
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mitochondria-targeting fluorescent molecules for high efficiency cancer growth inhibition and imaging†
Hao
Chen‡
ac,
Jing
Wang‡
ab,
Xin
Feng
bd,
Mark
Zhu
b,
Simon
Hoffmann
b,
Alex
Hsu
b,
Kun
Qian
b,
Daijuan
Huang
b,
Feng
Zhao
b,
Wei
Liu
a,
Huimao
Zhang
*a and
Zhen
Cheng
 *b
*b
aDepartment of Radiology, The First Hospital of Jilin University, Changchun, 130021, China. E-mail: huimaozhanglinda@163.com
bMolecular Imaging Program at Stanford (MIPS), Bio-X Program, Department of Radiology, Canary Center at Stanford for Cancer Early Detection, Stanford University, California 94305-5344, USA. E-mail: zcheng@stanford.edu
cCenter for Molecular Imaging Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China
dThe College of Veterinary Medicine, Jilin University, Changchun, 130021, China
First published on 8th July 2019
Abstract
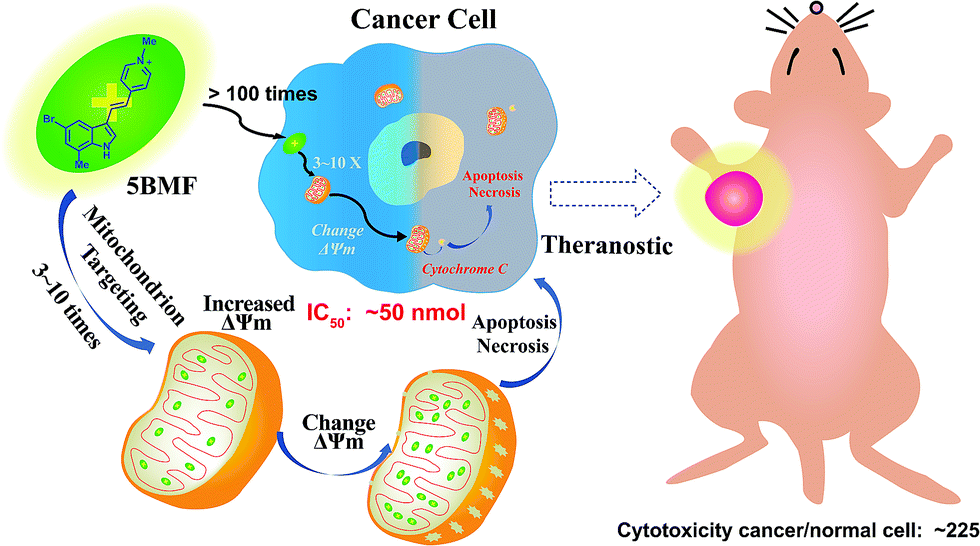
Fluorescent mitochondria-accumulating delocalized lipophilic cations (DLCs) for cancer therapy have drawn significant attention in the field of cancer theranostics. One of the most promising fluorescent DLCs, F16, can selectively trigger the apoptosis and necrosis of cancer cells, making it an attractive targeted theranostic drug candidate. However, it suffers from low clinical translation potential, largely due to its inefficient anti-cancer activity (IC50 in the μM range) and poorly understood structure–activity relationship (SAR). In this report, eleven indole-ring substituted F16 derivatives (F16s) were synthesized. Among these derivatives, 5BMF was identified as a highly effective theranostic agent, with in vitro studies showing a low IC50 of ∼50 nM (to H2228 cells) and high cancer to normal cell selectivity index of 225. In vivo studies revealed that tumors treated with 5BMF were significantly suppressed (almost no growth over the treatment period) compared to the PBS treated control group, and also no obvious toxicity to mice was found. In addition, the tumor imaging capability of 5BMF was demonstrated by in vivo fluorescence imaging. Finally, we report for the first time a proposed SAR for F16 DLCs. Our work lays down a solid foundation for translating 5BMF into a novel and highly promising DLC for cancer theranostics.
Introduction
Development of chemotherapeutic drugs for cancer-based on mitochondria-targeting molecules has been an intensely explored field.1 Delocalized lipophilic cations (DLCs) can accumulate in cancer cell mitochondria with >100 fold selectivity over normal cell mitochondria2 because of the natural mitochondrial membrane potential difference between cancer cells (ΔΨc, ∼−220 mV)3 and normal cells (ΔΨn, ∼−140 mV),4,5 representing one class of the most attractive mitochondria-targeting anti-cancer agents. Many DLCs also function as fluorescent dyes6,7 due to their rigid structure, such as the first DLC discovered – rhodamine 123 (Fig. S1†).8–10 As a result, plenty of DLCs are naturally theranostic agents, having potential applications in chemotherapy, photothermal/photodynamic therapy, optical imaging and fluorescence image-guided surgery.11,12 Furthermore, some DLCs such as rhodamine 123 exhibit avidity to tumor hypoxia. When these mitochondrial-targeting agents are combined with 2-deoxy-D-glucose (2-DG, Phase II clinical trial), enhanced toxicity against drug-resistant tumors and improved normal cell compatibility can be achieved compared to either drug alone.13–15 Such new DLCs can offer a powerful regimen for cancer therapy.Currently, rhodamine 123, MKT-077, and F16 are the most representative DLCs (Fig. S1†), and they have drawn great interest for cancer drug discovery. MKT-077 is the first DLC tested in a clinical trial, though the study was halted at Phase II due to limited anti-tumor effect and mild nephrotoxicity.10,16,17 Another DLC, F16, was identified through a high-throughput chemical library screen. It selectively accumulates in the mitochondria of cancer cells, disrupting normal function by dissipating the proton gradient across the inner mitochondrial membrane (decrease of ΔΨm, Scheme 1), triggering apoptosis or necrosis depending on the genetic background.18 However, F16's therapeutic efficacy has often been thought to be insufficient for drug development,19 as previous studies on structural modifications of F16 resulted in a limited improvement on their anti-cancer activity.20,21 A general opinion arose that DLCs naturally have limited anti-cancer activity because they often fail to cause large disruption of mitochondrial membrane potential (decrease of ATP production) to kill cancer cells.19,22 As a result, DLCs have been used simply as a cargo group to deliver functional molecules to the mitochondria selectively.19,23–25
 | ||
| Scheme 1 Mitochondria-targeting F16 derivative 5BMF for high efficiency cancer treatment and imaging. | ||
Herein, we synthesized a small library of F16 derivatives (F16s, 11 compounds), each with different substitutions at the indole position. Optical spectrum measurement studies indicated that they could be used for both in vitro and in vivo fluorescence imaging. In vitro cell studies of F16s showed strong anti-cancer cell activity, with an IC50 as low as ∼50 nM, and up to 225 fold higher selectivity for cancer cells over normal cells (Scheme 1). The most promising analog, 5BMF, was chosen for further in vivo studies using non-small-cell lung cancer (NSCLC) mouse models. After 21 days of treatment, the tumor volume of the 5BMF treated mice decreased dramatically compared to that of the control, with no obvious toxicity. Moreover, the in vivo fluorescence imaging results indicated that 5BMF could also be used as a targeted fluorescent probe for tumor diagnosis and potentially image-guided surgery. This study reports the first high efficiency indole-substituted F16 developed as a DLC based mitochondria-targeting tumor theranostic agent, as well as the first proposal of the structure–activity relationship (SAR) of F16. The results of this work provide a new perspective to high-efficiency anti-cancer DLC development.
Results and discussion
The key points for designing an effective F16 compound for cancer theranostic are: (1) a single positive charge on one part of the compound; (2) a whole-molecule π-conjugated system to spread the positive charge all over the compound; (3) to adjust the substituent group to fine-tune the charge distribution and polarity for efficient accumulation in mitochondria. F16 tautomerization studies show how positive charge spreads over the rigid structure (Fig. S2†). Positions 1, 2, 5, and 7 are the ideal positions to bear a positive charge (Fig. 1a). Changing the substituted group at those positions can affect the polarity and positive charge distribution of the compounds. These changes can also theoretically affect cross-membrane activity, resulting in further variations in anti-tumor activity. Keeping this hypothesis in mind, we designed and synthesized eleven F16 derivatives with methyl, phenyl, halogen, nitro, and nitrile substitutions at the 1, 2, 5, 6, and 7 positions of the indole ring according to a reported method,21 with yields between 50 and 86% (Fig. 1b). Recrystallization and semi-preparative HPLC purification methods were used to achieve a final purity for all F16 products of above 98% (Fig. S3†). The maximum solubility of F16s in water at room temperature is around 1 mM (Fig. S4†).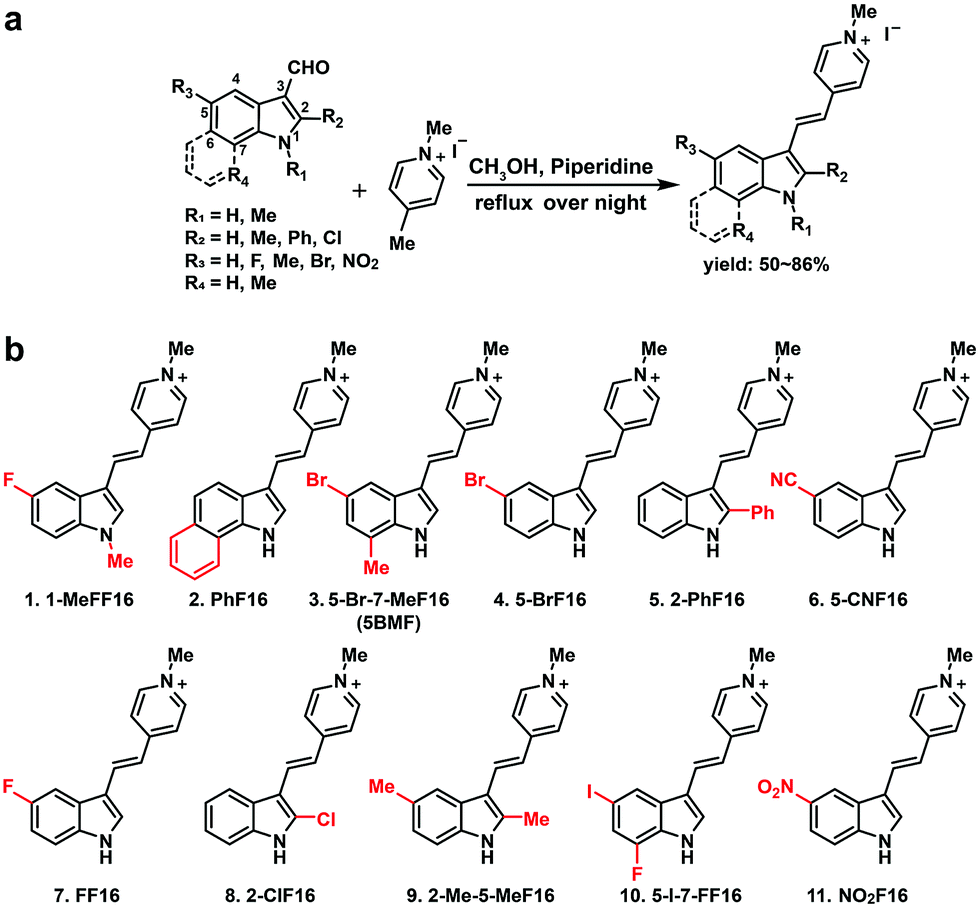 | ||
| Fig. 1 Synthesis of F16 derivatives (F16s) (a) and their chemical structures (b). | ||
The absorbance, fluorescence, and photostability of all F16 derivatives were measured in aqueous solution with concentrations of 31 μM, 7.8 μM, and 10 μM, respectively. The absorption peaks of all F16s were around 425 nm, except compounds 5 and 9 for which they were about 450 nm. Compound 4 shows the greatest absorbance at 425 nm. All F16s' fluorescence peaks were around 525 nm (Fig. 2b). Quantum yields were measured in ethanol using rhodamine 6G as a reference. Compound 4 shows the highest quantum yield (49.1%). The quantum yield of compounds 3, 7, and 9 is around 20%, 1 and 2 around 12%, and 5, 6, 8, 10, and 11 under 10%. Compound 2, 3, 4, 5, 7, and 11 have similar photostability to rhodamine 6G and MitoTracker® Green, while 1, 8, and 9 bleached quite easily (Fig. S5†).
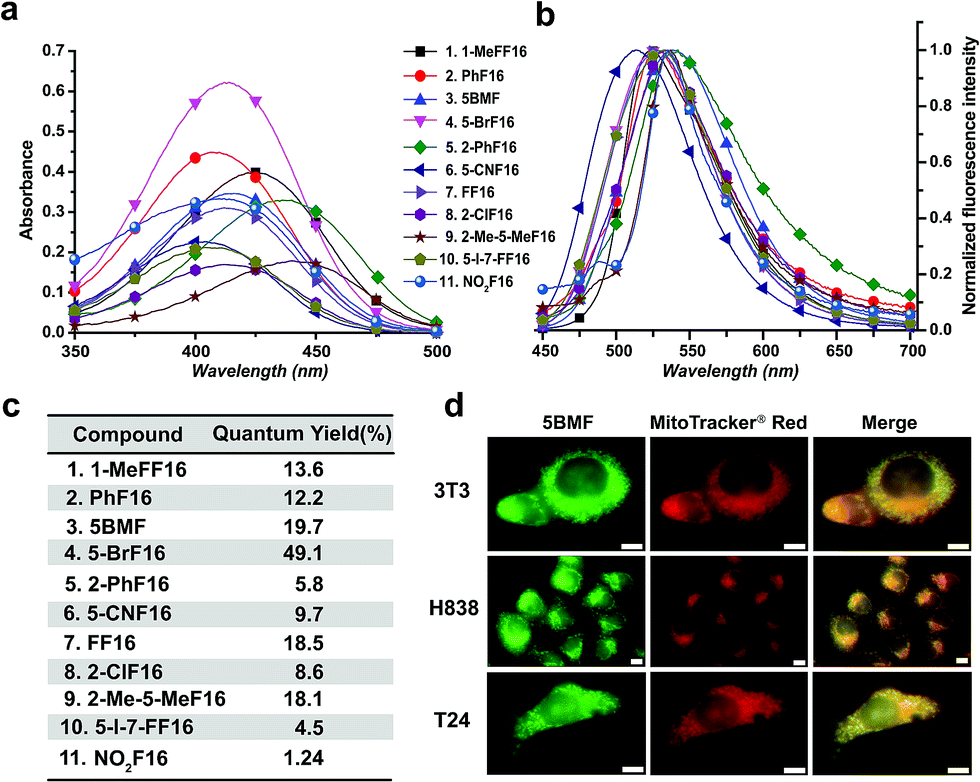 | ||
| Fig. 2 Optical properties of F16s. The absorption (a) and normalized emission (b) spectra of F16s were measured in water with a concentration of 31 μM and 7.8 μM, respectively (legend applies to both a and b). (c) F16s' relative quantum yields. EtOH as the solvent, compared with rhodamine 6G (QY: 95%). (d) Colocalization of 5BMF (GFP channel) with mitochondrial specific probe MitoTracker® red (microscopy RFP channel). The Pearson's correlation coefficient (γ) for 3T3, H838 and T24 is 0.978, 0.924, and 0.929, respectively (calculated using ImageJ, JACoP plugin). 63× oil objective lens. White scale bar: 10 μm. | ||
R1 (Fig. 1a) methyl substituted 1 revealed the highest absorption (compared to 7), but dramatically decreased photostability (Fig. S5,† comparing 1 with 7). This implies the importance of the indole nitrogen alkyl substitution in the fluorescent dye structure design. R2 chlorinated and methylated 8 and 9 demonstrated poor photostability maybe because the R2 position of F16s is an electron-deficient position (Fig. S2†) which can easily react with an electron-rich group. In aqueous solution, under laser excitation, the electron-rich −OH can attack the R2 position, destroy the delocalized π bond, and quench the fluorescence. Notably, R3 bromine substituted 4 showed double the molar absorptivity compared to fluorine substituted 7 (Fig. 2a). This implies that substitutions of different electronegativities can significantly impact the molar absorptivity, which may inspire the design of photoacoustic imaging probes and organic photoelectric materials.26
To test F16's mitochondrial targeting properties, the synthesized F16 derivatives were co-stained with MitoTracker® red. The results showed that all F16s were able to stain the mitochondria of all tested cell lines (Fig. S6†). As a representative, 5BMF (3) colocalized with MitoTracker in NIH-3T3, H838, and T24 cell lines is shown in Fig. 2d. Its Pearson's correlation coefficient (γ) is 0.978, 0.924, and 0.929, respectively.27
Previous studies show that F16 is driven into the mitochondria by their membrane potential (ΔΨm).18 To verify that F16 derivatives preserve the same property, H838 cells were preincubated in high K+ medium (137 mM) to depolarize the plasma membrane, then 5BMF (3 μM) was added and incubated for 30 min. Compared to low K+ (3.6 mM) medium, high K+ medium H838 resulted in much less 5BMF accumulation in mitochondria (Fig. 3a and c). In addition, when carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP), a protonophore that dissipates the ΔΨm, was added to cells preloaded with 5BMF, the mitochondrial staining immediately decayed and diffused into the cytoplasm (Fig. 3b and d). As with other DLCs, the selectivity of F16s for mitochondria is driven by the negative transmembrane potential.18
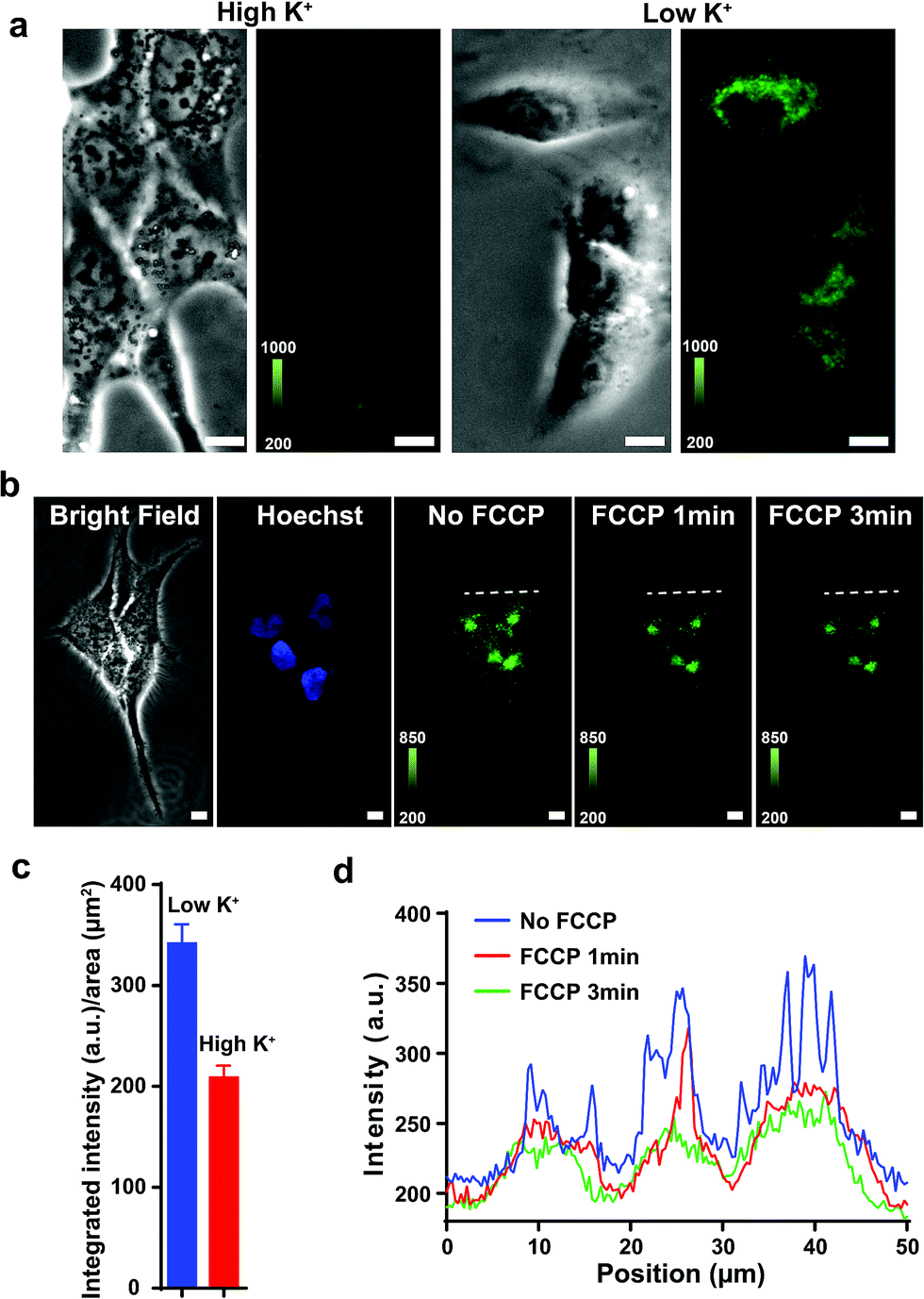 | ||
| Fig. 3 (a) Fluorescent signal in H838 cells' mitochondria in high K+ and low K+ buffers after incubation with 5BMF for 30 min. Left: bright field image. Right: fluorescence image. (b) 5BMF fluorescent signal in H838 cells' mitochondria before and after FCCP was added. From left to right, H838 cells bright field imaging, Hoechst staining of the nucleus, 5BMF staining of mitochondria (GFP channel, green) without FCCP, and 5BMF staining after adding FCCP for 1 and 3 min. All images were taken with a 20× objective lens. (c) Mean fluorescence density of cells in low K+ and high K+ buffers. (d) Cross-sectional fluorescence intensity profiles taken along white-dashed lines of (b). White scale bar: 10 μm. | ||
The cytotoxicity of F16 derivatives was tested in vitro with the bladder cancer T24 cell, NSCLC H838 cell, and normal cell NIH-3T3. Six of the eleven F16s displayed a strong anti-tumor activity, with IC50 values within 0.36–6.2 μM (2, 3, 4, 5, 7, and 10). Two of them showed selectivity indices larger than 10 (3 and 7). 5BMF had the best anti-tumor activity and relatively high selectivity (Fig. 4a). The 5BMF's IC50 for T24 and H838 was 0.82 and 0.36 μM, compared to that of F16,28 which was 18.8 and 46.6 μM (Fig. S7†), 23 and 129 times lower, respectively. From the results of the in vitro study, we propose the SAR of F16s as follows (Fig. 4b): R1 substitution with the methyl group decreases anti-tumor cell activity (ATCA) and normal cell toxicity (NCT) (compared to 1 and 7); R2 substitution increases NCT (compared to 5 and 8); R3 substitution by an electron withdrawing group increases ATCA, and was the biggest factor in ATCA (compared to 4, 6, 7, and 11); R4 substitution by an electron donating group increases ATCA (compared to 3, 4, and 10); indole ring conjugation with an extended π system increases the NCT and ATCA (compared to 2, F16).
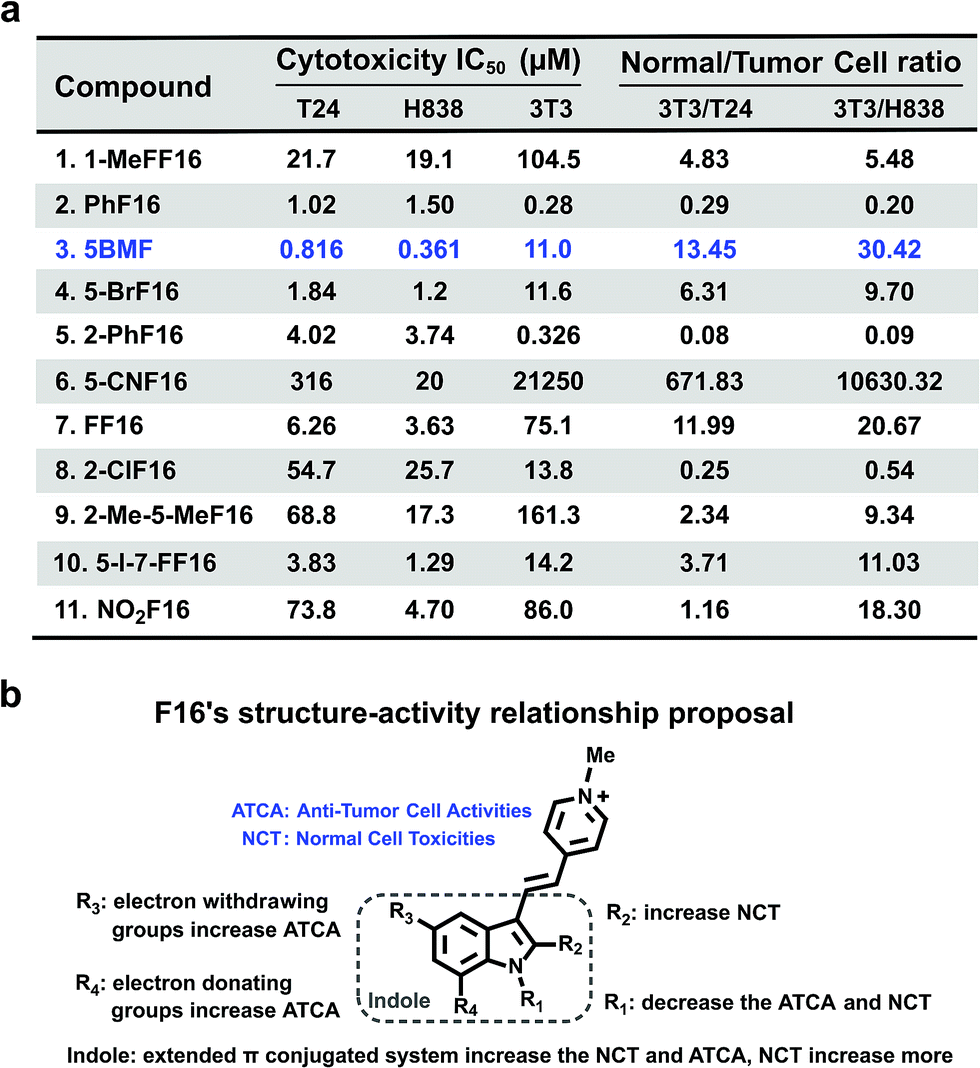 | ||
| Fig. 4 (a) F16s' cytotoxicity IC50 of T24, H838, and NIH-3T3 cell lines and their ratio. (b) F16s structure–anti-tumor activity relationship proposal. | ||
To further prove that an acceptor at the R3 position and a donor at the R4 position would increase the cell toxicity, another F16 analog 5-Cl-7-MeF16 with an acceptor at the R3 position (–Cl) and a donor group at the R4 position (–CH3) was synthesized (Fig. S8a†). Its cytotoxicity IC50 and normal cell to tumor cell ratio were also tested using T24, H838, and 3T3 cells (Fig. S8b†). Compared with 5BMF, the 5-Cl-7-MeF16's ATCA is slightly better (T24: 0.390 μM; H838: 0.274 μM) but its NCT is higher (3T3: 0.964 μM). This demonstrates that an acceptor group at the R3 position and a donor group at the R4 position surely increase the ATCA. But suitable substituent groups are needed to maintain a good normal cell to tumor cell toxicity ratio.
For years, most of the research regarding F16 based anti-cancer agents targeted the N-methylpyridinium group for modifications (Fig. S1†). But changing the N-methylpyridinium's substituted position was found to have limited effects on increasing F16's anti-cancer activity.20 In addition, our previous study showed that conjugating triphenylphosphonium (TPP) salts (Fig. S1†) to the N-methylpyridinium position leads to anti-cancer activity loss.29 Thus, it seems that the indole ring plays a key role in determining F16's anti-tumor activity.
The NSCLC mouse model was then chosen as an especially valuable route for further in vivo study of F16s' anti-tumor activity. Lung cancer is the most common malignancy and is the leading cause of cancer-related deaths worldwide.30 85% of lung cancers are NSCLC.31 For most patients with advanced or recurrent NSCLC, tumor recurrence and progression caused by drug resistance is a major cause of therapeutic failure.32 New anti-NSCLC drug development is urgent and imperative.33 Here, the cytotoxicity of the promising 5BMF was tested with 9 NSCLC cell lines including H838, HCC4006, HCC827, H1693, H2030, H2228, A549, H1437, and H1944. 5BMF showed impressively high ATCA in all these cell lines. In particular the H2228 cell line showed an IC50 of 48.9 nM, in addition to an anti-cancer cell to anti-normal cell (3T3, Fig. 4a) ratio around 225 (Fig. 5a).
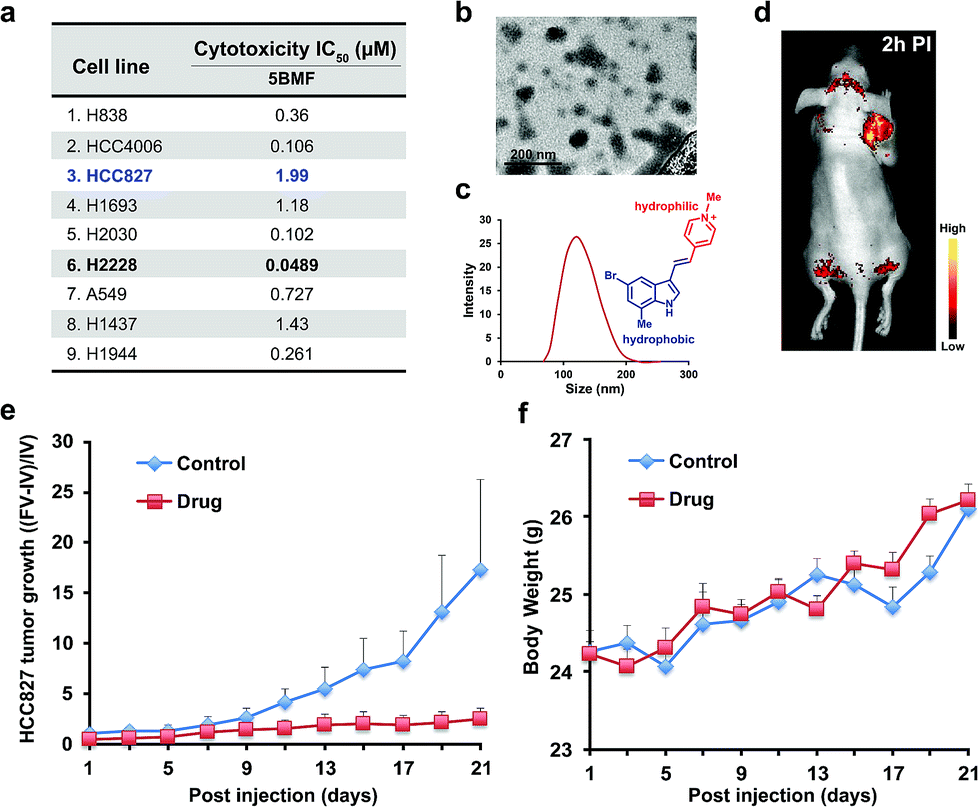 | ||
| Fig. 5 (a) 5BMF's IC50 of 9 lung cancer cell lines. (b) TEM image of 5BMF, scale bar: 200 nm. (c) DLS curve of 5BMF in PBS buffer, inset image: chemical structure of the amphiphilic 5BMF with the hydrophilic N-methylpyridinium group (red) and hydrophobic indole group (blue). (d) A representative in vivo fluorescence image of a human lung cancer HCC827 bearing mouse 2 h post-injection (PI) of 5BMF (15 mg kg−1, 0.306 M kg−1; for a 20 g mouse, inject 6.11 mM 5BMF PBS solution 150 μL; fluorescence imaging was performed with 450 nm excitation light, and the emission light was collected using a 550 nm long-pass filter) (n = 4). (e) Effects of 5BMF on the tumor growth of HCC827 in nude mice (n = 10 per group). FV, final volume. IV, initial volume. (f) Effects of 5BMF on the tumor-bearing nude mice's body weight (n = 10 per group). The treatment was started when the tumors reached ∼3 mm in diameter (day 1). Control group: PBS. Drug group: 5BMF, 15 mg kg−1, 0.306 M kg−1; for a 20 g mouse, inject 6.11 mM 5BMF PBS solution 150 μL; IV injections were given on day 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, and 21. | ||
The amphiphilic F16s were composed of the hydrophilic N-methylpyridinium group and hydrophobic indole group, which have the potential to self-assemble into nanoparticles in aqueous solution (Fig. 5c). 5BMF was found to form a self-assembly of ∼120 nm nanoparticles in 1× pH 7.4 PBS from 5 mM to 0.125 mM concentration (Fig. 5b and c). And it decayed less than 20% in pH 3.0–10.0 buffers for 24 h, which demonstrated its stability at different pH (Fig. S9†). Usually, ∼120 nm nanoparticles could accumulate in solid tumors by the enhanced permeability and retention (EPR) effect.34 In order to verify 5BMF's tumor accumulation ability, the well-explored HCC827 cell was chosen for in vivo imaging study.35,36Fig. 5d shows typical fluorescence images taken 2 h post intravenous injection (PI) of 15 mg kg−15BMF into nude mice bearing subcutaneous HCC827 tumor. The tumor was clearly visualized from the surrounding background tissue with a tumor to background ratio of ∼2, which was sufficient for fluorescence image-guided surgery. Quantitative analysis of ex vivo tumor imaging at 40 min and 2 h PI is shown in Fig. S10.† The tumor, kidney, bone, stomach, bowel, pancreas, and skin accumulated more 5BMF compared with the liver, spleen, heart, lung, and brain (Fig. S10a†). The tumor to most normal organ ratios (2 h PI, ∼2 to ∼7, Fig. S10b†) were high, and especially high tumor/lung ratios were observed (∼7, Fig. S10b†), implying potential low lung tissue toxicity and use for tumor detection and fluorescence image-guided lung cancer surgery applications.
In order to prove the anti-tumor effect of 5BMF persuasively, the highest IC50 (Fig. 5a) HCC827 cell was chosen for the further in vivo treatment study. The treatment was started when the subcutaneous HCC827 tumors reached ∼3 mm in diameter (day 1). The dosage and administration frequency were 15 mg kg−1, IV injections on day 1, 3, 5, 7, 9, 11, 13, 15, 17, 19, 21, which were referenced from a MKT-077 study.37Fig. 5e shows the treatment response of the HCC827 xenograft to intravenous (IV) injection with 5BMF. As indicated by the mean tumor growth rate in both control and treated groups, the inhibitory tumor growth effects for 5BMF appeared from the 9th day after treatment and exhibited significant ATCA at 21 days PI (Fig. 5e). The tumor growth of the 5BMF treated group (drug group) was suppressed dramatically compared to the PBS treated group (control group), with a statistically significant result (p = 0.0426, statistical comparison at the 21st day of the treatment). Approximately the mean tumor volume of the drug group increased ∼2 times after the 21 day-treatment period, while that of the control group increased ∼16 times. Meanwhile the body weight between the control and treated groups showed no significant difference (p = 0.477) (Fig. 5f), implying low in vivo toxicity of 5BMF. Tumor pathologic section H & E staining results displayed that 5BMF caused tumor inflammatory cell infiltration, fibrous tissue proliferation, and cell apoptosis (Fig. S11†) similar to the parent compound F16.28 The major organs' histologic examination results did not reveal any significant microscopic lesions in the 5BMF treated mouse group over four weeks, compared to the control group (Fig. S12†).
Conclusions
In summary, this study demonstrates that various substitutions in F16's indole ring significantly affect the absorbance, fluorescence and anti-tumor or normal cell toxicity activity. By applying this SAR, researchers can achieve high-efficiency, low-toxicity anti-tumor DLCs through rational design. More importantly, our study discovered the compound 5BMF that displays significant anti-tumor activity not only in 10 cancer cell lines but also in nude mice implanted with human NSCLC. In addition to cancer treatment, 5BMF can also be used as a fluorescence imaging probe for tumor imaging. Thus, F16s have a bright future for use as a platform for developing targeted theranostic agents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the Office of Science (BER), U.S. Department of Energy (DE-SC0008397) (ZC), the National Natural Science Foundation of China (81871406), the 6th youth development foundation (JDYY52015032) from the First Hospital of Jilin University, and the Jilin Provincial Health Special Project (2018SCZWSZX-018) from the finance department of Jilin Province. All animal experimentation was performed with the approval of Stanford University's Administrative Panel on Laboratory Animal Care. Moreover, all experiments were performed in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals.Notes and references
- L. Biasutto, L. F. Dong, M. Zoratti and J. Neuzil, Mitochondrion, 2010, 10, 670–681 CrossRef CAS.
- E. D. Tseligka, A. Rova, E. P. Amanatiadou, G. Calabrese, J. Tsibouklis, D. G. Fatouros and I. S. Vizirianakis, Pharm. Res., 2016, 33, 1945–1958 CrossRef CAS.
- V. R. Fantin, J. St-Pierre and P. Leder, Cancer Cell, 2006, 10, 172 CrossRef CAS.
- G. Bagkos, K. Koufopoulos and C. Piperi, Med. Hypotheses, 2014, 83, 175–181 CrossRef CAS.
- G. Bagkos, K. Koufopoulos and C. Piperi, Curr. Pharm. Des., 2014, 20, 4570–4579 CrossRef CAS.
- S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS.
- J. Wang, C. Xiang, F. F. Tian, Z. Q. Xu, F. L. Jiang and Y. Liu, RSC Adv., 2014, 4, 18205–18216 RSC.
- L. V. Johnson, M. L. Walsh and L. B. Chen, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 990–994 CrossRef CAS.
- S. D. Bernal, T. J. Lampidis, R. M. McIsaac and L. B. Chen, Science, 1983, 222, 169–172 CrossRef CAS PubMed.
- M. Kurtoglu and T. J. Lampidis, Mol. Nutr. Food Res., 2009, 53, 68–75 CrossRef CAS PubMed.
- X. Yan, Y. Zhou and S. Liu, Theranostics, 2012, 2, 988–998 CrossRef CAS.
- J. Morgan and A. R. Oseroff, Adv. Drug Delivery Rev., 2001, 49, 71–86 CrossRef CAS.
- G. Cheng, J. Zielonka, B. P. Dranka, D. McAllister, A. C. Mackinnon, J. Joseph and B. Kalyanaraman, Cancer Res., 2012, 72, 2634–2644 CrossRef CAS.
- J. C. Wang, Z. Jiang, L. P. Xiang, Y. F. Li, M. R. Ou, X. Yang, J. W. Shao, Y. S. Lu, L. F. Lin, J. Z. Chen, Y. Dai and L. Jia, Sci. Rep., 2014, 4, 5006 CrossRef.
- B. Kalyanaraman, G. Cheng, M. Hardy, O. Ouari, M. Lopez, J. Joseph, J. Zielonka and M. B. Dwinell, Redox Biol., 2018, 14, 316–327 CrossRef CAS PubMed.
- C. D. Britten, E. K. Rowinsky, S. D. Baker, G. R. Weiss, L. Smith, J. Stephenson, M. Rothenberg, L. Smetzer, J. Cramer, W. Collins, D. D. Von Hoff and S. G. Eckhardt, Clin. Cancer Res., 2000, 6, 42–49 CAS.
- R. Wadhwa, L. Colgin, T. Yaguchi, K. Taira, R. R. Reddel and S. C. Kaul, Cancer Res., 2002, 62, 4434–4438 CAS.
- V. R. Fantin, M. J. Berardi, L. Scorrano, S. J. Korsmeyer and P. Leder, Cancer Cell, 2002, 2, 29–42 CrossRef CAS.
- V. Reshetnikov, S. Daum, C. Janko, W. Karawacka, R. Tietze, C. Alexiou, S. Paryzhak, T. Dumych, R. Bilyy, P. Tripal, B. Schmid, R. Palmisano and A. Mokhir, Angew. Chem., 2018, 130, 12119–12122 CrossRef.
- D. W. Li, Z. D. Qi, X. L. Ding, J. H. Li, F. L. Jiang, Y. Liu, D. W. J. Kwong and W. K. Wong, J. Lumin., 2013, 134, 830–836 CrossRef CAS.
- S. Wu, Q. Z. Cao, X. L. Wang, K. Cheng and Z. Cheng, Chem. Commun., 2014, 50, 8919–8922 RSC.
- J. S. Modica-Napolitano and J. R. Aprille, Adv. Drug Delivery Rev., 2001, 49, 63–70 CrossRef CAS.
- H. He, D. W. Li, L. Y. Yang, L. Fu, X. J. Zhu, W. K. Wong, F. L. Jiang and Y. Liu, Sci. Rep., 2015, 5, 13543 CrossRef.
- C. Xiang, D. W. Li, Z. D. Qi, F. L. Jiang, Y. S. Ge and Y. Liu, Luminescence, 2013, 28, 865–872 CrossRef CAS.
- J. A. Jara, V. Castro-Castillo, J. Saavedra-Olavarria, L. Peredo, M. Pavanni, F. Jana, M. E. Letelier, E. Parra, M. I. Becker, A. Morello, U. Kemmerling, J. D. Maya and J. Ferreira, J. Med. Chem., 2014, 57, 2440–2454 CrossRef CAS.
- T. Uemura, N. Yanai, S. Watanabe, H. Tanaka, R. Numaguchi, M. T. Miyahara, Y. Ohta, M. Nagaoka and S. Kitagawa, Nat. Commun., 2010, 1, 83 CrossRef.
- S. Bolte and F. P. Cordelieres, J. Microsc. (Oxford, U. K.), 2006, 224, 213–232 CrossRef CAS.
- V. R. Fantin and P. Leder, Cancer Res., 2004, 64, 329–336 CrossRef CAS.
- S. Wu, Q. Cao, X. Wang, K. Cheng and Z. Cheng, Chem. Commun., 2014, 50, 8919–8922 RSC.
- J. R. Molina, P. Yang, S. D. Cassivi, S. E. Schild and A. A. Adjei, Mayo Clin. Proc., 2008, 83, 584–594 CrossRef.
- E. Tsvetkova and G. D. Goss, Curr. Oncol., 2012, 19, S45–S51 CAS.
- F. R. Hirsch, G. V. Scagliotti, J. L. Mulshine, R. Kwon, W. J. Curran, Jr., Y. L. Wu and L. Paz-Ares, Lancet, 2017, 389, 299–311 CrossRef CAS.
- T. Reungwetwattana and G. K. Dy, J. Carcinog., 2013, 12, 22 CrossRef.
- T. Stylianopoulos, Ther. Delivery, 2013, 4, 421–423 CrossRef CAS.
- T. G. Bivona, H. Hieronymus, J. Parker, K. Chang, M. Taron, R. Rosell, P. Moonsamy, K. Dahlman, V. A. Miller, C. Costa, G. Hannon and C. L. Sawyers, Nature, 2011, 471, 523–526 CrossRef CAS.
- K. R. Jakobsen, C. Demuth, A. T. Madsen, D. Hussmann, J. Vad-Nielsen, A. L. Nielsen and B. S. Sorensen, Oncogenesis, 2017, 6, e307 CrossRef CAS.
- K. Koya, Y. Li, H. Wang, T. Ukai, N. Tatsuta, M. Kawakami, T. Shishido and L. B. Chen, Cancer Res., 1996, 56, 538–543 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01410a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |