
Ligandless nickel-catalyzed transfer hydrogenation of alkenes and alkynes using water as the hydrogen donor†
Xiaoping
Hu‡
a,
Gaonan
Wang‡
a,
Chunxiang
Qin
b,
Xin
Xie
a,
Chunli
Zhang
a,
Wei
Xu
a and
Yuanhong
Liu
 *a
*a
aState Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, People's Republic of China. E-mail: yhliu@sioc.ac.cn
bDepartment of Chemistry, Shanghai University, 99 Shang-Da Road, Shanghai 200444, P. R. China
First published on 11th June 2019
Abstract
The first general route for nickel-catalyzed transfer hydrogenation reaction of alkenes and alkynes using water as the hydrogen source has been developed. The method features the use of inexpensive and air-stable nickel(II) salt as the pre-catalyst and zinc powder as a reducing agent, allowing the TH reaction to occur under mild reaction conditions with a wide substrate scope and functional group tolerance. No ligand was required for this reaction. The reaction has also been applied successfully to the reduction of nitrogen-containing heterocycles.
Introduction
Transition-metal-catalyzed hydrogenation of unsaturated π-systems is one of the most fundamental and important reactions in organic chemistry.1 These reactions usually proceed with hydrogen, a highly flammable and explosive gas. Recently, catalytic transfer hydrogenation (TH) has emerged as a promising and powerful strategy because it is operationally simple and much safer than direct hydrogenation using hydrogen gas, as no gas containment or pressure vessel is necessary.2 Compared with the TH reactions of polarized substrates such as aldehydes,2 ketones,2 imines2 and α,β-unsaturated carbonyl compounds,3 the TH reactions of non-polarized unsaturated compounds such as normal alkenes or alkynes have been less developed. A variety of reagents including formic acid,4 ethanol,5 isopropanol,6 1,4-cyclohexadiene,7 amine–borane adducts8etc. have been used as possible in situ hydrogen sources in the TH reactions of alkenes or alkynes. However, there are a few examples using water as the hydrogen donor. The use of water as the hydrogen donor is highly attractive since it is cheap, non-toxic, and environmentally friendly, and allows the easy isotopic labeling of the products by D2O. It may also offer the opportunity to disclose new reaction patterns or selectivity. However, formidable challenges remain in water-mediated reactions catalyzed by a transition metal since the resulting organometallic intermediates are generally considered to be incompatible with water. It was known that in the presence of a reductant such as zinc, nickel salts could promote the reduction of unsaturated compounds in water; however, most of these reports were restricted to α,β-unsaturated carbonyls. In addition, a high catalyst loading (>30% to excess amounts of nickel salts) was required.9 It was also reported that a combination of a stoichiometric amount of NiCl2·2H2O with a large excess of lithium powder (8 equiv.) in the presence of an arene as an electron carrier was effective for the conversion of alkenes or alkynes into alkanes or alkenes.10 The hydrogen source came from the water content in the nickel salt in these cases.10 In these reactions, a high catalyst loading and a large excess of the reductant were employed. Therefore, a general and catalytic version of the Ni-catalyzed reduction of common alkenes and alkynes using water has not been reported to date. Recently, Pd/Ni or Pd-catalyzed transfer hydrogenation