isomers, J. Chem. Soc., Perkin Trans., 2, 2000, 1841 RSC.
- (a) E. Melliou, P. Magiatis, S. Mitaku, A.-L. Skaltsounis, E. Chinou and I. Chinou, J. Nat. Prod., 2005, 68, 78 CrossRef CAS PubMed; (b) T. Symeonidis, M. Chamilos, D. J. Hadjipavlou-Litina, M. Kallitsakis and K. E. Litinas, Bioorg. Med. Chem. Lett., 2009, 19, 1139 CrossRef CAS PubMed; (c) H. Zhao, A. C. Donnelly, B. R. Kusuma, G. E. L. Brandt, D. Brown, R. A. Rajewski, G. Vielhauer, J. Holzbeierlein, M. S. Cohen and B. S. J. Blagg, J. Med. Chem., 2011, 54, 3839 CrossRef CAS; (d) Y. Chen, S. Wang, X. Xu, X. Liu, M. Yu, S. Zhao, S. Liu, Y. Qiu, T. Zhang, B.-F. Liu and G. Zhang, J. Med. Chem., 2013, 56, 4671 CrossRef CAS PubMed; (e) T. Narumi, H. Takano, N. Ohashi, A. Suzuki, T. Furuta and H. Tamamura, Org. Lett., 2014, 16, 1184 CrossRef CAS PubMed; (f) B. Zhang, C. Ge, J. Yao, Y. Liu, H. Xie and J. Fang, J. Am. Chem. Soc., 2015, 137, 757 CrossRef CAS.
- K. Sasano, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2013, 135, 10954 CrossRef CAS PubMed.
- W. Z. Zhang, S. Liu and X. B. Lu, Beilstein J. Org. Chem., 2015, 11, 906 CrossRef CAS PubMed.
- W.-Z. Zhang, M.-W. Yang and X.-B. Lu, Green Chem., 2016, 18, 4181 RSC.
- Z. Zhang, T. Ju, M. Miao, J.-L. Han, Y.-H. Zhang, X.-Y. Zhu, J.-H. Ye, D.-G. Yu and Y.-G. Zhi, Org. Lett., 2017, 19, 396 CrossRef CAS PubMed.
- (a) M. C. Kim, J. H. Lee, B. Shin, L. Subedi, J. W. Cha, J.-S. Park, D.-C. Oh, S. Y. Kim and H. C. Kwon, Org. Lett., 2015, 17, 5024 CrossRef CAS PubMed; (b) P. Fu, S. La and J. B. MacMillan, J. Nat. Prod., 2016, 79, 455 CrossRef CAS PubMed.
- Z. Zhang, C.-J. Zhu, M. Miao, J.-L. Han, T. Ju, L. Song, J.-H. Ye, J. Li and D.-G. Yu, Chin. J. Chem., 2018, 36, 430 CrossRef CAS.
- L. Song, L. Zhu, Z. Zhang, J.-H. Ye, S. Yan, J. Han, Z. Yin, Y. Lan and D.-G. Yu, Org. Lett., 2018, 20, 3776 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |