
Nickel-catalyzed highly regioselective hydrocyanation of alkenes with Zn(CN)2†
Gaonan
Wang
,
Xin
Xie
,
Wei
Xu
and
Yuanhong
Liu
 *
*
State Key Laboratory of Organometallic Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: yhliu@sioc.ac.cn
First published on 1st May 2019
Abstract
The first general and regioselective nickel-catalyzed hydrocyanation of terminal alkenes with Zn(CN)2 using an air-stable and inexpensive nickel(II) salt as the precatalyst has been established. The strategy avoids the use of the volatile and hazardous reagent HCN. Aryl/heteroaryl alkenes are effectively converted to branched nitrile derivatives, while aliphatic alkenes or active alkenes are transformed to linear nitriles with good to excellent regioselectivity.
Introduction
Nitriles are versatile building blocks that can be converted into amines, isocyanates, amides, aldehydes, carboxylic acids, esters and heterocycles, not only in the laboratory but also on the industrial scale.1 They have also found many applications in materials, pharmaceuticals, cosmetics, and agrochemicals.2 In particular, due to the important properties such as hydrogen binding affinity to target proteins and metabolic stability of the nitrile functional group in pharmaceuticals, currently, over 30 nitrile-containing drugs are marketed, and more than 20 additional nitrile-containing leads are tested in clinical trials.3Transition-metal-catalyzed hydrocyanation of alkenes (addition of HCN to alkenes) provides a concise and highly atom-economical access to alkyl nitriles (Scheme 1).4–7 The most remarkable application of this reaction is the DuPont adiponitrile (a precursor of Nylon 6,6) process from 1,3-butadiene by nickel catalysis with a production of 1 million tons per year.8 However, these methods usually have the following drawbacks: (1) the hydrogen cyanide gas is extremely toxic, and is not easy to handle in a normal laboratory. Recently, acetone cyanohydrin7h–i and TMSCN/MeOH7j have been explored as less volatile precursors for generating HCN in situ, but they still pose a significant risk. (2) The substrate scope in these reactions was quite limited. Most of the studies concentrated on the hydrocyanation of aryl alkenes, while there are few examples with non-activated aliphatic alkenes9 as the substrates, possibly due to the low reactivity of these alkenes, and the high instability of the alkyl-metal intermediates which can be deactivated by β-hydride elimination, or forming the inactive dicyanonickel(II) complexes, etc.4c,7d (3) Although Markovnikov selectivity can be usually achieved for aryl alkenes, the selectivity control of aliphatic alkenes still remains a challenge.4c Therefore, it is of high importance to develop much safer, efficient, and regioselective catalyst systems. Recently, Morandi et al.10 reported elegant Ni-catalyzed transfer hydrocyanation of alkenes using isovaleronitrile as a HCN donor. Low to high linear-to-branched regioselectivities were observed for both aryl and aliphatic terminal alkenes. In this system, the air sensitive Ni(COD)2 and AlMe2Cl (as a Lewis acidic additive) were used. During our manuscript preparation, Pd-catalyzed transfer hydrocyanation of alkenes with anti-Markovnikov selectivity using 1-methylcyclohexa-2,5-diene-1-carbonitrile as a HCN source was reported.6b Nevertheless, the highly regioselective hydrocyanation by utilization of inexpensive and Earth-abundant metal catalysts is still quite rare and highly desirable. Recently, we have reported a highly efficient nickel-catalyzed hydrocyanation reaction of alkynes with Zn(CN)2 using Ni(acac)2/(neocuproine)/Mn as the catalyst system in the presence of water.11 However, when we applied these reaction conditions to alkenes, no desired products were observed. In order to achieve the hydrocyanation of alkenes, we made great efforts. Herein we report the first nickel-catalyzed hydrocyanation of terminal alkenes that (a) utilizes relatively less toxic Zn(CN)2 as the cyano source while obviating the use of the hazardous HCN, (b) accommodates both aryl and aliphatic alkenes with wide functional group tolerance, (c) displays high regioselectivity for both aryl and aliphatic alkenes in which the branched or linear regioselectivity was found to be strongly influenced by the substitution patterns of the substrates, and (d) proceeds under mild reaction conditions (80 °C) using the air stable and inexpensive nickel(II) salts as the precatalyst12 without the addition of air-sensitive additives (Scheme 1).
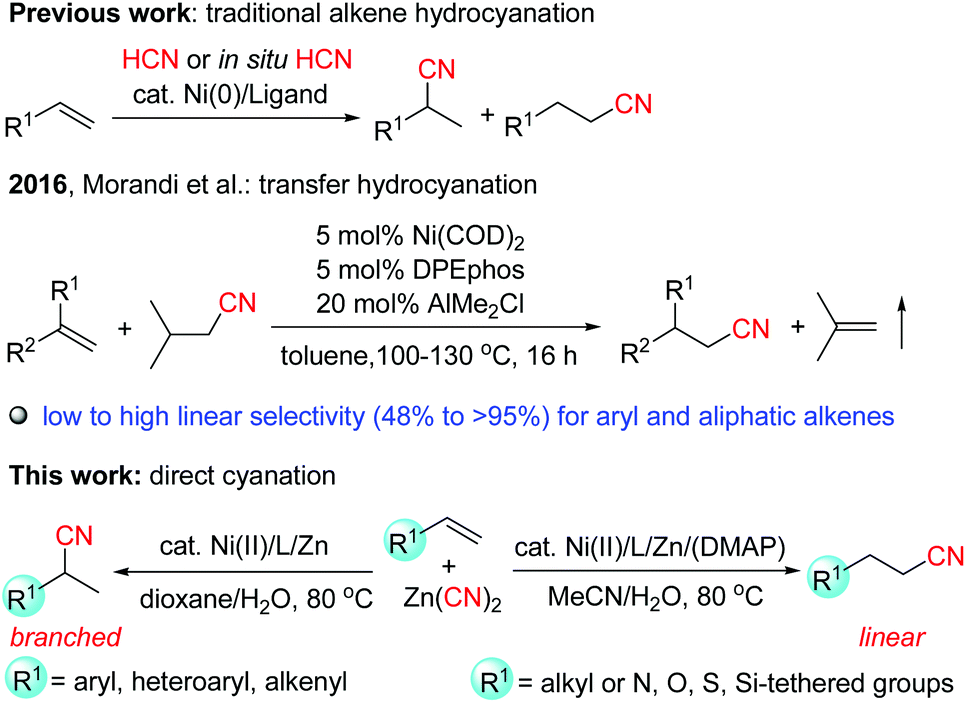 | ||
| Scheme 1 Nickel-catalyzed cyanation reactions of alkenes. | ||
Results and discussion
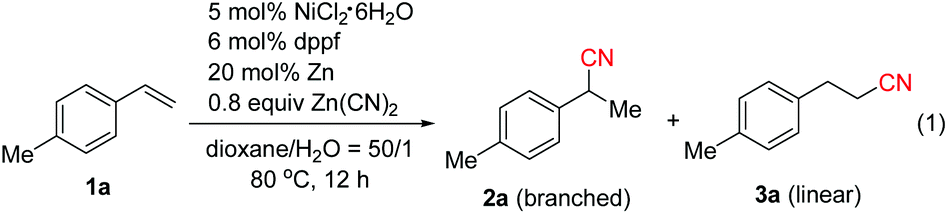
We initially investigated the cyanation reaction of aryl alkene 1a. After a thorough optimization study of the nickel catalysts, ligands, additives and solvents,13 we discovered that the use of NiCl2·6H2O (5 mol%), dppf (6 mol%) and Zn powder (20 mol%) enabled the reaction to take place in dioxane/H2O (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 80 °C. The desired product 2a with Markovnikov selectivity was formed efficiently in 89% yield, along with a linear product 3a in 3% yield (Table 1, entry 1). The high regioselectivity is possibly attributed to the formation of the more stable η3-benzylnickel cyanide (π-allyl nickel complex) intermediate. Subsequently, the effect of the amount of water was investigated. Neither 2a nor 3a was obtained when increasing the amount of water (Table 1, entry 2). Possibly, higher cyanide concentration in this case caused catalyst poisoning. The addition of one or two equivalents of water resulted in lower product yields and regioselectivity (Table 1, entries 3 and 4). Screening of other nickel catalysts showed that NiCl2(DME) displayed higher catalytic activity (Table 1, entries 5 and 6). As expected, PPh3 was not effective in this reaction. It was known that phosphite type ligands displayed the best performance in Ni(0)-catalyzed hydrocyanation of alkenes using HCN. However, in our case, the use of P(OPh)3 failed to give any desired products, possibly owing to its lower stability in water (Table 1, entry 7). It was suggested that bidentate ligands with larger bite angles promote the reductive elimination and diminish the formation of a dicyanonickel(II) complex4b,7d in the hydrocyanation reaction. In our case, bidentate phosphine ligands with small bite angles such as dppp and dppb,14 or DPEphos with a bite angle of 105°4b afforded the products in low yields and selectivity (Table 1, entry 8), while Xantphos with a large bite angle (β = 114°)4b showed higher efficiency than dppf (Table 1, entry 9). After brief screening of the solvents, THF was found to give products with comparable yields and selectivity to those obtained with dioxane (Table 1, entries 10 and 11). Lowering the amount of Zn(CN)2 to 0.6 equivalents could also afford 2a in excellent yield within 4 h, and only a trace of linear isomer 3a was detected (Table 1, entry 12). Control experiments indicated that the reaction could not take place without either NiCl2·6H2O, Xantphos or Zn (entry 13). It is worth noting that the yield of 2a decreased to 25% without the addition of water, emphasizing again the importance of water (Table 1, entry 14).
:1) at 80 °C. The desired product 2a with Markovnikov selectivity was formed efficiently in 89% yield, along with a linear product 3a in 3% yield (Table 1, entry 1). The high regioselectivity is possibly attributed to the formation of the more stable η3-benzylnickel cyanide (π-allyl nickel complex) intermediate. Subsequently, the effect of the amount of water was investigated. Neither 2a nor 3a was obtained when increasing the amount of water (Table 1, entry 2). Possibly, higher cyanide concentration in this case caused catalyst poisoning. The addition of one or two equivalents of water resulted in lower product yields and regioselectivity (Table 1, entries 3 and 4). Screening of other nickel catalysts showed that NiCl2(DME) displayed higher catalytic activity (Table 1, entries 5 and 6). As expected, PPh3 was not effective in this reaction. It was known that phosphite type ligands displayed the best performance in Ni(0)-catalyzed hydrocyanation of alkenes using HCN. However, in our case, the use of P(OPh)3 failed to give any desired products, possibly owing to its lower stability in water (Table 1, entry 7). It was suggested that bidentate ligands with larger bite angles promote the reductive elimination and diminish the formation of a dicyanonickel(II) complex4b,7d in the hydrocyanation reaction. In our case, bidentate phosphine ligands with small bite angles such as dppp and dppb,14 or DPEphos with a bite angle of 105°4b afforded the products in low yields and selectivity (Table 1, entry 8), while Xantphos with a large bite angle (β = 114°)4b showed higher efficiency than dppf (Table 1, entry 9). After brief screening of the solvents, THF was found to give products with comparable yields and selectivity to those obtained with dioxane (Table 1, entries 10 and 11). Lowering the amount of Zn(CN)2 to 0.6 equivalents could also afford 2a in excellent yield within 4 h, and only a trace of linear isomer 3a was detected (Table 1, entry 12). Control experiments indicated that the reaction could not take place without either NiCl2·6H2O, Xantphos or Zn (entry 13). It is worth noting that the yield of 2a decreased to 25% without the addition of water, emphasizing again the importance of water (Table 1, entry 14).
|
|
|||
|---|---|---|---|
| Entry | Deviation from the conditions shown in eqn (1) | Yielda (%) | |
| 2a | 3a | ||
| a Determined by 1H NMR of the crude reaction mixture using mesitylene as an internal standard. b The reaction time was 4 h. c 0.6 equiv. Zn(CN)2 were used. | |||
| 1 | None | 89 | 3 |
| 2 | Dioxane/H2O = 25/1 | 0 | 0 |
| 3 | 1.0 equiv. H2O was used | 33 | 9.5 |
| 4 | 2.0 equiv. H2O were used | 75 | 10 |
| 5 | NiCl2(DME) instead of NiCl2·6H2O | 87 | 4 |
| 6 | Ni(acac)2 instead of NiCl2·6H2O | 0 | 0 |
| 7 | 10 mol% PPh3 or P(OPh)3 instead of 6 mol% dppf | 0 | 0 |
| 8 | dppp, dppb or DPEPhos instead of dppf | 3–37 | 0–8 |
| 9 | Xantphos instead of dppf | 96 | 1 |
| 10 | Xantphos instead of dppf, MeCN or DMF instead of dioxane | 26–49 | 0–33 |
| 11 | Xantphos instead of dppf, THF instead of dioxane | 95 | 1 |
| 12 | Xantphos instead of dppf, 0.6 equiv. Zn(CN) 2 were used | 97 | 1 |
| 13b,c | Without NiCl2·6H2O, Xantphos or Zn | 0 | 0 |
| 14b,c | Without H2O | 25 | 0 |
With the optimized reaction conditions in hand (Table 1, entry 12), the scope of the aryl alkenes was first evaluated (Scheme 2). Gratifyingly, a wide range of styrenes were suitable for this reaction, and generally the corresponding branched nitriles were obtained in good to high yields with excellent regioselectivity. Electrically neutral styrene provided 2b in 88% yield. Only small amounts of product 2c derived from sterically hindered 2-methylstyrene were formed. However, replacing Xantphos by dppf and increasing the amount of Zn to one equivalent enhanced the reaction efficiency significantly, leading to 2c and linear product 3c in 63% and 10% yields, respectively. We envisioned that the use of a stoichiometric amount of Zn might cause a facile reduction of inactive Ni(II) species generated during the reaction process to Ni(0), and using adequate amounts of Ni(0) should be the key to the success of this reaction. Alkenes bearing electron-donating groups on the aryl ring such as p-iBu, p-MeO, and 3,5-(MeO)2 or even with the highly challenging free hydroxyl and amino groups turned out to be efficient substrates (2d–2h). Notably, product 2d, a precursor of the famous drug ibuprofen, could be obtained in 75% yield. Electron-deficient aryl alkenes bearing a p-F, p-CF3 or p-CN group also worked well (2i–2k). Boron-substituted aryl alkene 1l was well tolerated in the reaction, while the Bpin moiety remained intact. The reaction could be applied to biaryl or naphthyl substituted alkenes [2m, 2n and 2o (a precursor of the drug naproxen)]. 1,1-Disubstituted styrene transformed to 2p smoothly in 75% yield under modified reaction conditions with ZnI2 as the additive. ZnI2 possibly behaves as a Lewis acid to coordinate with the CN group which promotes the reductive elimination step.4c,7a,15 The substrate scope could also be extended to heteroaryl-substituted alkenes (2q, 2s–2t). To our surprise, the use of 2-vinylpyridine led to linear product 2r in 91% yield exclusively. We envisioned that pyridyl may act as a coordinating group to form a five-membered metal chelate ring16 that stabilizes the C(sp3)–Ni intermediate and facilitates the linear product formation. In addition, the high efficiency of this method in the hydrocyanation of formononetin (2u) and estrone (2v) derivatives demonstrates its potential value in late-stage functionalization of medicinally relevant compounds. Hydrocyanation of 1,3-diene was also successful (2w, 80% yield), which could not be cyanated by the transfer hydrocyanation reaction.10 The practicality of this method was elucidated by the gram-scale synthesis of 2a (86% yield).
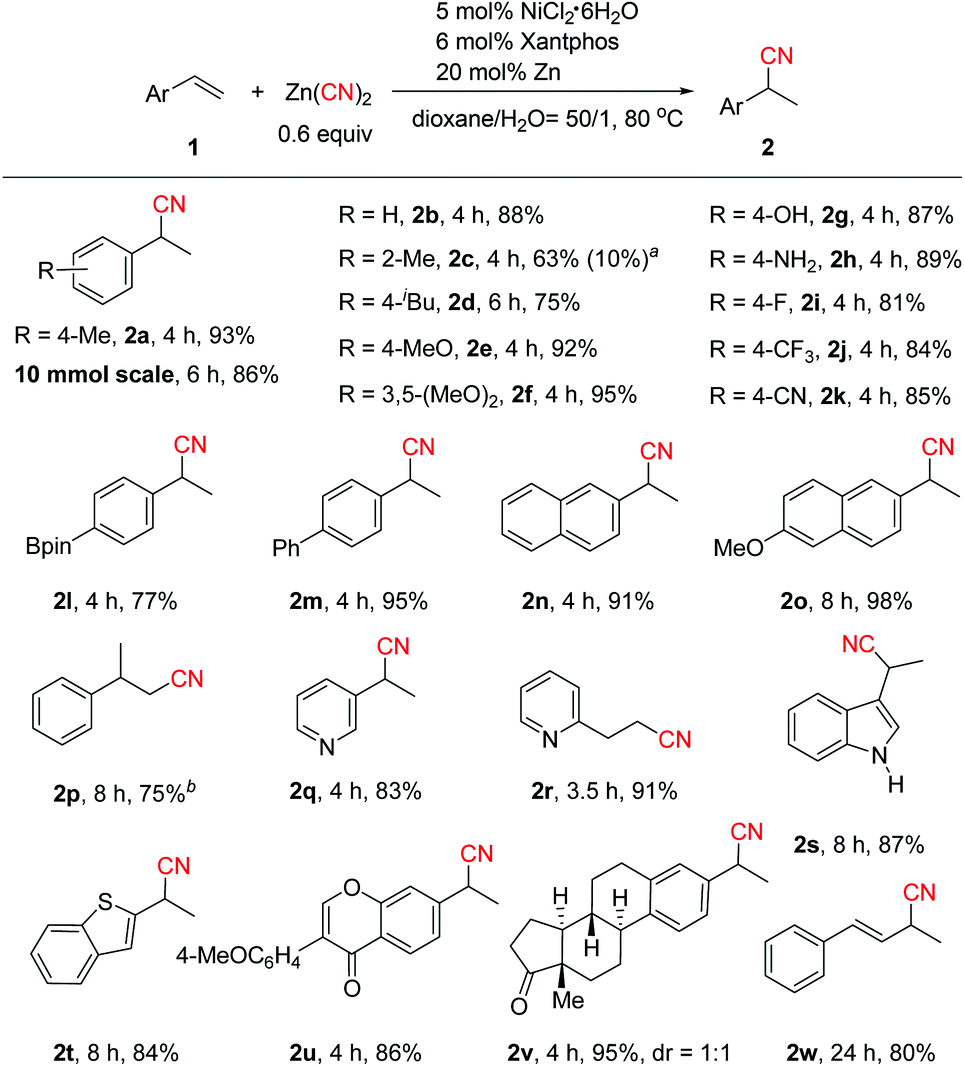 | ||
| Scheme 2 Scope of aryl/heteroaryl alkenes. Isolated yields. a6 mol% dppf and 1.0 equiv. Zn were used. The isolated yield of the minor regioisomer is shown in parentheses. b5 mol% NiCl2·6H2O, 6 mol% dppf, 1.0 equiv. Zn, 0.5 equiv. ZnI2 and 2.0 equiv. H2O were used in MeCN. | ||
Next, the substrate scope of aliphatic alkenes was examined (Scheme 3). Under the standard conditions for aryl alkenes, unexpectedly, 2-allylnaphthalene was converted to branched nitrile 5 in 88% yield, which was likely formed through alkene isomerization followed by hydrocyanation. In fact, the alkene isomer of 2-(prop-1-enyl)naphthalene 4a′ (E/Z = 33.3:1) could be isolated in 94% yield at the early stage of the reaction. The results clearly indicated that nickel hydride was formed during the process since it was known that Ni-H species could catalyze the isomerization of alkenes.17 In order to avoid the alkene isomerization, we re-optimized the reaction conditions.13 Inspired by our recent work on nickel-catalyzed cyanation of aryl halides with Zn(CN)218a using DMAP as a co-ligand,18 after many trials, we found that the reaction proceeded with good to high linear selectivity in the presence of 5 mol% Ni(ClO4)2·6H2O, 6 mol% dppp, 1.0 equiv. of Zn, 2.0 equiv. of H2O and 1.0 equiv. of DMAP in MeCN at 80 °C. In particular, DMAP was found to play an important role in improving the regioselectivity, since without DMAP, branched product 5 was formed in high yield (89%) in the case of 2-allylnaphthalene.13 The large change in regioselectivity might be attributed to the steric effect around the alkylnickel cyanide intermediate in which DMAP coordinates to the metal and increases the steric bulkiness around nickel. Thus the less hindered linear alkylnickel cyanide intermediate is favored leading to the linear product. The scope of aliphatic alkenes was then investigated under the revised conditions. For example, the reaction of the same substrate of 2-allylnaphthalene with Zn(CN)2 afforded linear product 6a in 78% yield, along with 9% of branched product 7a. Alkenes with alkyl side chains bearing aryl functional groups were transformed to 6b–6c in high yields with good linear selectivity. Common alkenes without any functional groups such as 1-decene and sterically congested vinylcyclohexane could also be cyanated efficiently (6d–6e). Excellent linear selectivity was observed in the latter case due to the steric effect of the cyclohexyl group. Allylic amine derivatives were confirmed to be valuable substrates for this reaction to afford the desired linear nitriles in 78%-81% yields (6f–6g). TBS-protected secondary allylic alcohol was compatible and showed excellent linear selectivity (6h). Unexpectedly, when allylic alcohol 5-phenylpent-1-en-3-ol 4t was employed as the substrate, lactone product 8 was obtained via sequential hydrocyanation/cyclization. Thus, the reaction provided a concise and efficient route to lactones. Protected and unprotected primary alcohols were well tolerated (6i–6j). It was noted that unprotected alcohols were not suitable for the transfer hydrocyanation reaction.10 Excellent linear selectivity (6k–6m) was also achieved with aliphatic alkenes containing ester, keto and aldehyde groups. In the case of diethyl allylphosphonate, we found that the reaction proceeded more efficiently to afford 6n in 85% yield using 5 mol% NiCl2·6H2O, 6 mol% dppf, 20 mol% Zn, and 2.0 equiv. H2O without the addition of DMAP. Encouraged by this result, we further explored the hydrocyanation of a series of activated alkenes under the conditions for the formation of 6n. Notably, all these reactions occurred with excellent anti-Markovnikov selectivity, and various N, O, Si or S-containing groups were well tolerated (6o–6s).
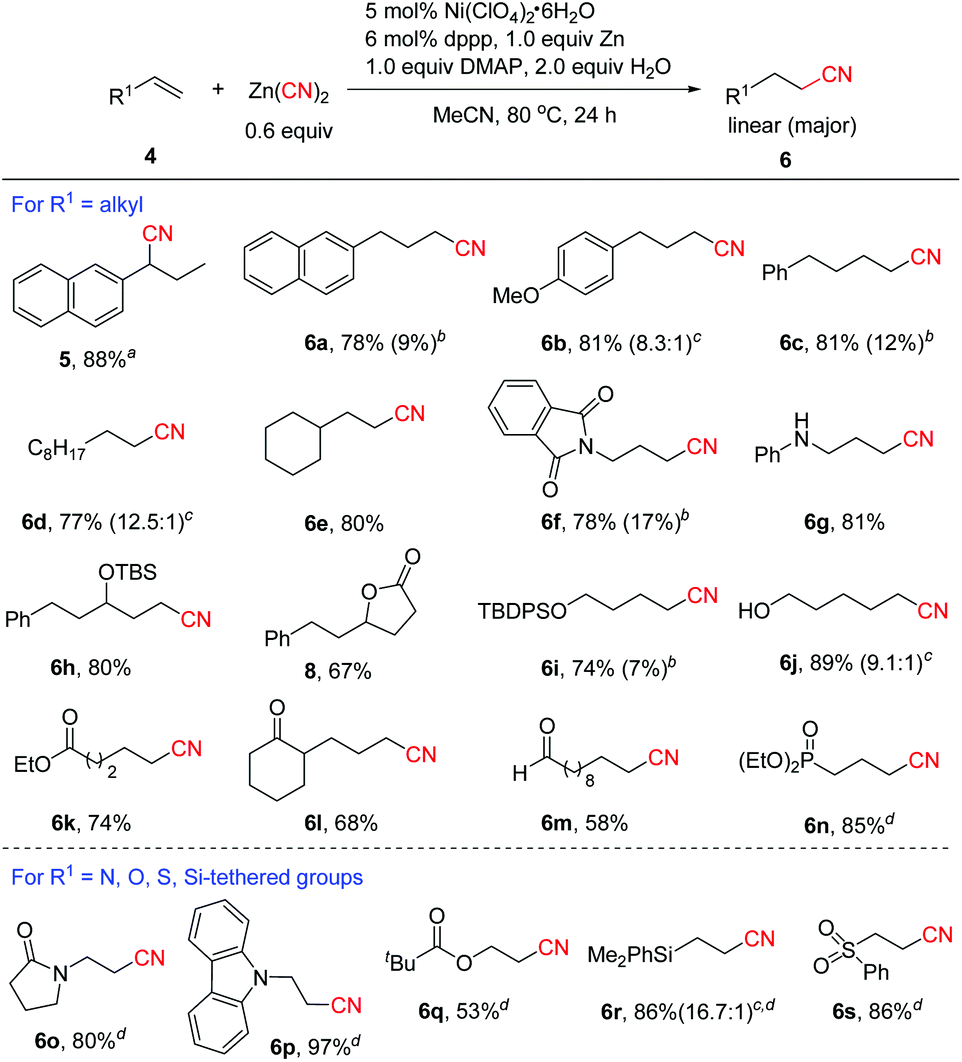 | ||
| Scheme 3 Scope of aliphatic alkenes. Isolated yields. a5 mol% NiCl2·6H2O, 6 mol% Xantphos, 20 mol% Zn, dioxane/H2O = 50/1. bThe yields of the branched nitrile 7 are shown in parentheses. cOverall yields of 6 and 7, and the ratio of 6:7 is shown in parentheses. d5 mol% NiCl2·6H2O, 6 mol% dppf, 20 mol% Zn and 2.0 equiv. H2O were used in MeCN at 80 °C for 12 h. | ||
To probe the reaction mechanism, we carried out various control experiments.13 It was found that no desired product was formed from 1a using NiCl2(DME) as the catalyst in the absence of water (Scheme 4, eqn (1)). Deuterium labeling experiments using D2O produced deuterated 2a-d in 89% yield in which deuterium is found in both the benzylic position and methyl group (Scheme 4, eqn (2)). The results implied that the hydrogen source for hydrocyanation comes from water. The scrambling of the deuterium between the benzylic and homobenzylic carbon atoms of 2a-d indicated that nickel hydride addition to the alkene is reversible, and it is possible that the resulting η3-benzyl nickel species equilibrates with a linear alkylnickel intermediate.7g The Ni(cod)2 or Ni(I) complex of NiCl(dppf) could also catalyze the reaction efficiently in the absence of the reductant (Scheme 4, eqn (3) and (4)). It is unlikely that the reaction goes through Ni(I) species involving transmetalation of Ni–Cl with Zn(CN)2 to form a Ni(I)–CN species followed by addition to the alkene and protonation, since it is hard to explain the observed regioselectivity and deuterium labeling results.13 We suggest that in the case of the Ni(I)-catalyzed reaction, the Ni(I) species might undergo a disproportionation reaction to afford Ni(0) and Ni(II) complexes, in which Ni(0) is the real active species for the reaction. Employing TMSCN/MeOH as the in situ HCN donor resulted in no formation of 2a under the standard conditions for 2a (Scheme 4, eqn (5)). Additionally, the mercury poisoning experiments indicated that the reaction was not inhibited, implying that a heterogeneous system might not be involved (Scheme 4, eqn (6)).
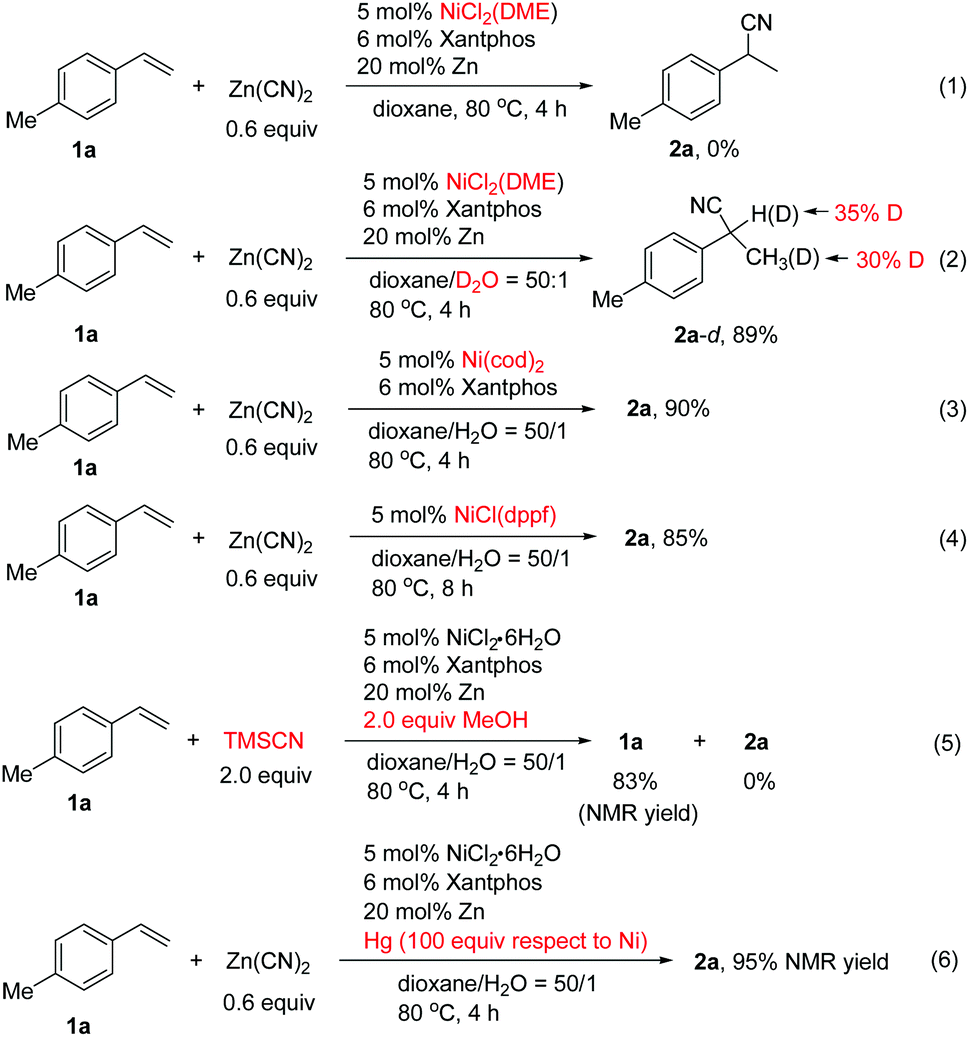 | ||
| Scheme 4 Control experiments. | ||
Although more efforts should be made to make the reaction mechanism clear, based on the above results, we tentatively propose the following reaction mechanism represented by hydrocyanation of styrene shown in Scheme 5. In path a, Ni(0) complex 9, formed via reduction of NiCl2·6H2O with Zn, reacts with water to give a LnNi(II)–H intermediate 10 either by oxidative addition of water19,20 or by an unclear process. Migratory insertion of styrene into the Ni–H bond affords η3-benzyl nickel(II) complex 11. A fast equilibrium between 11 and 15via intermediate 14 exists, but 11 is more favored due to its higher stability. Subsequent transmetalation of 11 with Zn(CN)2 followed by reductive elimination provides branched nitrile 2 selectively. Alternatively, complex 10 may first react with Zn(CN)2 to generate a H–Ni–CN species followed by insertion and reductive elimination to give 2. The reaction of nickel π–alkene complex 12/13 with H2O may also afford the same intermediate 11 (path b). In the case of aliphatic alkenes, linear products 6 were observed as the major products, which were formed through the less hindered alkyl nickel complex (type 15).
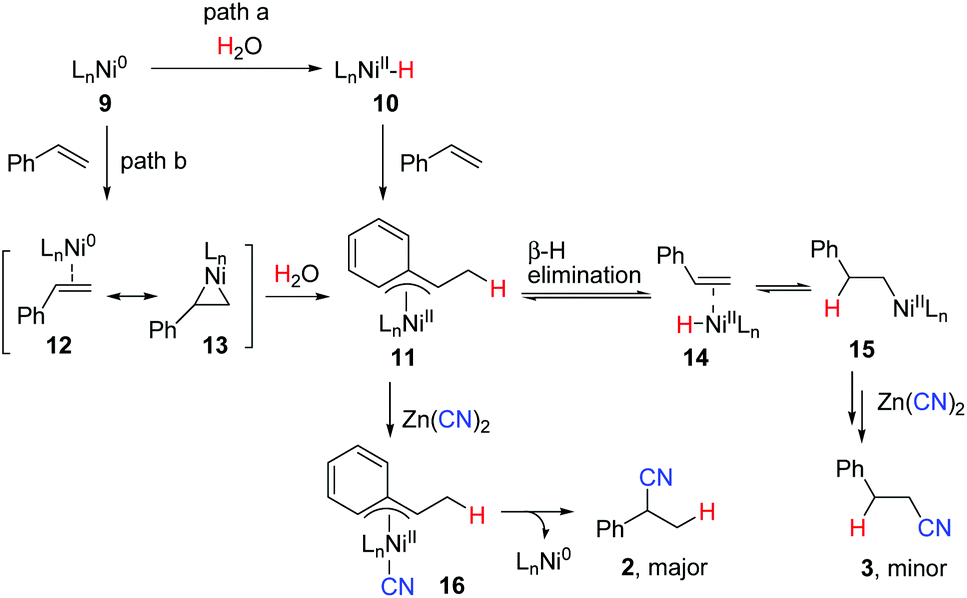 | ||
| Scheme 5 Possible reaction mechanism. | ||
Conclusions
In summary, we have described a first general and highly regioselective method for alkene hydrocyanation using relatively less toxic Zn(CN)2 as the cyano source and the air-stable nickel(II) salt as the precatalyst. The regioselectivity of these reactions was found to depend remarkably on the nature of terminal alkenes. Preliminary mechanistic studies indicate that the Ni(COD)2 or diphosphine-ligated nickel(I) chloride complex also displays high catalytic activity without the need for the reductant for this transformation. This method might find wide utility in pharmaceutical chemistry for drug discovery and development. Further investigations on the detailed reaction mechanism and application of this chemistry are in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Key Research and Development Program (2016YFA0202900), the National Natural Science Foundation of China (21772217), Science and Technology Commission of Shanghai Municipality (18XD1405000), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000), and Shanghai Institute of Organic Chemistry (sioczz201807) for financial support.Notes and references
- R. C. Larcok, Comprehensive Organic Transformations: A guide to Functional Group Preparations, John Wiley & Sons, New York, 1989 Search PubMed.
- P. Pollak, G. Romeder, F. Hagedorn and H. Gelbke, in Ullman's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 1985, vol. A17, pp. 363–376 Search PubMed.
- F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef PubMed.
- For reviews, see: (a) M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368 CrossRef CAS PubMed; (b) L. Bini, C. Müller and D. Vogt, Chem. Commun., 2010, 46, 8325 RSC; (c) L. Bini, C. Müller and D. Vogt, ChemCatChem, 2010, 2, 590 CrossRef CAS; (d) T. V. RajanBabu, Org. React., 2011, 75, 1 Search PubMed.
- For Co-catalyzed hydrocyanation of alkenes, see: (a) P. Arthur Jr., D. C. England, B. C. Pratt and G. M. Whitman, J. Am. Chem. Soc., 1954, 76, 5364 CrossRef; (b) B. Gaspar and E. M. Carreira, Angew. Chem., Int. Ed., 2007, 46, 4519 CrossRef CAS PubMed.
- For Pd-catalyzed hydrocyanation of alkenes, see: (a) P. S. Elmes and W. R. Jackson, J. Am. Chem. Soc., 1979, 101, 6128 CrossRef CAS; (b) A. Bhunia, K. Bergander and A. Studer, J. Am. Chem. Soc., 2018, 140, 16353 CrossRef CAS PubMed.
- For selected papers of Ni(0)-catalyzed hydrocyanation of alkenes with HCN, see: (a) C. A. Tolman, W. C. Seidel, J. D. Druliner and P. J. Domaille, Organometallics, 1984, 3, 33 CrossRef CAS; (b) T. A. Tolman, R. J. McKinney, W. C. Seidel, J. D. Druliner and W. R. Stevens, Adv. Catal., 1985, 33, 1 Search PubMed; (c) R. J. McKinney and D. C. Roe, J. Am. Chem. Soc., 1986, 108, 5167 CrossRef CAS; (d) M. Kranenburg, P. C. J. Kamer, P. W. N. M. van Leeuwen, D. Vogt and W. Keim, J. Chem. Soc., Chem. Commun., 1995, 2177 RSC; (e) W. Goertz, P. C. J. Kamer, P. W. N. M. van Leeuwen and D. Vogt, Chem. – Eur. J., 2001, 7, 1614 CrossRef CAS PubMed; (f) B. Saha and T. V. RajanBabu, Org. Lett., 2006, 8, 4657 CrossRef CAS PubMed; (g) L. Bini, E. A. Pidko, C. Müller, R. A. van Santen and D. Vogt, Chem. – Eur. J., 2009, 15, 8768 CrossRef CAS PubMed. For selected papers of Ni(0)-catalyzed hydrocyanation of alkenes with acetone cyanohydrin, see: (h) M. de Greef and B. Breit, Angew. Chem., Int. Ed., 2009, 48, 551 CrossRef CAS PubMed; (i) S. Arai, H. Hori, Y. Amako and A. Nishida, Chem. Commun., 2015, 51, 7493 RSC. For Ni(0)-catalyzed hydrocyanation of alkenes with TMSCN/MeOH, see: (j) A. Falk, A.-L. Göderz and H. G. Schmalz, Angew. Chem., Int. Ed., 2013, 52, 1576 CrossRef CAS PubMed.
- T. A. Tolman, J. Chem. Educ., 1986, 63, 19 CrossRef.
- (a) B. W. Taylor and H. E. Swift, J. Catal., 1972, 26, 254 CrossRef CAS; (b) W. Goertz, P. C. J. Kamer, P. W. N. M. van Leeuwen and D. Vogt, Chem. Commun., 1997, 1521 RSC.
- X. Fang, P. Yu and B. Morandi, Science, 2016, 351, 832 CrossRef CAS PubMed.
- X. Zhang, X. Xie and Y. Liu, J. Am. Chem. Soc., 2018, 140, 7385 CrossRef CAS PubMed.
- For a Ni(II)-catalyzed hydrocyanation of alkenes using acetone cyanohydrin with low regioselectivity, see: K. Nemoto, T. Nagafuchi, K. Tominaga and K. Sato, Tetrahedron Lett., 2016, 57, 3199 CrossRef CAS.
- For details, see ESI.†.
- P. C. J. Kamer, P. W. N. M. van Leeuwen and J. N. H. Reek, Acc. Chem. Res., 2001, 34, 895 CrossRef CAS PubMed.
- J. Huang, C. M. Haar and S. P. Nolan, Organometallics, 1999, 18, 297 CrossRef CAS.
- (a) W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600 RSC; (b) S. Thapa, R. K. Dhungana, R. T. Magar, B. Shrestha, S. KC and R. Giri, Chem. Sci., 2018, 9, 904 RSC.
- H. J. Lim, C. R. Smith and T. V. RajanBabu, J. Org. Chem., 2009, 74, 4565 CrossRef CAS PubMed.
- (a) X. Zhang, A. Xia, H. Chen and Y. Liu, Org. Lett., 2017, 19, 2118 CrossRef CAS PubMed. For related papers involving DMAP as a co-ligand, see: (b) Y.-L. Xiao, Q.-Q. Min, C. Xu, R.-W. Wang and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 5837 CrossRef CAS PubMed; (c) X. Wang, S. Wang, W. Xue and H. Gong, J. Am. Chem. Soc., 2015, 137, 11562 CrossRef CAS PubMed.
- For a review, see: O. V. Ozerov, Chem. Soc. Rev., 2009, 38, 83 RSC.
- For proposed Ni–H formation from nickel-catalyzed reactions in the presence of water, see: (a) K. Takai, S. Sakamoto and T. Isshiki, Org. Lett., 2003, 5, 653 CrossRef CAS PubMed; (b) M. Gaydou, T. Moragas, F. Juliá-Hernández and R. Martin, J. Am. Chem. Soc., 2017, 139, 12161 CrossRef CAS PubMed; (c) Ref. 11 .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qo00396g |
| This journal is © the Partner Organisations 2019 |