of alkenes or alkynes using water11 in the presence of magnesium,11a Cp2TiCl,11b Me3SiSiMe3,11c B2(OH)4,11d or B2Pin2,11e or Rh-catalyzed reaction in the presence of zinc12 has been reported. During our manuscript preparation, a Pd/Mn-catalyzed transfer hydrogenation of alkynes using water was reported.13 Nevertheless, the catalytic TH reactions by utilization of the inexpensive and earth-abundant metal catalysts are quite rare and will be highly desirable in organic chemistry. Recently, we focused our efforts on the Ni-catalyzed hydrocyanation reactions of alkynes14 and alkenes15 using Zn(CN)2 as the cyanide source and water as the hydrogen source. During the course of the mechanistic study, we occasionally found that alkynes could be reduced by nickel-catalyzed transfer hydrogenation in the presence of water.14 Inspired by our work, we decided to conduct an in-depth study on this topic. We now discovered that the simple combination of an air-stable nickel(II) pre-catalyst with a reducing metal under ligandless conditions is highly effective for transfer hydrogenation of alkenes and alkynes with water as the hydrogen donor (Scheme 1). The method proceeds with high generality, a wide substrate scope and good functional group tolerance. Remarkably, the reaction is also applicable to the reduction of nitrogen-containing heterocycles, which has not been achieved in nickel-catalyzed TH reactions using water.16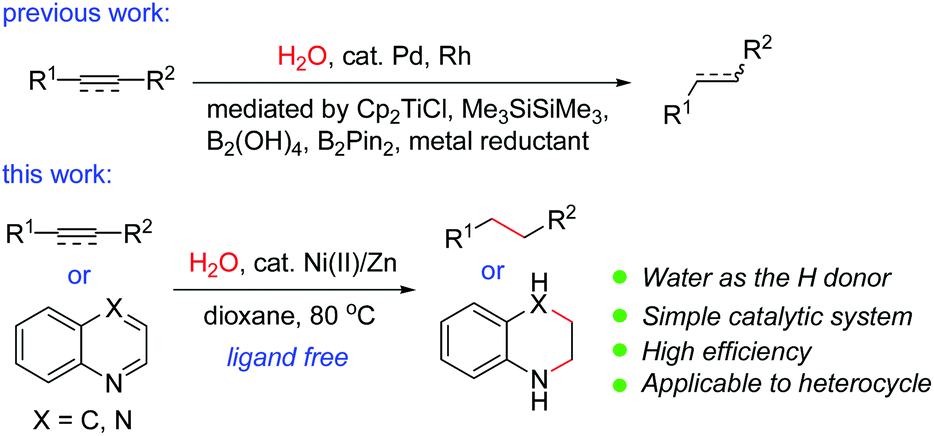 | ||
| Scheme 1 Transition-metal-catalyzed transfer hydrogenation reactions. | ||
Results and discussion
We initially investigated the nickel-catalyzed transfer hydrogenation of 2-vinylnaphthalene 1a with H2O. After a thorough screening of the nickel catalysts, ligands, reductants, solvents, additives etc., a highly efficient catalyst system featuring NiCl2·6H2O (5 mol%) and zinc powder (2.0 equiv.) in dioxane/H2O (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 56 equiv. H2O) at 80 °C was established. To our surprise, the reaction proceeded without the need for a ligand. Under these reaction conditions, 1a was reduced smoothly to afford 2-ethylnaphthalene 2a in 88% yield (Table 1, entry 1). The use of other nickel salts such as NiCl2, NiBr2 or NiI2 as the catalyst was also effective for this reaction (entries 2–4). However, the use of NiF2 or Ni(acac)2 led to either a trace amount of 2a or no conversion (entries 5 and 6). Inferior results were found when other reducing agents such as Al, Mn, and Mg were employed (entries 7–9). The reaction proceeded smoothly in DMF/H2O and THF/H2O, with the formation of 2a in 90% and 95% yields, respectively (entries 10 and 11). Unexpectedly, the use of MeCN/H2O significantly lowered the conversion, and only a trace amount of 2a was observed (entry 12). The amount of H2O also played an important role in this reaction, since decreasing the amount of H2O (dioxane/H2O = 25:1, 11 equiv. H2O) afforded 2a in only 33% yield (entry 13). The best result was achieved by using dioxane/H2O = 3:1 (entry 14, 93 equiv. H2O). The results indicated that a sufficient amount of water was required for this reaction. A high yield of 2a could also be observed when reducing the reaction time to 5 h (entry 15). Decreasing the amount of Zn or lowering the reaction temperature resulted in lower yields of 2a (entries 16 and 17). Control experiments indicated that both the nickel catalyst and Zn were essential for the reaction (entries 18 and 19).
:1, 56 equiv. H2O) at 80 °C was established. To our surprise, the reaction proceeded without the need for a ligand. Under these reaction conditions, 1a was reduced smoothly to afford 2-ethylnaphthalene 2a in 88% yield (Table 1, entry 1). The use of other nickel salts such as NiCl2, NiBr2 or NiI2 as the catalyst was also effective for this reaction (entries 2–4). However, the use of NiF2 or Ni(acac)2 led to either a trace amount of 2a or no conversion (entries 5 and 6). Inferior results were found when other reducing agents such as Al, Mn, and Mg were employed (entries 7–9). The reaction proceeded smoothly in DMF/H2O and THF/H2O, with the formation of 2a in 90% and 95% yields, respectively (entries 10 and 11). Unexpectedly, the use of MeCN/H2O significantly lowered the conversion, and only a trace amount of 2a was observed (entry 12). The amount of H2O also played an important role in this reaction, since decreasing the amount of H2O (dioxane/H2O = 25:1, 11 equiv. H2O) afforded 2a in only 33% yield (entry 13). The best result was achieved by using dioxane/H2O = 3:1 (entry 14, 93 equiv. H2O). The results indicated that a sufficient amount of water was required for this reaction. A high yield of 2a could also be observed when reducing the reaction time to 5 h (entry 15). Decreasing the amount of Zn or lowering the reaction temperature resulted in lower yields of 2a (entries 16 and 17). Control experiments indicated that both the nickel catalyst and Zn were essential for the reaction (entries 18 and 19).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Reductant | Solvent | Time (h) | Yielda (%) |
| a Determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. The yields of the unreacted 1a are shown in parentheses. b 1.0 equiv. of Zn was used. c 50 °C. | |||||
| 1 | NiCl2·6H2O | Zn | Dioxane/H2O (5/1) | 10 | 88 |
| 2 | NiCl2 | Zn | Dioxane/H2O (5/1) | 10 | 89 |
| 3 | NiBr2 | Zn | Dioxane/H2O (5/1) | 10 | 89 |
| 4 | NiI2 | Zn | Dioxane/H2O (5/1) | 10 | 79 (7) |
| 5 | NiF2 | Zn | Dioxane/H2O (5/1) | 10 | 2 (85) |
| 6 | Ni(acac)2 | Zn | Dioxane/H2O (5/1) | 10 | — (89) |
| 7 | NiCl2·6H2O | Al | Dioxane/H2O (5/1) | 10 | 24 (68) |
| 8 | NiCl2·6H2O | Mn | Dioxane/H2O (5/1) | 10 | 13 (81) |
| 9 | NiCl2·6H2O | Mg | Dioxane/H2O (5/1) | 10 | — (96) |
| 10 | NiCl2·6H2O | Zn | DMF/H2O (5/1) | 10 | 90 (1) |
| 11 | NiCl2·6H2O | Zn | THF/H2O (5/1) | 10 | 95 |
| 12 | NiCl2·6H2O | Zn | CH3CN/H2O (5/1) | 10 | 1 (58) |
| 13 | NiCl2·6H2O | Zn | Dioxane/H2O (25/1) | 10 | 33 (59) |
| 14 | NiCl 2 ·6H 2 O | Zn | Dioxane/H 2 O (3/1) | 10 | 96 |
| 15 | NiCl2·6H2O | Zn | Dioxane/H2O (3/1) | 5 | 95 |
| 16b | NiCl2·6H2O | Zn | Dioxane/H2O (3/1) | 5 | 48 (48) |
| 17c | NiCl2·6H2O | Zn | Dioxane/H2O (3/1) | 5 | 32 (63) |
| 18 | — | Zn | Dioxane/H2O (3/1) | 5 | — (94) |
| 19 | NiCl2·6H2O | — | Dioxane/H2O (3/1) | 5 | — (94) |

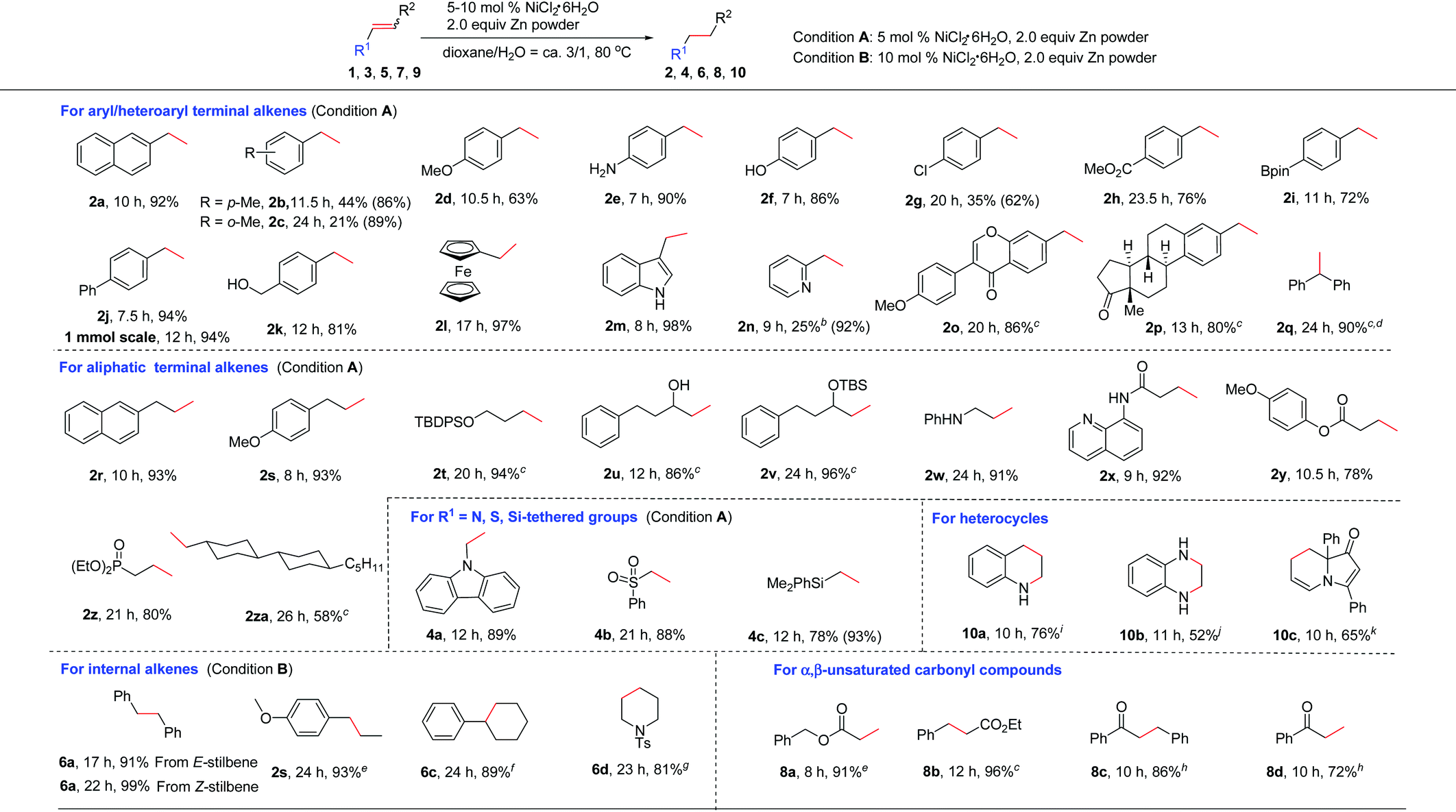
Next, the generality of this nickel-catalyzed transfer hydrogenation protocol was investigated. The scope of aryl alkenes was first studied under the conditions shown in Table 1, entry 14. A wide range of aryl/heteroaryl alkenes were compatible for this reaction (Table 2). Aryl alkenes bearing a methyl group at the para- or ortho-position afforded high yields of 2b and 2c (86–89%); however, a longer reaction time was needed for 2c. The results indicated that steric hindrance has a certain influence on this reaction. Electron-donating groups such as p-MeO, free amino and OH groups as well as electron-withdrawing groups such as p-Cl, p-CO2Me on the aryl rings were well tolerated, and the corresponding products of 2d–2h were obtained in 62–90% yields. Notably, a chlorine substituent was well suited in this reaction (2g). Special alkenes such as p-boron-, p-phenyl-, p-CH2OH-aryl alkenes and ferroceneyl-alkenes turned out to be efficient substrates (2i–2l). Heteroaryl alkenes such as indolyl and pyridyl alkenes reacted with water very well to form 2m and 2n in excellent yields. The reaction could be readily applied to natural product derivatives, for example estrone or formononetin derived alkenes transformed to 2o and 2p in high efficiency, while the ketone moiety in these substrates remained intact. In these cases, increasing the catalyst loading to 10 mol% was required to consume the substrates completely. When sterically hindered 1,1-diphenylethylene 1q was employed as the substrate, a high yield of the desired 2q (ca. 94%) was also observed; however, a small amount of the starting material (5%) remained, which could not be separated from 2q by column chromatography. 1q was reduced completely and cleanly by replacing zinc powder by zinc flakes in the presence of 10 mol% NiCl2·6H2O. Possibly, zinc flakes may reduce Ni(II)-salt or related nickel intermediates more efficiently than zinc powder.
|
a Isolated yields. The 1H NMR yields of the volatile products are shown in parentheses.
b Containing a small amount of stabilizer and solvent.
c Condition B.
d 2.0 equiv. of Zn flakes (325 mesh) were used.
e Condition A.
f 3.0 equiv. of Zn flakes (325 mesh) were used.
g 3.0 equiv. of Zn powder were used.
h 20 mol% NiCl2·6H2O and 4.0 equiv. of Al powder (200 mesh) in dioxane/H2O (5:1).
i 10 mol% NiCl2·6H2O, 3.0 equiv. of Zn powder and 1.0 equiv. of Et3N at 100 °C in dioxane/H2O (5:1).
j 10 mol% NiCl2·6H2O and 3.0 equiv. of Zn powder at 100 °C in dioxane/H2O (3:1).
k 10 mol% NiCl2·6H2O, 2.0 equiv. of Zn powder and 1.0 equiv. of Et3N at 100 °C in dioxane/H2O (5:1).
|
|---|
|
Aliphatic alkenes were also easily reduced to give the corresponding alkanes in good to excellent yields. As expected, allylic-naphthalene (1r) and -benzene (1s) gave 2r and 2s in high yields. An alkyl side chain bearing an OTBDPS group was also suitable (2t). Non-protected or protected allylic alcohols transformed to 2u and 2v efficiently. In the case of 2u, the isomerization to the corresponding ketone product was not observed.17 Allylic amine 1w was also confirmed to be valuable substrate for this reaction to afford the desired alkane 2w in 91% yield. The possible deallylation product was not observed although it was known that Ni could catalyze the C–N bond cleavage reaction in allylic amines.18 In addition, a series of allylic substrates bearing various functional groups such as amide, ester or phosphate groups were converted to desired 2x–2z successfully. Alkene 1za without any functional group was reduced smoothly (2za). The reactivity of various activated alkenes was also examined, and alkenes tethered with carbazolyl, sulfonyl, and silyl groups were all compatible with this system (4a–4c).
As for internal alkenes, high conversions were achieved from both cis-(Z-5a) and trans-stilbene (E-5a), indicating that the reaction was not sensitive to the geometry of the double bond. A common naturally occurring trans-anethole worked well (2s). The reaction with tri-substituted alkenes such as 2,3,4,5-tetrahydro-1,1′-biphenyl required the use of 3.0 equiv. of zinc flakes as the reductant (6c). In the case of cyclic alkene 5d, double bond migration (chain walking) to the closer position of the NTs group was observed during the reaction. A high yield of the alkane 6d was achieved by increasing the amount of Zn to 3.0 equiv. The use of an α,β-unsaturated ester revealed a faster and clean reduction (8a and 8b). When the reaction was applied to an α,β-unsaturated ketone such as chalcone, only a low yield (23%) of the double bond reduction product 8c was obtained due to the competing side reactions such as over-reduction or alkene oligomerization. To our delight, highly chemoselective reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond over the CO bond could be achieved by switching the reductant of Zn to Al and using 20 mol% NiCl2·6H2O as the catalyst (8c).19 Under this condition, a terminal unsaturated ketone was also reduced selectively (8d).
C bond over the CO bond could be achieved by switching the reductant of Zn to Al and using 20 mol% NiCl2·6H2O as the catalyst (8c).19 Under this condition, a terminal unsaturated ketone was also reduced selectively (8d).
Reduction of heteroaromatic compounds is generally more difficult than alkenes and alkynes due to the resonance stabilization and the possible deactivation of the catalyst by either the substrate or the product. The transfer hydrogenation of heterocycles has been much less developed.20 When the reaction of quinoline was carried out in the presence of 10 mol% NiCl2·6H2O/3.0 equiv. Zn, only a little product was observed, along with various byproducts. After many efforts, we found that the addition of 1.0 equiv. of Et3N significantly improved the efficiency of this reaction to provide the desired 10a in 76% yield. The role of Et3N is not clear yet. It might help to stabilize the catalytically active species by a weak coordination.21 Quinoxaline was reduced to 10b in 52% yield, and in this case, the addition of Et3N resulted in a complex mixture. Notably, selective reduction of one of the CC bonds was observed with indolizinone (10c).
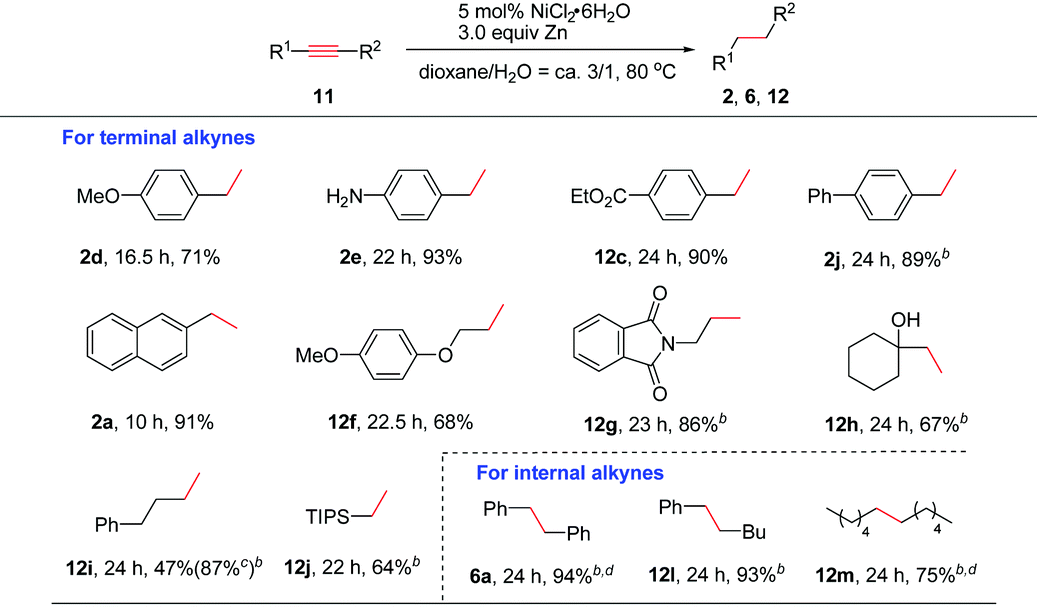
The reactivity of alkynes was also evaluated (Table 3). In these cases, 3.0 equiv. of Zn were required in order to reduce alkynes completely to alkanes. Terminal arylacetylenes bearing electron-withdrawing groups or electron-rich groups were well compatible (2d–2e and 12c). Alkynes with a biphenyl (2j) or 2-naphthyl group (2a) were also smoothly reduced. Aliphatic terminal alkynes bearing various functional groups such as ether, amide, OH, or phenyl groups and silyl alkyne worked well to afford the corresponding 12f–12j in moderate to good yields. Particularly, propargyl amide (12g) and tertiary propargyl alcohol (12h) were tolerable. Bis(aryl)alkynes such as diphenylacetylene were cleanly reduced (6a). Reduction of phenyl, alkyl-acetylene and dialkyl acetylene also proceeded smoothly (12l and 12m). In alkyne reductions, we could observe the formation of alkenes at the early stage of the reaction in some cases. Thus, the reaction proceeds via generation of an alkene intermediate which undergoes further reduction to deliver the alkane product.
In order to understand the mechanism, various control experiments were performed. Reduction of diphenylacetylene with D2O using NiCl2 as the catalyst afforded the deuterated product 6a-d in 86% yield with high deuterium incorporation (Scheme 2, eqn (1)), indicating that water acts as the hydrogen source for this reaction. A kinetic isotope effect was examined by employing an equimolar mixture of H2O and D2O as the H and D donor, which revealed a primary kinetic isotope effect (kH/kD = 2.3). Parallel experiments indicated that kH/kD = 3.22 The results imply that the O–H bond cleavage of H2O is likely involved in the rate-determining step (Scheme 2, eqn (2)).11d In the reaction of alkene 1a with D2O, deuterium was found in both the benzylic position and methyl group (Scheme 2, eqn (3)). The results indicated that a Ni–H species might be generated, and addition of Ni–H to the alkene is reversible under the reaction conditions. The yield of the product 2a was decreased significantly upon addition of mercury (Scheme 2, eqn (4)), indicating that a heterogeneous system might be involved in this transfer hydrogenation reaction. We also noted that hydrogen gas evolution could be detected at 80 °C regardless of the presence or absence of a substrate.9d The detailed reaction mechanism was not clear yet at the moment. Possibly, a Ni(0) species is first generated through reduction of the Ni(II) salt by Zn. This is followed by a catalytic hydrogenation process involving reduction of the substrate on the nickel surface with the absorbed H2 gas.9d,23
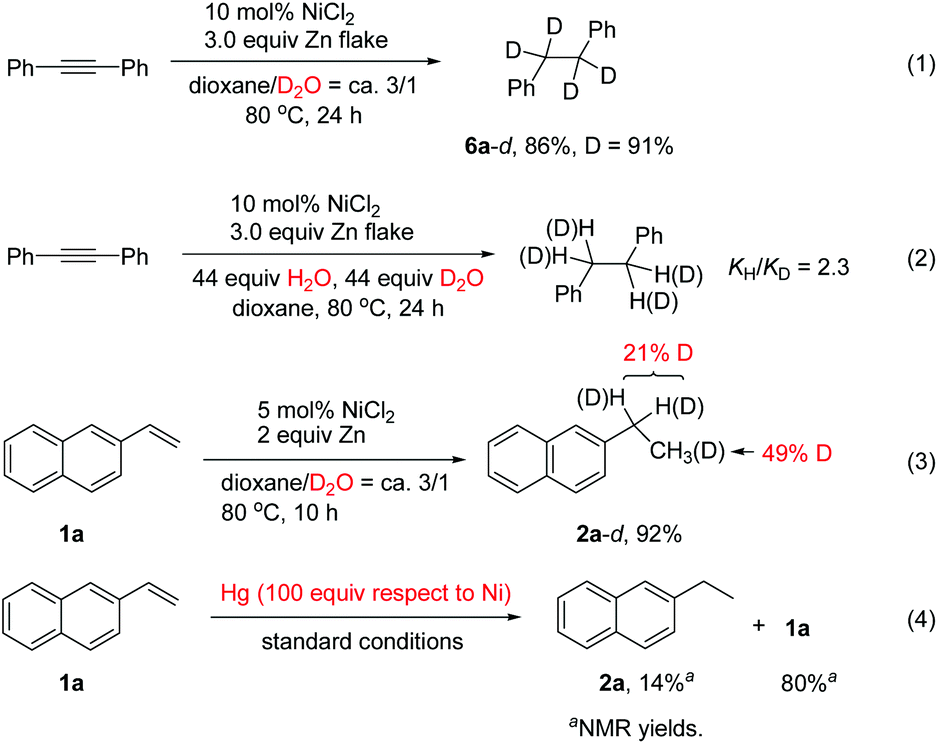 | ||
| Scheme 2 Control experiments. | ||
Conclusions
In summary, we have developed the first general and operationally simple protocol for nickel-catalyzed transfer hydrogenation reaction of alkenes and alkynes using water as the hydrogen source. The method features the use of inexpensive and air-stable nickel(II) salt as the pre-catalyst and zinc as a reducing agent, allowing the TH reaction to occur under mild reaction conditions with a wide substrate scope and functional group tolerance. The method does not require the use of a ligand. The reaction has also been applied successfully to the reduction of nitrogen-containing heterocycles. Further investigations on the detailed reaction mechanism and application of this chemistry are in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Key R&D Program of China (2016YFA0202900), the National Natural Science Foundation of China (21772217), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000), the Science and Technology Commission of Shanghai Municipality (18XD1405000) and the Shanghai Institute of Organic Chemistry (sioczz201807) for financial support.Notes and references
- (a) The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007 Search PubMed; (b) Modern Reduction Methods, ed. P. G. Andersson and I. J. Munslow, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008 Search PubMed.
- For reviews, see: (a) C. Wang, X. Wu and J. Xiao, Chem. – Asian J., 2008, 3, 1750 CrossRef CAS PubMed; (b) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621 CrossRef CAS PubMed; (c) S. Werkmeister, J. Neumann, K. Junge and M. Beller, Chem. – Eur. J., 2015, 21, 12226 CrossRef CAS PubMed.
- For selected papers, see: (a) A. D. Kosal and B. L. Ashfeld, Org. Lett., 2010, 12, 44 CrossRef CAS PubMed; (b) J.-Y. Shang, F. Li, X.-F. Bai, J.-X. Jiang, K.-F. Yang, G.-Q. Lai and L.-W. Xu, Eur. J. Org. Chem., 2012, 2809 CrossRef CAS; (c) B. Ding, Z. Zhang, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Org. Lett., 2013, 15, 3690 CrossRef CAS PubMed.
- (a) J. M. Brunel, Tetrahedron, 2007, 63, 3899 CrossRef CAS; (b) J. Broggi, V. Jurčík, O. Songis, A. Poater, L. Cavallo, A. M. Z. Slawin and C. S. J. Cazin, J. Am. Chem. Soc., 2013, 135, 4588 CrossRef CAS PubMed.
- Y. Wang, Z. Huang, X. Leng, H. Zhu, G. Liu and Z. Huang, J. Am. Chem. Soc., 2018, 140, 4417 CrossRef CAS PubMed.
- (a) S. Horn and M. Albrecht, Chem. Commun., 2011, 47, 8802 RSC; (b) G. Zhang, Z. Yin and J. Tan, RSC Adv., 2016, 6, 22419 RSC; (c) Z. Mazloomi, R. Pretorius, O. Pàmies, M. Albrecht and M. Diéguez, Inorg. Chem., 2017, 56, 11282 CrossRef CAS PubMed.
- J. F. Quinn, D. A. Razzano, K. C. Golden and B. T. Gregg, Tetrahedron Lett., 2008, 49, 6137 CrossRef CAS.
- H. Dong and H. Berke, J. Organomet. Chem., 2011, 696, 1803 CrossRef CAS.
- (a) K. Sakai, M. Ishige, H. Kono, I. Motoyama, K. Watanabe and K. Hata, Bull. Chem. Soc. Jpn., 1968, 41, 1902 CrossRef CAS; (b) C. Petrier and J.-L. Luche, Tetrahedron Lett., 1987, 28, 2347 CrossRef CAS; (c) C. Petrier and J.-L. Luche, Tetrahedron Lett., 1987, 28, 2351 CrossRef CAS; (d) C. Petrier, J.-L. Luche, S. Lavaitte and C. Morat, J. Org. Chem., 1989, 54, 5313 CrossRef CAS; (e) M. J. Hazarika and N. C. Barua, Tetrahedron Lett., 1989, 30, 6567 CrossRef CAS; (f) C. Pétrier, S. Lavaitte and C. Morat, J. Org. Chem., 1990, 55, 1664 CrossRef; (g) H. IIikti, T. Benabdallah, K. Bentayeb, A. A. Othman and Z. Derriche, S. Afr. J. Chem., 2008, 61, 31 Search PubMed; (h) Y.-G. Chen, B. Shuai, C. Ma, X.-J. Zhang, P. Fang and T.-S. Mei, Org. Lett., 2017, 19, 2969 CrossRef CAS PubMed.
- (a) F. Alonso and M. Yus, Tetrahedron Lett., 1996, 37, 6925 CrossRef CAS; (b) F. Alonso and M. Yus, Tetrahedron Lett., 1997, 38, 149 CrossRef CAS; (c) F. Alonso and M. Yus, Tetrahedron Lett., 1998, 54, 1921 CrossRef CAS. For related FeCl2·4H2O/Li/arene mediated reduction, see: (d) Y. Moglie, F. Alonso, C. Vitale, M. Yus and G. Radivoy, Tetrahedron, 2006, 62, 2812 CrossRef CAS.
- (a) O. Muhammad, S. U. Sonavane, Y. Sasson and M. Chidambaram, Catal. Lett., 2008, 125, 46 CrossRef CAS; (b) A. G. Campaña, R. E. Estévez, N. Fuentes, R. Robles, J. M. Cuerva, E. Buñuel, D. Cárdenas and J. E. Oltra, Org. Lett., 2007, 9, 2195 CrossRef PubMed; (c) E. Shirakawa, H. Otsuka and T. Hayashi, Chem. Commun., 2005, 5885 RSC; (d) S. P. Cummings, T.-N. Le, G. E. Fernandez, L. G. Quiambao and B. J. Stokes, J. Am. Chem. Soc., 2016, 138, 6107 CrossRef CAS PubMed; (e) D. P. Ojha, K. Gadde and K. R. Prabhu, Org. Lett., 2016, 18, 5062 CrossRef CAS PubMed.
- T. Sato, S. Watanabe, H. Kiuchi, S. Oi and Y. Inoue, Tetrahedron Lett., 2006, 47, 7703 CrossRef CAS.
- C.-Q. Zhao, Y.-G. Chen, H. Qiu, L. Wei, P. Fang and T.-S. Mei, Org. Lett., 2019, 21, 1412 CrossRef CAS PubMed.
- X. Zhang, X. Xie and Y. Liu, J. Am. Chem. Soc., 2018, 140, 7385 CrossRef CAS PubMed.
- G. Wang, X. Xie, W. Xu and Y. Liu, Org. Chem. Front., 2019, 6, 2037 RSC.
- For reduction of nitrogen-containing heterocycles by NiCl2·2H2O–Li–arene combination with a stoichiometric amount of Ni salt and a large excess of lithium powder, see: (a) G. Radivoy, F. Alonso and M. Yus, Tetrahedron, 1999, 55, 14479 CrossRef CAS. For other metal-catalyzed TH reactions using water, see: (b) C. Wang, C. Li, X. Wu, A. Pettman and J. Xiao, Angew. Chem., Int. Ed., 2009, 48, 6524 CrossRef CAS PubMed; (c) D. Talwar, H. Y. Li, E. Durham and J. Xiao, Chem. – Eur. J., 2015, 21, 5370 CrossRef CAS PubMed.
- R. Uma, C. Crévisy and R. Grée, Chem. Rev., 2003, 103, 27 CrossRef CAS PubMed.
- T. Taniguchi and K. Ogasawara, Tetrahedron Lett., 1998, 39, 4679 CrossRef CAS.
- An Al/NiCl2·6H2O system with high loading (10 equiv.) for both metals has been used to reduce α,β-unsaturated compounds, see ref. 9e.
- For other metal-catalyzed TH reactions using water or alcohol as the hydrogen source, see: (a) V. H. Mai and G. I. Nikonov, Organometallics, 2016, 35, 943 CrossRef CAS; (b) Ref. 16b and c ; (c) Ref. 5 .
- J. Yu and J. B. Spencer, Chem. Commun., 1998, 1103 RSC.
- See the ESI.†.
- We found that in several cases, the yields of the desired products were influenced by the volume of the reaction vessel. The yield of the product was decreased by using a larger tube. The results indicated that the H2 gas concentration may have an important impact on the reaction.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1908616. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qo00616h |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |